Reduction in Mitochondrial Superoxide Dismutase Modulates Alzheimer's Disease-Like Pathology and Accelerates the Onset of Behavioral Changes in Human Amyloid Precursor Protein Transgenic Mice (original) (raw)
Abstract
Alzheimer's disease (AD) is associated with accumulations of amyloid-β (Aβ) peptides, oxidative damage, mitochondrial dysfunction, neurodegeneration, and dementia. The mitochondrial antioxidant manganese superoxide dismutase-2 (Sod2) might protect against these alterations. To test this hypothesis, we inactivated one Sod2 allele (Sod2+/−) in human amyloid precursor protein (hAPP) transgenic mice, reducing Sod2 activity to ∼50% of that in Sod2 wild-type (Sod2+/+) mice. A reduction in Sod2 activity did not obviously impair mice without hAPP/Aβ expression. In hAPP mice, however, it accelerated the onset of behavioral alterations and of deficits in prepulse inhibition of acoustic startle, a measure of sensorimotor gating. In these mice, it also worsened hAPP/Aβ-dependent depletion of microtubule-associated protein 2, a marker of neuronal dendrites. Sod2 reduction decreased amyloid plaques in the brain parenchyma but promoted the development of cerebrovascular amyloidosis, gliosis, and plaque-independent neuritic dystrophy. Sod2 reduction also increased the DNA binding activity of the transcription factor nuclear factor κB. These results suggest that Sod2 protects the aging brain against hAPP/Aβ-induced impairments. Whereas reductions in Sod2 would be expected to trigger or exacerbate neuronal and vascular pathology in AD, increasing Sod2 activity might be of therapeutic benefit.
Keywords: amyloid precursor protein, transgenic mice, superoxide dismutase-2, Alzheimer's disease, mitochondria, behavior
Introduction
Aging is an established risk factor for sporadic Alzheimer's disease (AD) (Braak and Braak, 1997; Munoz and Feldman, 2000; Lindsay et al., 2002), and age-associated oxidative damage and mitochondrial decline may contribute to its pathogenesis (Mutisya et al., 1994; Butterfield et al., 2001; Hirai et al., 2001; Praticò, 2002; Eckert et al., 2003; Coskun et al., 2004). Although multiple lines of evidence support a causal role of amyloid-β (Aβ) peptides in AD (Selkoe and Schenk, 2003; Tanzi and Bertram, 2005), direct in vivo evidence that mitochondrial dysfunction or redox imbalance triggers or worsens AD-related neuronal deficits and behavioral changes is only beginning to emerge (Calon et al., 2004; Lustbader et al., 2004; Beal, 2005).
The Aβ cascade hypothesis and mitochondrial stress hypothesis are not mutually exclusive. Aβ, which is derived from the human amyloid precursor protein (hAPP), may itself promote oxidation and compromise mitochondrial function (Hensley et al., 1994; Cardoso et al., 2004; Cutler et al., 2004; Lustbader et al., 2004; Wang et al., 2004; Crouch et al., 2005). Consistent with a copathogenic interaction between Aβ and mitochondria, patients with trisomy 21 (Down's syndrome), who typically develop early-onset AD because they have an extra copy of the APP gene, have mitochondrial dysfunction (Busciglio et al., 2002). Moreover, APP, Aβ, and γ-secretase have been detected in mitochondria (Anandatheerthavarada et al., 2003; Hansson et al., 2004; Keil et al., 2004; Lustbader et al., 2004).
To investigate links between mitochondrial pathophysiology and Aβ-induced neuronal alterations, we focused on superoxide dismutase-2 (Sod2), the main superoxide scavenger in mitochondria (Li et al., 1995; Lebovitz et al., 1996). Sod2 is enriched around amyloid plaques (Blanchard et al., 2003; Aucoin et al., 2005) and brain microvessels (Tong et al., 2005) in hAPP transgenic mice but decreased in AD brains overall (Omar et al., 1999). Depending on their genetic background, homozygous Sod2 knock-out (_Sod2_−/−) mice die in late embryonic development or postnatally within 3 weeks (Li et al., 1995; Lebovitz et al., 1996; Huang et al., 2001). When the mice are treated with a catalytic antioxidant that cannot enter the brain, systemic pathology is attenuated, survival is extended, and the mice die from a severe movement disorder characterized by cortical and brain stem neurodegeneration (Melov et al., 1998). In contrast, Sod2+/− mice are viable, fertile, and have a normal lifespan (Kokoszka et al., 2001; Van Remmen et al., 2003).
On the Sod2+/+ background, transgenic mice expressing hAPP/Aβ in neurons develop many features of AD, including age-dependent accumulation of amyloid plaques, neuritic dystrophy, reductions in presynaptic and postsynaptic proteins involved in neurotransmission, deficits in learning and memory, and increased risk of premature death (Chin et al., 2005; Kobayashi and Chen, 2005; Palop et al., 2005). Here we assessed whether Sod2 reduction can worsen these alterations. Our study was based on the hypotheses that in most cases of sporadic AD, Aβ may be necessary, but not sufficient, to elicit neuronal impairments and that pathogenic cofactors, such as reduced Sod2 activity, can determine the onset, severity, and specific nature of clinical and pathological manifestations. The results we obtained in our mouse models support these hypotheses and pinpoint Sod2 as a potentially important modulator of AD pathogenesis.
Materials and Methods
Transgenic mice and preparation of tissues.
hAPP mice were from our line J20 (Mucke et al., 2000), in which the platelet-derived growth factor (PDGF) β chain promoter directs primarily neuronal expression of an alternatively spliced minigene. This minigene encodes hAPP695, hAPP751, and hAPP770 carrying the familial AD-linked Swedish (K670N, M671L) and Indiana (V717F) mutations (hAPP770 numbering). Originally established in C57BL/6xDBA F1 hybrids, the line had been crossed for more than five generations onto a C57BL/6J inbred background using nontransgenic mice from The Jackson Laboratory (Bar Harbor, ME). Sod2+/− mice [strain C57BL/6-_Sod2tm1Leb_, stock 002973 (Lebovitz et al., 1996)] were from The Jackson Laboratory, where they had been crossed for more than five generations onto a C57BL/6J background from a 129/B6 hybrid background. Male heterozygous hAPP transgenic mice on the Sod2+/+ background were mated with female Sod2+/− mice that lacked the hAPP transgene. All mice were wild type for murine APP. Genotypes of the offspring (Sod2+/+, Sod2+/−, hAPP/Sod2+/+, and hAPP/Sod2+/−) were determined by PCR analysis of tail DNA.
For immunohistochemistry, electron microscopy, biochemistry, and gene expression analyses, mice at different ages were anesthetized and flush-perfused transcardially with 0.9% saline. Hemibrains were fixed in 4% paraformaldehyde in PBS for 48 h or microdissected into the hippocampus, entorhinal cortex, and neocortex, frozen in liquid nitrogen, and stored at −80°C. For electron microscopy, 25- to 30-month-old mice were perfused first with ice-cold heparinized (10 U/ml) saline and then with 21 ml of 2% paraformaldehyde and 0.1% glutaraldehyde in PBS. The left hemibrain was postfixed in 2% paraformaldehyde and 2% glutaraldehyde in 0.10 m sodium cacodylate for 24 h.
To decrease variability, only male mice underwent behavioral testing; all other measurements were performed on groups containing both sexes. Mice were behaviorally tested at some or all of the following ages as indicated in Results and in the figure legends: 4–7 months (adult), 10–12 months (middle-aged), and 21–23 months (old). To increase sample sizes and to ensure reproducibility of findings, most behavioral tests were performed on two separate age-matched and genotype-balanced groups of mice, typically at different time points, and the data were pooled for analysis. All protocols involving the use of mice were approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco.
Elevated plus maze.
Emotional and exploratory behaviors were assessed with an elevated plus-shaped maze consisting of two open arms and two closed arms equipped with rows of infrared photocells interfaced with a computer (Hamilton-Kinder, Poway, CA) as described previously (Raber et al., 2000a). Briefly, mice were placed individually in the center of the maze and allowed ad libitum access for 10 min. The time spent and distance moved in each of the arms were calculated from recorded beam breaks. After testing of each mouse, the equipment was thoroughly cleaned to standardize odors.
Startle reactivity and prepulse inhibition.
Acoustic startle reactivity was measured with two identical startle chambers (Hamilton-Kinder) containing a transparent nonrestrictive Plexiglas box resting on a platform inside a sound-proof, ventilated box. A high-frequency speaker mounted 15 cm above the box produced all of the acoustic stimuli. Mouse movements were detected and transduced by a piezoelectric accelerometer mounted under each cylinder. Movements were digitized and stored by a computer and interface assembly. Movements were monitored for 100 ms after the onset of each stimulus, and the maximum amplitude response (Newton) was used to determine the startle response.
For testing, mice were placed inside the chamber and exposed to a background noise of 65 dB. After a 5 min acclimation period, each session consisted of 80 trials of five types: 24 trials of a 40 ms, 120 dB (55 dB above background) startle stimulus alone (to measure maximum acoustic startle amplitude); 14 trials without startle stimulus (to measure baseline movements in the chamber); and 14 trials each of a 40 ms stimulus at 4, 8, or 16 dB above background (prepulse), followed by a 100 ms interval and a 40 ms 120 dB startle stimulus. In the first five and the last five trials of each session, the startle stimulus alone was presented to determine the degree of habituation to the startle stimulus. The other trials of startle stimulus alone and prepulse plus startle stimulus were presented in pseudorandom order with an average intertrial interval of 15 s (range, 7–23 s).
The percentage of prepulse inhibition of the startle response was calculated as follows: 100 − [(average response to prepulse plus startle stimulus/average response to startle stimulus alone) × 100]. Thus, a high value indicates high prepulse inhibition reflected by a large reduction in startle response when the prepulse preceded the startle stimulus.
Water-maze test.
Mice were tested as described previously (Raber et al., 2002) with minor modifications. Briefly, the water-maze pool (diameter, 122 cm) contained opaque water (21–22°C) with a platform (diameter, 10 cm) submerged 1.5 cm. Mice were trained to locate first a cued platform (days 1 and 2) and then a hidden platform (days 3–5 or 3–7) in two daily sessions 3.5 h apart, each consisting of three trials. To increase the likelihood that this test would detect subtle differences between genotypes, and to account for potential age-related decline in performance (Benice et al., 2006), we extended its acquisition phase by four sessions in mice that were ≥10 months. The maximum trial length was 60 s, with an intertrial interval of ∼10 min. Mice that failed to find the hidden platform were led to it and put on it for 15 s. During the cued training, the platform was moved to a different quadrant for each session. During the hidden platform training, the platform location was kept constant throughout training (in the center of the target quadrant). The starting point at which each mouse was placed into the water was changed between trials. The time to reach the platform (latency), path length, and swim speed were recorded with an EthoVision video tracking system (Noldus, Wageningen, The Netherlands). Because there were no significant genotype-dependent differences in average swim speeds in the last hidden platform training session, even in the 21- to 23-month-old mice (Sod2+/+, 16.8 ± 0.6 cm/s; Sod2+/−, 17.0 ± 0.7 cm/s; hAPP/Sod2+/+, 15.1 ± 0.8 cm/s; hAPP/Sod2+/−, 15.1 ± 0.7 cm/s), escape latency was used as the main measure for analysis. A 60 s probe trial (platform removed) was performed 1 h after the last hidden-platform session.
Rotarod test and other behavioral tests.
Motor coordination and balance were assessed by placing mice individually on a rotating drum 4 cm in diameter suspended 42 cm over a foam pad (San Diego Instruments, San Diego, CA) and measuring the time each mouse was able to maintain its balance on the rod (fall latency). The speed of the rotarod was accelerated from 5 to 35 rpm in increments of 5 rpm every 15 s. Mice that failed to stay on for at least 5 s in the first trial were placed on the rotarod for a second trial. hAPP expression and Sod2 reduction did not affect the performance of mice in the rotarod test or in the open field (tested only in mice ≥10 months of age; data not shown), indicating that behavioral changes were not attributable to impaired motor control or altered activity.
mRNA analysis.
Total RNA was extracted from brain tissues with Trizol reagent (Invitrogen, Indianapolis, IN) and further purified with RNeasy columns (Qiagen, Valencia, CA) according to the manufacturer's recommendations. To remove genomic DNA, column-bound RNAs were treated for 15 min at room temperature with RNase-free DNase (27 Kunitz units; Qiagen). RNA concentrations were determined by spectrophotometric analysis of the ratio of absorbances at 260 and 280 nm. Sod2 mRNA levels in each sample were determined in duplicate by quantitative fluorogenic reverse transcription-PCR (qfRT-PCR) using cyclophilin mRNA as a loading control. RT reactions were performed with Reverse Transcription kits (Applied Biosystems, Foster City, CA) and random hexamers plus oligo-dT (2.5 μm each) priming for first-strand cDNA synthesis in a 50 μl reaction containing 500 ng of total RNA, 1× reaction buffer, 5.5 mm MgCl2, 500 μm dNTPs, 0.4 U/μl RNase inhibitor, and 1.25 U/μl Multiscribe RT (or H2O as a “no RT control”). Reactions were incubated at 25°C for 10 min, 48°C for 30 min, and 95°C for 5 min. qfRT-PCR reactions consisted of 10 μl of diluted cDNA reaction mixture (equivalent to 4.0 ng of RNA template), 12.5 μl of SYBR Green PCR Master Mix (Applied Biosystems), and 300 nm of each forward and reverse primer in a final volume of 25 μl. The following primers (5′–3′) were used: mouse Sod2, CACAAGCACAGCCTCCCAG (forward) and CGCGTTAATGTGTGGCTCC (reverse); mouse cyclophilin, CGATGACGAGCCCTTGG (forward) and TCTGCTGTCTTTGGAACTTTGTC (reverse). Serial dilutions of pooled cDNA samples from all mice analyzed were used to generate standard curves to determine the relative levels of specific mRNAs in individual samples.
For the analysis of transforming growth factor β1 (TGF-β1) and apolipoprotein E (apoE) mRNA levels, qfRT-PCR reactions were set up as above, except the following primers (5′–3′) were used: mouse TGF-β1, AGTACAGCAAGGTCCTTGCCC (forward) and GCAGCACGGTGACGCC (reverse); mouse apoE, AAGCAACCAACCCTGGGAG (forward) and AGGCCCTGAACCGCTTCTGGGA (reverse).
Western blot analysis of Sod2 levels.
Brain tissue was homogenized with a hand-held motorized pestle (Kontes, Vineland, NJ) in 6 vol of radioimmunoprecipitation assay buffer (150 mm NaCl, 50 mm Tris-HCl, pH 8.0, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS), containing Complete Protease Inhibitor Cocktail (Roche, Indianapolis, IN) and 1 μg/ml pepstatin. Homogenates were sonicated on ice five times for 1 s, with a 5 s pause between pulses, and flash-frozen. Lysates were then thawed on ice and centrifuged at 21,000 × g for 10 min at 4°C. Protein concentrations of cleared lysates were determined with the Coomassie Plus assay (Pierce, Rockford, IL). Protein (10 μg) was electrophoresed through 4–12% NuPAGE Bis-Tris SDS-polyacrylamide gels (Invitrogen), followed by transfer to nitrocellulose membranes (Amersham Biosciences, Piscataway, NJ), Western blotting with anti-Sod2 (0.2 μg/ml final concentration; Sod110; Stressgen, Victoria, British Columbia, Canada), and detection of immunoreactive bands with Super Signal West Pico Chemiluminescent substrate (Pierce). Protein loading was assessed with anti-α-tubulin (1:500,000; Sigma, St. Louis, MO). Specificity of the antibody for Sod2 was confirmed by the absence of signal after probing whole-brain extract from an _Sod2_−/− mouse (see Fig. 1B).
Figure 1.
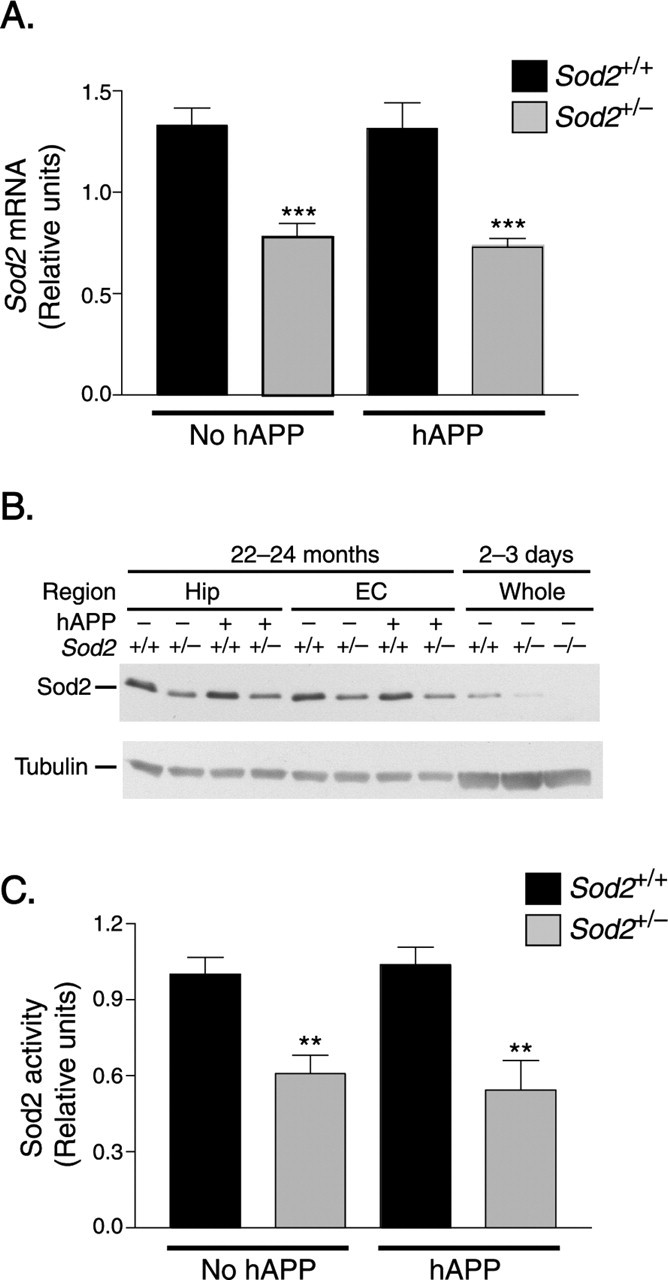
Ablation of one Sod2 allele reduces Sod2 mRNA, protein, and activity levels. A, Reduction in hippocampal Sod2 mRNA levels in 6- to 8-month-old Sod2+/− mice compared with age-matched Sod2+/+ mice, as determined by qfRT-PCR. n = 10 mice per group. B, Reduction in Sod2 protein levels in the hippocampus (Hip) and entorhinal cortex (EC) of 22- to 24-month-old and 2- to 3-day-old Sod2+/− mice compared with age-matched Sod2+/+ mice, demonstrated by Western blot analysis. C, Reduction in Sod2 activity in the entorhinal cortex of 16- to 19-month-old Sod2+/− mice compared with age-matched Sod2+/+ mice, as determined by SOD assay. n = 7–10 mice per group. **p < 0.01; ***p < 0.001 versus either Sod2+/+ group (Tukey's HSD test). Error bars indicate SEM.
Sod2 activity assay.
Sod2 activity was measured with the SOD assay kit-WST (Dojindo Molecular Technologies, Gaithersburg, MD). This assay is based on the ability of superoxide dismutases to inhibit the xanthine/xanthine oxidase-derived superoxide anion reduction in WST-1 formazan, the production of which is monitored by the absorbance at 440 nm. The procedure was performed according to the manufacturer's recommendations with the following modifications. Tissue lysates were sonicated on ice using 20 1 s pulses with a 5 s pause between pulses to break open mitochondrial membranes and centrifuged at 10,000 × g for 15 min at 4°C. Protein concentrations of supernatants were determined with the Coomassie Plus assay. Sod2 activity was measured after preincubating samples (34 μg of protein per reaction) in 20 mm potassium cyanide for 40 min at room temperature to inhibit Sod1 activity. Results were expressed as a proportion of activity (percentage of inhibition) relative to samples from Sod2+/+ mice. The linear range of the assay was determined by serially diluting samples and comparing the percentage of inhibition to a standard curve generated with serial dilutions of bovine erythrocyte (total) SOD (Sigma) and Bacillus sp. Sod2 (Wako, Tokyo, Japan).
Mitochondrial aconitase activity.
Fresh tissues were gently homogenized in 3 vol (w/v) of lysis buffer (in mm: 50 Tris HCl, pH 8.0, 0.6 MnCl2, and 2 citric acid) containing Complete Protease Inhibitor Cocktail (Roche) with a microscale hand-held tissue grinder (Kontes). Lysates were flash-frozen and freeze-thawed three times in liquid nitrogen and water at room temperature, respectively, to break open the mitochondrial membranes. Lysates were centrifuged at 10,000 × g at 4°C for 10 min, and protein in the supernatant was quantified using the Micro BCA protein assay kit (Pierce). Mitochondrial and cytosolic aconitases were separated by electrophoresis on 16.5 × 14 cm, 300-μm-thick cellulose gels (Cellogel; Accurate Chemical and Scientific Company, Westbury, NY) in 20 mm potassium phosphate buffer and 3.6 mm citric acid at pH 7.8 in a horizontal gel chamber run cathode to anode. Activity was visualized by staining for 7 min at room temperature in a solution containing 100 mm potassium phosphate buffer, 1 mm NADP+, 5 mm MgCl2, 2.5 mm _cis-_aconitic acid, 0.5 mg/ml MTT (2,3-bis-(2-methoxy-4-nitro-5-sulfenyl)-2_H_-tetrazolium-5-carboxanilide disodium salt), 0.1 mg/ml phenazine methosulfate, and 100 U (1 ml) of isocitrate dehydrogenase. Band intensities were quantified by densitometry with Gel Expert software (Nucleotech, San Mateo, CA). The linear range of the assay was established by comparing 18, 9, and 4.5 μg of protein for each sample.
Aβ ELISA and plaque quantitation.
Snap-frozen hippocampi were homogenized in 5 m guanidine hydrochloride buffer (Sigma), and Aβ peptides were quantitated by ELISA as described previously (Johnson-Wood et al., 1997). For plaque quantitations, vibratome sections were immunoperoxidase labeled with an Elite kit (Vector Laboratories, Burlingame, CA), using biotinylated anti-Aβ1–5 (5 μg/ml, 3D6; Elan Pharmaceuticals, South San Francisco, CA) (Johnson-Wood et al., 1997) and DAB, and counterstained with 1% hematoxylin. The percentage of area of the hippocampus covered by 3D6 immunoreactivity (-IR) was determined with a Quantimet 570C (Leica, Deerfield, IL). Three sections were analyzed per mouse, and the average was used to calculate group means. The vascular amyloid score was determined by visual inspection of 3D6-stained brain sections: 0, no vessels affected; 0.5, occasional vessels affected; 1.0–3.0, multiple vessels affected mildly (1.0–1.5), moderately (2.0–2.5), or severely (3.0). The experimenter was blinded to the genotype of the mice.
Acid–urea gel and Western blot analysis of Aβ.
Snap-frozen hippocampi and cortex were homogenized in 80% formic acid, and Aβ1–42, Aβ1–40, and other C-terminally truncated Aβ species were detected by high-resolution acid–urea PAGE and Western blot analysis with the 3D6 antibody (3.1 μg/ml) as described previously (Rockenstein et al., 2005) with minor modifications. Briefly, 800 μg (hippocampus) or 1.5 mg (cortex) of total protein or 2 ng each of synthetic Aβ1–42 and Aβ1–40 standards were analyzed per lane. Specificity of the 3D6 antibody for human Aβ species was confirmed by the absence of signal after probing whole-brain extract from a nontransgenic control mouse. Densitometric quantitation of Aβ1–42 and Aβ1–40 was performed and reported as Aβ1–42/Aβ1–40.
Immunohistochemical analyses.
Vibratome sections were immunostained for microtubule-associated protein-2 (MAP2), and the percentage of area occupied by immunoreactive neuronal structures of defined signal intensity was determined by computer-aided image analysis as described previously (Buttini et al., 1999; Van Uden et al., 2002). Vibratome sections were immunostained with an anti-Iba-1 antibody (1 μg/ml; Wako) to detect activated microglia (Kanazawa et al., 2002) or with an anti-glial fibrillary acid protein (GFAP) antibody (2 μg/ml; Chemicon, Temecula, CA) to detect activated astrocytes. The extent of neuritic dystrophy and its relationship to amyloid deposits in the brains of hAPP/Sod2+/+ and hAPP/Sod2+/− mice were assessed in double-immunolabeled sections as described previously (Masliah et al., 2000; Rockenstein et al., 2001). Briefly, vibratome sections (40 μm) were labeled with mouse monoclonal antibodies against heavy and intermediate molecular weight phosphorylated neurofilaments (1:1000, SMI312; Sternberger Immunocytochemicals, Baltimore, MD), which were detected with FITC-conjugated secondary antibodies (1:75; Vector Laboratories), and with an anti-Aβ monoclonal antibody (1:2000, 4G8; Signet, Dedham, MA), which was detected with the Tyramide Signal Amplification-Direct (Red) system (1:100; NEN Life Sciences, Boston, MA). Sections from mice of the different genotypes were processed simultaneously under the same conditions, and results were confirmed in an independent experiment. Controls included the development of sections that had been incubated overnight in the absence of one or the other of the primary antibodies. Sections were imaged with a Zeiss (Oberkochen, Germany) 63× (numerical aperture, 1.4) objective on an Axiovert 35 microscope (Zeiss) with an MRC1024 laser-scanning confocal microscope system (Bio-Rad, Wattford, UK) (Masliah et al., 2000). For each mouse, we obtained four series of five optical sections (0.2 μm in thickness in the _z_-axis covering a total depth of 10 μm) of the neocortex. These images were used to determine the proportion of dystrophic neurites associated with amyloid and the percentage of area of the neuropil occupied by dystrophic neurites that were or were not associated with amyloid deposits.
Electron microscopy.
Ultrastructural analysis was performed as described previously (Wyss-Coray et al., 2000; Rockenstein et al., 2001). Briefly, brains were prepared as described above. Vibratome sections of this tissue were further postfixed in 1% glutaraldehyde and 0.1% osmium tetroxide in 0.1 m sodium cacodylate buffer and embedded in Epoxy. Blocks were sectioned with an Ultracut E ultramicrotome (Leica, Nussloch, Germany) and analyzed with a Zeiss EM10 electron microscope. For each mouse and brain region (neocortex and hippocampus), 10 electron micrographs were prepared at 5000× and 10 electron micrographs were prepared at 15,000×. Three mice per genotype were analyzed, and representative images are shown.
Nuclear factor κB activity assay.
Nuclear extracts were prepared from freshly isolated hippocampi as described previously (Sun et al., 1994). Briefly, tissues were gently triturated in cytoplasmic extraction buffer [10 mm HEPES, pH 7.4, 10 mm KCl, 0.1 mm EDTA, 0.1 mm EGTA, 1 mm dithiothreitol (DTT), 0.5 mm phenylmethylsulfonyl fluoride (PMSF), 1 mm Na3VO4, and protease inhibitor cocktail 1 (Calbiochem, San Diego, CA)]. After 10 min on ice, 23 μl of ice-cold 10% NP-40 was added, the samples were mixed for 10 s at low speed on a vortex, and the nuclei were pelleted by centrifugation at 13,000 × g at 4°C for 20 s. The supernatant was removed, and the nuclei were resuspended and solubilized for 30 min on ice in nuclear extraction buffer: [20 mm HEPES, pH 7.4, 0.4 m NaCl, 1 mm EDTA, 1 mm EGTA, 0.5 mm PMSF, 1 mm DTT, 1 mm Na3VO4, and protease inhibitor cocktail 1 (Calbiochem)]. The lysate was centrifuged for 5 min at 13,000 × g at 4°C, and the supernatant was collected (nuclear extract). For DNA binding activity, oligonucleotide probes containing either a nuclear factor κB (NF-κB) or Oct1 consensus sequence (Promega, Madison, WI) were radiolabeled with [γ-32P] ATP (Amersham Biosciences) and incubated for 10 min at room temperature with 10 μg of nuclear extract in a solution containing 5 mm HEPES, pH 7.8, 5 mm MgCl2, 50 mm KCl, 0.5 mm DTT, 10% glycerol, 0.083 mg/ml salmon testes DNA (Sigma), and 0.33 mg/ml poly dI-dC (Amersham Biosciences) and separated on 5% nondenaturing polyacrylamide Criterion gels (Bio-Rad, Hercules, CA). Gels were dried and exposed to film. Autoradiographs were analyzed densitometrically with Image Quant (Molecular Dynamics, Sunnyvale, CA).
Statistical analysis.
Statistical analyses were performed with SPSS 10.0 (SPSS, Chicago, IL). Unless indicated otherwise, data represent group means ± SEM. Differences between means (two samples) were assessed by the Student's t test or Mann–Whitney U test. Differences among multiple means were assessed by two-factor ANOVA followed, as indicated, by Tukey's honestly significant difference (HSD) test or least significant difference (LSD) test. Differences in learning curves were assessed by repeated-measures ANOVA. For the probe trials, main effects and interactions between genotype and preference for the target quadrant were assessed by two-way ANOVA. Preference for the target quadrant versus each of the other three quadrants was assessed by paired t tests, comparing the target quadrant to all of the other three quadrants.
Results
Ablation of one Sod2 allele reduces Sod2 activity to 50% of wild-type levels
Heterozygous hAPP transgenic mice from line J20 (Mucke et al., 2000) were crossed with mice in which one Sod2 gene had been inactivated by homologous recombination (Sod2+/−) (Lebovitz et al., 1996), resulting in hAPP/Aβ-expressing mice with two (hAPP/Sod2+/+) or one (hAPP/Sod2+/−) functional Sod2 alleles and Sod2+/+ and Sod2+/− littermates that did not inherit the hAPP transgene.
In adult mice with or without hAPP expression, ablation of one Sod2 allele reduced the levels of Sod2 mRNA, protein, and activity to approximately half of those in nontransgenic wild-type controls, as determined by qfRT-PCR, Western blot analysis, and enzymatic activity assay, respectively (Fig. 1). Expression of hAPP/Aβ did not alter Sod2 expression at the mRNA or protein level in Sod2+/+ and Sod2+/− mice (Fig. 1A,B). Thus, reduction in Sod2 activity and expression of hAPP/Aβ did not increase expression of the remaining wild-type Sod2 allele.
To determine whether reduction in Sod2 elicits redox alterations in the brain, we measured the activity of the redox-sensitive, iron-sulfur-center-containing mitochondrial aconitase that has been used as an indicator of superoxide toxicity (Gardner et al., 1995; Van Remmen et al., 2001). In 9- to 11-month-old mice with or without hAPP/Aβ expression, mitochondrial aconitase activity in the hippocampus was, on average, 15–17% lower on the Sod2+/− background than on the Sod2+/+ background (Sod2+/−, 0.96 ± 0.11 vs Sod2+/+, 1.15 ± 0.12, n = 7–8 mice per group; hAPP/Sod2+/−, 1.02 ± 0.07 vs hAPP/Sod2+/+, 1.20 ± 0.37, n = 3–4 mice per group), but this trend did not reach statistical significance. Thus, even when combined, high levels of Aβ and reduction in Sod2 activity by 50% did not inactivate aconitase in hippocampal mitochondria in this strain of mice. Western blot analysis of carbonylated proteins also revealed no clear evidence for increased oxidative damage in brains of Sod2+/− mice with or without hAPP expression (data not shown). Nonetheless, Sod2 reduction modulated multiple aspects of the hAPP/Aβ-dependent phenotype.
Sod2 reduction accelerates the onset of hAPP/Aβ-dependent behavioral alterations
hAPP mice on the Sod2+/+ background develop a range of behavioral alterations (Raber et al., 2000b; Higgins and Jacobsen, 2003; Palop et al., 2003; Götz et al., 2004; Chin et al., 2005; Kobayashi and Chen, 2005). To determine whether any of these alterations are worsened by Sod2 reduction, we compared the behavior of hAPP/Sod2+/+ and hAPP/Sod2+/− mice in a battery of behavioral tests; Sod2+/+ and Sod2+/− mice without hAPP were included as additional controls. Sod2 reduction had the most striking impact on the performance of hAPP mice in the elevated plus maze and on prepulse inhibition of acoustic startle. The plus maze provides putative measures of emotionality and exploratory behavior (Pellow et al., 1985; Lister, 1987) and was recently shown to correlate with antioxidant gene expression levels in mice (Hovatta et al., 2005). Prepulse inhibition is widely used as a measure of sensorimotor gating and has been shown previously to be deficient in hAPP mice (Geyer et al., 2002; McCool et al., 2003; Savonenko et al., 2003).
At 4–7 months of age, only hAPP/Sod2+/− mice, but not hAPP/Sod2+/+ mice, spent more time in the open arms of the elevated plus maze than the control groups (Fig. 2A), whereas by 10–12 months, both hAPP/Sod2+/− and hAPP/Sod2+/+ mice showed this behavioral alteration (Fig. 2B), suggesting that Sod2 reduction promotes an earlier manifestation of this hAPP/Aβ-dependent phenotype. The increased time young adult hAPP/Sod2+/− mice spent in the open arms did not result from generalized hyperactivity, because the distance moved in the closed arms was similar in all four groups of mice, and total basic movements were not different in hAPP/Sod2+/− and hAPP/Sod2+/+ mice (data not shown). Similar to mice at 10–12 months, 21- to 23-month-old hAPP-expressing mice on both Sod2 backgrounds tended to spend more time in the open arms of the elevated plus maze than the control groups. However, at 21–23 months, even Sod2+/+ and Sod2+/− mice without hAPP/Aβ expression spent more time in the open arms than younger mice of the same genotype (p < 0.001 for age effect by ANOVA), diminishing differences between mice with and without hAPP expression. hAPP/Sod2+/− mice still spent significantly more time in the open arms relative to Sod2+/− mice without hAPP expression (Fig. 2C).
Figure 2.

Sod2 reduction accelerates the onset of hAPP/Aβ-dependent behavioral alterations in the elevated plus maze. A, At 4–7 months, only hAPP mice on the Sod2+/− background spent significantly more time in the open arms of the maze than the control groups. n = 12–19 mice per group. B, At 10–12 months, hAPP mice showed this alteration on both the Sod2+/− and the Sod2+/+ backgrounds. n = 13–17 mice per group. C, By 21–23 months, time in the open arms had increased also in the control groups, diminishing the relative differences between the groups. n = 13–16 mice per group. *p < 0.05 versus mice without hAPP on the same Sod2 background (LSD test); ***p < 0.001 versus any of the other groups (LSD test and Tukey's HSD test). Error bars indicate SEM.
Sod2 reduction also accelerated the onset of hAPP/Aβ-dependent deficits in prepulse inhibition of acoustic startle (Fig. 3A,B). A 120 dB pulse superimposed on a 65 dB background noise elicited marked startle responses in all mice tested (Fig. 3C). To inhibit this response, prepulses of three different intensities were selected in pilot experiments (4, 8, and 16 dB above background). At 10–12 months of age, hAPP/Sod2+/− mice, but not hAPP/Sod2+/+ mice, had impaired prepulse inhibition compared with Sod2+/+ mice (4 dB prepulse) or with Sod2+/+ and Sod2+/− mice (8 dB prepulse) (Fig. 3A). At 21–23 months, both hAPP/Sod2+/− and hAPP/Sod2+/+ mice showed deficits in prepulse inhibition (8 dB prepulse) (Fig. 3B).
Figure 3.
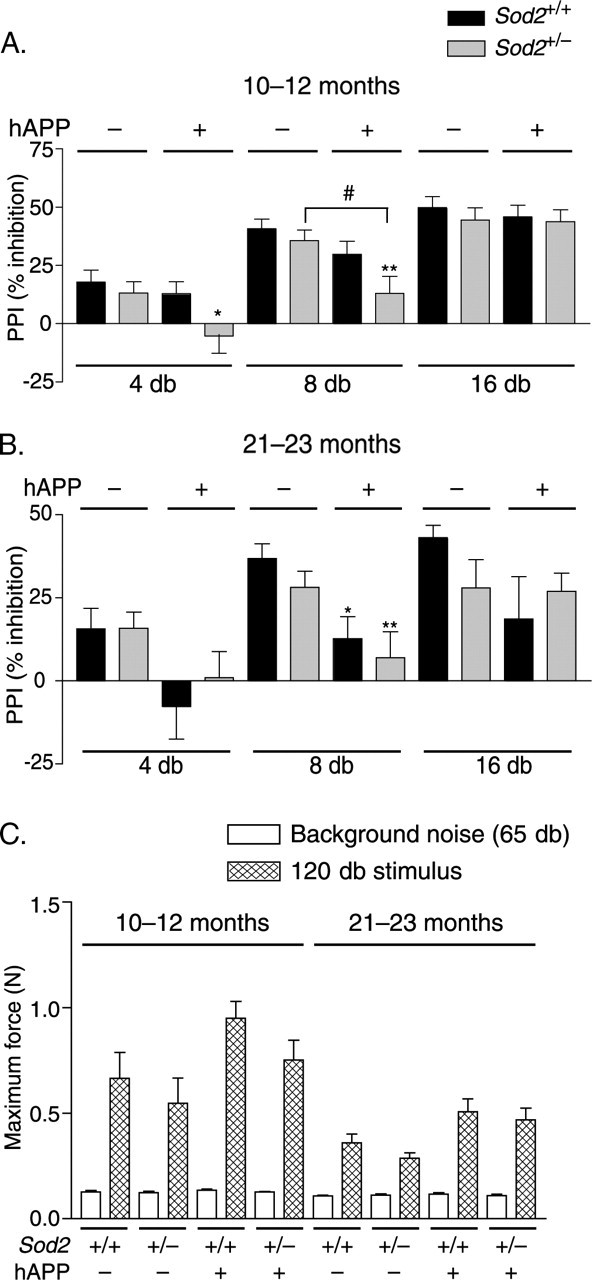
Sod2 reduction accelerates the onset of hAPP/Aβ-dependent deficits in prepulse inhibition (PPI) of acoustic startle. Mice were tested for sensorimotor gating by measuring PPI of acoustic startle. A positive prepulse inhibition value indicates that the response to a startle stimulus was lower when the stimulus was preceded by a prepulse (4, 8, or 16 dB) than when it was presented alone. A, At 10–12 months, only hAPP mice on the Sod2+/− background had impaired PPI. n = 13–17 mice per group. B, At 21–23 months, hAPP mice showed this alteration on both the Sod2+/− and the Sod2+/+ backgrounds. n = 13–16 mice per group. C, At both ages, all groups of mice exhibit robust startle reactivity, as indicated by comparing baseline activity in the startle chamber (background noise) to the response to a 120 dB stimulus (55 dB over background). Although the magnitude of this response is lower in the 21- to 23-month-old mice relative to the 10- to 12-month-old mice (p < 0.05 for a main effect of age for all four genotypes), there is no age-by-genotype interaction. ANOVA revealed a main effect of hAPP genotype (p < 0.05) at both ages but not of the Sod2 genotype at either age, although Tukey's HSD post hoc test revealed no significant differences between Sod2+/+ and hAPP/Sod2+/+ mice or Sod2+/− and hAPP/Sod2+/− mice at either age (p = 0.051 for hAPP/Sod2+/− vs Sod2+/− at 21–23 months of age). ∗p < 0.05; ∗∗p < 0.01 versus Sod2+/+ mice without hAPP; #p < 0.05 (Tukey's HSD test). Error bars indicate SEM.
At both ages, the magnitude of the startle response to pulses 55 dB above background was slightly increased in hAPP mice relative to mice without hAPP expression (Fig. 3C). This finding is consistent with observations in a different line of hAPP/Sod2+/+ mice, which also had impaired prepulse inhibition and exaggerated startle responses (McCool et al., 2003). For all genotypes, the magnitude of the startle response was lower at 21–23 months than at 10–12 months (p < 0.05 for a main effect of age for all four genotypes). However, there was no Sod2 genotype effect on the magnitude of the startle response at either age, nor was there any age-by-genotype interaction, confirming that specific deficits in prepulse inhibition in hAPP/Sod2+/− mice at 10–12 months of age did not result from decreased startle reactivity.
Sod2 reduction had little or no impact on other behavioral measures. For example, hAPP mice from line J20 have profound learning and memory deficits in the Morris water maze even at 5–7 months of age (Palop et al., 2003), and these deficits were not significantly increased by the deletion of one Sod2 allele (Fig. 4).
Figure 4.
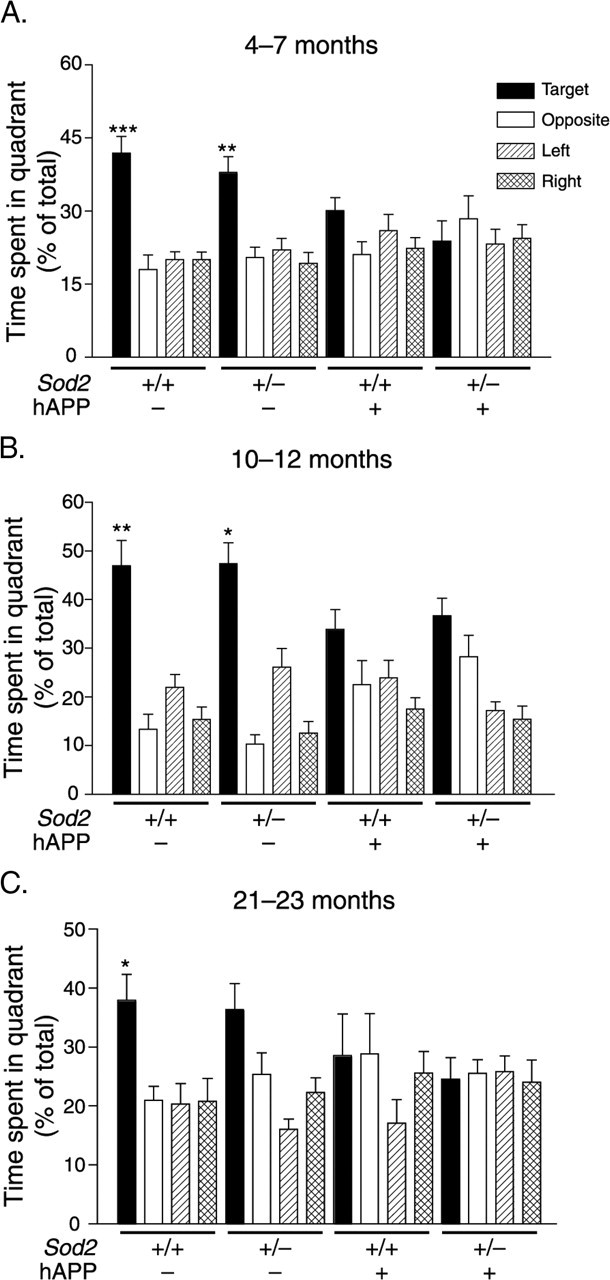
Expression of hAPP/Aβ alone is sufficient to elicit severe probe trial deficits in the Morris water maze. Mice were first trained in two daily sessions to locate a visually cued platform and then a hidden platform. After the last hidden-platform session, the platform was removed for a probe trial to assess whether mice favored the target quadrant in which the platform had been during hidden platform training. At 4–7 (A), 10–12 (B), and 21–23 (C) months of age, Aβ-expressing mice showed no preference for the target quadrant relative to the other three quadrants, whereas mice without hAPP/Aβ expression preferred the target quadrant. n = 11–19 (A, B) and n = 7–9 (C) mice per group. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus all of the other three quadrants for that group of mice (paired t test). Two-way ANOVA revealed a main effect of hAPP (p < 0.05), but not Sod2, on time spent in the target quadrant at all three ages. Error bars indicate SEM.
Sod2 reduction augments hAPP/Aβ-dependent depletion of MAP2
PDGF-hAPP mice with high levels of Aβ expression develop age-dependent depletions of MAP2 (Games et al., 1995; Hsia et al., 1999), which is normally most abundant in neuronal dendrites. Consistent with these findings, 9- to 11-month-old hAPP/Sod2+/+ mice had decreased levels of MAP2 in the hippocampus and neocortex (data not shown). Sod2 reduction had no effect on the severity of these alterations at this age, although cortical MAP2 levels tended (p = 0.074) to be lower in hAPP/Sod2+/− mice than in hAPP/Sod2+/+ mice (data not shown). However, by 21–24 months, Sod2 reduction significantly worsened both the hippocampal and the cortical MAP2 depletion in hAPP mice (Fig. 5).
Figure 5.

Sod2 reduction worsens the extent of hAPP/Aβ-dependent depletion of MAP2. Brain sections were immunostained for MAP2. At 21–24 months, reductions in MAP2-immunoreactive neuronal structures in the hippocampus (A) and neocortex (B) were worse in hAPP/Sod2+/− mice. n = 6–13 mice per group. ∗p < 0.05, ∗∗p < 0.01 as indicated by brackets; ∗∗p < 0.01, ∗∗∗p < 0.001 versus Sod2+/+ mice without hAPP (Tukey's HSD test). Error bars indicate SEM. IR, Immunoreactivity.
Sod2 reduction elicits an aggressive cerebrovascular amyloidosis in hAPP mice
Even at 21–24 months of age, Sod2+/+ mice (Fig. 6A) and Sod2+/− mice (Fig. 6B) without hAPP/Aβ expression had a normal cerebrovascular architecture characterized by discrete endothelial cell layers, smooth basement membranes, and well defined, undisturbed smooth muscle cell layers. In age-matched hAPP/Sod2+/+ mice, the smooth muscle cell layer of some cerebral blood vessels contained large deposits of amyloid fibrils, although the basic structural organization of the vessel walls remained relatively intact (Fig. 6C). hAPP/Sod2+/− mice had a more prominent and widespread cerebrovascular amyloidosis that was accompanied by striking disruptions of the vascular architecture (Fig. 6D). The extensive amyloid deposits in vessel walls of hAPP/Sod2+/− mice were associated with a marked curvature and waviness of the basement membrane (Fig. 6E). These vascular changes in hAPP/Sod2+/− mice were sometimes accompanied by the presence of foamy macrophage-like cells containing lipid vacuoles (Fig. 6F), which were seen only rarely, and without lipid vacuoles, in hAPP/Sod2+/+ mice (data not shown).
Figure 6.
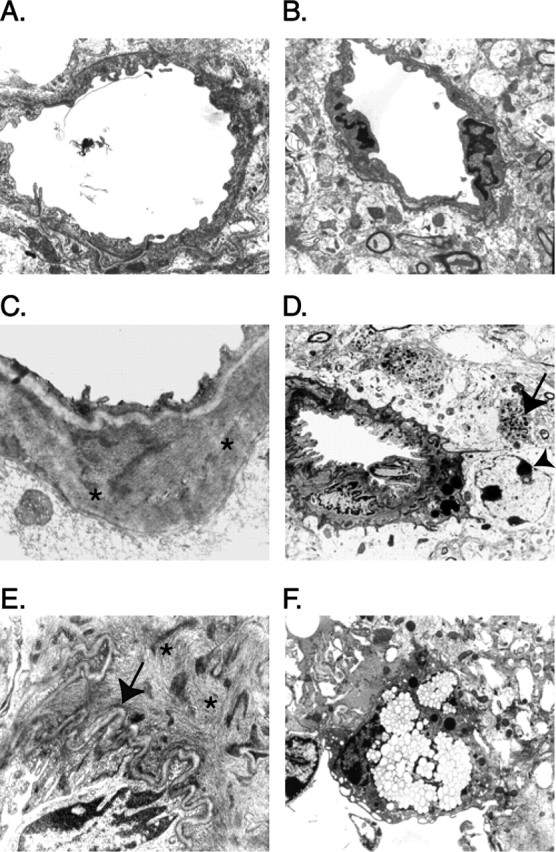
Sod2 reduction alters amyloid deposition and worsens hAPP/Aβ-dependent neurodegenerative alterations. Electron microscopy was used to detect ultrastructural alterations in the frontal cortex of mice at 21–24 months of age. A–C, In Sod2+/+ (A) and Sod2+/− (B) mice without hAPP, cerebral blood vessels had a normal appearance, but in hAPP/Sod2+/+ mice (C), some cerebral blood vessels had amyloid deposits in the smooth muscle cell layer (asterisks). However, hAPP/Sod2+/+ mice showed little or no disruption of the vascular architecture. D, hAPP/Sod2+/− mice, on the other hand, had more prominent vascular amyloid deposits that were frequently associated with gliosis (arrowhead) and dystrophic neurites (arrow). E, Some hAPP/Sod2+/− mice had extensive amyloid (asterisks) in the smooth muscle cell layer associated with prominent waviness of the basement membrane (arrow). F, Blood vessels in hAPP/Sod2+/− mice were also sometimes associated with perivascular foamy macrophage-like cells containing abundant lipid vacuoles.
Cerebrovascular amyloidosis in hAPP mice was also detectable by labeling of brain sections with antibodies against human Aβ. Quantitation of the resulting immunoreactivity revealed significantly greater vascular Aβ deposition in hAPP/Sod2+/− mice than in hAPP/Sod2+/+ mice at 9–11 and 21–24 months of age (Fig. 7A,C; and data not shown) and a positive correlation between age (from 9 to 24 months) and vascular amyloid deposition in both groups of mice (p < 0.05 for hAPP/Sod2+/+ and p < 0.01 for hAPP/Sod2+/− mice; Spearman rank test). Notably, the slope of the line generated by this correlation, a measure of the rate of accumulation, was greater in the hAPP/Sod2+/− group (data not shown). No amyloid deposits in the parenchyma or the vasculature were detected in 3-month-old mice from either group (data not shown), and thus these mice were excluded from the correlation analysis.
Figure 7.

Sod2 reduction promotes cerebrovascular amyloidosis but reduces the parenchymal plaque burden. A, hAPP/Sod2+/− mice had more vascular amyloid deposits than hAPP/Sod2+/+ mice at both 9–11 and 21–24 months of age. B, In contrast, parenchymal plaque loads were similar at 9–11 months but lower in hAPP/Sod2+/− mice at 21–24 months. n = 8–13 mice per group. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 relative to Sod2+/+ mice at the same age (Mann–Whitney U test). Error bars indicate SEM. C, Representative images of frontal cortex sections immunostained for Aβ with the 3D6 antibody depicting more prominent parenchymal plaques in hAPP/Sod2+/+ mice and more prominent vascular amyloidosis in hAPP/Sod2+/− mice at 9–11 and 21–24 months of age.
Sod2 reduction decreases parenchymal plaque burden in old mice but does not affect levels of soluble Aβ in young mice without plaques
At 9–11 months, hAPP/Sod2+/+ and hAPP/Sod2+/− mice had a similar overall burden of parenchymal amyloid plaques in the frontal cortex (Fig. 7B) and hippocampus (data not shown), although plaques appeared to be smaller in hAPP/Sod2+/− mice than in hAPP/Sod2+/+ mice at 9–11 months of age (Fig. 7C). However, by 21–24 months, the parenchymal plaque load in hAPP/Sod2+/− mice was less than half of that in hAPP/Sod2+/+ mice, a striking contrast to the prominent vascular amyloidosis in hAPP/Sod2+/− mice (Fig. 7).
The distribution of amyloid deposits in old hAPP/Sod2+/− mice is remarkably similar to that elicited in hAPP mice by astroglial overexpression of TGF-β1 (Wyss-Coray et al., 1997, 2001). Moreover, TGF-β1 mRNA levels in postmortem brain tissues from humans with AD correlate positively with vascular amyloidosis and negatively with parenchymal plaque load (Wyss-Coray et al., 1997). We therefore compared TGF-β1 mRNA levels by qfRT-PCR in the hippocampus of 6- to 8-month-old mice. Although relative TGF-β1 mRNA levels tended to be somewhat higher in hAPP/Sod2+/− mice than in hAPP/Sod2+/+ mice (161.5 ± 16.0 vs 142.2 ± 10.8; mean ± SEM; n = 10 mice per group), this trend was not statistically significant.
Because deposition of amyloid in the vasculature is also influenced by expression of apoE (Fryer et al., 2005), we measured mouse apoE mRNA levels in the hippocampus of 6- to 8-month-old mice. hAPP/Aβ expression significantly lowered apoE mRNA levels, but Sod2 reduction did not further augment this effect (Sod2+/+, 1.70 ± 0.14 vs hAPP/Sod2+/+, 1.28 ± 0.04, and hAPP/Sod2+/−, 1.26 ± 0.08; p < 0.05; n = 10 mice per group). In the absence of hAPP/Aβ expression, Sod2 reduction tended to lower apoE mRNA levels (Sod2+/+, 1.70 ± 0.14 vs Sod2+/−, 1.40 ± 0.08; n = 10 mice per group), but this trend did not reach statistical significance.
Another factor that has been implicated in the redistribution of amyloid deposits from the parenchyma to the vasculature is the relative concentration of Aβ peptides ending at residue 40 versus residue 42 (Herzig et al., 2004). Vascular amyloid deposits are typically enriched in Aβ40 and parenchymal plaques in Aβ42 (Van Dorpe et al., 2000; Herzig et al., 2004; Fryer et al., 2005). Thus, in mice with established differences in vascular and parenchymal amyloid burden, it is impossible to determine whether differences in their Aβ42/Aβ40 ratio are a cause or a consequence of their different amyloid distributions. Measurement of soluble Aβ levels in the hippocampus of 3- to 4-month-old mice that had not yet formed detectable amyloid deposits revealed no differences between hAPP/Sod2+/+ mice (n = 19) and hAPP/Sod2+/− mice (n = 8) in Aβ1–x (212.0 ± 11.3 vs 227.1 ± 12.3 pg/mg tissue), Aβ1–42 (54.8 ± 5.3 vs 61.7 ± 10.6 pg/mg tissue), and Aβ1–42/Aβ1–x (0.25 ± 0.01 vs 0.26 ± 0.03) ratios. Because Aβ1–42/Aβ1–40 ratios may be the key determinant of vascular amyloid deposition (Van Dorpe et al., 2000; Herzig et al., 2004; Fryer et al., 2005) and ELISAs using the 266 antibody do not distinguish between Aβ1–40 and other C-terminally truncated forms, we performed Western blot analysis with the 3D6 antibody after separating the various Aβ species by high-resolution, highly denaturing acetic acid–urea PAGE. This method reliably detects Aβ1–42 as well as C-terminally truncated Aβ species (DeMattos et al., 2001; Rockenstein et al., 2005). Western blot analysis in tissues from 3- to 4-month-old mice revealed no differences in Aβ1–42/Aβ1–40 ratios between hAPP/Sod2+/+ and hAPP/Sod2+/− mice (n = 5 per group) in the hippocampus (0.39 ± 0.09 vs 0.51 ± 0.14) and cortex (0.45 ± 0.12 vs 0.34 ± 0.05).
Sod2 reduction worsens neuritic dystrophy
Because Sod2 reduction decreased the parenchymal plaque burden in old hAPP/Sod2+/− mice (Fig. 7B), we wondered whether Sod2 reduction would also decrease the extent of plaque-associated neuronal alterations. We focused on dystrophic neurites, marked bulbous dilations, and curvy trajectories of neuronal processes that typically, although not always, colocalize with parenchymal plaques (Onorato et al., 1989; Masliah et al., 1993; Knowles et al., 1999; Brendza et al., 2005). Despite their decrease in parenchymal plaques, hAPP/Sod2+/− mice showed a marked increase in neuritic dystrophy. Compared with hAPP/Sod2+/+ mice (Fig. 8A), hAPP/Sod2+/− mice had many more dystrophic neurites containing thickened, electrodense material (Fig. 8B,C). Notably, much of this neuritic dystrophy in hAPP/Sod2+/− mice did not appear to colocalize with amyloid deposits. To more directly assess plaque-independent neuritic dystrophy, confocal image analysis of sections from 21- to 24-month-old mice were double-labeled with antibodies against neurofilaments (SMI312) and Aβ (4G8). Dystrophic neurites that were at least 10 μm away from amyloid deposits, as determined by confocal analysis of four series of five optical sections (0.2 μm in thickness in the z_-axis covering a total depth of 10 μm) and reported as percentage of area covered, were more abundant in hAPP/Sod2+/− mice than in hAPP/Sod2+/+ mice (0.73 ± 0.11 vs 0.33 ± 0.06; n = 8–13 mice per group; ∗∗_p < 0.01, Mann–Whitney U test).
Figure 8.

Sod2 reduction promotes plaque-independent neuritic dystrophy. Dystrophic neurites in the frontal cortex were detected at 25–30 months by electron microscopy. A, Neuritic dystrophy (arrow) without evidence for parenchymal amyloid deposits in an hAPP/Sod2+/+ mouse. B, C, This type of neuritic dystrophy (arrows) was increased in hAPP/Sod2+/− mice.
Sod2 reduction increases microgliosis and activation of the injury-responsive transcription factor NF-κB but decreases astrocytosis in old hAPP mice
Compared with Sod2+/+ mice, hAPP/Sod2+/+ mice had increased levels of Iba-1-IR, a marker of microglial activation in the hippocampus and cortex (Fig. 9A,B). In both brain regions, Sod2 reduction significantly enhanced the microgliosis in hAPP mice but did not affect microglial activation in the absence of hAPP/Aβ expression. Compared with Sod2+/+ mice, hAPP/Sod2+/+ mice also had increased levels of GFAP-IR, a marker of astroglial activation, in the hippocampus and cortex (Fig. 9C). Sod2 reduction decreased the astrocytosis in the brain parenchyma of hAPP mice but did not affect astrocytosis in the absence of hAPP/Aβ expression. hAPP/Sod2+/− mice still had a prominent perivascular astocytosis (data not shown).
Figure 9.
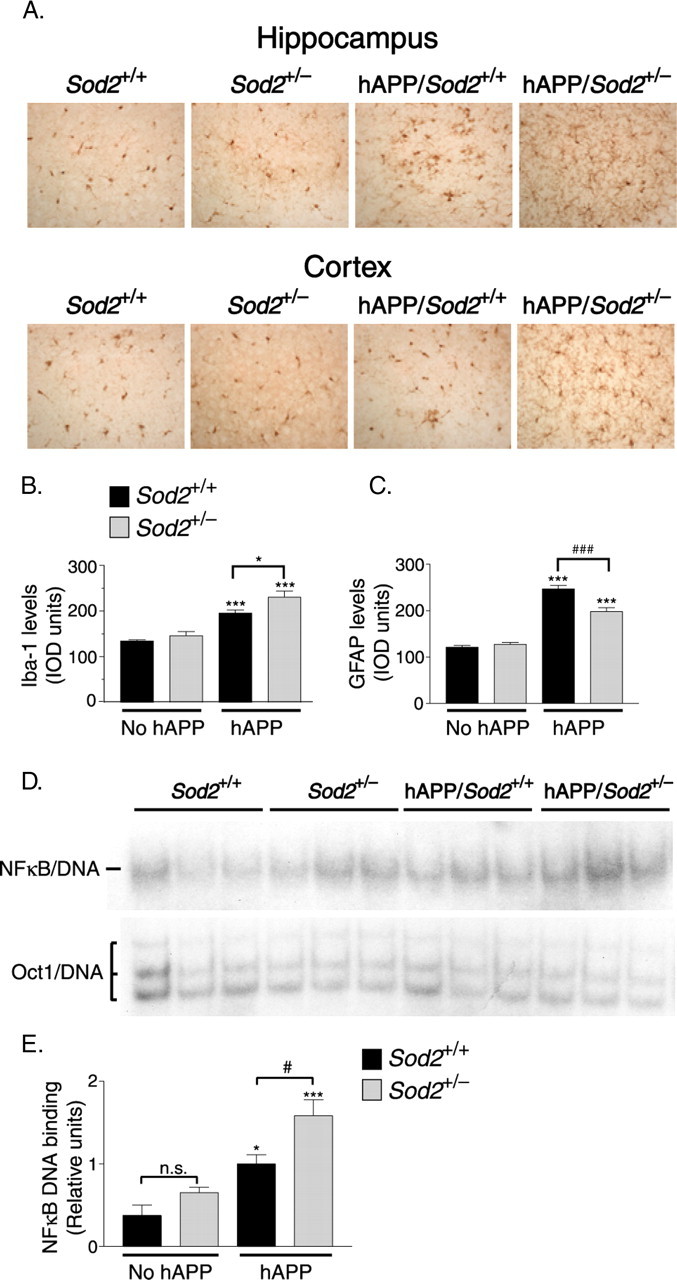
hAPP/Aβ expression and Sod2 reduction result in the activation of microglia and NF-κB. A, Sections of the hippocampus (top) and frontal cortex (bottom) of 21- to 24-month-old mice were immunostained for the microglial activation marker Iba-1. B, Quantitation of Iba-1-IR in the frontal cortex revealed low levels of microglial labeling in mice without hAPP on either Sod2 background and microgliosis in hAPP/Aβ-expressing mice that was exacerbated by Sod2 reduction. n = 7–13 mice per group. ∗p < 0.05; ∗∗∗p < 0.001 versus Sod2+/+ without hAPP (Tukey's HSD test). C, Quantitation of GFAP-IR in the frontal cortex revealed low levels of astrocyte labeling in mice without hAPP on either Sod2 background and astrocytosis in hAPP/Aβ- expressing mice that was lessened by Sod2 reduction. ∗∗∗p < 0.001 versus any of the other groups; ### p < 0.001 as indicated by bracket (Tukey's HSD test). D, NF-κB DNA binding activity (top) was determined by EMSA using nuclear extracts from the hippocampi of 25- to 30-month-old mice. Competition with unlabeled “cold” oligonucleotide abolished the signal and established the specificity of the assay for NF-κB (data not shown). A separate EMSA was performed to measure the DNA binding activity of Oct1 (bottom) as a control for the quality of the nuclear extract preparation. E, NF-κB and Oct1 DNA binding activity were quantitated by densitometric analysis; NF-κB activity was normalized against Oct1 activity and plotted as a proportion of activity in hAPP/Sod2+/+ mice. n = 6–9 mice per group. ∗p < 0.05, ∗∗∗p < 0.001 versus Sod2+/+ mice without hAPP; #p < 0.05; n.s., not significant (Tukey's HSD test). Error bars indicate SEM. IOD, Integrated optical density.
Finally, we assessed the activity of NF-κB, an injury-response transcription factor, the DNA binding activity of which is activated by numerous stimuli, including reactive oxygen species and inflammatory mediators (Staal et al., 1990; García-Ruiz et al., 1995; May and Ghosh, 1998; Nadeau and Rivest, 2000; Kabe et al., 2005). To determine NF-κB activity, we performed electrophoretic mobility shift assays (EMSAs) on nuclear extracts from hippocampi of 25- to 30-month-old mice. hAPP/Aβ expression significantly increased NF-κB DNA binding, and this effect was augmented by Sod2 reduction, resulting in the highest levels of NF-κB activity in hAPP/Sod2+/− mice (Fig. 9D,E).
Discussion
Most common neurodegenerative disorders likely result from complex interactions between genetic and environmental risk factors (Farrer et al., 1997; Munoz and Feldman, 2000; Lindsay et al., 2002). One such factor may be aging-related alterations in the redox state of mitochondria (Hirai et al., 2001; Eckert et al., 2003; Beal, 2005) Our study shows that partial reduction in the main mitochondrial superoxide scavenger Sod2 accelerates the onset of hAPP/Aβ-dependent behavioral abnormalities and worsens a range of AD-related molecular and pathological alterations.
How might Sod2 reduction modulate the phenotype of hAPP mice or AD? An obvious mechanism is increased mitochondrial levels of superoxide radicals and resultant oxidative damage. To assess this possibility, we measured mitochondrial aconitase activity, a well-established mitochondrial sensor of superoxide that is lower in the heart and liver of adult Sod2+/− mice than in Sod2+/+ controls (Williams et al., 1998; Van Remmen et al., 2001). In the hippocampus of Sod2+/− mice with or without hAPP expression, there was only a trend of reduced mitochondrial aconitase activity compared with Sod2+/+ controls, and we are unaware of any reports documenting more significant mitochondrial impairments in brain tissues of adult Sod2+/− or hAPP mice under baseline conditions. Because long-lived cells with high levels of energy consumption have evolved effective combinations of antioxidant defense systems (Halliwell and Gutteridge, 1985), neuronal mitochondria may be better protected against Sod2 reduction than mitochondria in peripheral organs.
Nonetheless, over prolonged periods, even subtle redox alterations in mitochondria might impair redox-sensitive signaling pathways and increase the susceptibility of neurons to other challenges. Sod2+/− mice are more sensitive than Sod2+/+ mice to kainate (Liang and Patel, 2004), ischemia/reperfusion (Kim et al., 2002), mitochondrial toxins (Andreassen et al., 2001), overexpression of familial amyotrophic lateral sclerosis mutant Sod1 (Andreassen et al., 2000), and hAPP/Aβ (present study). Aβ may, in turn, sensitize mice to superoxide-induced mitochondrial alterations [e.g., through the redox activity of its Met-35 (Butterfield and Bush, 2004; Lustbader et al., 2004) or through interactions with the mitochondrial enzyme Aβ-binding alcohol dehydrogenase (Lustbader et al., 2004)].
Although the precise mechanisms by which Sod2 reduction promotes Aβ-induced neuronal deficits remain to be determined, our study clearly highlights the impact this reduction can have on neuronal integrity and function. Neuronal expression of hAPP/Aβ in transgenic mice results in cognitive and behavioral alterations (Higgins and Jacobsen, 2003; Palop et al., 2003; Götz et al., 2004; Kobayashi and Chen, 2005). Sod2 reduction accelerated the onset of hAPP/Aβ-dependent behavioral alterations in the elevated plus maze, which provides putative measures of anxiety-like behavior. hAPP mice with reduced Sod2 exhibited less anxiety-like behavior (or more disinhibition) in the plus maze, consistent with recently identified correlations between anxiety-like behavior and antioxidant levels (Hovatta et al., 2005; Krömer et al., 2005). Although the exact relationship between these alterations in mice and AD in humans is tentative, AD patients clearly have altered emotional behaviors and activity levels (Chung and Cummings, 2000; Harper et al., 2004; Hatfield et al., 2004).
Sod2 reduction also accelerated the onset of hAPP/Aβ-dependent alterations in prepulse inhibition of acoustic startle, a measure of sensorimotor gating (Geyer et al., 2002). Prepulse inhibition could be affected by age-dependent hearing deficits, which have been documented in C57BL/6 mice [http://jaxmice.jax.org/strain/000664.html (Walton et al., 1995)]. Indeed, the intensity of startle response declined with age in all groups of mice, suggesting an age-dependent decrease in hearing and/or a loss of muscle strength. However, hearing loss is unlikely to account for the effect of Sod2 reduction on prepulse inhibition at 10–12 months. At this age, all groups of mice exhibited a robust startle response to the 120 dB stimulus that was partially inhibited by a lower intensity prepulse, and there was no difference in startle reactivity between hAPP/Sod2+/− and hAPP/Sod2+/+ mice. In addition, prepulse inhibition increased with prepulse stimulus intensity in all groups, suggesting that the mice discriminated between the different prepulse intensities. Finally, prepulse inhibition deficits were present only at selected prepulse intensities. Therefore, it is likely that Sod2 reduction accelerates the onset of bona fide prepulse inhibition deficits in hAPP mice. These results may relate closely to prepulse inhibition deficits in patients with early AD (Veki et al., 2006). That Sod2 reduction did not worsen impairments in spatial learning and memory may be attributable to the fact that they were already very severe in hAPP/Sod2+/+ mice. Whether Sod2 reduction can modulate these cognitive functions in hAPP mice with lower levels of Aβ production and less severe deficits remains to be determined.
Sod2 reduction had striking effects on Aβ deposition, increasing vascular amyloidosis and decreasing parenchymal plaques. In a different line of hAPP transgenic mice, Sod2 reduction increased the overall amyloid burden (Li et al., 2004). A side-by-side comparison of the models may be required to explain these differences. Although many hAPP transgenic lines with moderate to high-level Aβ production show some vascular amyloidosis by 12–15 months, vascular amyloidosis is usually less prominent than parenchymal plaques (Calhoun et al., 1999; Wyss-Coray et al., 1999; Van Dorpe et al., 2000; Christie et al., 2001; Kuo et al., 2001; Castellani et al., 2004). Because vascular amyloid increased before parenchymal amyloid decreased in our hAPP/Sod2+/− mice (relative to hAPP/Sod2+/+ mice), it is unlikely that Sod reduction caused a direct redistribution of Aβ from plaques into the vasculature. Factors that can tip the balance from parenchymal to vascular amyloid formation include high Aβ40/Aβ42 ratios (Mucke et al., 2000; Herzig et al., 2004), increased astroglial production of TGF-β1 (Wyss-Coray et al., 1997, 2001), and apolipoprotein E4 (Fryer et al., 2005; Mazur-Kolecka et al., 2006). We found no significant effects of Sod2 reduction on Aβ40/Aβ42 ratios or on TGF-β1 and apoE mRNA levels in hAPP mice. Thus, Sod2 reduction may promote vascular over parenchymal Aβ deposition by an alternative mechanism. Interestingly, Aβ42 and Aβ40 differ in the way they self-aggregate, and oxidation of Met-35 in Aβ42 makes Aβ42 aggregate like Aβ40 (Bitan et al., 2003). Thus, Sod2 reduction might create a prooxidative milieu in which Aβ42 is deposited like Aβ40, promoting vascular over parenchymal amyloid formation.
Aβ deposition in the vasculature, known as cerebral amyloid angiopathy (CAA), occurs in 80–90% of AD patients (Castellani et al., 2004; Greenberg et al., 2004; Attems, 2005). Yet, the relationship between CAA and neuronal deficits remains a matter of active study and debate (Iadecola et al., 1999; Castellani et al., 2004; Greenberg et al., 2004; Attems, 2005; Mazur-Kolecka et al., 2006). Interestingly, Sod2 reduction enhanced CAA in our hAPP mice before it enhanced MAP2 deficits, raising the possibility that CAA may actually contribute to neuronal impairments. Furthermore, aged hAPP/Sod2+/− mice had a prominent vasculopathy and a marked increase in dystrophic neurites, most of which were not colocalized with parenchymal amyloid deposits. These findings highlight a potential cooperation between Aβ and mitochondrial superoxide in the pathogenesis of vasculopathy and neuritic dystrophy. They are consistent with recent studies demonstrating an association between neuritic beading and mitochondrial impairments induced by activated microglia in primary cell cultures (Takeuchi et al., 2005).
Additional studies are needed to determine whether the glial alterations we observed in hAPP/Sod2+/− mice, compared with hAPP/Sod2+/+ mice, are protective or harmful and whether they result directly from Sod2 reduction within glial cells or represent innate immune responses to neuronal injury and vascular amyloid. Increased activation of microglia/macrophages could promote the degradation of parenchymal Aβ (Wyss-Coray et al., 2001; Simard et al., 2006). However, microglial activation by Aβ may also contribute to neuronal injury (Wyss-Coray and Mucke, 2002). Interestingly, NFκ-B expression in microglia was necessary for nonfibrillar Aβ to elicit microglia-dependent neurotoxicity in mixed cultures of primary neurons and glia (Chen et al., 2005). In the current study, Sod2 reduction significantly worsened the hAPP/Aβ-dependent activation of microglia and NF-κB. Thus, NF-κB activity may both reflect and mediate the increased neuronal impairments in hAPP/Sod2+/− mice. Because reduction in Sod2 activity increased the severity of diverse AD-related neuronal impairments and accelerated the onset of hAPP/Aβ-dependent behavioral changes, preventing or reversing Sod2 reductions may be of therapeutic benefit.
Footnotes
J. Raber's present address: Departments of Behavioral Neuroscience and Neurology, Division of Neuroscience, Oregon National Primate Research Center, Oregon Health and Science University, Portland, OR.
L. Kekonius's present address: Karolinska Institute, Stockholm, Sweden.
J. Puoliväli's present address: Cerebricon Ltd., Kuopio, Finland.
This work was supported by National Institutes of Health Grants AG011385 and AG022074 (L.M.), by a University of California San Francisco Neuroscience Program Postdoctoral Training Grant, and by a grant from the Giannini Family Foundation (L.E.). We thank S. Mantha, T.-T. Huang, and C. Epstein for advice on the aconitase assay; A. O'Mahony and M. Montano for advice on the NF-κB assay; M. Hafiz and D.A. Butterfield for independent confirmation of oxidative damage assessment; C. McCulloch for advice on statistical analyses; A. LeFevour, H. Ordanza, X. Wang, and K. Bielewicz for technical assistance; I. Cheng and J. Palop for helpful discussions; S. Ordway and G. Howard for editorial assistance; and D. McPherson and L. Manuntag for help in preparing this manuscript.
References
- Anandatheerthavarada HK, Biswas G, Robin MA, Avadhani NG (2003). Mitochondrial targeting and a novel transmembrane arrest of Alzheimer's amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol 161:41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreassen OA, Ferrante RJ, Klivenyi P, Klein AM, Shinobu LA, Epstein CJ, Beal MF (2000). Partial deficiency of manganese superoxide dismutase exacerbates a transgenic mouse model of amyotrophic lateral sclerosis. Ann Neurol 47:447–455. [PubMed] [Google Scholar]
- Andreassen OA, Ferrante RJ, Dedeoglu A, Albers DW, Klivenyi P, Carlson EJ, Epstein CJ, Beal MF (2001). Mice with a partial deficiency of manganese superoxide dismutase show increased vulnerability to the mitochondrial toxins malonate, 3-nitropropionic acid, and MPTP. Exp Neurol 167:189–195. [DOI] [PubMed] [Google Scholar]
- Attems J (2005). Sporadic cerebral amyloid angiopathy: Pathology, clinical implications, and possible pathomechanisms. Acta Neuropathol (Berl) 110:345–359. [DOI] [PubMed] [Google Scholar]
- Aucoin JS, Jiang P, Aznavour N, Tong XK, Buttini M, Descarries L, Hamel E (2005). Selective cholinergic denervation, independent from oxidative stress, in a mouse model of Alzheimer's disease. Neuroscience 132:73–86. [DOI] [PubMed] [Google Scholar]
- Beal MF (2005). Mitochondria take center stage in aging and neurodegeneration. Ann Neurol 58:495–505. [DOI] [PubMed] [Google Scholar]
- Benice T, Rizk A, Kohama S, Pfankuch T, Raber J (2006). Sex-differences in age-related cognitive decline in C57BL/6J mice associated with increased brain microtubule-associated protein 2 and synaptophysin immunoreactivity. Neuroscience 137:413–423. [DOI] [PubMed] [Google Scholar]
- Bitan G, Tarus B, Vollers SS, Lashuel HA, Condron MM, Straub JE, Teplow DB (2003). A molecular switch in amyloid assembly: Met35 and amyloid β-protein oligomerization. J Am Chem Soc 125:15359–15365. [DOI] [PubMed] [Google Scholar]
- Blanchard V, Moussaoui S, Czech C, Touchet N, Bonici B, Planche M, Canton T, Jedidi I, Gohin M, Wirths O, Bayer TA, Langui D, Duyckaerts C, Tremp G, Pradier L (2003). Time sequence of maturation of dystrophic neurites associated with Aβ deposits in APP/PS1 transgenic mice. Exp Neurol 184:247–263. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E (1997). Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 18:351–357. [DOI] [PubMed] [Google Scholar]
- Brendza RP, Bacskai BJ, Cirrito JR, Simmons KA, Skoch JM, Klunk WE, Mathis CA, Bales KR, Paul SM, Hyman BT, Holtzman DM (2005). Anti-Aβ antibody treatment promotes the rapid recovery of amyloid-associated neuritic dystrophy in PDAPP transgenic mice. J Clin Invest 115:428–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busciglio J, Pelsman A, Wong C, Pigino G, Yuan M, Mori H, Yankner BA (2002). Altered metabolism of the amyloid β precursor protein is associated with mitochondrial dysfunction in Down's syndrome. Neuron 33:677–688. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Bush AI (2004). Alzheimer's amyloid β-peptide (1–42): Involvement of methionine residue 35 in the oxidative stress and neurotoxicity properties of this peptide. Neurobiol Aging 25:563–568. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Drake J, Pocernich C, Castegna A (2001). Evidence of oxidative damage in Alzheimer's disease brain: central role for amyloid β-peptide. Trends Mol Med 7:548–554. [DOI] [PubMed] [Google Scholar]
- Buttini M, Orth M, Bellosta S, Akeefe H, Pitas RE, Wyss-Coray T, Mucke L, Mahley RW (1999). Expression of human apolipoprotein E3 or E4 in the brains of _Apoe_−/− mice: isoform-specific effects on neurodegeneration. J Neurosci 19:4867–4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Weiderhold KH, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, Jucker M (1999). Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci USA 96:14088–14093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calon F, Lim G, Yang F, Morihara T, Teter B, Ubeda O, Rostaing P, Triller A, Salem N Jr, Ashe K, Frautschy S, Cole G (2004). Docosahexaenoic acid protects from dendritic pathology in an Alzheimer's disease mouse model. Neuron 43:633–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso SM, Santana I, Swerdlow RH, Oliveira CR (2004). Mitochondria dysfunction of Alzheimer's disease cybrids enhances Aβ toxicity. J Neurochem 89:1417–1426. [DOI] [PubMed] [Google Scholar]
- Castellani RJ, Smith MA, Perry G, Friedland RP (2004). Cerebral amyloid angiopathy: major contributor or decorative response to Alzheimer's disease pathogenesis. Neurobiol Aging 25:599–602. [DOI] [PubMed] [Google Scholar]
- Chen J, Zhou Y, Mueller-Steiner S, Chen LF, Kwon H, Yi S, Mucke L, Gan L (2005). SIRT1 protects against microglia-dependent amyloid-β toxicity through inhibiting NF-κB signaling. J Biol Chem 280:40364–40374. [DOI] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Puoliväli J, Massaro C, Bien-Ly N, Gerstein H, Scearce-Levie K, Masliah E, Mucke L (2005). Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer's disease. J Neurosci 25:9694–9703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie R, Yamada M, Moskowitz M, Hyman B (2001). Structural and functional disruption of vascular smooth muscle cells in a transgenic mouse model of amyloid angiopathy. Am J Pathol 158:1065–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung JA, Cummings JL (2000). Neurobehavioral and neuropsychiatric symptoms in Alzheimer's disease: characteristics and treatment. Neurol Clin 18:829–846. [DOI] [PubMed] [Google Scholar]
- Coskun PE, Beal MF, Wallace DC (2004). Alzheimer's brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc Natl Acad Sci USA 101:10726–10731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch PJ, Blake R, Duce JA, Ciccotosto GD, Li QX, Barnham KJ, Curtain CC, Cherny RA, Cappai R, Dyrks T, Masters CL, Trounce IA (2005). Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-β1–42J Neurosci 25:672–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, Troncoso JC, Mattson MP (2004). Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer's disease. Proc Natl Acad Sci USA 101:2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Dodart J-C, Paul SM, Holtzman DM (2001). Peripheral anti-Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 98:8850–8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert A, Keil U, Marques CA, Bonert A, Frey C, Schussel K, Muller WE (2003). Mitochondrial dysfunction, apoptotic cell death, and Alzheimer's disease. Biochem Pharmacol 66:1627–1634. [DOI] [PubMed] [Google Scholar]
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM (1997). Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. J Am Med Assoc 278:1349–1356. [PubMed] [Google Scholar]
- Fryer JD, Simmons K, Parsadanian M, Bales KR, Paul SM, Sullivan PM, Holtzman DM (2005). Human apolipoprotein E4 alters the amyloid-β40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J Neurosci 25:2803–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L (1995). Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 373:523–527. [DOI] [PubMed] [Google Scholar]
- García-Ruiz C, Colell A, Morales A, Kaplowitz N, Fernández-Checa JC (1995). Role of oxidative stress generated from the mitochondrial electron transport chain and mitochondrial glutathione status in loss of mitochondrial function and activation of transcription factor nuclear factor-κB: studies with isolated mitochondria and rat hepatocytes. Mol Pharmacol 48:825–834. [PubMed] [Google Scholar]
- Gardner PR, Raineri I, Epstein LB, White CW (1995). Superoxide radical and iron modulate aconitase activity in mammalian cells. J Biol Chem 270:13399–13405. [DOI] [PubMed] [Google Scholar]
- Geyer MA, McIlwain KL, Paylor R (2002). Mouse genetic models for prepulse inhibition: an early review. Mol Psychiatry 7:1039–1053. [DOI] [PubMed] [Google Scholar]
- Götz J, Streffer J, David D, Schild A, Hoerndli F, Pennanen L, Kurosinski P, Chen F (2004). Transgenic animal models of Alzheimer's disease and related disorders: histopathology, behavior and therapy. Mol Psychiatry 9:664–683. [DOI] [PubMed] [Google Scholar]
- Greenberg SM, Gurol ME, Rosand J, Smith EE (2004). Amyloid angiopathy-related vascular cognitive impairment. Stroke 35:2616–2619. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JMC (1985). In: Free radicals in biology and medicine Oxford: Clarendon. [DOI] [PubMed]
- Hansson CA, Frykman S, Farmery MR, Tjernberg LO, Nilsberth C, Pursglove SE, Ito A, Winblad B, Cowburn RF, Thyberg J, Ankarcrona M (2004). Nicastrin, presenilin, APH-1, and PEN-2 form active γ-secretase complexes in mitochondria. J Biol Chem 279:51654–51660. [DOI] [PubMed] [Google Scholar]
- Harper DG, Stopa EG, McKee AC, Satlin A, Fish D, Volicer L (2004). Dementia severity and Lewy bodies affect circadian rhythms in Alzheimer disease. Neurobiol Aging 25:771–781. [DOI] [PubMed] [Google Scholar]
- Hatfield CF, Herbert J, van Someren EJ, Hodges JR, Hastings MH (2004). Disrupted daily activity/rest cycles in relation to daily cortisol rhythms of home-dwelling patients with early Alzheimer's dementia. Brain 127:1061–1074. [DOI] [PubMed] [Google Scholar]
- Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, Butterfield DA (1994). A model for β-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci USA 91:3270–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig MC, Winkler DT, Burgermeister P, Pfeifer M, Kohler E, Schmidt SD, Danner S, Abramowski D, Sturchler-Pierrat C, Burki K, Van Duinen SG, Maat-Schieman ML, Staufenbiel M, Mathews PM, Jucker M (2004). Aβ is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci 7:954–960. [DOI] [PubMed] [Google Scholar]
- Higgins GA, Jacobsen H (2003). Transgenic mouse models of Alzheimer's disease: phenotype and application. Behav Pharmacol 14:419–438. [DOI] [PubMed] [Google Scholar]
- Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA (2001). Mitochondrial abnormalities in Alzheimer's disease. J Neurosci 21:3017–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovatta I, Tennant RS, Helton R, Marr RA, Singer O, Redwine JM, Ellison JA, Schadt EE, Verma IM, Lockhart DJ, Barlow C (2005). Glyoxalase 1 and glutathione reductase 1 regulate anxiety in mice. Nature 438:662–666. [DOI] [PubMed] [Google Scholar]
- Hsia A, Masliah E, McConlogue L, Yu G, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L (1999). Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci USA 96:3228–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TT, Carlson EJ, Kozy HM, Mantha S, Goodman SI, Ursell PC, Epstein CJ (2001). Genetic modification of prenatal lethality and dilated cardiomyopathy in Mn superoxide dismutase mutant mice. Free Radic Biol Med 31:1101–1110. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Zhang F, Niwa K, Eckman C, Turner SK, Fischer E, Younkin S, Borchelt DR, Hsiao KK, Carlson GA (1999). SOD 1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat Neurosci 2:157–161. [DOI] [PubMed] [Google Scholar]
- Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L (1997). Amyloid precursor protein processing and Aβ42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci USA 94:1550–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabe Y, Ando K, Hirao S, Yoshida M, Handa H (2005). Redox regulation of NF-κB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal 7:395–403. [DOI] [PubMed] [Google Scholar]
- Kanazawa H, Ohsawa K, Sasaki Y, Kohsaka S, Imai Y (2002). Macrophage/microglia-specific protein Iba1 enhances membrane ruffling and Rac activation via phospholipase C-γ-dependent pathway. J Biol Chem 277:20026–20032. [DOI] [PubMed] [Google Scholar]
- Keil U, Bonert A, Marques CA, Scherping I, Weyermann J, Strosznajder JB, Muller-Spahn F, Haass C, Czech C, Pradier L, Muller WE, Eckert A (2004). Amyloid β-induced changes in nitric oxide production and mitochondrial activity lead to apoptosis. J Biol Chem 279:50310–50320. [DOI] [PubMed] [Google Scholar]
- Kim GW, Kondo T, Noshita N, Chan PH (2002). Manganese superoxide dismutase deficiency exacerbates cerebral infarction after focal cerebral ischemia/reperfusion in mice: implications for the production and role of superoxide radicals. Stroke 33:809–815. [DOI] [PubMed] [Google Scholar]
- Knowles RB, Wyart C, Buldyrev SV, Cruz L, Urbanc B, Hasselmo ME, Stanley HE, Hyman BT (1999). Plaque-induced neurite abnormalities: Implications for disruption of neural networks in Alzheimer's disease. Proc Natl Acad Sci USA 96:5274–5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi DT, Chen KS (2005). Behavioral phenotypes of amyloid-based genetically modified mouse models of Alzheimer's disease. Genes Brain Behav 4:173–196. [DOI] [PubMed] [Google Scholar]
- Kokoszka JE, Coskun P, Esposito LA, Wallace DC (2001). Increased mitochondrial oxidative stress in the Sod2 (+/−) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc Natl Acad Sci USA 98:2278–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krömer SA, Kessler MS, Milfay D, Birg IN, Bunck M, Czibere L, Panhuysen M, Pütz B, Deussing JM, Holsboer F, Landgraf R, Turck CW (2005). Identification of glyoxalase-I as a protein marker in a mouse model of extremes in trait anxiety. J Neurosci 25:4375–4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo YM, Beach TG, Sue LI, Scott S, Layne KJ, Kokjohn TA, Kalback WM, Luehrs DC, Vishnivetskaya TA, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, Weller RO, Roher AE (2001). The evolution of A beta peptide burden in the APP23 transgenic mice: implications for A beta deposition in Alzheimer disease. Mol Med 7:609–618. [PMC free article] [PubMed] [Google Scholar]
- Lebovitz RM, Zhang HJ, Vogel H, Cartwright JJ, Dionne L, Lu NF, Huang S, Matzuk MM (1996). Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci USA 93:9782–9787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Calingasan NY, Yu F, Mauck WM, Toidze M, Almeida CG, Takahashi RH, Carlson GA, Flint Beal M, Lin MT, Gouras GK (2004). Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. J Neurochem 89:1308–1312. [DOI] [PubMed] [Google Scholar]
- Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ (1995). Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet 11:376–381. [DOI] [PubMed] [Google Scholar]
- Liang LP, Patel M (2004). Mitochondrial oxidative stress and increased seizure susceptibility in Sod2−/+ mice. Free Radic Biol Med 36:542–554. [DOI] [PubMed] [Google Scholar]
- Lindsay J, Laurin D, Verreault R, Hebert R, Helliwell B, Hill GB, McDowell I (2002). Risk factors for Alzheimer's disease: a prospective analysis from the Canadian Study of Health and Aging. Am J Epidemiol 156:445–453. [DOI] [PubMed] [Google Scholar]
- Lister RG (1987). The use of a plus-maze to measure anxiety in the mouse. Psychopharmacology 92:180–185. [DOI] [PubMed] [Google Scholar]
- Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS (2004). ABAD directly links Aβ to mitochondrial toxicity in Alzheimer's disease. Science 304:448–452. [DOI] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Hansen L, Alford M, DeTeresa R, Terry R (1993). An antibody against phosphorylated neurofilaments identifies a subset of damaged association axons in Alzheimer's disease. Am J Pathol 142:871–882. [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Alford M, Mallory M, Rockenstein E, Moechars D, Van Leuven F (2000). Abnormal glutamate transport function in mutant amyloid precursor protein transgenic mice. Exp Neurol 163:381–387. [DOI] [PubMed] [Google Scholar]
- May MJ, Ghosh S (1998). Signal transduction through NF-κB. Immunol Today 19:80–88. [DOI] [PubMed] [Google Scholar]
- Mazur-Kolecka B, Dickson D, Frackowiak J (2006). Induction of vascular amyloidosis-β by oxidative stress depends on APOE genotype. Neurobiol Aging in press. [DOI] [PubMed]
- McCool MF, Varty GB, Del Vecchio RA, Kazdoba TM, Parker EM, Hunter JC, Hyde LA (2003). Increased auditory startle response and reduced prepulse inhibition of startle in transgenic mice expressing a double mutant form of amyloid precursor protein. Brain Res 994:99–106. [DOI] [PubMed] [Google Scholar]
- Melov S, Schneider JA, Day BJ, Hinerfeld D, Coskun P, Mirra SS, Crapo JD, Wallace DC (1998). A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genet 18:159–163. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu G-Q, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L (2000). High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 20:4050–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz DG, Feldman H (2000). Causes of Alzheimer's disease. CMAJ 162:65–72. [PMC free article] [PubMed] [Google Scholar]
- Mutisya EM, Bowling AC, Beal MF (1994). Cortical cytochrome oxidase activity is reduced in Alzheimer's disease. J Neurochem 63:2179–2184. [DOI] [PubMed] [Google Scholar]
- Nadeau S, Rivest S (2000). Role of microglial-derived tumor necrosis factor in mediating CD14 transcription and nuclear factor κB activity in the brain during endotoxemia. J Neurosci 20:3456–3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omar RA, Chyan YJ, Andorn AC, Poeggeler B, Robakis NK, Pappolla MA (1999). Increased expression but reduced activity of antioxidant enzymes in Alzheimer's disease. J Alzheimers Dis 1:139–145. [DOI] [PubMed] [Google Scholar]
- Onorato M, Mulvihill P, Connolly J, Galloway P, Whitehouse P, Perry G (1989). Alteration of neuritic cytoarchitecture in Alzheimer disease. Prog Clin Biol Res 317:781–789. [PubMed] [Google Scholar]
- Palop JJ, Jones B, Kekonius L, Chin J, Yu G-Q, Raber J, Masliah E, Mucke L (2003). Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer's disease-related cognitive deficits. Proc Natl Acad Sci USA 100:9572–9577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Bien-Ly N, Massaro C, Yeung BZ, Yu G-Q, Mucke L (2005). Vulnerability of dentate granule cells to disruption of Arc expression in human amyloid precursor protein transgenic mice. J Neurosci 25:9686–9693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellow S, Chopin R, File SE, Briley M (1985). Validation of open:closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. J Neurosci Methods 14:149–167. [DOI] [PubMed] [Google Scholar]
- Praticò D (2002). Alzheimer's disease and oxygen radicals: new insights. Biochem Pharmacol 63:563–567. [DOI] [PubMed] [Google Scholar]
- Raber J, Akana SF, Bhatnagar S, Dallman MF, Wong D, Mucke L (2000a). Hypothalamic-pituitary-adrenal function in _Apoe_−/− mice: Possible role in behavioral and metabolic alterations. J Neurosci 20:2064–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raber J, Wong D, Yu G-Q, Buttini M, Mahley RW, Pitas RE, Mucke L (2000b). Alzheimer's disease: apolipoprotein E and cognitive performance. Nature 404:352–354. [DOI] [PubMed] [Google Scholar]
- Raber J, LeFevour A, Buttini M, Mucke L (2002). Androgens protect against apolipoprotein E4-induced cognitive deficits. J Neurosci 22:5204–5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Mante M, Sisk A, Masliah E (2001). Early formation of mature amyloid-β protein deposits in a mutant APP transgenic model depends on levels of Aβ1–42J Neurosci Res 66:573–582. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Mante M, Alford M, Adame A, Crews L, Hashimoto M, Esposito L, Mucke L, Masliah E (2005). High β-Secretase activity elicits neurodegeneration in transgenic mice despite reductions in amyloid-β levels: Implications for the treatment of Alzheimer's disease. J Biol Chem 280:32957–32967. [DOI] [PubMed] [Google Scholar]
- Savonenko AV, Xu GM, Price DL, Borchelt DR, Markowska AL (2003). Normal cognitive behavior in two distinct congenic lines of transgenic mice hyperexpressing mutant APP(SWE)Neurobiol Dis 12:194–211. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Schenk D (2003). Alzheimer's disease: Molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol 43:545–584. [DOI] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S (2006). Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron 49:489–502. [DOI] [PubMed] [Google Scholar]
- Staal FJ, Roederer M, Herzenberg LA (1990). Intracellular thiols regulate activation of nuclear factor κB and transcription of human immunodeficiency virus. Proc Natl Acad Sci USA 87:9943–9947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun SC, Elwood J, Beraud C, Greene WC (1994). Human T-cell leukemia virus type I Tax activation of NF-κB/Rel involves phosphorylation and degradation of IκBα and RelA (p65)-mediated induction of the c-rel gene. Mol Cell Biol 14:7377–7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H, Mizuno T, Zhang G, Wang J, Kawanokuchi J, Kuro R, Suzumura A (2005). Neuritic beading induced by activated microglia is an early feature of neuronal dysfunction toward neuronal death by inhibition of mitochondrial respiration and axonal transport. J Biol Chem 280:10444–10454. [DOI] [PubMed] [Google Scholar]
- Tanzi R, Bertram L (2005). Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell 120:545–555. [DOI] [PubMed] [Google Scholar]
- Tong XK, Nicolakakis N, Kocharyan A, Hamel E (2005). Vascular remodeling versus amyloid β-induced oxidative stress in the cerebrovascular dysfunctions associated with Alzheimer's disease. J Neurosci 25:11165–11174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dorpe J, Smeijers L, Dewachter I, Nuyens D, Spittaels K, Van den Haute C, Mercken M, Moechars D, Laenen I, Kuiperi C, Bruynseels K, Tesseur I, Loos R, Vanderstichele H, Checler F, Sciot R, Van Leuven F (2000). Prominent cerebral amyloid angiopathy in transgenic mice overexpressing the London mutant of human APP in neurons. Am J Pathol 157:1283–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Remmen H, Williams MD, Guo Z, Estlack L, Yang H, Carlson EJ, Epstein CJ, Huang TT, Richardson A (2001). Knockout mice heterozygous for Sod2 show alterations in cardiac mitochondrial function and apoptosis. Am J Physiol Heart Circ Physiol 281:H1422–H1432. [DOI] [PubMed] [Google Scholar]
- Van Remmen H, Ikeno Y, Hamilton M, Pahlavani M, Wolf N, Thorpe SR, Alderson NL, Baynes JW, Epstein CJ, Huang TT, Nelson J, Strong R, Richardson A (2003). Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol Genomics 16:29–37. [DOI] [PubMed] [Google Scholar]
- Van Uden E, Mallory M, Veinbergs I, Alford M, Rockenstein E, Masliah E (2002). Increased extracellular amyloid deposition and neurodegeneration in human amyloid precursor protein transgenic mice deficient in receptor-associated protein. J Neurosci 22:9298–9304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veki A, Goto K, Sato N, Iso H, Morita Y (2006). Prepulse inhibition of acoustic startle response in mild cognitive impairment and mild dementia of Alzheimer type. Psychiatry Clin Neurosci 60:55–62. [DOI] [PubMed] [Google Scholar]
- Walton JP, Frisina RD, Meierhans LR (1995). Sensorineural hearing loss alters recovery from short-term adaptation in the C57BL/6 mouse. Hear Res 88:19–26. [DOI] [PubMed] [Google Scholar]
- Wang Q, Rowan MJ, Anwyl R (2004). β-amyloid-mediated inhibition of NMDA receptor-dependent long-term potentiation induction involves activation of microglia and stimulation of inducible nitric oxide synthase and superoxide. J Neurosci 24:6049–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MD, Van Remmen H, Conrad CC, Huang TT, Epstein CJ, Richardson A (1998). Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J Biol Chem 273:28510–28515. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Mucke L (2002). Inflammation in neurodegenerative disease–a double-edged sword. Neuron 35:419–432. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Masliah E, Mallory M, McConlogue L, Johnson-Wood K, Lin C, Mucke L (1997). Amyloidogenic role of cytokine TGF-β1 in transgenic mice and Alzheimer's disease. Nature 389:603–606. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Von Euw D, Masliah E, Mucke L, Lacombe P (1999). Alzheimer's disease-like cerebrovascular pathology in transforming growth factor-β1 transgenic mice and functional metabolic correlates. Ann NY Acad Sci 903:317–323. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Sanan D, Mucke L, Masliah E (2000). Chronic overproduction of TGF-β1 in astrocytes promotes Alzheimer's disease-like microvascular degeneration in transgenic mice. Am J Pathol 156:139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Yan F, Yu G-Q, Rohde M, McConlogue L, Masliah E, Mucke L (2001). TGF-β1 promotes microglial amyloid-β clearance and reduces plaque burden in transgenic mice. Nat Med 7:612–618. [DOI] [PubMed] [Google Scholar]