Targeted gene addition to a predetermined site in the human genome using a ZFN-based nicking enzyme (original) (raw)
Abstract
Zinc-finger nucleases (ZFNs) drive highly efficient genome editing by generating a site-specific DNA double-strand break (DSB) at a predetermined site in the genome. Subsequent repair of this break via the nonhomologous end-joining (NHEJ) or homology-directed repair (HDR) pathways results in targeted gene disruption or gene addition, respectively. Here, we report that ZFNs can be engineered to induce a site-specific DNA single-strand break (SSB) or nick. Using the _CCR5-_specific ZFNs as a model system, we show that introduction of a nick at this target site stimulates gene addition using a homologous donor template but fails to induce significant levels of the small insertions and deletions (indels) characteristic of repair via NHEJ. Gene addition by these _CCR5_-targeted zinc finger nickases (ZFNickases) occurs in both transformed and primary human cells at efficiencies of up to ∼1%–8%. Interestingly, ZFNickases targeting the AAVS1 “safe harbor” locus revealed similar in vitro nicking activity, a marked reduction of indels characteristic of NHEJ, but stimulated far lower levels of gene addition—suggesting that other, yet to be identified mediators of nick-induced gene targeting exist. Introduction of site-specific nicks at distinct endogenous loci provide an important tool for the study of DNA repair. Moreover, the potential for a SSB to direct repair pathway choice (i.e., HDR but not NHEJ) may prove advantageous for certain therapeutic applications such as the targeted correction of human disease-causing mutations.
Zinc-finger nucleases (ZFNs) are chimeric endonucleases created by fusing the cleavage domain from the type IIS restriction enzyme FokI to a designed zinc-finger protein (ZFP). In order to generate a DNA double strand break (DSB) at a predetermined genomic site, two engineered ZFNs must be designed to bind DNA with the appropriate orientation and spacing (i.e., on opposite sides of the DNA with 5 or 6 bp of sequence separating them) (Carroll 2008; Cathomen and Joung 2008). This architecture (analogous to that depicted in Fig. 1A) permits the two FokI cleavage domains to heterodimerize—a prerequisite for subsequent cleavage of the targeted DNA (Bitinaite et al. 1998).
Figure 1.

Mutation of the catalytic domain of a ZFN monomer generates a ZFN heterodimer with nicking activity. (A) Illustration of the ZFN heterodimer architecture for single-stranded break (SSB) generation. The FokI catalytic domain of ZFN (left, blue) is mutated to generate a catalytically inactivated form. This mutant productively heterodimerizes with the WT ZFN (right, red), resulting in cleavage of a single DNA strand. (B) Illustration of the DNA products following in vitro cleavage with the indicated WT and mutant ZFN combinations relative to the 5′ end-labeled DNA substrate. The position of the WT FokI ZFN binding site(s) is shown as a raised solid block and the anticipated cleavage events indicated. (C) Illustration of the expected digestion patterns following strand-specific nicking when resolved under nondenaturing and denaturing conditions. Strand-specificity is revealed since the cleavage site is off-center and the DNA substrate is labeled at the 5′ end only (see panel B above). (D) In vitro assessment of double-strand DNA cleavage with the D450N/WT and D467A/WT ZFN variants under nondenaturing conditions. (E) In vitro assessment of nicking activity with the D450N/WT and D467A/WT ZFN variants under denaturing conditions. The WT/WT combination provides a positive control for nicking of both strands. Numbers at the bottom of each lane indicate % of cleaved products (indicated by arrows). Numbers in parentheses shown are % cleavage activity (Ave ± SD) relative to the WT/WT combination (set as 100%).
Repair of the ZFN-induced site-specific DSB provides the molecular basis for gene disruption, gene correction, or gene addition—the different potential outcomes of ZFN-mediated genome editing (for review, see Urnov et al. 2010). Specifically, a given DSB may be repaired via either the nonhomologous end joining (NHEJ) or homology-directed repair (HDR) pathways (Kowalczykowski 2000; Sung and Klein 2006; Wyman and Kanaar 2006; Brugmans et al. 2007). HDR utilizes a homologous donor sequence as template for the conservative repair of the DNA break. Provision of a suitably designed donor DNA molecule can, therefore, specify gene addition or gene correction at the ZFN-induced DSB. NHEJ, on the other hand, simply catalyzes the rejoining of the two DNA ends—a process that can result in the deletion or insertion of nucleotides at the repair junction. DNA repair via NHEJ is, therefore, mutagenic. To date, ZFN-induced DNA repair via HDR or NHEJ has been utilized to target modifications to the genomes of numerous species (Le Provost et al. 2010) including Caenorhabditis elegans (Morton et al. 2006), Drosophila melanogaster (Bibikova et al. 2002), silkworms (Takasu et al. 2010), zebrafish (Doyon et al. 2008; Meng et al. 2008), sea urchins (Ochiai et al. 2010), Arabidopsis thaliana (Osakabe et al. 2010; Zhang et al. 2010), tobacco (Maeder et al. 2008; Cai et al. 2009; Townsend et al. 2009), corn (Shukla et al. 2009), mice (Carbery et al. 2010; Meyer et al. 2010), and rats (Geurts et al. 2009). ZFNs have also been used to efficiently engineer a diverse range of mammalian cell types (Urnov et al. 2010), including human stem cells, with subsequent retention of full potency and normal growth characteristics (Hockemeyer et al. 2009; Zou et al. 2009; Holt et al. 2010). Moreover, the therapeutic potential of ZFNs is currently under evaluation in clinical trials (clinicaltrials.gov identifiers NCT00842634, NCT01252641, NCT01044654).
Given the markedly different genome editing outcomes mediated by NHEJ and HDR, it would be desirable to have control over the choice of pathway used and thus specify the outcome of repair. In practice, for gene disruption this can already be achieved, since NHEJ is the predominant pathway in mammalian cells (Lieber 2008) and codelivery of donor DNA is unnecessary. For gene correction and addition, however, NHEJ events represent competition to the desired homology-directed outcome at the target locus. One potential strategy for enforcing DNA repair pathway bias would be the targeted introduction of a DNA single strand break (SSB) or nick. Like the DSB, a DNA SSB/nick can theoretically stimulate homology-directed repair pathways (Holliday 1964; Meselson and Radding 1975; Radding 1982). For example, SSBs induced by the bacteriophage fd gene II protein (gIIp) stimulated HDR in yeast (Galli and Schiestl 1998), while nicks generated by specific mutants of the RAG proteins—critical components of the V(D)J recombination system—are repaired via initiation of homologous recombination (Lee et al. 2004). Moreover, experiments using a nick-inducing variant of the I-Anil LAGLIDADG homing endonuclease showed that the SSB/nicks induced by these engineered enzymes are also repaired via HDR (Smith et al. 2009). Importantly, and in contrast to a DSB, a DNA nick or SSB is not a bona fide substrate for repair via the NHEJ pathway. Thus, a targeted SSB has the potential to restrict repair to the homology-directed pathway.
Here, we describe the engineering of a zinc finger nickase (ZFNickase) by mutation of a residue critical for FokI cleavage activity in one monomer of the ZFN heterodimer, thus allowing the ZFNs to heterodimerize on DNA but restricting cleavage to a single DNA strand. We show that targeting of these ZFNickases to the endogenous CCR5 locus stimulates gene correction and gene addition via HDR at frequencies of between ∼1%–8% in both transformed and primary human lines. Importantly, we show that the CCR5 ZFNickase-induced gene addition occurs without a marked increase in detectable mutagenesis by the error-prone NHEJ repair pathway. Specific outcomes of either gene addition/correction or gene disruption may, therefore, be modulated via the targeted introduction of a single- or double-strand break, respectively. Moreover, beyond the targeted event itself, elimination of DNA repair via NHEJ has the potential to eradicate mutagenesis at putative off-target cleavage sites, further improving the fidelity of genome editing.
Results
Mutation of the catalytic domain of a ZFN monomer generates a ZFN heterodimer with nicking activity
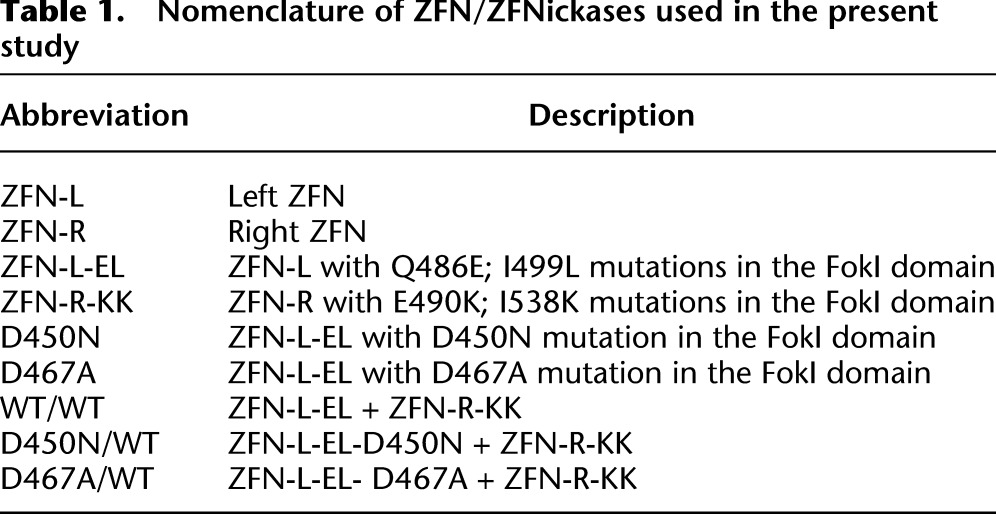
To generate ZFNs with strand-specific nicking activity, we mutated the FokI catalytic domain in one of the two ZFNs (Fig. 1A) necessary for dimerization and subsequent DNA cleavage (Bitinaite et al. 1998). Specifically, we focused on amino acids D450 and D467, both previously shown to result in catalytically inactive FokI variants when mutated (Waugh and Sauer 1993; Sanders et al. 2009). To eliminate the possibility of activity from homodimers of the catalytically active ZFN, we performed our nicking studies in the context of an obligate heterodimer ZFN architecture (Miller et al. 2007; Szczepek et al. 2007). Specifically, we used ZFN heterodimers encoding the Q486E; I499L variant partnered with the E490K; I538K variant—referred to below as EL or KK, respectively. A detailed description of the different WT and mutant ZFN combinations employed below is provided in Table 1.
Table 1.
Nomenclature of ZFN/ZFNickases used in the present study
To test the activity of ZFNs bearing D450N or D467A point mutations, digests were performed using a radiolabeled linear 292-bp PCR fragment of the CCR5 gene arranged to contain the target site for _CCR5_-specific ZFNs off center (Fig. 1B,C). Provision of in vitro synthesized _CCR5_-specific ZFNs (ZFN-L-EL and ZFN-R-KK) resulted in efficient double-strand cleavage of the template DNA into two smaller fragments (>82% cleavage efficiency) regardless of whether the reaction was resolved by PAGE under nondenaturing (Fig. 1D, WT/WT) or denaturing (Fig. 1E, WT/WT) conditions. When coupled with the WT right-hand ZFN (ZFN-R-KK), introduction of the D450N mutation into ZFN-L-EL eliminated double-strand cleavage (Fig. 1D, D450N/WT), while the D467A mutant retained very weak DNA cleavage activity (Fig. 1D, D467A/WT), resulting in a detectable activity (3.4%) in one out of four repeated experiments. To confirm strand-specific nicking activity, the same cleavage products were resolved under denaturing conditions to separate the two single-strand DNA molecules. Strikingly, both the D450N and D467A mutations largely eliminated cleavage of one of the two DNA strands, observed as the persistence of the full-length, linear single-strand template (Fig. 1E). Asymmetry of the ZFN site within the target DNA duplex revealed a digestion pattern consistent with only the WT ZFN (ZFN-R-KK) retaining cleavage activity (Fig. 1B–E). Relative to the WT/WT ZFN-mediated DNA cleavage (set as 100% here), the cleavage efficiency of D450N/WT and D467A/WT variants are 41.1 ± 10.8% and 53.2 ± 6.1% (Ave ± SD, n = 4), respectively, i.e., >80% cleavage of one of the two DNA strands (complete nicking of all double-stranded DNAs will show a read-out of 50% cleavage efficiency in this assay system). Together, these data demonstrate that elimination of cleavage activity in one half of a ZFN pair by introduction of either the D450N or D467A mutations results in the generation of a potent, strand-specific ZFNickase.
Targeted nicks stimulate homologous repeat deletion
As an initial test of nick-induced genome editing in living cells, we investigated whether a ZFNickase-induced SSB would stimulate intra-molecular recombination (homologous repeat deletion) in an integrated reporter in haploid Saccharomyces cerevisiae (Fig. 2A). The MEL1 reporter assays were performed as described (Doyon et al. 2008) using a reporter construct containing the CCR5 ZFN target sites. DNA repair of a direct or converted DSB by single-strand annealing (SSA) will result in the elimination of a sequence that interrupts and disables the MEL1 reporter gene via annealing of two flanking short direct repeats (Fig. 2A). The frequency of restoration of the secreted α-galactosidase reporter gene (MEL1) can be measured by direct sampling of the media. Induction of ZFNickase expression resulted in a significant increase in α-galactosidase activity with both the D450N/WT (40.0–75.5 mU) and D467A/WT (51.7–96.0 mU) ZFNickase combinations compared to cultures treated with control vectors (2.7–4.1 mU) (Fig. 2B). In the same experiments, the DSB-inducing WT/WT ZFN combination stimulated approximately fourfold higher levels of α-galactosidase activity (data not shown). This reduction in the levels of restored reporter gene expression is consistent with the assumed need for the nick to first be converted into a DSB during S phase DNA replication in order to initiate the SSA process (Galli and Schiestl 1998; Kuzminov 2001). These data suggest that a ZFNickase-induced SSB is sufficient to stimulate intra-molecular recombination resulting in homologous repeat deletion in yeast.
Figure 2.

Targeted nicks stimulate homologous repeat deletion. (A) Illustration of the MEL1 reporter assay. A MEL1 reporter yeast strain, carrying the CCR5 ZFN target site introduced between two overlaping fragments of the MEL1 gene (MEL and EL1, respectively), was transformed with ZFN expression plasmids for ZFN-L-EL ([*] catalytically inactive FokI domain) and ZFN-R-KK, whose expression are under the control of galactose-inducible promoter (P_GAL_). ZFN expression was induced by the addition of galactose to the growth media and a DNA break or nick generated (depending on the constructs used). The repaired MEL1 gene directs the expression of α-galactosidase that can then be measured to determine the relative efficiencies of DNA break- or nick-induced repair. (CYC1_t) Transcriptional terminator of CYC1 (cytochrome c1) gene, (HIS3) the gene encoding yeast imidazoleglycerol-phosphate dehydratase required for histidine biosynthesis, (LEU2) the gene encoding yeast beta-isopropylmalate dehydrogenase (IMDH) required for leucine biosynthesis; HIS3 and LEU2 are used for selection of transformants, (P_PGK1) promoter of the phosphoglycerate kinase 1 (PGK1) gene, (KanMX) dominant resistance module for selection of S. cerevisiae transformants against geneticin (G418) (Voth et al. 2001). (B) Induction of a targeted nick in yeast stimulates homologous repeat deletion and restoration of the reporter gene (MEL1). ZFN expression was induced for 2 to 6 h, and reporter activity was determined as previously described (Doyon et al. 2008). (C) Evaluating direct double-strand DNA cleavage activity of the ZFNickases in yeast. A haploid yeast strain harboring the CCR5 ZFN target site integrated into the HO locus was transformed with the indicated galactose-inducible ZFNickase expression plasmids (D450N/WT, D467A/WT), DSB-inducing ZFN vectors (WT/WT), or a no ZFN control (Control). Tenfold serial dilutions of each cell line were plated on minimal media containing either glucose (ZFN expression off) or galactose (ZFN expression on) and incubated for 3 d. Direct DSB-induction results in a severe reduction in yeast cell viability (compare WT/WT+Glucose with WT/WT+galactose). No loss of viability is observed with the nickases. (D) Western blot to assess the expression levels of the various ZFN variants in panel C. Aliquots of the ZFN-induced cultures were harvested after 6 h of galactose induction. Cell lysates were run on SDS-PAGE, transferred, and blotted with either an anti-FLAG monoclonal antibody (Sigma) to detect FLAG-tagged ZFN or anti-H3 monoclonal antibody (Millipore) as loading control. No significant differences in ZFN expression were observed. (E) Induction of a targeted nick in mammalian cells stimulates homologous repeat deletion mediated restoration of reporter gene expression. K562 cells were either untreated or transfected with a GFP reporter DNA plasmid (Supplemental Fig. S1) in the absence (No ZFN) or presence of the indicated ZFN expression plasmids. Cells were collected 3 d post-transfection and subjected to flow cytometry analysis after a 5-min incubation with propidium iodide (PI) to stain nonviable cells (PI+). GFP+PI− cells (gate at top left) represent the number of viable cells which have restored marker gene expression. One representative example from three independent experiments is shown.
To rule out the possibility that ZFNickase-induced MEL1 expression was a result of direct DSB induction, we exploited the fact that a single persistent DSB induced by a nuclease is lethal to haploid yeast cells (Moore and Haber 1996). The CCR5 ZFN target site was integrated into the HO locus of the haploid BY4741 strain. As previously described for the HO endonuclease (Moore and Haber 1996), inducing expression of a DSB-generating WT/WT ZFN resulted in a survival rate of ∼0.1–0.2% (Fig. 2C, WT/WT—compare glucose [ZFN expression off] and galactose [ZFN expression on] plates). This effect is the consequence of a single targeted DSB, since elimination of the CCR5 target site abolishes ZFN-induced lethality (data not shown). In contrast, yeast transformed with inducible expression constructs encoding either of the ZFNickase variants (D450N/WT or D467A/WT) grew indistinguishably from cultures treated with a control vector (Fig. 2C), despite similar ZFN expression levels for all variants tested (Fig. 2D). Thus, in the context of a living cell, a targeted DNA nick can drive an intra-molecular repair event resulting in homologous repeat deletion. Moreover, these data suggest that in yeast ZFNickases do not cause direct DSB generation with measurable efficiency.
To extend these observations to mammalian cells, we tested whether a ZFNickase-induced SSB could drive homologous repeat deletion in the human myelogenous leukemia cell line K562. In this co-transfection assay, homologous repeat deletion is measured by reconstitution of GFP expression, where the CCR5 ZFN binding sites are present between two repeated partial eGFP sequences (Supplemental Fig. S1A). In agreement with the yeast studies, we observed a significant increase in GFP+ cells in samples transfected with the ZFNickases D450N/WT (3.55%) and D467A/WT (3.40%), compared to a control sample treated with the GFP reporter alone (0.67%). Consistent with the data from yeast, ZFNickase-induced reconstitution of GFP expression in K562 cells was ∼2.5-fold less efficient than that obtained by the use of a direct DSB-inducing WT/WT ZFN pair (8.58%) (Fig. 2E). Together with the yeast data, these studies show that the CCR5 ZFNickase can stimulate homologous repeat deletion in mammalian cells via the induction of a DNA nick.
Targeted nicks promote HDR-mediated gene modification at an endogenous locus
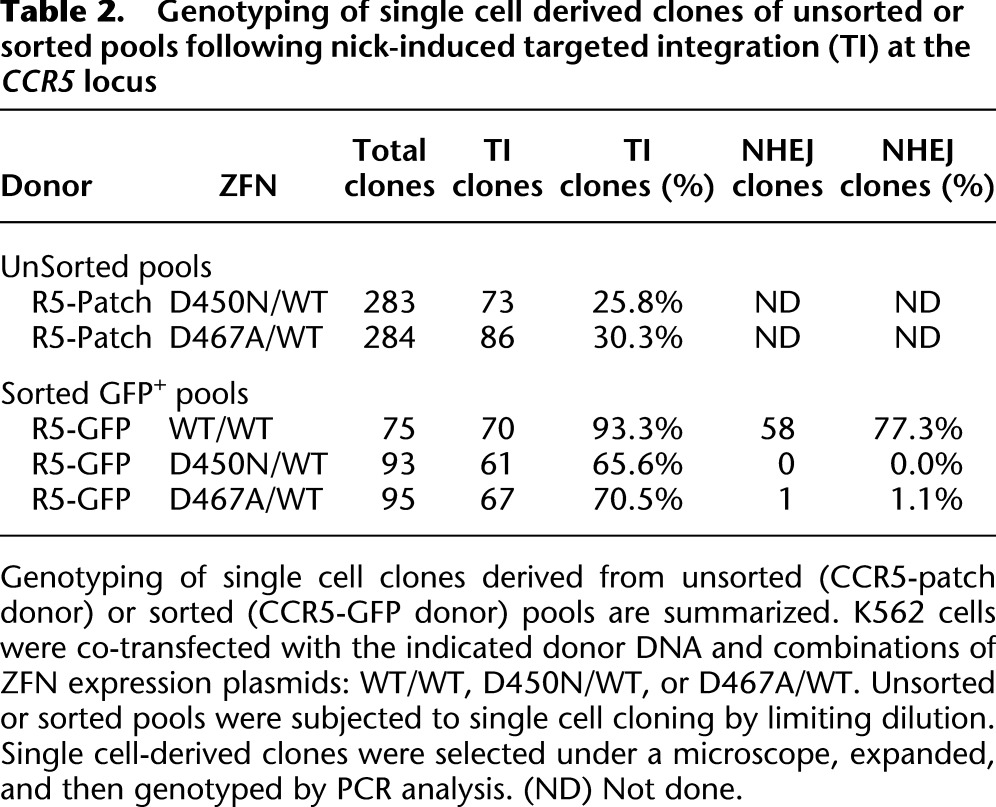
Next, we wished to evaluate whether ZFNickases could promote efficient addition of a small DNA patch (as might be performed for gene correction of a specific mutation) at a predetermined endogenous locus in mammalian cells. K562 cells were cotransfected with the _CCR5_-ZFN/ZFNickase variants (above) and a homologous donor molecule comprising a 51-bp patch of novel sequence containing a BglI site (generating a restriction fragment length polymorphism [RFLP]) flanked by homology arms identical to the sequence immediately surrounding the CCR5 ZFN target site (CCR5-patch donor) (Supplemental Fig. S2A; Fig. 3A). Cotransfection of the CCR5-patch donor with the WT/WT ZFN pair (DSB-inducing) resulted in the transfer of the RFLP to the chromosome at a frequency of 30.6% of the endogenous CCR5 alleles in the absence of selection for the desired event (Fig. 3B). Importantly, K562 cells cotransfected with the D450N/WT or D467A/WT ZFNickase pairs also led to the introduction of the RFLP to the endogenous CCR5 locus at frequencies of 7.3 and 8.0%, respectively. RFLP addition could not be detected in any of the samples treated with a single ZFN (Fig. 3B, ZFN-L-EL WT, D450N, D467A, or ZFN-R-KK). Analysis of single cell-derived clones derived by limiting dilution from the ZFNickase-treated cell pools revealed that >25% of the expanded clones were heterozygous for the RFLP (Table 2; Supplemental Fig. S2B,C)—a result consistent with the presence of at least three CCR5 alleles in the largely triploid K562 cell line (karyotype data not shown). Additional experiments (total n = 9) revealed that the D450N/WT ZFNickase is ∼sixfold less efficient than the DSB-generating WT/WT ZFN at inducing DNA patch addition into the CCR5 locus in K562 cells (4.0 ± 2.6% vs. 29.6 ± 5.8%, respectively).
Figure 3.

Targeted nicks stimulate gene addition by homology-directed repair (HDR) without inducing mutations characteristic of nonhomologous end joining (NHEJ). (A) Illustration of experimental procedure. Cells were transfected or transduced with the indicated ZFN variants in the presence of a RFLP-containing donor DNA. Genomic DNA was collected 3–10 d post-transfection or -transduction and subjected to a RFLP assay to confirm DNA repair via HDR in panel B or a Surveyor nuclease assay to detect the induction of indels characteristic of repair via NHEJ in panel C. The integration of a patch sequence introduces an additional BglI site to the CCR5 locus. Upon BglI digestion, 86-bp, 926-bp, and 1553-bp products will be generated from the patch integrated allele, whereas an 86-bp and 2519-bp product will be generated from the wild-type allele. Note that the 86-bp product is not visible (it has been run off the gel). For the Surveyor nuclease assay, two additional products (estimated sizes are ∼66 bp and 97 bp, respectively) will be generated by the induction of indels caused by NHEJ repair at the ZFN target site. The uncleaved “parental” PCR product is 163 bp. (B) K562 cells were transfected with the indicated ZFN combinations in the presence of a CCR5-patch donor (Supplemental Fig. S2A). Genomic DNA was collected 10 d post-transfection and subjected to a RFLP assay. The percentage of BglI-sensitive DNA (indicated by arrows) resulting from targeted integration of the patch sequence is indicated below each lane. (C) The samples in panel B were also analyzed for the presence of indels using the Surveyor nuclease assay. (Arrows) Specific cleavage products. Numbers at the bottom of each lane indicate the percentage of CCR5 alleles containing indels, i.e., % of Surveyor nuclease cleaved products (indicated by arrows). (D) Nick-induced targeted gene addition in primary human cells. Primary human fibroblast cells were transduced with a lenti-CCR5-patch vector (30 MOI) in the absence (No ZFN) or presence of indicated Ad5/F35 ZFN vectors (300 MOI). Cells were collected 3 d post-transduction, genomic DNA isolated, and a RFLP assay performed. The percentage of BglI-sensitive DNA (indicated by arrows) resulting from targeted integration of the patch sequence is indicated below each lane.
Table 2.
Genotyping of single cell derived clones of unsorted or sorted pools following nick-induced targeted integration (TI) at the CCR5 locus
To understand whether the CCR5 ZFNickase-induced SSBs are also repaired by the NHEJ pathway, we attempted to detect the small insertions and deletions (indels) characteristic of DSB repair via error-prone NHEJ at the ZFN target site. Analysis of the identical ZFN/ZFNickase-treated K562 cell pools (Fig. 3B) using the Surveyor nuclease assay revealed indels in 40.6% of the CCR5 alleles when the DSB-inducing WT/WT ZFN pair was used (Fig. 3C). In contrast, no NHEJ events were detected in samples treated with the D450N/WT or D467A/WT ZFNickases or any of the single ZFN constructs (assay detection limit ∼0.5%). Taken together, these results suggest that a targeted DNA nick at the CCR5 locus can be repaired via a homology-directed process (exploiting the RFLP-containing donor DNA), but that such nicks do not generate the indels characteristic of the error-prone NHEJ repair pathway.
Targeted nicks promote HDR-mediated site-specific gene addition
Next, we asked whether ZFNickases can successfully insert a larger fragment of heterologous DNA. A CCR5-GFP donor was constructed (Supplemental Fig. S3A) in which a 1.6-kb autonomous PGK promoter-driven eGFP expression cassette was flanked by homology arms identical to the sequence immediately surrounding the CCR5 ZFN target site. K562 cells were cotransfected with the CCR5-GFP donor and the indicated CCR5 ZFN or ZFNickases pairs, GFP+ cells were isolated by fluorescence-activated cell sorting (FACS), and this sorted population was used to confirm bona fide targeted gene addition via Southern blotting (Supplemental Fig. S3) and generate single cell-derived clones by limiting dilution. Analysis of all 75 clones derived from the sample treated with the WT/WT ZFNs uncovered 70 clones (93.3%) demonstrating HDR-driven targeted gene addition to the CCR5 locus (Table 2). Importantly, 58 of these clones (77.3%) also harbored CCR5 alleles with indels characteristic of repair via NHEJ. In contrast, the clones derived from cotransfection with the D450N/WT ZFNickase pair revealed no indels in any clone, yet 61 (65.6%) of the total 93 clones had undergone addition of the GFP expression cassette to the CCR5 locus (Table 2). Only one out of 95 clones (1.1%) derived from the cells treated with D467A/WT ZFNickase showed indel characteristic of repair via NHEJ at CCR5, whereas 67 clones (70.5%) exhibited target gene addition (Table 2). This may be explained by the residual DSB activity that we observed with the D467A/WT ZFNickase (Fig. 1). Together, these data demonstrate that a ZFNickase-induced SSB is sufficient to induce site-specific insertion of a gene-sized piece of DNA at the CCR5 locus. Moreover, this homology-driven gene addition process can occur in the absence of detectable repair via NHEJ.
Nick-induced gene addition is portable to primary human cells
To determine the utility of nick-induced targeted gene addition in primary human cells, we attempted to modify primary human fibroblasts, often employed as the starting cell for the generation of induced pluripotent stem cells (iPSCs). Using the _CCR5_-targeted D450N/WT pair and the CCR5-patch donor (Supplemental Fig. S2A), we observed ∼1.4% site-specific gene addition (Fig. 3D, D450N/WT) in the absence of selection for the desired events. Nick-induced targeted gene addition was markedly lower than the WT/WT DSB-inducing ZFNs (Fig. 3D, WT/WT) in this cell type, whereas site-specific gene addition was undetectable in cells treated with the CCR5-patch donor alone (Fig. 3D, No ZFN). These data suggest that the strategy of using targeted ZFNickases to mediate HDR-driven genome editing may be applicable to a variety of different cell types, including primary cells, albeit with lower efficiency.
ZFNickases-induced SSB bias repair against error-prone NHEJ
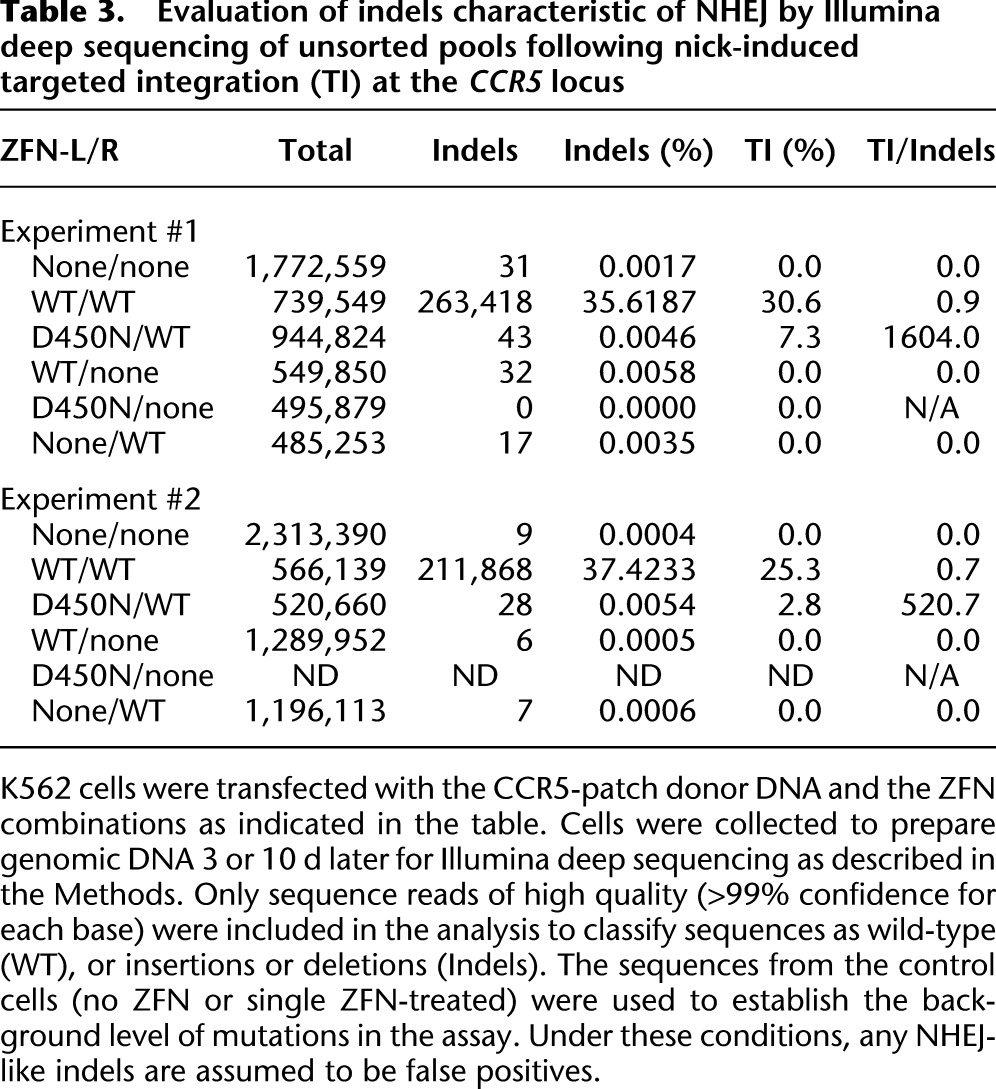
The results above suggest that a targeted nick may represent a pathway-specific stimulator of DNA repair. To confirm this result, we sought to maximize our assay sensitivity for the detection of mutations caused by NHEJ. We, therefore, performed Illumina deep sequencing of the CCR5 target locus from ZFN (WT/WT)-, ZFNickase (D450/WT)-, or control (no ZFN or single ZFN)-treated K562 cells (Table 3). In the first experiment, we obtained between 485,000 and 1.7 million high quality sequence reads (>99% confidence for each base) per sample. The WT/WT ZFN-treated sample contained 263,418 of 739,549 sequences (35.6%), with indels characteristic of repair by NHEJ (Table 3; Supplemental Table S1). In sharp contrast, treatment with the D450N/WT ZFNickase revealed just 43 of 944,824 sequences (0.0046%), with indels consistent with repair by NHEJ representing a >7800-fold reduction. The low frequency of indels detected in the D450N/WT ZFNickase-treated sample is similar to that detected in single WT ZFN monomer-treated samples (WT/none, 0.0058%; and none/WT, 0.0035%) and slightly above the background present in the non-ZFN-treated (none/none, 0.0017%) or single D450N ZFN-treated (D450N/none, 0.0000%) samples. Similar data were obtained in a second experiment in which the D450N/WT vs. single ZFN-treated sample revealed a very low frequency of indels (0.0054% vs. ∼0.0006%). Thus, the D450N/WT ZFNickase biased repair away from the error-prone NHEJ pathway, resulting in a 1600- or 520-fold preference for HDR over NHEJ (Fig. 3B; Table 3). In contrast, cells treated with the WT/WT ZFN preferentially utilized NHEJ over HDR (measured by targeted integration [TI]) for DNA repair ( Table 3, TI/Indels < 1) under similar experimental conditions. Together, these data demonstrate that a targeted nick is not a preferred substrate for repair by NHEJ. Thus, the CCR5 ZFNickases result in a stringent pathway choice enabling DNA repair to proceed preferentially via the homology-directed repair pathways.
Table 3.
Evaluation of indels characteristic of NHEJ by Illumina deep sequencing of unsorted pools following nick-induced targeted integration (TI) at the CCR5 locus
Genome-wide assessment of DSB induction following ZFNickase treatment
To further assess the fidelity of ZFNickase-mediated genome editing, we determined DSB levels genome-wide following treatment with either WT/WT ZFNs or the two different ZFNickase variants (D450N/WT or D467A/WT). We used two well-validated assays for visualizing DNA double-strand breaks that involve antibody-mediated detection of proteins associated with sites of DNA damage. Our initial studies used detection of the phosphorylated histone H2AFX (γH2AFX), a marker of DNA damage that forms foci at the sites of double-strand breaks (Rogakou et al. 1998, 1999; Stiff et al. 2004). FACS-analysis revealed significant phosphorylated histone H2AFX signal in cells treated with WT/WT ZFNs (14.70% γH2AFX+) at 2 d post-transfection (Fig. 4A, WT/WT), while the D450N/WT samples showed only 0.33% γH2AFX+ cells (Fig. 4A, D450N/WT). Slightly higher γH2AFX staining (4.34% γH2AFX+) was observed in cells treated with D467A/WT (Fig. 4A, D467A/WT), consistent with our previous observations, suggesting that the D467A mutant retains low DNA cleavage activity (Fig. 1; Table 2).
Figure 4.

Genome-wide evaluation of DSB formation in ZFNickase-treated cells by γH2AFX and TP53BP1 staining. K562 cells were transfected with the CCR5-patch donor in the absence (No ZFN) or presence of the indicated ZFN/ZFNickase combinations. Cells were collected at 2 d (A,B) or at the indicated time (C) post-transfection and subjected to anti-γH2AFX (A) or anti-TP53BP1 (B,C) antibody staining for flow cytometry analysis (A) or immunofluorescence microscopy (B,C). Numbers of TP53BP1+ loci were counted using the SimplePCI6 software (Compix) and expressed as average TP53BP1+ foci/nucleus (C) based on counting of at least three randomly selected fields. (*) A more than twofold difference in average TP53BP1+ foci/nucleus and p < 0.05 in comparison to the non-ZFN-treated control sample (Student's _t_-test).
To confirm the γH2AFX results, we repeated these studies using antibodies for an alternative DNA damage marker, tumor protein p53 binding protein 1 (TP53BP1), which localizes to sites of DNA damage and forms foci that may be visualized by immunofluorescence microscopy (Schultz et al. 2000; Anderson et al. 2001; Rappold et al. 2001). The numbers of TP53BP1 foci per nucleus were counted after immunostaining at various time points post-transfection (Fig. 4B,C). Again, we observed an increase in TP53BP1 foci in cells treated with WT/WT ZFNs (6.09 ± 1.07 foci/cell) compared to cells treated with the D450N/WT pair (0.84 ± 0.17 foci/cell) or no ZFN control samples (0.82 ± 0.22 foci/cell). The D467A/WT ZFNickase pair, as expected, revealed a small but measurable increase in the number of TP53BP1 foci (1.77 ± 0.33 foci/cell). Across all samples tested, the γH2AFX and TP53BP1 levels returned to background within 7 d post-transfection (Fig. 4C; Supplemental Fig. S4). Together, the absence of markedly increased levels of γH2AFX and TP53BP1 confirms that the D450N/WT ZFNickase does not drive an increase in the number of DSBs genome-wide. These data support the notion that CCR5 ZFNickases achieve targeted gene addition by directly inducing a SSB. Moreover, the absence of direct DSB generation further highlights the repair pathway bias obtained with an induced nick, thus avoiding potential mutations driven by error-prone NHEJ-mediated repair.
Discussion
We show here that a targeted DNA nickase built upon the zinc-finger nuclease obligate heterodimer architecture (Miller et al. 2007) supports homology-directed site-specific gene addition (and correction) at an endogenous human gene in transformed and primary human cells. Importantly, gene addition occurs in the absence of a significant increase in the number of indels characteristic of error-prone NHEJ-mediated DNA repair. Application of ZFNickases thus results in marked repair pathway bias, effectively forcing the cell to employ a homology-directed repair pathway. Given the vastly different genome editing outcomes supported by HDR (e.g., gene addition/correction) and NHEJ (e.g., gene disruption), the ability to restrict the products of repair to bias the desired editing event has the potential to further improve the precision of nuclease-induced genome engineering.
DNA SSB/nicks may be repaired by a rapid process involving: SSB detection by poly (ADP-ribose) polymerase-1 (PARP1), DNA end processing by various enzymes, DNA gap filling by DNA polymerases, and DNA ligation by DNA ligases (for review, see Caldecott 2008). However, firm evidence for a different role for SSB/nicks has also emerged over the years. In yeast, SSBs induced by the gene II protein (gIIp) can stimulate HDR (Galli and Schiestl 1998). In mammalian cells, the “nick-only” mutants of the RAG proteins for V(D)J recombination were shown to be potent stimulators of homologous recombination (Lee et al. 2004). More recently, it was demonstrated that mammalian cells can efficiently utilize HDR to repair single-strand DNA gaps opposite an abasic site or benzo[a]pyrene adduct (Adar et al. 2009). DNA nicks generated by adeno-associated virus (AAV) Rep78 and Rep68 proteins can greatly enhance HDR (van Nierop et al. 2009). Furthermore, a “nick-only” version of the I-Anil homing endonuclease was also shown to stimulate HDR-mediated DNA repair in mammalian cells using a reporter assay system engineered to contain the I-Anil target site (Smith et al. 2009; Metzger et al. 2011). The ability to introduce a site-specific DNA nick at a predetermined site in the human genome as presented here will be an important tool for the further molecular dissection of these repair pathways.
Mechanistically, we present several lines of evidence that the homology-directed repair process proceeds via an initial SSB. First, ZFNs carrying the D450N mutation demonstrate no measurable DNA cleavage activity in vitro (Fig. 1), while the D467A mutant retains a small amount of catalytic activity. Second, measurement of γH2AFX and TP53BP1 foci (Fig. 4) reveals no increase in genome-wide DSB formation for the D450N mutant (and a low but measurable increase for D467A). Third, expression of either the D450N/WT or D467A/WT ZFN combinations in yeast does not result in the loss of cell viability observed for direct DSB-generating WT/WT ZFNs (Fig. 2C). Finally, using an ultradeep sequencing approach capable of detecting mutations at a frequency of ∼1 in 20,000 events, we observe vanishing low levels of indels characteristic of DSB repair via NHEJ in the D450N/WT ZFNickase-treated sample (Table 3). Compared to the WT/WT ZFNs (direct DSB-inducing), this represents a ∼7000-fold reduction of indel mutations at the target locus. Last but not least, the levels of targeted gene addition driven by the ZFNickases (i.e., 4.0 ± 2.6% of alleles without selection for the desired event) in the absence of significant levels of indels (NHEJ) are difficult to explain through direct DSB generation since NHEJ is generally more efficient than HDR for DSB repair in human cells (Mao et al. 2008). Thus, instead of direct DSB generation, it is more likely that the ZFNickase initially induces only SSBs to stimulate DNA repair.
While the current studies do not provide direct information on the mechanism by which an induced SSB results in the stimulation of homologous repeat deletion or targeted gene addition, the most obvious models progress via the conversion of the SSB to either a single DSB end (DSE) (Cox 2001; Michel et al. 2001; Helleday 2003; Delacote and Lopez 2008) or a DSB (Kuzminov 2001; Saleh-Gohari et al. 2005) during DNA replication due to collapsed replication forks. Perhaps, therefore, the most surprising feature of the data we present here is the extent to which such a DSE or DSB is not a substrate for NHEJ (at least as determined by indel frequency).
The fact that DSB repair is so highly coordinated with the cell cycle, i.e., NHEJ occurring primarily in G1, while HDR takes place predominantly during S and G2/M (Aylon and Kupiec 2005; Barlow and Rothstein 2010), combined with the replication-dependent generation of the DSE or DSB (from a targeted SSB), provides at least one potential explanation for the marked preference for HDR that we observe. Independent of mechanism, the degree of repair pathway bias obtained with a targeted DNA nick adds to the tool box of methods for genome editing. ZFNickases based upon the D450N/WT combination support HDR—yet we were unable to drive any significant increase in the frequency of indels characteristic of DSB repair by NHEJ. This result has particular relevance in settings where the avoidance of off-target ZFN action is of paramount importance, such as the potential therapeutic use of a genetically modified cell. To this end, we show that ZFNickases are functional in primary human fibroblasts, a popular starting cell for the generation of iPSCs (Fig. 3D). This said, we have found that a ZFNickase-induced SSB is less efficient at stimulating homologous recombination than its ZFN-induced DSB relative, likely reflecting the need for SSB to DSB conversion during S phase before proceeding via the HDR pathway.
To begin to determine the potential generality of the results with the _CCR5_-specific ZFNickases, we generated a second pair of ZFNickases targeting a distinct target site, namely the PPP1R12C gene (also known as the AAVS1 locus). D450N/WT variants of the _AAVS1_-specific ZFNs were able to generate a nick but not a DSB when presented with AAVS1 target DNA in vitro (Supplemental Fig. S5A). Moreover, the AAVS1 D450/WT ZFNickases also stimulated HDR (resulting in the addition of a novel HindIII site) at the endogenous AAVS1 locus with a significantly (20-fold) lower efficiency (Supplemental Fig. S5B). Illumina deep sequencing results confirmed that the AAVS1 D450N/WT ZFNickase-treated sample had a 1160-fold reduction in the percentage of NHEJ-driven deletions compared to the WT/WT ZFN-treated samples under conditions of repeat transfection (Supplemental Table S3). Interestingly, detailed analysis of the WT/WT ZFN-treated samples revealed a preferred repair event “GCCA-duplication” that comprised 82% of all NHEJ-driven insertions in the WT/WT ZFN-treated sample. In contrast to the deletion data, ZFNickase-treated samples revealed a 45-fold reduction in insertions compared to the WT/WT ZFN-treated sample, perhaps suggesting that such insertions may be created through a different DNA repair mechanism more compatible with initiation via an SSB (Kvikstad et al. 2007; Russell and Hirata 2008). Excluding these “jackpot” GGCA-duplication events, deep sequencing analysis of samples treated with AAVS1 D450/WT ZFNickases revealed that the HDR repair pathway is markedly preferred over the classic NHEJ pathway causing undefined insertions and deletions (TI/und_indel = 9.76 compared to 1.08 in the WT/WT ZFN-treated sample) (Supplemental Table S3).
Taken together, these studies demonstrate that the D450N mutant described here can be used as a general approach to the generation of ZFNickases with in vitro DNA-nicking activities. Application of these enzymes in vivo demonstrates the anticipated reduction/elimination of indel generation. Interestingly, however, the in vivo efficiency of ZFNickase-induced gene addition through the HDR pathway was markedly dependent on the specific ZFNickase used. While beyond the scope of the current study, these data suggest that, in contrast to ZFN deployment, the general application of nickases will require the elucidation of additional factors (beyond the choice of FokI domain itself) that gate the activity of nick-driven targeted gene insertion.
In summary, the data reported here demonstrate that ZFNickase-stimulated gene addition can occur at endogenous human loci in the absence of significant mutagenesis by the error-prone NHEJ repair pathway. These data provide a first step toward controlling the outcomes of either gene addition/correction or gene disruption via the appropriate use of homologous donor DNA and the targeted introduction of either a single- or double-strand break, respectively.
Methods
Zinc-finger nuclease constructs
The amino acid sequences of the base CCR5 ZFN-L and ZFN-R are as described in Perez et al. (2008), and the full amino acid sequences are included in Supplemental Figure S6. The indicated mutations were introduced into the FokI cleavage domain using the QuikChange mutagenesis kit (Stratagene/Agilent Technologies). The residue number of each mutation refers to its position in the full-length FokI endonuclease.
In vitro DNA cleavage assay
A 2.5-kb fragment of the CCR5 locus containing the ZFN target site was cloned into the pCR4-Topo vector (Invitrogen), and used as a template for PCR amplification of a 292-bp fragment of CCR5 using primers: 5′-AAGATGGATTATCAAGTGTCAAGTCC-3′ and 5′-CAAAGTCCCACTGGGCG-3′. The resulting PCR product was end-labeled with [γ-32P]ATP and T4 polynucleotide kinase (New England Biolabs) as per the manufacturer's protocols. Unincorporated nucleotides were removed with a G50 spin column (GE Healthcare), and the resulting mixture was diluted to 1 ng/μl in FokI buffer consisting of 20 mM Tris-HCl pH 8.5, 150 mM NaCl, 2 mM MgCl2, 5% (v/v) glycerol, 10 μM ZnCl2, 0.5 mg/mL BSA, and 1 mM DTT. The ZFNs were in vitro synthesized using the TNT-Quick coupled transcription/translation system (Promega) according to the manufacturer's recommendations, except that the incubation time was increased to 2 h. The appropriate ZFNs and the radiolabeled 292-bp target DNA were mixed in FokI buffer and incubated at 37°C for 2 h. The radiolabeled DNA was then extracted with a phenol/chloroform mixture and either untreated (double-stranded DNA) or treated (single-stranded DNA) with a glyoxal/DMSO solution [1.0 M glyoxal, 10 mM NaH2PO4/Na2HPO4, pH 7.0, ∼50% vol/vol DMSO] at 50°C for 1 h (McMaster and Carmichael 1977). Double-stranded or single-stranded DNAs were then separated on a 10% polyacrylamide gel, the gel was dried, and cleavage quantified by phosphorimager (Molecular Dynamics).
Yeast assays
MEL1 reporter assays were performed as described (Doyon et al. 2008, 2011) using a MEL1 reporter construct containing a 1-kb EcoRI fragment of the CCR5 gene containing the CCR5 ZFN target site (Fig. 2A). The left arm (MEL) is 750 bp long, whereas the right arm (EL1) is 1810 bp long. There is a 450-bp overlap (identical sequence). To assay direct DSB-inducing activity of ZFNs in yeast (Fig. 2C), the CCR5 ZFN target site was integrated into the HO locus of the BY4741 strain using HO-poly-KanMX4-HO (Voth et al. 2001). The yeast strain was transformed with the various ZFN expression constructs, individual colonies were picked and grown in raffinose minimal media until they reached a density of 1 OD600nm. Tenfold serial dilutions of the cells were then plated on minimal media containing glucose (repressing) or galactose (inducing) and incubated for 3 d at 30°C.
Endogenous gene modification
The donor plasmids used to introduce either a BglI restriction enzyme site (CCR5-Patch) or eGFP (CCR5-GFP) at the endogenous CCR5 gene are shown in Supplemental Figures S2A and S3A. K562 cells were cultured in RPMI1640 media (Invitrogen) supplemented with 10% (v/v) FBS, 2 mM L-glutamine, 100 U/mL penicillin, and 100 mg/mL streptomycin. Cells were nucleofected with ZFN and donor plasmids using either the Amaxa nucleofector or 96-well shuttle system (Amaxa Biosystems/Lonza) according to the manufacturer's instructions. For nucleofection of 1–2 × 106 cells with the Amaxa nucleofector, 1.25–2.5 μg of each ZFN expression plasmid and 12.5–25.0 μg of the donor constructs were used. For nucleofection of 1–2 × 105 cells with the Amaxa 96-well shuttle system, 0.4 μg of each ZFN expression plasmid and 0.8 μg of the donor constructs were used. Cells were collected 3 d post-transfection or as indicated in the figure legends. Genomic DNA was extracted with the DNeasy Tissue kit (Qiagen) according to the supplier's instructions. Frequency of gene modification by NHEJ was evaluated by the Surveyor nuclease assay as described previously (Miller et al. 2007; Perez et al. 2008; Guschin et al. 2010), except when samples were analyzed after also being exposed to the CCR5-patch donor. For these samples, a modified protocol was exploited in which we first gel-purified a 2.5-kb CCR5 “outside” PCR product which uses primers that bind to sequences in CCR5 that are outside the region present in the donor DNA. This parental PCR product was then used as a template to amplify a 163-bp fragment of CCR5 using the inside primers (5′-CAATGTGAAGCAAATCGCAGC-3′and 5′-CAGGTCAGAGATGGCCAGGTTG-3′). Samples with the patch integrated at CCR5 also contain an additional 209-bp PCR product. The 163-bp product resulting from amplification with the “inside” PCR primers were gel-purified and then processed as described previously (Miller et al. 2007; Perez et al. 2008; Guschin et al. 2010).
Frequency of gene modification by homologous recombination was evaluated by a restriction fragment length polymorphism assay and flow cytometric analysis as described below. The RFLP assay was performed based on the insertion of a unique BglI site between the CCR5 ZFN binding sequences within the CCR5 gene. Briefly, a pair of CCR5 primers (5′-CTGCCTCATAAGGTTGCCCTAAG-3′ and 5′-CCAGCAATAGATGATCCAACTCAAATTCC-3′) located outside the region of homology encoded by the CCR5 donor molecules was used to PCR-amplify a 2.5-kb CCR5 fragment in the presence of [α-32P]dATP and [α-32P]dCTP. PCR products were passed through a G-50 column and digested with BglI. The products were resolved on a 10% polyacrylamide gel; the gel was dried and RFLP knock-in quantified using a phosphorimager (Molecular Dynamics). The frequency of targeted integration was calculated by measuring the ratio of cleaved to total product.
To construct the GFP reporter for testing homologous repeat deletion, two fragments of the eGFP open reading frame were interrupted via the insertion of a stretch of DNA containing the CCR5 ZFN target site such that 430 bp of direct repeat of the eGFP coding sequence flanked the insertion (Supplemental Fig. S1A). K562 cells were transfected with the GFP reporter with or without ZFN expression constructs, and cells were collected 2 or 3 d post-transfection and subjected to flow cytometric analysis after 5 min incubation with 5 μg/mL propidium iodide (PI) (Invitrogen) to facilitate exclusion of nonviable cells (PI+) from the analysis.
Illumina deep sequencing
Genomic DNA was amplified using the CCR5 primers (5′-CTGCCTCATAAGGTTGCCCTAAG-3′ and 5′-CCAGCAATAGATGATCCAACTCAAATTCC-3′) located outside the CCR5 homology region contained within the donor DNA molecules. The amplified 2.5-kb CCR5 fragments were gel-purified and used as templates for PCR reactions using a nested set of primers containing BpmI and XhoI restriction enzyme sites (5′-CTACTCACTGGTGTTCATCTGGAGTTTTGTGGGCAACATGC-3′ and 5′-CCATAACTGGAGACTGGGCGGCAGCATAGTGACTCGAGAAGGGGACAGTAAGAAGG-3′). The resulting amplicons were digested with BpmI and XhoI to remove 16 bp at the 5′ end of the PCR products, thus allowing sequencing to begin as close to the putative ZFN cleavage sites as possible. The digested products were gel-purified and ligated to adaptors (with or without 3-nt “bar code” unique to the experiment) which also exploits the BpmI- or XhoI-DNA overhangs (Supplemental Table S2). Adaptor-ligated PCR products were then gel-purified and PCR amplified using Illumina Genomic DNA Primers (Illumina). The resulting PCR products were subjected to Illumina deep sequencing using an Illumina Genome Analayzer instrument located at the California Institute for Quantitative Biosciences, University of California (Berkeley, CA). A read length of 36 bp was used and a custom-written computer script was used to extract all high-quality sequence reads and then classify high-quality reads based on their alignment with the WT template sequence. A quality score cutoff of 20 was used to exclude sequences with at least one base call with a score below 20. Sequences that did not contain any insertions or deletions and matched the wild-type sequences at 33 or more of the 36-bp read length were classified as wild type. Sequences that contained a deletion of two or more contiguous bases and no other changes relative to the wild-type sequence were classified as NHEJ-mediated deletions. Sequences that contained an insertion of two or more contiguous bases consistent with NHEJ-mediated insertion (i.e., a duplication of nearby sequence resulting from fill-in of single-stranded overhangs) and no other changes relative to wild type were classified as NHEJ-mediated insertions.
Transduction of primary human fibroblast cells
Primary human fibroblast cells derived from normal neonatal foreskin were purchased from ATCC (Manassas, VA) and cultured in Minimum Essential Medium (Invitrogen) supplemented with 10% (v/v) FBS, 2 mM L-glutamine, 1.0 mM sodium pyruvate, 100 U/mL penicillin, and 100 mg/mL streptomycin. For delivery to primary cells, we employed a replication-defective chimeric adenoviral vector (Ad5/F35) to express the ZFNs/ZFNickases (as described previously in Perez et al. 2008), combined with a lentiviral vector (LV) for delivery of the donor DNA (CCR5-patch), also generated as previously described (Lombardo et al. 2007), in which the CCR5 homologous arms and patch sequence are the same as described in Supplemental Figure S2A. This combination of vectors has previously proven very effective in mesenchymal stem cells (Benabdallah et al. 2010). For targeted integration, cells were incubated with the lenti-CCR5-patch vector for 6 h, followed by addition of Ad5/F35 ZFN vectors. Cells were collected 3–5 d later, and genomic DNA was prepared for the RFLP and Surveyor nuclease assay as above.
Measurement of genome-wide DSB formation by γH2AFX and TP53BP1 staining
For intracellular staining of phosphorylated histone H2AFX, cells collected at the indicated time points post-nucleofection were permeabilized with perm/wash buffer (0.05% Saponin, 2.5% FBS, and 0.02% NaN3 in PBS) and then incubated with anti-γH2AFX monoclonal antibody (Upstate), followed by incubation with Alexa Fluor488-conjugated goat anti-mouse immunoglobulin (Ig) (Invitrogen). Cells were then analyzed using a Guava Easycyte single cell analysis system (Guava Technologies). For TP53BP1 immunocytochemistry, cells were collected to prepare slides by cytospin (Thermo Scientific) and stained with anti-TP53BP1 rabbit polyclonal antibodies (Bethyl Laboratories), followed by photography using a CCD camera connected to an immunofluorescence microscope (Nikon) as described before in detail (Perez et al. 2008).
Data access
All Illumina deep sequencing data have been submitted to the NCBI Sequence Read Archive (SRA) (http://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi) under accession number SRA050213.
Acknowledgments
We thank Fyodor Urnov, Gary Lee, Joshua Kahn, James Li, and Bryan Zeitler (Sangamo Biosciences Inc.) for generation of some of the ZFN and donor constructs, Lei Zhang (Sangamo Biosciences Inc.) for critical advice, Leath Tonkin (Vincent J. Coates Genomics Sequencing Laboratory, California Institute for Quantitative Biosciences, University of California, Berkeley, CA) and Fyodor Urnov for Illumina deep sequencing, Anureet Tiwana and Samuel T. Test (Children's Hospital Oakland Research Institute) for performing cell sorting, and Maria Jasin (Memorial Sloan-Kettering Cancer Center, New York, NY) and the anonymous referees for careful reading of the manuscript.
Footnotes
[Supplemental material is available for this article.]
References
- Adar S, Izhar L, Hendel A, Geacintov N, Livneh Z 2009. Repair of gaps opposite lesions by homologous recombination in mammalian cells. Nucleic Acids Res 37: 5737–5748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson L, Henderson C, Adachi Y 2001. Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol Cell Biol 21: 1719–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylon Y, Kupiec M 2005. Cell cycle-dependent regulation of double-strand break repair: A role for the CDK. Cell Cycle 4: 259–261 [PubMed] [Google Scholar]
- Barlow JH, Rothstein R 2010. Timing is everything: Cell cycle control of Rad52. Cell Div 5: 7 doi: 10.1186/1747-1028-5-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benabdallah BF, Allard E, Yao S, Friedman G, Gregory PD, Eliopoulos N, Fradette J, Spees JL, Haddad E, Holmes MC, et al. 2010. Targeted gene addition to human mesenchymal stromal cells as a cell-based plasma-soluble protein delivery platform. Cytotherapy 12: 394–399 [DOI] [PubMed] [Google Scholar]
- Bibikova M, Golic M, Golic KG, Carroll D 2002. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics 161: 1169–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitinaite J, Wah DA, Aggarwal AK, Schildkraut I 1998. FokI dimerization is required for DNA cleavage. Proc Natl Acad Sci 95: 10570–10575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugmans L, Kanaar R, Essers J 2007. Analysis of DNA double-strand break repair pathways in mice. Mutat Res 614: 95–108 [DOI] [PubMed] [Google Scholar]
- Cai CQ, Doyon Y, Ainley WM, Miller JC, Dekelver RC, Moehle EA, Rock JM, Lee YL, Garrison R, Schulenberg L, et al. 2009. Targeted transgene integration in plant cells using designed zinc finger nucleases. Plant Mol Biol 69: 699–709 [DOI] [PubMed] [Google Scholar]
- Caldecott KW 2008. Single-strand break repair and genetic disease. Nat Rev Genet 9: 619–631 [DOI] [PubMed] [Google Scholar]
- Carbery ID, Ji D, Harrington A, Brown V, Weinstein EJ, Liaw L, Cui X 2010. Targeted genome modification in mice using zinc finger nucleases. Genetics 186: 451–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll D 2008. Progress and prospects: Zinc-finger nucleases as gene therapy agents. Gene Ther 15: 1463–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathomen T, Joung JK 2008. Zinc-finger nucleases: The next generation emerges. Mol Ther 16: 1200–1207 [DOI] [PubMed] [Google Scholar]
- Cox MM 2001. Historical overview: Searching for replication help in all of the rec places. Proc Natl Acad Sci 98: 8173–8180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacote F, Lopez BS 2008. Importance of the cell cycle phase for the choice of the appropriate DSB repair pathway, for genome stability maintenance: The _trans_-S double-strand break repair model. Cell Cycle 7: 33–38 [DOI] [PubMed] [Google Scholar]
- Doyon Y, McCammon JM, Miller JC, Faraji F, Ngo C, Katibah GE, Amora R, Hocking TD, Zhang L, Rebar EJ, et al. 2008. Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat Biotechnol 26: 702–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon Y, Vo TD, Mendel MC, Greenberg SG, Wang J, Xia DF, Miller JC, Urnov FD, Gregory PD, Holmes MC 2011. Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nat Methods 8: 74–79 [DOI] [PubMed] [Google Scholar]
- Galli A, Schiestl RH 1998. Effects of DNA double-strand and single-strand breaks on intrachromosomal recombination events in cell-cycle-arrested yeast cells. Genetics 149: 1235–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geurts AM, Cost GJ, Freyvert Y, Zeitler B, Miller JC, Choi VM, Jenkins SS, Wood A, Cui X, Meng X, et al. 2009. Knockout rats via embryo microinjection of zinc-finger nucleases. Science 325: 433 doi: 10.1126/science.1172447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guschin DY, Waite AJ, Katibah GE, Miller JC, Holmes MC, Rebar EJ 2010. A rapid and general assay for monitoring endogenous gene modification. Methods Mol Biol 649: 247–256 [DOI] [PubMed] [Google Scholar]
- Helleday T 2003. Pathways for mitotic homologous recombination in mammalian cells. Mutat Res 532: 103–115 [DOI] [PubMed] [Google Scholar]
- Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC, Katibah GE, Amora R, Boydston EA, Zeitler B, et al. 2009. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol 27: 851–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday R 1964. The induction of mitotic recombination by mitomycin C in Ustilago and Saccharomyces. Genetics 50: 323–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt N, Wang J, Kim K, Friedman G, Wang X, Taupin V, Crooks GM, Kohn DB, Gregory PD, Holmes MC, et al. 2010. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol 28: 839–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczykowski SC 2000. Initiation of genetic recombination and recombination-dependent replication. Trends Biochem Sci 25: 156–165 [DOI] [PubMed] [Google Scholar]
- Kuzminov A 2001. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc Natl Acad Sci 98: 8241–8246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvikstad EM, Tyekucheva S, Chiaromonte F, Makova KD 2007. A macaque's-eye view of human insertions and deletions: Differences in mechanisms. PLoS Comput Biol 3: 1772–1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Provost F, Lillico S, Passet B, Young R, Whitelaw B, Vilotte JL 2010. Zinc finger nuclease technology heralds a new era in mammalian transgenesis. Trends Biotechnol 28: 134–141 [DOI] [PubMed] [Google Scholar]
- Lee GS, Neiditch MB, Salus SS, Roth DB 2004. RAG proteins shepherd double-strand breaks to a specific pathway, suppressing error-prone repair, but RAG nicking initiates homologous recombination. Cell 117: 171–184 [DOI] [PubMed] [Google Scholar]
- Lieber MR 2008. The mechanism of human nonhomologous DNA end joining. J Biol Chem 283: 1–5 [DOI] [PubMed] [Google Scholar]
- Lombardo A, Genovese P, Beausejour CM, Colleoni S, Lee YL, Kim KA, Ando D, Urnov FD, Galli C, Gregory PD, et al. 2007. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol 25: 1298–1306 [DOI] [PubMed] [Google Scholar]
- Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM, Eichtinger M, Jiang T, Foley JE, Winfrey RJ, Townsend JA, et al. 2008. Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell 31: 294–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Bozzella M, Seluanov A, Gorbunova V 2008. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst) 7: 1765–1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMaster GK, Carmichael GG 1977. Analysis of single- and double-stranded nucleic acids on polyacrylamide and agarose gels by using glyoxal and acridine orange. Proc Natl Acad Sci 74: 4835–4838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Noyes MB, Zhu LJ, Lawson ND, Wolfe SA 2008. Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nat Biotechnol 26: 695–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meselson MS, Radding CM 1975. A general model for genetic recombination. Proc Natl Acad Sci 72: 358–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger MJ, McConnell-Smith A, Stoddard BL, Miller AD 2011. Single-strand nicks induce homologous recombination with less toxicity than double-strand breaks using an AAV vector template. Nucleic Acids Res 39: 926–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, de Angelis MH, Wurst W, Kuhn R 2010. Gene targeting by homologous recombination in mouse zygotes mediated by zinc-finger nucleases. Proc Natl Acad Sci 107: 15022–15026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel B, Flores MJ, Viguera E, Grompone G, Seigneur M, Bidnenko V 2001. Rescue of arrested replication forks by homologous recombination. Proc Natl Acad Sci 98: 8181–8188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JC, Holmes MC, Wang J, Guschin DY, Lee YL, Rupniewski I, Beausejour CM, Waite AJ, Wang NS, Kim KA, et al. 2007. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol 25: 778–785 [DOI] [PubMed] [Google Scholar]
- Moore JK, Haber JE 1996. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol 16: 2164–2173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton J, Davis MW, Jorgensen EM, Carroll D 2006. Induction and repair of zinc-finger nuclease-targeted double-strand breaks in Caenorhabditis elegans somatic cells. Proc Natl Acad Sci 103: 16370–16375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochiai H, Fujita K, Suzuki K, Nishikawa M, Shibata T, Sakamoto N, Yamamoto T 2010. Targeted mutagenesis in the sea urchin embryo using zinc-finger nucleases. Genes Cells 15: 875–885 [DOI] [PubMed] [Google Scholar]
- Osakabe K, Osakabe Y, Toki S 2010. Site-directed mutagenesis in Arabidopsis using custom-designed zinc finger nucleases. Proc Natl Acad Sci 107: 12034–12039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, Wang N, Lee G, Bartsevich VV, Lee YL, et al. 2008. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol 26: 808–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radding CM 1982. Homologous pairing and strand exchange in genetic recombination. Annu Rev Genet 16: 405–437 [DOI] [PubMed] [Google Scholar]
- Rappold I, Iwabuchi K, Date T, Chen J 2001. Tumor suppressor p53 binding protein 1 (53BP1) is involved in DNA damage-signaling pathways. J Cell Biol 153: 613–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM 1998. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 273: 5858–5868 [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Boon C, Redon C, Bonner WM 1999. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol 146: 905–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell DW, Hirata RK 2008. Human gene targeting favors insertions over deletions. Hum Gene Ther 19: 907–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh-Gohari N, Bryant HE, Schultz N, Parker KM, Cassel TN, Helleday T 2005. Spontaneous homologous recombination is induced by collapsed replication forks that are caused by endogenous DNA single-strand breaks. Mol Cell Biol 25: 7158–7169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders KL, Catto LE, Bellamy SR, Halford SE 2009. Targeting individual subunits of the FokI restriction endonuclease to specific DNA strands. Nucleic Acids Res 37: 2105–2115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz LB, Chehab NH, Malikzay A, Halazonetis TD 2000. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J Cell Biol 151: 1381–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla VK, Doyon Y, Miller JC, DeKelver RC, Moehle EA, Worden SE, Mitchell JC, Arnold NL, Gopalan S, Meng X, et al. 2009. Precise genome modification in the crop species Zea mays using zinc-finger nucleases. Nature 459: 437–441 [DOI] [PubMed] [Google Scholar]
- Smith AM, Takeuchi R, Pellenz S, Davis L, Maizels N, Monnat RJ Jr, Stoddard BL 2009. Generation of a nicking enzyme that stimulates site-specific gene conversion from the I-AniI LAGLIDADG homing endonuclease. Proc Natl Acad Sci 106: 5099–5104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiff T, O'Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA 2004. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res 64: 2390–2396 [DOI] [PubMed] [Google Scholar]
- Sung P, Klein H 2006. Mechanism of homologous recombination: Mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol 7: 739–750 [DOI] [PubMed] [Google Scholar]
- Szczepek M, Brondani V, Buchel J, Serrano L, Segal DJ, Cathomen T 2007. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat Biotechnol 25: 786–793 [DOI] [PubMed] [Google Scholar]
- Takasu Y, Kobayashi I, Beumer K, Uchino K, Sezutsu H, Sajwan S, Carroll D, Tamura T, Zurovec M 2010. Targeted mutagenesis in the silkworm Bombyx mori using zinc finger nuclease mRNA injection. Insect Biochem Mol Biol 40: 759–765 [DOI] [PubMed] [Google Scholar]
- Townsend JA, Wright DA, Winfrey RJ, Fu F, Maeder ML, Joung JK, Voytas DF 2009. High-frequency modification of plant genes using engineered zinc-finger nucleases. Nature 459: 442–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD 2010. Genome editing with engineered zinc finger nucleases. Nat Rev Genet 11: 636–646 [DOI] [PubMed] [Google Scholar]
- van Nierop GP, de Vries AA, Holkers M, Vrijsen KR, Goncalves MA 2009. Stimulation of homology-directed gene targeting at an endogenous human locus by a nicking endonuclease. Nucleic Acids Res 37: 5725–5736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voth WP, Richards JD, Shaw JM, Stillman DJ 2001. Yeast vectors for integration at the HO locus. Nucleic Acids Res 29: E59 doi: 10.1093/nar/29.12.e59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waugh DS, Sauer RT 1993. Single amino acid substitutions uncouple the DNA binding and strand scission activities of Fok I endonuclease. Proc Natl Acad Sci 90: 9596–9600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyman C, Kanaar R 2006. DNA double-strand break repair: All's well that ends well. Annu Rev Genet 40: 363–383 [DOI] [PubMed] [Google Scholar]
- Zhang F, Maeder ML, Unger-Wallace E, Hoshaw JP, Reyon D, Christian M, Li X, Pierick CJ, Dobbs D, Peterson T, et al. 2010. High frequency targeted mutagenesis in Arabidopsis thaliana using zinc finger nucleases. Proc Natl Acad Sci 107: 12028–12033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Maeder ML, Mali P, Pruett-Miller SM, Thibodeau-Beganny S, Chou BK, Chen G, Ye Z, Park IH, Daley GQ, et al. 2009. Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell 5: 97–110 [DOI] [PMC free article] [PubMed] [Google Scholar]