Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing (original) (raw)
Abstract
DNA methylation is crucial for a wide variety of biological processes, yet no technique suitable for the methylome analysis of DNA methylation at single-cell resolution is available. Here, we describe a methylome analysis technique that enables single-cell and single-base resolution DNA methylation analysis based on reduced representation bisulfite sequencing (scRRBS). The technique is highly sensitive and can detect the methylation status of up to 1.5 million CpG sites within the genome of an individual mouse embryonic stem cell (mESC). Moreover, we show that the technique can detect the methylation status of individual CpG sites in a haploid sperm cell in a digitized manner as either unmethylated or fully methylated. Furthermore, we show that the demethylation dynamics of maternal and paternal genomes after fertilization can be traced within the individual pronuclei of mouse zygotes. The demethylation process of the genic regions is faster than that of the intergenic regions in both male and female pronuclei. Our method paves the way for the exploration of the dynamic methylome landscapes of individual cells at single-base resolution during physiological processes such as embryonic development, or during pathological processes such as tumorigenesis.
Gene transcription is crucial for a cell to maintain its identity and physiological function and is regulated within individual cells. Epigenetic status is important in transcriptional regulation and is potentially heterogeneous even within a relatively homogeneous cell type due to the different cell subpopulations present (Jaenisch and Bird 2003; Toyooka et al. 2008). This is especially prominent in tumors in which both the genomes and epigenomes of the individual cells are heterogeneous (Rodriguez-Paredes and Esteller 2011; Marusyk et al. 2012). Moreover, it is very difficult to obtain large numbers of cells for epigenome analysis in some circumstances, such as for mammalian early embryos (Smallwood et al. 2011; Tang et al. 2011a; Smith et al. 2012). It is highly desirable to develop a single-cell epigenome analysis technique, ideally one that provides single-base resolution. As one of the most important epigenetic modifications, DNA methylation is critical for a wide variety of biological processes, including the regulation of genomic imprinting and X-chromosome inactivation, as well as the repression of transposable elements within the genome (Bird 2002; Lister et al. 2009; Hackett et al. 2012). DNA is methylated at the carbon atom occupying the fifth position of the cytosine ring (5mC) and is catalyzed by the DNA cytosine methyltransferases, Dnmt1, Dnmt3a, and Dnmt3b (Reik 2007). DNA methylation is functionally important for mammalian development because both Dnmt1 and Dnmt3b knockout mice are embryonic lethal, whereas Dnmt3a mutant mice die within 1 mo after birth (Okano et al. 1999; Li 2002). The reduced representation bisulfite sequencing (RRBS) technique has been developed to dissect the methylomes of mammalian cells (Meissner et al. 2005). RRBS is based on the lack of even distribution for CpG sites within the mammalian genome; these sites tend to cluster together as CpG islands (CGIs) that are usually located near the promoter regions of annotated genes (Deaton and Bird 2011). Thus, after cutting the genome into short fragments via a restriction enzyme that recognizes CpG and its flanking sequences, a majority of the CGIs will be recovered and sequenced with high coverage even with relatively low numbers of total sequencing reads. RRBS has been shown to be effective for as few as 60 mouse early embryonic cells (Chan et al. 2012; Smallwood and Kelsey 2012) and has led to significant findings regarding global demethylation and remethylation processes during the early cleavage and post-implantation stages of mouse embryonic development, respectively (Smith et al. 2012). Recently, a method for the epigenetic analysis of histone modifications for an individual locus at single-cell resolution has been developed (Gomez et al. 2013). However, single-cell epigenome analysis at whole-genome scale has never been achieved.
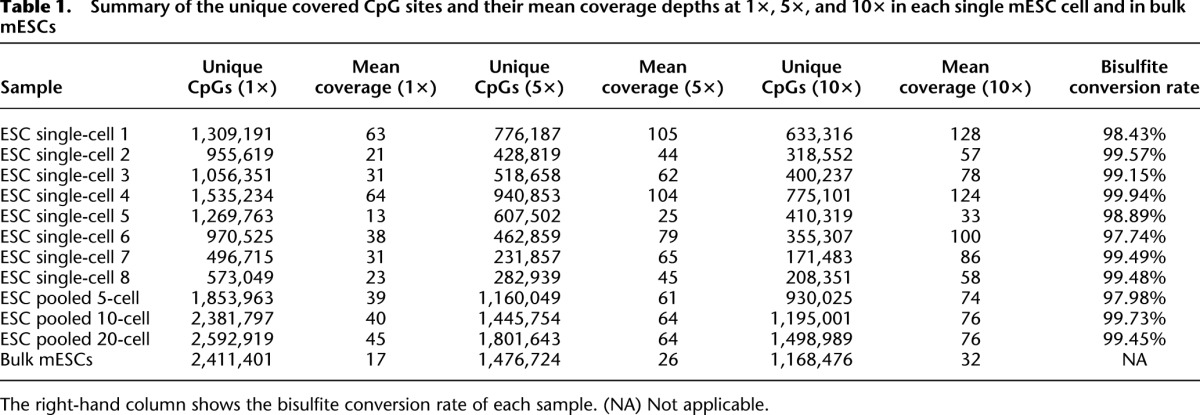
We report the development of a single-cell methylome analysis technique based on RRBS (scRRBS) and demonstrate its effective use for mouse embryonic stem cells (mESCs), sperm, and oocytes, as well as for male and female pronuclei of the zygotes. We were able to recover 0.5 to 1.5 million CpG sites from a single mESC, and the methylation levels for all analyzed genomic regions were comparable to those obtained from bulk mESCs (Table 1; Supplemental Table 1). Furthermore, we show for the first time that the methylome of the first polar body is comparable with that of the metaphase II oocyte within the same gamete. Finally, we used our method to prove that the demethylation process of the male pronucleus occurs more quickly than that of the female pronucleus in the same zygote.
Table 1.
Summary of the unique covered CpG sites and their mean coverage depths at 1×, 5×, and 10× in each single mESC cell and in bulk mESCs
Results
Characterization of the single-cell RRBS methylome analysis technique
To improve the suitability of the RRBS method for single-cell analysis, we reasoned that one of the primary obstacles to success was the massive loss of DNA during multiple purification steps. Thus, we thoroughly modified the original protocol (Smith et al. 2009; Gu et al. 2011a) and integrated all of the experimental processes in a single-tube reaction without including any purification steps prior to the bisulfite conversion process. That is, all of the following steps were completed within the same reaction tube: the lysis of an individual cell; the release of naked, double-stranded genomic DNA; adding a spike-in of lambda DNA; digestion of the genomic DNA using a restriction enzyme; the end-repair and dA-tailing of the DNA fragments; ligation of the adaptors to the DNA fragments; and the bisulfite conversion of the ligated DNA. After this procedure, the converted DNA was purified using Zymo spin columns with 10 ng tRNA as a carrier. The purified DNA was then enriched via two rounds of PCR amplification and subjected to deep sequencing (for details, see Methods) (Fig. 1).
Figure 1.

A schematic of the single-cell RRBS (scRRBS) technique. Note the completion of all of the following steps within the same reaction tube: lysis of an individual cell, release of the naked double-stranded genomic DNA, spiking with lambda DNA, digestion of the genomic DNA using a restriction enzyme, end-repair and dA-tailing of the DNA fragments, ligation of the adaptors to the DNA fragments, and bisulfite conversion of the ligated DNA.
Whether the conversion rate for the trace amounts of DNA obtained from a single cell after being treated with bisulfite is comparable to that of bulk DNA had not previously been determined. By spiking trace amounts of unmethylated lambda DNA into the single-cell samples, we determined that the conversion rate resulting from bisulfite treatment is 99.2% on average (ranging from 97.7% to 99.9%), indicating that the DNA from each single cell was converted highly efficiently under our bisulfite treatment condition (Table 1; Supplemental Table 1).
We then determined how many CpG sites could be recovered using our scRRBS approach. We analyzed eight individual mESCs and determined the presence of 496,715 to 1,535,234 CpG sites in each cell (Table 1). That is, our approach recovered on average 1.02 million (40%) of the total 2.5 million CpG sites that could be detected using RRBS with bulk cells (Supplemental Table 2; Smith et al. 2012). As expected, when we merged the RRBS data for individual cells together, additional CpG sites were recovered (e.g., 1.53 million CpG sites were recovered by merging eight individual mESCs) (Fig. 2A). Similarly, if we merged the scRRBS data of five single sperm cells together, we were able to capture 0.73 million CpG sites (Supplemental Fig. 2; also see below). Moreover, our method is also applicable to small numbers of cells rather than individual cells. We showed that using 20 mESCs as the starting material, our method could detect 63% (1.52 million) of the CpG sites that are recovered using RRBS with bulk mESCs (Fig. 2A; Supplemental Table 3).
Figure 2.

The sensitivity and reproducibility of the single-cell RRBS technique. (A) The number and proportion of CpG sites detected in the merged single mESC RRBS data set overlapped with those from the RRBS of the bulk mESCs. (B) Pearson correlation heatmap among the methylomes of all RRBS samples of single cells, pooled cells, or bulk cells. The color key from green to red indicates low to high correlation, respectively. (C) DNA methylation map of the CpG sites at a representative locus in the RRBS data from eight single cells and bulk mESCs. The upward blue bars and downward red bars indicate methylated CpGs and unmethylated CpGs, respectively. (D) The methylation levels of different genomic regions of single mESCs; pooled mESCs of five cells, 10 cells, and 20 cells; and bulk mESCs.
We then determined the accuracy of our method and compared the data obtained using individual mESCs with that obtained using bulk mESCs, and found that the correlation coefficient was reasonably high (R = 0.77 on average) when comparing the CpG sites recovered using both methods (Fig. 2B; Supplemental Fig. 3). Moreover, when we merged the single-cell RRBS data from all eight individual mESCs, the correlation coefficient between the merged single-cell data and the data from the bulk mESCs obtained was 0.90 (Supplemental Fig. 1). Furthermore, unsupervised hierarchical clustering analysis showed that the methylomes of individual mESCs clearly clustered together with those of bulk mESCs but remained separate from those of oocytes, sperm, and male and female pronuclei (Fig. 2B; also see below). Figure 2C and Supplemental Figure 3 show several loci that are representative of the methylation status of the individual and bulk mESCs. Furthermore, the methylation levels measured for specific genomic regions of individual mESCs are similar to those of bulk mESCs (Fig. 2D; Supplemental Fig. 4). These results indicate that our method can be used to accurately analyze the global DNA methylation status within individual cells. We then sought to determine whether the method is robust by comparing the methylome of different individual mESCs; the resulting correlation coefficient was reasonably high among individual mESCs (R = 0.67 on average), verifying that our method was clearly reproducible (Fig. 2B).
We then sought to determine whether the scRRBS method enables us to obtain the absolute methylation status of a CpG site. In theory, our method should only detect a CpG locus as either fully methylated (100%) or unmethylated (0%) but not as an intermediate methylation value (e.g., 30% methylation) in a haploid cell such as sperm, as can be detected in bulk analysis due to the heterogeneity within the cell population. To investigate this aspect, we applied our method to single sperm cells. We found that 88%–94% of the CpG sites detected using our method within an individual sperm cell are either fully methylated (100%) or unmethylated (0%) (Fig. 3A; Supplemental Fig. 5), indicating that our method is accurate and digitized. By applying the same strategy, we found that 84%–90% of the CpG sites are either fully methylated or unmethylated in single mESCs (Supplemental Fig. 6). Figure 3B and Supplemental Figure 5 show several loci that are representative of the methylation status in individual sperm cells and bulk sperm. To verify this result using an independent approach, we analyzed one fully methylated locus and one unmethylated locus in single sperm cells and validated their methylation status via methylation-sensitive restriction digestion coupled with nested PCR within individual sperm cells. We found that the methylated locus can be digested using MspI (methylation-insensitive restriction enzyme) but not by HpaII (methylation-sensitive restriction enzyme), whereas the unmethylated locus can be digested by both enzymes (Fig. 3C). This indicates that the methylation status obtained using scRRBS is accurate and can be verified using an independent approach within individual cells.
Figure 3.

The methylation status of single sperm cells. (A) The proportion of fully methylated (≥90% methylated with read depths greater than or equal to three) and unmethylated (≤10% methylated with read depths greater than or equal to three) CpG sites within the total CpG sites covered in the scRRBS of an individual sperm cell. (B) The methylation status of a representative locus on chromosome 1 showing that most of the detected CpG sites were either methylated or unmethylated. Filled black circles represent methylated CpG sites, whereas open circles represent unmethylated CpG sites. Gaps in the methylation profiles represent CpG sites that were not recovered in the single-cell RRBS data. The filled brown circles represent all of the CpG sites in the corresponding region of the genome. (C) Agarose gel analysis of the methylation-sensitive restriction digestion coupled with nested PCR in single sperm cells. (Top) A methylated locus. (Bottom) An unmethylated locus. The first five lanes (excluding the marker lane) indicate five individual single sperm cells digested with MspI, MspI, HpaII, HpaII, and no enzyme, respectively. The next two lanes indicate 1 ng of bulk sperm genomic DNA treated with MspI or HpaII, respectively, as positive controls. A weaker band (468 bp) at the upward side of the strong band (320 bp) in the fourth lane (excluding the marker lane) of the top panel is the amplification product of the first-round PCR.
Applying the analysis to male and female pronuclei
After fertilization, the maternal and paternal genomes of the zygote underwent different types of global demethylation; the former was passively demethylated during DNA replication, whereas the latter was primarily actively demethylated by TET3 oxidation of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) (Mayer et al. 2000; Okada et al. 2010; Gu et al. 2011b; Wossidlo et al. 2011; Hackett et al. 2013). However, this biological process has not been investigated at the single-cell level on a genome-wide scale. We addressed this issue by applying our scRRBS method to individual female and male pronuclei isolated from the same zygotes. First, we sought to determine whether the methylomes of the first polar bodies were similar to those of the metaphase II oocytes; in previous studies, the methylomes of the polar bodies were never analyzed separately from those of the metaphase II oocytes. We compared the first polar body and the metaphase II oocyte within the same female gamete and found that their methylomes were very similar and clustered together in the unsupervised hierarchical clustering analysis (Fig. 2B). Furthermore, when we analyzed DNA methylation for different genomic regions, we found that the methylation levels of specific genomic regions, such as the intragenic or intergenic regions measured in the first polar bodies, were comparable to those measured in metaphase II oocytes (Fig. 4A).
Figure 4.

Global demethylation in male and female pronuclei during pronucleus stages within individual zygotes. (A) The DNA methylation levels of different genomic regions of metaphase II oocytes and the first polar bodies within the same gametes. (B) Hoechst 33342 staining of pronuclei in the zygotes, indicating the distance between each pair of male and female pronuclei in individual zygotes. From zygote 1 to zygote 5, the distance between the male and female pronuclei gradually decreases. (C) Global methylation levels in male and female pronuclei within the same zygotes. Note that the methylation levels decrease significantly in both male (red line) and female (blue line) pronuclei.
We then chose zygotes at different pronucleus stages and performed single-cell RRBS analyses separately for both the male and female pronuclei. The female pronuclei were derived from metaphase II oocytes, whereas the male pronuclei were derived from sperm. Unsupervised hierarchical clustering analysis showed that all of the female pronuclei clustered properly with metaphase II oocytes, whereas all of the male pronuclei clustered together with sperm (Fig. 2B). This shows that our scRRBS analysis can be used to clearly determine the methylome of individual female and male pronuclei. Furthermore, we found that during zygotic development, the methylation levels of the pronuclei decreased significantly as they became closer to each other (Fig. 4B,C). More importantly, the demethylation of the male pronuclei was more dramatic than that of the female pronuclei, which is consistent with previous immunostaining results and bisulfite sequencing results of several individual loci that showed faster demethylation of male pronuclei than female pronuclei (Oswald et al. 2000; Santos et al. 2002; Farthing et al. 2008; Iqbal et al. 2011; Inoue et al. 2012). This is also consistent with the fact that both the male and female pronuclei passively demethylated their genomes by dilution due to DNA replication during the pronucleus stages, whereas the male pronuclei also underwent active global demethylation via TET3 at the same time (Ferreira and Carmo-Fonseca 1997; Gu et al. 2011b).
Finally, we sought to determine whether the demethylation process was synchronized in the genome or whether some genomic regions would demethylate faster than other regions. We found that during the development of the zygotes through the pronucleus stages, the methylation levels of the genic regions decreased faster than those of the intergenic regions in both the female (from 20% to 15%) and male pronuclei (from 21% to 10%) (Fig. 5A; Supplemental Table 4). This is consistent with the possibility that the genic regions were replicated earlier during the replication of the maternal and paternal genomes and were therefore passively demethylated earlier than the intergenic regions (Ferreira and Carmo-Fonseca 1997; Iqbal et al. 2011). Several representative loci indicating significant demethylation of the male pronuclei are shown in Figure 5B and Supplemental Figure 7. For the repeat elements in the male pronucleus, the methylation levels of the short interspersed nuclear elements (SINEs) decreased more quickly, from 60% in sperm to 35% in the late pronucleus stage; in contrast, the methylation levels of the long interspersed nuclear elements (LINEs) decreased more slowly, from 81% to 67% (Fig. 5C; Supplemental Table 4). This indicates that during the demethylation process of repeat elements in the paternal genome, SINEs were demethylated faster than LINEs and long terminal repeats (LTRs) (Xu et al. 2011). During the pronucleus stages, high-density CpG promoters (HCP), intermediate-density CpG promoters (ICP), and low-density CpG promoters (LCP) do not experience significant demethylation in either male or female pronuclei (Supplemental Fig. 8). The results clearly show that our scRRBS method can be used to trace the methylome of mammalian cells with single-cell and single-base resolution.
Figure 5.

The demethylation patterns in various genomic regions in male and female pronuclei during zygotic development. (A) DNA methylation dynamics of male and female pronuclei in intragenic and intergenic regions. The methylation levels in the male (red line) and female (blue line) pronuclei decreased, whereas the demethylation in the male pronuclei was more dramatic than that in the female pronuclei. (B) The methylation profile of a representative Meox2 locus on chromosome 12 in the male pronuclei. The upward blue bars in the left panel represent fully methylated CpG sites, whereas the downward red bars represent unmethylated CpG sites. The green bar in the panel on the right shows the average methylation levels of the CpG sites in this region. (C) DNA methylation dynamics of male and female pronuclei in repeat regions. The left, middle, and right panels display the methylation levels of male and female pronuclei in the SINE, LINE, and LTR regions, respectively. Red and blue lines indicate male and female pronuclei, respectively.
Discussion
Single-cell genomics is crucial to understanding the gene regulation networks within individual cells, which are the fundamental biological units of organisms. We and others developed single-cell RNA-seq transcriptome analysis techniques several years ago that enable gene expression dynamics to be analyzed within an individual cell (Kurimoto et al. 2006; Tang et al. 2009, 2011b). Recently, single-cell genome sequencing techniques have also been developed that enable researchers to analyze the heterogeneity of single-cell genomes (Kalisky and Quake 2011; Navin et al. 2011; Zong et al. 2012). Here, we report the development of a single-cell methylome analysis technique based on RRBS that enables us to dissect the complexity of the methylomes within an individual cell. To our knowledge, this represents the development of the first single-cell epigenome analysis technique. Our single-cell RRBS technique can be directly applied either to a single cell or to a pool of a small number of cells. Both strategies have advantages and disadvantages. The RRBS of a single cell is a digitized method that can exclude the effect of the heterogeneity of a population of cells. However, the coverage of this method is relatively low, and the cost for sequencing all of the libraries of these single cells is relatively high. On the other hand, the RRBS of a pool of a few cells is not a digitized method, and the unknown heterogeneity of a population of cells will obscure the interpretation of the methylation status of these loci within individual cells. However, the RRBS of a pool of a few cells has relatively high coverage, and the sequencing cost is relatively low because only one or two sequencing libraries in the population need to be sequenced.
Our scRRBS method has several advantages. First, we spiked trace amounts of unmethylated lambda DNA into the samples to accurately measure the bisulfite conversion rate for each single-cell sample. This enabled us to maintain a very low percentage (<0.8%) of false-positive determinations of DNA methylation due to the non-conversion of unmethylated cytosines that were perceived falsely as methylated cytosines. Second, our method is highly sensitive and can detect 40% of the CpG sites from a single cell compared with RRBS using bulk cells (Supplemental Table 2). Third, our method has intrinsic controls with a cytosine called as fully methylated, unmethylated, or undetected. This is crucial for single-cell epigenome analysis; if we can only call a cytosine as methylated but cannot discriminate unmethylated cytosines from undetected cytosines due to sample losses, a very high rate of false negatives will appear in the assay. This is especially relevant for techniques based on ChIP-seq. For accurate measurements to be obtained using a single-cell ChIP-seq technique, the method must be able to discriminate between the histone marks of the unmodified status and the undetected status due to sample losses. Fourth, our method is flexible and permits both single-cell methylome analysis and the pooling of a small amount of cells to obtain measurements for the population as a whole. Fifth, our method is based on the RRBS technique, which can strongly enrich CpG-dense sites in the genome; thus, a relatively low number of sequence reads is required to detect these target CpG sites at high coverage (Gu et al. 2010, 2011a). This makes it feasible to sequence the methylomes of hundreds or even thousands of single-cell samples. Sixth, our method performs a digital count of the methylation status of the CpG sites within a single cell. For the haploid sperm cells, we determined that 91.6% of all detected CpG sites were either unmethylated or fully methylated (Fig. 3A). For the diploid mESCs, the unmethylated or fully methylated CpG sites accounted for 86.9% of all detected loci (Supplemental Figure 6). One possible explanation for this finding is that ∼4.7% of the CpG sites within a single mESC are likely to be present as one unmethylated allele together with one fully methylated allele. This phenomenon deserves further study.
Our method has limitations. Because it is based on the RRBS technique, it can detect 10% of the CpG sites in the entire genome at most, leaving 90% of the CpG sites as intractable (Gu et al. 2010, 2011a). By combining all of the steps prior to PCR amplification into a one-tube reaction, we maximally reduce the sample losses arising from the use of multiple purification steps. However, the dramatic DNA degradation that occurs during bisulfite conversion (another major potential hurdle to single-cell RRBS) remains unresolved. In the future, additional strategies are needed to overcome this problem and to further improve the coverage of single-cell methylome analysis techniques. Moreover, when 20 mESCs were pooled together, we recovered only 63% of the CpG sites compared with those recovered using RRBS with bulk mESCs (Fig. 2A). It is possible that some CpG sites are more difficult for our scRRBS technique to detect. It is also possible that bias existed from cell to cell when a small number of cells were pooled together for RRBS using our method. Second, we cannot discriminate between 5mC and 5hmC using bisulfite sequencing alone (Wossidlo et al. 2011; Bock 2012), although in the majority of somatic cell types, the frequency of 5hmC modifications is significantly lower than that of 5mC (Wu and Zhang 2011). In the future, it may be possible to combine our single-cell RRBS method with the TAB-seq or oxBS-seq technique to develop a method that can detect single-cell hydroxymethylomes (Booth et al. 2012; Yu et al. 2012). Third, the RRBS technique provides relatively poor coverage for imprinting loci in general. Thus, our scRRBS method cannot clearly identify the imprinting status of these loci within an individual cell. To our knowledge, no DNA methylation assay for even just an individual imprinting locus within an individual cell is currently available.
Methods
Isolation of zygotes, oocytes, and sperm
Four- to five-week-old female C57BL/6N-strain mice were injected initially with 5 IU PMSG (Sigma), followed by 5 IU hCG (Sigma) 45 h later to super-ovulate the mature oocytes. These super-ovulated mice were either euthanized to collect oocytes or mated with 129S2/Sv male mice to obtain male and female pronuclei from the zygotes. The metaphase II oocytes were collected from the oviduct ampulla, and the naked oocytes and polar bodies were obtained via treatment with acidic Tyrode's solution (Sigma) to remove the zona pellucida. The spermatozoa were obtained from the caudal epididymides of adult 129S2/Sv male mice. Only spermatozoa that swam up in HTF medium (Quinn's Advantage) with vigorous motility were collected for further RRBS library constructions. All isolated cells were washed several times in 0.1% PBS-BSA solution to avoid any possible somatic contaminants. Pronuclei at different stages were collected from the zygotes precisely via a daily vaginal plug check. The zygotes were obtained by treating with hyaluronidase (Sigma) to remove any attached cumulus cells. After staining with 5 μg/mL Hoechst 33342 (Invitrogen) for 10 min, visible pronuclei were isolated by applying a Piezo Micromanipulator (PrimeTech)–assisted biopsy (Supplemental Fig. 9). Male and female pronuclei were distinguished based on their relative distances to the polar bodies.
Culture of mESCs
The mESCs were maintained without feeders on gelatinized dishes in the presence of 1000 units/mL leukemia inhibitory factor (LIF; Millipore) in Dulbecco's modified eagle's medium (DMEM/F-12; GIBCO) supplemented with 20% fetal calf serum (FCS; GIBCO) for routine passage without any modifications, as previously described (Bao et al. 2009).
Construction of single-cell RRBS sequencing libraries
Single cells were transferred into 5 μL of lysis buffer (20 mM Tris-EDTA [pH 8.0], 20 mM KCl, and 0.3% Triton X-100) using a mouth pipette, 1 mg/mL protease (Qiagen) was added, and 60 fg unmethylated lambda DNA (Fermentas) was then spiked in. The cells were then lysed for 3 h at 50°C and then heat-inactivated for 30 min at 75°C. The released naked DNA was then incubated with 9 units MspI (Fermentas) in an 18 μL reaction for 3 h at 37°C; this enzyme specifically recognizes and cuts unique DNA sequences (C^CGG). The digested DNA was then filled-in and tailed with an extra A to the 3′ blunted ends in a 20 μL reaction in the presence of 5 units of Klenow fragment (exo-; Fermentas), supplemented with 1 mM dATP (New England Biolabs), 0.1 mM dGTP (New England Biolabs), and 0.1 mM dCTP (New England Biolabs). Illumina standard premethylated indexed adaptors were then ligated with the dA-tailed DNA fragments in the presence of 30 units of highly concentrated T4 DNA ligase (Fermentas) in a 25 μL reaction. Bisulfite conversion was performed in a 150 μL reaction using the MethyCode bisulfite conversion kit (Invitrogen) following the manufacturer's standard protocol: First, we denatured the double-stranded DNA for 10 min at 98°C; then we incubated the reaction for 2.5 h at 64°C to ensure full bisulfite conversion. All of these steps were performed in a PCR thermocycler. After this step, the converted DNA was subjected to on-column desulfonation and purified using Zymo-Spin columns (Zymo) with 10 ng tRNA (Roche) as a carrier; the DNA was finally eluted in 30 μL of elution buffer. The purified DNA was then subjected to two rounds of amplification in 50 μL reactions using 1 unit of uracil stalling-free _Pfu_Turbo Cx polymerase (Stratagene) in the first round of PCR and 1 unit of Phusion high-fidelity DNA polymerase (New England Biolabs) in the second round of PCR. The conditions for the first round of PCR were as follows: 2 min at 95°C, followed by 25 cycles of 20 sec at 95°C, 30 sec at 60°C, and 1 min at 72°C. The conditions for the second round of PCR were as follows: 2 min at 98°C, followed by 22 cycles of 10 sec at 98°C, 30 sec at 60°C, and 1 min at 72°C. After PCR enrichment, DNA fragments between 200 and 500 bp were size-selected and recovered after resolving on a 12% native polyacrylamide TBE gel. The final libraries were assessed using Fragment Analyzer (Advanced Analytical Technologies) to check size distributions (Supplemental Fig. 10) and quantified using a standard curve-based qPCR assay (Agilent). To exclude the possibility of significant contamination of the scRRBS, we performed negative controls by omitting the single cell during the cell-picking step. That is, we only transferred the carryover buffer into the lysis buffer and performed all of the following steps in the same way as for the scRRBS samples. Three samples of negative controls were used and prepared as sequencing libraries, and these were sequenced at a depth comparable to that of the single-cell samples. We mapped the data to the mouse genome using the same parameters as the single-cell samples and found that the contamination in these negative control samples was negligible (Supplemental Fig. 11). This rigorously proved that our scRRBS method is generally free of contamination. The final quality-ensured libraries were used for pair-ended deep sequencing on an Illumina HiSeq2000 Sequencer, and all clusters that passed the filter were converted into FASTQ files using a standard Illumina pipeline. When starting with bulk cells, we constructed bulk-cell RRBS libraries according to previously published protocols (Gu et al. 2011a).
Single-cell methylation-sensitive digestion coupled with nested PCR
Spermatozoa were isolated from the caudal epididymides of adult 129S2/Sv male mice, and single sperm cells were picked and lysed in 5 μL of lysis buffer (the same as for scRRBS) and then treated with 9 units of MspI (Fermentas) or 9 units of HpaII (Fermentas) in an 18 μL reaction volume for 3 h at 37°C, respectively. The samples were then subjected to nested PCR without purification. The conditions for the first round of PCR were as follows: 5 min at 94°C followed by 30 cycles of 30 sec at 94°C, 30 sec at 55°C, and 45 sec at 72°C in 100 μL reaction volumes. One microliter of the first-round PCR product was then used as a template for a 20 μL second-round PCR. The conditions for the second round of PCR were as follows: 5 min at 94°C followed by 30 cycles of 30 sec at 94°C, 30 sec at 60°C, and 45 sec at 72°C. The final PCR products were visualized on a 1.5% agarose gel (Fig. 3C). For a positive control, we used 1 ng of bulk sperm genomic DNA as the template for one round of PCR amplification (the PCR conditions were the same as those used for the second round of the single-cell sample PCR). The primers used for the nested PCR are listed in Supplemental Table 5.
Data processing
First, the raw pair-end FASTQ reads were trimmed to remove the adapter sequences and low-quality bases. The remaining truncated reads were then aligned to the mm9 mouse reference genome (downloaded from the UCSC Genome Browser) using the Bismark tool (http://www.bioinformatics.bbsrc.ac.uk/projects/bismark/) (Krueger and Andrews 2011) with the default parameters and applying a customized pairwise alignment Perl script (Supplemental Table 6). The 48,502-bp lambda DNA genome was built as an extra reference to calculate the bisulfite conversion rate. When we estimated the CpG coverage in the merged groups of two to eight ESCs, we simply added the CpG sites only if these CpG sites were captured at least once in any one of these single cells. However, when the methylation level of the merged eight single cells was computed, only CpG sites that were covered in at least six single-cell samples with no less than 3× coverage in each single cell were considered. The subsequent statistical computing and graphics were performed with customized Perl scripts and R packages (http://www.r-project.org/).
Annotation of genomic regions
High-density CpG promoter (HCP), intermediate-density CpG promoter (ICP), and low-density CpG promoter (LCP) annotations were all taken from the reference by Mikkelsen et al. (2007) without any modifications. In detail, three promoter types were defined based on the transcription start sites (TSS) of known RefSeq genes. In detail, HCP, which indicated the “CpG-rich” promoters, was identified as having a GC density ≥0.55 and the observed to expected CpG ratio (CpG O/E) ≥ 0.6; promoters with CpG O/E ≤ 0.4 were classified as LCP; the remaining nonoverlapping promoter populations (0.4 < CpG O/E < 0.6) were classified as ICP. The annotated repeat elements such as LINEs, SINEs, and LTRs were downloaded directly from the RepeatMasker track of the UCSC Genome Browser. Other regions such as CGIs, exons, and introns were downloaded from the UCSC Genome Browser. Intragenic regions were included from the TSS to the transcription termination sites (TTS), whereas the intergenic regions were defined as the complement of the intragenic regions.
Calculation of CpG site methylation levels
The methylation level of each single CpG site was estimated as the number of reported Cs (methylated) divided by the total number of reported Cs (methylated) or Ts (unmethylated) at the same position of the reference genome. For the single-cell data, we only selected CpG sites that were covered by no less than three reads in depth for the subsequent analysis, regardless of the amplification bias and errors introduced in the preparation of the libraries or high-throughput sequencing workflow. Theoretically, every covered CpG site in our single-cell RRBS method should be defined digitally as either fully methylated (100%) or unmethylated (0%), respectively. Considering the potential amplification and sequencing errors, the methylation level of ≥90% or ≤10% CpG sites was reassigned as fully methylated (100%) or unmethylated (0%), respectively. CpG sites with methylation levels ranging from 10% to 90% were discarded in the subsequent analysis. The methylation level of the sampled single cell was further described as the proportion of fully methylated CpG sites to the total CpG sites that we covered. Regarding the other RRBS data sets (using more than two cells or bulk cells as starting materials; for example, the pooled groups of five, 10, and 20 ESCs or bulk ESCs), the following cutoffs were applied: CpG sites with less than 10-fold coverage were discarded, and the remaining informative CpG sites were retained for further analysis.
Calculation of the methylation levels of the annotated genomic regions
The methylation level of each annotated genomic region in each sample was measured as the sum of the methylation level of every CpG site divided by the total number of the CpG sites that we covered in the given region. CpG sites with less than 3× coverage in the single-cell RRBS data set or less than 10× coverage in the pooled groups of five, 10, and 20 ESCs or bulk ESCs RRBS data sets were discarded.
Data reproducibility
To estimate the reproducibility of our method, the Pearson correlation coefficients for all of the samples were computed using the R command “cor” with “pairwise.complete.obs” as the value of the parameter “use.” An unsupervised hierarchical clustering analysis was performed using the “hclust” function in R software and was further integrated with a customized correlation heatmap (Fig. 2B).
5mC and 5hmC immunostaining
Zygotes collected from the mouse oviduct ampulla were fixed with 4% paraformaldehyde (Sigma) for 15 min at room temperature and washed three times in PBST, followed by permeabilization with 0.5% Triton X-100 (Sigma) for 15 min. The DNA was then denatured with 4 M HCl for 10 min and neutralized with 100 mM Tris-HCl (pH 8.5) for 15 min at room temperature. The zygotes were then blocked with 0.1% PBS-BSA (Sigma) overnight at 4°C and incubated with anti-5mc antibody (1/200, BIMECY-0500; Eurogentec) or anti-5-hmC antibody (1/500, 39769; Active Motif) for 1 h at 37°C. After washing in PBST three times, the zygotes were incubated with Alexa Fluor 568 goat anti-mouse IgG (1/500, A-11004; Invitrogen) or donkey anti-rabbit IgG-FITC (1/100, sc-2012; Santa Cruz) for 1 h at 37°C. Finally, the zygotes were mounted with 5 μg/mL DAPI (Sigma), and fluorescence was detected under an inverted fluorescence microscope (Nikon) using an EM-CCD camera. All images were acquired and analyzed using NIS-Elements BR Microscope Imaging Software (Nikon) (Supplemental Fig. 9B).
Data access
All sequencing data have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE47343.
Acknowledgments
We thank C. He, J. Qiao, X.S. Xie, and Y. Huang for their insightful discussion and useful assistance. The project was supported by grants from the National Basic Research Program of China (2012CB966704 and 2011CB966303) and the National Natural Science Foundation of China (31271543).
Footnotes
[Supplemental material is available for this article.]
References
- Bao S, Tang F, Li X, Hayashi K, Gillich A, Lao K, Surani MA 2009. Epigenetic reversion of post-implantation epiblast to pluripotent embryonic stem cells. Nature 461: 1292–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A 2002. DNA methylation patterns and epigenetic memory. Genes Dev 16: 6–21 [DOI] [PubMed] [Google Scholar]
- Bock C 2012. Analysing and interpreting DNA methylation data. Nat Rev Genet 13: 705–719 [DOI] [PubMed] [Google Scholar]
- Booth MJ, Branco MR, Ficz G, Oxley D, Krueger F, Reik W, Balasubramanian S 2012. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 336: 934–937 [DOI] [PubMed] [Google Scholar]
- Chan MM, Smith ZD, Egli D, Regev A, Meissner A 2012. Mouse ooplasm confers context-specific reprogramming capacity. Nat Genet 44: 978–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaton AM, Bird A 2011. CpG islands and the regulation of transcription. Genes Dev 25: 1010–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farthing CR, Ficz G, Ng RK, Chan CF, Andrews S, Dean W, Hemberger M, Reik W 2008. Global mapping of DNA methylation in mouse promoters reveals epigenetic reprogramming of pluripotency genes. PLoS Genet 4: e1000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira J, Carmo-Fonseca M 1997. Genome replication in early mouse embryos follows a defined temporal and spatial order. J Cell Sci 110: 889–897 [DOI] [PubMed] [Google Scholar]
- Gomez D, Shankman LS, Nguyen AT, Owens GK 2013. Detection of histone modifications at specific gene loci in single cells in histological sections. Nat Methods 10: 171–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Bock C, Mikkelsen TS, Jäger N, Smith ZD, Tomazou E, Gnirke A, Lander ES, Meissner A 2010. Genome-scale DNA methylation mapping of clinical samples at single-nucleotide resolution. Nat Methods 7: 133–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Smith ZD, Bock C, Boyle P, Gnirke A, Meissner A 2011a. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc 6: 468–481 [DOI] [PubMed] [Google Scholar]
- Gu TP, Guo F, Yang H, Wu HP, Xu GF, Liu W, Xie ZG, Shi L, He X, Jin SG, et al. 2011b. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 477: 606–610 [DOI] [PubMed] [Google Scholar]
- Hackett JA, Reddington JP, Nestor CE, Dunican DS, Branco MR, Reichmann J, Reik W, Surani MA, Adams IR, Meehan RR 2012. Promoter DNA methylation couples genome-defence mechanisms to epigenetic reprogramming in the mouse germline. Development 139: 3623–3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett JA, Sengupta R, Zylicz JJ, Murakami K, Lee C, Down TA, Surani MA 2013. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science 339: 448–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Matoba S, Zhang Y 2012. Transcriptional activation of transposable elements in mouse zygotes is independent of Tet3-mediated 5-methylcytosine oxidation. Cell Res 22: 1640–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Jin SG, Pfeifer GP, Szabó PE 2011. Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc Natl Acad Sci 108: 3642–3647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch R, Bird A 2003. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat Genet 33 (Suppl):245–254 [DOI] [PubMed] [Google Scholar]
- Kalisky T, Quake SR 2011. Single-cell genomics. Nat Methods 8: 311–314 [DOI] [PubMed] [Google Scholar]
- Krueger F, Andrews SR 2011. Bismark: A flexible aligner and methylation caller for bisulfite-seq applications. Bioinformatics 27: 1571–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurimoto K, Yabuta Y, Ohinata Y, Ono Y, Uno KD, Yamada RG, Ueda HR, Saitou M 2006. An improved single-cell cDNA amplification method for efficient high-density oligonucleotide microarray analysis. Nucleic Acids Res 34: e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E 2002. Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet 3: 662–673 [DOI] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. 2009. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462: 315–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marusyk A, Almendro V, Polyak K 2012. Intra-tumour heterogeneity: A looking glass for cancer? Nat Rev Cancer 12: 323–334 [DOI] [PubMed] [Google Scholar]
- Mayer W, Niveleau A, Walter J, Fundele R, Haaf T 2000. Demethylation of the zygotic paternal genome. Nature 403: 501–502 [DOI] [PubMed] [Google Scholar]
- Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R 2005. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res 33: 5868–5877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. 2007. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448: 553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, et al. 2011. Tumour evolution inferred by single-cell sequencing. Nature 472: 90–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Yamagata K, Hong K, Wakayama T, Zhang Y 2010. A role for the elongator complex in zygotic paternal genome demethylation. Nature 463: 554–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E 1999. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99: 247–257 [DOI] [PubMed] [Google Scholar]
- Oswald J, Engemann S, Lane N, Mayer W, Olek A, Fundele R, Dean W, Reik W, Walter J 2000. Active demethylation of the paternal genome in the mouse zygote. Curr Biol 10: 475–478 [DOI] [PubMed] [Google Scholar]
- Reik W 2007. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 447: 425–432 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Paredes M, Esteller M 2011. Cancer epigenetics reaches mainstream oncology. Nat Med 17: 330–339 [DOI] [PubMed] [Google Scholar]
- Santos F, Hendrich B, Reik W, Dean W 2002. Dynamic reprogramming of DNA methylation in the early mouse embryo. Dev Biol 241: 172–182 [DOI] [PubMed] [Google Scholar]
- Smallwood SA, Kelsey G 2012. Genome-wide analysis of DNA methylation in low cell numbers by reduced representation bisulfite sequencing. Methods Mol Biol 925: 187–197 [DOI] [PubMed] [Google Scholar]
- Smallwood SA, Tomizawa S, Krueger F, Ruf N, Carli N, Segonds-Pichon A, Sato S, Hata K, Andrews SR, Kelsey G 2011. Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nat Genet 43: 811–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, Gu H, Bock C, Gnirke A, Meissner A 2009. High-throughput bisulfite sequencing in mammalian genomes. Methods 48: 226–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZD, Chan MM, Mikkelsen TS, Gu H, Gnirke A, Regev A, Meissner A 2012. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature 484: 339–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A, et al. 2009. mRNA-seq whole-transcriptome analysis of a single cell. Nat Methods 6: 377–382 [DOI] [PubMed] [Google Scholar]
- Tang F, Barbacioru C, Nordman E, Bao S, Lee C, Wang X, Tuch BB, Heard E, Lao K, Surani MA 2011a. Deterministic and stochastic allele specific gene expression in single mouse blastomeres. PLoS ONE 6: e21208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F, Lao K, Surani MA 2011b. Development and applications of single-cell transcriptome analysis. Nat Methods 8: S6–S11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyooka Y, Shimosato D, Murakami K, Takahashi K, Niwa H 2008. Identification and characterization of subpopulations in undifferentiated ES cell culture. Development 135: 909–918 [DOI] [PubMed] [Google Scholar]
- Wossidlo M, Nakamura T, Lepikhov K, Marques CJ, Zakhartchenko V, Boiani M, Arand J, Nakano T, Reik W, Walter J 2011. 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat Commun 2: 241. [DOI] [PubMed] [Google Scholar]
- Wu H, Zhang Y 2011. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev 25: 2436–2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YN, Cui XS, Tae JC, Jin YX, Kim NH 2011. DNA synthesis and epigenetic modification during mouse oocyte fertilization by human or hamster sperm injection. J Assist Reprod Genet 28: 325–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Hon GC, Szulwach KE, Song CX, Zhang L, Kim A, Li X, Dai Q, Shen Y, Park B, et al. 2012. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell 149: 1368–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong C, Lu S, Chapman AR, Xie XS 2012. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 338: 1622–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]