Constitutive Cytokine mRNAs Mark Natural Killer (NK) and NK T Cells Poised for Rapid Effector Function (original) (raw)
Abstract
Natural killer (NK) and NK T cells are tissue lymphocytes that secrete cytokines rapidly upon stimulation. Here, we show that these cells maintain distinct patterns of constitutive cytokine mRNAs. Unlike conventional T cells, NK T cells activate interleukin (IL)-4 and interferon (IFN)-γ transcription during thymic development and populate the periphery with both cytokine loci previously modified by histone acetylation. Similarly, NK cells transcribe and modify the IFN-γ gene, but not IL-4, during developmental maturation in the bone marrow. Lineage-specific patterns of cytokine transcripts predate infection and suggest evolutionary selection for invariant but distinct types of effector responses among the earliest responding lymphocytes.
Keywords: NK T cells, NK cells, cytokine mRNAs, IL-4, IFN-γ
Introduction
Effective immunity involves regulated cytokine production by lymphocytes. Conventional T cells are an important source of cytokines during infection, but they must clonally expand, differentiate into effector cells, and migrate to the site of infection over the course of several days to perform their function. To fill the lag before antigen-specific cytokine production during the activation and expansion of adaptive immunity, several lymphocyte populations reside at sites of pathogen entry where they are able to secrete cytokines within minutes to hours of infection. These cell types include NK T and NK cells in the blood, liver, spleen, and bone marrow (1, 2). NK cells are required for effective host defense against herpes viruses in mice and humans (3–5). Although the precise evolutionary niche subserved by NK T cells is not completely clear, the capacity of NK T cells to activate rapid cytokine expression has been exploited to manipulate the outcomes of autoimmunity and cancer (6).
Aside from their expression of common NK-associated surface antigens, such as NK1.1, NK T and NK cells share developmental requirements. Deficiencies in certain cytokines, such as IL-15 or lymphotoxin αβ, or transcription factors such as Ets-1 or Irf-1, lead to loss of both cell lineages (6). Recent studies suggest their capacity to express cytokines rapidly may also be developmentally acquired (7, 8). Although other studies elegantly demonstrate how these cells become activated (6, 9), the mechanisms underlying their rapid cytokine production or their distinct cytokine patterns, IFN-γ in the case of NK cells and both IL-4 and IFN-γ in the case of NK T cells, remain unknown. Elucidation of such mechanisms may have important implications for understanding polarized cytokine production by T cells in adaptive immune responses.
We recently generated IL-4 gene knockin bicistronic reporter mice that allow the faithful tracking of IL-4 expression using an enhanced green fluorescent protein (eGFP) reporter while preserving intact endogenous IL-4 (10). Here, we report the generation of similarly configured mice to enumerate the expression of IFN-γ using enhanced yellow fluorescent protein (eYFP). Naive CD4 T cells from both mice, designated 4get (IL-4 GFP-enhanced transcript) and Yeti (YFP-enhanced transcript for IFN-γ), are initially nonfluorescent and faithfully activate eGFP or eYFP under Th2 or Th1 conditions, respectively. Unexpectedly, tissue NK and NK T cells were spontaneously fluorescent, consistent with basal gene activation. Further study in wild-type mice confirmed that NK and NK T cells, in contrast to naive T cells, contain constitutive cytokine transcripts that correlate with chromatin modifications at the respective cytokine loci and have the capacity for rapid cytokine production.
Materials and Methods
Generation of IFN-γ Reporter Mice.
A 6-kb ClaI-BamHI fragment was isolated from a 129/SvJ BAC clone (Research Genetics) containing exons 2–4 and 2.5 kb of 3′ untranslated sequence of the Ifng gene. BamHI and SalI sites were introduced downstream of the translational stop and upstream of the endogenous polyadenylation site using PCR-mediated mutagenesis, and the mutated fragment was inserted into pgkTK containing herpes simplex thymidine kinase for negative selection (11). A bicistronic reporter cassette containing an encephalomyocarditis virus (EMCV) internal ribosome entry site (IRES) element was modified as previously described (10), and was cloned 5′ of eYFP followed by a bovine growth hormone polyadenylation signal (CLONTECH Laboratories, Inc.). A loxP-flanked neomycin resistance cassette derived from pL2new2 (12) was placed at the 3′ end to generate the final selectable cassette, which was cloned into the BamHI and SalI sites in the mutated Ifng gene to generate the final targeting construct. The NotI-linearized construct was electroporated into PrmCre ES cells, which express Cre recombinase under control of the protamine promoter (13). Selection of resistant clones and generation of mice was performed as previously described (10). Targeted mice were designated Yeti, for yellow-enhanced transcript for IFN-γ.
Flow Cytometry and T Cell Polarization.
Naive, resting CD4 T cells were purified from lymph nodes of the indicated mice using a MoFlo® high speed cell sorter (DakoCytomation) as previously described (14). Th1 and Th2 conditions for polarizing cells stimulated with 100 nM ovalbumin peptide, antigen-presenting cells, and cytokines were as previously described (14).
For phenotypic analysis, livers were perfused with PBS and dispersed, and lymphocytes were isolated from the interface of a 40%/60% percoll step gradient, washed, and resuspended in staining buffer. CD1-α-galactosylceramide (αGalCer) tetramers were generated as previously described (15), and used at optimal concentration to stain cells for 20 min at room temperature. After cooling on ice, cells were stained for an additional 25 min with the indicated antibodies. Antibodies to the following markers were used to identify lymphocyte populations, and were purchased from BD Biosciences, unless otherwise indicated: TCRβ-APC, CD122-PE, CD122-biotin, NK1.1-PE, NK1.1-biotin, DX5-APC, Ly49A/D-PE, Ly49G2-APC, CD11b-APC, CD62L-APC, CD8-biotin, CD19-biotin, CD24-biotin, CD4-TriColor (Caltag), CD4-APC-Cy7 (Caltag), CD8-APC-Cy7 (Caltag), and streptavidin-APC (Molecular Probes). Samples were analyzed and/or sorted on a MoFlo® high speed cell sorter (DakoCytomation) and data was analyzed using FlowJo® software (Tree Star).
Immunohistochemistry.
4get mice were injected with 1.33 μg anti-CD3 mAb (2C11; BD Biosciences) intravenously in PBS via the tail vein. After 90 min, the spleens were harvested, incubated at 4°C for 2 h in 4% paraformaldehyde, rinsed overnight in PBS, and frozen in OCT embedding compound. 7-μm sections were cut with a cryomicrotome (Leica) and placed onto charged glass slides (Fisher Scientific). Endogenous peroxidase activity was quenched in 1% H2O2 and 0.1% azide for 1 h, followed by Fc-block (BD Biosciences) with 1% mouse and rat serum, and avidin and biotin (Vector Laboratories). Sections were then incubated with rabbit anti–GFP polyclonal antibody (Ab 6556; Novus Biologicals), followed by biotinylated donkey anti–rabbit F(ab′)2 (Jackson ImmunoResearch Laboratories), streptavidin-peroxidase, and FITC tyramide from the TSATM-fluorescein kit according to the manufacturer's instructions (PerkinElmer). Sections were quenched and blocked as described above and incubated with biotinylated anti-TCRβ antibody (H57; BD Biosciences), followed by streptavidin-Cy5 (Caltag). Biotin was blocked as described above, and sections were incubated sequentially with biotinylated anti–IFN-γ antibody (XMG1.2; BD Biosciences), streptavidin-peroxidase, and biotinyl-tyramide. Deposited biotin was detected by streptavidin-Cy3 (Caltag). Nuclei were counterstained for 5 min with a 10 μg/ml solution of 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI; Boehringer) in PBS. Slides were mounted in Vectashield (Vector Laboratories). Digital images in the DAPI, FITC, Cy3, and Cy5 channels were collected using a deconvolution fluorescence microscope equipped with Slidebook software (Intelligent Imaging Innovations). Images were converted to RGB, colored, and overlaid using Adobe Photoshop 5.5 software. IFN-γ levels and GFP levels were set against isotype control (biotinylated rat IgG1) and wild-type BALB/c tissue, respectively.
Quantitative RT-PCR and Chromatin Immunoprecipitations.
NK T and NK cells were enriched from the spleens of C57BL/6 mice by magnetic bead depletion of CD8-, CD19-, and CD24-expressing cells (QIAGEN), and then sorted to >99% purity. Th1 and Th2 cells were generated by stimulating DO11.10 × TCR Cα−/− transgenic T cells for 5 d as previously described (14). Total RNA was isolated and quantitative RT-PCR was performed using primer/probe sets specific for IL-4, IFN-γ, and HPRT as previously described (14). For chromatin immunoprecipitations, 107 cells of the indicated populations were sorted, fixed, and sonicated to shear genomic DNA. Chromatin immunoprecipitations using control rabbit IgG or antibodies to the acetylated form of histone 3 (H3) were performed according to the manufacturer's instructions (Upstate Biotechnology). Serial fivefold dilutions of recovered DNA were used to template a 35-cycle PCR using primers to the IL-4 promoter (16) or the IFN-γ promoter (17). Dilutions of DNA recovered from control immunoprecipitations were amplified using IFN-γ promoter primers to verify equal amounts of starting material for each cell population.
Results
Generation of Yeti Mice and Characterization of CD4 T Cells.
Using the same strategy used to create 4get mice (10), homologous recombination in 129/SvJ-derived PrmCre embryonic stem (ES) cells (13) was used to generate a bicistronic IFN-γ/eYFP gene linked by a modified EMCV IRES to replace the endogenous IFN-γ gene (Fig. 1 , a and b). Chimeric males were bred to wild-type C57BL/6 or BALB/c mice, and offspring were selected using Southern blot for the presence of the knockin allele and deletion of the neomycin selection cassette. Subsequently backcrossed mice were screened for the presence of the knockin allele and loss of the Cre transgene. Mice used in this study were heterozygous Yeti/wild-type mice backcrossed six generations to C57BL/6 or to DO11.10 TCR transgenic mice on a BALB/c background.
Figure 1.

Characterization of eYFP expression in Yeti mice. (a) Map of the murine ifng locus, the targeting construct, and the mutated gene. A genomic fragment of the ifng gene was mutated by the addition of an IRES element, eYFP, and a polyadenylation signal (pA), followed by a loxP-flanked neomycin resistance (neo) cassette. Thymidine kinase (tk) was added upstream for counterselection. Cre-mediated recombination of the neomycin cassette in chimeric males resulted in the final mutated locus (bottom). Ifng exons are numbered and depicted as filled boxes. Southern probe (a) spans the first exon. BamHI (B), ClaI (C), and SacI (S) sites are indicated. (b) Genomic DNA from wild-type and targeted ES cell clones was digested with BamHI and blotted with probe (a) to detect the appearance of a 9-kb fragment corresponding to the mutated allele. (c) Naive T cells from DO11.10 Yeti (left) and DO11.10 4get mice (right) were stimulated with 100 nM ovalbumin peptide for 4 d under Th1 (top) or Th2 (bottom) conditions. Histograms with bold lines indicate eYFP expression in Yeti Th1 cells and eGFP expression in 4get Th2 cells. Wild-type DO11.10 controls are shown in gray.
Naive CD4 T cells (CD4+ CD62Lhi) from lymphoid tissues of Yeti mice were uniformly negative for eYFP expression. Activation of DO11.10 × Yeti TCR transgenic T cells with ovalbumin peptide revealed robust eYFP expression under Th1 conditions, but not Th2 conditions (Fig. 1 c, left). In contrast, activation of DO11.10 × 4get TCR transgenic T cells with ovalbumin peptide revealed robust eGFP expression under Th2 conditions, but not Th1 conditions (Fig. 1 c, right). Fluorescence under both conditions was first detected by 36 h and peaked between 4 and 5 d after stimulation (unpublished data). Thus, naive CD4 T cells from Yeti and 4get mice reliably report the activation of polarized cytokines in vitro.
NK T and NK Cells Are Spontaneously Fluorescent in 4get and Yeti Mice.
The design of the reporter constructs should allow detection of the respective cytokines from any appropriately activated cells in the mice. Indeed, infection of 4get mice with the helminth, Nippostrongylus brasiliensis, was used to identify eosinophils as a major component of the innate cytokine response to worms (18). To begin analysis of NK and NK T cells, we first examined the livers and spleens of naive 4get and Yeti mice, as these organs contain significant populations of both NK and NK T cells that can be readily recognized using appropriate surface markers. Unexpectedly, most resident liver lymphocytes were spontaneously fluorescent in the absence of specific immunization (Fig. 2) . NK T cells with an invariant Vα14 T cell receptor, which recognize the glycolipid αGalCer presented by CD1d, were the predominant eGFP-expressing cells in the livers of 4get mice (Fig. 2 a). >80% of liver NK T cells expressed the IL-4 reporter (Fig. 2 b). CD1 −/− 4get mice lack NK T cells, and few residual eGFP+ cells remained in the liver of such mice (Fig. 2 a). In contrast, Yeti mice had several populations of cells that spontaneously expressed eYFP, including NK T cells, NK cells, and tissue CD8 T cells, as well as small numbers of tissue CD4 T cells (Fig. 2, a and b). Because few CD8 T cells in the spleen expressed high level eYFP fluorescence (Fig. 1 b), we speculate that the high frequency of hepatic eYFP+ CD8 T cells represents differentiated effector cells that entered the liver in response to environmental antigens, or alternatively, cells marked for death and removal in hepatic tissue (19). This notion is supported by the fact that eYFP+ CD8 T cells in the liver, lungs, and lymph nodes of uninfected Yeti mice were uniformly CD62Llo, consistent with an effector/memory phenotype (unpublished data). In contrast to CD8 T cells, the cytokine profiles of NK T cells and NK cells were similar in the spleen as assessed using the indicator mice. The majority of NK T cells were positive for both IL-4 and IFN-γ reporters, and NK cells expressed IFN-γ but not IL-4 (Fig. 2 b). The majority of conventional CD4 and CD8 T cells in the spleen expressed neither IL-4 nor IFN-γ markers (Fig. 2 b). These data demonstrate that the majority of tissue effector cells in the liver, a critical organ for surveying pathogens from both systemic and portal blood circulations, spontaneously express cytokine reporters.
Figure 2.
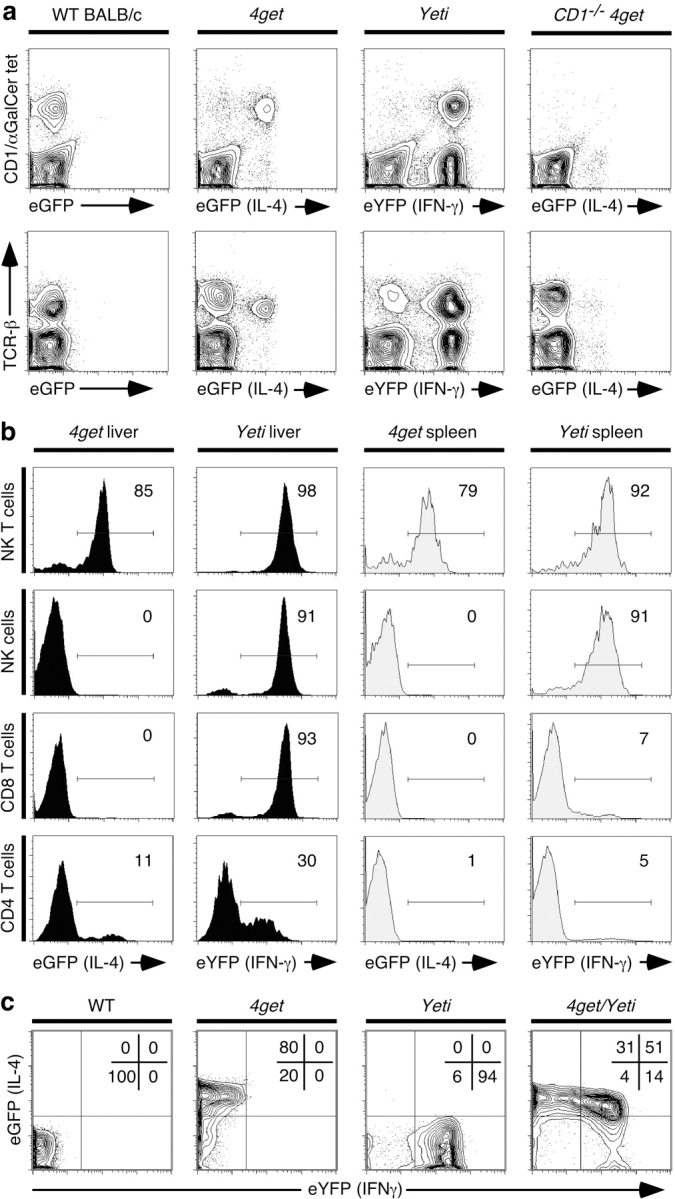
Spontaneous fluorescence in liver lymphocytes of naive cytokine reporter mice. (a) Liver cells isolated from wild-type, 4get, Yeti, and CD1d − / − 4get mice were stained with CD1d/αGalCer tetramers and antibodies to TCRβ. The top displays CD1d/αGalCer tetramer staining versus the fluorescent cytokine reporter, and the bottom depicts TCRβ expression versus the fluorescent marker. FACS® plots represent 5% contours with outliers and are representative of 7–10 mice per group. (b) Liver and spleen cells were stained with phenotypic markers to identify major lymphocyte populations. The following phenotypes were used to identify the different cell populations: NK T cells: CD8− TCRβ1 CD1d/αGalCer tetramer+; NK cells: CD3− CD8− CD4− CD122+; CD8 T cells: CD4− CD1d/αGalCer tetramer− TCRβ1 CD8+; and CD4 T cells: CD8− CD1d/αGalCer tetramer− TCRβ1 CD4+. Histograms are representative of three experiments. The electronic gates were set to contain <1% of wild-type, non(auto)-fluorescent controls. (c) Liver NK T cells from wild-type, 4get, Yeti, and 4get/Yeti intercrossed mice were examined for eGFP versus eYFP fluorescence. Data are representative of four mice.
NK T cells were fluorescent for both IL-4 and IFN-γ reporters in 4get and Yeti mice, suggesting that in contrast to conventional Th1 or Th2 cells, NK T cells contain constitutive mRNAs for both cytokines. Therefore, we examined liver NK T cells from 4get/Yeti intercrossed mice to confirm the fluorescence profiles from the individual reporter mice. Despite difficulties in compensating electronically for the closely related emission spectra of eGFP and eYFP, the data in Fig. 2 c show that the majority of NK T cells in 4get/Yeti mice were spontaneously fluorescent for both IL-4 and IFN-γ reporters. The fact that the percentage of double positive cells does not exactly match the values obtained from individual reporter mice (51% empirically in 4get/Yeti vs. 80% 4get × 94% Yeti = 75%) may reflect the difficulties in compensation.
These observations were puzzling given that neither NK T cells nor NK cells store or secrete detectable cytokine protein before cognate stimulation (7, 15). So why are the cells from the reporter mice so uniformly bright for the fluorescent markers, which require translation of the bicistronic mRNA? We considered the design of the reporter mice, which contain an IRES derived from EMCV driving translation of the fluorescent marker. IRES elements facilitate translation of viral RNA under conditions where host cell translation is repressed, as in the case of poliovirus infection (20). Moreover, a subset of endogenous cellular mRNAs also contain IRES elements, which allow their translation during times of repressed protein synthesis (21). IRES-mediated translation involves initiation factors distinct from those required for conventional, cap-mediated translation (21). We reasoned that the IRES element inserted into the IL-4 and IFN-γ genes might allow constitutive translation of eGFP in 4get mice and eYFP in Yeti mice under conditions where 5′ cap-dependent translation of the cytokine mRNAs might be more carefully regulated. In support of this idea, eGFP fluorescence in 4get Th2 cells required active translation and decayed with a half-life of 16 h after incubation with the protein synthesis inhibitors puromycin or cycloheximide (unpublished data). Therefore, 4get and Yeti mice faithfully report the presence of cytokine mRNAs, but translation of the downstream fluorescent reporter and the upstream endogenous cytokine can be dissociated.
In Situ Localization of the NK T Cell Response.
To assay the capacity for rapid cytokine protein production in fluorescent cells of the reporter mice, we examined the response of NK T cells in vivo. Activation of NK T cells by systemically administered anti-CD3 causes a burst of both IL-4 and IFN-γ detected in tissues and serum that is lost in the absence of these cells (15, 22). The vast majority of spontaneously eGFP+ cells in the spleens of unimmunized 4get mice are NK T cells, and analysis of Yeti and 4get/Yeti mice suggests that these cells also contain IFN-γ transcripts (Fig. 2). Therefore, we injected 4get mice with a dose of anti-CD3 mAb demonstrated to activate cytokine secretion from NK T cells (22) and used immunohistochemistry to examine IFN-γ protein expression from the unmodified, wild-type ifng gene in eGFP+ cells. This strategy eliminated any potential artificial activation of NK T cells that might occur during tissue dissociation and cell sorting. In resting 4get spleen, eGFP-expressing cells were distributed outside the T cell zones, which were delineated by TCRβ staining (Fig. 3 a). The low level of spontaneous IFN-γ protein did not colocalize with eGFP-expressing cells in untreated mice (Fig. 3 a). After anti-CD3 injection, a ring of IFN-γ protein–expressing cells appeared at the periphery of the periarteriolar lymphoid sheath (Fig. 3 b). In contrast to untreated mice, the majority of this IFN-γ protein signal colocalized with eGFP-expressing cells, consistent with their identification as NK T cells (Fig. 3 c, arrows). An isotype control antibody for IFN-γ did not stain eGFP-expressing cells from anti-CD3–treated 4get mice (Fig. 3 d), and IFN-γ1 splenocytes from treated wild-type mice were not eGFP+ (unpublished data), confirming the specificity of the staining. These in situ data localize the NK T cell response to the periphery of the periarteriolar lymphoid sheath, and support a model of constitutive mRNA transcripts nucleating a rapid cytokine response after stimulation in vivo.
Figure 3.
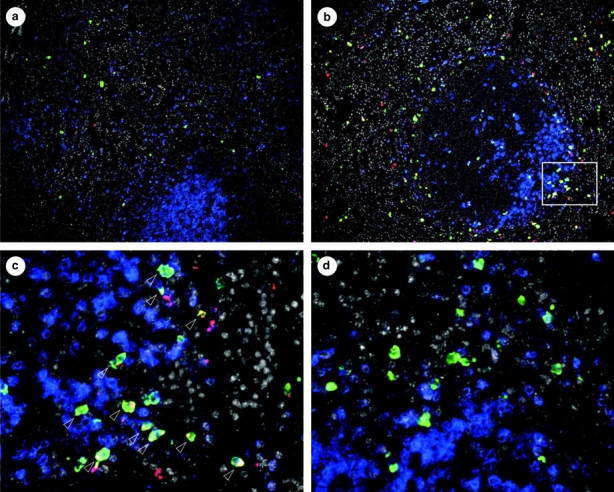
In situ localization of the NK T cell cytokine response. Spleen sections from untreated or anti-CD3–injected 4get mice were stained with antibodies to eGFP (green), IFN-γ or isotype control (red), and TCRβ (blue). DAPI fluorescence is in gray. (a) Untreated 4get spleen. ×100. (b) Anti-CD3–treated 4get spleen. ×100. (c) ×400 image of area indicated by the white box in b. Arrows indicate cells with costaining for eGFP and IFN-γ. (d) Isotype control staining for IFN-γ in the spleen of an anti-CD3–treated 4get mouse. Magnification of a and b is ×100 and ×400 c and d. Data are representative of two independent experiments.
Transcription and Chromatin Modification of Cytokine Genes in NK T and NK Cells.
To confirm the results obtained with the reporter mice, we examined cytokine mRNAs in NK T and NK cells purified by cell sorting from spleens of wild-type C57BL/6 mice. We compared the abundance of IL-4 and IFN-γ mRNA using quantitative RT-PCR to that of highly purified, naive CD4 T cells sorted from lymph nodes of the same mice. Consistent with the results from the reporter mice, NK T cells from wild-type mice contained >1,000-fold more IL-4 message and 200-fold more IFN-γ message than naive CD4 T cells (Fig. 4 a). NK cells also contained >200-fold more IFN-γ transcripts than naive CD4 T cells, but negligible levels of IL-4 mRNA (Fig. 4 a). We performed a similar analysis comparing cytokine mRNA abundance among wild-type, 4get, and Yeti NK and NK T cells sorted from unimmunized mice (Fig. 4 b). Cells from all three strains contained comparable amounts of cytokine mRNAs, confirming the fluorescence profiles in Fig. 2. The lack of cytokine mRNA in naive CD4 T cells suggests that these transcripts are not indiscriminately induced by the isolation and sorting procedure, which was done in the cold. The RT-PCR assay detected only mature mRNAs that were spliced and polyadenylated because we used oligo-dT primers for the RT reaction, and amplified with PCR primers that spanned exon/exon junctions (14). The abundance of cytokine message in these cells approached that of Th1 and Th2 cells produced in vitro (Fig. 4 a). Upon in vivo activation of NK T and NK cells with anti-CD3 or poly I/C, respectively, cytokine transcription increased even higher, consistent with previous observations (22) and similar to the induction seen upon in vitro restimulation of Th1 and Th2 cells (Fig. 4 a). These data demonstrate that NK T cells and NK cells reside in tissues poised with constitutive cytokine transcripts, which are induced further upon stimulation.
Figure 4.

Analysis of spontaneous cytokine mRNAs and chromatin modifications in wild-type NK T and NK cells. (a) NK T and NK cells (see Fig. 1 b for gating) were sorted from spleens of unimmunized C57BL/6 mice, from mice immunized intravenously 90 min earlier with 1.33 μg anti-CD3, or from mice injected intraperitoneally 4 h earlier with 150 μg poly I/C. 5′-nuclease fluorogenic RT-PCR was performed on isolated total RNA and the abundance of cytokine mRNA was normalized to HPRT message. This ratio was compared with the cytokine/HPRT ratio of highly purified, naive CD4 T cells sorted from lymph nodes of the same animals. Th1 and Th2 cells were generated by in vitro stimulation and restimulated with PMA and ionomycin for 3 h. Data are the average of three independently processed samples per group and are representative of two experiments. (b) NK T and NK cells were sorted from spleens of unimmunized wild-type, 4get, or Yeti mice. Cytokine mRNA abundance was determined as for Fig. 4 a. (c) 107 cells of the indicated populations were sorted from the spleens and lymph nodes of C57BL/6 mice. Chromatin immunoprecipitations were performed using control rabbit IgG (C) or antibodies to acetylated H3. PCR using primers to the IL-4 and IFN-γ promoters was performed on serial fivefold dilutions of DNA recovered from the experimental immunoprecipitation. Dilutions of input DNA were amplified with primers to the IFN-γ promoter to demonstrate comparable amounts of starting material.
In CD4 T cells, commitment to the Th1 or Th2 subset is accompanied by chromatin remodeling at the respective cytokine loci. During Th1 differentiation, histone proteins surrounding the IFN-γ promoter become acetylated to promote accessibility to transcription factors. Conversely, Th2 cells demonstrate acetylated histones at the IL-4 locus but not the IFN-γ gene (16, 17). Because the amount of cytokine mRNA in NK T and NK cells is comparable to that of differentiated CD4 effector T cells (Fig. 4 a), we analyzed the status of the chromatin surrounding the IL-4 and IFN-γ promoters. Sorted NK T and NK cells from unimmunized wild-type mice were used for chromatin immunoprecipitations with an antibody to the acetylated form of H3, a marker of accessible chromatin (23). Naive CD8 and CD4 T cells had low basal levels of acetylated H3 at either cytokine promoter (Fig. 4 c). In contrast, NK cells had high levels of acetylated H3 at the IFN-γ promoter, but not the IL-4 promoter (Fig. 4 c). NK T cells demonstrated high levels of H3 acetylation at both the IL-4 and IFN-γ promoters (Fig. 4 c). These data show that NK T and NK cells in wild-type mice contain chromatin modifications at cytokine genes that correlate with the presence of abundant cytokine mRNAs in the respective lineages. The low levels of chromatin modifications found in conventional lymphocytes isolated at the same time suggest these changes are not induced by the procedures used for cell purification.
NK T and NK Cells Activate Cytokine Transcription during Development.
Because peripheral NK T and NK cells are spontaneously fluorescent in the reporter mice, we tracked their development in the thymus and bone marrow, respectively, to determine when these cytokine transcript profiles become established. After stochastic rearrangement of the canonical Vα14 TCR and selection on double positive precursor thymocytes (1), NK T cells undergo a maturation process characterized by sequential up-regulation of CD44 and NK1.1 (8). We examined the thymi of 2-wk-old 4get and Yeti mice, which have populations of both immature and mature NK T cells. Benlagha et al. (8) recently demonstrated that NK T cells undergo a shift in αGalCer-stimulated cytokine production from IL-4 to IFN-γ as they mature to CD44hi NK1.1hi cells. In support of this model, we found that the fluorescence profile in 4get and Yeti thymic NK T cells precisely mirrored this shift (Fig. 5 a). CD44lo NK1.1lo cells expressed high levels of eGFP (IL-4) and low levels of eYFP (IFN-γ). Concomitant with maturation to CD44hi NK1.1hi cells, eGFP (IL-4) expression was progressively down-regulated and eYFP (IFN-γ) expression increased (Fig. 5 a). The IL-4 reporter signal in mature thymic NK T cells was low, but still higher than conventional CD4 T cells, which do not activate detectable IL-4 during thymic development (unpublished data). These data suggest that the developmental shift in cytokine production defined functionally by Benlagha et al. results from dynamic transcriptional regulation of IL-4 and IFN-γ during NK T cell maturation. Moreover, because the majority of NK T cells in the periphery express both IL-4 and IFN-γ mRNA (Fig. 2), these data support a model whereby NK T cells exit the thymus before terminal differentiation, which then occurs in peripheral tissues (8).
Figure 5.
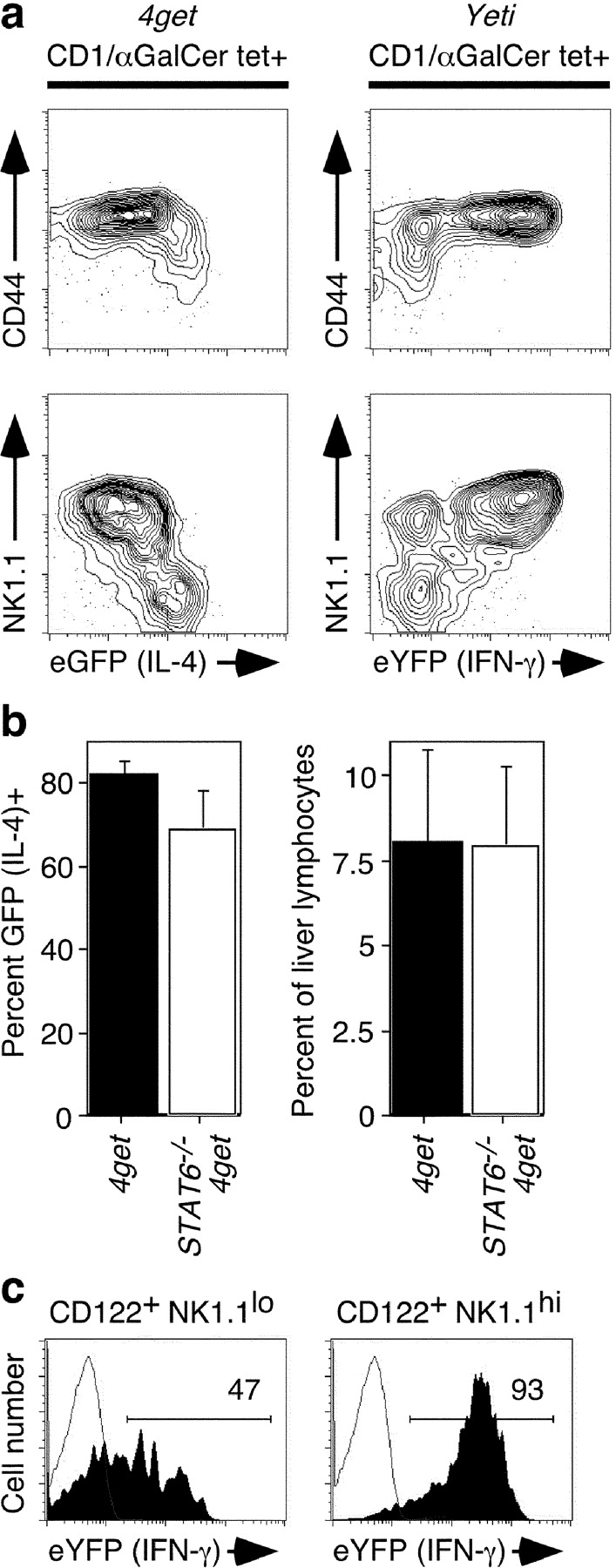
Developmental activation of cytokine mRNAs in NK T and NK cells. (a) Thymi were isolated from 2-wk-old 4get and Yeti mice, and gated CD1d/αGalCer tetramer+ cells were examined for expression of CD44, NK1.1, and the fluorescent cytokine reporter. FACS® plots depict 5% probability contours with outliers and are representative of three mice from each group. (b) Liver lymphocytes were isolated from 4get mice or 4get × Stat6 −/− mice. On the left, the percentage of CD1d/αGalCer tetramer+ cells expressing eGFP (IL-4) was compared. On the right, the percentage of NK T cells in the liver was enumerated. Means and standard deviations are depicted for four mice per group. (c) Bone marrow cells were isolated from the femurs of 2-wk-old Yeti mice. NK lineage cells were identified as [CD3, TCRβ, CD4, CD8, B220]− CD122+. eYFP (IFN-γ) expression was examined in NK1.1− and NK1.1+ NK lineage cells. Data are representative of five animals.
In conventional CD4 T cells, Stat6 signals are required to stabilize IL-4 production during Th2 differentiation (24). We examined Stat6 − / − 4get NK T cells to determine if Stat6 plays a similar role in NK T development. However, neither the percentage of NK T cells that expressed eGFP nor the accumulation of these cells in the liver was affected by the absence of Stat6 (Fig. 5 b). Similar results were observed in the spleens of Stat6 − / − 4get mice and IL-4 receptor α−/− 4get mice (unpublished data). Thus, IL-4's biologic activity is not required for developmental activation of IL-4 transcription in NK T cells.
Next, we examined NK cell development in the bone marrow of 2-wk-old Yeti mice. The earliest committed NK precursor is CD3− and expresses the β chain of the IL-2/15 receptor (CD122), but none of the other markers associated with the NK lineage, including NK1.1 and the immunomodulatory Ly49 and NKG2 receptors (7, 25). We found that almost half of these earliest identifiable NK precursors (CD122+ NK1.1lo) had already begun to express eYFP (IFN-γ; Fig. 5 c). Upon up-regulation of NK1.1, bone marrow NK lineage cells became indistinguishable from their mature peripheral counterparts at the level of IFN-γ expression (Figs. 5 c and 2 a). Up-regulation of IFN-γ transcription preceded acquisition of other NK lineage markers, including α2 integrin, CD11b, and Ly49 receptors (unpublished data). We also examined NK lineage cells in the thymi of embryonic day 16 Yeti mice and found that the vast majority of fetal NK precursors already expressed abundant eYFP, supporting a developmentally regulated process (unpublished data).
Discussion
We demonstrate that NK T cells and NK cells, distinguished by their ability to mobilize effector cytokines rapidly after immunization or infection, reside in the periphery spontaneously poised with constitutive cytokine transcripts. Modification of the respective cytokine loci in a manner promoting access by transcription factors correlates with the presence of cytokine mRNAs. Unlike conventional T cells, NK T and NK cells activate transcription of cytokine genes during early development in the thymus and bone marrow, respectively. In the case of IL-4 for NK T cells, neither the percentage of IL-4+ cells nor the tissue localization of NK T cells was affected by the absence of Stat6 or the IL-4 receptor. These data suggest that the lineage-specific developmental modification of the cytokine loci proceeds by a process independent of these pathways, which, for IL-4, seem critical for the efficient generation of conventional tissue Th2 cells (17).
Although we and others have shown that NK T cells and NK cells do not produce cytokine protein until cognate stimulation (Fig. 3; 7, 15), we cannot formally exclude the possibility that the abundant cytokine mRNAs we document are being translated below the threshold of detection. Additionally, we have not determined the turnover rates for these constitutive mRNAs in vivo. Does their abundance reflect an equilibrium between basal transcription and turnover, or are they generated during development and then maintained in the absence of new transcription? Further work will be required to distinguish between these two possibilities and to explore the relative contributions of cytokine transcription and translation to the rapid effector function of these cells. As shown here, however, cells marked by the presence of these constitutive cytokine mRNAs are precisely the same cells that produce cytokines rapidly after challenge, even when measured at genetically unmanipulated, wild-type alleles (Fig. 3).
We favor a model whereby NK T and NK cells activate distinct cytokine transcription profiles as a result of lineage-specific developmental cues, rather than pathogen recognition. These effector lymphocytes, by virtue of constitutive cytokine mRNAs and accessible, modified cytokine loci, arm the periphery with the capacity for rapid, programmed responses that may serve two important functions. First, their immediate, stereotyped effector function could provide protection at sites of pathogen entry during the time required for the expansion and migration of antigen-specific, conventional T cells. Second, the cytokines elaborated by these cells may in turn affect conventional T cell differentiation (6). We speculate that conventional T cells acquire a similar fate after differentiation and migration into peripheral tissues. We note that many of the hepatic CD8 T cells, in contrast to spleen CD8 T cells, were spontaneously fluorescent in the Yeti mice, consistent with the presence of constitutive IFN-γ mRNA (Fig. 2). Indeed, memory CD8 T cells were recently found to contain constitutive mRNAs for IFN-γ and the chemokine RANTES (26, 27), suggesting that a similar mechanism may contribute to the rapid memory response of adaptive immune cells.
Acknowledgments
The authors thank C. McArthur, K. Mohrs, N. Flores, L. Gardiner, J. Lin, and L. Stowring for technical support, O. Naidenko and S. Sidobre for production of CD1d tetramers, and J.G. Cyster for helpful comments.
Supported in part by National Institutes of Health grants AI30663 and HL56385 (to R.M. Locksley), and CA52511 (to M. Kronenberg). L. Gapin is supported by a Cancer Research Institute Fellowship. R.M. Locksley is an Ellison Medical Foundation Senior Scholar in Global Infectious Diseases. The authors declare that they have no competing financial interests.
D.B. Stetson and M. Mohrs contributed equally to this work.
M. Mohrs' present address is The Trudeau Institute, 100 Algonquin Ave., Saranac Lake, NY 12983.
L. Gapin's present address is University of Colorado Health Sciences Center, National Jewish Medical and Research Center, 1400 Jackson St., Denver, CO 80206.
Abbreviations used in this paper: αGalCer, α-galactosylceramide; DAPI, 4′,6-diamidine-2′-phenylindole dihydrochloride; eGFP, enhanced green fluorescent protein; EMCV, encephalomyocarditis virus; ES, embryonic stem; eYFP, enhanced yellow fluorescent protein; H3, histone 3; IRES, internal ribosome entry site.
References
- 1.Bendelac, A., M.N. Rivera, S.H. Park, and J.H. Roark. 1997. Mouse CD1-specific NK1 T cells: development, specificity, and function. Annu. Rev. Immunol. 15:535–562. [DOI] [PubMed] [Google Scholar]
- 2.Cerwenka, A., and L.L. Lanier. 2001. Natural killer cells, viruses and cancer. Nat. Rev. Immunol. 1:41–49. [DOI] [PubMed] [Google Scholar]
- 3.Arase, H., E.S. Mocarski, A.E. Campbell, A.B. Hill, and L.L. Lanier. 2002. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science. 296:1323–1326. [DOI] [PubMed] [Google Scholar]
- 4.French, A.R., and W.M. Yokoyama. 2003. Natural killer cells and viral infections. Curr. Opin. Immunol. 15:45–51. [DOI] [PubMed] [Google Scholar]
- 5.Biron, C.A., K.S. Byron, and J.L. Sullivan. 1989. Severe herpesvirus infections in an adolescent without natural killer cells. N. Engl. J. Med. 320:1731–1735. [DOI] [PubMed] [Google Scholar]
- 6.Kronenberg, M., and L. Gapin. 2002. The unconventional lifestyle of NKT cells. Nat. Rev. Immunol. 2:557–568. [DOI] [PubMed] [Google Scholar]
- 7.Kim, S., K. Iizuka, H.S. Kang, A. Dokun, A.R. French, S. Greco, and W.M. Yokoyama. 2002. In vivo developmental stages in murine natural killer cell maturation. Nat. Immunol. 3:523–528. [DOI] [PubMed] [Google Scholar]
- 8.Benlagha, K., T. Kyin, A. Beavis, L. Teyton, and A. Bendelac. 2002. A thymic precursor to the NK T cell lineage. Science. 296:553–555. [DOI] [PubMed] [Google Scholar]
- 9.Natarajan, K., N. Dimasi, J. Wang, R.A. Mariuzza, and D.H. Margulies. 2002. Structure and function of natural killer cell receptors: multiple molecular solutions to self, nonself discrimination. Annu. Rev. Immunol. 20:853–885. [DOI] [PubMed] [Google Scholar]
- 10.Mohrs, M., K. Shinkai, K. Mohrs, and R.M. Locksley. 2001. Analysis of type 2 immunity in vivo with a bicistronic IL-4 reporter. Immunity. 15:303–311. [DOI] [PubMed] [Google Scholar]
- 11.Tybulewicz, V.L., C.E. Crawford, P.K. Jackson, R.T. Bronson, and R.C. Mulligan. 1991. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 65:1153–1163. [DOI] [PubMed] [Google Scholar]
- 12.Gu, H., Y.R. Zou, and K. Rajewsky. 1993. Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell. 73:1155–1164. [DOI] [PubMed] [Google Scholar]
- 13.O'Gorman, S., N.A. Dagenais, M. Qian, and Y. Marchuk. 1997. Protamine-Cre recombinase transgenes efficiently recombine target sequences in the male germ line of mice, but not in embryonic stem cells. Proc. Natl. Acad. Sci. USA. 94:14602–14607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grogan, J.L., M. Mohrs, B. Harmon, D.A. Lacy, J.W. Sedat, and R.M. Locksley. 2001. Early transcription and silencing of cytokine genes underlie polarization of T helper cell subsets. Immunity. 14:205–215. [DOI] [PubMed] [Google Scholar]
- 15.Matsuda, J.L., O.V. Naidenko, L. Gapin, T. Nakayama, M. Taniguchi, C.R. Wang, Y. Koezuka, and M. Kronenberg. 2000. Tracking the response of natural killer T cells to a glycolipid antigen using CD1d tetramers. J. Exp. Med. 192:741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Avni, O., D. Lee, F. Macian, S.J. Szabo, L.H. Glimcher, and A. Rao. 2002. T(H) cell differentiation is accompanied by dynamic changes in histone acetylation of cytokine genes. Nat. Immunol. 3:643–651. [DOI] [PubMed] [Google Scholar]
- 17.Fields, P.E., S.T. Kim, and R.A. Flavell. 2002. Cutting edge: changes in histone acetylation at the IL-4 and IFN-gamma loci accompany Th1/Th2 differentiation. J. Immunol. 169:647–650. [DOI] [PubMed] [Google Scholar]
- 18.Shinkai, K., M. Mohrs, and R.M. Locksley. 2002. Helper T cells regulate type-2 innate immunity in vivo. Nature. 420:825–829. [DOI] [PubMed] [Google Scholar]
- 19.Crispe, I.N., T. Dao, K. Klugewitz, W.Z. Mehal, and D.P. Metz. 2000. The liver as a site of T-cell apoptosis: graveyard, or killing field? Immunol. Rev. 174:47–62. [DOI] [PubMed] [Google Scholar]
- 20.Johannes, G., M.S. Carter, M.B. Eisen, P.O. Brown, and P. Sarnow. 1999. Identification of eukaryotic mRNAs that are translated at reduced cap binding complex eIF4F concentrations using a cDNA microarray. Proc. Natl. Acad. Sci. USA. 96:13118–13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sachs, A.B., P. Sarnow, and M.W. Hentze. 1997. Starting at the beginning, middle, and end: translation initiation in eukaryotes. Cell. 89:831–838. [DOI] [PubMed] [Google Scholar]
- 22.Yoshimoto, T., and W.E. Paul. 1994. CD4pos, NK1.1pos T cells promptly produce interleukin 4 in response to in vivo challenge with anti-CD3. J. Exp. Med. 179:1285–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strahl, B.D., and C.D. Allis. 2000. The language of covalent histone modifications. Nature. 403:41–45. [DOI] [PubMed] [Google Scholar]
- 24.Grogan, J.L., and R.M. Locksley. 2002. T helper cell differentiation: on again, off again. Curr. Opin. Immunol. 14:366–372. [DOI] [PubMed] [Google Scholar]
- 25.Rosmaraki, E.E., I. Douagi, C. Roth, F. Colucci, A. Cumano, and J.P. Di Santo. 2001. Identification of committed NK cell progenitors in adult murine bone marrow. Eur. J. Immunol. 31:1900–1909. [DOI] [PubMed] [Google Scholar]
- 26.Kaech, S.M., S. Hemby, E. Kersh, and R. Ahmed. 2002. Molecular and functional profiling of memory CD8 T cell differentiation. Cell. 111:837–851. [DOI] [PubMed] [Google Scholar]
- 27.Swanson, B.J., M. Murakami, T.C. Mitchell, J. Kappler, and P. Marrack. 2002. RANTES production by memory phenotype T cells is controlled by a posttranscriptional, TCR-dependent process. Immunity. 17:605–615. [DOI] [PubMed] [Google Scholar]