Hypothetical review: thymic aberrations and type-I interferons; attempts to deduce autoimmunizing mechanisms from unexpected clues in monogenic and paraneoplastic syndromes (original) (raw)
Abstract
In sporadic autoimmune disorders, dendritic cells are increasingly being incriminated as agents provocateurs. However, the mechanisms and any ‘danger signals’ that induce them to autoimmunize remain enigmatic. Here, we focus on unexpected clues from two prototypic/ highly informative autoimmune syndromes, acquired thymoma-associated myasthenia gravis and the monogenic autoimmune polyendocrine syndrome type-1 (APS1), caused by mutations in the AutoImmune Regulator (AIRE). Both involve the thymus, and in both we find early, persistent, highly prevalent and high-titre neutralizing autoantibodies against type-I interferons, regardless of the exact AIRE genotype or the characteristically variable clinical phenotype in APS1. Thus these key innate↔adaptive immune intermediaries are now implicated in APS1 and paraneoplastic myasthenia as well as in systemic lupus erythematosus and other sporadic autoimmune disorders. The currently accepted notion that autoimmunization proceeds automatically (by ‘default’) does not explain how, when or where autoimmune responses are initiated against which targets in APS1, or whether exogenous or internal danger signals are involved, or predict whether the primary auto-immunogenic targets are AIRE-dependent. As the parallels between these syndromes must hold novel clues to these puzzles, they demand explanations. To unify these and other findings, we propose that autoimmunization occurs centrally in aberrant thymic environments rendered ‘dangerous’ by AIRE-deficiency (possibly by excess undegraded nucleic acids/dead cell debris). The ensuing autoreactivity focuses early on the locally abundant type I interferons and then on other peripheral tissue autoantigens that are still expressed despite the absence of AIRE. These ideas raise numerous questions that others may already have the materials to address.
Keywords: AIRE, APECED, autoimmunity, autoimmunization, autoantibody, myasthenia gravis, self-tolerance, thymic epithelial cells, thymoma, thymus, type I interferon
I.Human autoimmune syndromes: highly informative experiments of nature
Even in the best animal models, the initiation of autoimmune responses is hard to study. In humans, we should treasure any rare clues, such as the recurring associations between particular autoimmune disorders and certain tumour types or single gene defects. Such monogenic diseases include the Immune dysregulation, PolyEndocrino-pathy X-linked syndrome (IPEX) and autoimmune polyendocrine syndrome type-1 (APS1). Here we discuss a provocative new finding in APS1 that also suggests links between genetic, paraneoplastic and acquired idiopathic autoimmunity.
Monogenic defects have revealed secrets about natural tolerance to peripheral tissue-restricted antigens (TRAgs). For instance, mutations in FOXP3, the master transcription factor in regulatory T-cells (T-reg), lead to the devastating autoimmune syndrome IPEX [1,2]. Though variable, it typically manifests at/soon after birth with both autoimmune and immunodeficiency disorders, including diabetes, severe enteropathy, hypothyroidism and/or skin disorders, and recurrent viral, bacterial and yeast infections (Table 1). Both the patients and _FoxP3_- deficient mice usually die very young [1,2]. This precocious onset (relative to APS1) and grave prognosis suggest to us that many cells that should normally become T-regs must instead ‘flip’ into becoming actively auto-aggressive in IPEX, rather than merely failing to regulate; indeed, in mouse models, some apparently convert to pathogenic TH2 or TH17 profiles [3,4].
Table 1.
The tissues targeted, and the hierarchies of cytokines recognized, by autoantibodies, in autoimmune syndromes.
| ← | Autoantibodies: | → | ||||
|---|---|---|---|---|---|---|
| Associated infections | ← | anti-cytokine NAAbs | → | |||
| Syndrome | Defect(s): | Target (s) | organ-specific | ← | % seropositive against: | → |
| MG/thymoma | cTEC neoplasm | motor endplates, BM progenitors; | CMC* (rare) | AChR; titin | 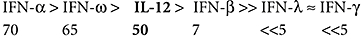 |
|
| APS1 | ↓expression of some TRAgs by mTECs: altered APC activity | adr, parathyr, skin, nails, teeth, gut, liver, gonads, pancreatic β cells, thyroid; | CMC (∼95%) | P450 cytochromes, GAD |  |
|
| IPEX | ↓FOXP3 expression by T-regs | panc β, thyroid, gut, skin, haemopoietic lineages; | viral, bacterial, yeast | P450 cytochromes et al. | demand to be tested in IPEX |
There are interesting clinical parallels and contrasts with APS1 (Table 1). Caused by mutations in the AutoImmune Regulator (AIRE; see below), it is also rare, but most intensively studied in Nordic populations [5–7]. Slightly less drastic, its effects are even more wide-ranging, protracted and, above all, highly variable [5]. By definition, the patients have at least two of the diagnostic ‘APS1 triad’ of chronic mucocutaneous candidiasis (CMC), adreno-cortical failure and hypoparathyroidism [5,6]. Onset is usually with CMC in infancy/childhood; the autoimmunity subsequently targets ectodermal and endocrine tissues (Table 1) in variable combinations. Patients with any of the associated endocrine disorders may develop the corresponding autoantibodies several years before each is diagnosed [5–7]. Many features are unexplained, including the CMC-susceptibility [8], the autoimmune focus on endocrine tissues/its hierarchy of unusual preferences/their timing [5].
In nearly all APS1 patients (>90%), mutations are detected in both alleles encoding AIRE, a zinc finger protein showing many hallmarks of transcriptional regulators [6]. Most abundant in medullary thymic epithelial cells (mTECs) [6,9–13], wild-type AIRE governs the focal expression of one subset of TRAgs [9–15], apparently in small clonelets of mTECs [9], whose development depends on the alternative NF-κB pathway [16,17]. Expression of different TRAg subsets is presumably controlled by other analogous unidentified factors [9,10,12–16]. This thymic TRAg expression normally favours deletion of potentially auto-aggressive T-cells as they mature; that deletion is clearly defective in _Aire_−/− mice [9–14] and presumably also in _AIRE_-mutant APS1 patients. Though expressed focally, TRAgs might globally censor all maturing T-cells if: (a) roving dendritic cells (DCs) take-up TRAgs from mTECs and present their epitopes to any T-cells they encounter whilst wandering through the medulla [9]. Importantly, wild-type AIRE is also expressed, though at low levels, in mononuclear/DCs [6,9,13], in both the periphery and the thymus. Indeed, it apparently also has wide-ranging influences on antigen-presenting activity [13,18–20]; we suggest that these normally create a tolerogenic ‘climate’, especially in the thymic medulla, and that some of them concomitantly affect handling of Candida in the periphery [8]; (b) TRAgs induce ‘dominant’ tolerance by favouring selection of FOXP3+T-reg, as is now emerging both in humans [21,22] and even in _Aire_−/− mice [23]. Notably, these mice only replicate the human APS1 disease partially. Their autoimmune manifestations differ considerably [10,13,24], and also vary with the genetic background [13], which may yet prove to affect Candida susceptibility.
Quantitative influences can be crucial in predisposing to sporadic autoimmune diseases, even from two- to four-fold changes in thymic expression levels of such TRAgs as insulin [25,26], of AIRE itself [27], and also of IL-2, on which the selection of T-reg critically depends [28]. Indeed, many ‘Aire-dependent’ TRAgs are still expressed in _Aire_−/− mice, though at reduced levels [6,9,10,13]. There is much discussion about how AIRE normally acts [6,9–13], but very little about how/where autoimmune responses are initiated in APS1; here we discuss unexpected new clues.
II.Involvement of type-I interferons
These novel clues particularly involve type-I interferons (IFNs). Secreted early in response to viral and some bacterial stimuli, these cytokines are front-line defenders against infections [29–32] (Table 2). They are also key linkers between innate and adaptive immune responses, helping to activate DC and other antigen-presenting cells (APCs), as well as favouring specific TH1 T-cell responses in humans [32]. They are produced at low levels by many cell types in response to diverse infections, but at much higher levels by plasmacytoid DCs (PDCs) [33] and other APCs when stimulated via various Toll-like receptors (TLRs) [34–36], and possibly by endogenous stimuli (see section Vc).
Table 2.
Interferon types and subtypes.
| IFN | Homology with IFN-α con | Producer cell type(s) | Receptor | ← | Actions | → |
|---|---|---|---|---|---|---|
| Inhibition of | Promotion of | |||||
| Type I: | IFN-α (12 subtypes) | >80% | Any: esp PDC | IFN-αR1/ IFN-αR2 | Viral replication, Tumour growth, Cell proliferation | Inflammation, MHC-class I and FcR expression |
| IFN-ω | ∼60% | … … … … | DC maturation, macrophage activation | |||
| IFN-β | ∼30% | Any; esp fibroblasts | … … … … | TH development, antibody responses | ||
| Type III: | IFN-λ | 12% | PBMC: esp PDC | IFN-LR/ IL-10R2 | ← As type I but generally weaker [29–32] → | |
| Type II: | IFN-γ | 10% | TH1, cytotoxic T and NK cells | IFN-γR1/ IFN-γR2 | Viral replication | Inflammation, DC and macrophage activation ↑ expression of MHC complex and adhesion mols |
Type I IFNs are implicated generally in autoimmunization by assorted apparently confusing findings [35–46], and especially in SLE (Box 1; see Section VII). Indeed, several autoimmune disorders may follow prolonged anti-viral therapy with recombinant IFN-α2, (e.g. for hepatitis C) (Box 1). Possibly this causes immune dysregulation and unmasks pre-existing autoimmune tendencies [38]. Such treatments may also evoke neutralizing autoantibodies (NAAbs) that block IFN-α2 actions both in vitro and _in vivo_[47]. Conversely, ‘spontaneous’ anti-IFN-α antibodies have been noted in some autoimmune disorders (including SLE) [44–46], and even in pooled therapeutic human Ig preparations [48]. Evidently, type-I IFNs are significantly immunogenic.
Box 1
Findings implicating Type I IFNs in autoimmunization
- IFN-α treatment increases risk of thyroid disease, type 1 diabetes, SLE, MG, psoriasis, etc. [37,38].
- In SLE
- Predisposition by:
* high serum IFN-α levels [39];
* trisomy for type I IFN gene cluster [40];
* IFRF5 polymorphisms [41,42]. - Increased activation of PDCs [35];
- Early elevated serum IFN-α levels [35] + IFN-α signature [36,43];
- Stimulation of IFN-α production by DNA:Ab complexes [35,36];
- Neutralizing anti-IFN-α autoantibodies in occasional patients [44–46].
- Predisposition by:
- High titre neutralizing anti-IFN autoantibodies in certain autoimmune diseases (see Table 3).
Anti-IFN antibodies in thymoma patients
Our involvement grew out of our experience with IFNs [34,46,48], and with myasthenia gravis (MG) associated with thymomas [49]. These neoplasms of cortical TEC often under-express HLA-class II [49,50], and generate abundant maturing thymocytes, but in such a disorganized environment that self-tolerance induction may well be defective [50], especially as AIRE expression is usually undetectable too [51,52]. However, whereas they rarely have other autoimmune diseases, at least 30% of all thymoma patients develop MG, which suggests to us some more selective autosensitization within these tumours [49].
Surprisingly, we found broadly-reactive NAAbs against the IFN-αs, usually at high titres, in ∼70% of these patients [46] (Tables 1 and 3). Most sera neutralize all 12 IFN-α subtypes and the antigenically distinct IFN-ω, and ∼7% of them the distantly related IFN-β too (Table 1). They also bind immobilized IFNs strongly/specifically in ELISAs, and are mainly IgG1. Already elevated at diagnosis, titres consistently remain high for years afterwards. They change surprisingly little after thymomas are removed, despite concomitant immunosuppressive drug therapy for the MG [46] (Section Va).
Table 3.
Neutralizing anti-cytokine autoantibodies in patient groups.
| Patient subgroup | Autoantibodies vs→ | AChR | ←IFN-α/ω | NAAb→ | IL-12 | Reference |
|---|---|---|---|---|---|---|
| (n) | prev | prev | max titre | prev | ||
| Thymoma: no MG | (21) | 5–20%§ | ∼50% | 0·3 × 10−6 | ∼25% | [46] |
| MG/thymoma | (118) | 100% | 64% | 0·5 × 10−6 | 52% | |
| MG/no thymoma* (late-onset) | (28) | 100% | ∼30% | 0·2 × 10−6 | ∼20% | |
| Other MG subgroups* | (49) | ∼70% | 0% | – | ∼5% | |
| Other autoimmune disorders | – | v. rare; <10% in SLE | v. rare | |||
| APS1† | (127) | –¶ | 100% | 4 × 10−6 | <5% | [53] |
| Non-syndromic†,‡ APS1 triad members | (16) | n.d. | <10% | – | <10% | |
| APS2 | (9) | n.d. | <10% | – | <10% |
Clearly, these NAAbs must be evoked by cell type(s) producing a wide range of native type-I IFNs. Interestingly, ∼50% of MG/thymoma patients independently have NAAbs against IL-12 (Tables 1 and 3), some of which recognize the p40 subunit it shares with IL-23. Although these also proved to inhibit TH1 polarization of uncommitted T-cells _in vitro_[54], intractable infections are surprisingly rare even in patients taking corticosteroids (Table 1 legend), though other type I IFN-deficiencies predispose to Herpes simplex virus (HSV) infections [55]. We almost never find NAAbs against numerous other cytokines (Table 1 legend), although anti-IFN-γ NAAbs have been noted in totally separate patients with atypical mycobacterial infections [56].
These anti-IFN-α responses appear tightly restricted to patients with MG plus thymoma, or thymoma or late-onset MG alone (Table 3). Otherwise, NAAbs proved to be very rare after various infections, and in a range of neoplastic and other autoimmune diseases, with some exceptions in SLE [44,46]. Their very similar serology may hint that some late-onset MG patients previously had occult thymomas that were subsequently rejected by anti-tumour autoimmune reactions, a rare clue in this puzzling subgroup.
Although these anti-IFN NAAbs show very few clinical correlations, even with the (highly variable) thymoma histology [46,51], titres against both IFN-α and IL-12 usually rise strikingly when these tumours recur [46,54]. Cells cultured from them spontaneously produce NAAbs against IFN-α and/or IL-12, but not against AChR [54]. To us, that again implies selective boosting by cell type(s) expressing native IFN-α (or IL-12) in thymomas, perhaps macrophages or DCs, which are present regardless of the exact histology (see Section Va).
III.New clues in APS1
Recalling that occasional MG/thymoma patients also develop CMC, we tested for anti-cytokine antibodies in APS1, in Professor Perheentupa's classic collection of ∼80 Finnish patients [5] and in 17 Norwegians [53]. We immediately found even higher-titre NAAbs against IFN-α and IFN-ω[53] (Tables 1 and 3). Moreover, they were present: (a) in 100% of the patients with mutations in both AIRE alleles, whatever the exact change [53,57], even in one family with a dominant negative G228W AIRE mutation [58] (A Meloni et al., in revision), but not in unaffected relatives; (b) in the earliest to the last sample in almost every patient tested (over decades), titres reaching 1:4 × 106 in one 3·3-year-old; (c) regardless of the exact clinical phenotype. They preceded each member of the diagnostic APS1 triad and/or the other manifestations in every (unusual) informative case, so they were clearly not a result of any one of them. (d) Their prevalences and titres were even higher than in MG/thymoma patients. While a few sera were largely specific for IFN-ω, most neutralized all 12 subtypes of IFN-α (Tables 1 and 3); also IFN-β in ∼20% of cases (at lower titres), and even, in around two-thirds of these, the distantly related type-III IFN-λ1 (alias IL-29). Much less obvious in MG/thymoma patients, such predilections for IFN-ω, or IFN-λ1, seem remarkable in view of their modest or weak homology with the IFN-αs (Table 2).
Clearly, the hierarchy of preferences differs between these syndromes although both involve the thymus (Table 1). Indeed, like several other cytokines, IL-12 (and IFN-γ) were recognized rarely in APS1 [53] (Table 1 legend).
(e) These NAAbs were highly restricted to APS1; they were undetectable in patients with each of its component diseases in isolation (Table 3), implying distinct pathways in them.
IV.Implications for clinicians
(a)The diagnostic criteria for APS1 – ripe for review [57–59]
These early, persistent, highly prevalent NAAbs against IFN-ω have >95% diagnostic specificity and sensitivity for APS1, especially in early/atypical cases without the full triad, or even with <2 AIRE mutations detected [53,57; Meloni et al., in revision]. Much simpler and cheaper than detecting AIRE mutations, the assays for these NAAbs are robust and readily applicable in reference microbiology laboratories. We suggest that they might also have prognostic value if they appear before onset of APS1, e.g. in siblings of known APS1 patients followed prospectively. Analogous studies in type-1 diabetes have led to predictive algorithms [60].
(b)For protection by type I IFNs against infections
Because CMC is so rare in MG/ thymoma patients, we doubt that these NAAbs predispose to it, at least in adults. They could, instead, be markers of upstream defect(s) in AIRE signalling in APC [8] that cause ineffective handling of Candida. In addition, recent reports implicate type I IFNs (plus IL-27) in regulating production of IL-17 by T-cells [61]. IL-17 is apparently a two-edged sword. While it can protect against infections including CMC [62], unbridled TH17 responses can reportedly antagonize both protection by TH1s and neutrophil killing of yeasts [63]. If so, the NAAbs in APS1 children might predispose to CMC by thus dysregulating TH17 reactivity; indeed, that might also promote autoimmune responses or damage.
More broadly, other infections seem surprisingly rare in APS1, as also in MG/thymoma patients [46,54] (Table 1 legend). However, reports of recurrent HSV infections in one APS1 patient [64], and of HSV encephalitis in genetic type I IFN deficiencies [55], warrant further scrutiny in APS1. We suspect that there is compensation by IFN-β, which is rarely neutralized [46,53]; also that type I IFNs act locally in APS1, e.g. in mucosal sites, before they encounter the NAAbs. These can nevertheless neutralize systemically; in preliminary experiments, the patients' blood APC show reduced expression of several IFN-dependent genes [65], presumably reflecting prior effects of anti-IFN NAAbs.
(c)For therapy
The involvement of the thymus in APS1 pathogenesis hypothesized in Section VI could have one important practical consequence. Early thymectomy should prevent at least some later-appearing autoimmune manifestations if our thinking is correct (possibly even if not). In MG, by contrast, thymomas are removed too late to affect the myasthenia.
V.Implications for autoimmunizing mechanisms
(a)Potential thymic connections
Spontaneous heritable antibody responses are rare, even in inbred mice. Amidst such clinical, genetic and serological diversity in APS1, the heritability, high penetrance, precocity and persistence of the anti-IFN autoantibodies in humans are doubly remarkable. At the very least, they must be markers of crucial early event(s); IFNs could conceivably be involved directly in pathogenesis as well (or even in normal thymic tolerogenesis). In view of the provocative parallels with MG/thymoma, we propose unifying central autoimmunization mechanisms involving DCs activated in aberrant thymic environments (see Section VI).
Pursuing these ideas, we double-labelled normal infant thymus sections for IFN-α and the type-I/type-III IFN-inducible protein MxA [66]. We found numerous IFN-α-producing cells in both cortex and medulla [see also 67], which all appeared to be CD68+ macrophages or DCs and not TECs [53]. However, MxA expression was negligible in the cortex (where type I IFNs might block thymopoiesis) [68]. By contrast, it was abundant in the medulla, further implying secretion of type-I/type-III IFNs physiologically, and therefore some endogenous stimulation (see Section Vc).
In sections of MG/thymomas, we found numerous IFN-α+ macrophages (CD68+) and some CD68+DCs [69]. The nearby MxA-labelling again implies local IFN secretion. However, we could not pinpoint any obvious autoimmunizing micro-environment(s), but these might nonetheless be transient (as in psoriasis) [70], or focal, involving elusive/ minority cell type(s) (e.g. killer DCs) [71]. We suggest that using mAbs specific for IFN-ω, IFN-α8 or IL-12 might be more informative; so should scrutiny of any available APS1 thymi.
Testing reactive tonsils as controls did reveal an explanation for the striking persistence of these NAAbs. We noted numerous IFN-α+macrophages in the germinal centres (GC). The clear MxA expression by other adjacent GC cells again indicates local secretion of IFNs [53], which thus must normally be available to re-stimulate/ perpetuate any NAAb responses once initiated (e.g. in thymomas).
Curiously, AIRE expression is almost undetectable in >90% of all thymomas [51,52]. Paradoxically, however, APS1-type diseases, and autoantibodies, are generally rare in thymoma patients [51]; the one striking exception is the anti-IFN response. Conversely, MG has not been reported in APS1 patients, even though expression by normal human mTECs of one key muscle TRAg is apparently AIRE-dependent [27]. If a thymoma merely fails to tolerize the numerous T-cells it generates and exports, those specific for other TRAgs must be restrained subsequently, possibly by their AIRE-controlled expression in lymph nodes [72]. However, the strong bias towards MG in such patients makes us suspect active selection/autoimmunization against muscle targets within thymomas. So does the autoimmunization against muscle, IFNs and IL-12 in patients with non-thymopoietic thymomas [46,49,54].
(b)Potential implication of DCs
Dendritic cells are the producers par excellence of the wide range of native type-I IFNs recognized in APS1 and MG/thymoma patients. They are also crucial agents provocateurs in sporadic autoimmune disorders, including human SLE [35,36], psoriasis [70], giant cell arteritis [72] and the diabetes in NOD mice [74]. However, any contributions they make to the anti-IFN responses must be highly selective, as we find no such NAAbs in juvenile diabetes [46] or giant cell arteritis patients [69]. The crucial difference may be that these diseases do not primarily involve the thymus, where DCs and PDCs can be generated from common pre-T/NK/DC progenitors [68]. We also suggest that these might well behave abnormally in thymomas, where thymopoiesis is often grossly aberrant [49].
(c)Possible danger signals
Viral or bacterial infections seem unlikely provoking factors in view of: the strong connections with the thymus/thymoma, both apparently sterile environments [67]; our failure to find any clear environmental effects on APS1 (A. Meloni et al., in revision); the very early onset of IPEX (even in utero); and the high prevalence of model ‘SLE’ even in germ-free mice [75]. As summarized in Table 4, a wide variety of endogenous stimuli, especially nucleic acids derived from dying cells [35,81,87,90], can evoke type I IFN secretion by DCs via various TLR-dependent and -independent signalling pathways [29,34,35,84–86,91], particularly when directed to the correct cellular compartments (e.g. endosomes).
Table 4.
Potential endogenous IFN-α-inducing danger signals in APS1 thymus and/or MG/thymomas.
| Source of signal | Signal recognized via: – | Further evidence implicating such signals in autoimmunization |
|---|---|---|
| Dying/apoptotic cells: | ||
| Systemic | C1q | •C1q is an autoantibody target in SLE [76] so are SR-A and MARCO [77] |
| Scavenger receptors (SR-A and MARCO) | ||
| In tumours | Involves calreticulin [78] | •Some tumours regularly autoimmunize, e.g. SCLC, thymoma [49] |
| •IFN-α enhances response [79] | ||
| In germinal centres (GC) | MFG-E8 | •Autoantibody-mediated glomerulonephritis in MFG-E8−/− mice [80] |
| •Some SLE patients' GC contain excess apoptotic cells [81] | ||
| Autologous nucleic acids: | ||
| RNAs/RNPs [29,35,36,81] | TLR-7 [82]; TLR8 | •TLR7 gene duplication biases to anti-nucleolar autoimmunity [83,84] |
| DNA [29,35] | TLR9 [82] | •DNAs are targets of autoantibodies in SLE |
| TLR-independent [85,86] | •Impaired DNA degradation predisposes to autoimmunity [87] | |
| RNA/DNA hybrids [87] | Not known | •RNAse 2 defect in AGS → IFN-α over-production [88]→ SLE-like vasculitis [88] |
| DNA- or RNA-containing immune complexes | TLR9 or TLR7 [82] | •Induce IFN-α over-production by PDC in SLE [35,36] |
| •PDC over-activity in SLE [35,36] | ||
| intracellular DNA or RNA | RIG I and MDA-5 | •Induce IFN-α over-production by mDC [89] |
We propose that, rather than increasing normal type I IFN secretion, AIRE defects in thymus/thymoma cause: (i) inappropriate handling concomitantly of dead cell derivatives and of Candida in the periphery; (ii) the well-known APC over-activation [9,19,20,92], together leading to (iii) enhanced autoimmunogenicity of the IFN-producing cells and perhaps also of TRAgs from _AIRE_-mutant mTECs as they apoptose physiologically [93], thanks to the newly recognized caspase-recruitment CARD domain in AIRE [94]. Collectively, (i–iii) create a dangerous ‘climate’ where autoantigens are likelier to autoimmunize than tolerize (Fig. 1). That seems plausible in the APS1 thymus, in thymomas, and in other autoimmunizing neoplasms where even apoptotic death can be dangerous [78,91], particularly in the presence of IFN-α[79]; apparently also in GC, where macrophages are needed to dispose of dying cells/prevent SLE-like disorders [80]. Among other corroborative evidence (Table 4), impaired degradation of DNA or RNA/DNA hybrids can predispose to autoimmunity [87,88]. Intriguingly also, in DNAse II−/− mice, IFN-β released by overloaded macrophages causes defects in both erythro- [95] and thymo-poiesis [68,96], further parallels with other unexplained thymoma associations [49].
Fig. 1.
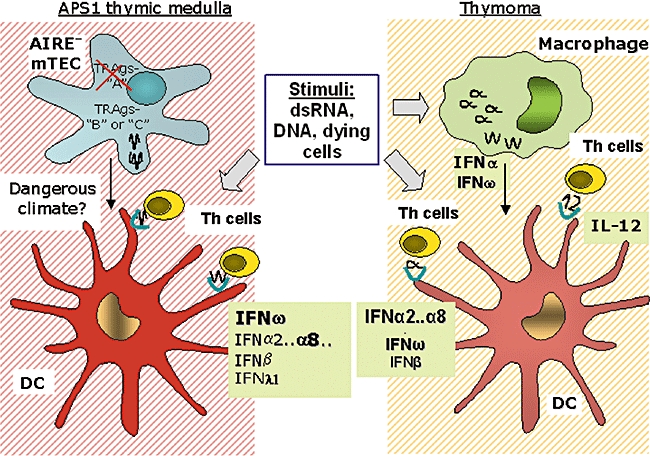
Proposed scenarios for central autoimmunization: parallels between APS1 and thymomas. In the APS1 thymus, we propose that: together with undegraded RNA,DNA or RNA/DNA hybrids, defects in AIRE/ APC activation change the ‘climate’ from tolerogenic to dangerous (symbolized by shaded backgrounds); locally abundant IFNs (especially IFN-ω) then start to autoimmunize (against themselves); so (later) do AIRE-down-regulated or -independent TRAgs (subsets B or C) that are still produced by medullary TECs but then perhaps cross-presented by over-efficient DCs (red). In thymomas, cell death and AIRE-deficiency create a similarly dangerous climate. Type I IFNs secreted by macrophages are likewise cross-presented by DCs, as are some of the muscle and neuronal autoantigens known to be expressed by the TECs [100; not shown here]. Some of the DCs also autoimmunize against IL-12 that they themselves produce.
VI.‘Central’ autoimmunization by subsets of TRAgs in APS1?
Many researchers tacitly assume that, when AIRE is defective, autoimmunization is the default option and proceeds automatically/‘stochastically’[97], requiring no further explanation. That is challenged by several characteristics of APS1: (a) the very variable onset-age, usually substantially later than in IPEX; (b) the even greater variability in targeting of different organs/its relative timing, which partly reflects the patient's HLA type [5,6]; (c) the consistent focus on the parathyroids (and adrenals), unusual targets of sporadic autoimmunity; (d) the anti-IFN responses, which also seem much too strong/early/omni-present to be ‘stochastic’; and (e) their apparent absence in non-syndromic parathyroid or adrenal autoimmunity, which argues for some entirely different, more generic, central (thymic) autoimmunizing process in APS1, which seems much neater than an assortment of separate mechanisms in each target organ.
The primary targets in APS1 are not yet known, nor whether they are normally (a) up- or (b) down-regulated by AIRE or (c) AIRE-independent. In general, many so-called ‘Aire-dependent’ (subset A) TRAgs are still expressed, but at reduced levels, in the _Aire_−/− thymus [9,10,13], while insulin and others are undetectable [13–15]. Whereas dominant auto-reactivity has been reported against two clearly Aire-independent antigens (a pancreatic enzyme [15] and α-fodrin [18]), responses against one retinal TRAg clearly do not depend on its expression in the _Aire_−/− thymus [14]. We suggest that autoimmunization takes place centrally against dominant TRAgs and peripherally against others.
Proposed scenarios for central autoimmunization: parallels between APS1 and thymomas
To us, these close parallels between APS1 and thymoma implicate thymic aberrations in autoimmunization. Indeed, we propose that: thymic expression of self-antigens in the absence of wild-type AIRE is highly dangerous; AIRE-down-regulated or AIRE-independent TRAgs (subsets B or C) are still available in APS1 and become primary targets of this central initiating mechanism (Fig. 1); in the normal thymus (not shown), wild-type AIRE not only up- or down-regulates subset-A or -B TRAgs but also creates a tolerogenic climate, via its effects on DC behaviour [9,18–20]; and in the APS1 thymus (Fig. 1), by contrast, products of physiological cell death become ‘dangerous’ because AIRE-deficient APCs over-react to them, and the climate ‘flips’ so that they autoimmunize T-cells instead of tolerizing them. These T-cells focus initially on the type-I/III IFNs that are normally available locally, and can enhance the danger [79,98], but then on any subset-B or -C TRAgs (from the mTECs). That must take more time than the direct attack we propose in IPEX; in thymomas, excessive cell death in the absence of AIRE has similar consequences. However: (i) the IFN-secreting cells may be macrophages, but (ii) the cells that (cross-)prime against IFNs might nonetheless be DCs; and (iii) some DCs independently autoimmunize against the IL-12 that they themselves produce [99].
If there are, indeed, parallel mechanisms of autoimmunization in APS1 and MG/thymomas, why is the outcome so different? We propose that it reflects partly their childhood- versus adult-onset, and partly the autoantigens available. While these evidently include type I IFNs in both cases, thymoma epithelial cells are mainly cortical; though also AIRE-negative [51,52], they are apparently biased towards expressing muscle and neuronal autoantigens [100], whereas the mTECs in APS1 may instead have endocrine leanings.
VII.Further parallels in the periphery?
Parallel mechanisms could operate in peripheral lymph nodes. Indeed, their recently reported Aire-dependent expression of some TRAgs/presentation to CD8+ T-cells [72] could be relevant in SLE, though in a very different context. Much evidence there again strongly implicates type I IFNs in both initiating and amplifying the autoimmune responses [35,36,84] (Box 1). We also find NAAbs that prefer IFN-α8 in occasional SLE patients [46]. Remarkably, assorted auto-antigens recognized in SLE not only congregate in apoptotic cell blebs but also have clip-sites for granzyme-B; such clipping could create neo-epitopes to which there is no natural T-cell tolerance and so favour autoimmunization [101]. Intriguingly, we find one plausible clip-site, in IFN-α8, close to an important serological epitope (A. Meager, D. Isenberg, in preparation). Cytotoxicity might seem unexpected in SLE: it could be involved at early stages via the granzyme B expressed by some DCs [71,102], and later, during epitope spreading, through the activation of granzyme B+ cytotoxic T-cells during SLE flares [103].
In conclusion, these parallels, combined with recent evidence from idiopathic auto-immune disorders [35,36,70,73], and from the potential involvement of IFN-α8/Granzyme-B in SLE, together implicate DCs strongly in igniting autoimmune responses, and type-I IFNs, with their adjuvant actions, in fanning the flames, e.g. by promoting DC and plasma cell maturation [35,36,79,98]. They may also help to explain other puzzling associations (e.g. bone marrow aplasias with thymoma). Finally, the striking species differences in actions of some key players (e.g. type-I IFNs [32]) as well as AIRE [24]) demand continuing studies in humans as well as mice (Table 5).
Table 5.
Outstanding questions.
| 1. | Does AIRE normally regulate expression of type I or III IFNs in DC (or even mTEC)? |
|---|---|
| 2. | Are the initiating abnormalities in APS1 in the thymus, lymph nodes or target tissues? |
| 3. | Do these involve APC in either location? If so, do they concomitantly affect the disposal of Candida or even of host RNAs, RNPs, DNAs or RNA/DNA hybrids? |
| 4. | Are the hierarchies of IFNs recognized by these autoantibodies clues to the cell types that produce them, to the activating stimuli or merely to their immunogenicity? |
| 5. | Do type I IFNs promote autoimmunization in APS1? |
| 6. | Is there a prodromal phase of DC over-activation/innate immune responses in APS1? If so, what are the stimuli, and do they influence the onset-age? |
| 7. | Are AIRE-mutant mTEC biased towards expressing endocrine or ectodermal TRAgs? |
| 8. | Might early thymectomy prevent the later appearance of new autoimmune features in APS1? |
| 9. | Are any of the above abnormalities also seen in thymomas, or in IPEX? |
| 10. | Could aberrant type I IFN production explain the defective erythro- or thymo-poiesis that sometimes associates with certain thymoma types? |
| 11. | Do the patients' anti-IFN NAAbs affect the balance between TH1, TH2 and TH-17 responses? |
| 12. | Do any Aire-mutant mouse strains show elevated anti-type I IFN antibodies or _Candida_-susceptibility? |
Acknowledgments
We are extremely grateful to the patients for their samples, and for the pioneering work of Professors J. Perheentupa, E. Husebye, K. Krohn and F. Cetani, and numerous other colleagues with whom it is a delight to collaborate and exchange ideas.
References
- 1.Ochs H D, Ziegler S F, Torgerson TR. Foxp3 acts as a rheostat of the immune response. Immunol Revs. 2005;203:156–64. doi: 10.1111/j.0105-2896.2005.00231.x. [DOI] [PubMed] [Google Scholar]
- 2.Wildin RS, Freitas A. IPEX and FOXP3: clinical and research perspectives. J Autoimmun. 2005;25(Suppl):56–62. doi: 10.1016/j.jaut.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 3.Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature. 2007;445:766–70. doi: 10.1038/nature05479. [DOI] [PubMed] [Google Scholar]
- 4.Zheng Y, Rudensky AY. Foxp3 in control of the regulatory T cell lineage. Nat Immunol. 2007;8:457–62. doi: 10.1038/ni1455. [DOI] [PubMed] [Google Scholar]
- 5.Perheentupa J. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J Clin Endocrinol Metab. 2006;91:2843–50. doi: 10.1210/jc.2005-2611. [DOI] [PubMed] [Google Scholar]
- 6.Peterson P, Uibo AR, Kampe O. Polyendocrine syndromes. In: Rose N, Mackay IR, editors. The autoimmune diseases. 4. Amsterdam: Elsevier, Academic press; 2007. pp. 515–26. [Google Scholar]
- 7.Söderbergh A, Myhre AG, Ekwall O, et al. Prevalence and clinical association of 10 defined autoantibodies in autoimmune polyendocrine syndrome type 1. J Clin Endocrinol Metab. 2004;89:557–62. doi: 10.1210/jc.2003-030279. [DOI] [PubMed] [Google Scholar]
- 8.Brännström J, Hässler S, Peltonen L, Herrmann B, Winqvist O. Defect internalization and tyrosine kinase activation in Aire deficient antigen presenting cells exposed to Candida albicans antigens. Clin Immunol. 2006;121:265–73. doi: 10.1016/j.clim.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 9.Kyewski B, Klein L. A central role for central tolerance. Annu. Revs. Immunol. 2006;24:571–606. doi: 10.1146/annurev.immunol.23.021704.115601. [DOI] [PubMed] [Google Scholar]
- 10.Anderson MS, Venanzi ES, Klein L, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science. 2002;298:1395–401. doi: 10.1126/science.1075958. [DOI] [PubMed] [Google Scholar]
- 11.Liston A, Gray DH, Lesage S, et al. Gene dosage – limiting role of Aire in thymic expression, clonal deletion, and organ-specific auto-immunity. J Exp Med. 2004;200:1015–26. doi: 10.1084/jem.20040581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goodnow CC, Sprent J, Fazekas de St Groth B, Vinuesa CG. Cellular and genetic mechanisms of self-tolerance and autoimmunity. Nature. 2005;435:590–7. doi: 10.1038/nature03724. [DOI] [PubMed] [Google Scholar]
- 13.Mathis D, Benoist C. A decade of AIRE. Nat Revs Immunol. 2007;7:645–50. doi: 10.1038/nri2136. [DOI] [PubMed] [Google Scholar]
- 14.DeVoss J, Hou Y, Johannes K, et al. Spontaneous autoimmunity prevented by thymic expression of a single self-antigen. J Exp Med. 2006;203:2727–35. doi: 10.1084/jem.20061864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niki S, Oshikawa K, Mouri Y, et al. Alteration of intra-pancreatic target-organ specificity by abrogating Aire in NOD mice. J Clin Invest. 2006;116:1292–301. doi: 10.1172/JCI26971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chin RK, Zhu M, Christiansen PA, et al. Lymphotoxin pathway-directed, autoimmune regulator-independent central tolerance to arthritogenic collagen. J Immunol. 2006;177:290–7. doi: 10.4049/jimmunol.177.1.290. [DOI] [PubMed] [Google Scholar]
- 17.Rossi SW, Kim MY, Leibbrandt A, et al. RANK signals from CD4(+)3(−) inducer cells regulate development of Aire-expressing epithelial cells in the thymic medulla. J Exp Med. 2007;204:1267–72. doi: 10.1084/jem.20062497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuroda N, Mitani T, Takeda N, et al. Development of autoimmunity against transcriptionally unrepressed target antigen in the thymus of Aire-deficient mice. J Immunol. 2005;174:1862–70. doi: 10.4049/jimmunol.174.4.1862. [DOI] [PubMed] [Google Scholar]
- 19.Anderson MS, Venanzi ES, Chen Z, Berzins SP, Benoist C, Mathis D. The cellular mechanism of Aire control of T cell tolerance. Immunity. 2005;23:227–39. doi: 10.1016/j.immuni.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 20.Ramsey C, Hassler S, Marits P, et al. Increased antigen presenting cell-mediated T cell activation in mice and patients without the autoimmune regulator. Eur J Immunol. 2006;36:305–17. doi: 10.1002/eji.200535240. [DOI] [PubMed] [Google Scholar]
- 21.Ryan KR, Lawson CA, Lorenzi AR, Arkwright PD, Isaacs JD, Lilic D. CD4+CD25+ T-regulatory cells are decreased in patients with autoimmune polyendocrinopathy candidiasis ectodermal dystrophy. J All Clin Immunol. 2005;116:1158–9. doi: 10.1016/j.jaci.2005.08.036. [DOI] [PubMed] [Google Scholar]
- 22.Kekalainen E, Tuovinen H, Joensuu J, et al. A defect of regulatory T cells in patients with auto-immune polyendocrinopathy-candidiasis-ectodermal dystrophy. J Immunol. 2007;178:1208–15. doi: 10.4049/jimmunol.178.2.1208. [DOI] [PubMed] [Google Scholar]
- 23.Aschenbrenner K, D'Cruz LM, Vollmann EH, et al. Selection of Foxp3(+) regulatory T cells specific for self antigen expressed and presented by Aire(+) medullary thymic epithelial cells. Nat Immunol. 2007;8:351–8. doi: 10.1038/ni1444. [DOI] [PubMed] [Google Scholar]
- 24.Pöntynen N, Miettinen A, Arstila TP, et al. Aire- deficient mice do not develop the same profile of tissue-specific autoantibodies as APECED patients. J Autoimmun. 2006;27:96–104. doi: 10.1016/j.jaut.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 25.Vafiadis P, Bennett ST, Todd JA, et al. Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat Genet. 1997;15:289–92. doi: 10.1038/ng0397-289. [DOI] [PubMed] [Google Scholar]
- 26.Pugliese A, Zeller M, Fernandez A, et al. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat Genet. 1997;15:293–7. doi: 10.1038/ng0397-293. [DOI] [PubMed] [Google Scholar]
- 27.Giraud M, Taubert R, Vandiedonck C, et al. An IRF8-binding promoter variant and AIRE control CHRNA1 promiscuous expression in thymus. Nature. 2007;448:934–7. doi: 10.1038/nature06066. [DOI] [PubMed] [Google Scholar]
- 28.Yamanouchi J, Rainbow D, Serra P, et al. Interleukin-2 gene variation impairs regulatory T cell function and causes autoimmunity. Nat Genet. 2007;39:329–37. doi: 10.1038/ng1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Revs. 2004;202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x. [DOI] [PubMed] [Google Scholar]
- 30.Sheppard P, Kindsvogel W, Xu W, et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol. 2003;4:63–8. doi: 10.1038/ni873. [DOI] [PubMed] [Google Scholar]
- 31.Kotenko S, Gallagher G, Baurin VV, et al. IFN-λs mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- 32.Berenson LS, Ota N, Murphy KM. Issues in t-helper 1 development – resolved and unresolved. Immunol Rev. 2004;202:157–74. doi: 10.1111/j.0105-2896.2004.00208.x. [DOI] [PubMed] [Google Scholar]
- 33.Cella M, Jarrossay D, Facchetti F, et al. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med. 1999;5:919–23. doi: 10.1038/11360. [DOI] [PubMed] [Google Scholar]
- 34.Nakhaei P, Paz S, Hiscott J. Activation of interferon gene expression through Toll-like receptor-dependent and -independent pathways. In: Meager A, editor. The interferons: characterization and application. Weinheim: Wiley VCH; 2004. pp. 35–61. [Google Scholar]
- 35.Ronnblom L, Eloranta ML, Alm GV. The type I interferon system in systemic lupus erythematosus. Arthritis Rheum. 2006;54:408–20. doi: 10.1002/art.21571. [DOI] [PubMed] [Google Scholar]
- 36.Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006;25:383–92. doi: 10.1016/j.immuni.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 37.Gota C, Calabrese L. Induction of clinical autoimmune disease by therapeutic interferon alpha. Autoimmunity. 2003;36:511–18. doi: 10.1080/08916930310001605873. [DOI] [PubMed] [Google Scholar]
- 38.Borg FA, Isenberg DA. Syndromes and complications of interferon therapy. Curr Opin Rheumatol. 2007;19:61–6. doi: 10.1097/BOR.0b013e328010c547. [DOI] [PubMed] [Google Scholar]
- 39.Niewold TB, Hua J, Lehman TJ, Harley JB, Crow MK. High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun. 2007;8:492–502. doi: 10.1038/sj.gene.6364408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhuang H, Kosboth M, Lee P, et al. Lupus-like disease and high interferon levels corresponding to trisomy of the type I interferon cluster on chromosome 9p. Arthritis Rheum. 2006;54:1573–9. doi: 10.1002/art.21800. [DOI] [PubMed] [Google Scholar]
- 41.Sigurdsson S, Göring HH, Kristjansdottir G, et al. Comprehensive evaluation of the genetic variants of interferon regulatory factor 5 reveals a novel 5bp length polymorphism as strong risk factor for systemic lupus erythematosus. Hum Mol Genet. 2008;17:872–81. doi: 10.1093/hmg/ddm359. [DOI] [PubMed] [Google Scholar]
- 42.Graham RR, Kozyrev SV, Baechler EC, et al. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet. 2006;38:550–5. doi: 10.1038/ng1782. [DOI] [PubMed] [Google Scholar]
- 43.Bauer JW, Baechler EC, Petri M, et al. Elevated serum levels of interferon-regulated chemokines are biomarkers for active human systemic lupus erythematosus. PLoS Med. 2006;3:e491. doi: 10.1371/journal.pmed.0030491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Panem S, Check IJ, Henriksen D, Vilcek J. Antibodies to alpha-interferon in a patient with systemic lupus erythematosus. J Immunol. 1982;129:1–3. [PubMed] [Google Scholar]
- 45.Slavikova M, Schmeisser H, Kontsekova E, Mateicka F, Borecky L, Kontsek P. Incidence of autoantibodies against type I and type II interferons in a cohort of systemic lupus erythematosus patients in Slovakia. J Interferon Cytokine Res. 2003;23:143–7. doi: 10.1089/107999003321532475. [DOI] [PubMed] [Google Scholar]
- 46.Meager A, Wadhwa M, Dilger P, et al. Anti-cytokine autoantibodies in autoimmunity: preponderance of neutralizing autoantibodies against interferon-alpha, interferon-omega and interleukin-12 in patients with thymoma and/or myasthenia gravis. Clin Exp Immunol. 2003;132:128–36. doi: 10.1046/j.1365-2249.2003.02113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Antonelli G. In vivo development of antibody to interferons: an up-date to 1996. J Interferon Cytokine Res. 1997;17(Suppl)(1):S39–46. [PubMed] [Google Scholar]
- 48.Wadhwa M, Meager A, Dilger P, et al. Neutralizing antibodies to granulocyte-macrophage colony-stimulating factor, interleukin-1α and interferon-α but not other cytokines in human immunoglobulin preparations. Immunology. 2000;99:113–23. doi: 10.1046/j.1365-2567.2000.00949.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Willcox N, Sheppard M, Loehrer PJ. Thymic tumours and their autoimmune associations. In: Souhami RL, Tannock I, Hohenberger P, Horiot J-C, editors. Oxford textbook of oncology. 2. Oxford: Oxford University Press; 2002. pp. 2143–52. [Google Scholar]
- 50.Kadota Y, Okumura M, Miyoshi S, et al. Altered T cell development in human thymoma is related to impairment of MHC class II transactivator expression induced by interferon-gamma (IFN-gamma) Clin Exp Immunol. 2000;121:59–68. doi: 10.1046/j.1365-2249.2000.01256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ströbel P, Murumagi A, Klein R, et al. Deficiency of the autoimmune regulator AIRE in thymomas is insufficient to elicit autoimmune polyendocrinopathy syndrome type 1 (APS-1) J Pathol. 2007;211:563–71. doi: 10.1002/path.2141. [DOI] [PubMed] [Google Scholar]
- 52.Scarpino S, Di Napoli A, Stoppacciaro A, et al. Expression of autoimmune regulator gene (AIRE) and T regulatory cells in human thymomas. Clin Exp Immunol. 2007;149:504–12. doi: 10.1111/j.1365-2249.2007.03442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meager A, Visvalingam K, Peterson P, et al. Anti-interferon autoantibodies in autoimmune polyendocrinopathy syndrome type-1. PloS Med. 2006;3:e289. doi: 10.1371/journal.pmed.0030289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shiono H, Roxanis I, Zhang W, et al. Scenarios for auto-immunization of T and B cells in myasthenia gravis. Ann NY Acad Sci. 2003;998:237–56. doi: 10.1196/annals.1254.026. [DOI] [PubMed] [Google Scholar]
- 55.Conley ME. Immunodeficiency: UNC-93B gets a toll call. Trends Immunol. 2007;28:99–100. doi: 10.1016/j.it.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 56.Patel SY, Ding L, Brown MR, et al. Anti-IFN-gamma autoantibodies in disseminated non-tuberculous mycobacterial infections. J Immunol. 2005;175:4769–76. doi: 10.4049/jimmunol.175.7.4769. [DOI] [PubMed] [Google Scholar]
- 57.Wolff AS, Erichsen MM, Meager A, et al. Autoimmune polyendocrine syndrome type 1 in Norway: phenotypic variation, autoantibodies, and novel mutations in the autoimmune regulator gene. J Clin Endocrinol Metab. 2007;92:595–603. doi: 10.1210/jc.2006-1873. [DOI] [PubMed] [Google Scholar]
- 58.Cetani F, Barbesino G, Borsari S, et al. A novel mutation of the autoimmune regulator gene in an Italian kindred with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy, acting in a dominant fashion and strongly co-segregating with hypothyroid autoimmune thyroiditis. J Clin Endocrinol Metab. 2001;86:4747–52. doi: 10.1210/jcem.86.10.7884. [DOI] [PubMed] [Google Scholar]
- 59.Buzi F, Badolato R, Mazza C, et al. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome: time to review diagnostic criteria? J Clin Endocrinol Metab. 2003;88:3146–8. doi: 10.1210/jc.2002-021495. [DOI] [PubMed] [Google Scholar]
- 60.Barker JM, Barriga KJ, Yu L, et al. Prediction of autoantibody positivity and progression to type I diabetes. Diabetes autoimmunity study in the young (DAISY) J. Clin Endocrinol Metab. 2004;89:3896–902. doi: 10.1210/jc.2003-031887. [DOI] [PubMed] [Google Scholar]
- 61.Guo B, Chang EY, Cheng G. The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. J Clin Invest. 2008;118:1680–90. doi: 10.1172/JCI33342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Milner JD, Brenchley JM, Laurence A, et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature. 2008;452:773–6. doi: 10.1038/nature06764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Romani L, Zelante T, De Luca A, Fallarino F, Puccetti P. IL-17 and therapeutic kynurenines in pathogenic inflammation to fungi. J Immunol. 2008;180:5157–62. doi: 10.4049/jimmunol.180.8.5157. [DOI] [PubMed] [Google Scholar]
- 64.Nagafuchi S, Umene K, Yamanaka F, et al. Recurrent herpes simplex virus infection in a patient with auto-immune polyendocrinopathy-candidiasis-ectodermal dystrophy associated with L29P and IVS9-1G>C compound heterozygous autoimmune regulator gene mutations. J Intern Med. 2007;261:605–10. doi: 10.1111/j.1365-2796.2007.01786.x. [DOI] [PubMed] [Google Scholar]
- 65.Kisand K, Link M, Wolff AS, et al. Interferon autoantibodies associated with AIRE-deficiency decrease the expression of IFN-stimulated genes. Blood. 2008 doi: 10.1182/blood-2008-03-144634. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Flohr F, Schneider-Schaulies S, Haller O, Kochs G. The central interactive region of human MxA GTPase is involved in GTPase activation and interaction with viral target structures. FEBS Lett. 1999;463:24–8. doi: 10.1016/s0014-5793(99)01598-7. [DOI] [PubMed] [Google Scholar]
- 67.Khan NU, Gibson A, Foulis AK. The distribution of immunoreactive interferon-alpha in formalin-fixed paraffin-embedded normal human foetal and infant tissues. Immunology. 1990;71:230–5. [PMC free article] [PubMed] [Google Scholar]
- 68.Schmidlin H, Dontje W, Groot F, et al. Stimulated plasmacytoid dendritic cells impair human T-cell development. Blood. 2006;108:3792–800. doi: 10.1182/blood-2006-02-004978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Willcox N, Leite MI, Kadota Y, et al. Autoimmunizing mechanisms in thymoma and thymus. Ann N Y Acad Sci. 2008;1132:163–173. doi: 10.1196/annals.1405.021. [DOI] [PubMed] [Google Scholar]
- 70.Nestlé FO, Conrad C, Tun-Kyi A, et al. Plasmacytoid predendritic cells initiate psoriasis through interferon-{alpha} production. J Exp Med. 2005;202:135–43. doi: 10.1084/jem.20050500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chan CW, Crafton E, Fan HN, et al. Interferon-producing killer dendritic cells provide a link between innate and adaptive immunity. Nat Med. 2006;12:207–13. doi: 10.1038/nm1352. [DOI] [PubMed] [Google Scholar]
- 72.Lee JW, Epardaud M, Sun J, et al. Peripheral antigen display by lymph node stroma promotes T cell tolerance to intestinal self. Nat Immunol. 2007;8:181–90. doi: 10.1038/ni1427. [DOI] [PubMed] [Google Scholar]
- 73.Ma-Krupa W, Jeon MS, Spoerl S, Tedder TF, Goronzy JJ, Weyand CM. Activation of arterial wall dendritic cells and breakdown of self-tolerance in giant cell arteritis. J Exp Med. 2004;199:173–83. doi: 10.1084/jem.20030850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Turley S, Poirot L, Hattori M, Benoist C, Mathis D. Physiological beta cell death triggers priming of self-reactive T cells by dendritic cells in a type-1 diabetes model. J Exp Med. 2003;198:1527–37. doi: 10.1084/jem.20030966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maldonado MA, Kakkanaiah V, MacDonald GC, et al. The role of environmental antigens in the spontaneous development of autoimmunity in MRL-lpr mice. J Immunol. 1999;162:6322–30. [PubMed] [Google Scholar]
- 76.Trouw LA, Daha MR. Role of anti-C1q autoantibodies in the pathogenesis of lupus nephritis. Expert Opin Biol Ther. 2005;5:243–51. doi: 10.1517/14712598.5.2.243. [DOI] [PubMed] [Google Scholar]
- 77.Wermeling F, Chen Y, Pikkarainen T, et al. Class A scavenger receptors regulate tolerance against apoptotic cells, and autoantibodies against these receptors are predictive of systemic lupus. J Exp Med. 2007;204:2259–65. doi: 10.1084/jem.20070600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Obeid M, Tesniere A, Ghiringhelli F, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- 79.Ferrantini M, Capone I, Belardelli F. Interferon-alpha and cancer: mechanisms of action and new perspectives of clinical use. Biochimie. 2007;89:884–93. doi: 10.1016/j.biochi.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 80.Hanayama R, Tanaka M, Miyasaka K, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. 2004;304:1147–50. doi: 10.1126/science.1094359. [DOI] [PubMed] [Google Scholar]
- 81.Baumann I, Kolowos W, Voll RE, et al. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46:191–201. doi: 10.1002/1529-0131(200201)46:1<191::AID-ART10027>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 82.Barrat FJ, Meeker T, Gregorio J, et al. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med. 2005;202:1131–9. doi: 10.1084/jem.20050914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312:1669–72. doi: 10.1126/science.1124978. [DOI] [PubMed] [Google Scholar]
- 84.Baccala R, Hoebe K, Kono DH, Beutler B, Theofilopoulos AN. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat Med. 2007;13:543–51. doi: 10.1038/nm1590. [DOI] [PubMed] [Google Scholar]
- 85.Martin DA, Elkon KB. Intracellular mammalian DNA stimulates myeloid dendritic cells to produce type I IFNs predominantly through a toll-like receptor 9-independent pathway. Arthritis Rheum. 2006;54:951–62. doi: 10.1002/art.21677. [DOI] [PubMed] [Google Scholar]
- 86.Wagner H, Bauer S. All is not Toll: new pathways in DNA recognition. J Exp Med. 2006;203:265–8. doi: 10.1084/jem.20052191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nagata S. DNA degradation in development and programmed cell death. Annu Revs Immunol. 2005;23:853–75. doi: 10.1146/annurev.immunol.23.021704.115811. [DOI] [PubMed] [Google Scholar]
- 88.Crow YJ, Leitch A, Hayward BE, et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutières syndrome and mimic congenital viral brain infection. Nat Genet. 2006;38:910–6. doi: 10.1038/ng1842. [DOI] [PubMed] [Google Scholar]
- 89.Diebold SS, Massacrier C, Akira S, Paturel C, Morel Y, Reis e Sousa C. Nucleic acid agonists for Toll-like receptor 7 are defined by the presence of uridine ribonucleotides. Eur J Immunol. 2006;36:3256–67. doi: 10.1002/eji.200636617. [DOI] [PubMed] [Google Scholar]
- 90.Gallucci S, Matzinger P. Natural adjuvants: endogenous activators of dendritic cells. Nat Med. 1999;5:1249–55. doi: 10.1038/15200. [DOI] [PubMed] [Google Scholar]
- 91.Apetoh L, Ghiringhelli F, Tesniere A, et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev. 2007;220:47–59. doi: 10.1111/j.1600-065X.2007.00573.x. [DOI] [PubMed] [Google Scholar]
- 92.Sato K, Sato U, Tateishi S, et al. Aire downregulates multiple molecules that have contradicting immune-enhancing and immune-suppressive functions. Biochem Biophys Res Commun. 2004;318:935–40. doi: 10.1016/j.bbrc.2004.04.116. [DOI] [PubMed] [Google Scholar]
- 93.Gray D, Abramson J, Benoist C, Mathis D. Proliferative arrest and rapid turnover of thymic epithelial cells expressing Aire. J Exp Med. 2007;204:2521–8. doi: 10.1084/jem.20070795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ferguson BJ, Alexander CE, Rossi SW, et al. AIRE's card revealed; a new structure for central tolerance provokes transcriptional plasticity. J Biol Chem. 2008;283:1723–31. doi: 10.1074/jbc.M707211200. [DOI] [PubMed] [Google Scholar]
- 95.Yoshida H, Okabe Y, Kawane K, Fukuyama H, Nagata S. Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat Immunol. 2005;6:49–56. doi: 10.1038/ni1146. [DOI] [PubMed] [Google Scholar]
- 96.Kawane K, Fukuyama H, Yoshida H, et al. Impaired thymic development in mouse embryos deficient in apoptotic DNA degradation. Nat Immunol. 2003;4:138–44. doi: 10.1038/ni881. [DOI] [PubMed] [Google Scholar]
- 97.Goodnow CC. Multistep pathogenesis of autoimmune disease. Cell. 2007;130:25–35. doi: 10.1016/j.cell.2007.06.033. [DOI] [PubMed] [Google Scholar]
- 98.Haeryfar SM. The importance of being a pDC in antiviral immunity: the IFN mission versus Ag presentation? Trends Immunol. 2005;26:311–7. doi: 10.1016/j.it.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 99.Matsumoto M, Funami K, Tanabe M, et al. Subcellular localization of Toll-like R 3 in human DC. J Immunol. 2003;171:3154–62. doi: 10.4049/jimmunol.171.6.3154. [DOI] [PubMed] [Google Scholar]
- 100.Romi F, Bø L, Skeie GO, Myking A, Aarli JA, Gilhus NE. Titin and ryanodine receptor epitopes are expressed in cortical thymoma along with costimulatory molecules. J Neuroimmunol. 2002;128:82–9. doi: 10.1016/s0165-5728(02)00145-5. [DOI] [PubMed] [Google Scholar]
- 101.Casciola-Rosen L, Andrade F, Ulanet D, Wong WB, Rosen A. Cleavage by granzyme B is strongly predictive of autoantigen status: implications for initiation of autoimmunity. J Exp Med. 1999;190:815–26. doi: 10.1084/jem.190.6.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vermi W, Facchetti F, Rosati S, et al. Nodal and extra-nodal tumor-forming accumulation of plasmacytoid monocytes/interferon-producing cells associated with myeloid disorders. Am J Surg Pathol. 2004;28:585–95. doi: 10.1097/00000478-200405000-00004. [DOI] [PubMed] [Google Scholar]
- 103.Blanco P, Pitard V, Viallard JF, Taupin JL, Pellegrin JL, Moreau JF. Increase in activated CD8+ T lymphocytes expressing perforin and granzyme B correlates with disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2005;52:201–11. doi: 10.1002/art.20745. [DOI] [PubMed] [Google Scholar]