Structure of a complex of the ATPase SecA and the protein-translocation channel (original) (raw)
. Author manuscript; available in PMC: 2020 Apr 17.
Published in final edited form as: Nature. 2008 Oct 16;455(7215):936–943. doi: 10.1038/nature07335
Abstract
Most proteins are secreted from bacteria by the interplay of the cytoplasmic ATPase SecA and a membrane channel, formed from the heterotrimeric SecY complex. We report crystal structures of SecA bound to the SecY complex, both isolated from different species, with a maximum resolution of 4.5Å. One copy of SecA in its transition state of ATP hydrolysis is bound to one SecY molecule. Both partners undergo major conformational changes upon interaction. The polypeptide-crosslinking domain of SecA makes a large conformational change that could capture the translocation substrate in a “clamp”. Polypeptide movement through the SecY channel could be achieved by the motion of a “two-helix finger” of SecA inside the cytoplasmic funnel of SecY, and the coordinated tightening and widening of SecA’s “clamp”. SecA binding generates a “window” at the lateral gate of the SecY channel and it displaces the plug domain, preparing the channel for signal sequence binding and channel opening.
Introduction
Many bacterial proteins are transported across or are integrated into the plasma membrane, a process that is similar to protein translocation across the endoplasmic reticulum (ER) membrane in eukaryotes (for review, see ref.1). Translocation begins with a targeting phase, during which proteins are directed to the membrane by hydrophobic sequences, which are either cleavable signal sequences or trans-membrane (TM) segments of membrane proteins. Subsequently, soluble proteins, such as secretory proteins, are transported through a hydrophilic channel across the membrane, whereas hydrophobic TM segments of membrane proteins exit sideways through a lateral gate of the channel into the surrounding lipid phase. The channel is formed from a conserved heterotrimeric membrane protein complex, called the SecY complex in bacteria and archaea, and the Sec61 complex in eukaryotes1. The complex consists of a large 〈- subunit (SecY or Sec61p), and two smaller ®- and ©-subunits (called SecG and SecE in bacteria). The channel can associate with different partners that provide a driving force for translocation. In bacteria, the SecY channel can bind either to the ribosome to translocate polypeptides during their synthesis (co-translational translocation), or it can associate with the cytoplasmic ATPase SecA to transport secretory proteins after completion of their synthesis (post-translational translocation). SecA is also required for the biosynthesis of some membrane proteins2–4. How the channel connects to its different partners and how polypeptides are moved through the channel is unclear.
The crystal structure of an archaeal SecY complex (from Methanococcus jannaschii) and biochemical experiments show that a single SecY copy forms the active channel5–7. SecY consists of two linked halves, TMs 1–5 and 6–10, which form a lateral gate at the front and are clamped together at the back by SecE5. The helices of SecY form an hourglass-shaped pore that consists of cytoplasmic and external funnels. The constriction of the pore is located about half way across the membrane and is formed from a ring of hydrophobic amino acids that project their side chains radially inward (pore ring). The cytoplasmic funnel is empty, whereas the external funnel is plugged by a short helix that interacts with the pore residues. This crystal structure therefore represents a closed channel and its opening involves plug displacement8, 9.
Post-translational translocation in bacteria requires only the SecY complex and the RecA-like ATPase SecA, which “pushes” polypeptides through the SecY channel10–12. SecA consists of two nucleotide-binding domains (NBD1 and NBD2), which bind the nucleotide between them, a polypeptide-crosslinking domain (PPXD), a helical scaffolding domain (HSD), and a helical wing domain (HWD)13. Although several crystal structures of isolated SecA have been determined, the function of the different domains and the mechanism by which SecA moves polypeptides through the channel remain unknown. Disulfide crosslinking experiments suggest that SecA binds through its NBD1 domain to a non-translocating SecY copy, and moves the polypeptide chain through a neighboring SecY molecule6. These and other experiments indicate that SecA functions as a monomer during translocation7, 14–16, but the issue is controversial17–19.
Here, we report crystal structures of SecA bound in its transition state of nucleotide hydrolysis to the SecY channel. The structures suggest mechanisms for how the channel is opened and prepared for the arrival of a translocation substrate, and how SecA moves polypeptides through the channel.
Structure determination of SecA-SecY complexes
We crystallized complexes containing Bacillus subtilis SecA without its non-essential C-terminal domain and either Thermotoga maritima SecYE or Aquifex aeolicus SecYEG. These crystals diffracted X-rays to a maximum resolution of 6.5 Å and 7.5 Å, respectively. A higher resolution dataset (4.5 Å) was obtained for a complex in which both partners were from T. maritima and the SecYEG complex was Se-Met derivatized. All complexes were crystallized in the detergent Cymal-6 in the presence of ADP and BeF3-. The structure of the B. subtilis SecA-T. maritima SecYE complex was determined by molecular replacement with a B. subtilis SecA structure20 and served as an initial model for the other complexes. The building of a 4.5Å resolution model of the T. maritima SecA-SecY complex was facilitated by the Se-Met positions (Supplementary Fig. S1), and by the high quality of the electron density map that allowed the identification of large amino acid side chains (Fig. 1a, b). Model building also took into account conserved interactions between amino acids in previously determined SecY and SecA structures5, 20, 21. The final structure was refined to working and free R factors of 28.0% and 32.2% (Table 1), respectively, and was used for all interpretations. It comprises all residues of SecA and most residues of SecYEG. No model could be built for the periplasmic loop between TMs 1 and 2a of SecY (residues 42–54), as well as for residues of some termini (SecY 427–436; SecE 1–9; SecG 1–8 and 74–76). In addition, there are uncertainties about the tip of the loop between TMs 6 and 7. An ADP*BeF3- complex was placed in the electron density observed in the nucleotide binding pocket of SecA. A detailed description of the methods is given in the Supplementary material.
Figure 1.
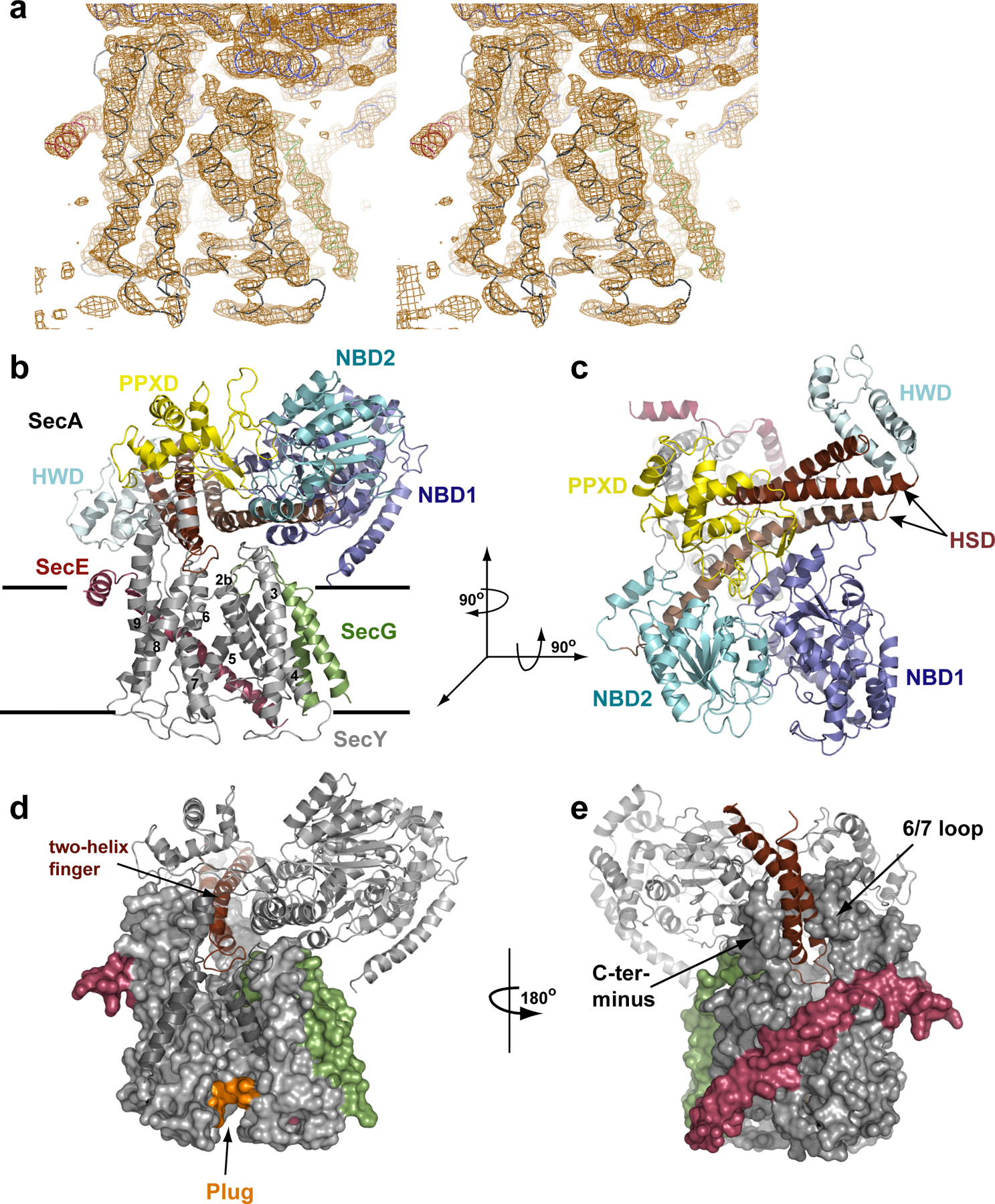
Architecture of the T. maritima SecA–SecYEG complex. a, Stereo view of a σA-weighted, phase combined, NCS averaged, and B-factor sharpened 2Fo - Fc electron density map (contoured at 1σ). The view of the lateral gate of SecY is shown, with the Cα-trace of SecY in grey, SecE in red, SecG in green and SecA in blue. b, Cartoon of the complex viewed from the side. The lines indicate the membrane boundaries. c, As in b, but viewed from the cytoplasm. d, The two-helix finger of SecA inside the cytoplasmic funnel of SecY. TM2b and TM8, as well as the tip of the 6–7 loop, are shown as cartoons for clarity. Plug residues are coloured in orange. e, As in d, but shown from the back.
Table 1.
Crystallographic statistics
| Data Collection | ||||
|---|---|---|---|---|
| Data set | Tm$ SecA-SecYEG | Tm SecA-SecYEG | Bs SecA-Tm SecYE | Bs SecA-Aa SecYEG |
| Compound | Se-Met SecYEG | native | native | native |
| Resolution limit [Å] | 4.5 | 5.7 | 6.2 | 7.5 |
| Space group | P212121 | P21 | P65 | P212121 |
| Cell constants [Å]/[°] | 101.6, 156.0,358.1 | 172.5, 102.5, 181.9 | 240.3,240.3, 179.5 | 146.4, 168.0, 187.7 |
| (a, b, c, / α, β, γ) | 90, 90, 90 | 90, 100.9, 90 | 90,90, 120 | 90,90, 90 |
| Measured reflections | 76164 | 39014 | 28484 | 6238 |
| Unique reflections | 34733 | 18704 | 13510 | 5416 |
| Mean I/σ(I) | 16.1 (2.0)# | 16.5(1.5) | 26.2 (2.5) | 16.2(1.6) |
| Rsym (%)a | 0.065 (0.85) | 0.055 (0.87) | 0.075 (>1.0) | 0.072 (0.76) |
| Completeness (%) | 97.7 (89.7) | 93.3 (81.8) | 100.0(100.0) | 99.1 (99.8) |
| Redundancy | 11.6 (8.8) | 3.8 (2.5) | 9.4 (9.0) | 3.5(3.6) |
| SAD-Phasingb | ||||
| FOM (from SOLVE) | 0.25 (0.12) | |||
| FOM (post NCS averag.) | 0.72 (0.27) | |||
| Refinement | Rigid body | |||
| Resolution range [Å] | 15–4.5 | 15–6.2 | 15–7.5 | |
| NCSc | 2 | 2 | 2 | 2 |
| Rwork(%)‖ | 28.0 | 42.0 | 36.4 | |
| Rfree (%)¶ | 32.2 | 42.7 | 38.4 | |
| Number of Reflections | ||||
| Total | 32721 | 9176 | 4884 | |
| Rfree | 3269 | 1012 | 532 | |
| Number of atomsd | ||||
| Protein | 21312 | |||
| Nonprotein | 62 | |||
| Model geometry | ||||
| r.m.s.d. bond length [Å] | 0.0097 | |||
| r.m.s.d. bond angle [°] | 1.34 | |||
| Average B-factor [Å2] | ||||
| Main chain | 374 | |||
| Side chain | 338 | |||
| Ramachandran analvsise (for 1333 residues) | ||||
| Most favored | 65.3 | |||
| Allowed | 27.9 | |||
| Generously allowed | 5.3 | |||
| Disallowed | 1.5 |
Architecture of the SecA-SecYEG complex
The structures obtained with SecA and SecY from three different species all have the same architecture (Fig. 1c, d, and Supplementary Fig. S2, S3), despite the complexes forming different crystal contacts. Proteoliposomes containing purified T. maritima SecYEG can translocate a substrate in the presence of B. subtilis or T. maritima SecA (S. Miller, Y. N., and J. Z., unpublished results). Together, these data suggest that the structures correspond to a physiological state of SecA-SecY interaction. Crystals were only obtained in the presence of ADP and BeF3- or AlF4-, corresponding to a transition state of ATP hydrolysis. Gel filtration experiments indicated that the interaction between SecA and SecY is strongest in this state (Supplementary Fig. S4).
The structure shows one SecA molecule bound to one copy of the SecY complex (Fig. 1c, d). The flat SecA molecule is oriented approximately parallel to the plane of the membrane (Fig. 1c). Viewed from the cytosol, NBD1, NBD2, PPXD, and HSD sit at the corners of an approximate square (Fig. 1d). NBD1 is linked diagonally with the PPXD. Likewise, the linked NBD2 and HSD are located in opposite corners of the square.
The extensive interactions of SecA with the SecY channel cover approximately 6,800Å2 of surface area. The most crucial interactions occur between the PPXD of SecA and the 8/9 and the 6/7 loops of SecY. SecA constructs lacking the PPXD indeed fail to form stable complexes with SecY (data not shown), and mutations in the two SecY loops affect SecA-mediated translocation9, 22 The HSD of SecA also makes extensive contacts with SecY, accounting for almost 50% of the buried surface area. Its interaction with the C-terminal tail of SecY is probably functionally important, as indicated by the effect of SecY truncations and point mutations23, 24 and by the interference of a His-tag with SecA-SecY interaction (Y.N., unpublished results). The long helix of the HSD rests on the 2b/3 loop of SecY and the SecG loop. These interactions are probably not strong, as the contact surfaces are small, the 2b/3 loop contains only few conserved amino acids, and SecG is not essential for translocation. Minor interactions also exist between SecG and the NBD1 of SecA (residues 420 to 430 and the N-terminus). NBD2 makes no contacts with the SecY channel.
A “two-helix finger”, which corresponds to the shorter helices of the HSD and was not previously considered to be a separate domain, is inserted into the cytoplasmic funnel of the channel (Fig. 1e,f). The loop between the two helices is located in the cytoplasmic funnel of the channel, right above the entrance to the SecY pore. The finger has an angle of ~45o with respect to the plane of the membrane and rests in a V-shaped opening between the 6/7 loop and the C-terminal tail of SecY (Fig. 1f).
The structure supports our previous proposal that SecA functions as a monomer during translocation14. The single SecY copy seen in the structure must be the one through which polypeptide chains are moved during translocation. Disulfide-bridge crosslinking shows that the 6/7 loop of the non-translocating SecY copy contacts a face of NBD1 that is exposed in our structure6. This copy could bind in an approximate back-to-back orientation, although its exact location is difficult to predict. The non-translocating SecY is lost upon detergent solubilization7, along with the ability of SecY to stimulate the translocation-substrate dependent ATPase activity of SecA (unpublished results). In a membrane, the interaction between the two SecY complexes might be stabilized by negatively charged lipids, which are known to be essential for translocation25, 26. SecA itself does not seem to make strong contacts with lipids, as only its extreme N-terminus comes close to the membrane surface.
Conformational changes in SecA
Overall, the architecture of T. maritima SecA closely resembles the structure of B. subtilis SecA with which it shares ~45% sequence identity (Fig. 2a–c). The most notable difference is the insertion of a mainly 〈-helical loop at the periphery of NBD1 (residue 156 to 206) (Fig. 2c). Crystal structures of SecA alone from different organisms show the molecule in two different conformations, with the PPXD either packed against the HWD or rotated away from it (Fig. 2a, b)13, 20. When SecA binds to the SecY complex, the PPXD rotates even further, making contact with NBD2 (Fig. 2c). With respect to the closed conformation, the overall movement of the PPXD is a large rigid body rotation by 80o and a tilt by 10o towards the NBDs, with the hinge formed by two short ®-strands connecting the PPXD and NBD1. An evolutionarily conserved loop in the PPXD now contacts both NBD1 and NBD2 and comes close to the nucleotide-binding site (Fig. 2c).
Figure 2.
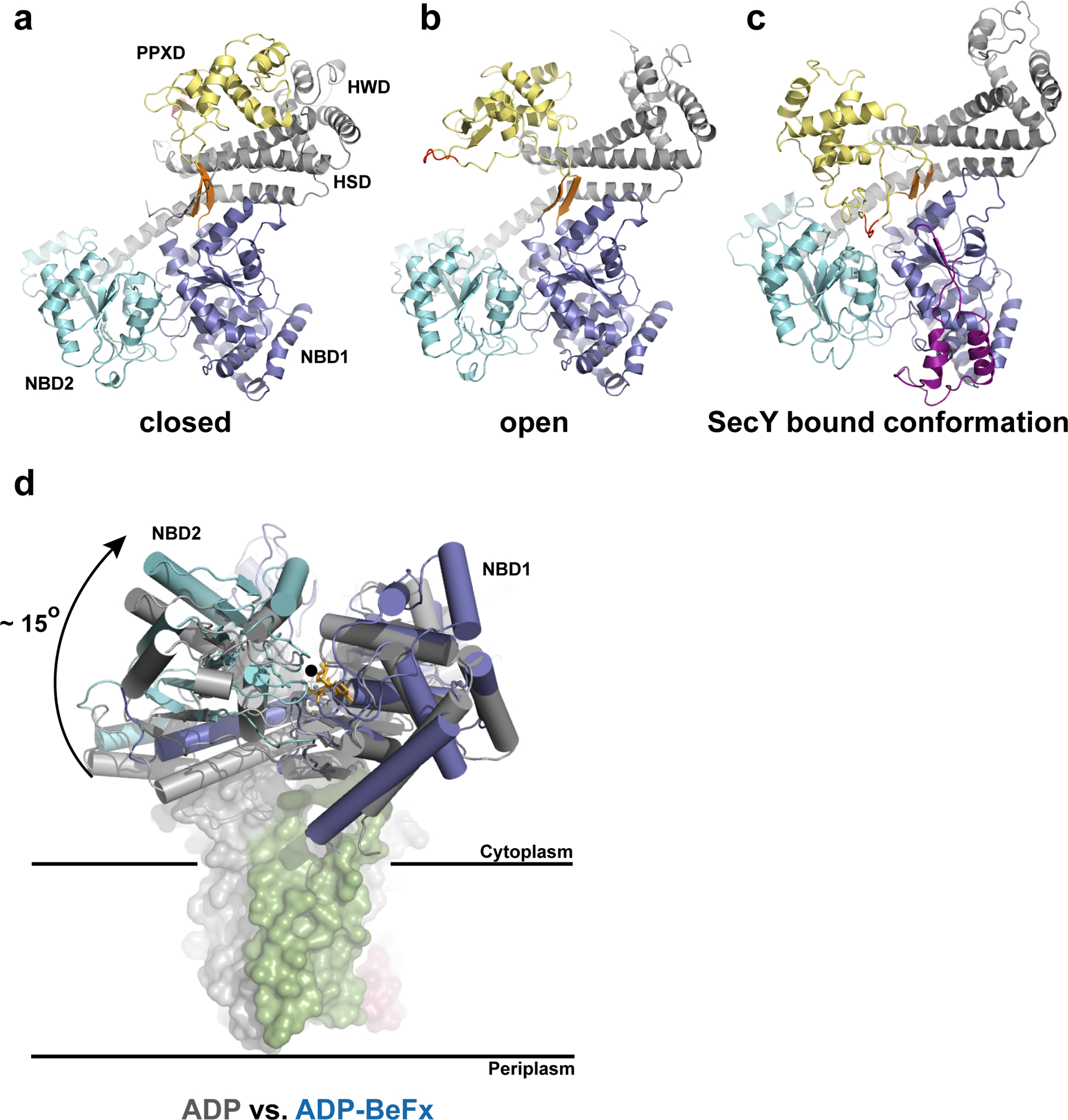
Conformational changes of SecA. a–c, Different orientations of the PPXD relative to the NBDs. The view is from the cytosol, as in Fig. 1c. The closed (a) and open (b) conformations refer to the B. subtilis SecA structures (PDB accessions 1M6N and 1TF2, respectively). The loop of the PPXD contacting NBD1 and NBD2 in the SecA–SecY complex is highlighted in red. The insertion in the T. maritima NBD1 is shown in purple. The conserved β-strands connecting NBD1 with the PPXD are shown in orange. d, Position of the NBDs in the ADP-bound state of B. subtilis SecA (grey) versus the ADP–BeF x bound state of T. maritima SecA (blue) when associated with SecY. The structures were aligned with respect to NBD1. The rotation axis indicated by a black circle is parallel to the plane of the membrane. The modelled ADP–BeF3-is shown as orange sticks.
NBD1 and NBD2 are closer to each other than in any reported structure of isolated SecA (Fig. 2d), all of which correspond to nucleotide free or ADP bound states. The relative positions of NBD1 and NBD2 in our structure are characteristic of the ATP or transition states of Rec-like ATPases, as shown for the PcrA helicase27. With NBD1 as a reference, the progression from the transition state of ATP hydrolysis to the ADP state would cause an ~15o rigid body rotation of NBD2 along an axis running parallel to the plane of the membrane and through the nucleotide binding cleft (Fig. 2d). The similarity with related ATPases suggests that the movement of the Arg finger of NBD2 (Arg570) towards the ©-phosphate triggers ATP hydrolysis.
Conformational changes in the SecY complex
As expected from sequence similarities, the T. maritima SecY complex has a similar overall structure as the one from M. jannaschii5. The major difference is the replacement of Sec® by SecG. SecG has two TMs with both termini located on the external side of the membrane, in agreement with previous topology determinations (Fig. 1c)28, 29. The N-terminal TM was assigned on the basis of Met24, which is seen in an anomalous difference Fourier map of the Se-Met derivatized SecY complex (Supplementary Fig. S1). The C-terminal TM has the same orientation and position as the single TM of Sec® in M. jannaschii (Fig. 3a), suggesting that these TMs may have analogous functions. The SecG loop is sandwiched between SecA and SecY (Fig. 1c). Our structure and other data30 make it unlikely that the TM segments of SecG invert their orientation during translocation28 (for further discussion, see Supplementary Material).
Figure 3.
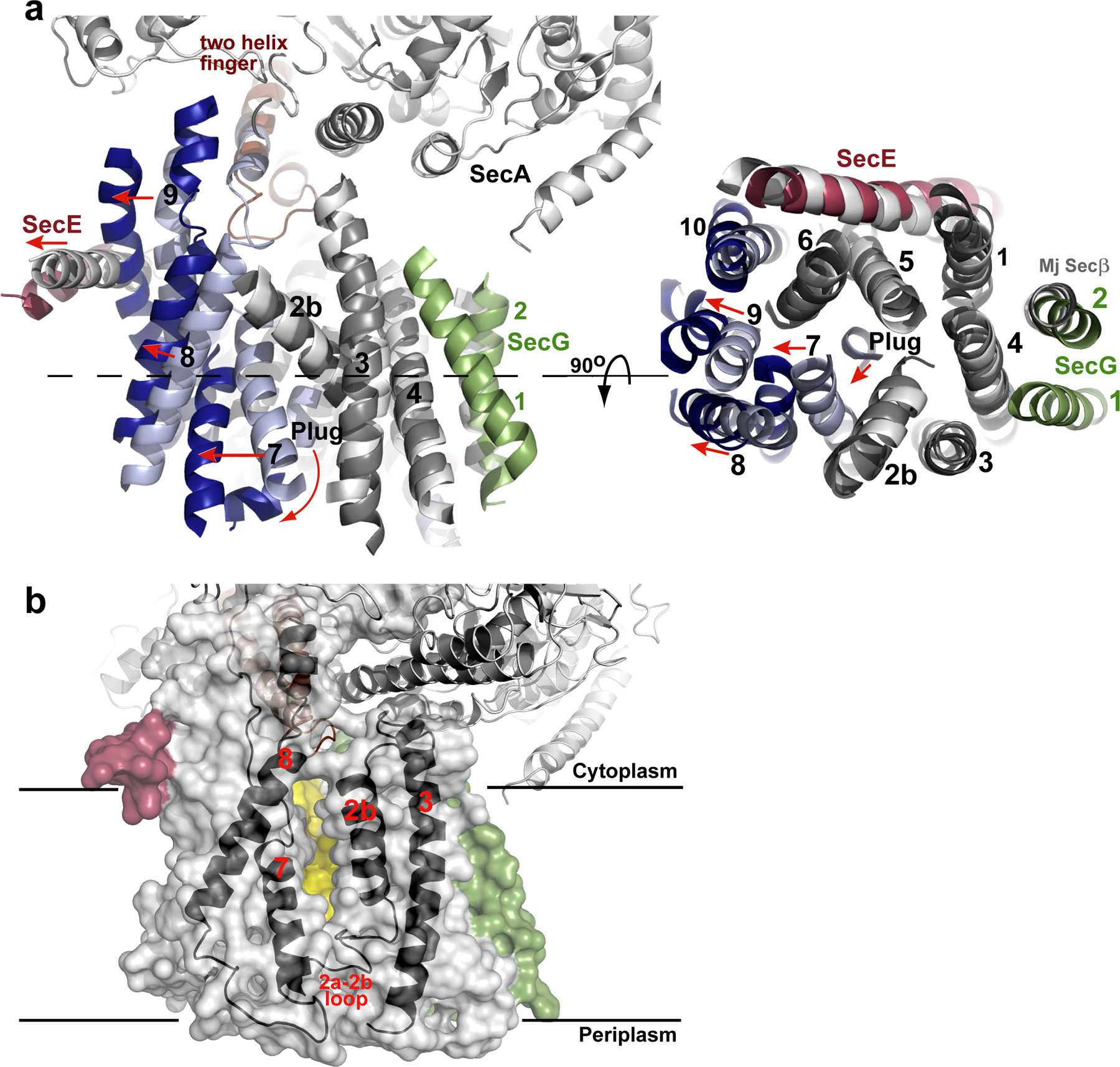
Conformational changes of SecY. a, Comparison of the T. maritima SecA–SecYEG complex with the SecY channel from M. jannaschii (Mj). The alignment is on the basis of TM2b–TM5 of SecY. The view on the right corresponds to a slice through the SecY channel (the plane is indicated by the dashed line on the left). TMs are numbered, and loops are omitted, for clarity. Similarly positioned (root mean squared deviation (r.m.s.d.) ≤1.8 Å) and deviating TMs of T. maritima SecY are shown in grey and blue, respectively. Helix movements are indicated by red arrows. b, Semi-transparent surface of the SecY channel. The window in the lateral gate of SecY is indicated by a view on the back wall of the channel, formed by TM5 and TM10 (in yellow). TM2b, TM3, TM7 and TM8 are shown as black cartoons.
The SecA-induced conformational changes of the SecY complex can best be appreciated by comparison with the crystal structure of the M. jannaschii SecY complex5. Although archaea lack SecA, the structure of this SecY complex is remarkably similar to the one in SecA-containing organisms, represented by the ~8Å structure of the E. coli SecY complex31, 32. While the N-terminal half of T. maritima SecY aligns well with the corresponding region of M. jannaschii SecY, all helices in the C-terminal half shift outwards, with TMs 8 and 9 showing the most dramatic changes (Fig. 3a). Without this conformational change, SecA’s two-helix finger would clash with the 〈-helical region of the 8/9 loop. The shift of TM 9 causes the amphipathic helix of SecE, which lies on the cytoplasmic surface of the membrane, to also move outwards.
The displacement of TM 7 generates a “window” in the lateral gate towards the center of the lipid bilayer (Fig. 3b). The gap between the side chains of TM7 and TM2b has a width of about 5Å. On the cytoplasmic side of the membrane, the lateral gate appears to be closed towards the head groups of the phospholipids, as TM2b and TM8 remain in close proximity. Tyr321 at this position could serve as a gatekeeper. The lateral gate is also closed towards the external side by the loop following TM2a.
TM2a, the plug, has moved away from the centre of the SecY channel towards the external side of the membrane (Fig. 3a; Fig. S2). It no longer contacts the pore residues, and instead packs against the C-terminus of TM 7. Despite its displacement, the plug still closes the channel (Fig. 1f), likely preventing even the permeation of water and ions. The plug sits at the front of the channel and is not close to the TM segment of SecE, as would be expected from disulfide crosslinking experiments performed in the presence of a translocating chain8, 9.
Implications for protein translocation
Translocation starts in the cytosol with an interaction of SecA with a signal-sequence containing polypeptide chain, a process that is often mediated by the chaperone SecB (for review, see ref.1). Next, SecA binds to the SecY complex, leading to a pre-activated state that must prepare the channel for the arrival of the substrate; this state is likely represented by our structure. Finally, the polypeptide chain inserts into the SecY channel as a loop, with the signal sequence intercalated into the lateral gate between TM2b and 7, and the following region in the pore proper33 (Fig. 5a). Although our structure lacks a translocating polypeptide, it suggests how each of these different steps might occur.
Figure 5.
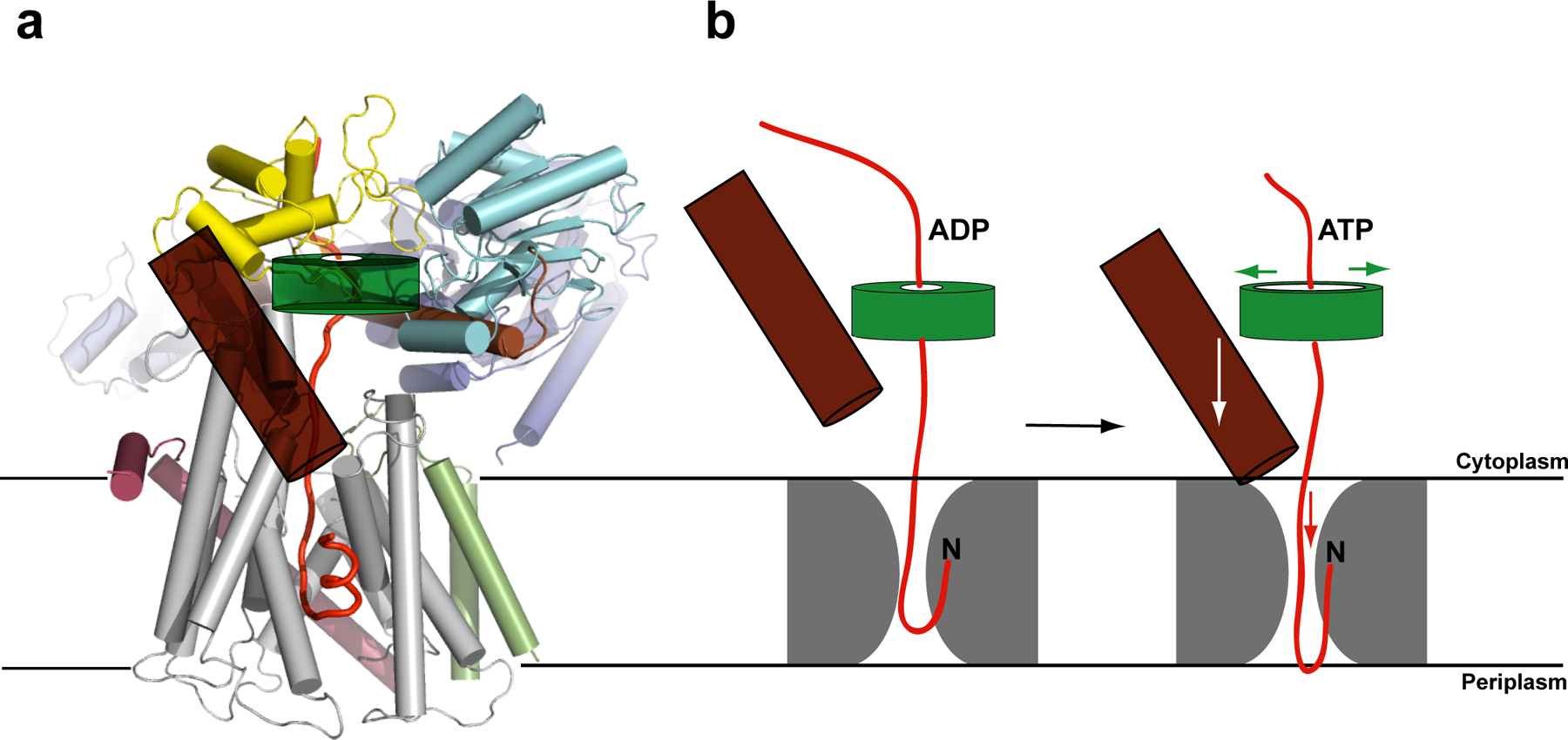
Model for SecA-mediated protein translocation. a, Proposed polypeptide translocation pathway through the SecA–SecYEG complex. The signal sequence of a hypothetical polypeptide (red trace) is intercalated into the lateral gate of SecY. The clamp of SecA is highlighted as a semi-transparent green torus, and the two-helix finger is shown as a brown cylinder. b, Upon ATP binding, the clamp of SecA would widen, allowing the two-helix finger to bind to the polypeptide and move it into the channel. After ATP hydrolysis, the clamp would tighten and the finger would reset. SecA probably adopts more than two conformational states during ATP hydrolysis. The structure might represent a situation in which the two-helix finger is in its ‘down-state’ and the clamp is already closed, although the conformation of the clamp may be different in the presence of a translocating polypeptide.
Capturing a polypeptide by SecA
The striking rotation of the PPXD (Figs. 2a–c) suggests that SecA captures its substrates in a groove formed between the PPXD, NBD2, and parts of the HSD. The groove is accessible in previously observed conformations (Fig. 2a, b)13, 20, whereas it would close around the polypeptide in the SecY bound state (Fig. 2c). Viewed from the cytosol, the open groove has the perfect dimensions to bind a polypeptide chain (Fig. 4a). The inside is lined with conserved residues and contains a hydrophobic patch (Fig. 4a, b). The lower part of the groove is formed by the two-helix finger and its back by the strictly conserved ®-strands connecting NBD1 with the PPXD (Fig. 2a–c). Disulfide bridge crosslinking experiments confirm the postulated site of polypeptide binding (parallel paper). The rotation of the PPXD may allow SecA to continuously bind and release a substrate in the cytosol, before the complex eventually associates with SecY. Once bound, the closed clamp would align the polypeptide chain with the SecY pore (Fig. 1c, 4c).
Figure 4.
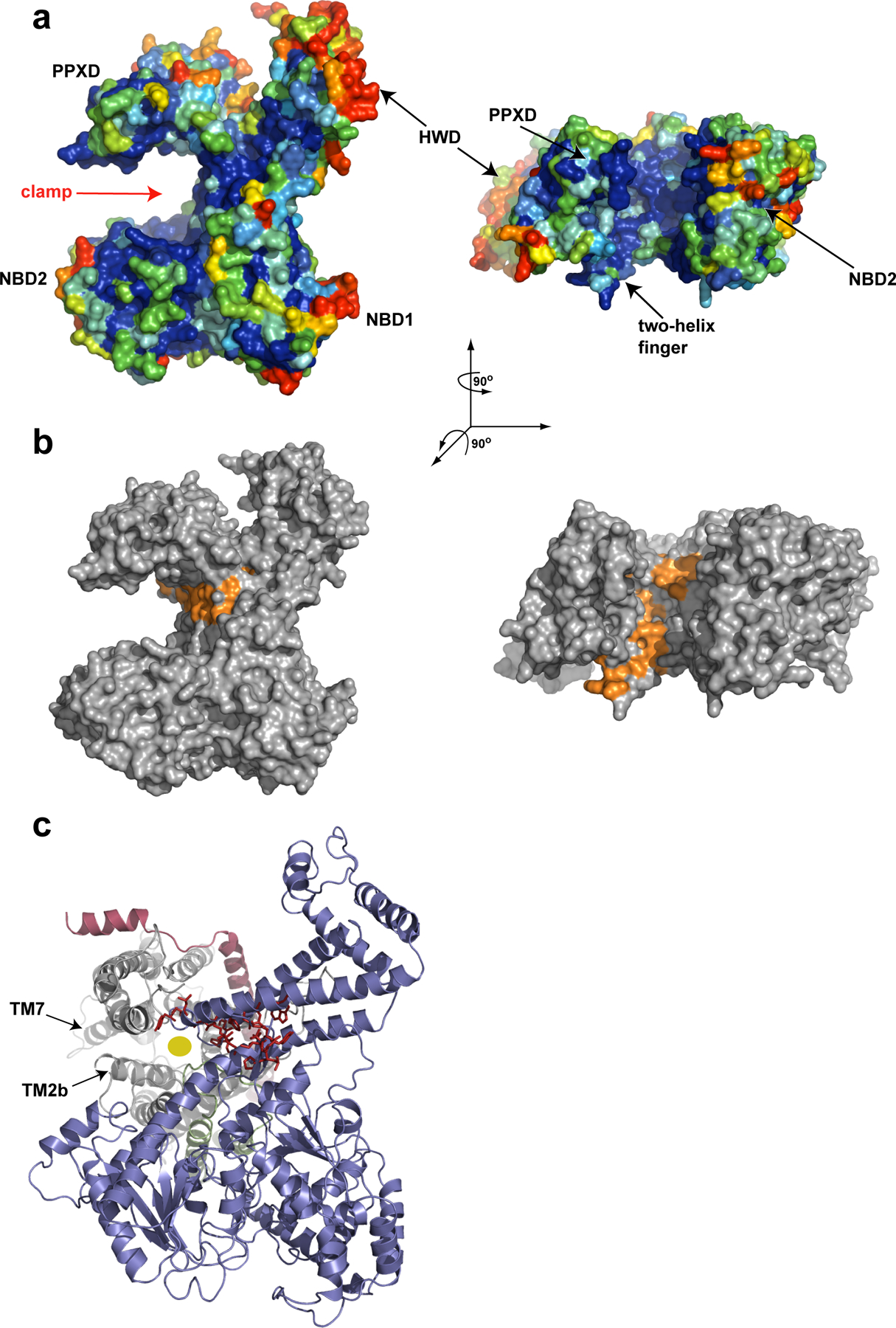
The polypeptide clamp of SecA. a, Surface representation of B. subtilis SecA (PDB accession 1TF2) with conserved amino acid residues colour coded (blue denotes the highest, and red denotes the lowest conservation). b, As in a, except that the conserved hydrophobic residues along the proposed polypeptide clamp of SecA are shown in orange. c, The T. maritima SecA–SecYEG complex with SecA shown in blue, and SecY, SecE and SecG shown in grey, red and green, respectively. The PPXD was removed for clarity. The residues that were labelled orange in b are shown as red sticks. The yellow circle indicates the translocation pore in SecY.
Although a recent NMR study reported that a signal peptide binds to the outside of the clamp (Supplementary Fig. S5)33, our structure suggests that the signal sequence could also bind to the same hydrophobic patch that interacts with the succeeding polypeptide segments. This would explain why the transfer of a signal sequence from SecA into the SecY channel requires a full cycle of ATP hydrolysis as for any other polypeptide segment20, and why SecA can translocate polypeptides lacking a signal sequence when associating with a signal-sequence suppressor mutant of SecY (prlA)35.
The cytosolic chaperone SecB interacts with the C-terminal domain of SecA36, which is missing in our structures. However, the C-terminus of SecA is located on the membrane-distal side, indicating that SecB would bind “on top” of SecA, opposite to the SecY channel. The spatial arrangement of the three components supports the stepwise transfer of a polypeptide from SecB to SecA to SecY37.
Signal sequence binding by SecY
Once released from SecA, a signal sequence of a translocation substrate is recognized by its intercalation into the lateral gate of SecY (Fig. 5a). The hydrophobic region of a signal sequence forms a helix of approximately two turns33 whose accommodation into the SecA-induced “window” would require only a slight further opening. The signal sequence would insert as a loop from the inside of the cytoplasmic funnel of SecY. The positively charged N-terminus might be retained on the cytosolic side of the “window” by an interaction with negatively charged phospholipid head groups. Intercalation of the signal sequence would cause the plug (TM2a) to be released from the channel. Insertion of the segment following the signal sequence into the pore proper would fix the open state of the channel.
Assuming that a “window” in the lateral gate is maintained even when the plug is fully displaced, a polypeptide segment inside the pore could contact the hydrocarbon chains of the surrounding lipids. If a segment is sufficiently hydrophobic, it would partition into the lipid phase, becoming a TM segment of a membrane protein38. Seams of the lateral gate on either side of the membrane would prevent phospholipid head groups from moving into the pore, explaining how the channel can be stable despite a window towards the center of the lipid bilayer.
Movement of the polypeptide by SecA
We propose that the tip of the two-helix finger of SecA moves up and down inside the cytoplasmic funnel of SecY during the ATP hydrolysis cycle, thereby pushing the polypeptide chain into the pore (Fig. 5b). The finger tip indeed contacts a translocating polypeptide (parallel paper). The finger would move the polypeptide when SecA binds ATP, and it would release it and reset upon ATP hydrolysis. This mechanism is analogous to the one proposed for hexameric RecA-like ATPases, such as ClpX, ClpA, HslU, and p9739–43. In these cases, each monomer has a loop with an essential Tyr (or Trp in the case of p97) at its tip, which moves inside the central pore and contacts the polypeptide. Most SecAs have a Tyr and a Gln in the loop at the tip of the two-helix finger. The movement of the two-helix finger could also cause the cyclic widening and constriction of the SecY pore. Our structure makes it unlikely that any part of SecA crosses the channel during translocation12.
Coordinated with movements of the two-helix finger, the clamp of SecA would hold and release the translocating polypeptide chain during the ATP hydrolysis cycle (Fig. 5b). In addition, a translocating polypeptide might transiently form a short ®-sheet with the two ®-strands connecting the NBD1 with the PPXD. This sequence-independent mode of interaction has been observed in some SecA structures, in which either the weakly structured C-terminal tail of SecA or a segment of a neighboring SecA molecule forms the additional ®-strand13, 21. The translocating polypeptide chain might thus stabilize the interaction of the PPXD with the NBDs, which in turn would be required for high-affinity binding of SecA to SecY. At the end of translocation, when the polypeptide chain has moved past the ®-strands, the PPXD would be free to rotate away from the NBD2, resulting in the lateral release of the last residues of the polypeptide and the disengagement of SecA from the SecY channel.
The perfect alignment of the mouth of the clamp with the lateral gate of the SecY channel offers an explanation for the participation of SecA in the biosynthesis of some membrane proteins2–4. Initially, membrane proteins would be translocated co-translationally, but the translating ribosome may detach occasionally from SecY. The polypeptide segment between the channel and the ribosome may then be recognized and moved into the channel by SecA. When the next TM segment is synthesized by the ribosome and arrives in the channel, it would exit the lateral gate of SecY into the lipid phase and drag the following polypeptide segment out of the SecA clamp. This would result in the release of SecA from SecY and allow the ribosome to rebind to the SecY channel.
How could the relative movement of the NBDs during the nucleotide hydrolysis cycle cause movements of the PPXD and the two-helix finger? We postulate that, during the transition from the ATP to the ADP state, both NBDs rotate outwards and towards the membrane. Because the PPXD is fixed by its interaction with the SecY channel, this could result in the transient widening of the clamp of SecA. The movement of the NBDs would also be propagated by the long helix of the HSD to the two-helix finger. In one scenario, the NBDs would maintain their interactions with the long helix of the HSD, resulting in its bending. This could change the tilt of the associated two-helix finger, such that its tip would move away from the SecY pore. An alternative possibility is that the interactions of the long helix of the HSD with the NBD1 are broken, perhaps by association of the NBD1 with the non-translocating SecY copy. In this case, the long helix would serve as a lever arm with a pivot point on the 2b/3 loop of SecY, and this could move the finger away from the SecY pore. The structures now provide the basis for experimental tests of these proposals.
Supplementary Material
SupInfo
Acknowledgements
We thank B. van den Berg and P. Bendapudi for initial experiments with the B. subtilis SecA-T. maritima SecY complex, G. Skiniotis for electron microscopy analysis, W. Li for help with data processing, the staff at APS beamline X24 and at BNL beamline X29, and the SBGrid consortium at Harvard Medical School. We thank S. Harrison and A. Brunger for helpful comments, and S. Harrison, B. van den Berg, and A. Osborne for critical reading of the manuscript.
References
- 1.Rapoport TA Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 450, 663–669 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Neumann-Haefelin C, Schafer U, Muller M & Koch HG SRP-dependent co-translational targeting and SecA-dependent translocation analyzed as individual steps in the export of a bacterial protein. EMBO J 19, 6419–6426 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qi HY & Bernstein HD SecA is required for the insertion of inner membrane proteins targeted by the Escherichia coli signal recognition particle. J Biol Chem 274, 8993–8997 (1999). [DOI] [PubMed] [Google Scholar]
- 4.Duong F & Wickner W Sec-dependent membrane protein biogenesis: SecYEG, preprotein hydrophobicity and translocation kinetics control the stop-transfer function. Embo J 17, 696–705 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van den Berg B et al. X-ray structure of a protein-conducting channel. Nature 427, 36–44 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Osborne AR & Rapoport TA Protein translocation is mediated by oligomers of the SecY complex with one SecY copy forming the channel. Cell 129, 97–110 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Duong F Binding, activation and dissociation of the dimeric SecA ATPase at the dimeric SecYEG translocase. EMBO J. 22, 4375–4384 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harris CR & Silhavy TJ Mapping an interface of SecY (PrlA) and SecE (PrlG) by using synthetic phenotypes and in vivo cross-linking. J. Bacteriol 181, 3438–3444 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tam PC, Maillard AP, Chan KK & Duong F Investigating the SecY plug movement at the SecYEG translocation channel. EMBO J 24, 3380–3388 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brundage L, Hendrick JP, Schiebel E, Driessen AJM & Wickner W The purified E. coli integral membrane protein SecY/E Is sufficient for reconstitution of SecA-dependent precursor proteintranslocation. Cell 62, 649–657 (1990). [DOI] [PubMed] [Google Scholar]
- 11.Akimaru J, Matsuyama SI, Tokuda H & Mizushima S Reconstitution of a Protein Translocation System Containing Purified SecY, SecE, and SecA from Escherichia-Coli. Proc. Natl. Acad. Sci. USA 88, 6545–6549 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Economou A & Wickner W SecA promotes preprotein translocation by undergoing ATP-driven cycles of membrane insertion and deinsertion. Cell 78, 835–843 (1994). [DOI] [PubMed] [Google Scholar]
- 13.Hunt JF et al. Nucleotide control of interdomain interactions in the conformational reaction cycle of SecA. Science 297, 2018–2026 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Or E, Navon A & Rapoport T Dissociation of the dimeric SecA ATPase during protein translocation across the bacterial membrane. EMBO J. 21, 4470–4479 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Or E, Boyd D, Gon S, Beckwith J & Rapoport T The bacterial ATPase SecA functions as a monomer in protein translocation. J Biol Chem 280, 9097–9105 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Alami M, Dalal K, Lelj-Garolla B, Sligar SG & Duong F Nanodiscs unravel the interaction between the SecYEG channel and its cytosolic partner SecA. EMBO J 26, 1995–2004 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jilaveanu LB, Zito CR & Oliver D Dimeric SecA is essential for protein translocation. Proc Natl Acad Sci U S A 102, 7511–7516 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Keyzer J et al. Covalently dimerized SecA is functional in protein translocation. J Biol Chem 280, 35255–35260 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Mitra K, Frank J & Driessen A Co- and post-translational translocation through the protein-conducting channel: analogous mechanisms at work? Nat Struct Mol Biol 13, 957–964 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Osborne AR, Clemons WM Jr. & Rapoport TA A large conformational change of the translocation ATPase SecA. Proc Natl Acad Sci U S A 101, 10937–10942 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zimmer J, Li W & Rapoport TA A novel dimer interface and conformational changes revealed by an X-ray structure of B. subtilis SecA. J Mol Biol 364, 259–265 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Shiba K, Ito K, Yura T & Cerretti DP A defined mutation in the protein export gene within the spc ribosomal protein operon of Escherichia coli: isolation and characterization of a new temperature-sensitive secY mutant. EMBO J 3, 631–635 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chiba K, Mori H & Ito K Roles of the C-terminal end of SecY in protein translocation and viability of Escherichia coli. J Bacteriol 184, 2243–2250 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mori H, Shimizu Y & Ito K Superactive SecY variants that fulfill the essential translocation function with a reduced cellular quantity. J Biol Chem 277, 48550–48557 (2002). [DOI] [PubMed] [Google Scholar]
- 25.de Vrije T, de Swart R, Dowhan W, Tommassen J & de Kruijff B Phosphatidylglycerol is involved in protein translocation across Escherichia coli inner membranes. Nature 334, 173–175 (1988). [DOI] [PubMed] [Google Scholar]
- 26.Lill R, Dowhan W & Wickner W The ATPase Activity of SecA Is Regulated by Acidic Phospholipids, SecY, and the Leader and Mature Domains of Precursor Proteins. Cell 60, 271–280 (1990). [DOI] [PubMed] [Google Scholar]
- 27.Velankar SS, Soultanas P, Dillingham MS, Subramanya HS & Wigley DB Crystal structures of complexes of PcrA DNA helicase with a DNA substrate indicate an inchworm mechanism. Cell 97, 75–84 (1999). [DOI] [PubMed] [Google Scholar]
- 28.Nishiyama K, Suzuki T & Tokuda H Inversion of the membrane topology of SecG coupled with SecA-dependent preprotein translocation. Cell 85, 71–81 (1996). [DOI] [PubMed] [Google Scholar]
- 29.Satoh Y, Matsumoto G, Mori H & Ito K Nearest neighbor analysis of the SecYEG complex. 1. Identification of a SecY-SecG interface. Biochemistry 42, 7434–7441 (2003). [DOI] [PubMed] [Google Scholar]
- 30.van der Sluis EO, van der Vries E, Berrelkamp G, Nouwen N & Driessen AJ Topologically fixed SecG is fully functional. J Bacteriol 188, 1188–1190 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Breyton C, Haase W, Rapoport TA, Kuhlbrandt W & Collinson I Three- dimensional structure of the bacterial protein-translocation complex SecYEG. Nature 418, 662–665 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Bostina M, Mohsin B, Kuhlbrandt W & Collinson I Atomic model of the E. coli membrane-bound protein translocation complex SecYEG. J Mol Biol 352, 1035–1043 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Plath K, Mothes W, Wilkinson BM, Stirling CJ & Rapoport TA Signal sequence recognition in posttranslational protein transport across the yeast ER membrane. Cell 94, 795–807 (1998). [DOI] [PubMed] [Google Scholar]
- 34.Gelis I et al. Structural basis for signal-sequence recognition by the translocase motor SecA as determined by NMR. Cell 131, 756–769 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prinz WA, Spiess C, Ehrmann M, Schierle C & Beckwith J Targeting of signal sequenceless proteins for export in Escherichia coli with altered protein translocase. EMBO J. 15, 5209–5217 (1996). [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou J & Xu Z Structural determinants of SecB recognition by SecA in bacterial protein translocation. Nat Struct Biol 10, 942–947 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Hartl FU, Lecker S, Schiebel E, Hendrick JP & Wickner W The Binding Cascade of SecB to SecA to SecY/E Mediates Preprotein Targeting to the E. Coli Plasma Membrane. Cell 63, 269–279 (1990). [DOI] [PubMed] [Google Scholar]
- 38.Heinrich SU, Mothes W, Brunner J & Rapoport TA The Sec61p complex mediates the integration of a membrane protein by allowing lipid partitioning of the transmembrane domain. Cell 102, 233–244. (2000). [DOI] [PubMed] [Google Scholar]
- 39.Wang J et al. Crystal structures of the HslVU peptidase-ATPase complex reveal an ATP-dependent proteolysis mechanism. Structure 9, 177–184 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Siddiqui SM, Sauer RT & Baker TA Role of the processing pore of the ClpX AAA+ ATPase in the recognition and engagement of specific protein substrates. Genes Dev 18, 369–374 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hinnerwisch J, Fenton WA, Furtak KJ, Farr GW & Horwich AL Loops in the central channel of ClpA chaperone mediate protein binding, unfolding, and translocation. Cell 121, 1029–1041 (2005). [DOI] [PubMed] [Google Scholar]
- 42.DeLaBarre B, Christianson JC, Kopito RR & Brunger AT Central pore residues mediate the p97/VCP activity required for ERAD. Mol Cell 22, 451–462 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Martin A, Baker TA & Sauer RT Diverse pore loops of the AAA+ ClpX machine mediate unassisted and adaptor-dependent recognition of ssrA-tagged substrates. Mol Cell 29, 441–450 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SupInfo