Presynaptic Terminals Independently Regulate Synaptic Clustering and Autophagy of GABAA Receptors in Caenorhabditis elegans (original) (raw)
Abstract
Synaptic clustering of GABAA receptors is important for the function of inhibitory synapses, influencing synapse strength and, consequently, the balance of excitation and inhibition in the brain. Presynaptic terminals are known to induce GABAA receptor clustering during synaptogenesis, but the mechanisms of cluster formation and maintenance are not known. To study how presynaptic neurons direct the formation of GABAA receptor clusters, we have investigated GABAAreceptor localization in postsynaptic cells that fail to receive presynaptic contacts in_Caenorhabditis elegans_. Postsynaptic muscles in C. elegans receive acetylcholine and GABA motor innervation, and GABAAreceptors cluster opposite GABA terminals. Selective loss of GABA inputs caused GABAA receptors to be diffusely distributed at or near the muscle cell surface, confirming that GABA presynaptic terminals induce GABAA receptor clustering. In contrast, selective loss of acetylcholine innervation had no effect on GABAA receptor localization. However, loss of both GABA and acetylcholine inputs together caused GABAA receptors to traffic to intracellular autophagosomes. Autophagosomes normally transport bulk cytoplasm to the lysosome for degradation. However, we show that GABAA receptors traffic to autophagosomes after endocytic removal from the cell surface and that acetylcholine receptors in the same cells do not traffic to autophagosomes. Thus, autophagy can degrade cell-surface receptors and can do so selectively. Our results show that presynaptic terminals induce GABAA receptor clustering by independently controlling synaptic localization and surface stability of GABAA receptors. They also demonstrate a novel function for autophagy in GABAA receptor degradative trafficking.
Keywords: GABAA receptor, autophagy, synaptogenesis, UNC-49, nicotinic acetylcholine receptor, C. elegans
Introduction
The balance between neuronal excitation and inhibition is critical for information processing in the brain. When this balance is disrupted, epilepsy and anxiety disorders may result (Crestani et al., 1999; Baulac et al., 2001; Wallace et al., 2001; Cossette et al., 2002). The excitation–inhibition balance depends on inhibitory neurotransmission, mediated primarily by GABAA receptors (Liu, 2004). These receptors cluster in postsynaptic membranes opposite inhibitory presynaptic terminals. GABAA receptor clustering is important because it achieves high receptor densities in the immediate vicinity of GABA release sites, which profoundly affect the amplitude and kinetics of inhibitory synaptic currents (Semyanov et al., 2004). The mechanisms of GABAA receptor clustering in postsynaptic membranes are not well understood.
Clustering of neurotransmitter receptors is the end result of a developmental pathway initiated when presynaptic terminals contact the postsynaptic cell. At excitatory synapses, the pathway includes regulation of receptor synthesis, clustering of neurotransmitter receptors and scaffolding molecules, and elimination of nonsynaptic receptors (Broadie and Bate, 1993; Burden, 2002; Chih et al., 2005). At GABA synapses, postsynaptic GABAA receptors and scaffolding molecules also cluster where presynaptic terminals make contact (Levi et al., 1999; Rao et al., 2000; Brunig et al., 2002; Christie et al., 2002; Studler et al., 2002; Gally and Bessereau, 2003). The binding of presynaptic β-neurexin to postsynaptic neuroligin-2 molecules is an important first step in this pathway (Graf et al., 2004; Chih et al., 2005). Later events of the pathway leading to the formation and stable maintenance of GABAA receptor clusters remain to be discovered. One obstacle to dissecting the GABAA receptor clustering pathway has been the inability to experimentally eliminate presynaptic contacts. This experiment is important because it should reveal which aspects of GABAA receptor trafficking are influenced by presynaptic terminals.
The nematode Caenorhabditis elegans is a powerful model system for studying synaptogenesis because we can genetically manipulate presynaptic and postsynaptic cells and observe the results in vivo. The body-wall muscle is particularly useful to study inhibitory synaptogenesis. Body-wall muscles form synapses with GABA and non-GABA neurons (White et al., 1986). Thus, the influence of matched and nonmatched presynaptic contacts may be determined. GABAAreceptors in these cells are simple and uniform (Bamber et al., 2005), in contrast to the structurally complex mammalian GABAAreceptors. Finally, axon pathfinding can be manipulated, so presynaptic contacts to body-wall muscles may be selectively or completely eliminated. In this study, we eliminate presynaptic inputs and determine how GABAA receptor patterning is affected. We show that GABA terminals organize GABAA receptors into synaptic clusters, but that GABA and non-GABA terminals alike stabilize GABAA receptors on the cell surface. In the absence of presynaptic inputs, GABAA receptors are internalized and traffic to autophagosomes for degradation. Acetylcholine receptors (AChRs) in the same cells do not traffic to autophagosomes, indicating that autophagy is selective for GABAA receptors. These results are important because they help to delineate the separate steps in the GABAA receptor clustering pathway, and they identify autophagy as a novel degradative pathway for GABAA receptors.
Materials and Methods
C. elegans cultures
C. elegans were grown on nematode growth medium agar at 20–25°C. Bristol N2 was the wild-type strain; mutant alleles used in this study were unc-49(e407), unc-5(e53),unc-51(e369), rol-9(sc148),lin-15(n765ts), rme-8(b1023ts),unc-38(x20), snb-1(md247),unc-13(e1061), unc-31(e928),cha-1(p1152), cha-1(y226ts),acr-16(ok789), unc-64(x37), and_unc-25(e156)_.
Manipulation of motor axon pathfinding
Motor neurons must express the netrin receptor to project dorsally and innervate dorsal muscles. The netrin receptor is encoded by the unc-5 gene (Leung-Hagesteijn et al., 1992). In_unc-5(e53)_ mutants, the netrin receptor is defective, no motor neurons project dorsally, and dorsal muscles are non-innervated (Hedgecock et al., 1990). All investigation of non-innervated dorsal muscles were performed in the unc-5(e53) genetic background. We restored dorsal axon pathfinding individually to GABA neurons and ACh neurons in the_unc-5(e53)_ mutant background, by expressing the netrin receptor (UNC-5) under the control of promoters specific to GABA neurons [unc-47_(McIntire et al., 1997)] and ACh neurons [_acr-2_ (Hallam et al., 2000)]. In unc-5(e53) mutants carrying the P_unc-47::UNC-5 transgene, GABA axon pathfinding is normal, whereas ACh axon pathfinding remains defective. Thus, ACh innervation is effectively removed from the dorsal muscle cells in these strains. Likewise, GABA axon pathfinding is effectively removed in_unc-5(e53)_ mutants carrying P_acr-2_::UNC-5 transgenes. Experiments in which ACh innervation or GABA innervation was removed selectively from dorsal muscles were performed using these transgenes. We visualized GABA motor axons using the P_unc-47_::green fluorescent protein (GFP) construct, and we visualized ACh motor axons using the P_acr-2_::GFP construct (see below). We also used these constructs to verify that rescue of ACh neuron pathfinding did not affect GABA neuron pathfinding and vice versa (data not shown).
Transgenes
UNC-5 expression.
P_unc-47_::UNC-5 and P_acr-2_::UNC-5 transgenes were constructed by placing promoter fragments of unc-47 [GABA vesicular transporter (McIntire et al., 1997)] or_acr-2_ [a nicotinic acetylcholine receptor subunit expressed in DA and DB motor neurons (Hallam et al., 2000)] between the Xba_I and Nco_I of pYZ108, just upstream of the UNC-5 rescuing cDNA (Hamelin et al., 1993), generating pAR27.6 and pAR28.6, respectively. These plasmids were injected along with a plasmid containing rol-6sd, as a cotransformation marker (Kramer et al., 1990) (both at 40 ng/μl), and Rol lines were established. These arrays were integrated using X-irradiation, resulting in grIs2_[P_unc-47::UNC-5; rol-6sd_] and_grIs4 [P_acr-2_::UNC-5_; rol-6sd_], respectively.
Autophagosome markers.
Expression of monomeric red fluorescent protein (mRFP)-LGG-1 was achieved by swapping the GFP coding sequences for those of mRFP (Campbell et al., 2002) in the GFP-LGG-1 construct (Melendez et al., 2003), using the Kpn_I and_Nhe_I sites (resulting in a linker that was seven residues shorter than the linker between GFP and LGG-1), to generate the plasmid pAR40.1. pAR40.1 was coinjected with rol-6sd (both at 40 ng/μl), and Rol lines were established. This array was integrated using X-irradiation to generate_grIs16 [mRFP-LGG-1; _rol-6sd_]. To construct the tagged beclin constructs BEC-1-GFP and BEC-1-mRFP, we fused GFP or mRFP to the C terminus of the BEC-1 protein, encoded by the C. elegans beclin-1 homolog T19E7.3. GFP or mRFP were placed in-frame at the C terminus, between engineered PinA1 and Nhe_I restriction sites in the_Ce-bec-1 coding sequence. The entire BEC-1-GFP and BEC-1-mRFP coding sequence plus an additional 2500 bp of Ce-bec-1 5′ flanking DNA and 900 bp of Ce-bec-1 3′ flanking DNA were placed into a pBluescript-based vector, between Sph_I and_Bsi_W1 sites, generating pAR37.10 and pAR39.1, respectively. These plasmids were coinjected with pEK1 (both at 40 ng/μl) into_lin-15 worms to generate the _grEx115_[Ex(BEC-1-GFP;lin-15(+))] and _grEx129_[Ex(BEC-1-mRFP;lin-15(+))] arrays. The UNC-38-GFP transgene was constructed by inserting GFP in-frame at residue K430 in the intracellular loop between the third and fourth transmembrane domain (J. L. Bessereau, unpublished observation).
Other transgenes.
Other transgene arrays used in this study were as follows:oxIs12_[P_unc-47::GFP] (McIntire et al., 1997);juIs14_[P_acr-2::GFP] (Hallam et al., 2000);juIs1_[P_unc-25::SNB-GFP;_lin-15(+)_] (Hallam and Jin, 1998); GFP-LGG-1 array 5/3 (Melendez et al., 2003); and oxIs22_[UNC-49-GFP;_lin-15(+)_](Bamber et al., 1999). oxIs22 contains the full-length_unc-49 genomic rescuing fragment with GFP inserted, in-frame, into the intracellular loop of the UNC-49B subunit. This UNC-49B-GFP translational fusion protein fully rescues the uncoordinated phenotype of unc-49(e407) and localizes to synapses (Bamber et al., 1999).
Inhibition of endocytosis
To address the role of endocytosis in trafficking GABAA receptors to autophagosomes, we used the strains FY375 [_unc-5(e53); oxIs22_] and FY542 [rme-8(b1023ts); unc-5(e53); oxIs22_]. We maintained these at the permissive temperature (15°C). We first synchronized the populations of both strains by isolating eggs from a mixed-stage population and placing them on a plate lacking bacteria at 15°C, in which they arrested at the first larval stage. These animals were then transferred to a plate with bacteria at 15°C and were allowed to grow to the L4 larval stage. L4 larvae were washed from the 15°C plates with S-Basal and aliquoted by pipette to plates equilibrated at 26°C. At each time point, 7, 14, 26, 32, 40, 55, and 72.5 h after the shift to 26°C, animals were fixed, and UNC-49-GFP containing autophagosomes were counted. Counts were performed blind with respect to genotype. The groups were analyzed using a two-way ANOVA, using_post hoc t tests to compare significant differences at each time point (n = 10 for each time point, except for t = 0, in which only six worms with wild-type rme-8 were counted).
Electron microscopy
Serial thin sections were available from a previous study (Hedgecock et al., 1990) for eight wild-type and nine_unc-5_ adults covering the midbody region in transverse aspect. All animals had been immersion fixed, embedded in Epon, and post-stained with heavy metals in a parallel manner (Hedgecock et al., 1990;Hall, 1995). Sections were viewed on a Philips (Aachen, Germany) CM10 electron microscope, collecting micrographs on film. Film negatives were scanned into Adobe Photoshop (Adobe Systems, San Jose, CA) for data analysis and presentation. To efficiently gather statistics on the presence or absence of autophagosomes, large-muscle inclusions were scored on the microscope screen in well spaced thin sections of the midbody region, looking for organelles of ∼0.5 μm or larger. The dimensions of muscle sarcomeres served as an internal reference, independent of microscope magnification, in quickly measuring organelle size.
Staining and light microscopy
All epifluorescence imaging was performed on a Zeiss (Oberkochen, Germany) Axioskop 2 microscope equipped with a Princeton Instruments digital camera (Roper Scientific, Trenton, NJ). GFP was visualized using the Endow GFP filter set, and mRFP and Alexa Fluor 594 were visualized using the tetramethylrhodamine isothiocyanate (TRITC) filter set (Chroma Technology, Rockingham, VT). Live imaging was performed on worms paralyzed by 30 μm sodium azide. Alternatively, to reduce gut autofluorescence, worms were fixed before imaging (Bettinger et al., 1996). rol-9 was sometimes included in the genetic background to cause twisted body morphology, to aid photographing nerve cords. When quantifying the intracellular accumulation of UNC-49-GFP, we counted green fluorescent objects larger than ∼0.5 μm in diameter and routinely verified that they were not visible using the TRITC filter, to distinguish them from autofluorescent gut granules. Care was taken that double-labeled organelles (i.e., UNC-49-GFP and mRFP-LGG1) were clearly distant from gut to ensure they were not gut granules. For immunostaining, anti-UNC-49 antibodies (Gally and Bessereau, 2003) were used at a 1:500 dilution. Staining was performed using a protocol described by Bettinger et al. (1996), except that, after incubation with secondary antibodies, specimens were washed with buffer B for 1–3 h, transferred to the SlowFade equilibration buffer for 15 min, and mounted in SlowFade mounting medium (SlowFade kit; Invitrogen, Carlsbad, CA). Alexa Fluor 594-conjugated goat anti-rabbit IgG(heavy and light chains) secondary antibodies (Invitrogen) were used at 1:1000. Anti-UNC-29 staining was performed as described previously (Gally et al., 2004). Confocal images were taken on an Olympus Optical (Tokyo, Japan) FV300 using a 60× oil-immersion objective. Images were processed using Adobe Photoshop.
Electrophysiology
Electrophysiological analysis was performed as described previously (Richmond and Jorgensen, 1999). Briefly, animals were immobilized with cyanoacrylic glue, and a lateral cuticle incision was made exposing either the ventral or dorsal medial body-wall muscles. Muscle recordings were made in the whole-cell voltage-clamp configuration (holding potential of −60 mV) using an EPC-10 patch-clamp amplifier and digitized at 1 kHz. Data were acquired by Pulse software (HEKA Elektronik, Lambrecht/Pfalz, Germany) run on a Dell computer (Dell Computer Company, Round Rock, TX). The bath solution contained the following (in mm): 150 NaCl, 5 KCl, 5 CaCl2, 1 MgCl2, 10 glucose, and 15 HEPES, pH 7.35 (∼340 mOsm). The pipette solution contained the following (in mm): 120 KCl, 20 KOH, 4 MgCl2, 5 Tris, 0.25 CaCl2, 4 NaATP, 36 sucrose, and 5 EGTA, pH 7.2 (∼315 mOsm). Subsequent analysis and graphing were performed using Pulsefit (HEKA Elektronik) and Igor Pro (WaveMetrics, Lake Oswego, OR). GABA and levamisole were pressure ejected at a concentration of 500 μm for 100 ms during constant perfusion of the recording chamber.
Results
Strategy to disrupt innervation of postsynaptic muscle cells
Body-wall muscles on the dorsal side of the worm normally receive synaptic inputs from inhibitory GABA motor neurons and excitatory ACh motor neurons. Motor neuron cell bodies are located on the ventral side of the worm. Therefore, to make synaptic contact with dorsal muscles, motor axons must first grow dorsally (Fig. 1A). Once they reach the dorsal side, they extend longitudinally in a bundle called the dorsal nerve cord (Fig. 1A–C) and make synaptic contact with dorsal muscles (White et al., 1986). Dorsal axon growth requires the netrin receptor, encoded by the unc-5 gene (for a list of pertinent_C. elegans_ genes, their functions, and human homologs, see Table 1). By controlling which motor neurons express the netrin receptor (see Materials and Methods), we were able to control which motor neurons make synaptic contact with the dorsal muscles. To determine how the loss of presynaptic contact affects GABAA receptor localization patterns, we used a GFP-tagged C. elegans GABAA receptor subunit (UNC-49B-GFP, referred to here as GABAAR-GFP). GABAAR-GFP normally forms clusters opposite inhibitory presynaptic terminals (Fig. 1D) (Bamber et al., 1999;Gally and Bessereau, 2003).
Figure 1.
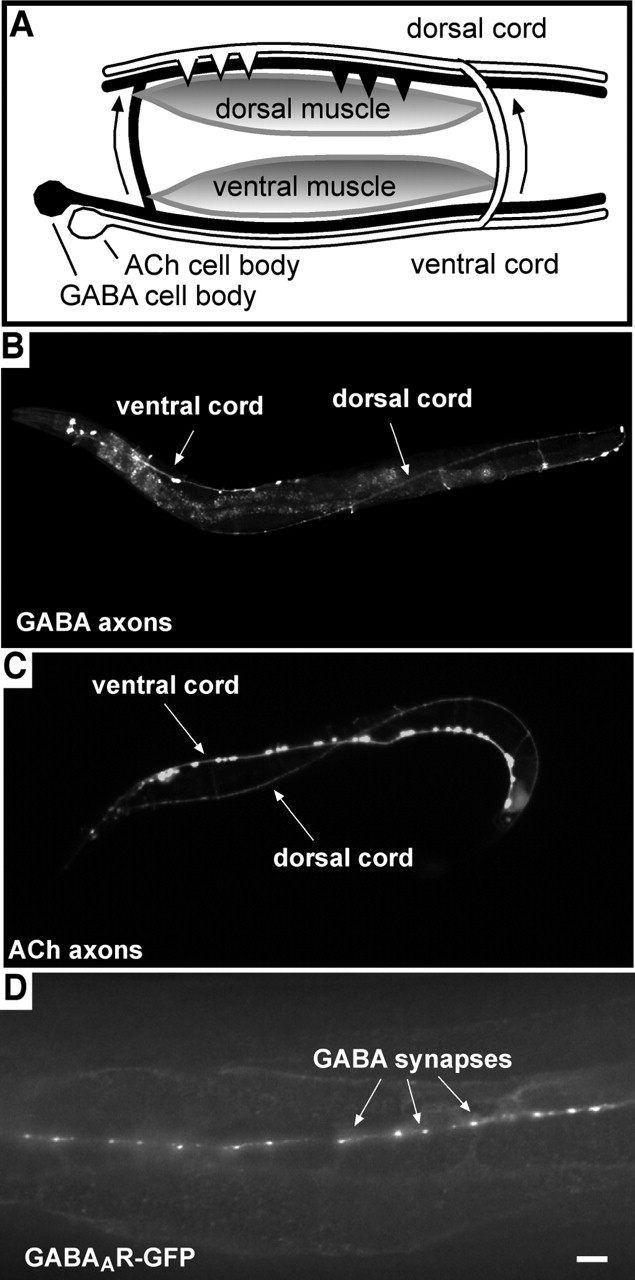
Normal neuromuscular anatomy in C. elegans.A, Illustration of normal C. elegans neuromuscular system. ACh (white) and GABA (black) motor neurons form synapses (triangles) on dorsal body-wall muscles (gray bodies). Cell bodies are ventral, so axons must grow dorsally (arrows) to reach dorsal muscles (neurons innervating ventral muscles not shown). B,C, Dorsal nerve cord contains GABA and ACh axons. Axons are visualized with GFP, expressed under the control of promoters specific for GABA (B) and ACh (C) neurons (see Materials and Methods). D, Normal synaptic localization of GABAAR-GFP in dorsal muscles. Scale bar, 5.0 μm.
Table 1.
Summary of C. elegans gene function and homologs
| C. elegans | gene | Yeast homolog | Mammalian homologs | ||
|---|---|---|---|---|---|
| Axon pathfinding | |||||
| unc-5 | Netrin receptor, dorsal axon guidancea | Unc5H1 | Netrin receptor, axon pathfindingb,c | ||
| Unc5H2 | |||||
| Unc5H3 | |||||
| Neurotransmitter receptors | |||||
| unc-49 | GABAAreceptord,e | GABAAR | |||
| unc-29 | ACh receptor subunite,f | AChR, non-α-subunit | |||
| unc-38 | ACh receptor subunite,f | AChR, α-subunit | |||
| Endocytosis | |||||
| rme-8 | Clathrin uncoating protein, endocytosisg | RME8 | Endosomal traffickingh | ||
| Autophagy | |||||
| unc-51 | Serine–threonine kinase, required for autophagyi | Atg1 | Induction of autophagyj | ULK1 | Function not reported, binds GABARAPk |
| bec-1 | Subunit of the phosphatidyl inositol-3kinase complex, required for autophagyh | Atg6 | Autophagy and vacuolar protein sortingi | beclin 1 | Autophagy, tumor suppressorl |
| lgg-1 | Required for autophagyh | Atg8 | Autophagosome associated, required for autophagyi | LC3 | Autophagym,n |
| GABARAP | GABAAR traffickingl,o | ||||
| GATE-16 | Intra-Golgi transportl,p |
Presynaptic innervation affects GABAA receptor clustering and trafficking
We first investigated the role of GABA presynaptic terminals in establishing postsynaptic GABAA receptor patterns. We examined worms in which GABA neurons could not project to dorsal body-wall muscles; therefore, the dorsal cord contained only ACh axons (Fig. 2A,D,G). GABAAreceptors did not form clusters in these animals but instead appeared to localize diffusely to the muscle cell surface (Fig. 2J). Therefore, GABA presynaptic terminals are necessary for GABAA receptor clustering in C. elegans, consistent with previous results (Gally and Bessereau, 2003).
Figure 2.

Loss of innervation affects GABAA receptor clustering and trafficking.A–C, Illustrations of dorsal muscles lacking motor inputs. D–F, GFP expressed in GABA presynaptic neurons. G–I, GFP expressed in ACh presynaptic neurons. J–L, Postsynaptic GABAAR-GFP. A,D, G,J, GABA dorsal pathfinding is disrupted, and muscle cells lack GABA inputs. B,E, H,K, ACh dorsal pathfinding is disrupted, and muscle cells lack ACh inputs. C,F, I,L, Both ACh and GABA dorsal pathfinding disrupted, and dorsal muscles lack all synaptic inputs. Arrowheads in**D, H, andI** indicate axons that extend dorsally a short distance but do not reach the dorsal cord. Arrows in **J**and L indicate dorsal midline. Scale bars, 5.0 μm.
To determine whether the GABA presynaptic terminals were sufficient to cluster GABAA receptors, we investigated whether ACh presynaptic terminals were required. When the dorsal nerve cord contained only GABA axons and not ACh axons (Fig. 2B,E,H), postsynaptic clustering of the GABAA receptor was normal (Fig. 2K). We confirmed that these clusters formed opposite GABA presynaptic terminals using a marker for GABA synaptic vesicles (data not shown). These data indicate that ACh presynaptic terminals are not required and that GABA terminals alone provide a signal to the postsynaptic cell to initiate synaptic GABAAreceptor clustering.
To test whether GABA and ACh motor axons have a redundant effect on postsynaptic GABAA receptors, we examined worms in which no neurons projected dorsally (Fig. 2C,F,I) and, consequently, dorsal muscles were non-innervated. GABAA receptors in the non-innervated cells accumulated in large intracellular organelles (Fig. 2L). The number of GABAAR-GFP-containing intracellular organelles was far higher in non-innervated muscles than when innervation of either type was present or in ventral muscles that receive normal presynaptic contacts (Table 2). These results indicate that presynaptic contact suppresses intracellular GABAA receptor accumulation. We conclude that presynaptic terminals play two roles in GABA synaptogenesis. GABA neurons specifically are required to organize GABAA receptors into synaptic clusters, and presynaptic contact by either type of motor neuron prevents GABAA receptors from accumulating within cells.
Table 2.
GABAAR-GFP accumulation in intracellular organelles
| Presynaptic contacts to dorsal muscles | Organelles per worm |
|---|---|
| Normal (wild-type worms) | 1.9 ± 0.3 (n = 66) |
| ACh only | 5.1 ± 1.1 (n = 15) |
| GABA only | 4.4 ± 1.6 (n = 18) |
| Absent (netrin receptor-defective mutant; unc-5) | 28.7 ± 1.6 (n = 65)* |
| 88 ± 2% dorsal (n = 15) | |
| Absent, autophagy defective (unc-5; unc-51 double mutant) | 4.5 ± 0.6 (n = 50) |
This finding suggests that presynaptic neurons provide a signal to regulate the trafficking of postsynaptic GABAA receptors. One possibility is that neurotransmitters released from the presynaptic cells serve as this signal. To test this hypothesis, we examined GABAAR-GFP localization in a variety of mutants with neurotransmission defects (supplemental Table S1, Fig. S1, available at www.jneurosci.org as supplemental material). We first determined that the neurotransmitter release machinery is not required. GABAAR-GFP clusters normally and does not accumulate inside cells in mutants defective for the release of synaptic vesicles (unc-13 and snb-1) or dense-core vesicles (unc-31). Second, we determined that neither ACh nor GABA signaling are required. Contact by ACh neurons still prevents the intracellular accumulation of GABAAR-GFP when ACh synthesis is defective or when postsynaptic ACh receptor function is eliminated. Likewise, contact by GABA neurons still prevents intracellular GABAAR-GFP accumulation when GABA synthesis is defective. Together, these results provide evidence that the presynaptic signal regulating GABAA receptor trafficking is not a neurotransmitter or a neuropeptide. It is more likely that the presynaptic cell expresses a cell-surface molecule or secretes a growth factor or extracellular matrix protein that serves as the signal.
GABAA receptors accumulate in autophagosomes
To identify the intracellular organelles in which GABAA receptors accumulate in the non-innervated cells, we examined electron micrographs of worms that lack the netrin receptor [unc-5_ mutants (Hedgecock et al., 1990)]. In these worms, motor neurons cannot extend axons dorsally, so dorsal muscles do not receive synaptic inputs. We observed elevated numbers of large structures, ranging from ∼0.5 to 2 μm in diameter, bounded by multiple membranes in non-innervated muscles (Fig. 3A,B). This morphology is typical of autophagosomes. We found examples of early autophagosomes [∼0.5 μm diameter, bounded by concentric rings of membrane (Fig. 3A)] and later-stage autophagosomes [larger bodies containing a uniform electron-opaque matrix, typical of autophagosomes undergoing fusion with lysosomes (Fig. 3B)]. These structures were increased fivefold in dorsal muscles of netrin receptor-deficient worms compared with wild type (Fig. 3C). Therefore, lack of innervation seems to cause increased autophagy. The ultrastructure of the muscles was otherwise normal. We did not observe loss of electron density indicative of autophagic cell death (Bursch, 2001), and we did not observe degenerative changes such as shrinkage of cytoplasm or sarcomeres that take place in aging_C. elegans muscle cells (Herndon et al., 2002) (Fig. 3D,E).
Figure 3.
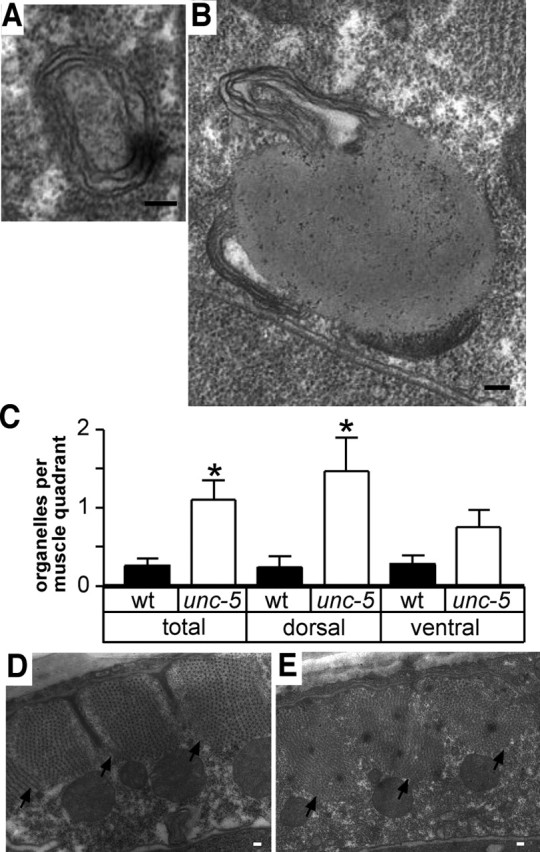
Autophagy is increased in non-innervated muscle cells.A, B, Electron micrographs of autophagosomes in the non-innervated dorsal body-wall muscles of netrin-defective (unc-5) worms. Autophagosomes typically constitute large membrane-bound organelles containing complex contents including additional membrane layers inside. Morphologies typical of early (A) and late (B) autophagosomes or autophagolysosomes were observed. C, Autophagy is significantly enhanced in netrin receptor-deficient (unc-5) mutants, mainly in dorsal muscles. Sarcomere volume and cytoplasm volume are comparable between normal (D) and non-innervated (E) dorsal muscles, indicating that lack of innervation does not cause muscle cell death or degeneration. Arrows indicate the boundary between sarcomeres and cytoplasm. Scale bars, 0.1 μm. *p < 0.05 compared with wild type, by Mann–Whitney_U_ test; n = 48 and 56 muscle quadrants in wild-type (wt) and unc-5 mutants, respectively.
This correlation between the lack of presynaptic contact and increased autophagy suggests that the GABAAR-GFP-positive organelles in non-innervated cells may be autophagosomes. To test this idea, we first determined whether formation of the GABAAR-GFP-positive organelles depended on the autophagy machinery. The_C. elegans_ UNC-51 protein is required for autophagy (Table 1) (Melendez et al., 2003). We observed greatly reduced numbers of GABAAR-GFP-positive intracellular organelles in non-innervated muscle cells when the UNC-51 protein was defective (Fig. 4A, Table 2). Thus, the autophagy machinery is required for organelle formation. Second, we tested whether intracellular GABA_A_ R-GFP fluorescence in non-innervated cells colocalized with markers of autophagosomes. GFP-tagged versions of the proteins LGG-1 and beclin-1 can be used to visualize autophagosomes in vivo in C. elegans and mammalian cells (Yue et al., 2002; Melendez et al., 2003). We tagged these proteins with mRFP to generate two red fluorescent autophagosome markers, called mRFP-LGG-1 and beclin-1-mRFP (see Materials and Methods). GABA_A_R-GFP and mRFP-LGG-1 colocalized in the intracellular organelles in non-innervated muscles (Fig. 4B). However, high background levels of mRFP-LGG-1 fluorescence in the muscle cell cytoplasm made the colocalization difficult to quantify. GABA_A_R-GFP and beclin-1-mRFP also colocalized in non-innervated muscles (Fig. 4C). Beclin-1-mRFP produced less background fluorescence, and we were able to determine that 100% of GABA_A_R-GFP-positive organelles were also positive for beclin-1-mRFP.
Figure 4.
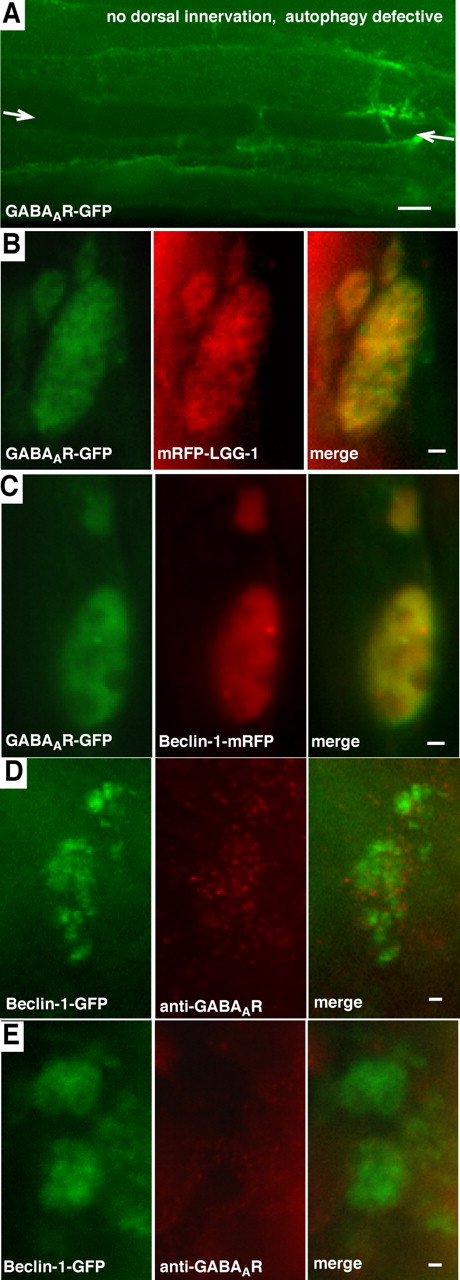
Evidence that GABAAR-GFP traffics to autophagosomes.A, GABAAR-GFP-containing organelles do not form in non-innervated muscles in an autophagy-defective mutant background (unc-5;unc-51 double mutant). GABAAR-GFP-containing organelles also contain the autophagosome markers mRFP-LGG-1 (B) and beclin-1-mRFP (C). D, GABAA receptor immunoreactivity is associated with beclin-1-GFP fluorescence in non-innervated muscle cells that do not overexpress GABAAR-GFP, indicating that endogenous GABAA receptors traffic to autophagosomes. E, In control worms lacking endogenous GABAA receptors (unc-49 mutants), autophagosomes in non-innervated muscle cells do not contain GABAAreceptor immunoreactivity. Arrows in A indicate the position of the dorsal midline. Scale bars: A, 5.0 μm; B–E, 1.0 μm.
To show that trafficking to autophagosomes is not caused by overexpression of a GFP-tagged GABA_A_ receptor, we also analyzed non-innervated muscles using an antibody against the endogenous GABA_A_receptor (Gally and Bessereau, 2003). We used beclin-1-GFP to visualize autophagosomes in this experiment because it provided the lowest background of any marker that we tested. We determined that 33 ± 8% of the autophagosomes were enriched for GABA_A_ receptor immunoreactivity in non-innervated cells (n_=10 worms, 94 organelles total) (Fig. 4D). In contrast, autophagosomes in control worms lacking endogenous GABA_A_receptors (i.e., unc-49 mutants) do not exhibit any increased GABA_A receptor immunoreactivity (Fig. 4E). Thus, trafficking of GABA_A_ receptors to autophagosomes is not an artifact of GFP tagging or overexpression. However, overexpression seems to result in increased accumulation of GABA_A_ receptors in autophagosomes, because GABA_A_R-GFP can be detected in ∼10-fold more autophagosomes than can endogenous GABA_A_R immunoreactivity.
The above results provide the first evidence for degradative trafficking of GABA_A_ receptors through the autophagy pathway. Does it take place under normal physiological conditions? We looked for colocalization of beclin-1-GFP and anti-GABA_A_R immunoreactivity in worms that had intact neuromuscular innervation. We occasionally detected organelles in muscle cells positive for both GABA_A_ receptor immunoreactivity and beclin-1-GFP (7 organelles in 10 worms) (Fig. 5). This result suggests that GABAA receptors can traffic to autophagosomes in normal muscles as well, but the pathway is negatively regulated by contact with presynaptic terminals.
Figure 5.

GABAA receptors traffic to autophagosomes in innervated muscles. GABAA receptor immunoreactivity associates with beclin-1-GFP in correctly innervated muscle cells in worms with functional netrin receptors. Scale bar, 1.0 μm.
GABAA receptors traffic to autophagosomes from the cell surface
We next investigated the mechanism of GABAA receptor trafficking to autophagosomes to better understand how the presynaptic signal patterns postsynaptic GABAA receptors. One possibility is that GABAA receptors reach the cell surface normally but then traffic to autophagosomes after removal by endocytosis, suggesting that presynaptic contact regulates receptor surface stability. Alternatively, GABAA receptors may fail to properly assemble when innervation is absent and traffic to autophagosomes as nonfunctional aggregates directly from the endoplasmic reticulum, suggesting that presynaptic contact regulates receptor assembly. To distinguish between these possibilities, we first looked at the role of endocytosis using a mutant with a temperature-sensitive endocytic defect. Endocytosis requires the RME-8 protein (Zhang et al., 2001; Chang et al., 2004). A mutant form of RME-8 functions normally at 15°C but becomes unstable and is degraded at 26°C (Zhang et al., 2001). In the presence of this mutation, intracellular GABAAR-GFP fluorescence in non-innervated muscles disappeared at 26°C. The same temperature shift had no effect when RME-8 was normal (Fig. 6A). This result shows that trafficking of GABAA receptors to autophagosomes requires endocytosis and implies that the receptors are correctly assembled and exported to the cell surface before they traffic to autophagosomes. To directly investigate surface expression of GABAA receptors, we performed electrophysiological recordings. The amplitude of the GABA response in non-innervated dorsal muscles was reduced by 30% compared with innervated cells (Fig. 6B). Although statistically significant, this reduction was modest, suggesting that GABAA receptor synthesis, assembly, and membrane insertion are normal despite the loss of presynaptic contact. Furthermore, GABA currents in non-innervated cells returned to their normal levels when autophagy was blocked by the unc-51 mutation (Fig. 6B), indicating that autophagic degradation, and not reduced GABAA receptor synthesis, is responsible for the 30% decrease. These results provide evidence that the autophagy pathway can function to reduce GABAA receptor surface expression and that presynaptic contact blocks GABAA receptor trafficking from the cell surface to the autophagosome.
Figure 6.
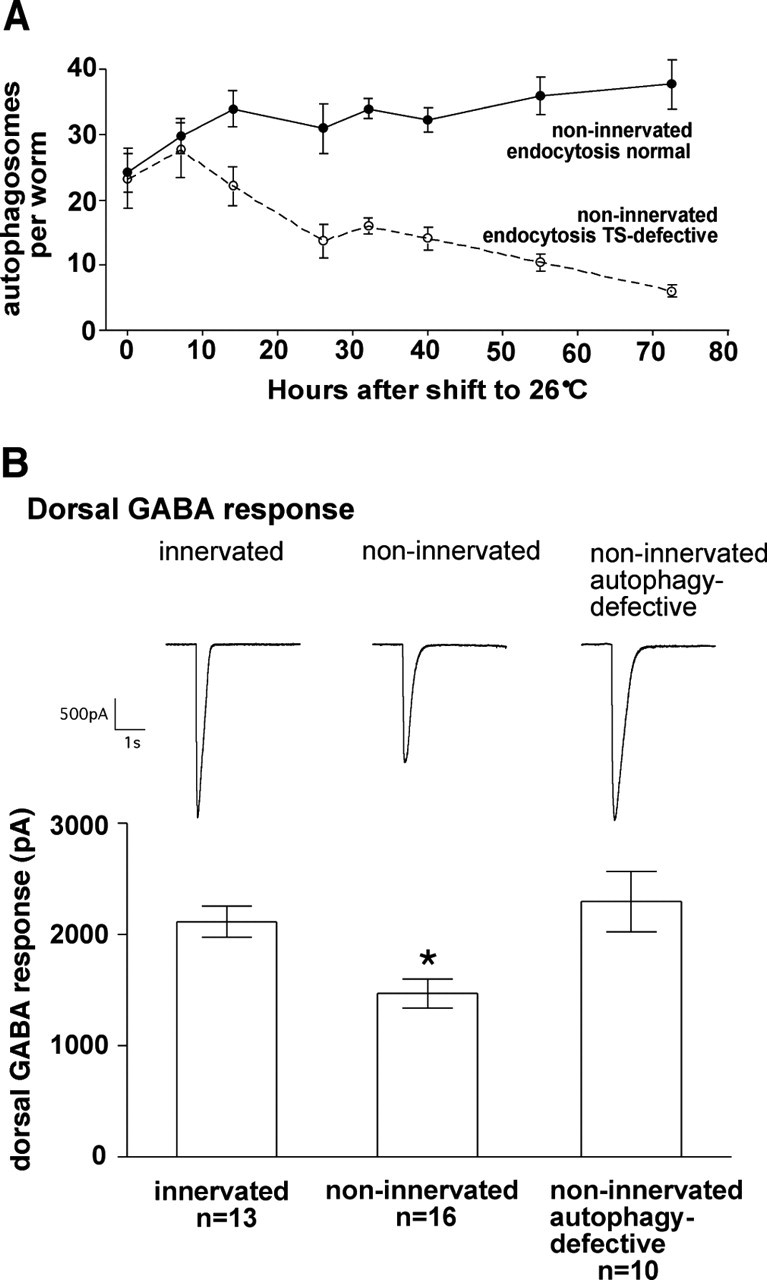
GABAA receptors are removed from the surface of non-innervated cells by trafficking to autophagosomes. A, Accumulation of GABAAR-GFP in autophagosomes requires ongoing endocytosis. In worms with a temperature-sensitive endocytosis defect (open symbols, dashed line), the number of GABAAR-GFP-containing autophagosomes decreases when animals are shifted from the permissive temperature (15°C) to the restrictive temperature (26°C). This decrease is not observed in control worms with normal endocytosis (filled symbols, solid line). Differences between endocytosis-defective and control strains are significant at 14 h and all subsequent time points (p< 0.05; n = 10 for each time point, except for_t_ = 0, in which n = 6 for the control strain).B, GABA currents are reduced in non-innervated dorsal cells as a result of autophagy. Left trace and bar, Wild type; middle trace and bar, netrin deficient (unc-5 mutant); right trace and bar, netrin deficient and autophagy deficient (unc-5;unc-51 double mutant). *p < 0.05 indicates that currents are significantly reduced in non-innervated dorsal cells compared with either normally innervated dorsal cells or non-innervated autophagy-defective dorsal cells (one-way ANOVA). Error bars are SEM.
Acetylcholine receptors do not traffic to autophagosomes
Finally, we examined acetylcholine receptors in non-innervated cells to determine whether autophagy is a general mechanism to degrade neurotransmitter receptors or whether it is used selectively for GABAA receptors. C. elegans body-wall muscles express two nicotinic ACh receptors. One of these is sensitive to the cholinergic agonist levamisole and contains the UNC-29 and UNC-38 subunits (Richmond and Jorgensen, 1999). We expressed a GFP-tagged UNC-38 subunit, resulting in a GFP-tagged acetylcholine receptor (AChR-GFP). AChR-GFP localizes to the ventral and dorsal nerve cords (Fig. 7A). In netrin-deficient worms, AChR-GFP is detectable only at the ventral nerve cord. We did not detect synaptic puncta at the dorsal midline, consistent with the absence of a dorsal cord in these mutants. We also did not observe intracellular fluorescence in dorsal muscles (Fig. 7B) (n = 40 worms), indicating that AChR-GFP does not accumulate in autophagosomes. We also characterized the other subunit, UNC-29, using an anti-UNC-29 antibody (Gally et al., 2004). In non-innervated muscles, GABAAR-GFP fluorescence was visible in autophagosomes, but UNC-29 immunoreactivity was not (Fig. 7C) (n = 15 worms). UNC-29 immunoreactivity was observed adjacent to GABAAR-GFP clusters along the ventral cord, indicating that the antibody staining was successful in this experiment. Thus, these AChRs do not accumulate in autophagosomes in non-innervated muscle cells. To confirm that they are still synthesized, we recorded levamisole currents (Fig. 7D). Currents were comparable in innervated and non-innervated dorsal muscles, suggesting that lack of innervation does not affect the levels of these ACh receptors. Furthermore, unlike GABA currents, levamisole currents were not increased when autophagy was blocked using the unc-51 mutation. Thus, under identical conditions, GABAA receptors traffic to autophagosomes, whereas an ACh receptor in the same cell does not, demonstrating that autophagy downregulates surface-expressed GABAA receptors selectively.
Figure 7.
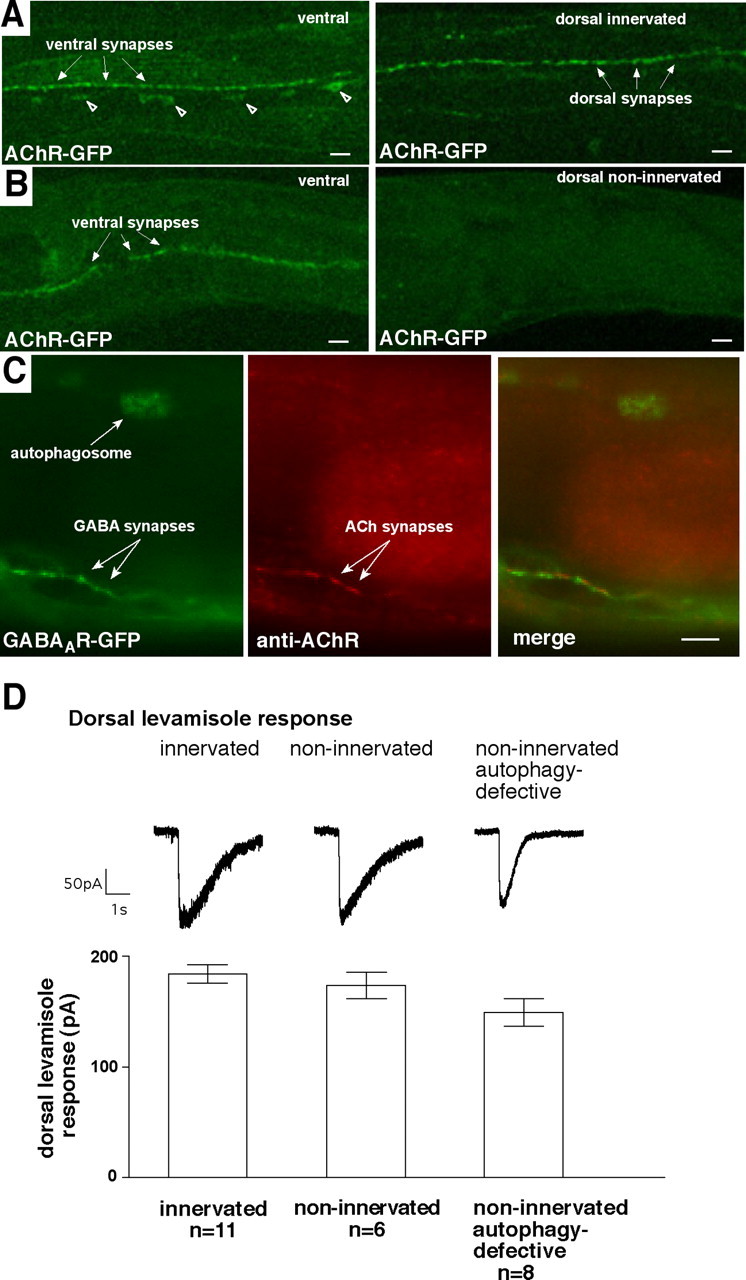
Acetylcholine receptors do not traffic to autophagosomes in non-innervated muscle cells. A, B, Localization pattern of GFP-tagged ACh receptors expressed in body-wall muscles.A, In a worm with functional netrin receptors, and therefore normal neuromuscular innervation, AChR-GFP is visible along the ventral (left) and dorsal (right) nerve cords. Cellular fluorescence in the ventral cord (open arrowheads) is probably neuronal (White et al., 1986). B, In a worm lacking functional netrin receptors, and therefore lacking dorsal motor innervation, AChR-GFP is visible along the ventral cord (left) but is not observed dorsally in either synaptic puncta or intracellular organelles (right).C, Immunoreactivity for another ACh receptor subunit, UNC-29, does not localize to autophagosomes. In a netrin-defective worm, GABAAR-GFP was visible along the ventral nerve cord and within an autophagosome in a dorsal muscle (left). UNC-29 immunoreactivity was also visible along the ventral nerve cord but not in the autophagosome (middle). Merged image shows localization of GABAAR-GFP and UNC-29 localization to adjacent synapses but no colocalization in the autophagosome (right).D, Electrophysiological response to the ACh agonist is the same in innervated and non-innervated cells and is not increased when autophagy is blocked (p > 0.05, one-way ANOVA). Left trace and bar, Wild type; middle trace and bar, netrin deficient (_unc-5_mutant); right trace and bar, netrin deficient and autophagy deficient (unc-5;unc-51 double mutant). Error bars are SEM. Scale bars, 5 μm.
Discussion
In this study, we characterized the roles of presynaptic innervation in the formation of postsynaptic GABAA receptor clusters. GABAA receptor localization patterns were compared in C. elegans body-wall muscles in the presence and absence of their normal contacts with GABA and ACh motor neurons. The results show that presynaptic innervation plays two roles. First, it promotes GABAA receptor surface stability. Receptors traffic from the cell surface to autophagosomes for degradation when presynaptic contacts are absent, suggesting that presynaptic neurons normally provide a signal that blocks autophagy. This signal is present on both GABA and ACh motor neurons and is not dependent on synaptic vesicle release. Second, innervation organizes GABAAreceptors into postsynaptic clusters. Clustering is induced specifically by GABA neurons, and the clustering signal is associated with synaptic vesicles (Gally and Bessereau, 2003). Our results corroborate studies in mammalian neurons that show that GABA terminals induce GABAA receptor synaptic clustering, but non-GABA terminals can also play a role (Levi et al., 1999; Rao et al., 2000; Brunig et al., 2002; Christie et al., 2002; Studler et al., 2002). They also provide two novel insights. First, presynaptic innervation independently regulates GABAA receptor surface stability and synaptic clustering. Second, autophagy can function as a selective degradation pathway for GABAAreceptors.
Although autophagy is often upregulated under stressful conditions, the autophagy observed in non-innervated C. elegans muscles seems to be different from a typical cellular stress response. A major function of autophagy is to alleviate nutrient stress by degrading and recycling cytoplasm and organelles (Klionsky and Emr, 2000). However, we observed far more autophagy in dorsal muscles than in ventral muscles, although cells on both sides of the animal had equal access to nutrients. Therefore, autophagy is not likely to be a secondary response to possible starvation in uncoordinated worms with defective motor axon pathfinding. Another important function of autophagy is to execute programmed cell death (Gozuacik and Kimchi, 2004). However, we did not observe the loss of electron density characteristic of autophagic cell death, and non-innervated cells did not die. The autophagy we observed is also distinct from the wasting, or atrophy, in denervated mammalian skeletal muscles. Muscle wasting is not autophagic but instead is attributable to the activity of the ubiquitin-proteasome system (Trout et al., 1981;Lecker et al., 1999). Instead, the role of autophagy in non-innervated C. elegans muscles may be to degrade postsynaptic proteins that would normally be incorporated into synapses. Autophagy can be stimulated by protein aggregates, as occurs in protein conformational disorders such as Huntington’s and Parkinson’s diseases (Ravikumar et al., 2002). Autophagy in non-innervated _C. elegans_muscle cells could be stimulated by synaptic scaffolding proteins that form ectopic cytoplasmic aggregates because their normal sites of assembly are absent. Once autophagosomes form, they may serve as a trafficking destination for GABAAreceptor-containing endocytic vesicles, thus allowing the coordinated degradation of cytoplasmic and membrane-bound postsynaptic proteins.
Our results demonstrate a novel role for autophagy in the degradation of cell-surface receptors. We showed that a properly folded and fully functional receptor can be trafficked from the cell surface to autophagosomes. Moreover, this pathway is used selectively: ACh receptors are expressed in the same cells and have similar structure and subcellular localization as GABAA receptors but do not traffic to autophagosomes under identical conditions. Selective degradation of surface-expressed receptors by autophagy could be important for the regulation of multiple aspects of cellular function. For example, it has been proposed that autophagy inhibits cancer progression by selectively degrading growth-promoting proteins such as cell-surface growth factor receptors (Qu et al., 2003; Yue et al., 2003). Our results support this idea by demonstrating that normal functional cell-surface receptors can selectively traffic to autophagosomes.
An important implication of these findings is that autophagy may be a mechanism to control the balance of neuronal excitation and inhibition. This balance depends on the relative strengths of inhibitory and excitatory synapses. At inhibitory synapses, GABAAreceptors are continually internalized by endocytosis. When the GABAA receptors recycle to the plasma membrane, synapse strength is maintained. When they traffic into a degradative pathway, synapse strength is reduced (Kittler and Moss, 2003). Autophagy of GABAA receptors satisfies two important criteria to serve as a mechanism to regulate the excitation–inhibition balance. First, autophagy functions as a degradative pathway for GABAA receptors: receptor surface levels were decreased because of autophagy, and GABAA receptors in autophagosomes gradually disappear when the influx of new receptors from the plasma membrane is blocked. Second, autophagy is selective for GABAA receptors and not ACh receptors, so it can control inhibitory synapse strength while leaving excitatory synapse strength unaffected. GABAA receptor autophagy is normally negatively regulated by a presynaptic signal. Modulating that signaling pathway provides a possible mechanism to control the excitation–inhibition balance.
Finally, our results provide evidence that the membrane dynamics of GABAAreceptor trafficking and autophagy may be similar at a biochemical level. We demonstrated the involvement of autophagy proteins in GABAA receptor degradative trafficking. Others have demonstrated that autophagy proteins are involved in GABAA receptor export to the plasma membrane. Specifically, GABAA receptor-associated protein (GABARAP) facilitates trafficking of GABAA receptors from the Golgi apparatus to the plasma membrane (Leil et al., 2004). GABARAP is a homolog of the yeast autophagy protein Atg8, and both proteins undergo C-terminal lipidation, which allows association with autophagosomal membranes (Kabeya et al., 2004). The involvement of autophagy proteins in both the biosynthesis and degradation of GABAA receptors raises the possibility that GABAA receptor trafficking and autophagy use overlapping biochemical mechanisms.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants MH64699 and NS43345 (B.A.B.), RR12596 (D.H.H.), and NS041477 (J.E.R.), the Whitehall Foundation, and the Primary Children’s Medical Center Foundation (Utah). A.M.R. was supported by NIH Genetics Training Grant GM007464 (University of Utah). We thank the C. elegans Genetics Center for providing strains, Y. Jin for providing the juIs14 array, B. Levine for providing the GFP-LGG-1 plasmid and strains, B. Grant for providing the rme-8 mutant, J.-L. Bessereau for providing anti-UNC-49 antibodies, anti-UNC-29 antibodies, and the UNC-38-GFP construct, B. Bass and S. Mango for generously providing access to equipment, J. Culotti for providing the unc-5 cDNA, K. Schuske, S. Mango for critical reading, and the Jorgensen Laboratory for strains and helpful discussion.
References
- Ackerman SL, Kozak LP, Przyborski SA, Rund LA, Boyer BB, Knowles BB (1997). The mouse rostral cerebellar malformation gene encodes an UNC-5-like protein. Nature 386:838–842. [DOI] [PubMed] [Google Scholar]
- Bamber BA, Beg AA, Twyman RE, Jorgensen EM (1999). The Caenorhabditis elegans unc-49 locus encodes multiple subunits of a heteromultimeric GABA receptor. J Neurosci 19:5348–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamber BA, Richmond JE, Otto JF, Jorgensen EM (2005). Composition of the GABA receptor at the Caenorhabditis elegans neuromuscular junction. Br J Pharmacol 144:502–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baulac S, Huberfeld G, Gourfinkel-An I, Mitropoulou G, Beranger A, Prud’homme JF, Baulac M, Brice A, Bruzzone R, LeGuern E (2001). First genetic evidence of GABAA receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat Genet 28:46–48. [DOI] [PubMed] [Google Scholar]
- Bettinger JC, Lee K, Rougvie AE (1996). Stage-specific accumulation of the terminal differentiation factor LIN-29 during Caenorhabditis elegans development. Development 122:2517–2527. [DOI] [PubMed] [Google Scholar]
- Broadie K, Bate M (1993). Innervation directs receptor synthesis and localization in Drosophila embryo synaptogenesis. Nature 361:350–353. [DOI] [PubMed] [Google Scholar]
- Brunig I, Scotti E, Sidler C, Fritschy JM (2002). Intact sorting, targeting, and clustering of gamma-aminobutyric acid A receptor subtypes in hippocampal neurons in vitro. J Comp Neurol 443:43–55. [DOI] [PubMed] [Google Scholar]
- Burden SJ (2002). Building the vertebrate neuromuscular synapse. J Neurobiol 53:501–511. [DOI] [PubMed] [Google Scholar]
- Bursch W (2001). The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ 8:569–581. [DOI] [PubMed] [Google Scholar]
- Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY (2002). A monomeric red fluorescent protein. Proc Natl Acad Sci USA 99:7877–7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HC, Hull M, Mellman I (2004). The J-domain protein Rme-8 interacts with Hsc70 to control clathrin-dependent endocytosis in Drosophila. J Cell Biol 164:1055–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chih B, Engelman H, Scheiffele P (2005). Control of excitatory and inhibitory synapse formation by neuroligins. Science 307:1324–1328. [DOI] [PubMed] [Google Scholar]
- Christie SB, Miralles CP, De Blas AL (2002). GABAergic innervation organizes synaptic and extrasynaptic GABAA receptor clustering in cultured hippocampal neurons. J Neurosci 22:684–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossette P, Liu L, Brisebois K, Dong H, Lortie A, Vanasse M, Saint-Hilaire JM, Carmant L, Verner A, Lu WY, Wang YT, Rouleau GA (2002). Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet 31:184–189. [DOI] [PubMed] [Google Scholar]
- Crestani F, Lorez M, Baer K, Essrich C, Benke D, Laurent JP, Belzung C, Fritschy JM, Luscher B, Mohler H (1999). Decreased GABAA-receptor clustering results in enhanced anxiety and a bias for threat cues. Nat Neurosci 2:833–839. [DOI] [PubMed] [Google Scholar]
- Finger JH, Bronson RT, Harris B, Johnson K, Przyborski SA, Ackerman SL (2002). The netrin 1 receptors Unc5h3 and Dcc are necessary at multiple choice points for the guidance of corticospinal tract axons. J Neurosci 22:10346–10356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming JT, Squire MD, Barnes TM, Tornoe C, Matsuda K, Ahnn J, Fire A, Sulston JE, Barnard EA, Sattelle DB, Lewis JA (1997). Caenorhabditis elegans levamisole resistance genes lev-1, unc-29, and unc-38 encode functional nicotinic acetylcholine receptor subunits. J Neurosci 17:5843–5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gally C, Bessereau JL (2003). GABA is dispensable for the formation of junctional GABA receptor clusters in Caenorhabditis elegans. J Neurosci 23:2591–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gally C, Eimer S, Richmond JE, Bessereau JL (2004). A transmembrane protein required for acetylcholine receptor clustering in Caenorhabditis elegans. Nature 431:578–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard M, Poupon V, Blondeau F, McPherson PS (2005). The DnaJ-domain protein RME-8 functions in endosomal trafficking. J Biol Chem 280:40135–40143. [DOI] [PubMed] [Google Scholar]
- Gozuacik D, Kimchi A (2004). Autophagy as a cell death and tumor suppressor mechanism. Oncogene 23:2891–2906. [DOI] [PubMed] [Google Scholar]
- Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM (2004). Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 119:1013–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DH (1995). Electron microscopy and three-dimensional image reconstruction. Methods Cell Biol 48:395–436. [DOI] [PubMed] [Google Scholar]
- Hallam SJ, Jin Y (1998). lin-14 regulates the timing of synaptic remodelling in Caenorhabditis elegans. Nature 395:78–82. [DOI] [PubMed] [Google Scholar]
- Hallam S, Singer E, Waring D, Jin Y (2000). The C. elegans NeuroD homolog cnd-1 functions in multiple aspects of motor neuron fate specification. Development 127:4239–4252. [DOI] [PubMed] [Google Scholar]
- Hamelin M, Zhou Y, Su MW, Scott IM, Culotti JG (1993). Expression of the UNC-5 guidance receptor in the touch neurons of C. elegans steers their axons dorsally. Nature 364:327–330. [DOI] [PubMed] [Google Scholar]
- Hedgecock EM, Culotti JG, Hall DH (1990). The unc-5, unc-6, and unc-40 genes guide circumferential migrations of pioneer axons and mesodermal cells on the epidermis in C. elegans. Neuron 4:61–85. [DOI] [PubMed] [Google Scholar]
- Herndon LA, Schmeissner PJ, Dudaronek JM, Brown PA, Listner KM, Sakano Y, Paupard MC, Hall DH, Driscoll M (2002). Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature 419:808–814. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T (2004). LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 117:2805–2812. [DOI] [PubMed] [Google Scholar]
- Kittler JT, Moss SJ (2003). Modulation of GABAA receptor activity by phosphorylation and receptor trafficking: implications for the efficacy of synaptic inhibition. Curr Opin Neurobiol 13:341–347. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ (2005). The molecular machinery of autophagy: unanswered questions. J Cell Sci 118:7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Emr SD (2000). Autophagy as a regulated pathway of cellular degradation. Science 290:1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer JM, French RP, Park EC, Johnson JJ (1990). The Caenorhabditis elegans rol-6 gene, which interacts with the sqt-1 collagen gene to determine organismal morphology, encodes a collagen. Mol Cell Biol 10:2081–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecker SH, Solomon V, Mitch WE, Goldberg AL (1999). Muscle protein breakdown and the critical role of the ubiquitin-proteasome pathway in normal and disease states. J Nutr 129:227S–237S. [DOI] [PubMed] [Google Scholar]
- Leil TA, Chen ZW, Chang CS, Olsen RW (2004). GABAAreceptor-associated protein traffics GABAA receptors to the plasma membrane in neurons. J Neurosci 24:11429–11438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung-Hagesteijn C, Spence AM, Stern BD, Zhou Y, Su MW, Hedgecock EM, Culotti JG (1992). UNC-5, a transmembrane protein with immunoglobulin and thrombospondin type 1 domains, guides cell and pioneer axon migrations in C. elegans. Cell 71:289–299. [DOI] [PubMed] [Google Scholar]
- Levi S, Chesnoy-Marchais D, Sieghart W, Triller A (1999). Synaptic control of glycine and GABAA receptors and gephyrin expression in cultured motoneurons. J Neurosci 19:7434–7449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B (1999). Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402:672–676. [DOI] [PubMed] [Google Scholar]
- Liu G (2004). Local structural balance and functional interaction of excitatory and inhibitory synapses in hippocampal dendrites. Nat Neurosci 7:373–379. [DOI] [PubMed] [Google Scholar]
- McIntire SL, Reimer RJ, Schuske K, Edwards RH, Jorgensen EM (1997). Identification and characterization of the vesicular GABA transporter. Nature 389:870–876. [DOI] [PubMed] [Google Scholar]
- Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B (2003). Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301:1387–1391. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y (2004). In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 15:1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki N, Yan J, Yuasa S, Ueno T, Kominami E, Masuho Y, Koga H, Muramatsu M (2000). Interaction of the Unc-51-like kinase and microtubule-associated protein light chain 3 related proteins in the brain: possible role of vesicular transport in axonal elongation. Brain Res Mol Brain Res 85:1–12. [DOI] [PubMed] [Google Scholar]
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B (2003). Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 112:1809–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Cha EM, Craig AM (2000). Mismatched appositions of presynaptic and postsynaptic components in isolated hippocampal neurons. J Neurosci 20:8344–8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Duden R, Rubinsztein DC (2002). Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet 11:1107–1117. [DOI] [PubMed] [Google Scholar]
- Richmond JE, Jorgensen EM (1999). One GABA and two acetylcholine receptors function at the C. elegans neuromuscular junction. Nat Neurosci 2:791–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagiv Y, Legesse-Miller A, Porat A, Elazar Z (2000). GATE-16, a membrane transport modulator, interacts with NSF and the Golgi v-SNARE GOS-28. EMBO J 19:1494–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semyanov A, Walker MC, Kullmann DM, Silver RA (2004). Tonically active GABA A receptors: modulating gain and maintaining the tone. Trends Neurosci 27:262–269. [DOI] [PubMed] [Google Scholar]
- Studler B, Fritschy JM, Brunig I (2002). GABAergic and glutamatergic terminals differentially influence the organization of GABAergic synapses in rat cerebellar granule cells in vitro. Neuroscience 114:123–133. [DOI] [PubMed] [Google Scholar]
- Trout JJ, Stauber WT, Schottelius BA (1981). Degeneration and regeneration in denervated tonic and phasic skeletal muscle: morphology and acid phosphatase cytochemistry. Virchows Arch B Cell Pathol Incl Mol Pathol 38:67–76. [DOI] [PubMed] [Google Scholar]
- Wallace RH, Marini C, Petrou S, Harkin LA, Bowser DN, Panchal RG, Williams DA, Sutherland GR, Mulley JC, Scheffer IE, Berkovic SF (2001). Mutant GABAA receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet 28:49–52. [DOI] [PubMed] [Google Scholar]
- Wang H, Bedford FK, Brandon NJ, Moss SJ, Olsen RW (1999). GABA(A)-receptor-associated protein links GABA(A) receptors and the cytoskeleton. Nature 397:69–72. [DOI] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, Brenner S (1986). The structure of the nervous system of Caenorhabditis elegans. Philos Trans R Soc Lond B Biol Sci 314:1–340. [DOI] [PubMed] [Google Scholar]
- Yue Z, Horton A, Bravin M, DeJager PL, Selimi F, Heintz N (2002). A novel protein complex linking the delta 2 glutamate receptor and autophagy: implications for neurodegeneration in lurcher mice. Neuron 35:921–933. [DOI] [PubMed] [Google Scholar]
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N (2003). Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA 100:15077–15082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Grant B, Hirsh D (2001). RME-8, a conserved J-domain protein, is required for endocytosis in Caenorhabditis elegans. Mol Biol Cell 12:2011–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]