Role for CD4+ CD25+ Regulatory T Cells in Reactivation of Persistent Leishmaniasis and Control of Concomitant Immunity (original) (raw)
Abstract
Reactivation of dormant infections causes an immense burden of morbidity and mortality in the world at large. Reactivation can occur as a result of immunosuppression, environmental insult, or aging; however, the cause of reactivation of such infections is often not clear. We have previously shown that persistence of the parasite Leishmania major is controlled by endogenous CD4+ CD25+ regulatory T (T reg) cells. In this report, we show that despite efficient parasite clearance at secondary sites of infection, Leishmania superinfection can cause disease reactivation at the primary site. Our results strongly suggest that T reg cells, whose numbers increase in sites of reactivation, are directly responsible for such reactivation. Depletion of CD25+ cells at the time of secondary challenge prevented disease reactivation at the site of persistent infection while strengthening the expression of immunity at the site of secondary challenge. Finally, transfer of T reg cells purified from infected mice into chronically infected mice was sufficient to trigger disease reactivation and prevent the expression of an effector memory response. Our results demonstrate that after persistence is achieved, an equilibrium between T reg cells and effector lymphocytes, which can be disturbed by superinfection, controls the efficiency of recall immune responses and disease reactivation.
Keywords: Leishmania, reactivation, CD4+ CD25+ regulatory, T cells, memory, chronicity
Introduction
A large number of infectious diseases that have severe consequences for human health (e.g., tuberculosis, leishmaniasis, toxoplasmosis, and herpes virus infections) are caused by pathogens that also produce persistent infections. Reactivation of such infections causes an immense burden of morbidity and mortality in the world at large. Reactivation commonly occurs under conditions of immunosuppression, but can also result from environmental insults and/or advancing age, presumably as a consequence of perturbing the immune system. However, a defined cause for reactivation or activation of infections is often not clear. More broadly, the mechanisms underlying the negotiation of latency by host and microbial organism, and particularly those underlying the breaking of latency, remain poorly understood. In human leishmaniasis, persistent infection follows self-cure or successful chemotherapy (1, 2). Such persistence often gives rise to disease reactivation including visceral leishmaniasis associated with HIV coinfection (3), post-kala-azar dermal leishmaniasis after cure of visceral leishmaniasis (4, 5), reactivation of localized, dormant skin lesions (Leishmania recidivans), and the development of destructive mucocutaneous leishmaniasis (espundia) months or years after healing of a localized cutaneous ulcer (6, 7).
C57BL/6 mice (B/6) provide a good murine model for cutaneous leishmaniasis because the pattern of infection closely mimics the human disease. After inoculation of Leishmania major parasites into B/6 mice, parasite expansion is controlled by cellular immune mechanisms that also mediate the development of a small cutaneous lesion that heals spontaneously after a few weeks (8, 9). During the primary infection, L. major can be transiently detected in a distal site (10). After reduction of the parasite burden and lesion healing, the persistence of a small number of parasites is associated with the establishment of powerful immunity against reinfection, a state referred to as concomitant immunity (1, 11, 12). Endogenous CD4+ CD25+ regulatory T (T reg) cells have been recently described as a unique population of CD4+ T cells that are able to prevent autoimmune diseases by suppressing the activation and expansion of self-reactive lymphocytes (13, 14). More generally, they can be defined by their capacity to control excessive or misdirected effector immune responses including responses against other pathogens or self-antigens (13, 15, 16). Using an intradermal low dose model of infection, we have previously demonstrated that T reg cells are essential for the development and maintenance of persistent cutaneous infection with L. major (17). T reg cells rapidly accumulate at sites of L. major infection, suppressing the ability of the immune response to completely eliminate the parasite. In this model, control of effector functions occurs through both IL-10–dependent and –independent mechanisms. Paradoxically, persistence of low numbers of parasites during latent infection is required for durable immunity against superinfection with Leishmania at secondary sites.
Although powerful and durable immunity to reinfection has been a consistent finding in mice and humans after healing of their primary lesions, and remains the rationale for live vaccination against cutaneous leishmaniasis, the consequences of secondary challenge to the conditions that maintain persistence in the primary site remain poorly understood. In this work, we show that when chronically infected mice are challenged at a distant site, despite powerful immunity to reinfection in the challenge site, transient reactivation of parasite replication and dermal pathology occurs at the primary site. Reactivation was associated with a local increase in the number of CD4+ CD25+ T cells. Depletion of CD25+ cells at the time of the challenge prevented disease reactivation at the primary site while strengthening the expression of immunity in the challenged site. Finally, the transfer of T reg cells purified from chronically infected mice into chronically infected mice was sufficient to trigger disease reactivation and prevent the expression of effector memory response. Our results demonstrate that although persistence is achieved, the equilibrium between T reg cells and effector lymphocytes, which could be disturbed in the case of superinfection, controls the efficiency of recall immune response and disease reactivation.
Materials and Methods
Mice.
C57BL/6 (B/6) mice were purchased from the Division of Cancer Treatment, National Cancer Institute or Charles River Laboratories. All mice were maintained in the National Institute of Allergy and Infectious Diseases (NIAID) Animal Care Facility or Cincinnati Children's Hospital Research Foundation under pathogen-free conditions.
Infection Protocol.
L. major clone V1 (MHOM/IL/80/Friedlin) promastigotes were grown at 26°C in medium 199 supplemented with 20% Hi-FCS (Hyclone), 100 U/ml penicillin, 100 μg/ml streptomycin, 2mM l-glutamine, 40 mM Hepes, 0.1 mM adenine (in 50 mM Hepes), 5 μg/ml hemin (in 50% triethanolamine), and 1 μg/ml 6-biotin (M199/S). Infective-stage promastigotes (metacyclics) of L. major were isolated from 4–5-d-old stationary cultures by negative selection of infective forms using peanut agglutinin (Vector Laboratories). Mice were infected in the ear dermis with 500 or 1,000 L. major metacyclic promastigotes using a 27 1/2 G needle in a volume of 10 μl.
Parasite Quantitation.
Parasite loads in the ears were determined as described previously (9). In brief, the ventral and dorsal sheets of the infected ears were separated and deposited dermal side down in RPMI containing 100 U/ml penicillin, 100 μg/ml streptomycin, and 50 μg/ml of liberase CI enzyme blend (Boehringer). Ears were incubated for 40 min at 37°C. The sheets were dissociated in RPMI with 10% serum and 0.05% DNase I (Sigma-Aldrich) using a medimachine (BD Biosciences) according to the manufacturer's instructions. The tissue homogenates were filtered using a 70-μm cell strainer (Falcon) and serially diluted in a 96-well flat-bottom microtiter plate containing biphasic medium, which was prepared using 50 μl NNN medium containing 20% of defibrinated rabbit blood overlaid with 100 μl M199/S. The number of viable parasites in each ear was determined from the highest dilution at which promastigotes could be grown out after 7 d of incubation at 26°C. The number of parasites was also determined in the local draining LNs (retromaxilar). The LNs were mechanically dissociated and parasite load in LN cells was determined by limiting dilution as described above.
Analysis of Dermal Lymphocytes.
Single cell suspensions from the ear dermis were obtained as described above. For the analysis of surface markers and intracytoplasmic staining for IFN-γ, cells were stimulated with _L. major_–infected bone marrow–derived dendritic cells (BMDC) as a source of antigen for 16 h. The cells were cultured for an additional 6 h with 10 μg/ml brefeldin A (18), and then fixed in 4% paraformaldehyde. Before staining, cells were incubated with an anti–Fcγ III/II receptor and 10% normal mouse serum in PBS containing 0.1% BSA, 0.01% NaN3. Cells were permeabilized and stained for the surface markers CD3 (145-2 C11, FITC-labeled), TCR-β (H57-597), CD4 (GK1.5 cychrome-conjugated), and CD8 (RM4-5 and 53-6.7, cychrome-conjugated), and for the cytokines IFN-γ and IL-10 (clones XNG1.2 and JE56-5H4). Incubations were performed for 30 min on ice. The isotype controls used were rat IgG2b (A95-1) and rat IgG2a (R35-95). All antibodies were purchased from BD Biosciences. The frequency of CD4+ and CD8+ T cells was determined by gating on CD3+ or TCR-β1 cells. For each sample, at least 100,000 cells were analyzed. The data were collected and analyzed using CELLQuest™ software and a FACSCalibur™ flow cytometer (Becton Dickinson).
Cytokine Measurements.
For cytokine measurements in culture supernatants, pooled cells from draining LNs were resuspended in RPMI containing FBS/penicillin/streptomycin at 6 × 106 cells/ml, and 0.1 ml was plated in 96-well U-bottom plates. Cells were incubated at 37°C in 5% CO2 with uninfected or _L. major_–infected BMDC for 48 h. In some experiments, dermal cell suspensions were stimulated for 12 h with 10 μg/ml anti-CD28 (37.51; BD Biosciences), 25 μg/ml soluble leishmanial antigen, and 5 ng/ml IL-2 (Endogen) as described previously (19). Cytokine IFN-γ, IL-10, IL-4, and IL-5 productions were analyzed using a mouse cytokine multiplex assay from Linco Research, Inc. according to the manufacturer's instructions. In brief, 25 μl of cell supernatant was incubated with antibody-coated bead mix for 16 h at 4°C. The beads were washed twice and incubated with secondary biotinylated antibody mix for 1 h at room temperature. Streptavidin-phycoerythrin was added and incubated for an additional 30 min at room temperature. The beads were washed three times, and then analyzed on a Luminex 100 platform (Luminex Corp.). Cytokine concentrations were calculated from standard curves.
T Cell Purification and Adoptive Cell Transfers.
CD4+ CD25+ or CD4+ CD25− T cells from mice chronically infected with L. major were purified from ears and local draining LNs by cell sorting as described previously (20). The T cell subsets were >98% pure as analyzed by flow cytometry. CD4+ CD25+ or CD4+ CD25− T cells (2–5 × 105/mouse) were transferred i.v. to mice that had been infected in one ear for >3 mo. In some experiments, mice were rechallenged with 1,000 metacyclic promastigotes in the opposite ear at the time of the transfer. Mice receiving no cells were included as controls.
CD25+ T Cell Depletion.
Mice were injected at the time of secondary challenge with 1 mg anti-CD25 (PC6C1; American Type Culture Collection [ATCC]) or isotype control (A1101-1; ATCC) antibodies. Antibodies were produced using serum-free medium (BD Biosciences) and a CELLine™ device (BD Biosciences) according to the manufacturer's instructions. Antibodies were purified by protein G affinity chromatography (Pierce Chemical Co.). The efficiency of depletion was monitored 2 d after depletion by flow cytometry analysis of LN, spleen, and dermal cells using 7D4 antibody (anti-CD25). The efficiency of depletion was >80%.
Statistical Analysis.
Statistical analyses were performed using GraphPad Prism software. Dual comparisons were made using the unpaired Student's t test. All data from parasite numbers were log transformed before statistical tests were conducted.
Results
Rechallenge in a Distal Site Induces Disease Reactivation at the Primary Site.
We have previously shown that during the chronic phase of the infection a high number of both effector lymphocytes (CD4+ CD25− T cells, producing IFN-γ) and T reg cells (CD4+ CD25+ T cells, producing IL-10) accumulate at sites of infection. A tight equilibrium between the two populations is responsible for the maintenance of parasite persistence (17). In this work, we examined the effect(s) of rechallenge in a distant site on the equilibrium between effector T cells and T reg cells at primary sites of infection and consequently on pathology and parasite burden.
B/6 mice were infected with 500 L. major metacyclics in one ear. This primary challenge resulted in a transient pathology (Fig. 1 B) that coincided with parasite control (Fig. 1 C). The acute phase of the infection was followed by establishment of a chronic phase, characterized by the stable maintenance of a low number of parasites at the inoculated site (latent site, ∼5 × 103 parasites per ear; Fig. 1 C) and its draining LN in the absence of overt pathology (17, 21). At 12 wk after challenge, mice were injected in the opposite ear (challenged ear) with 500 L. major metacyclics as shown in Fig. 1 A. Chronically infected mice were immune to reinfection, as evidenced by the rapid control of parasite number at the challenged site and by the fact that no pathology was detected (Fig. 1, B and C). Interestingly, not all of the parasites were eliminated from the dermis of the challenged site, as a low number of parasites were still present at 11 wk after challenge (250 parasites/ear; Fig. 1 C). Surprisingly, the expression of concomitant immunity was associated with reactivation at the primary site. Disease reactivation occurred between 3 and 6 wk after challenge and was manifested by (a) moderate increased lesion size (Fig. 1 B), (b) strong inflammation, as shown by the accumulation of neutrophils and eosinophils (not depicted), and (c) parasite expansion (from 2 × 103 to 6 × 105 at 3 wk after challenge; Fig. 1 C). At 6 wk after challenge, parasite persistence was reestablished in the reactivated site, with maintenance of a small number of parasites in absence of any detectable pathology.
Figure 1.

Challenge in a distal site with L. major induces disease reactivation of L. major latent site. (A) B/6 mice were infected in the ear with 500 L. major metacyclic promastigotes. At 12–15 wk, chronically infected mice were challenged in the other ear with 500 L. major metacyclic promastigotes. The disease outcome was followed up to 12 wk after secondary challenge. (B) Lesion size, expressed as mean induration ± SD, 3–10 mice with 6–20 ears per group. (C) Parasite number per ear, expressed as geometric mean ± SD, six ears per group. **, statistically significant between primary and secondary ear (P < 0.01); •, primary site; ○, secondary site.
Reactivation of the Chronic Site upon Secondary Challenge Correlates with a Local Increase in CD4+ CD25+ T Cells.
The numbers of CD4+ CD25− and CD4+ CD25+ T cells were analyzed at different time points in both primary and secondary sites (Fig. 2 A). The number of CD4+ CD25+ T cells increased in the primary site during lesion reactivation (from 3 × 104 ± 5.7 × 103 before secondary challenge to 5.4 × 104 ± 6 × 103 at 3 wk after challenge). The number of CD4+ CD25+ T cells was sustained during reactivation at the primary site (between the third and sixth weeks after challenge), and then decreased at 12 wk after challenge. In contrast, the number of CD4+ CD25+ T cells did not increase significantly in the challenged site (Fig. 2 A).
Figure 2.
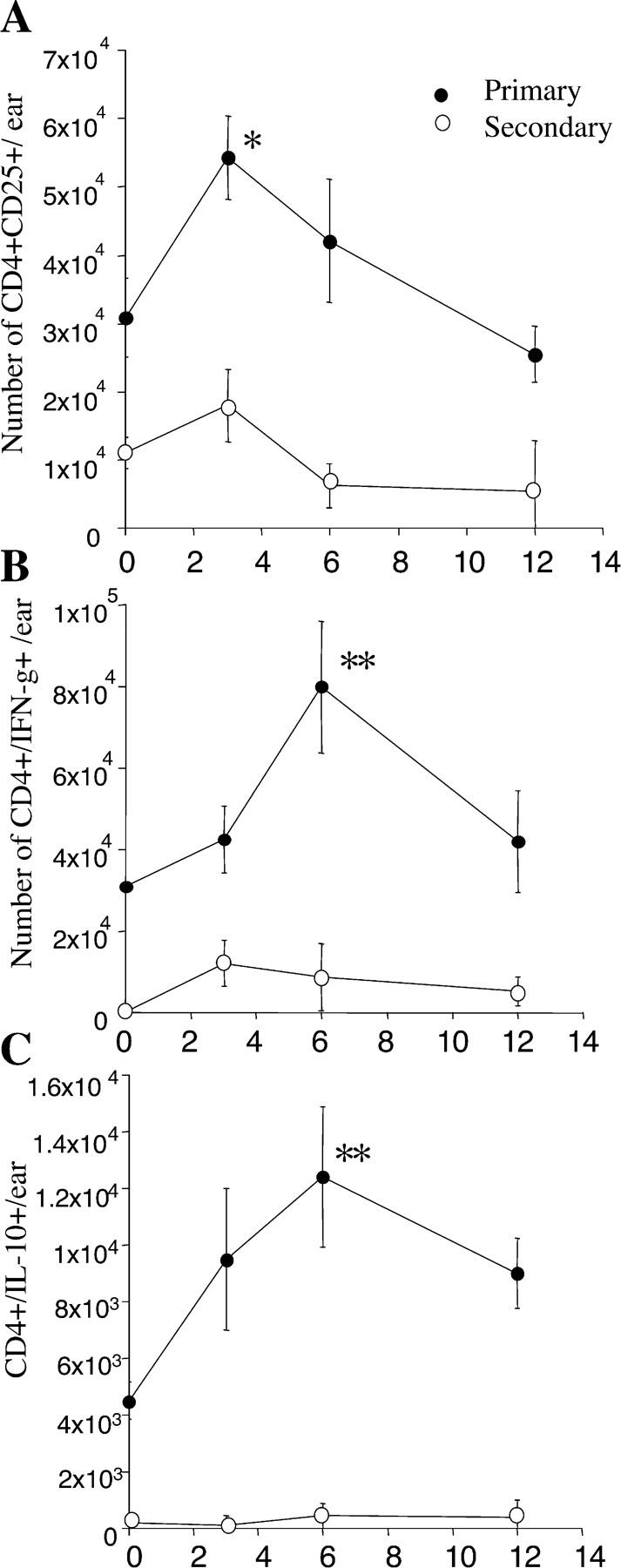
Increased frequency of dermal CD4+ CD25+ T cells, CD4+ IFN-γ, and CD4+ IL-10–producing cells during reactivation. Mice were infected in one ear with 500 L. major promastigotes. After clinical cure, mice were challenged in the other ear with 500 L. major promastigotes. Absolute number at primary (•) and secondary site (○) of (A) CD4+ CD25+ T cells, (B) CD4+ IFN-γ–producing cells, and (C) CD4+ IL-10–producing cells at different time points after secondary challenge. (A) Absolute number of CD3+ CD4+ cells per ear expressing CD25 compared with isotype control. Isolated cells were stained for surface markers and analyzed by flow cytometry. (B and C) Absolute number of CD3+ CD4+ cells per ear expressing IFN-γ and IL-10. Dermal cells were incubated overnight with _L. major_–infected BMDC before labeling for surface markers and intracellular cytokines. Values represent the mean of the absolute number of cells per ear ± SEM. Dermal cells from four mice per time point were analyzed individually. *, statistically significant between day 0 and the time point analyzed (*, P < 0.05; **, P < 0.01). This experiment is representative of three distinct experiments.
Before secondary challenge, CD4+ T cells producing IFN-γ in response to Leishmania antigen were only detectable in the primary site of infection (Fig. 2 B). After rechallenge, the number of CD4+ IFN-γ+ cells increased in the challenged site during parasite killing (threefold increase at 6 wk after infection; Fig. 2 B). Surprisingly, this accumulation of a higher number of IFN-γ–producing CD4+ T cells occurred at the primary site during reactivation as well (threefold increase at 6 wk after challenge; Fig. 2 B). The intradermal lymphocyte able to produce IFN-γ remained CD25 low or dim even after in vitro restimulation (not depicted). The IL-10–producing CD4+ T cells were only detectable in the primary site before secondary challenge (4.5 × 103 ± 6.2 × 102 cells per ear; Fig. 2 C). Reactivation also correlated with a significant increase in the number of IL-10–producing CD4+ T cells (threefold increase at 6 wk after challenge; Fig. 2 C). Interestingly, the accumulation of CD4+ IL-10+ paralleled the kinetics of accumulation of the IFN-γ–producing CD4+ cells. In contrast, the number of IL-10–producing CD4+ cells remained low in the secondary site (below 102 per ear at every time point analyzed; Fig. 2 C).
Transfer of T reg Cells Triggers Reactivation of Latent Infections.
We directly assessed the role of T reg cells in disease reactivation by transferring into chronically infected mice either CD4+ CD25+ or CD4+ CD25− T cells purified from previously infected, healed mice. This transfer was performed in the absence of additional parasitic challenge. The adoptive transfer of T reg cells into chronically infected mice led to a marked increase in CD4+ CD25+ T cell numbers at the site of primary infection (2.5-fold increase; not depicted). This increase consistently correlated with parasite replication and disease reactivation (Fig. 3 A). The transfer of CD4+ CD25− triggered reactivation in some animals and was inconsistent among the different experiments (Fig. 3 A). Reactivation induced by T reg cell transfer was accompanied by a significant increase of both IL-10 and IFN-γ production by dermal CD4+ T cells (over threefold increase at 4 wk after challenge; Fig. 3 B). A similar pattern was observed for cytokine production by local LN cells (not depicted). Such increases in cytokine production were not observed after the transfer of CD4+ CD25− T cells. Moreover, the level of IL-4 production by LN cells was negligible during disease reactivation (not depicted). These data indicate that adoptive transfer of T reg cells from infected mice was sufficient to trigger powerful reactivation of primary infection.
Figure 3.

Transfer of T reg cells triggers disease reactivation. Chronically infected B/6 mice were transferred i.v. (or not) with 5 × 105 T reg cells or CD4+ CD25− T cells from previously infected mice. (A) Parasite numbers at primary sites 4 wk after transfer, with four mice per group. **, statistically significant between control mice and mice transferred with T reg cells (P < 0.01). (B) Dermal CD4+ IFN-γ (open bars) and CD4+ IL-10–producing cells (closed bars) at 4 wk after transfer. Purified dermal cells were incubated overnight with _L. major_–infected BMDC before labeling for surface markers and intracytoplasmic staining for cytokines. Numbers represent the absolute number of CD4+ TCR-β+ IFN-γ+ or IL-10+ per ear. Dermal cells from six or eight ears were pooled. This experiment is representative of three distinct experiments.
CD25+ Cell Depletion Strengthens the Intensity of the Immune Response and Prevents Reactivation.
Because the adoptive transfer of T reg cells consistently induced disease reactivation, we treated chronically infected mice with a depleting anti-CD25 antibody at the time of challenge. The depletion of CD25+ cells at the time of the secondary challenge significantly prevented disease reactivation in the primary site (Fig. 4 A). Interestingly, such depletion also enhanced the efficiency of the effector immune response, with a better control of the parasite replication in the challenge site at 3 wk after challenge (1.1 × 102 for anti-CD25–treated mice compared with 5.1 × 104 parasites per ear for isotype-treated mice; Fig. 4 A). The higher dose of parasites used in this experiment induced a more rapid reactivation and immune response at the challenge site.
Figure 4.

Anti-CD25 treatment prevents disease reactivation and strengthens the intensity of immune response. Chronically infected B/6 mice were challenged in the opposite ear with 103 L. major promastigotes and treated at the same time with anti-CD25 or isotype control Abs. (A) Parasite number at 3 wk after challenge in mice treated with anti-CD25 or isotype Abs as indicated. **, statistically significant (P < 0.01). (B) Number of lymphocytes per ear in primary, latent, and rechallenged dermal site at 1.5 and 4.5 wk. Mice were treated with anti-CD25 or isotype control as indicated. Values represent the mean number of lymphocyte per ear ± SEM, four mice per group. *, statistically significant between isotype and anti-CD25 treatment (**, P < 0.01; ***, P < 0.001). (C) CD4+ IFN-γ+ cells in the dermis of challenged mice. Dermal cells were incubated with _L. major_–infected BMDC before labeling for surface markers and intracellular IFN-γ. Plots shown are gated on TCR-β+ lymphocytes. Numbers in the top right quadrant represent the percentage of CD4+ TCR-β+ T cells positive for IFN-γ compared with the isotype control. Ears from four animals were pooled. This experiment is representative of three distinct experiments.
As previously described, the chronic site is characterized by the accumulation of CD4+ T cells. Anti-CD25 treatment significantly increased the influx of CD4+ T cells at both primary and challenged sites (Fig. 4 B) after 1.5 wk. At 4.5 wk after challenge, a time when the isotype-treated mice had restored homeostatic conditions in both latent and challenged sites, CD25 depletion was accompanied with the maintenance of a high number of CD4+ T cells at both sites. In addition, CD25 depletion allowed for the sustained production of IFN-γ by intradermal CD4+ T cells in both latent (44% of CD4+ T cells compared with 17% in isotype-treated mice) and challenged sites (50% compared with 20%) at 4.5 wk after challenge (Fig. 4 C). Thus, a transient depletion of CD25+ cells at the time of the transfer was sufficient to prevent disease reactivation in the primary site and to intensify and prolong the local immune response at the challenged site.
T reg Cells from Chronically Infected Mice Suppress Immunity at Sites of Rechallenge.
To confirm that T reg cells are responsible for controlling the efficiency of the effector response after secondary challenge, we assessed the expression of immunity in mice transferred with T reg cells. Chronically infected B/6 mice were transferred with either CD4+ CD25+ or CD4+ CD25− T cells, each obtained from previously infected and healed mice, and challenged in the opposite ear with 1,000 L. major parasites. At 4 wk after challenge, parasite burden was evaluated at the challenged site. Mice transferred with T reg cells lost their capacity to control parasites in the challenged site, whereas mice transferred with CD4+ CD25− cells or not transferred were immune to secondary challenge as expected (Fig. 5 A). Analysis of the cytokine released by dermal cells from the challenge site revealed that at 1 wk after challenge, T reg cell transfer led to an increase of the IL-10 production at the challenged site (from 430 ± 57 to 1,230 ± 220 pg/ml). At 4 wk after transfer, the production of IFN-γ, IL-5, and IL-4 was significantly increased at the challenge site compared with control mice or mice transferred with CD4+ CD25− (Fig. 5, B and C). These results confirm that up-regulation of the number of T reg cells at the time of the challenge has detrimental consequences on the efficiency of the local effector immune response.
Figure 5.

Transfer of T reg cells prevents parasite control in immune mice. B/6 mice were infected in one ear with 500 L. major promastigotes. After clinical cure, mice were challenged in the other ear with 103 L. major promastigotes. At the time of secondary challenge, 5 × 105 T reg cells or CD4+ CD25− T cells purified from latently infected mice were adoptively transferred or not as noted. (A) At 4 wk after infection, parasite numbers were assessed in the challenged site. Numbers represent mean parasite burdens per ear ± SD, four mice per group. **, statistically significant compared with primary challenge (P < 0.01); n.s, nonsignificant compared with primary challenged. (B and C) Measure of cytokine release by dermal cells at 4 wk after challenge. IFN-γ, IL-10, IL-4, and IL-5 were measured in the supernatant of dermal cells activated with anti-CD28, IL-2, and soluble leishmanial antigen. Values represent the mean cytokine concentration ± SEM, four mice per group. *, statistically significant compared with secondary challenge control (*, P < 0.05; ***, P < 0.001). The data shown are representative of two distinct experiments.
T reg Cell and Effector Lymphocytes Migrate in Both the Primary and Immune Sites during Rechallenge.
We have shown that when effector immune response occurs, the number of T reg cells do not increases in the challenge site. On the other hand, T reg cells were able to interfere with concomitant immunity, suggesting that early after challenge they have the capacity to enter in the challenge site. To address this point, chronically infected B/6 mice were transferred with either CD4+ CD25+ or CD4+ CD25− T cells, each obtained from previously infected and healed congenic Ly5.1 mice, and challenged in the opposite ear with 1,000 L. major parasites. At 1 wk after transfer and challenge, a similar number of T reg cells and CD4+ CD25−, identified by their expression of congenic marker, were detected in the primary or secondary site of infection (Fig. 6). Interestingly, T reg cells from infected mice were able to enter in the primary site even in the absence of secondary challenge (3.2 × 104 ± 1.5 × 104 in the primary site, 1 wk after transfer of 5 × 105 T reg cells). T reg cells or CD4+ CD25− cells purified from naive mice were not detectable after transfer in the challenged or chronic site at 1 wk after infection (not depicted). Thus, early after challenge, T reg cells and effector cells entered with similar efficiency in both sites.
Figure 6.

T reg cells and effector cells migrate at sites of infection. B/6 mice were infected in one ear with 500 L. major promastigotes. After clinical cure, mice were challenged in the other ear with 103 L. major promastigotes. At the time of secondary challenge, 5 × 105 T reg cells or CD4+ CD25− T cells purified from healed congenic (Ly5.1) infected mice were adoptively transferred or not as noted. At 1 wk after infection, the number of Ly5.1+ cells in the primary or challenged site was evaluated by flow cytometry. Values represent the number of TCR-β, CD4, and Ly5.1+ cells per ear, with four mice per group. The data shown are representative of two distinct experiments. n.s, nonstatistically significant between the experimental groups.
Discussion
We have previously shown that the establishment of chronicity and the maintenance of a constant number of parasites at sites of infection were dependent on a tight equilibrium between effector lymphocytes and T reg cells (17). Absence or inhibition of T reg cells or IL-10 will promote complete clearance of the parasite, whereas depletion of effector cells or cytokines (e.g., IFN-γ, IL-12) will promote reactivation (1, 22, 23). In this work, we introduce an alternative and surprising condition that perturbs the maintenance of the chronic state. We show that when latency is achieved, the expression of immunity to a rechallenge infection in a distant site is associated with reactivation of disease at the primary site of infection.
A cross talk between the primary and secondary sites of infection has been previously reported. Preston and Dumonde (24), using various doses of infection, demonstrated that secondary challenge could impair the healing of the primary site during Leishmania infection. Furthermore, Muller (25) showed that a transient increase in footpad swelling of the healed primary site occurred after secondary challenge with the parasite. Our present results strongly suggest that the introduction of antigen in a secondary site initiates a disruption of the equilibrium between the two populations of lymphocytes in favor of T reg cells that are responsible for disease reactivation. This hypothesis is supported by the fact that reactivation (a) correlates with an increase in T reg cells at the primary site, (b) can be reproduced by the transfer of T reg cells from chronically infected mice, and (c) is prevented by the depletion of CD25+ cells. Furthermore, we showed that depletion of CD25+ cells strengthens the efficiency of effector mechanisms during memory response, whereas the transfer of T reg cells at the time of rechallenge prevented the expression of such immunity.
CD4+ CD25+ T reg cells, which constitute 5–10% of peripheral CD4+ T cells in normal rodents and humans at steady-state conditions, have been primarily described for their fundamental role in the control of autoimmunity. More generally, T reg cells can be defined by their role in the control of excessive or misdirected immune responses (for review see references 13 and 16). There is growing evidence that T reg cells play a significant role in microbial infection (26–30). Our demonstration that T reg cells are increased in number and play a role in the reactivation of dormant infection after reinfection seems highly relevant to clinical settings because individuals with persistent Leishmania infections will, in many instances, be reexposed to the parasite. The data seems especially relevant to certain reactivation diseases like Leishmania recidivans or post-kala-azar dermal leishmaniasis that in each case represent spontaneous reactivation of cutaneous lesions in previously cured individuals (4, 6). Although CD25 is also expressed on recently activated T cells, the observation that the number of CD4+ CD25+ T cells do not increase during the expression of immunity at the secondary site strongly suggests that in the cutaneous site, only T reg cells express high level of CD25. This is further supported by our previous observation that the vast majority of the cells expressing CD25 at sites of infection were derived from endogenous T reg cells (17). During the early phase of L. major infection, the parasite is transiently detected in distant cutaneous sites (10). Such even may occur in the context of a secondary challenge and contribute to the initiation of the reactivation process. Although we cannot rule out the possibility that the increase of T reg cells observed in the context of superinfection is not the cause but a consequence of the reactivation process, the transfer of T reg cells was sufficient to consistently trigger disease reactivation. Furthermore, the depletion of CD25+ cells at the time of the rechallenge prevented such reactivation.
Our results suggest that the numbers of T reg cells that are present at the time of the recall response are directly responsible for the efficiency of the effector immune responses. A depletion of CD25+ T cells at the time of the challenge was sufficient to enhance the efficiency of the recall response. The rapid control of parasite charge after depletion of CD25+ T cells was associated with a significant increase of the early influx of CD4+ T cells in the challenged site as well as their sustained accumulation compared with control mice. Furthermore, we observed an enhanced and sustained capacity of CD4+ T cells to produce IFN-γ in response to Leishmania antigen after CD25+ cell depletion. Although a decrease of T reg cells led to enhanced parasite control, the adoptive transfer of exogenous T reg cells from latently infected mice was able to hamper the efficiency of the recall responses. The mechanism by which an excess of T reg cells control concomitant immunity is not fully understood, but might be at least partially explained by their capacity to rapidly accumulate at challenge site and promote local IL-10 production. Although T reg cells are not a significant source of IL-4 or IL-5, or IFN-γ in response to Leishmania antigen (17), their transfer also led to a marked increase in the production of those cytokines at the challenge site. Thus, T reg cell transfer led to a profound modification of the local microenvironment in favor of cytokines known to promote parasite expansion (31, 32). All together, those results demonstrate that even in immune mice, in which effector mechanisms are more efficient than during primary challenge, T reg cells can control the intensity of the recall response.
We have shown that during recall responses, the number of T reg cells increased at reactivated sites while it decreased in challenged sites. This result is consistent with our previous report indicating that T reg cells, compared with effector cells, are preferentially attracted to sites of L. major infection during parasite expansion and after clinical cure. In contrast, the proportion of effector cells increased during the effector immune response (17). Differences in chemokine responsiveness or receptor expression between T reg cells and effector T cells have been shown previously in various models (33–35). Therefore, a plausible explanation for the coexpression of immunity and reactivation would be that upon pathogen reexposure, both populations of lymphocytes expand, but that their pattern of trafficking is distinct. To maintain homeostasis, the chronic site would preferentially attract and favor the survival of T reg cells, whereas effector T cells would be preferentially attracted to the proinflammatory challenged site. A nonexclusive possibility would be that the migration of T reg cells or effector cells in distinct sites produce a distinct outcome. In the chronic site, effector cells would not be able to overwrite T reg cell–mediated deactivation of infected APCs, whereas T reg cells will further deactivate the site favoring parasite replication. In the challenged site, the rapid arrival/expansion of effector cells could more efficiently activate local APCs, leading to an amplification of the effector immune response, inhibition of T reg cell suppressive functions (36–38), and parasite killing. Consistently with this hypothesis, using adoptive transfer model we have shown that early after challenge, both populations were able to migrate to primary and secondary sites of infection. The production of IFN-γ was increased in both sites, suggesting that effector T cells were recruited at latent and challenged sites. Furthermore, CD25+ cell depletion enhanced the efficiency of the recall response, an effect that supports their presence at the challenged site. The signals to which T reg cells are actually responding upon arrival in infected tissue remain to be addressed. A proportion of T reg cells may respond to Leishmania antigen, as our previous studies showed that a significant proportion of T reg cells in latent sites were Leishmania antigen specific (17). On the other hand, some T reg cells could also respond to cytokines such as IL-2 (a cytokine that is required for their generation and survival; 39, 40), which memory effector cells would rapidly produce after antigen exposure.
During the reactivation process that occurs after superinfection or after T reg cell transfer, the production of IL-10 by dermal and LN CD4+ T cells was significantly increased. A role for IL-10 in mediating Leishmania susceptibility in both susceptible and resistant strains is well established (19, 41–44). We have previously shown that IL-10, which is produced by T reg cells, contributes directly to parasite persistence (17). The mechanism(s) by which IL-10 allows parasite survival and expansion are not fully understood, but it is likely due to its potent deactivating role of infected APCs that would become unresponsive to activation by IFN-γ (45). During disease reactivation, IFN-γ production was increased, suggesting that effector T cell functions are not suppressed. In the presence of deactivating cytokines or in the absence of secondary signals, IFN-γ can itself promote parasite expansion (32). These results support the idea that during the reactivation process, deactivation mediated by IL-10–producing T reg cells prevails over effector mechanisms. Such principals appear to apply to various models of infection including Leishmania donovani and Schistosoma mansoni, in which the levels of a deactivating cytokine such as IL-10 (but not IFN-γ) predict the outcome of the infection (46–48).
Reactivation or activation of latent infections with bacteria (e.g., Mycobacterium tuberculosis), protozoa (e.g., Leishmania spp., Toxoplasma), and viruses (e.g., herpes viruses) causes an immense burden of morbidity and mortality in the world at large. Reactivation can occur as a result of immunosuppression, environmental insults, or with advancing age (49, 50); however, a definite cause for the reactivation or primary activation of dormant infections is often not clear. In this work we have shown that despite efficient parasite clearance at the secondary site of infection, Leishmania superinfection can also cause disease reactivation at the primary site. Such coexpression in the same animal of immunity versus disease reactivation may provide for a more general mechanism by which some dormant infections are activated or reactivated.
Acknowledgments
We thank Calvin Eigsti, Kevin Holmes (NIAID), Dan Marmer and Sue Vergamini (Cincinnati Children's Hospital Research Foundation) of the flow cytometry unit for FACS® sorting, Sandra Cooper (NIAD) and Dr. Keller (Cincinnati Children's Hospital Research Foundation) for help with the mouse care, and Dr. C. Chougnet and J. Suffia for critical reading of the manuscript.
This work was supported by the NIAID (National Institutes of Health), Cincinnati Children's Hospital Medical Center, and the Ellison Medical foundation.
S. Mendez's present address is Dept. of Microbiology and Tropical Medicine, George Washington University, Washington, DC 20037.
C.A. Piccirillo's present address is Dept. of Microbiology and Immunology, Host Resistance Laboratory, McGill University, Montreal QC H3A2B4, Canada.
Abbreviations used in this paper: BMDC, bone marrow–derived dendritic cells; T reg, regulatory T.
References
- 1.Stenger, S., N. Donhauser, H. Thuring, M. Rollinghoff, and C. Bogdan. 1996. Reactivation of latent leishmaniasis by inhibition of inducible nitric oxide synthase. J. Exp. Med. 183:1501–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vardy, D.A., A. Cohen, L. Kachko, A. Zvulunov, and S. Frankenburg. 1999. Relapse of cutaneous leishmaniasis in a patient with an infected subcutaneous rheumatoid nodule. Br. J. Dermatol. 141:914–917. [DOI] [PubMed] [Google Scholar]
- 3.Alvar, J., C. Canavate, B. Gutierrez-Solar, M. Jimenez, F. Laguna, R. Lopez-Velez, R. Molina, and J. Moreno. 1997. Leishmania and human immunodeficiency virus coinfection: the first 10 years. Clin. Microbiol. Rev. 10:298–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.el Hassan, A.M., H.W. Ghalib, E.E. Zijlstra, I.A. Eltoum, M. Satti, M.S. Ali, and H.M. Ali. 1992. Post kala-azar dermal leishmaniasis in the Sudan: clinical features, pathology and treatment. Trans. R. Soc. Trop. Med. Hyg. 86:245–248. [DOI] [PubMed] [Google Scholar]
- 5.Choi, C.M., and E.A. Lerner. 2001. Leishmaniasis as an emerging infection. J. Invest. Dermatol. 6:175–182. [DOI] [PubMed] [Google Scholar]
- 6.Saravia, N.G., A.F. Holguin, D. McMahon-Pratt, and A. D'Alessandro. 1985. Mucocutaneous leishmaniasis in Colombia: Leishmania braziliensis subspecies diversity. Am. J. Trop. Med. Hyg. 34:714–720. [DOI] [PubMed] [Google Scholar]
- 7.Da-Cruz, A.M., D.V. Filgueiras, Z. Coutinho, W. Mayrink, G. Grimaldi, Jr., P.M. De Luca, S.C. Mendonca, and S.G. Coutinho. 1999. Atypical mucocutaneous leishmaniasis caused by Leishmania braziliensis in an acquired immunodeficiency syndrome patient: T-cell responses and remission of lesions associated with antigen immunotherapy. Mem. Inst. Oswaldo Cruz. 94:537–542. [DOI] [PubMed] [Google Scholar]
- 8.Reiner, S.L., and R.M. Locksley. 1995. The regulation of immunity to Leishmania major. Annu. Rev. Immunol. 13:151–177. [DOI] [PubMed] [Google Scholar]
- 9.Belkaid, Y., S. Kamhawi, G. Modi, J. Valenzuela, N. Noben-Trauth, E. Rowton, J. Ribeiro, and D.L. Sacks. 1998. Development of a natural model of cutaneous leishmaniasis: powerful effects of vector saliva and saliva preexposure on the long-term outcome of Leishmania major infection in the mouse ear dermis. J. Exp. Med. 188:1941–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nicolas, L., S. Sidjanski, J.H. Colle, and G. Milon. 2000. Leishmania major reaches distant cutaneous sites where it persists transiently while persisting durably in the primary dermal site and its draining lymph node: a study with laboratory mice. Infect. Immun. 68:6561–6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aebischer, T., S.F. Moody, and E. Handman. 1993. Persistence of virulent Leishmania major in murine cutaneous leishmaniasis: a possible hazard for the host. Infect. Immun. 61:220–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramirez, J.L., and P. Guevara. 1997. Persistent infections by Leishmania (Viannia) braziliensis. Mem. Inst. Oswaldo Cruz. 92:333–338. [DOI] [PubMed] [Google Scholar]
- 13.Shevach, E.M. 2002. CD4+ CD25+ suppressor T cells: more questions than answers. Nat. Rev. Immunol. 2:389–400. [DOI] [PubMed] [Google Scholar]
- 14.Gavin, M.A., S.R. Clarke, E. Negrou, A. Gallegos, and A. Rudensky. 2002. Homeostasis and anergy of CD4(+) CD25(+) suppressor T cells in vivo. Nat. Immunol. 3:33–41. [DOI] [PubMed] [Google Scholar]
- 15.Maloy, K.J., and F. Powrie. 2001. Regulatory T cells in the control of immune pathology. Nat. Immunol. 2:816–822. [DOI] [PubMed] [Google Scholar]
- 16.Asseman, C., and M. von Herrath. 2002. About CD4pos CD25pos regulatory cells. Autoimmun. Rev. 1:190–197. [DOI] [PubMed] [Google Scholar]
- 17.Belkaid, Y., A.C. Piccirilo, S. Mendez, E. Shevack, and D.L. Sacks. 2002. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature. 420:502–507. [DOI] [PubMed] [Google Scholar]
- 18.Belkaid, Y., E. Von Stebut, S. Mendez, R. Lira, E. Caler, S. Bertholet, M.C. Udey, and D. Sacks. 2002. CD8(+) T cells are required for primary immunity in C57BL/6 mice following low-dose, intradermal challenge with Leishmania major. J. Immunol. 168:3992–4000. [DOI] [PubMed] [Google Scholar]
- 19.Belkaid, Y., K.F. Hoffmann, S. Mendez, S. Kamhawi, M.C. Udey, T.A. Wynn, and D.L. Sacks. 2001. The role of interleukin (IL)-10 in the persistence of Leishmania major in the skin after healing and the therapeutic potential of anti–IL-10 receptor antibody for sterile cure. J. Exp. Med. 194:1497–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thornton, A.M., and E.M. Shevach. 1998. CD4+ CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med. 188:287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Belkaid, Y., S. Mendez, R. Lira, N. Kadambi, G. Milon, and D. Sacks. 2000. A natural model of Leishmania major infection reveals a prolonged “silent” phase of parasite amplification in the skin before the onset of lesion formation and immunity. J. Immunol. 165:969–977. [DOI] [PubMed] [Google Scholar]
- 22.Muller, I., and J.A. Louis. 1989. Immunity to experimental infection with Leishmania major: generation of protective L3T4+ T cell clones recognizing antigen(s) associated with live parasites. Eur. J. Immunol. 19:865–871. [DOI] [PubMed] [Google Scholar]
- 23.Stobie, L., S. Gurunathan, C. Prussin, D.L. Sacks, N. Glaichenhaus, C.Y. Wu, and R.A. Seder. 2000. The role of antigen and IL-12 in sustaining Th1 memory cells in vivo: IL-12 is required to maintain memory/effector Th1 cells sufficient to mediate protection to an infectious parasite challenge. Proc. Natl. Acad. Sci. USA. 97:8427–8432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Preston, P.M., and D.C. Dumonde. 1976. Experimental cutaneous leishmaniasis. V. Protective immunity in subclinical and self-healing infection in the mouse. Clin. Exp. Immunol. 23:126–138. [PMC free article] [PubMed] [Google Scholar]
- 25.Muller, I. 1992. Role of T cell subsets during the recall of immunologic memory to Leishmania major. Eur. J. Immunol. 22:3063–3069. [DOI] [PubMed] [Google Scholar]
- 26.Aseffa, A., A. Gumy, P. Launois, H.R. MacDonald, J.A. Louis, and F. Tacchini-Cottier. 2002. The early IL-4 response to Leishmania major and the resulting Th2 cell maturation steering progressive disease in BALB/c mice are subject to the control of regulatory CD4+CD25+ T cells. J. Immunol. 169:3232–3241. [DOI] [PubMed] [Google Scholar]
- 27.Hori, S., T.L. Carvalho, and J. Demengeot. 2002. CD25+ CD4+ regulatory T cells suppress CD4+ T cell-mediated pulmonary hyperinflammation driven by Pneumocystis carinii in immunodeficient mice. Eur. J. Immunol. 32:1282–1291. [DOI] [PubMed] [Google Scholar]
- 28.Xu, D., L.H. Komai-Koma, M. Campbell, C. Mc, C. Sharry, J. Alexander, and F.Y. Liew. 2003. CD4(+) CD25(+) regulatory T cells suppress differentiation and functions of Th1 and Th2 cells, Leishmania major infection, and colitis in mice. J. Immunol. 170:394–399. [DOI] [PubMed] [Google Scholar]
- 29.Suvas, S., U. Kumaraguru, C.D. Pack, S. Lee, and B.T. Rouse. 2003. CD4+ CD25+ T cells regulate virus-specific primary and memory CD8+ T cell responses. J. Exp. Med. 198:889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hisaeda, H., Y. Maekawa, D. Iwakawa, H. Okada, K. Himeno, K. Kishihara, S. Tsukumo, and K. Yasutomo. 2004. Escape of malaria parasites from host immunity requires CD4(+)CD25(+) regulatory T cells. Nat. Med. 10:29–30. [DOI] [PubMed] [Google Scholar]
- 31.Sacks, D., and N. Noben-Trauth. 2002. The immunology of susceptibility and resistance to Leishmania major in mice. Nat. Rev. Immunol. 2:845–858. [DOI] [PubMed] [Google Scholar]
- 32.Qi, H., J. Ji, N. Wanasen, and L. Soong. 2004. Enhanced replication of Leishmania amazonensis amastigotes in gamma interferon-stimulated murine macrophages: implications for the pathogenesis of cutaneous leishmaniasis. Infect. Immun. 72:988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bystry, R.S., V. Aluvihare, K.A. Welch, M. Kallikourdis, and A.G. Betz. 2001. B cells and professional APCs recruit regulatory T cells via CCL4. Nat. Immunol. 2:1126–1132. [DOI] [PubMed] [Google Scholar]
- 34.Iellem, A., M. Mariani, R. Lang, H. Recalde, P. Panina-Bordignon, F. Sinigaglia, and D. D'Ambrosio. 2001. Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4+ CD25+ regulatory T cells. J. Exp. Med. 194:847–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szanya, V., J. Ermann, C. Taylor, C. Holness, and C.G. Fathman. 2002. The subpopulation of CD4+CD25+ splenocytes that delays adoptive transfer of diabetes expresses l-selectin and high levels of CCR7. J. Immunol. 169:2461–2465. [DOI] [PubMed] [Google Scholar]
- 36.Pasare, C., and R. Medzhitov. 2003. Toll pathway-dependent blockade of CD4+ CD25+ T cell-mediated suppression by dendritic cells. Science. 299:1033–1036. [DOI] [PubMed] [Google Scholar]
- 37.Serra, P., A. Amrani, J. Yamanouchi, B. Han, S. Thiessen, T. Utsugi, J. Verdaguer, and P. Santamaria. 2003. CD40 ligation releases immature dendritic cells from the control of regulatory CD4+CD25+ T cells. Immunity. 19:877–889. [DOI] [PubMed] [Google Scholar]
- 38.Choi, B.K., J.S. Bae, E.M. Choi, W.J. Kang, S. Sakaguchi, D.S. Vinay, and B.S. Kwon. 2003. 4-1BB-dependent inhibition of immunosuppression by activated CD4+CD25+ T cells. J. Leukoc. Biol. 75:785–791. [DOI] [PubMed] [Google Scholar]
- 39.Malek, T.R. 2003. The main function of IL-2 is to promote the development of T regulatory cells. J. Leukoc. Biol. 74:961–965. [DOI] [PubMed] [Google Scholar]
- 40.Thornton, A., C. Piccirillo, and E. Shevach. 2004. Activation requirements for the induction of CD4+CD25+ T cell suppressor function. Eur. J. Immunol. 34:366–376. [DOI] [PubMed] [Google Scholar]
- 41.Groux, H., F. Cottrez, M. Rouleau, S. Mauze, S. Antonenko, S. Hurst, T. McNeil, M. Bigler, M.G. Roncarolo, and R.L. Coffman. 1999. A transgenic model to analyze the immunoregulatory role of IL-10 secreted by antigen-presenting cells. J. Immunol. 162:1723–1729. [PubMed] [Google Scholar]
- 42.Kane, M.M., and D.M. Mosser. 2001. The role of IL-10 in promoting disease progression in leishmaniasis. J. Immunol. 166:1141–1147. [DOI] [PubMed] [Google Scholar]
- 43.Yamakami, K., S. Akao, T. Tadakuma, Y. Nitta, J. Miyazaki, and N. Yoshizawa. 2002. Administration of plasmids expressing interleukin-4 and interleukin-10 causes BALB/c mice to induce a T helper 2-type response despite the expected T helper 1-type response with a low-dose infection of Leishmania major. Immunology. 105:515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Noben-Trauth, N., R. Lira, H. Nagase, W.E. Paul, and D.L. Sacks. 2003. The relative contribution of IL-4 receptor signaling and IL-10 to susceptibility to Leishmania major. J. Immunol. 170:5152–5158. [DOI] [PubMed] [Google Scholar]
- 45.Gazzinelli, R.T., I.P. Oswald, S.L. James, and A. Sher. 1992. IL-10 inhibits parasite killing and nitrogen oxide production by IFN-gamma-activated macrophages. J. Immunol. 148:1792–1796. [PubMed] [Google Scholar]
- 46.Schopf, L.R., K.F. Hoffmann, A.W. Cheever, J.F. Urban, Jr., and T.A. Wynn. 2002. IL-10 is critical for host resistance and survival during gastrointestinal helminth infection. J. Immunol. 168:2383–2392. [DOI] [PubMed] [Google Scholar]
- 47.Murray, H.W., C.M. Lu, S. Mauze, S. Freeman, A.L. Moreira, G. Kaplan, and R.L. Coffman. 2002. Interleukin-10 (IL-10) in experimental visceral leishmaniasis and IL-10 receptor blockade as immunotherapy. Infect. Immun. 70:6284–6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Murray, H.W., A.L. Moreira, C.M. Lu, J.L. DeVecchio, M. Matsuhashi, X. Ma, and F.P. Heinzel. 2003. Determinants of response to interleukin-10 receptor blockade immunotherapy in experimental visceral leishmaniasis. J. Infect. Dis. 188:458–464. [DOI] [PubMed] [Google Scholar]
- 49.Rajagopalan, S. 2001. Tuberculosis and aging: a global health problem. Clin. Infect. Dis. 33:1034–1039. [DOI] [PubMed] [Google Scholar]
- 50.Gane, E., and H. Pilmore. 2002. Management of chronic viral hepatitis before and after renal transplantation. Transplantation. 74:427–437. [DOI] [PubMed] [Google Scholar]