Leukocyte arrest: Biomechanics and molecular mechanisms of β2 integrin activation (original) (raw)
. Author manuscript; available in PMC: 2016 May 17.
Published in final edited form as: Biorheology. 2015 Dec 16;52(5-6):353–377. doi: 10.3233/BIR-15085
Abstract
Integrins are a group of heterodimeric transmembrane receptors that play essential roles in cell–cell and cell–matrix interaction. Integrins are important in many physiological processes and diseases. Integrins acquire affinity to their ligand by undergoing molecular conformational changes called activation. Here we review the molecular biomechanics during conformational changes of integrins, integrin functions in leukocyte biorheology (adhesive functions during rolling and arrest) and molecules involved in integrin activation.
Keywords: Talin, RIAM, kindlin-3, PSGL-1, GPCR, Rap-1
1. Integrins
Integrins are αβ heterodimers that function as activation-dependent adhesion molecules at the interface between cells and immobilized ligands in the extracellular matrix or other cell surfaces [1–3]. The interactions of integrins with their ligands are broadly relevant to a multitude of physiological and disease situations, such as inflammation [4–7], immune responses [8–11], thrombosis and hemostasis [12–15], extracellular matrix assembly [1–3,16–18], tumor metastasis [15,19–22] and other cellular processes. This review is focused on the four _β_2 integrins (Table 1) among the known 24 integrins [9,23]: _α_L_β_2 (LFA-1, lymphocyte function-associated antigen 1), _α_M_β_2 (Mac-1, macrophage-1 antigen), _α_X_β_2 (p150,95), _α_D_β_2.
Table 1. _β_2 integrins expressed on leukocytes.
| α subunit | β subunit | Alternative name | Expressed in cells | Main ligands |
|---|---|---|---|---|
| _α_L (CD11a) | _β_2 (CD18) | LFA-1 | Neutrophils [24–26], T-lymphocytes [11,27,28], B-lymphocytes [28,29], Monocyte [26], Macrophage [30], Dendritic cells [31], Natural killer cells [32] | ICAM-1 [28,33–36], ICAM-2 [25,37], ICAM-3 [38], ICAM-4 [39,40], ICAM-5 [41,42], Collagen [34] |
| _α_M (CD11b) | _β_2 (CD18) | Mac-1 | Neutrophils [24,26,43], T-lymphocytes [27], B-lymphocytes [44], Monocytes [26,45], Macrophages [46], Dendritic cells [47], Natural killer cells [32] | ICAM-1 [48–50], ICAM-2 [51], ICAM-4 [39], Fibrinogen [43,45,52], Collagen [34], iC3b [34,53], Heparin [54], GPIb_α_ [55], JAM- 3 [56], Thy-1 [57], Plasminogen [58] |
| _α_X (CD11c) | _β_2 (CD18) | p150,95 | Neutrophils [59,60], T-lymphocytes [27], B-lymphocytes [61,62], Monocytes [59,63], Macrophages [59], Dendritic cells [46], Natural killer cells [32] | ICAM-1 [64,65], ICAM-2 [63], ICAM-4 [66], VCAM-1 [63], Fibrinogen [60,62,65], Collagen [34], iC3b [34,59,65,67], Heparin [68], GPIb_α_ [69], Thy-1 [70], Plasminogen [71] |
| _α_D (CD11d) | _β_2 (CD18) | Neutrophils [72,73], T-lymphocytes [27], Monocytes [72,73], Macrophages [72–74], Dendritic cells [73] | ICAM-3 [72], VCAM-1 [75,76], Fibrinogen [77], Vitronectin [77], Cyr61 [77], Plasminogen [77] |
Excellent reviews cover the structures of integrins [9,78–81]. Both α and β subunits of integrins have large ectodomains, a single membrane-spanning helix (transmembrane, TM) and, usually, a short unstructured cytoplasmic tail (Fig. 1). Typically the α and β subunits contain around 1000 and 750 amino acids, respectively [78]. Specifically, human _α_L has 1170 amino acids, _α_M has 1152 amino acids, _α_X has 1163 amino acids, _α_D has 1161 amino acids and _β_2 has 769 amino acids. All α chains of the _β_2 integrins contain an I domain with homology to von Willebrand factor (VWF) A domain. The ectodomains are composed of several domains with flexible linkers between them. Each α ectodomain has (from C to N terminus) calf-1 and 2 domains, a thigh domain, a β propeller domain, and an α I domain. The _β_2 ectodomains have a _β_-tail domain, integrin epidermal growth factor like repeat domains (I-EGF-1 to 4), a plexin-semaphorin-integrin (PSI) domain, a hybrid domain, and a β I-like domain. The ectodomains can be divided into headpiece and tailpiece as shown in Fig. 1. The α and β cytoplasmic tails of integrins are extended and flexible and can directly bind several adapter proteins with different functional effects [82–88] (Table 2).
Fig. 1.
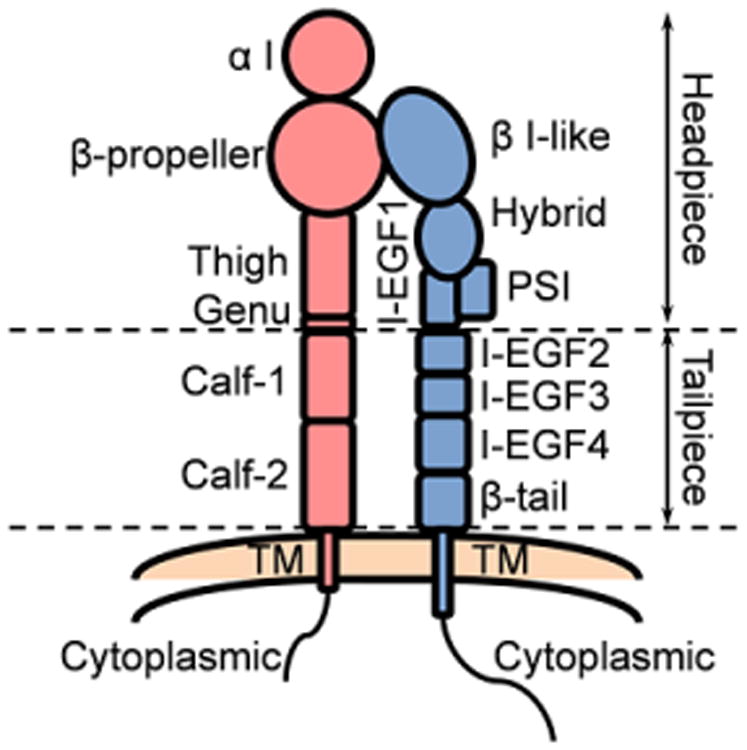
Structural schematic of the extended _β_2 integrin. α chain red, β chain blue. Subdomains and headpiece/tailpiece portions labeled.
Table 2. Cytoplasmic tail binding proteins of integrins*.
| Tail of subunits | Activating | Inactivating | Signaling | Recycling |
|---|---|---|---|---|
| α | RapL [89–92] | SHARPIN [21], Nischarin [95,96], MDGI [97], Paxillin [98,99] | PP2A [93] | Rab21 [94], p120RasGAP [94] |
| β | Talin [100–104], Kindlin [100,109–111], Cytohesin [117,118] | Dok1 [105,106], Filamin [112–114], ICAP1 [119] | _α_-actinin [103,107], 14-3-3 [115], Arg [120], Src [122,123] | Numb [108], SNX17 [116], DAB2 [121] |
_β_2 integrins, also known as leukocyte integrins [4,27], are the most important molecules in recruiting leukocytes, especially neutrophils and naïve lymphocytes, from the blood stream to sites of immune responses and inflammation. _β_2 integrins are involved in slowing down rolling [24,100,124–128], promoting arrest [100,129–137], supporting spreading and migration [128,136–143], homotypic and heterotypic cell–cell interactions [8,10,11,144–148] and phagocytosis [6,137,149–153]. Unlike other integrins, leukocyte integrins have few extracellular matrix ligands.
2. Conformational activation
Integrins regulate their adhesiveness through changes in the conformation of their ectodomain [154], which can increase ligand affinity by ∼10,000 fold [155]. The first integrin structure was solved for _α_V_β_3 integrin [156,157] starting a rich stream of publications with information about integrin ectodomain structures. It is widely accepted that _β_2 integrins have at least three conformations with different ligand binding affinities [8,9,88,158–160] (Fig. 2(A)–(C)): bent ectodomain with closed headpiece (E−H−, resting state, low affinity), extended ectodomain with closed headpiece (E+H−, intermediate [88,100,135,161–163] or low [159] affinity), and extended ectodomain with open headpiece (E+H+, high affinity). Furthermore, a structure of bent ectodomain with open headpiece (E−H+, Fig. 2(D)), which can bind small soluble ligands [164,165], was found in structures of _α_v_β_3 [164] (Electron Microscopy, EM) and _α_x_β_2 [67] (X-ray crystallography), but it is not clear whether this structure exists on the cell surface and whether it has any function.
Fig. 2.
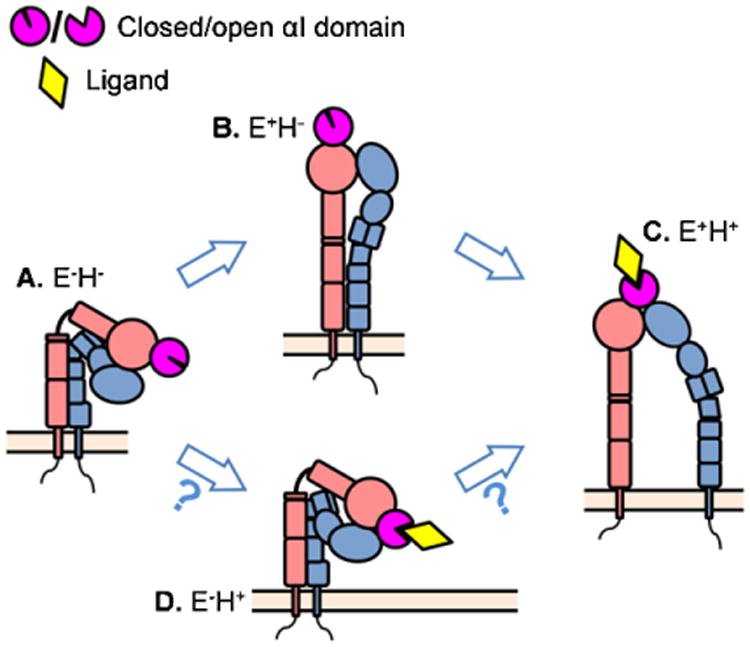
The existing conformations of the _β_2 integrins and the proposed consequences of conformational changes. (A) The resting state of _β_2 integrin has bent ectodomain with closed headpiece. (B) The resting _β_2 integrin can extend its ectodomain. This conformation may have intermediate affinity to the ligand and mediate leukocyte slow rolling. (C) Further conformational changes can induce headpiece-opening and acquire fully activated _β_2 integrin. This conformation has high affinity to the ligands and is thought to support arrest and leukocyte adhesion. (D) The structure of bent ectodomain with open headpiece was also crystallized in _β_2 integrin.
2.1. Models of integrin activation
Most studies on conformational integrin activation are limited to the truncated ectodomains, often modified by site-directed mutagenesis to stabilize conformations. In these studies, amino acids thought to be in close proximity in one of the conformations are replaced by cysteines to form disulfide bonds. By necessity, such stabilized integrins must be studied outside their natural cellular context. These experiments suggested two different models of integrin conformational activation. The “switchblade” model proposes that integrin extension is required for headpiece opening and the bent integrin first undergoes extension followed by rearrangements in the metal ion-dependent adhesion site (MIDAS) motif leading to headpiece-opening and high affinity ligand binding [8,9,154,161,162,166,167] (Fig. 2, (A) to (B) to (C)). An alternative model is the “deadbolt” model, where interactions between the headpiece and the legs keep the integrin in the closed, bent conformation and extension can only occur after the “deadbolt” is released. Two studies have shown that E−H+ integrin can exist, suggesting that integrins can open their headpiece prior to extending their ectodomain (Fig. 2, (A) to (D) to (C)). It is not known whether this conformation exists on living cells. A third model is extension first, then force-facilitated headpiece opening [10,11,135].
2.2. Extension
The resting state of _β_2 integrins shows a bent structure shaped like an inverted “V” with the low affinity headpiece closely approaching the plasma membrane [154,168], experimentally verified in live leukocytes by Förster resonance energy transfer (FRET) [169,170], in which FRET from α I domain (FITC-conjugated antibodies) to plasma membrane (Octadecyl rhodamine B, ORB) was observed in resting leukocytes and disappeared when the cells were activated. The bent ectodomain of _β_2 integrins is about 11 nm above the plasma membrane, whereas the extended ectodomain is about 23 nm (with α I domain) [78]. To allow the headpiece to bind ligands on other cells or surfaces in trans, the ectodomain needs to be extended. Integrin extension is initiated by inside-out signaling [9]. EM and FRET studies show that the α and β feet of extended integrins are more separated than those of bent integrins [154,160]. This could be achieved by lateral displacement of the cytoplasmic tails or by a change of the angle between the α and β transmembrane domains, or both. Such molecular rearrangements could conceivably provide the force necessary to extend the ectodomain. There is good evidence that _β_2 integrin extension is mediated by talin binding in the β cytoplasmic tail of integrin [88,100], thus causing the conformational changes of cytoplasmic tail and transmembrane domain [171].
2.3. Headpiece opening
The integrin headpiece includes the α I domain, the β propeller domain and the thigh domain of the α subunit and the β I-like domain, the hybrid domain, the PSI domain and the I-EGF-1 domain of the β subunit [9]. In _β_2 integrins, the ligand-binding pocket is located in the α I domain. During integrin activation, the headpiece undergoes conformational changes allowing two ligand binding sites to be exposed, one for the external ligand like ICAM-1 and one for an internal ligand formed by the α I domain, binding to the β I-like domain. On _α_M or _α_L, the MIDAS is formed by the metal ion (such as Mg2+) and the residues T209, D242 and the D140XSXS motif of the _α_M I domain [9,172], or the residues T206, D239 and the D137XSXS motif of _α_L I domain [155], respectively. It has been demonstrated by introducing disulfide bonds that wild-type isolated _α_L I-domain has low affinity for ICAM-1, whereas pulling down the _α_7 helix of the I-domain partially or completely will include the stabilized intermediate or high affinity _α_L I-domain [155,173]. The α I-domain sits on top of the β propeller domain, in close proximity to the β I-like domain. In natural integrin without disulfide bonds, it is thought that upon integrin activation, the β I-like domain binds an internal ligand (amino acid residue G310) of the _α_L I domain. This binding pulls down the _α_7 helix and stabilizes the high affinity conformation of α I [9,67,154,174]. The internal ligand binding requires that the MIDAS in the β I-like domain is open, which is thought to be induced by hybrid domain swing-out [175,176]. In the “switchblade” model, it is suggested that integrin extension enables hybrid domain swing-out [175,176], thus inducing further conformational changes of the α and β I and I-like domains and acquiring high affinity for ligand [9,174]. However, in cell-free systems it has also been observed that bent integrin can have swung-out hybrid domain and open headpiece [154,174]. This bent conformation with open headpiece [67,164,177] (E−H+) can bind (small) soluble ligands [164,165,177] prior to extension, suggesting that integrin extension is not necessary for headpiece-opening. These observations are difficult to reconcile with the switchblade model. Kindlin-3 (another important adapter protein) deficient murine neutrophils or kindlin-3 knock down HL-60 cells show a defect in headpiece-opening as reported by conformation-specific antibodies [100]. A mutant talin-1 (L325R) [178] was also demonstrated to prevent headpiece-opening of _β_2 integrin on neutrophils, which exhibit a similar phenotype as kindlin-3 knock out neutrophils [100]: both show normal slow rolling but deficient arrest.
2.4. Cytoplasmic tail separation
In live cells, the cytoplasmic domains of the integrin α and β subunits are close to one another [171] in the resting (bent) state, close enough so FRET occurs between fluorescent proteins fused to the α and β cytoplasmic domains [171]. Replacement of the α and β cytoplasmic domains with acidic and basic amino acids that form a heterodimeric _α_-helical coiled-coil forces the two cytoplasmic domain together and stabilizes integrins in their inactive state [179]. The natural integrin cytoplasmic tails have flexible structures and several binding sites for different regulatory adaptor proteins (Table 2). Important regions of the cytoplasmic tails are the NPxY motifs in the _β_2 tail, which bind talin (membrane-proximal NPxY) [83,101] and kindlin (membrane-distal NPxY) [109,110], respectively. The integrin “off” state is stabilized by binding of filamin or other phosphotyrosine-binding (PTB) domain-containing proteins. It is thought that filamin competes with talin. Tyrosine phosphorylation of the _β_-tail by some Src family kinases (SFK) has also been suggested to be involved in stabilizing the bent conformation through inhibiting talin binding [180]. β tail phosphorylation promotes the binding of PTB-containing proteins such as Dok1 (docking protein 1), thus preventing the binding of talin. The binding of other PTB-containing talin competitors, such as ICAP1 (integrin cytoplasmic domain associated protein 1), is not dependent on integrin tyrosine phosphorylation [84]. Upon talin binding, cytoplasmic tails separation occurred, resulting in diminished FRET between fluorescent proteins fused to the α and β cytoplasmic domains [171]. This separation of α and β cytoplasmic tails is thought to be critical for integrin extension and, in the switchblade model, also for headpiece opening [179].
2.5. Transmembrane domain (TMD) structure changes
The TMD of α and β are both α helices. The α and β helices cross at specific angles relative to the plane of the plasma membrane. This is stabilized by in-register alignment of side-chain arrays (Fig. 3) [181]. Nuclear magnetic resonance (NMR) spectroscopy showed the structures of the _α_IIb_β_3 TMD complex – the _α_IIb TMD helix is roughly perpendicular to the membrane while the _β_3 TMD helix is tilted [182,183]. A specific _α_-helical interface (salt-bridge) between the α and β transmembrane domains stabilizes integrin in the resting state [181,184,185]. Three models have been proposed for the movement of the transmembrane domains during integrin activation: moving apart [184], pistoning up and down [181], or an angle change [181] between α and β. The pistoning model is related to the angle change model, because it suggests that intracellular activating signals could shorten the portion of helix which is buried within the lipid bilayer, thereby changing membrane tilt angle and register with the neighboring helix to avoid hydrophobic mismatch with the fixed width of the membrane bilayer. This change in tilt angle could be the critical event in disrupting transmembrane interactions that stabilize the low-affinity conformation, leading to integrin activation [181,186]. Structural studies of a complex between talin and the integrin _β_3 cytoplasmic domain [102] suggested two talin binding sites on the _β_3 cytoplasmic domain – membrane-proximal (MP) and membrane-distal (MD) sites. Talin binding to the MD part of the β cytoplasmic domain subsequently engages the MP binding site, resulting in reorientation of the TMD (Fig. 3(A)). The interaction of the MP part with talin results in the position and angle change of MP part, thus leading to the pistoning [102] and moving apart [187] of the transmembrane helices. A recent study showed that phosphatidylinositol 4,5-bisphosphate (PIP2) can interact with residue Arg995 within the TMD and break the salt-bridge when talin is bound, thus helping integrin activation [188].
Fig. 3.
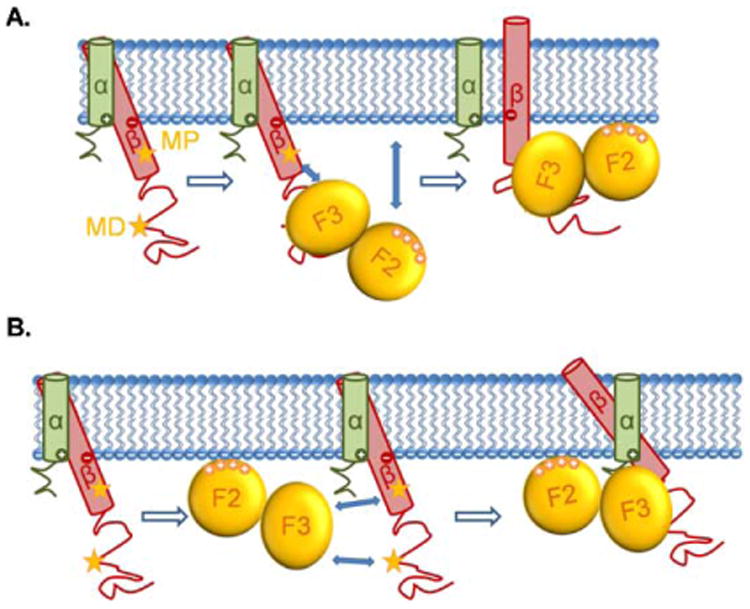
The movements of _β_2 integrins' TMD induced by talin binding. The α and β TMDs of _β_2 integrins cross at an angle at rest. The two stars indicate the talin binding sites (MP and MD) of the β cytoplasmic domain. (A) Upon binding to the talin F3 domain through the MD binding site of the β cytoplasmic domain, the attractions between talin F3 domain and the MP binding site of β cytoplasmic domain, as well as the attractions between the talin F2 domain and the plasma membrane, force the β TMD to move, resulting in an angle change between α and β TMDs, along with the pistoning of β TMD and the dissociation of α and β TMDs. (B) A more recent study suggested that talin first interacts with the plasma membrane through its F2 domain, then the attractions between talin F3 domain and the two β cytoplasmic binding sites force the β TMD to move and change their crossing angle, along with the pistoning of β TMD and the dissociation of α and β TMDs.
3. Adaptor proteins and integrin activation
The main adaptor molecules regulating integrin activation include talin, Rap1-GTP interacting adapter molecule (RIAM) and kindlin (kindlin-3 in leukocytes). Talin and kindlin are reported to bind the integrin β cytoplasmic tail directly, whereas RIAM is thought to bind and recruit talin to integrin.
3.1. Talin
Talin is a high-molecular-weight cytoskeletal protein concentrated at regions of cell-substratum contact [189]. There are two genes, talin-1 and talin-2. Talin-1 contains an N-terminal 47-kD head domain and a ∼220-kD C-terminal flexible rod domain [190]. Talin-1 interacts with both actin and integrin [103]. The head domain consists of an N-terminal FERM domain (protein 4.1, ezrin, radixin and moesin) with three subdomains (F1, F2, F3) and an F0 subdomain with no homology to known domains [191]. It is suggested that the talin head domain has a unique extended structure different from the typical cloverleaf structure seen in other FERM domains [192]. The F3 subdomain contains a PTB domain that binds the integrin β subunit tail at the membrane-proximal NxxY site and promotes integrin activation [101–104]. The talin rod domain is composed of a series of helical bundles (R1 to 13) and a C-terminal single helix dimerization domain (DD). The rod domain contains multiple binding sites for RIAM [193–195], vinculin [195,196], deleted in liver cancer 1 (DLC1) [197], synemin [198], a second binding site for integrin [199,200], binding sites for actin [87,201,202] and internal binding sites for the talin F3 subdomain [203]. It is not clear whether and how all these binding sites are occupied when the attendant integrins are resting or activated.
Talin binding to integrin β cytoplasmic tail leads to spatial changes of the tail and TMD. Two models have been proposed for these changes: the “moving apart” model, in which talin binding leads to the moving apart of the α/β “legs” as demonstrated by FRET assay [171]; and the “piston and angle change” model, in which talin binding leads to angle change, pistoning up and down along with moving apart of the α and β TMDs (Fig. 3), supported by NMR structure studies using short peptides including the TMDs [102,181,187,204,205]. It is thought that the change of the position of the α and β chains relative to each other induces the conformation changes in the ectodomain, but it is not known how the forces are transduced.
The interaction of the talin F3 subdomain with the _β_2 cytoplasmic tail is necessary but not sufficient for integrin activation. The interaction of talin and the membrane is also important [87]. Some basic residues in the F2 subdomain (Fig. 3), which is located close to the plasma membrane during talin-integrin binding, can bind acidic phospholipids in the plasma membrane [187]. This interaction is also thought to be necessary for integrin activation [204], because mutations of the membrane binding residues on F2 domain significantly decrease the binding of PAC-1 antibody, which is specific for activated _α_IIb_β_3 integrin. A recent molecular dynamics study using atomistic molecular stimulation demonstrated that PIP2 and 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) are important for talin-mediated salt bridge breaking, which leads to spatial changes of the β TMD and leads to integrin activation [188].
3.2. RIAM
RIAM is a broadly expressed adapter protein that contains an RA (Ras association)-like domain, a pleckstrin homology (PH) domain, and several proline-rich sequences. RIAM is important for talin recruitment and integrin activation in leukocytes [206,207], but not platelets [208]. A short amphipathic helix (residues 6–30) in the N-terminal region of RIAM binds talin. A fusion protein containing this helix fused to the membrane-targeting sequence of Rap1A mimics recruitment of talin to the plasma membrane by the RIAM-Rap1 complex and supports integrin activation [193]. This result shows that RIAM is sufficient to recruit talin, but does not address the question whether it is required. RIAM and vinculin compete for the same binding site on talin-1 and their binding to talin is mutually exclusive [194]. RIAM is located in the newly formed protrusions of adherent cells, whereas vinculin is located in mature focal adhesions [194]. This suggests a model where RIAM binding to talin initializes the talin recruitment and integrin activation, then RIAM is replaced by vinculin to further stabilize the activated state of integrin. Interestingly, RIAM was also shown to immunoprecipitate with kindlin-3, even before it immunoprecipitates with talin [209]. However, whether RIAM directly interact with kindlin-3 is unknown.
3.3. Kindlin-3
Kindlin is a family of focal adhesion proteins including three subtypes – kindlin-1, 2, and 3, which are also known as fermitin family homolog 1, 2 and 3 (FERMT1, FERMT2, FERMT3) respectively [210]. Kindlin-3 is expressed in leukocytes and platelets and is another essential player that binds to the integrin _β_2 subunit tail at the membrane-distal NxxY site [109,110]. Kindlin binding to _β_2 does not compete with talin binding. Kindlin shows high levels of sequence similarity to talin FERM domain [211]. The main difference is that kindlin contains a PH domain that may be involved in membrane recruitment by phosphoinositide binding [190]. The subdomain 3 in kindlin binds the distal NPxY motif of integrin β tails [109,212,213]. Kindlin-3 is expressed exclusively in cells of haematopoietic origin. Mutations in kindlin-3 are found in leukocyte adhesion deficiency III (LAD-III) patients, which have severe bleeding and immune deficiency caused by defective integrin activation in leukocytes and platelets [109,214– 217]. These findings demonstrate a critical role for kindlin-3 in leukocyte integrin activation, but the specific mechanisms remain controversial. Kindlin-3 knockout mice show a defect in neutrophil arrest but not slow rolling, which suggests that kindlin-3 is required for headpiece-opening but not extension of _β_2 integrin [100]. Kindlin-3 knockdown in the HL-60 promyeloid cell line results in a binding defect of mAb24 [218–221], which is specific for open _β_2 headpiece, but not KIM127 [222,223], which reports _β_2 integrin extension. These results suggest essential roles of kindlin-3 in headpiece-opening of _β_2 integrin [100]. An alternative hypothesis suggests that kindlin can promote talin binding to integrin cytoplasmic tail [224]. However, although kindlin, talin and integrin cytoplasmic tail can form a ternary complex, the binding of kindlin to integrins neither enhances talin–integrin binding nor increases talin targeting to the membrane [225,226], and no direct talin–kindlin interaction has been reported. Another hypothesis suggests that kindlin-binding may induce integrin clustering to enhance binding of multivalent ligands via increased avidity, rather than through conformational changes that lead to increased affinity for monovalent ligand [190,227], which is experimentally supported [228].
4. Leukocyte arrest and signaling of integrin activation
During infection or inflammation, leukocytes in blood, including granulocytes, monocytes and lymphocytes are recruited to the site of immune responses in a cascade-like fashion [17,27,136,229–234] that includes rolling, arrest, crawling, transendothelial migration and migration in tissues. Integrins play vital roles in all these processes. From rolling to arrest, leukocytes need to adhere with sufficient force to balance the force imposed by the drag and torque exerted by the flowing blood. Arrest is thought to be triggered by rapid changes in _β_2 integrin conformation through inside-out signaling, which may be triggered from surface receptors P-selectin glycoprotein ligand-1 (PSGL-1) or chemokine receptors. This requires regulation of several kinases and assembly and disassembly of multiprotein complexes that form around the cytoplasmic tails of integrins.
4.1. Rolling
The dominant molecules involved in leukocyte rolling are selectins and their ligands, whose biomechanics has been reviewed before [24,25,125,127,235–238]. The interaction of endothelial selectins with leukocyte PSGL-1 primes integrin activation, specifically promoting extension of _β_2 integrins [24,88,125,126,239]. Assays using conformation-specific antibodies have confirmed that rolling on P-or E-selectins can induce integrin extension on leukocytes within human blood [24]. Thus, _β_2 integrins also serve as “rolling receptors” [27]: they transiently engage their ligand ICAM-1, thereby reducing rolling velocity (slow rolling). Slow rolling has been proposed to be mediated by extended integrin with intermediate affinity [24,100,125–127,135,161–163,240]. This mechanism of integrin-dependent slow rolling was confirmed by using an allosteric inhibitor [125,163] that allows integrin to assume the extended, but not high affinity conformation, and by using kindlin-3 knocked out mice, in which integrin can be extended but the headpiece cannot be opened [100].
4.2. PSGL-1 signaling (Fig. 4)
Fig. 4.
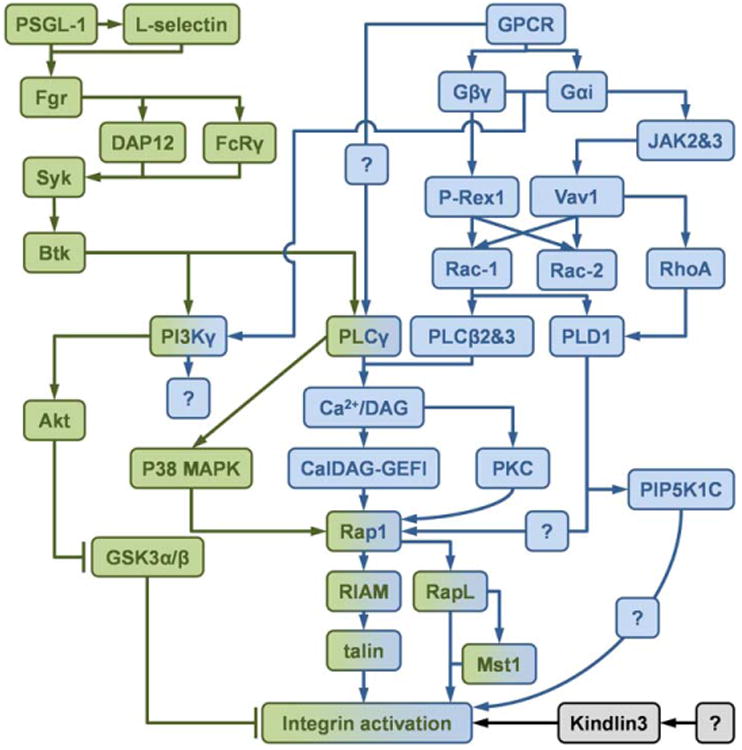
Schematics of the inside-out signaling of integrin activation. The integrin activation through inside-out signaling can be divided into PSGL-1 signaling (green) and chemokine receptor (GPCR) signaling (blue). Molecules involved in both two signaling pathway are shown in both colors. The signaling source of kindlin-3 (black) is unknown.
During leukocyte rolling, interactions of endothelial E- and P-selectins with PSGL-1 activate a key signaling pathway. PSGL-1 is the major ligand for P-selectin [239,241,242] and also binds E-selectin [236,243] and L-selectin [237] under flow conditions. PSGL-1 is preferentially located on the tips of leukocyte microvilli [25,244], which enables interactions with selectins during rolling. Granulocytes rolling on P- or E-selectin partially activate _β_2 integrin through PSGL-1 induced inside-out signaling, which was demonstrated by observation of slow rolling on ICAM-1 [24,125,126] accompanied by increased staining with the _β_2 extension reporter antibody KIM127 [24]. A mutational study of PSGL-1 showed that the cytoplasmic tail was crucial for this signal transduction, whereas the extracellular domain is responsible for leukocyte rolling [238].
Under no-flow conditions, soluble P-selectin-Fc chimeric protein was shown to induce SFK-dependent phosphorylation of Naf-1, followed by recruitment of the phosphoinositide-3-OH kinase (PI3K) p85-p110_δ_ (PI3K_δ_) heterodimer and priming of integrin activation [245]. This pathway may be important when leukocytes bind platelets, which have much higher density of P-selectin than endothelial cells. This pathway is not operative in rolling leukocytes, because PI3K_δ_ deficient leukocytes showed normal slow rolling on P-selectin and ICAM-1 under flow conditions [246].
On the neutrophil surface, PSGL-1 is closely associated with L-selectin and this does not require interaction of the L-selectin lectin domain with the known selectin-binding domain of PSGL-1 [127]. Engagement of P- or E-selectin with PSGL-1 triggers signaling through lateral interaction with L-selectin [127], followed by phosphorylation of the SFKs Fgr, Hck and Lyn [126,127,246,247]. Further studies indicated that Fgr is the main SFK required for slow rolling [126]. The phosphorylation of Fgr induces phosphorylation of the ITAM-containing adaptor proteins DAP12 or FcR_γ_ [126], which subsequently interact with and phosphorylate the tyrosine kinase Syk [126,246]. Bruton tyrosine kinase (Btk) [246,248], which is one of Tec family kinase, acts downstream from Syk, where the signaling appears to divide into two pathways: one through phospholipase C-γ_2 (PLC_γ_2); the other through PI3K_γ [248]. The downstream molecules of PLC_γ_2 are CalDAG-GEFI and p38 MAPK, which are known to activate the small GTPase Rap1a [240]. Rap-1a activation is associated with LFA-1 activation, but the details are unknown. Rap-1 has been proposed to bring talin-1 to the β_2 subunit, or it may be involved in kindlin-3 recruitment. In the PI3K_γ pathway, Akt is phosphorylated [127,248], which was reported to inhibit the integrin negative regulator GSK3_α_/β in platelets [249]. Again, the details are unknown. The PI3K_γ_ pathway could also be involved in integrin clustering.
4.3. Chemokine receptor signaling (Fig. 4)
Although PSGL-1 signaling can prime integrin activation by promoting integrin extension and cause slow rolling of leukocytes [24,100,125,126,250], chemokine receptor signaling is thought to be required for full integrin activation (probably associated with headpiece opening) and leukocyte arrest [125,130,135,251]. Chemokine receptors are G-protein coupled receptors (GPCRs), which are seven-transmembrane proteins that are identified by the coupling with the heterotrimeric G protein containing a particular α_-subunit (G_α_i) paired with a βγ -complex (G_βγ) [252]. Several chemokine receptors are expressed on leukocytes, and many of them can trigger arrest [253]. The best-studied chemokine receptor in this context is the C-X-C chemokine receptor type 2 (CXCR2), which binds the chemokine (C-X-C motif) ligands (CXCL) 1, 2, 3, 5, 6 and 7.
The chemokine receptor signaling pathway triggering integrin activation is incompletely understood [88,234]. In neutrophils, the most relevant chemokine receptor responsible for arrest is CXCR2 [254]. In vitro flow chamber assays show that immobilized CXCL1 stimulating CXCR2 leads to LFA-1 activation and neutrophil arrest on an E-selectin/intercellular adhesion molecule 1 (ICAM-1) substrate through G_α_i depended signaling [125]. Specifically, G_α_i2 but not G_α_i3 is required. G_α_i-dependent Ras activation leads to the activation of phosphoinositide 3-kinase (PI3K) by binding to its catalytic subunit [255]. The dissociated G_βγ_ subunit has been shown to interact with a number of molecules, including PI3K isoforms [256,257], PDZ proteins [258], guanine nucleotide exchange factors (GEFs) such as PIP3-dependent Rac exchanger (P-Rex) [259] and protein kinase D [260]. A recent study showed that the β subunits 1, 2, 4, and 5 in G_βγ_ are indispensable for GPCR-mediated integrin activation and leukocyte recruitment, in which Ras-related C3 botulinum toxin substrate 1 (Rac-1) and phosphoinositide phospholipase C (PLC) β_2 and β_3 are involved in downstream signaling pathway [261]. In vivo, PI3K_γ [262,263] and protein kinase C (PKC) θ [264] deficient neutrophils show no defect in arrest, but they revert to rolling again, suggesting that there is a defect in integrin bond maturation. Blockade of p44/42 and p38 mitogen-activated protein kinases (MAPK), PI3K, or PKC signaling does not affect chemokine triggered integrin activation on monocytes. The role of inositol triphosphate (IP3) and intracellular free calcium (Ca2+), the calcium-binding messenger protein calmodulin, and inositol-1,4,5-triphosphate receptors as downstream events of PLC activation is controversial [265,266]. Ca2+/diacylglycerol (DAG)-regulated guanine nucleotide exchange factor I (CalDAG-GEFI) is required for activation of Ras-related protein 1 (Rap1) and integrin in neutrophils [267]. In contrast, when the neutrophils were stimulated by fMLP, integrins were activated by activation of Rac through proto-oncogene vav (Vav1) and P-Rex 1 [268]. Two (incomplete) chemokine receptor pathways leading to activation of different integrins were demonstrated in T-lymphocytes stimulated by CXCL12 or PMA: PLC_γ → Ca2+/DAG → CalDAG-GEFI → Rap1 → LFA-1 and PLC_γ_ → Ca2+/diacylglycerol → PKC → very late antigen-4 (VLA-4) [269] respectively. Another GTPase, cell division control protein 42 (Cdc42), may be a negative regulator of integrin activation upon chemokine stimulation [270]. Phospholipase D1 (PLD-1) was demonstrated as the downstream of Rac1 and RhoA [270]. Phosphatidylinositol-4-phosphate 5-kinase type I gamma (PIP5KC) is downstream of PLD-1 and found to specifically control the transition of LFA-1 from an extended low-intermediate state to a high-affinity state [270]. In a recent study [271] CXCL12 was shown to induce Janus kinase (JAK) 2 and 3 activation in a G_α_i-independent manner. JAK2 and 3 were upstream of Vav1, which then activates Ras homolog gene family member A (RhoA) and Rac-1. Blockade of RhoA and PLD-1 inhibits the activation of Rap1a. Another study on _α_IIb_β_3 suggests that Rap1 is downstream of PKC [272].
In conclusion, the proximal (GPCR to G_α_i2 to PLC_β_) and distal (GALDAG-GEFI to Rap1 to talin-1 to integrin) parts of the pathway are clear, but the middle parts are not clear. It is also unknown whether integrin activation is local, i.e. in the microvillus where the leukocyte is in touch with chemokine, or global (whole cell).
4.4. Rap-1
As reviewed, the pathways of PSGL-1 signaling and chemokine receptor signaling may converge on Rap1. However, PSGL-1 ligation triggers only extension and chemokine receptor ligation triggers high affinity (open headpiece). This discrepancy requires that molecules other than Rap-1 are involved. During integrin activation, activated Rap1 needs to be recruited to the plasma membrane by a signaling module comprising the cytosolic adapter proteins ADAP (adhesion and degranulation promoting adapter protein) and SKAP55 (src kinase-associated protein of 55 kDa) [273]. RapL is one of the Rap1 downstream effectors, which was identified by a yeast two-hybrid screen [89,274]. RapL binds to a site consisting of two lysine residues (K1097/K1099) following the GFFKR motif, which is found only in the _α_L [90] subunit. RapL forms a complex with the serine/threonine kinase Mst1. Rap1 activation recruits both RapL and Mst1 to LFA-1 and actives Mst1 kinase activity. RapL-deficiency impairs Mst1 activation in cells. Knocking down Mst1 expression reduces integrin activation in response to chemokine stimulation [91], but it is not known how Mst1 is involved in extension or high affinity.
RIAM is another important Rap1 binding protein, which is very important in integrin activation [275]. RIAM has been reported to interact with SKAP-55 and play key roles in the recruitment of Rap1/RIAM complex to the plasma membrane [276]. The ability of Rap1 to activate integrins depends on talin binding to integrin cytoplasmic tail [272]. Rap1 induces the formation of an integrin-activation complex containing talin in combination with RIAM [272,277]. RIAM deficient mice have no defect in platelet arrest [206,208], but a severe defect in neutrophil arrest [207], macrophage adhesion [207], and lymphocyte adhesion [206,207]. A recent study shows that SLAT (SWAP-70-like adaptor of T cells) can interact with Rap-1 through its PH domain and promotes TCR-mediated, Rap1-dependent LFA-1 activation and adhesion [278]. Since SLAT is activated by T cell receptor engagement, this mechanism is unlikely to be involved in triggering arrest.
5. Conclusions and open questions
_β_2 integrins are fascinating molecular machines that translate intracellular adaptor binding (kindlin-3, talin-1, RIAM) into conformation changes of the ectodomains. This may be transmitted through moving the cytoplasmic and transmembrane domains apart, or changing their crossing angle. The molecular mechanics by which these movements are translated to ectodomain changes are unknown. _β_2 integrin activation is triggered by PSGL-1 ligation or chemokine receptor ligation or both. Under physiologic conditions, PSGL-1 ligation alone reduces the rolling velocity in a _β_2 integrin-dependent fashion. When (soluble or immobilized) chemokines are available, full integrin activation (E+H+) ensues and leads to arrest of the rolling cell. Both the PSGL-1and the chemokine receptor signaling pathways to _β_2 integrins are only partially understood. The initial hypothesis that all integrins are activated in the same way was refuted: not only are different integrins activated in different ways, but activation mechanisms even appear to be cell type-specific. The methods used to study integrin activation include crystallography of conformationally stabilized integrins, rotary shadowing EM, NMR, reporter antibodies, and functional studies of rolling and arrest. It is very challenging to integrate findings obtained with these fundamentally different and mutually exclusive methods.
Acknowledgments
This research was supported by funding from National Institutes of Health, USA (NIH, HL078784).
References
- 1.Hynes RO. Integrins: Bidirectional, allosteric signaling machines. Cell. 2002;110(6):673–87. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 2.Ruoslahti E. Integrins. The Journal of Clinical Investigation. 1991;87(1):1–5. doi: 10.1172/JCI114957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwartz MA. Transmembrane signalling by integrins. Trends in Cell Biology. 1992;2(10):304–8. doi: 10.1016/0962-8924(92)90120-c. [DOI] [PubMed] [Google Scholar]
- 4.Gahmberg CG, Valmu L, Fagerholm S, Kotovuori P, Ihanus E, Tian L, et al. Leukocyte integrins and inflammation. Cellular and Molecular Life Sciences: CMLS. 1998;54(6):549–55. doi: 10.1007/s000180050183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Savinko TS, Morrison VL, Uotila LM, Wolff CH, Alenius HT, Fagerholm SC. Functional Beta2-integrins restrict skin inflammation in vivo. The Journal of Investigative Dermatology. 2015;135(9):2249–57. doi: 10.1038/jid.2015.164. [DOI] [PubMed] [Google Scholar]
- 6.El Kebir D, Filep JG. Modulation of neutrophil apoptosis and the resolution of inflammation through β2 integrins. Frontiers in Immunology. 2013;4:60. doi: 10.3389/fimmu.2013.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zarbock A, Kempf T, Wollert KC, Vestweber D. Leukocyte integrin activation and deactivation: Novel mechanisms of balancing inflammation. Journal of Molecular Medicine. 2012;90(4):353–9. doi: 10.1007/s00109-011-0835-2. [DOI] [PubMed] [Google Scholar]
- 8.Springer TA, Dustin ML. Integrin inside-out signaling and the immunological synapse. Current Opinion in Cell Biology. 2012;24(1):107–15. doi: 10.1016/j.ceb.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annual Review of Immunology. 2007;25:619–47. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Comrie WA, Babich A, Burkhardt JK. F-actin flow drives affinity maturation and spatial organization of LFA-1 at the immunological synapse. The Journal of Cell Biology. 2015;208(4):475–91. doi: 10.1083/jcb.201406121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Comrie WA, Li S, Boyle S, Burkhardt JK. The dendritic cell cytoskeleton promotes T cell adhesion and activation by constraining ICAM-1 mobility. The Journal of Cell Biology. 2015;208(4):457–73. doi: 10.1083/jcb.201406120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shattil SJ, Newman PJ. Integrins: Dynamic scaffolds for adhesion and signaling in platelets. Blood. 2004;104(6):1606–15. doi: 10.1182/blood-2004-04-1257. [DOI] [PubMed] [Google Scholar]
- 13.Coller BS, Shattil SJ. The GPIIb/IIIa (integrin αIIbβ3) odyssey: A technology-driven saga of a receptor with twists, turns, and even a bend. Blood. 2008;112(8):3011–25. doi: 10.1182/blood-2008-06-077891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chico TJ, Chamberlain J, Gunn J, Arnold N, Bullens SL, Gadek TR, et al. Effect of selective or combined inhibition of integrins αIIbβ3 and αvβ3 on thrombosis and neointima after oversized porcine coronary angioplasty. Circulation. 2001;103(8):1135–41. doi: 10.1161/01.cir.103.8.1135. [DOI] [PubMed] [Google Scholar]
- 15.Cheresh DA. Integrins in thrombosis, wound healing and cancer. Biochemical Society Transactions. 1991;19(4):835–8. doi: 10.1042/bst0190835. [DOI] [PubMed] [Google Scholar]
- 16.Phillipson M, Kubes P. The neutrophil in vascular inflammation. Nature Medicine. 2011;17(11):1381–90. doi: 10.1038/nm.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nature Reviews Immunology. 2013;13(3):159–75. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 18.Vecino E, Heller JP, Veiga-Crespo P, Martin KR, Fawcett JW. Influence of extracellular matrix components on the expression of integrins and regeneration of adult retinal ganglion cells. PLoS ONE. 2015;10(5):e0125250. doi: 10.1371/journal.pone.0125250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Foubert P, Varner JA. Integrins in tumor angiogenesis and lymphangiogenesis. Methods in Molecular Biology. 2012;757:471–86. doi: 10.1007/978-1-61779-166-6_27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ruoslahti E. Specialization of tumour vasculature. Nature Reviews Cancer. 2002;2(2):83–90. doi: 10.1038/nrc724. [DOI] [PubMed] [Google Scholar]
- 21.Rantala JK, Pouwels J, Pellinen T, Veltel S, Laasola P, Mattila E, et al. SHARPIN is an endogenous inhibitor of β1-integrin activation. Nature Cell Biology. 2011;13(11):1315–24. doi: 10.1038/ncb2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Desgrosellier JS, Cheresh DA. Integrins in cancer: Biological implications and therapeutic opportunities. Nature Reviews Cancer. 2010;10(1):9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Byron A, Humphries JD, Askari JA, Craig SE, Mould AP, Humphries MJ. Anti-integrin monoclonal antibodies. Journal of Cell Science. 2009;122(22):4009–11. doi: 10.1242/jcs.056770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuwano Y, Spelten O, Zhang H, Ley K, Zarbock A. Rolling on E-or P-selectin induces the extended but not high-affinity conformation of LFA-1 in neutrophils. Blood. 2010;116(4):617–24. doi: 10.1182/blood-2010-01-266122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sundd P, Gutierrez E, Koltsova EK, Kuwano Y, Fukuda S, Pospieszalska MK, et al. ‘Slings’ enable neutrophil rolling at high shear. Nature. 2012;488(7411):399–403. doi: 10.1038/nature11248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sumagin R, Prizant H, Lomakina E, Waugh RE, Sarelius IH. LFA-1 and Mac-1 define characteristically different intralumenal crawling and emigration patterns for monocytes and neutrophils in situ. Journal of Immunology. 2010;185(11):7057–66. doi: 10.4049/jimmunol.1001638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hogg N, Laschinger M, Giles K, McDowall A. T-cell integrins: More than just sticking points. J Cell Sci. 2003;116(Pt 23):4695–705. doi: 10.1242/jcs.00876. [DOI] [PubMed] [Google Scholar]
- 28.Makgoba MW, Sanders ME, Ginther Luce GE, Dustin ML, Springer TA, Clark EA, et al. ICAM-1 a ligand for LFA-1-dependent adhesion of B, T and myeloid cells. Nature. 1988;331(6151):86–8. doi: 10.1038/331086a0. [DOI] [PubMed] [Google Scholar]
- 29.Arana E, Harwood NE, Batista FD. Regulation of integrin activation through the B-cell receptor. Journal of Cell Science. 2008;121(14):2279–86. doi: 10.1242/jcs.017905. [DOI] [PubMed] [Google Scholar]
- 30.Shrivastava A, Shishodia S, Sodhi A. Expression of LFA-1 adhesion molecules on cisplatin-treated macrophages. Biochimica et Biophysica Acta (BBA) – Molecular Cell Research. 1998;1402(3):269–76. doi: 10.1016/s0167-4889(98)00025-1. [DOI] [PubMed] [Google Scholar]
- 31.Balkow S, Heinz S, Schmidbauer P, Kolanus W, Holzmann B, Grabbe S, et al. LFA-1 activity state on dendritic cells regulates contact duration with T cells and promotes T-cell priming. Blood. 2010;116(11):1885–94. doi: 10.1182/blood-2009-05-224428. [DOI] [PubMed] [Google Scholar]
- 32.Morris MA, Ley K. Trafficking of natural killer cells. Current Molecular Medicine. 2004;4(4):431–8. doi: 10.2174/1566524043360609. [DOI] [PubMed] [Google Scholar]
- 33.Fisher KL, Lu J, Riddle L, Kim KJ, Presta LG, Bodary SC. Identification of the binding site in intercellular adhesion molecule 1 for its receptor, leukocyte function-associated antigen 1. Molecular Biology of the Cell. 1997;8(3):501–15. doi: 10.1091/mbc.8.3.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lahti M, Heino J, Kapyla J. Leukocyte integrins αLβ2, αMβ2 and αXβ2 as collagen receptors–receptor activation and recognition of GFOGER motif. The International Journal of Biochemistry & Cell Biology. 2013;45(7):1204–11. doi: 10.1016/j.biocel.2013.03.016. [DOI] [PubMed] [Google Scholar]
- 35.Rothlein R, Dustin ML, Marlin SD, Springer TA. A human intercellular adhesion molecule (ICAM-1) distinct from LFA-1. Journal of Immunology. 1986;137(4):1270–4. [PubMed] [Google Scholar]
- 36.Stanley P, Hogg N. The I domain of integrin LFA-1 interacts with ICAM-1 domain 1 at residue Glu-34 but not Gln-73. Journal of Biological Chemistry. 1998;273(6):3358–62. doi: 10.1074/jbc.273.6.3358. [DOI] [PubMed] [Google Scholar]
- 37.Staunton DE, Dustin ML, Springer TA. Functional cloning of ICAM-2, a cell adhesion ligand for LFA-1 homologous to ICAM-1. Nature. 1989;339(6219):61–4. doi: 10.1038/339061a0. [DOI] [PubMed] [Google Scholar]
- 38.Klickstein LB, York MR, Fougerolles AR, Springer TA. Localization of the binding site on intercellular adhesion molecule-3 (ICAM-3) for lymphocyte function-associated antigen 1 (LFA-1) The Journal of Biological Chemistry. 1996;271(39):23920–7. doi: 10.1074/jbc.271.39.23920. [DOI] [PubMed] [Google Scholar]
- 39.Ihanus E, Uotila L, Toivanen A, Stefanidakis M, Bailly P, Cartron JP, et al. Characterization of ICAM-4 binding to the I domains of the CD11a/CD18 and CD11b/CD18 leukocyte integrins. European Journal of Biochemistry/FEBS. 2003;270(8):1710–23. doi: 10.1046/j.1432-1033.2003.03528.x. [DOI] [PubMed] [Google Scholar]
- 40.Bailly P, Tontti E, Hermand P, Cartron JP, Gahmberg CG. The red cell LW blood group protein is an intercellular adhesion molecule which binds to CD11/CD18 leukocyte integrins. European Journal of Immunology. 1995;25(12):3316–20. doi: 10.1002/eji.1830251217. [DOI] [PubMed] [Google Scholar]
- 41.Tian L, Kilgannon P, Yoshihara Y, Mori K, Gallatin WM, Carpén O, et al. Binding of T lymphocytes to hippocampal neurons through ICAM-5 (telencephalin) and characterization of its interaction with the leukocyte integrin CD11a/CD18. European Journal of Immunology. 2000;30(3):810–8. doi: 10.1002/1521-4141(200003)30:3<810::AID-IMMU810>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 42.Tian L, Lappalainen J, Autero M, Hanninen S, Rauvala H, Gahmberg CG. Shedded neuronal ICAM-5 suppresses T-cell activation. Blood. 2008;111(7):3615–25. doi: 10.1182/blood-2007-09-111179. [DOI] [PubMed] [Google Scholar]
- 43.Kim HY, Skokos EA, Myer DJ, Agaba P, Gonzalez AL. αVβ3 integrin regulation of respiratory burst in fibrinogen adherent human neutrophils. Cellular and Molecular Bioengineering. 2014;7(2):231–42. doi: 10.1007/s12195-014-0322-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ding C, Ma Y, Chen X, Liu M, Cai Y, Hu X, et al. Integrin CD11b negatively regulates BCR signalling to maintain autoreactive B cell tolerance. Nature Communications. 2013;4:2813. doi: 10.1038/ncomms3813. [DOI] [PubMed] [Google Scholar]
- 45.Altieri DC, Bader R, Mannucci PM, Edgington TS. Oligospecificity of the cellular adhesion receptor Mac-1 encompasses an inducible recognition specificity for fibrinogen. The Journal of Cell Biology. 1988;107(5):1893–900. doi: 10.1083/jcb.107.5.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Metlay JP, Witmer-Pack MD, Agger R, Crowley MT, Lawless D, Steinman RM. The distinct leukocyte integrins of mouse spleen dendritic cells as identified with new hamster monoclonal antibodies. The Journal of Experimental Medicine. 1990;171(5):1753–71. doi: 10.1084/jem.171.5.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Varga G, Balkow S, Wild MK, Stadtbaeumer A, Krummen M, Rothoeft T, et al. Active MAC-1 (CD11b/CD18) on DCs inhibits full T-cell activation. Blood. 2007;109(2):661–9. doi: 10.1182/blood-2005-12-023044. [DOI] [PubMed] [Google Scholar]
- 48.Parkos CA, Colgan SP, Diamond MS, Nusrat A, Liang TW, Springer TA, et al. Expression and polarization of intercellular adhesion molecule-1 on human intestinal epithelia: Consequences for CD11b/CD18-mediated interactions with neutrophils. Molecular Medicine. 1996;2(4):489–505. [PMC free article] [PubMed] [Google Scholar]
- 49.Diamond MS, Staunton DE, de Fougerolles AR, Stacker SA, Garcia-Aguilar J, Hibbs ML, et al. ICAM-1 (CD54): A counter-receptor for Mac-1 (CD11b/CD18) The Journal of Cell Biology. 1990;111(6 Pt 2):3129–39. doi: 10.1083/jcb.111.6.3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Diamond MS, Staunton DE, Marlin SD, Springer TA. Binding of the integrin Mac-1 (CD11b/CD18) to the third immunoglobulin-like domain of ICAM-1 (CD54) and its regulation by glycosylation. Cell. 1991;65(6):961–71. doi: 10.1016/0092-8674(91)90548-d. [DOI] [PubMed] [Google Scholar]
- 51.Xie J, Li R, Kotovuori P, Vermot-Desroches C, Wijdenes J, Arnaout MA, et al. Intercellular adhesion molecule-2 (CD102) binds to the leukocyte integrin CD11b/CD18 through the A domain. Journal of Immunology. 1995;155(7):3619–28. [PubMed] [Google Scholar]
- 52.Wright SD, Weitz JI, Huang AJ, Levin SM, Silverstein SC, Loike JD. Complement receptor type three (CD11b/CD18) of human polymorphonuclear leukocytes recognizes fibrinogen. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(20):7734–8. doi: 10.1073/pnas.85.20.7734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beller DI, Springer TA, Schreiber RD. Anti-Mac-1 selectively inhibits the mouse and human type three complement receptor. The Journal of Experimental Medicine. 1982;156(4):1000–9. doi: 10.1084/jem.156.4.1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Diamond MS, Alon R, Parkos CA, Quinn MT, Springer TA. Heparin is an adhesive ligand for the leukocyte integrin Mac-1 (CD11b/CD18) The Journal of Cell Biology. 1995;130(6):1473–82. doi: 10.1083/jcb.130.6.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Simon DI, Chen Z, Xu H, Li CQ, Dong J, McIntire LV, et al. Platelet glycoprotein ibα is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18) The Journal of Experimental Medicine. 2000;192(2):193–204. doi: 10.1084/jem.192.2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Santoso S, Sachs UJ, Kroll H, Linder M, Ruf A, Preissner KT, et al. The junctional adhesion molecule 3 (JAM-3) on human platelets is a counterreceptor for the leukocyte integrin Mac-1. The Journal of Experimental Medicine. 2002;196(5):679–91. doi: 10.1084/jem.20020267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wetzel A, Chavakis T, Preissner KT, Sticherling M, Haustein UF, Anderegg U, et al. Human Thy-1 (CD90) on activated endothelial cells is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18) Journal of Immunology. 2004;172(6):3850–9. doi: 10.4049/jimmunol.172.6.3850. [DOI] [PubMed] [Google Scholar]
- 58.Lishko VK, Novokhatny VV, Yakubenko VP, Skomorovska-Prokvolit HV, Ugarova TP. Characterization of plasmino-gen as an adhesive ligand for integrins αMβ2 (Mac-1) and α5β1 (VLA-5) Blood. 2004;104(3):719–26. doi: 10.1182/blood-2003-09-3016. [DOI] [PubMed] [Google Scholar]
- 59.Myones BL, Dalzell JG, Hogg N, Ross GD. Neutrophil and monocyte cell surface p150,95 has iC3b-receptor (CR4) activity resembling CR3. The Journal of Clinical Investigation. 1988;82(2):640–51. doi: 10.1172/JCI113643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Loike JD, Sodeik B, Cao L, Leucona S, Weitz JI, Detmers PA, et al. CD11c/CD18 on neutrophils recognizes a domain at the N terminus of the Aα chain of fibrinogen. Proceedings of the National Academy of Sciences. 1991;88(3):1044–8. doi: 10.1073/pnas.88.3.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, et al. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c+ B-cell population is important for the development of autoimmunity. Blood. 2011;118(5):1305–15. doi: 10.1182/blood-2011-01-331462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Postigo AA, Corbí AL, Sánchez-Madrid F, de Landázuri MO. Regulated expression and function of CD11c/CD18 in-tegrin on human B lymphocytes. Relation between attachment to fibrinogen and triggering of proliferation through CD11c/CD18. The Journal of Experimental Medicine. 1991;174(6):1313–22. doi: 10.1084/jem.174.6.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sadhu C, Ting HJ, Lipsky B, Hensley K, Garcia-Martinez LF, Simon SI, et al. CD11c/CD18: Novel ligands and a role in delayed-type hypersensitivity. Journal of Leukocyte Biology. 2007;81(6):1395–403. doi: 10.1189/jlb.1106680. [DOI] [PubMed] [Google Scholar]
- 64.Frick C, Odermatt A, Zen K, Mandell KJ, Edens H, Portmann R, et al. Interaction of ICAM-1 with β2-integrin CD11c/CD18: Characterization of a peptide ligand that mimics a putative binding site on domain D4 of ICAM-1. European Journal of Immunology. 2005;35(12):3610–21. doi: 10.1002/eji.200425914. [DOI] [PubMed] [Google Scholar]
- 65.Choi J, Choi J, Nham SU. Characterization of the residues of αX I-domain and ICAM-1 mediating their interactions. Mol Cells. 2010;30(3):227–34. doi: 10.1007/s10059-010-0111-2. in English. [DOI] [PubMed] [Google Scholar]
- 66.Ihanus E, Uotila LM, Toivanen A, Varis M, Gahmberg CG. Red-cell ICAM-4 is a ligand for the monocyte/macrophage integrin CD11c/CD18: Characterization of the binding sites on ICAM-4. Blood. 2007;109(2):802–10. doi: 10.1182/blood-2006-04-014878. [DOI] [PubMed] [Google Scholar]
- 67.Sen M, Yuki K, Springer TA. An internal ligand-bound, metastable state of a leukocyte integrin, αXβ2. The Journal of Cell Biology. 2013;203(4):629–42. doi: 10.1083/jcb.201308083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vorup-Jensen T, Chi L, Gjelstrup LC, Jensen UB, Jewett CA, Xie C, et al. Binding between the integrin αXβ2 (CD11c/CD18) and heparin. Journal of Biological Chemistry. 2007;282(42):30869–77. doi: 10.1074/jbc.M706114200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yakubenko VP, Ustinov V, Pluskota E, López JA, Wang Y, Simon DI. Platelet-leukocyte conjugation mediated by GPIbα–αXβ2 (CD11c/CD18) interaction. Blood. 2013;122(21):1029. [Google Scholar]
- 70.Choi J, Leyton L, Nham SU. Characterization of αX I-domain binding to Thy-1. Biochemical and Biophysical Research Communications. 2005;331(2):557–61. doi: 10.1016/j.bbrc.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 71.Jongyun G, Jeongsuk C, Joo Hee L, Sang-Uk N. Identification of critical residues for plasminogen binding by the αX I-domain of the β2 integrin, αXβ2. Mol Cells. 2007;24(2):240–6. [PubMed] [Google Scholar]
- 72.Van der Vieren M, Le Trong H, Wood CL, Moore PF, John TS, Staunton DE, et al. A novel leukointegrin, αdβ2, binds preferentially to ICAM-3. Immunity. 1995;3(6):683–90. doi: 10.1016/1074-7613(95)90058-6. [DOI] [PubMed] [Google Scholar]
- 73.Miyazaki Y, Vieira-de-Abreu A, Harris ES, Shah AM, Weyrich AS, Castro-Faria-Neto HC, et al. Integrin αDβ2 (CD11d/CD18) is expressed by human circulating and tissue myeloid leukocytes and mediates inflammatory signaling. PLoS ONE. 2014;9(11):e112770. doi: 10.1371/journal.pone.0112770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Miyazaki Y, Bunting M, Stafforini DM, Harris ES, McIntyre TM, Prescott SM, et al. Integrin αDβ2 is dynamically expressed by inflamed macrophages and alters the natural history of lethal systemic infections. Journal of Immunology. 2008;180(1):590–600. doi: 10.4049/jimmunol.180.1.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Grayson MH, Van der Vieren M, Sterbinsky SA, Michael Gallatin W, Hoffman PA, Staunton DE, et al. αdβ2 integrin is expressed on human eosinophils and functions as an alternative ligand for vascular cell adhesion molecule 1 (VCAM-1) The Journal of Experimental Medicine. 1998;188(11):2187–91. doi: 10.1084/jem.188.11.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Van der Vieren M, Crowe DT, Hoekstra D, Vazeux R, Hoffman PA, Grayson MH, et al. The leukocyte integrin αDβ2 binds VCAM-1: Evidence for a binding interface between I domain and VCAM-1. Journal of Immunology. 1999;163(4):1984–90. [PubMed] [Google Scholar]
- 77.Yakubenko VP, Yadav SP, Ugarova TP. Integrin αDβ2, an adhesion receptor up-regulated on macrophage foam cells, exhibits multiligand-binding properties. Blood. 2006;107(4):1643–50. doi: 10.1182/blood-2005-06-2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Campbell ID, Humphries MJ. Integrin structure, activation, and interactions. Cold Spring Harbor Perspectives in Biology. 2011;3(3):a004994. doi: 10.1101/cshperspect.a004994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Arnaout MA, Goodman SL, Xiong JP. Structure and mechanics of integrin-based cell adhesion. Current Opinion in Cell Biology. 2007;19(5):495–507. doi: 10.1016/j.ceb.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Askari JA, Buckley PA, Mould AP, Humphries MJ. Linking integrin conformation to function. J Cell Sci. 2009;122(Pt 2):165–70. doi: 10.1242/jcs.018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bennett JS, Berger BW, Billings PC. The structure and function of platelet integrins. Journal of Thrombosis and Haemostasis: JTH. 2009;7(Suppl 1):200–5. doi: 10.1111/j.1538-7836.2009.03378.x. [DOI] [PubMed] [Google Scholar]
- 82.Morse EM, Brahme NN, Calderwood DA. Integrin cytoplasmic tail interactions. Biochemistry. 2014;53(5):810–20. doi: 10.1021/bi401596q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Legate KR, Fässler R. Mechanisms that regulate adaptor binding to β-integrin cytoplasmic tails. Journal of Cell Science. 2009;122(2):187–98. doi: 10.1242/jcs.041624. [DOI] [PubMed] [Google Scholar]
- 84.Anthis NJ, Campbell ID. The tail of integrin activation. Trends in Biochemical Sciences. 2011;36(4):191–8. doi: 10.1016/j.tibs.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Abram CL, Lowell CA. The ins and outs of leukocyte integrin signaling. Annual Review of Immunology. 2009;27:339–62. doi: 10.1146/annurev.immunol.021908.132554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pouwels J, Nevo J, Pellinen T, Ylanne J, Ivaska J. Negative regulators of integrin activity. J Cell Sci. 2012;125(Pt 14):3271–80. doi: 10.1242/jcs.093641. [DOI] [PubMed] [Google Scholar]
- 87.Calderwood DA, Campbell ID, Critchley DR. Talins and kindlins: Partners in integrin-mediated adhesion. Nature Reviews Molecular Cell Biology. 2013;14(8):503–17. doi: 10.1038/nrm3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lefort CT, Ley K. Neutrophil arrest by LFA-1 activation. Frontiers in Immunology. 2012;3:157. doi: 10.3389/fimmu.2012.00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Katagiri K, Maeda A, Shimonaka M, Kinashi T. RAPL, a Rap1-binding molecule that mediates Rap1-induced adhesion through spatial regulation of LFA-1. Nature Immunology. 2003;4(8):741–8. doi: 10.1038/ni950. [DOI] [PubMed] [Google Scholar]
- 90.Tohyama Y, Katagiri K, Pardi R, Lu C, Springer TA, Kinashi T. The critical cytoplasmic regions of the αL/β2 integrin in Rap1-induced adhesion and migration. Molecular Biology of the Cell. 2003;14(6):2570–82. doi: 10.1091/mbc.E02-09-0615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Katagiri K, Imamura M, Kinashi T. Spatiotemporal regulation of the kinase Mst1 by binding protein RAPL is critical for lymphocyte polarity and adhesion. Nature Immunology. 2006;7(9):919–28. doi: 10.1038/ni1374. [DOI] [PubMed] [Google Scholar]
- 92.Katagiri K, Ohnishi N, Kabashima K, Iyoda T, Takeda N, Shinkai Y, et al. Crucial functions of the Rap1 effector molecule RAPL in lymphocyte and dendritic cell trafficking. Nature Immunology. 2004;5(10):1045–51. doi: 10.1038/ni1111. [DOI] [PubMed] [Google Scholar]
- 93.Nho RS, Kahm J. β1-integrin-collagen interaction suppresses FoxO3a by the coordination of Akt and PP2A. The Journal of Biological Chemistry. 2010;285(19):14195–209. doi: 10.1074/jbc.M109.052845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mai A, Veltel S, Pellinen T, Padzik A, Coffey E, Marjomaki V, et al. Competitive binding of Rab21 and p120RasGAP to integrins regulates receptor traffic and migration. The Journal of Cell Biology. 2011;194(2):291–306. doi: 10.1083/jcb.201012126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Alahari SK, Reddig PJ, Juliano RL. The integrin-binding protein Nischarin regulates cell migration by inhibiting PAK. The EMBO Journal. 2004;23(14):2777–88. doi: 10.1038/sj.emboj.7600291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Alahari SK, Lee JW, Juliano RL. Nischarin, a novel protein that interacts with the integrin α5 subunit and inhibits cell migration. The Journal of Cell Biology. 2000;151(6):1141–54. doi: 10.1083/jcb.151.6.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nevo J, Mai A, Tuomi S, Pellinen T, Pentikainen OT, Heikkila P, et al. Mammary-derived growth inhibitor (MDGI) interacts with integrin α-subunits and suppresses integrin activity and invasion. Oncogene. 2010;29(49):6452–63. doi: 10.1038/onc.2010.376. [DOI] [PubMed] [Google Scholar]
- 98.Han J, Rose DM, Woodside DG, Goldfinger LE, Ginsberg MH. Integrin α4β1-dependent T cell migration requires both phosphorylation and dephosphorylation of the α4 cytoplasmic domain to regulate the reversible binding of paxillin. The Journal of Biological Chemistry. 2003;278(37):34845–53. doi: 10.1074/jbc.M304691200. [DOI] [PubMed] [Google Scholar]
- 99.Feral CC, Rose DM, Han J, Fox N, Silverman GJ, Kaushansky K, et al. Blocking the α4 integrin-paxillin interaction selectively impairs mononuclear leukocyte recruitment to an inflammatory site. The Journal of Clinical Investigation. 2006;116(3):715–23. doi: 10.1172/JCI26091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lefort CT, Rossaint J, Moser M, Petrich BG, Zarbock A, Monkley SJ, et al. Distinct roles for talin-1 and kindlin-3 in LFA-1 extension and affinity regulation. Blood. 2012;119(18):4275–82. doi: 10.1182/blood-2011-08-373118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Calderwood DA, Zent R, Grant R, Rees DJ, Hynes RO, Ginsberg MH. The talin head domain binds to integrin β subunit cytoplasmic tails and regulates integrin activation. The Journal of Biological Chemistry. 1999;274(40):28071–4. doi: 10.1074/jbc.274.40.28071. [DOI] [PubMed] [Google Scholar]
- 102.Wegener KL, Partridge AW, Han J, Pickford AR, Liddington RC, Ginsberg MH, et al. Structural basis of integrin activation by talin. Cell. 2007;128(1):171–82. doi: 10.1016/j.cell.2006.10.048. [DOI] [PubMed] [Google Scholar]
- 103.Sampath R, Gallagher PJ, Pavalko FM. Cytoskeletal interactions with the leukocyte integrin β2 cytoplasmic tail. Activation-dependent regulation of associations with talin and α-actinin. The Journal of Biological Chemistry. 1998;273(50):33588–94. doi: 10.1074/jbc.273.50.33588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Garcia-Alvarez B, de Pereda JM, Calderwood DA, Ulmer TS, Critchley D, Campbell ID, et al. Structural determinants of integrin recognition by talin. Molecular Cell. 2003;11(1):49–58. doi: 10.1016/s1097-2765(02)00823-7. [DOI] [PubMed] [Google Scholar]
- 105.Geier F, Fengos G, Iber D. A computational analysis of the dynamic roles of talin, Dok1, and PIPKI for integrin activation. PLoS ONE. 2011;6(11):e24808. doi: 10.1371/journal.pone.0024808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Gupta S, Chit JCY, Feng C, Bhunia A, Tan SM, Bhattacharjya S. An alternative phosphorylation switch in integrin β2 (CD18) tail for Dok1 binding. Scientific Reports. 2015;5:11630. doi: 10.1038/srep11630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stanley P, Smith A, McDowall A, Nicol A, Zicha D, Hogg N. Intermediate-affinity LFA-1 binds α-actinin-1 to control migration at the leading edge of the T cell. The EMBO Journal. 2008;27(1):62–75. doi: 10.1038/sj.emboj.7601959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bogdanovic O, Delfino-Machin M, Nicolas-Perez M, Gavilan MP, Gago-Rodrigues I, Fernandez-Minan A, et al. Numb/Numbl-Opo antagonism controls retinal epithelium morphogenesis by regulating integrin endocytosis. Developmental Cell. 2012;23(4):782–95. doi: 10.1016/j.devcel.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 109.Moser M, Nieswandt B, Ussar S, Pozgajova M, Fassler R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nature Medicine. 2008;14(3):325–30. doi: 10.1038/nm1722. [DOI] [PubMed] [Google Scholar]
- 110.Montanez E, Ussar S, Schifferer M, Bosl M, Zent R, Moser M, et al. Kindlin-2 controls bidirectional signaling of inte-grins. Genes & Development. 2008;22(10):1325–30. doi: 10.1101/gad.469408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ma YQ, Qin J, Wu C, Plow EF. Kindlin-2 (Mig-2): A co-activator of β3 integrins. The Journal of Cell Biology. 2008;181(3):439–46. doi: 10.1083/jcb.200710196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu J, Das M, Yang J, Ithychanda SS, Yakubenko VP, Plow EF, et al. Structural mechanism of integrin inactivation by filamin. Nature Structural & Molecular Biology. 2015;22(5):383–9. doi: 10.1038/nsmb.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Truong T, Shams H, Mofrad MR. Mechanisms of integrin and filamin binding and their interplay with talin during early focal adhesion formation. Integrative Biology. 2015;7(10):1285–96. doi: 10.1039/c5ib00133a. [DOI] [PubMed] [Google Scholar]
- 114.Sharma CP, Ezzell RM, Arnaout MA. Direct interaction of filamin (ABP-280) with the beta 2-integrin subunit CD18. Journal of Immunology. 1995;154(7):3461–70. [PubMed] [Google Scholar]
- 115.Bonet R, Vakonakis I, Campbell ID. Characterization of 14-3-3-zeta interactions with integrin tails. Journal of Molecular Biology. 2013;425(17):3060–72. doi: 10.1016/j.jmb.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bottcher RT, Stremmel C, Meves A, Meyer H, Widmaier M, Tseng HY, et al. Sorting nexin 17 prevents lysosomal degradation of β1 integrins by binding to the β1-integrin tail. Nature Cell Biology. 2012;14(6):584–92. doi: 10.1038/ncb2501. [DOI] [PubMed] [Google Scholar]
- 117.Geiger C, Nagel W, Boehm T, van Kooyk Y, Figdor CG, Kremmer E, et al. Cytohesin-1 regulates β-2 integrin-mediated adhesion through both ARF-GEF function and interaction with LFA-1. The EMBO Journal. 2000;19(11):2525–36. doi: 10.1093/emboj/19.11.2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kolanus W, Nagel W, Schiller B, Zeitlmann L, Godar S, Stockinger H, et al. αLβ2 integrin/LFA-1 binding to ICAM-1 induced by cytohesin-1, a cytoplasmic regulatory molecule. Cell. 1996;86(2):233–42. doi: 10.1016/s0092-8674(00)80095-1. [DOI] [PubMed] [Google Scholar]
- 119.Chang DD, Hoang BQ, Liu J, Springer TA. Molecular basis for interaction between Icap1α PTB domain and β1 integrin. The Journal of Biological Chemistry. 2002;277(10):8140–5. doi: 10.1074/jbc.M109031200. [DOI] [PubMed] [Google Scholar]
- 120.Simpson MA, Bradley WD, Harburger D, Parsons M, Calderwood DA, Koleske AJ. Direct interactions with the integrin β1 cytoplasmic tail activate the Abl2/Arg kinase. The Journal of Biological Chemistry. 2015;290(13):8360–72. doi: 10.1074/jbc.M115.638874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Teckchandani A, Mulkearns EE, Randolph TW, Toida N, Cooper JA. The clathrin adaptor Dab2 recruits EH domain scaffold proteins to regulate integrin β1 endocytosis. Molecular Biology of the Cell. 2012;23(15):2905–16. doi: 10.1091/mbc.E11-12-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Arias-Salgado EG, Lizano S, Sarkar S, Brugge JS, Ginsberg MH, Shattil SJ. Src kinase activation by direct interaction with the integrin β cytoplasmic domain. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(23):13298–302. doi: 10.1073/pnas.2336149100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Klinghoffer RA, Sachsenmaier C, Cooper JA, Soriano P. Src family kinases are required for integrin but not PDGFR signal transduction. The EMBO Journal. 1999;18(9):2459–71. doi: 10.1093/emboj/18.9.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dunne JL, Ballantyne CM, Beaudet AL, Ley K. Control of leukocyte rolling velocity in TNF-α-induced inflammation by LFA-1 and Mac-1. Blood. 2002;99(1):336–41. doi: 10.1182/blood.v99.1.336. [DOI] [PubMed] [Google Scholar]
- 125.Zarbock A, Lowell CA, Ley K. Spleen tyrosine kinase Syk is necessary for E-selectin-induced αLβ2 integrin-mediated rolling on intercellular adhesion molecule-1. Immunity. 2007;26(6):773–83. doi: 10.1016/j.immuni.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zarbock A, Abram CL, Hundt M, Altman A, Lowell CA, Ley K. PSGL-1 engagement by E-selectin signals through Src kinase Fgr and ITAM adapters DAP12 and FcRγ to induce slow leukocyte rolling. The Journal of Experimental Medicine. 2008;205(10):2339–47. doi: 10.1084/jem.20072660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Stadtmann A, Germena G, Block H, Boras M, Rossaint J, Sundd P, et al. The PSGL-1-L-selectin signaling complex regulates neutrophil adhesion under flow. The Journal of Experimental Medicine. 2013;210(11):2171–80. doi: 10.1084/jem.20130664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Herter JM, Rossaint J, Block H, Welch H, Zarbock A. Integrin activation by P-Rex1 is required for selectin-mediated slow leukocyte rolling and intravascular crawling. Blood. 2013;121(12):2301–10. doi: 10.1182/blood-2012-09-457085. [DOI] [PubMed] [Google Scholar]
- 129.Giagulli C, Scarpini E, Ottoboni L, Narumiya S, Butcher EC, Constantin G, et al. RhoA and zeta PKC control distinct modalities of LFA-1 activation by chemokines: Critical role of LFA-1 affinity triggering in lymphocyte in vivo homing. Immunity. 2004;20(1):25–35. doi: 10.1016/s1074-7613(03)00350-9. [DOI] [PubMed] [Google Scholar]
- 130.Lum AF, Green CE, Lee GR, Staunton DE, Simon SI. Dynamic regulation of LFA-1 activation and neutrophil arrest on intercellular adhesion molecule 1 (ICAM-1) in shear flow. The Journal of Biological Chemistry. 2002;277(23):20660–70. doi: 10.1074/jbc.M202223200. [DOI] [PubMed] [Google Scholar]
- 131.Ley K. Arrest chemokines. Microcirculation. 2003;10(3,4):289–95. doi: 10.1038/sj.mn.7800194. [DOI] [PubMed] [Google Scholar]
- 132.D'Ambrosio D, Albanesi C, Lang R, Girolomoni G, Sinigaglia F, Laudanna C. Quantitative differences in chemokine receptor engagement generate diversity in integrin-dependent lymphocyte adhesion. Journal of Immunology. 2002;169(5):2303–12. doi: 10.4049/jimmunol.169.5.2303. [DOI] [PubMed] [Google Scholar]
- 133.Constantin G, Majeed M, Giagulli C, Piccio L, Kim JY, Butcher EC, et al. Chemokines trigger immediate β2 integrin affinity and mobility changes: Differential regulation and roles in lymphocyte arrest under flow. Immunity. 2000;13(6):759–69. doi: 10.1016/s1074-7613(00)00074-1. [DOI] [PubMed] [Google Scholar]
- 134.DiVietro JA, Smith MJ, Smith BR, Petruzzelli L, Larson RS, Lawrence MB. Immobilized IL-8 triggers progressive activation of neutrophils rolling in vitro on P-selectin and intercellular adhesion molecule-1. Journal of Immunology. 2001;167(7):4017–25. doi: 10.4049/jimmunol.167.7.4017. [DOI] [PubMed] [Google Scholar]
- 135.Alon R, Feigelson SW. Chemokine-triggered leukocyte arrest: Force-regulated bi-directional integrin activation in quantal adhesive contacts. Current Opinion in Cell Biology. 2012;24(5):670–6. doi: 10.1016/j.ceb.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 136.Green CE, Schaff UY, Sarantos MR, Lum AF, Staunton DE, Simon SI. Dynamic shifts in LFA-1 affinity regulate neutrophil rolling, arrest, and transmigration on inflamed endothelium. Blood. 2006;107(5):2101–11. doi: 10.1182/blood-2005-06-2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Liu DQ, Li LM, Guo YL, Bai R, Wang C, Bian Z, et al. Signal regulatory protein α negatively regulates β2 integrin-mediated monocyte adhesion, transendothelial migration and phagocytosis. PLoS ONE. 2008;3(9):e3291. doi: 10.1371/journal.pone.0003291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Morrison VL, MacPherson M, Savinko T, San Lek H, Prescott A, Fagerholm SC. The β2 integrin-kindlin-3 interaction is essential for T-cell homing but dispensable for T-cell activation in vivo. Blood. 2013;122(8):1428–36. doi: 10.1182/blood-2013-02-484998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Constantin G, Laudanna C. A deadly migration. Immunity. 2010;32(2):147–9. doi: 10.1016/j.immuni.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 140.Schneider T, Issekutz TB, Issekutz AC. The role of α4 (CD49d) and β2 (CD18) integrins in eosinophil and neutrophil migration to allergic lung inflammation in the Brown Norway rat. American Journal of Respiratory Cell and Molecular Biology. 1999;20(3):448–57. doi: 10.1165/ajrcmb.20.3.3207. [DOI] [PubMed] [Google Scholar]
- 141.Thatte J, Dabak V, Williams MB, Braciale TJ, Ley K. LFA-1 is required for retention of effector CD8 T cells in mouse lungs. Blood. 2003;101(12):4916–22. doi: 10.1182/blood-2002-10-3159. [DOI] [PubMed] [Google Scholar]
- 142.Kim CH. Crawling of effector T cells on extracellular matrix: Role of integrins in interstitial migration in inflamed tissues. Cellular & Molecular Immunology. 2014;11(1):1–4. doi: 10.1038/cmi.2013.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Xu X, Jaeger ER, Wang X, Lagler-Ferrez E, Batalov S, Mathis NL, et al. Mst1 directs Myosin IIa partitioning of low and higher affinity integrins during T cell migration. PLoS ONE. 2014;9(8):e105561. doi: 10.1371/journal.pone.0105561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Dustin ML, Shaw AS. Costimulation: Building an immunological synapse. Science. 1999;283(5402):649–50. doi: 10.1126/science.283.5402.649. [DOI] [PubMed] [Google Scholar]
- 145.Lin W, Fan Z, Suo Y, Deng Y, Zhang M, Wang J, et al. The bullseye synapse formed between CD4+ T-cell and staphylococcal enterotoxin B-pulsed dendritic cell is a suppressive synapse in T-cell response. Immunology and Cell Biology. 2015;93(1):99–110. doi: 10.1038/icb.2014.76. [DOI] [PubMed] [Google Scholar]
- 146.Lin W, Suo Y, Deng Y, Fan Z, Zheng Y, Wei X, et al. Morphological change of CD4+ T cell during contact with DC modulates T-cell activation by accumulation of F-actin in the immunology synapse. BMC Immunology. 2015;16(1):49. doi: 10.1186/s12865-015-0108-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nolz JC, Medeiros RB, Mitchell JS, Zhu P, Freedman BD, Shimizu Y, et al. WAVE2 regulates high-affinity integrin binding by recruiting vinculin and talin to the immunological synapse. Molecular and Cellular Biology. 2007;27(17):5986–6000. doi: 10.1128/MCB.00136-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sumen C, Dustin ML, Davis MM. T cell receptor antagonism interferes with MHC clustering and integrin patterning during immunological synapse formation. The Journal of Cell Biology. 2004;166(4):579–90. doi: 10.1083/jcb.200404059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Schnitzler N, Haase G, Podbielski A, Lutticken R, Schweizer KG. A co-stimulatory signal through ICAM-f2 integrin-binding potentiates neutrophil phagocytosis. Nature Medicine. 1999;5(2):231–5. doi: 10.1038/5597. [DOI] [PubMed] [Google Scholar]
- 150.Jongstra-Bilen J, Harrison R, Grinstein S. Fcγ-receptors induce Mac-1 (CD11b/CD18) mobilization and accumulation in the phagocytic cup for optimal phagocytosis. The Journal of Biological Chemistry. 2003;278(46):45720–9. doi: 10.1074/jbc.M303704200. [DOI] [PubMed] [Google Scholar]
- 151.Dewitt S, Laffafian I, Hallett MB. Phagosomal oxidative activity during β2 integrin (CR3)-mediated phagocytosis by neutrophils is triggered by a non-restricted Ca2+ signal: Ca2+ controls time not space. J Cell Sci. 2003;116(Pt 14):2857–65. doi: 10.1242/jcs.00499. [DOI] [PubMed] [Google Scholar]
- 152.Wiedemann A, Patel JC, Lim J, Tsun A, van Kooyk Y, Caron E. Two distinct cytoplasmic regions of the β2 integrin chain regulate RhoA function during phagocytosis. The Journal of Cell Biology. 2006;172(7):1069–79. doi: 10.1083/jcb.200508075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.MacPherson M, Lek HS, Prescott A, Fagerholm SC. A systemic lupus erythematosus-associated R77H substitution in the CD11b chain of the Mac-1 integrin compromises leukocyte adhesion and phagocytosis. The Journal of Biological Chemistry. 2011;286(19):17303–10. doi: 10.1074/jbc.M110.182998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Nishida N, Xie C, Shimaoka M, Cheng Y, Walz T, Springer TA. Activation of leukocyte β2 integrins by conversion from bent to extended conformations. Immunity. 2006;25(4):583–94. doi: 10.1016/j.immuni.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 155.Shimaoka M, Xiao T, Liu JH, Yang Y, Dong Y, Jun CD, et al. Structures of the αL I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell. 2003;112(1):99–111. doi: 10.1016/s0092-8674(02)01257-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Xiong JP, Stehle T, Diefenbach B, Zhang R, Dunker R, Scott DL, et al. Crystal structure of the extracellular segment of integrin αVβ3. Science. 2001;294(5541):339–45. doi: 10.1126/science.1064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Xiong JP, Stehle T, Zhang R, Joachimiak A, Frech M, Goodman SL, et al. Crystal structure of the extracellular segment of integrin αVβ3 in complex with an Arg-Gly-Asp ligand. Science. 2002;296(5565):151–5. doi: 10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]
- 158.Chen W, Lou J, Zhu C. Forcing switch from short-to intermediate-and long-lived states of the αA domain generates LFA-1/ICAM-1 catch bonds. The Journal of Biological Chemistry. 2010;285(46):35967–78. doi: 10.1074/jbc.M110.155770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chen X, Xie C, Nishida N, Li Z, Walz T, Springer TA. Requirement of open headpiece conformation for activation of leukocyte integrin αXβ2. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(33):14727–32. doi: 10.1073/pnas.1008663107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Takagi J, Petre BM, Walz T, Springer TA. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 2002;110(5):599–611. doi: 10.1016/s0092-8674(02)00935-2. [DOI] [PubMed] [Google Scholar]
- 161.Schurpf T, Springer TA. Regulation of integrin affinity on cell surfaces. The EMBO Journal. 2011;30(23):4712–27. doi: 10.1038/emboj.2011.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zhu J, Luo BH, Xiao T, Zhang C, Nishida N, Springer TA. Structure of a complete integrin ectodomain in a physiologic resting state and activation and deactivation by applied forces. Molecular Cell. 2008;32(6):849–61. doi: 10.1016/j.molcel.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Salas A, Shimaoka M, Kogan AN, Harwood C, von Andrian UH, Springer TA. Rolling adhesion through an extended conformation of integrin αLβ2 and relation to α I and β I-like domain interaction. Immunity. 2004;20(4):393–406. doi: 10.1016/s1074-7613(04)00082-2. [DOI] [PubMed] [Google Scholar]
- 164.Adair BD, Xiong JP, Maddock C, Goodman SL, Arnaout MA, Yeager M. Three-dimensional EM structure of the ectodomain of integrin αVβ3 in a complex with fibronectin. The Journal of Cell Biology. 2005;168(7):1109–18. doi: 10.1083/jcb.200410068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zhu J, Boylan B, Luo BH, Newman PJ, Springer TA. Tests of the extension and deadbolt models of integrin activation. The Journal of Biological Chemistry. 2007;282(16):11914–20. doi: 10.1074/jbc.M700249200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Shimaoka M, Takagi J, Springer TA. Conformational regulation of integrin structure and function. Annual Review of Biophysics and Biomolecular Structure. 2002;31:485–516. doi: 10.1146/annurev.biophys.31.101101.140922. [DOI] [PubMed] [Google Scholar]
- 167.Ye F, Kim C, Ginsberg MH. Molecular mechanism of inside-out integrin regulation. Journal of Thrombosis and Haemostasis: JTH. 2011;9(Suppl 1):20–5. doi: 10.1111/j.1538-7836.2011.04355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Larson RS, Davis T, Bologa C, Semenuk G, Vijayan S, Li Y, et al. Dissociation of I domain and global conformational changes in LFA-1: Refinement of small molecule-I domain structure-activity relationships. Biochemistry. 2005;44(11):4322–31. doi: 10.1021/bi048187k. [DOI] [PubMed] [Google Scholar]
- 169.Lefort CT, Hyun YM, Kim M. Monitoring integrin activation by fluorescence resonance energy transfer. Methods in Molecular Biology. 2012;757:205–14. doi: 10.1007/978-1-61779-166-6_14. [DOI] [PubMed] [Google Scholar]
- 170.Lefort CT, Hyun YM, Schultz JB, Law FY, Waugh RE, Knauf PA, et al. Outside-in signal transmission by conformational changes in integrin Mac-1. The Journal of Immunology. 2009;183(10):6460–8. doi: 10.4049/jimmunol.0900983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kim M, Carman CV, Springer TA. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science. 2003;301(5640):1720–5. doi: 10.1126/science.1084174. [DOI] [PubMed] [Google Scholar]
- 172.Arnaout MA, Mahalingam B, Xiong JP. Integrin structure, allostery, and bidirectional signaling. Annual Review of Cell and Developmental Biology. 2005;21:381–410. doi: 10.1146/annurev.cellbio.21.090704.151217. [DOI] [PubMed] [Google Scholar]
- 173.Shimaoka M, Lu C, Palframan RT, von Andrian UH, McCormack A, Takagi J, et al. Reversibly locking a protein fold in an active conformation with a disulfide bond: Integrin αL I domains with high affinity and antagonist activity in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(11):6009–14. doi: 10.1073/pnas.101130498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Xie C, Zhu J, Chen X, Mi L, Nishida N, Springer TA. Structure of an integrin with an α I domain, complement receptor type 4. The EMBO Journal. 2010;29(3):666–79. doi: 10.1038/emboj.2009.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Takagi J, Strokovich K, Springer TA, Walz T. Structure of integrin α5β1 in complex with fibronectin. The EMBO Journal. 2003;22(18):4607–15. doi: 10.1093/emboj/cdg445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Xiao T, Takagi J, Coller BS, Wang JH, Springer TA. Structural basis for allostery in integrins and binding to fibrinogenmimetic therapeutics. Nature. 2004;432(7013):59–67. doi: 10.1038/nature02976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Gupta V, Gylling A, Alonso JL, Sugimori T, Ianakiev P, Xiong JP, et al. The β-tail domain (βTD) regulates physiologic ligand binding to integrin CD11b/CD18. Blood. 2007;109(8):3513–20. doi: 10.1182/blood-2005-11-056689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Yago T, Petrich BG, Zhang N, Liu Z, Shao B, Ginsberg MH, et al. Blocking neutrophil integrin activation prevents ischemia-reperfusion injury. The Journal of Experimental Medicine. 2015;212(8):1267–81. doi: 10.1084/jem.20142358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Lu C, Takagi J, Springer TA. Association of the membrane proximal regions of the α and β subunit cytoplasmic domains constrains an integrin in the inactive state. The Journal of Biological Chemistry. 2001;276(18):14642–8. doi: 10.1074/jbc.M100600200. [DOI] [PubMed] [Google Scholar]
- 180.Anthis NJ, Haling JR, Oxley CL, Memo M, Wegener KL, Lim CJ, et al. β integrin tyrosine phosphorylation is a conserved mechanism for regulating talin-induced integrin activation. The Journal of Biological Chemistry. 2009;284(52):36700–10. doi: 10.1074/jbc.M109.061275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Partridge AW, Liu S, Kim S, Bowie JU, Ginsberg MH. Transmembrane domain helix packing stabilizes integrin αIIbβ3 in the low affinity state. The Journal of Biological Chemistry. 2005;280(8):7294–300. doi: 10.1074/jbc.M412701200. [DOI] [PubMed] [Google Scholar]
- 182.Lau TL, Kim C, Ginsberg MH, Ulmer TS. The structure of the integrin αIIbβ3 transmembrane complex explains integrin transmembrane signalling. The EMBO Journal. 2009;28(9):1351–61. doi: 10.1038/emboj.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zhu J, Luo BH, Barth P, Schonbrun J, Baker D, Springer TA. The structure of a receptor with two associating trans-membrane domains on the cell surface: Integrin αIIbβ3. Molecular Cell. 2009;34(2):234–49. doi: 10.1016/j.molcel.2009.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Luo BH, Springer TA, Takagi J. A specific interface between integrin transmembrane helices and affinity for ligand. PLoS Biology. 2004;2(6):e153. doi: 10.1371/journal.pbio.0020153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Luo BH, Carman CV, Takagi J, Springer TA. Disrupting integrin transmembrane domain heterodimerization increases ligand binding affinity, not valency or clustering. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(10):3679–84. doi: 10.1073/pnas.0409440102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Liddington RC, Ginsberg MH. Integrin activation takes shape. The Journal of Cell Biology. 2002;158(5):833–9. doi: 10.1083/jcb.200206011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kalli AC, Campbell ID, Sansom MS. Multiscale simulations suggest a mechanism for integrin inside-out activation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(29):11890–5. doi: 10.1073/pnas.1104505108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Orłowski A, Kukkurainen S, Pöyry A, Rissanen S, Vattulainen I, Hytönen VP, et al. PIP2 and talin join forces to activate integrin. The Journal of Physical Chemistry B. 2015;119(38):12381–9. doi: 10.1021/acs.jpcb.5b06457. [DOI] [PubMed] [Google Scholar]
- 189.Burridge K, Connell L. A new protein of adhesion plaques and ruffling membranes. The Journal of Cell Biology. 1983;97(2):359–67. doi: 10.1083/jcb.97.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Moser M, Legate KR, Zent R, Fassler R. The tail of integrins, talin, and kindlins. Science. 2009;324(5929):895–9. doi: 10.1126/science.1163865. [DOI] [PubMed] [Google Scholar]
- 191.Goult BT, Bouaouina M, Elliott PR, Bate N, Patel B, Gingras AR, et al. Structure of a double ubiquitin-like domain in the talin head: A role in integrin activation. The EMBO Journal. 2010;29(6):1069–80. doi: 10.1038/emboj.2010.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Elliott PR, Goult BT, Kopp PM, Bate N, Grossmann JG, Roberts GC, et al. The structure of the talin head reveals a novel extended conformation of the FERM domain. Structure. 2010;18(10):1289–99. doi: 10.1016/j.str.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Lee HS, Lim CJ, Puzon-McLaughlin W, Shattil SJ, Ginsberg MH. RIAM activates integrins by linking talin to Ras GTPase membrane-targeting sequences. The Journal of Biological Chemistry. 2009;284(8):5119–27. doi: 10.1074/jbc.M807117200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Lee HS, Anekal P, Lim CJ, Liu CC, Ginsberg MH. Two modes of integrin activation form a binary molecular switch in adhesion maturation. Molecular Biology of the Cell. 2013;24(9):1354–62. doi: 10.1091/mbc.E12-09-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Goult BT, Zacharchenko T, Bate N, Tsang R, Hey F, Gingras AR, et al. RIAM and vinculin binding to talin are mutually exclusive and regulate adhesion assembly and turnover. The Journal of Biological Chemistry. 2013;288(12):8238–49. doi: 10.1074/jbc.M112.438119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Gingras AR, Ziegler WH, Frank R, Barsukov IL, Roberts GC, Critchley DR, et al. Mapping and consensus sequence identification for multiple vinculin binding sites within the talin rod. The Journal of Biological Chemistry. 2005;280(44):37217–24. doi: 10.1074/jbc.M508060200. [DOI] [PubMed] [Google Scholar]
- 197.Li G, Du X, Vass WC, Papageorge AG, Lowy DR, Qian X. Full activity of the deleted in liver cancer 1 (DLC1) tumor suppressor depends on an LD-like motif that binds talin and focal adhesion kinase (FAK) Proceedings of the National Academy of Sciences. 2011;108(41):17129–34. doi: 10.1073/pnas.1112122108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sun N, Critchley DR, Paulin D, Li Z, Robson RM. Identification of a repeated domain within mammalian α-synemin that interacts directly with talin. Exp Cell Res. 2008;314(8):1839–49. doi: 10.1016/j.yexcr.2008.01.034. [DOI] [PubMed] [Google Scholar]
- 199.Gingras AR, Ziegler WH, Bobkov AA, Joyce MG, Fasci D, Himmel M, et al. Structural determinants of integrin binding to the talin rod. The Journal of Biological Chemistry. 2009;284(13):8866–76. doi: 10.1074/jbc.M805937200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Rodius S, Chaloin O, Moes M, Schaffner-Reckinger E, Landrieu I, Lippens G, et al. The talin rod IBS2 α-helix interacts with the β3 integrin cytoplasmic tail membrane-proximal helix by establishing charge complementary salt bridges. Journal of Biological Chemistry. 2008;283(35):24212–23. doi: 10.1074/jbc.M709704200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Critchley DR. Biochemical and structural properties of the integrin-associated cytoskeletal protein talin. Annual Review of Biophysics. 2009;38:235–54. doi: 10.1146/annurev.biophys.050708.133744. [DOI] [PubMed] [Google Scholar]
- 202.Gingras AR, Bate N, Goult BT, Hazelwood L, Canestrelli I, Grossmann JG, et al. The structure of the C-terminal actin-binding domain of talin. The EMBO Journal. 2008;27(2):458–69. doi: 10.1038/sj.emboj.7601965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Song X, Yang J, Hirbawi J, Ye S, Perera HD, Goksoy E, et al. A novel membrane-dependent on/off switch mechanism of talin FERM domain at sites of cell adhesion. Cell Research. 2012;22(11):1533–45. doi: 10.1038/cr.2012.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Anthis NJ, Wegener KL, Ye F, Kim C, Goult BT, Lowe ED, et al. The structure of an integrin/talin complex reveals the basis of inside-out signal transduction. The EMBO Journal. 2009;28(22):3623–32. doi: 10.1038/emboj.2009.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Wegener KL, Campbell ID. Transmembrane and cytoplasmic domains in integrin activation and protein–protein interactions (review) Molecular Membrane Biology. 2008;25(5):376–87. doi: 10.1080/09687680802269886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Su W, Wynne J, Pinheiro EM, Strazza M, Mor A, Montenont E, et al. Rap1 and its effector RIAM are required for lymphocyte trafficking. Blood. 2015;126(25):2695–703. doi: 10.1182/blood-2015-05-644104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Sarah Klapproth MS, Klapproth S, Sperandio M, Fässler R, Moser M. RIAM controls bidirectional signaling of leukocyte integrins. Blood Forthcoming [Google Scholar]
- 208.Stritt S, Wolf K, Lorenz V, Vogtle T, Gupta S, Bosl MR, et al. Rap1-GTP-interacting adaptor molecule (RIAM) is dispensable for platelet integrin activation and function in mice. Blood. 2015;125(2):219–22. doi: 10.1182/blood-2014-08-597542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kliche S, Worbs T, Wang X, Degen J, Patzak I, Meineke B, et al. CCR7-mediated LFA-1 functions in T cells are regulated by 2 independent ADAP/SKAP55 modules. Blood. 2012;119(3):777–85. doi: 10.1182/blood-2011-06-362269. [DOI] [PubMed] [Google Scholar]
- 210.Karakose E, Schiller HB, Fassler R. The kindlins at a glance. J Cell Sci. 2010;123(Pt 14):2353–6. doi: 10.1242/jcs.064600. [DOI] [PubMed] [Google Scholar]
- 211.Kloeker S, Major MB, Calderwood DA, Ginsberg MH, Jones DA, Beckerle MC. The kindler syndrome protein is regulated by transforming growth factor-β and involved in integrin-mediated adhesion. Journal of Biological Chemistry. 2004;279(8):6824–33. doi: 10.1074/jbc.M307978200. [DOI] [PubMed] [Google Scholar]
- 212.Schmidt S, Nakchbandi I, Ruppert R, Kawelke N, Hess MW, Pfaller K, et al. Kindlin-3-mediated signaling from multiple integrin classes is required for osteoclast-mediated bone resorption. The Journal of Cell Biology. 2011;192(5):883–97. doi: 10.1083/jcb.201007141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Moser M, Bauer M, Schmid S, Ruppert R, Schmidt S, Sixt M, et al. Kindlin-3 is required for β2 integrin-mediated leukocyte adhesion to endothelial cells. Nature Medicine. 2009;15(3):300–5. doi: 10.1038/nm.1921. [DOI] [PubMed] [Google Scholar]
- 214.McDowall A, Svensson L, Stanley P, Patzak I, Chakravarty P, Howarth K, et al. Two mutations in the KINDLIN3 gene of a new leukocyte adhesion deficiency III patient reveal distinct effects on leukocyte function in vitro. Blood. 2010;115(23):4834–42. doi: 10.1182/blood-2009-08-238709. [DOI] [PubMed] [Google Scholar]
- 215.Crazzolara R, Maurer K, Schulze H, Zieger B, Zustin J, Schulz AS. A new mutation in the KINDLIN-3 gene ablates integrin-dependent leukocyte, platelet, and osteoclast function in a patient with leukocyte adhesion deficiency-III. Pediatric Blood & Cancer. 2015;62(9):1677–9. doi: 10.1002/pbc.25537. [DOI] [PubMed] [Google Scholar]
- 216.Gruda R, Brown AC, Grabovsky V, Mizrahi S, Gur C, Feigelson SW, et al. Loss of kindlin-3 alters the threshold for NK cell activation in human leukocyte adhesion deficiency-III. Blood. 2012;120(19):3915–24. doi: 10.1182/blood-2012-02-410795. [DOI] [PubMed] [Google Scholar]
- 217.Manevich-Mendelson E, Feigelson SW, Pasvolsky R, Aker M, Grabovsky V, Shulman Z, et al. Loss of Kindlin-3 in LAD-III eliminates LFA-1 but not VLA-4 adhesiveness developed under shear flow conditions. Blood. 2009;114(11):2344–53. doi: 10.1182/blood-2009-04-218636. [DOI] [PubMed] [Google Scholar]
- 218.Kamata T, Tieu KK, Tarui T, Puzon-McLaughlin W, Hogg N, Takada Y. The role of the CPNKEKEC sequence in the β2 subunit I domain in regulation of integrin αLβ2 (LFA-1) Journal of Immunology. 2002;168(5):2296–301. doi: 10.4049/jimmunol.168.5.2296. [DOI] [PubMed] [Google Scholar]
- 219.Lu C, Shimaoka M, Zang Q, Takagi J, Springer TA. Locking in alternate conformations of the integrin αLβ2 I domain with disulfide bonds reveals functional relationships among integrin domains. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(5):2393–8. doi: 10.1073/pnas.041618598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Yang W, Shimaoka M, Chen J, Springer TA. Activation of integrin β-subunit I-like domains by one-turn C-terminal α-helix deletions. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(8):2333–8. doi: 10.1073/pnas.0307291101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Dransfield I, Hogg N. Regulated expression of Mg2+ binding epitope on leukocyte integrin α subunits. The EMBO Journal. 1989;8(12):3759–65. doi: 10.1002/j.1460-2075.1989.tb08552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Lu C, Ferzly M, Takagi J, Springer TA. Epitope mapping of antibodies to the C-terminal region of the integrin β2 subunit reveals regions that become exposed upon receptor activation. Journal of Immunology. 2001;166(9):5629–37. doi: 10.4049/jimmunol.166.9.5629. [DOI] [PubMed] [Google Scholar]
- 223.Robinson MK, Andrew D, Rosen H, Brown D, Ortlepp S, Stephens P, et al. Antibody against the Leu-CAM β-chain (CD18) promotes both LFA-1-and CR3-dependent adhesion events. Journal of Immunology. 1992;148(4):1080–5. [PubMed] [Google Scholar]
- 224.Zhang Y, Wang H. Integrin signalling and function in immune cells. Immunology. 2012;135(4):268–75. doi: 10.1111/j.1365-2567.2011.03549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Bledzka K, Liu J, Xu Z, Perera HD, Yadav SP, Bialkowska K, et al. Spatial coordination of kindlin-2 with talin head domain in interaction with integrin β cytoplasmic tails. The Journal of Biological Chemistry. 2012;287(29):24585–94. doi: 10.1074/jbc.M111.336743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Kahner BN, Kato H, Banno A, Ginsberg MH, Shattil SJ, Ye F. Kindlins, integrin activation and the regulation of talin recruitment to αIIbβ3. PLoS ONE. 2012;7(3):e34056. doi: 10.1371/journal.pone.0034056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Ye F, Snider AK, Ginsberg MH. Talin and kindlin: The one-two punch in integrin activation. Frontiers of Medicine. 2014;8(1):6–16. doi: 10.1007/s11684-014-0317-3. [DOI] [PubMed] [Google Scholar]
- 228.Ye F, Petrich BG, Anekal P, Lefort CT, Kasirer-Friede A, Shattil SJ, et al. The mechanism of kindlin-mediated activation of integrin αIIbβ3. Current Biology: CB. 2013;23(22):2288–95. doi: 10.1016/j.cub.2013.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.McEver RP, Moore KL, Cummings RD. Leukocyte trafficking mediated by selectin-carbohydrate interactions. The Journal of Biological Chemistry. 1995;270(19):11025–8. doi: 10.1074/jbc.270.19.11025. [DOI] [PubMed] [Google Scholar]
- 230.Butcher EC. Leukocyte-endothelial cell recognition: Three (or more) steps to specificity and diversity. Cell. 1991;67(6):1033–6. doi: 10.1016/0092-8674(91)90279-8. [DOI] [PubMed] [Google Scholar]
- 231.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: The multistep paradigm. Cell. 1994;76(2):301–14. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 232.Ley K. Leukocyte adhesion to vascular endothelium. Journal of Reconstructive Microsurgery. 1992;8(6):495–503. doi: 10.1055/s-2007-1006736. [DOI] [PubMed] [Google Scholar]
- 233.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nature Reviews Immunology. 2007;7(9):678–89. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 234.Herter J, Zarbock A. Integrin regulation during leukocyte recruitment. Journal of Immunology. 2013;190(9):4451–7. doi: 10.4049/jimmunol.1203179. [DOI] [PubMed] [Google Scholar]
- 235.Sundd P, Pospieszalska MK, Cheung LS, Konstantopoulos K, Ley K. Biomechanics of leukocyte rolling. Biorheology. 2011;48(1):1–35. doi: 10.3233/BIR-2011-0579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Xia L, Sperandio M, Yago T, McDaniel JM, Cummings RD, Pearson-White S, et al. P-selectin glycoprotein ligand-1-deficient mice have impaired leukocyte tethering to E-selectin under flow. The Journal of Clinical Investigation. 2002;109(7):939–50. doi: 10.1172/JCI14151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Sperandio M, Smith ML, Forlow SB, Olson TS, Xia L, McEver RP, et al. P-selectin glycoprotein ligand-1 mediates L-selectin-dependent leukocyte rolling in venules. The Journal of Experimental Medicine. 2003;197(10):1355–63. doi: 10.1084/jem.20021854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Miner JJ, Xia L, Yago T, Kappelmayer J, Liu Z, Klopocki AG, et al. Separable requirements for cytoplasmic domain of PSGL-1 in leukocyte rolling and signaling under flow. Blood. 2008;112(5):2035–45. doi: 10.1182/blood-2008-04-149468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Zarbock A, Ley K, McEver RP, Hidalgo A. Leukocyte ligands for endothelial selectins: Specialized glycoconjugates that mediate rolling and signaling under flow. Blood. 2011;118(26):6743–51. doi: 10.1182/blood-2011-07-343566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Stadtmann A, Brinkhaus L, Mueller H, Rossaint J, Bolomini-Vittori M, Bergmeier W, et al. Rap1a activation by CalDAG-GEFI and p38 MAPK is involved in E-selectin-dependent slow leukocyte rolling. European Journal of Immunology. 2011;41(7):2074–85. doi: 10.1002/eji.201041196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Norman KE, Moore KL, McEver RP, Ley K. Leukocyte rolling in vivo is mediated by P-selectin glycoprotein ligand-1. Blood. 1995;86(12):4417–21. [PubMed] [Google Scholar]
- 242.Xia L, Ramachandran V, McDaniel JM, Nguyen KN, Cummings RD, McEver RP. N-terminal residues in murine P-selectin glycoprotein ligand-1 required for binding to murine P-selectin. Blood. 2003;101(2):552–9. doi: 10.1182/blood-2001-11-0036. [DOI] [PubMed] [Google Scholar]
- 243.Hirata T, Merrill-Skoloff G, Aab M, Yang J, Furie BC, Furie B. P-selectin glycoprotein ligand 1 (PSGL-1) is a physiological ligand for E-selectin in mediating T helper 1 lymphocyte migration. The Journal of Experimental Medicine. 2000;192(11):1669–76. doi: 10.1084/jem.192.11.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Moore KL, Patel KD, Bruehl RE, Li F, Johnson DA, Lichenstein HS, et al. P-selectin glycoprotein ligand-1 mediates rolling of human neutrophils on P-selectin. The Journal of Cell Biology. 1995;128(4):661–71. doi: 10.1083/jcb.128.4.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Wang HB, Wang JT, Zhang L, Geng ZH, Xu WL, Xu T, et al. P-selectin primes leukocyte integrin activation during inflammation. Nature Immunology. 2007;8(8):882–92. doi: 10.1038/ni1491. [DOI] [PubMed] [Google Scholar]
- 246.Yago T, Shao B, Miner JJ, Yao L, Klopocki AG, Maeda K, et al. E-selectin engages PSGL-1 and CD44 through a common signaling pathway to induce integrin αLβ2-mediated slow leukocyte rolling. Blood. 2010;116(3):485–94. doi: 10.1182/blood-2009-12-259556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Piccardoni P, Sideri R, Manarini S, Piccoli A, Martelli N, de Gaetano G, et al. Platelet/polymorphonuclear leukocyte adhesion: A new role for SRC kinases in Mac-1 adhesive function triggered by P-selectin. Blood. 2001;98(1):108–16. doi: 10.1182/blood.v98.1.108. [DOI] [PubMed] [Google Scholar]
- 248.Mueller H, Stadtmann A, Van Aken H, Hirsch E, Wang D, Ley K, et al. Tyrosine kinase Btk regulates E-selectin-mediated integrin activation and neutrophil recruitment by controlling phospholipase C (PLC) γ2 and PI3Kγ pathways. Blood. 2010;115(15):3118–27. doi: 10.1182/blood-2009-11-254185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Moore SF, van den Bosch MT, Hunter RW, Sakamoto K, Poole AW, Hers I. Dual regulation of glycogen synthase kinase 3 (GSK3)α/β by protein kinase C (PKC)α and Akt promotes thrombin-mediated integrin αIIbβ3 activation and granule secretion in platelets. The Journal of Biological Chemistry. 2013;288(6):3918–28. doi: 10.1074/jbc.M112.429936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Chesnutt BC, Smith DF, Raffler NA, Smith ML, White EJ, Ley K. Induction of LFA-1-dependent neutrophil rolling on ICAM-1 by engagement of E-selectin. Microcirculation. 2006;13(2):99–109. doi: 10.1080/10739680500466376. [DOI] [PubMed] [Google Scholar]
- 251.Rainger GE, Fisher AC, Nash GB. Endothelial-borne platelet-activating factor and interleukin-8 rapidly immobilize rolling neutrophils. The American Journal of Physiology. 1997;272(1 Pt 2):H114–22. doi: 10.1152/ajpheart.1997.272.1.H114. [DOI] [PubMed] [Google Scholar]
- 252.Qin K, Dong C, Wu G, Lambert NA. Inactive-state preassembly of Gq-coupled receptors and Gq heterotrimers. Nature Chemical Biology. 2011;7(10):740–7. doi: 10.1038/nchembio.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Ley K. Arrest chemokines. Frontiers in Immunology. 2014;5:150. doi: 10.3389/fimmu.2014.00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Smith ML, Olson TS, Ley K. CXCR2-and E-selectin-induced neutrophil arrest during inflammation in vivo. The Journal of Experimental Medicine. 2004;200(7):935–9. doi: 10.1084/jem.20040424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, et al. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370(6490):527–32. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- 256.Kerchner KR, Clay RL, McCleery G, Watson N, McIntire WE, Myung CS, et al. Differential sensitivity of phosphatidylinositol 3-kinase p110γ to isoforms of G protein βγ dimers. The Journal of Biological Chemistry. 2004;279(43):44554–62. doi: 10.1074/jbc.M406071200. [DOI] [PubMed] [Google Scholar]
- 257.Dbouk HA, Vadas O, Shymanets A, Burke JE, Salamon RS, Khalil BD, et al. G protein-coupled receptor-mediated activation of p110β by Gβγ is required for cellular transformation and invasiveness. Science Signaling. 2012;5(253):ra89. doi: 10.1126/scisignal.2003264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Li Z, Benard O, Margolskee RF. Gγ 13 interacts with PDZ domain-containing proteins. The Journal of Biological Chemistry. 2006;281(16):11066–73. doi: 10.1074/jbc.M600113200. [DOI] [PubMed] [Google Scholar]
- 259.Mayeenuddin LH, Garrison JC. Phosphorylation of P-Rex1 by the cyclic AMP-dependent protein kinase inhibits the phosphatidylinositiol (3,4,5)-trisphosphate and Gβγ-mediated regulation of its activity. The Journal of Biological Chemistry. 2006;281(4):1921–8. doi: 10.1074/jbc.M506035200. [DOI] [PubMed] [Google Scholar]
- 260.Jamora C, Yamanouye N, Van Lint J, Laudenslager J, Vandenheede JR, Faulkner DJ, et al. Gβγ-mediated regulation of Golgi organization is through the direct activation of protein kinase D. Cell. 1999;98(1):59–68. doi: 10.1016/S0092-8674(00)80606-6. [DOI] [PubMed] [Google Scholar]
- 261.Block H, Stadtmann A, Riad D, Rossaint J, Sohlbach C, Germena G, et al. Gnb isoforms orchestrate a signaling pathway comprising Rac1 and Plcβ2/3 leading to LFA-1 activation and neutrophil arrest in vivo. Blood. 2016;127(3):314–24. doi: 10.1182/blood-2015-06-651034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Smith DF, Deem TL, Bruce AC, Reutershan J, Wu D, Ley K. Leukocyte phosphoinositide-3 kinase {gamma} is required for chemokine-induced, sustained adhesion under flow in vivo. Journal of Leukocyte Biology. 2006;80(6):1491–9. doi: 10.1189/jlb.0306227. [DOI] [PubMed] [Google Scholar]
- 263.Liu L, Puri KD, Penninger JM, Kubes P. Leukocyte PI3Kγ and PI3Kδ have temporally distinct roles for leukocyte recruitment in vivo. Blood. 2007;110(4):1191–8. doi: 10.1182/blood-2006-11-060103. [DOI] [PubMed] [Google Scholar]
- 264.Bertram A, Zhang H, von Vietinghoff S, de Pablo C, Haller H, Shushakova N, et al. Protein kinase C-theta is required for murine neutrophil recruitment and adhesion strengthening under flow. Journal of Immunology. 2012;188(8):4043–51. doi: 10.4049/jimmunol.1101651. [DOI] [PubMed] [Google Scholar]
- 265.Hyduk SJ, Chan JR, Duffy ST, Chen M, Peterson MD, Waddell TK, et al. Phospholipase C, calcium, and calmodulin are critical for α4β1 integrin affinity up-regulation and monocyte arrest triggered by chemoattractants. Blood. 2007;109(1):176–84. doi: 10.1182/blood-2006-01-029199. [DOI] [PubMed] [Google Scholar]
- 266.Huo Y, Schober A, Forlow SB, Smith DF, Hyman MC, Jung S, et al. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nature Medicine. 2003;9(1):61–7. doi: 10.1038/nm810. [DOI] [PubMed] [Google Scholar]
- 267.Bergmeier W, Goerge T, Wang HW, Crittenden JR, Baldwin AC, Cifuni SM, et al. Mice lacking the signaling molecule CalDAG-GEFI represent a model for leukocyte adhesion deficiency type III. The Journal of Clinical Investigation. 2007;117(6):1699–707. doi: 10.1172/JCI30575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Lawson CD, Donald S, Anderson KE, Patton DT, Welch HC. P-Rex1 and Vav1 cooperate in the regulation of formyl-methionyl-leucyl-phenylalanine-dependent neutrophil responses. Journal of Immunology. 2011;186(3):1467–76. doi: 10.4049/jimmunol.1002738. [DOI] [PubMed] [Google Scholar]
- 269.Ghandour H, Cullere X, Alvarez A, Luscinskas FW, Mayadas TN. Essential role for Rap1 GTPase and its guanine exchange factor CalDAG-GEFI in LFA-1 but not VLA-4 integrin mediated human T-cell adhesion. Blood. 2007;110(10):3682–90. doi: 10.1182/blood-2007-03-077628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Bolomini-Vittori M, Montresor A, Giagulli C, Staunton D, Rossi B, Martinello M, et al. Regulation of conformer-specific activation of the integrin LFA-1 by a chemokine-triggered Rho signaling module. Nature Immunology. 2009;10(2):185–94. doi: 10.1038/ni.1691. [DOI] [PubMed] [Google Scholar]
- 271.Montresor A, Bolomini-Vittori M, Toffali L, Rossi B, Constantin G, Laudanna C. JAK tyrosine kinases promote hierarchical activation of Rho and Rap modules of integrin activation. The Journal of Cell Biology. 2013;203(6):1003–19. doi: 10.1083/jcb.201303067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Han J, Lim CJ, Watanabe N, Soriani A, Ratnikov B, Calderwood DA, et al. Reconstructing and deconstructing agonist-induced activation of integrin αIIbβ3. Current Biology: CB. 2006;16(18):1796–806. doi: 10.1016/j.cub.2006.08.035. [DOI] [PubMed] [Google Scholar]
- 273.Kliche S, Breitling D, Togni M, Pusch R, Heuer K, Wang X, et al. The ADAP/SKAP55 signaling module regulates T-cell receptor-mediated integrin activation through plasma membrane targeting of Rap1. Molecular and Cellular Biology. 2006;26(19):7130–44. doi: 10.1128/MCB.00331-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Carmona G, Gottig S, Orlandi A, Scheele J, Bauerle T, Jugold M, et al. Role of the small GTPase Rap1 for integrin activity regulation in endothelial cells and angiogenesis. Blood. 2009;113(2):488–97. doi: 10.1182/blood-2008-02-138438. [DOI] [PubMed] [Google Scholar]
- 275.Lafuente EM, van Puijenbroek AA, Krause M, Carman CV, Freeman GJ, Berezovskaya A, et al. RIAM, an Ena/VASP and Profilin ligand, interacts with Rap1-GTP and mediates Rap1-induced adhesion. Developmental Cell. 2004;7(4):585–95. doi: 10.1016/j.devcel.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 276.Menasche G, Kliche S, Chen EJ, Stradal TE, Schraven B, Koretzky G. RIAM links the ADAP/SKAP-55 signaling module to Rap1, facilitating T-cell-receptor-mediated integrin activation. Molecular and Cellular Biology. 2007;27(11):4070–81. doi: 10.1128/MCB.02011-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Watanabe N, Bodin L, Pandey M, Krause M, Coughlin S, Boussiotis VA, et al. Mechanisms and consequences of agonist-induced talin recruitment to platelet integrin αIIbβ3. The Journal of Cell Biology. 2008;181(7):1211–22. doi: 10.1083/jcb.200803094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Côte M, Fos C, Canonigo-Balancio AJ, Ley K, Bécart S, Altman A. SLAT promotes TCR-mediated, Rap1-dependent LFA-1 activation and adhesion through interaction of its PH domain with Rap1. Journal of Cell Science. 2015;128(23):4341–52. doi: 10.1242/jcs.172742. [DOI] [PMC free article] [PubMed] [Google Scholar]