The STING pathway and regulation of innate immune signaling in response to DNA pathogens (original) (raw)
- 6498 Accesses
- 91 Citations
- 19 Altmetric
- 1 Mention
- Explore all metrics
Abstract
The innate immune system has evolved a variety of sensing mechanisms to detect and counter microbial invasion. These include the Toll-like receptor (TLR), cytoplasmic, nucleotide binding oligomerization domain (NOD)-like receptor and RIG-I-like helicase (RLH) pathways. However, how the cell detects pathogen-associated DNA to trigger host defense, including the production of interferon, remains to be fully clarified. Understanding these processes could have profound implications into how we understand and treat a variety of microbial-related disease, including viral-associated cancers, as well as autoimmune disorders. Recently, an endoplasmic reticulum-associated molecule referred to as STING (for stimulator of interferon genes) was isolated and shown to be critical for regulating the production of IFN in response to cytoplasmic DNA. Here, we review recent discoveries relating to the detection of foreign DNA, including the importance of the STING and inflammasome pathways and the triggering of innate signaling processes.
Similar content being viewed by others
Host exposure to microbial pathogens such as viruses, bacteria, and fungi trigger the activation of innate immune responses that stimulate early host defense mechanisms such as the production of type I interferon (IFN), as well as invigorate adaptive immune responses involving cytotoxic T cell activity and antibody production [1–3]. The recognition of pathogenic microbes and the triggering of the innate immune cascade has become the subject of intense research over the past few years. Key activators of innate immune signaling pathways are now known to comprise pathogen-derived nucleic acid such as microbial genomic RNA and DNA (referred to as PAMPs—pathogen-associated molecular patterns) [1]. Recent findings indicate that a number of sensing systems have evolved in different cell types to detect pathogen-derived nucleic acid. Cellular invasion, for example by viruses, can directly trigger the production of type I IFN, or conversely cells of the hematopoietic lineage, such as macrophages and dendritic cells can engulf apoptotic cell debris comprising viral antigen and nucleic acid and produce IFN and other cytokines such as the interleukins, which can result in a pro-inflammatory response [4, 5].
The pattern recognition receptors (PRRs) responsible for sensing viral RNA have recently been isolated and have been found to involve DExD/H box RNA helicases called retinoic acid inducible gene I (RIG-I), melanoma differentiation-associated gene 5 (MDA5), and LGP2 [6, 7]. These helicases are responsible for the production of IFN in all cell types, in response to RNA virus infection, aside from plasmacytoid dendritic cells (pDCs) [8, 9]. Helicase interaction with viral RNA induces the recruitment of a molecule referred to as IPS-1 (also known as VISA, Cardif or MAVS) and TRAF3 and leads to the activation of mitogen activated protein (MAP) kinases, the IKK-related kinases, TBK1 (TANK binding kinase 1) and IKK-I (IκB kinase I) [10–13]. These kinases activate the transcription factors NF-κB, activator protein-1 (AP1) and interferon regulatory factor-3 (IRF3), which translocate into the nucleus and bind to and activate the IFNβ promoter [1–3]. IFN is secreted, binds to the type I IFN receptor (IFNAR) in an autocrine or paracrine fashion and activates the Jak/STAT pathway to initiate the production of numerous IFN-inducible genes which exert potent anti-viral activity through mechanisms that in many cases remain to be fully clarified [14].
In pDCs, another mechanism has evolved to trigger the production of type I IFN in response to RNA virus infection, which is essential for protection of the host. This involves the Toll-like receptor (TLR) pathway and specifically, TLR7, which associates with MyD88 (myeloid differentiation primary response gene 88) and members of the IRAK (interleukin-1 receptor associated kinase) and TRAF family [2, 3]. These events rapidly and potently lead to phosphorylation of IRF7, which, similar to IRF3, translocates to the nucleus to activate the production of type I IFN [9]. There are known to be approximately ten human TLRs, the different members of which recognize a variety of microbial PAMPs such as lipopolysaccharides (TLR4) common on bacterial cell walls and viral dsRNA (TLR3) that can similarly induce the production of IFN as well as other cytokines in a variety of cell types [1–3]. Finally, microbial PAMPs can be recognized by the NLR (nucleotide-binding domain leucine-rich repeat) family of sensors that can activate NF-κB and caspase-1 and trigger pro-inflammatory responses such as those involving production of IL-1β. For example, the NLRs NOD1 and 2 are known to recognize bacterial muramyl dipeptides to trigger the activation of NF-κB [3, 15].
However, while significant progress has been made in unraveling mechanisms responsible for recognizing bacteria cell wall components and RNA viruses, somewhat less is known about how microbial DNA is sensed by the cell to trigger innate immune responses. This is of profound interest since many pathogens such as cancer-causing viruses, bacteria, fungus, and parasites comprise DNA genomes, which are known to activate IFN production [1–3]. Further, endogenous self-DNA may be responsible for inadvertently activating our own innate immune pathways and mitigating autoimmune disease [5]. Recently a molecule, referred to as STING (for stimulator of interferon genes) was isolated that was shown to be pivotal to the production of type I IFN by DNA, in numerous cell types, including macrophages, DCs and fibroblasts [16, 17]. Here, we review the involvement of STING in this process, as well as illustrate what is presently known about innate signaling pathways triggered by DNA.
TLR-dependent DNA sensing mechanisms
A well-characterized DNA sensing receptor responsible for triggering innate immune responses is TLR9, which contains leucine-rich repeat (LRR) motifs, a Toll/IL-1R homology domain and is considered a type I integral membrane glycoprotein [3, 18]. TLR9 recognizes CpG (cytidine–phosphate–guanosine) DNA motifs that are commonly found in bacteria and viruses, but which is rare in vertebrates. Several studies using TLR9-deficient mice have emphasized a role for TLR9 in host innate immune responses against DNA viruses such as herpes simplex virus [3, 19, 20]. TLR9 is mainly expressed in pDCs, which, as mentioned, are a subset of DCs with a plasmacytoid morphology that produce IFN and cytokines in response to CpG DNA or RNA viruses [3, 21]. However, TLR9-deficient animals remain able to produce IFN following infection with DNA viruses, indicating the existence of key TLR-independent mechanisms responsible for activating DNA-mediated innate immune signaling [20, 22, 23].
Unprocessed TLR9 localizes on the endoplasmic reticulum (ER) in unstimulated pDCs. CpG DNA, internalized via a clathrin-dependent endocytic pathway, moves to endolysosomal compartments and associates with processed, active TLR9 that has trafficked to these regions from the ER [1–3, 24]. The trafficking of TLR9 is controlled by UNC93B, a 12-membrane-spanning ER protein that directly interacts with TLR9 [25, 26]. The proteolytic cleavage of endolysosomal TLR9 is required for TLR9 activation in response to CpG DNA [24].
Upon recognition of CpG DNA in endosomes, TLR9 interacts with MyD88, which contains a TIR domain and a death domain [1–3]. MyD88 interacts with IRAK-1 (IL-1R-associated kinase 1), IRAK-4, and IRF-7. This event leads to recruitment of TRAF6 (TNFR-associated factor 6), which activates the TAK1 (transforming growth factor β-activated kinase 1), MAPK and ultimately NF-κB. IRAK1 directly interacts with IRF7, and phosphorylates the C-terminal region of IRF7, which is required for transcriptional activity [1–3]. Recently, the rapamycin-sensitive PI(3)K-mTOR-p70S6K pathway has also been demonstrated as being important in regulating TLR9 activity [27].
DNA sensing pathways have been implicated in triggering innate and adaptive immune pathways mediated by plasmid-based vaccines. DNA-vector-based vaccines have been investigated in strategies to induce immunity against disease since they can be manufactured quickly and economically [28]. However, DNA vaccines exhibit low immunogenicity and so the enhancement of vaccine efficacy has become a key objective in the development of effective DNA vaccination protocols. Understanding the mechanisms of DNA-dependent innate signaling may therefore lead to improvements in effectiveness of DNA vaccine procedures. It has become apparent, however, that while TLR9 may indeed facilitate plasmid vaccination, a number of studies have indicated that TLR9 is not essential for the induction of immune responses following DNA immunization, again indicating that alternate DNA sensors may be critically important for this process, as we shall describe next [20, 22, 23]. However, the use of TLR ligands has indeed been explored to improve the immunogenicity of antigens used in vaccines. For example, synthetic oligodeoxynucleotide (ODN) ligands, such as CpG ODN for TLR9, have been reported to be a potent adjuvant that enhances the strength of immunization. Understanding these pathways may therefore lead to the design of new adjuvant-related concepts to improve vaccination strategies [28, 29].
It is noteworthy that DNA sensors such as TLR9 have been implicated in the development of autoimmune diseases such as systemic lupus erythematosus (SLE) [5, 15, 30]. It is considered that self-nucleic acids can act as endogenous ligands for DNA receptors, which raise serum levels of IFN and which correlate with both SLE disease activity and severity. Thus, the unraveling of the DNA-triggered innate immune signaling pathway may have important implications in our understanding of autoimmune disease and could lead to the development of new therapeutics to treat such disorders.
TLR-independent DNA-mediated innate immune signaling
Cytosolic DNA-mediated STING-dependent innate signaling
It became clear that cytosolic dsDNA derived from invading microbes or host cells could be recognized by TLR9-independent innate immune mechanisms to induce type I interferon responses. For example, infection of TLR9-deficient cells by DNA pathogens, such as herpes simplex virus 1 (HSV1), or intracellular bacteria such as Listeria monocytogenes, which is known to introduce DNA into cytosol during infection, remained able to produce type I IFNs [19, 20]. In addition, it has been reported that DNA derived from engulfed apoptotic cells can induce type I IFN expression in a TLR9-independent manner [31, 32]. Finally, the transfection of TLR9-lacking cells with synthetic double-stranded DNA (dsDNA) still resulted in the induction of type I IFN [33, 34]. These observations indicated the presence of a TLR-independent cytosolic DNA-mediated innate signaling, the components of which remained to be fully determined.
In an attempt to find key components of innate signaling, we utilized an expression cloning strategy to identify molecules that activated the IFN-β promoter [16]. This strategy was based on determining which genes, when overexpressed in 293T cells, were able to induce the transcription of a luciferase gene, which was controlled by the IFNβ promoter. Adopting this procedure, we identified a molecule referred to as STING (stimulator of interferon genes), which we demonstrated was essential for cytosolic DNA-mediated type I IFNs induction [16, 17]. STING, also known as MITA/MPYS/ERIS, contained multi-putative transmembrane regions in the amino terminal region, and was found to predominantly localize in the endoplasmic reticulum (ER) [16, 17, 35–37]. However, reports have also indicated that STING may reside in the mitochondria, although these studies were conducted using GFP-tagged STING in overexpression studies, which may have affected authentic localization. Overexpression of STING induced the activation of both NF-κB and IRF3 to stimulate type I IFN production [16, 35, 36]. Significantly, MEFs derived from STING-deficient mice failed to induce type I IFNs in response to infection with HSV-1 or Listeria monocytogenes, or transfection of interferon stimulatory DNA, ISD (which comprises double-stranded 45-base-pair oligonucleotides lacking CpG sequences) [16, 17]. Single-stranded DNA did not robustly activate type I IFN induction via the STING-regulatory pathway. Further investigation indicated that STING was essential for intracellular DNA-mediated activation of type I IFN in macrophages and in conventional DCs [16, 17]. Importantly, both HSV-1 and Listeria failed to induce type I IFN in DCs lacking STING either. While STING was found to be critical for the induction of type I IFN by intracellular DNA, it was not essential for IFN production stimulated by transfected dsRNA (poly IC) [16]. Further, while the production of type I IFN by transfected DNA was ablated in the absence of STING, IL1β production was not affected (which has been shown to be AIM2-dependent, see below) [17]. Thus, STING functions independently of AIM2 and is not critical for inflammasome activation. Finally, our studies indicated that STING was important for intracellular DNA-mediated and HSV-1-activated type I IFN production in pDCs, although the presence of TLR9 in these cells was found to partially compensate for the loss of STING in response to Listeria infection [17]. Given this, we observed that STING-deficient animals, which were viable, rapidly succumbed to lethal HSV-1 infection compared to control animals due to a lack of type I IFN production, which enabled robust virus replication [17].
Importantly, we noticed that STING −/− animals were also sensitive to negative-stranded virus infection, such as VSV. This would indicate that STING may facilitate the RIG-I pathway [16]. Immunoprecipitation experiments demonstrated that STING may associate with RIG-I complexes, but not with MDA5, perhaps explaining why loss of STING had no significant effect on poly IC signaling (which is largely mediated by MDA5) [1–3]. Our data indicated that ER-associated STING likely associates in close proximity with mitochondria associated with the ER (mitochondria associated membrane- MAM) and perhaps peroxisome complexes, both comprising IPS-1 required for RIG-I function [17, 38]. Interestingly, STING exhibited significant homology to flavivirus proteins such as yellow fever virus (YFV), dengue virus (DV), and hepatitis C virus (HCV) NS4 proteins, which are known to reside in the ER. DV NS4 was found to inhibit STING function [17]. Collectively, our data indicates that STING is an important component of innate immune signaling that governs not only intracellular DNA-mediated IFN signaling but also RIG-I signaling, and may be targeted by viruses for suppression.
Yeast two-hybrid and co-immunoprecipitation assays showed that STING associated with SSR2/TRAPβ, a member of the translocon-associated protein (TRAP) complex comprising four subunits (α–Δ) that facilitate translocation of proteins into the endoplasmic reticulum (ER) following translation [39, 40]. The translocon-associated protein (TRAP) complex is known to associate with the translocon, comprising three subunits, SEC61α, SEC61β, and SEC61γ [39, 41]. The translocon has been shown to be important for protein folding and secretion. RNA interference ablation of TRAPβ or SEC61β expression was found to inhibit IFNβ promoter activation following STING overexpression in 293T cells [16]. Although it is not clear how signaling from STING/translocon complexes to IRF3/NF-κB occurs, evidence indicates that the translocon may physically associate with the exocyst, an evolutionally conserved octameric complex that tethers secretory vesicles to membranes, and facilitates protein synthesis and secretion [42, 43]. Recently, the exocyst complex has been found to play a role in type I IFN production. For example, Sec5, one of the components of the exocyst complex, was found to recruit and activate TBK1 which, as mentioned, is essential for IRF3/7 activation and type I IFN induction in response to RNA and DNA pathogens [44]. Preliminary analysis indicated that STING could associate with TBK1 and that RNAi ablation of Sec5 reduced STING’s ability to activate the IFNβ promoter [16]. Thus, STING may link cytosolic DNA-mediated signaling to TBK1 activation through ER/translocon and exocyst interactions.
Following infection with HSV-1, STING was found to rapidly translocate from the ER, through the Golgi to a distinct perinuclear region, by mechanisms that presently remain unclear [17]. TBK1 was found to similarly traffic in response to HSV-1 infection, in a STING-dependent manner, and likely such complexes may be required for the activation of transcription factors such as IRF3 and 7 [17]. The endosomal compartments that comprise STING and TBK1 following trafficking in response to intracellular DNA remain to be fully characterized. However, the translocation process was found to involve autophagy-related gene 9a (Atg9a), and may involve STING ubiquitination [35, 45]. Thus, the translocon and STING trafficking to perinuclear endosomal compartments appear necessary for the stimulation of innate immune pathways involving the production of type I IFN, at least in response to intracellular DNA.
Finally, it is noteworthy that previous work has shown the importance of TBK-1 in stimulating plasmid DNA-based immune responses. Plausibly, TBK1-dependent pathways are triggered by intracellular plasmid B-form DNA, which augments immune responses by stimulating type I IFN and NF-κB-dependent pathways [46, 47]. Since we observed that STING functions upstream of TBK-1 and is required for TBK-1 activity in response to intracellular DNA, we investigated whether STING was important for the adjuvant effects of plasmid DNA. This study confirmed that STING is indeed required for effective DNA-mediated adaptive immune responses, such as those stimulated by plasmid DNA vaccination as well DNA virus infection such as with vaccinia virus [17]. Thus, the stimulation of STING-regulated pathways may provide a mechanism to augment DNA-based plasmid immunization strategies in the future.
While STING exhibits a number of putative transmembrane motifs, there do not appear to be any predicted DNA-binding motifs in this ER-resident molecule, suggesting that STING itself is unlikely to associate directly with DNA. However, a number of molecules have been reported to function as DNA sensors in the cell. For example, DAI [DNA-dependent activator of IRFs, also referred to as Z DNA binding-binding protein-1 (ZBP-1)] has been identified as a cytosolic DNA receptor to induce type I IFNs, at least in L929 cells [48]. Nevertheless, primary cells derived from DAI-deficient mice induce type I IFN normally in response to cytosolic DNA. DAI-deficient mice are also able to elicit an immune response to DNA-based vaccines, collectively suggesting the existence of alternate DNA sensors [46]. In this light, IFI16, a PYHIN protein similar to AIM2, has also been implicated in functioning as a cytoplasmic DNA sensor to induce IFN [49]. This activity was shown to be STING-dependent. The examination of IFI16 null mice may additionally emphasize the importance of IFI16 in innate immune signaling. Finally, the importance of high-mobility group box (HMGB) proteins in facilitating nucleic-acid-dependent innate immune signaling has been reported, emphasizing the complexity of DNA sensing pathways in the cell [50].
Cytosolic DNA-mediated RNA polymerase III/RIG-I/IPS-1-dependent innate signaling
Recent studies have also demonstrated the existence of a novel cytosolic DNA sensing mechanism that is able to induce type I IFN in a DNA-dependent RNA polymerase III manner [51, 52]. Transfected Poly(dA:dT) (synthetic AT-rich dsDNA), but not other types of DNA including poly(dG:dC) (synthetic GC-rich dsDNA), calf thymus DNA, PCR fragments, or plasmid DNA, were capable of producing dsRNA containing 5′ triphosphate, which activated RIG-I/IPS-1-dependent innate signaling [51, 52]. RNAi abrogation of POL3RF, a component of RNA polymerase III, or pharmacological inhibition of RNA polymerase III, was found to inhibit production of dsRNA from poly(dA:dT) [51, 52]. RIG-I recognizes 5′ triphosphate RNA species, and interacts with IPS-1, an essential adaptor molecule for RNA-inducing innate immune signaling [6, 10–13, 53, 54]. IPS-1 activates both NFκB and IRF-3 through FADD/RIP1 and TBK1, respectively [10–13, 55]. As we have mentioned, STING also appears to be involved in RIG-I/IPS-1-mediated signaling. However, STING-deficient cells still remain partially able to produce some IFNβ in response to infection with negative-stranded RNA viruses such as Sendai virus (SV) and VSV [16]. Moreover, while IFNβ production mediated by ISD, HSV-1, Listeria, or transfected plasmid DNA were completely abrogated in STING-deficient MEFs, such cells produced reduced but detectable amounts of IFNβ after transfection with poly(dA:dT) [17]. These observations indicate that AT-rich DNA may induce both STING-dependent and STING-independent RNA polymerase III/RIG-I/IPS-1-regulated innate immune signaling pathways.
Cytosolic DNA-mediated inflammasome-dependent innate signaling
It has been demonstrated that cytosolic DNA also triggers TBK1-independent inflammasome-dependent innate immune signaling [15, 56–59]. NALP3 (NACHT-leucine-rich repeat-PYD containing protein 3), also known as cryopyrin, recognizes Adenovirus infection and activates ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain)/caspase-1-mediated secretion of IL-1β [59]. Recently, four different groups identified the HIN-200 family member, AIM2 (absence in melanoma 2), as the cytoplasmic dsDNA sensor that activates ASC/caspase-1-mediated secretion of IL-1β [15, 56–59]. Importantly, AIM2 inflammasome is essential for caspase-1 activation but completely dispensable for type I IFNs production in response to cytosolic dsDNA, indicating cytosolic DNA-mediated AIM2 inflammasome-dependent signaling is distinct from type I IFNs-dependent innate signaling. As mentioned above, activation of the inflammasome and the production of IL-1β by intracellular DNA appear to be STING-independent. Thus, different DNA sensors exist to trigger pro-inflammatory or type I IFN production in response to infection.
DNA sensing mechanisms and autoimmunity
Type I IFN is an essential factor for host defense against evading pathogens, but inappropriate production of type I IFN leads to autoimmune diseases such as SLE, as mentioned earlier [60]. In eukaryotes, localization of self-DNA is restricted to the cell nucleus and mitochondria, thereby sequestering self-DNA from cytoplasmic DNA sensing mechanisms, which may activate pro-inflammatory cytokine pathways. Cellular DNases eliminate aberrant self-DNA found in apoptotic bodies, extracellular space, cytosol, and endosomes. Several studies have shown that defective clearance of self-DNA leads to inappropriate activation of type I IFNs production through a TLR-independent innate immune signaling, which is tightly linked to autoimmune diseases. For example, DNase I deficiency or mutations are associated with lupus-like syndrome in mice and humans [61, 62]. In addition, DNase II-deficient mice have shown the accumulation of incompletely digested DNA, which causes TLR-independent type I IFNs production, inflammatory responses, and early death, which are linked to autoimmune disease like chronic polyarthritis [63]. Crossing susceptible mice with mice deficient in the type I IFN receptor abrogated lethality, indicating the importance of excessive IFN production in pathogenesis [63].
Recently, several studies have reported that Trex1, 3′-repair exonuclease 1, regulates DNA homeostasis and its deficiency is linked to autoimmune diseases [64–66]. Mutations in the human Trex1 gene cause SLE and AGS (Aicardi-Goutieres syndrome) [64, 65]. In addition, it has been shown that Trex1-deficient mice develop lethal autoimmunity via elevated production of type I IFNs and auto-antibodies [66]. Interestingly, genetic ablation of IRF3 or IFNR rescued Trex1-deficient mice from mortality, suggesting IRF3-dependent IFN production was linked to autoimmune symptom in Trex1-deficient mice [66]. ssDNA derived from endogenous retroelements accumulated in cells derived from Trex1-deficient mice [66]. Collectively, these observations suggest that Trex1 is required for the prevention of autoimmunity that would otherwise develop during the activation of the cytosolic DNA-mediated innate immune signaling by cell-intrinsic substrates [66].
As described, TLR9 also recognizes self-DNA and induces type I IFNs. Several studies have suggested that the specialized localization of TLR9 is required for preventing recognition of self-DNA. For example, human DNA associated with LL37, an antimicrobial peptide produced in the skin of patients with psoriasis, is retained in early endosomes in pDC and facilitates type I IFN induction through the activation of TLR9. In addition, anti-DNA antibodies, found in serum of patients with systemic lupus erythematosus (SLE), bind self-DNA and induce type I IFN via a cooperative interaction between TLR9 and CD32 in pDC [67]. Moreover, it has been shown that chimeric TLR9, which localizes to the cell surface, can respond to synthetic TLR9 ligands but not to viral DNA, suggesting intracellular localization of TLR9 is required for preventing to recognize self-DNA [68].
Concluding remarks
While significant progress had been made into unraveling TLR9-dependent DNA recognition systems, little was known regarding cytosolic DNA-mediated innate immune signaling. However, recent studies have gradually shed light on the molecular mechanisms of intracellular DNA-mediated inflammatory responses involving AIM2 production of IL-1β as well as STING controlled DNA-mediated IFN production. In the latter case, the triggering of type I IFN production in response to select DNA viruses as well as intracellular bacteria appears STING-dependent [16, 17]. However, STING is also required for effective type I production in response to certain RNA virus infection such as VSV and Sendai virus. Given these observations, it will be of interest to evaluate the importance of STING in stimulating host defenses against other types of DNA pathogens including parasites such as malaria. Key issues that remain to be elucidated also include the identification of cytosolic DNA receptor(s) which activate the STING pathway to induce IFN expression. Finally, it will be intriguing to determine the role of STING-dependent and AIM2-dependent signaling in autoimmune diseases and to see if such pathways can be targeted to improve DNA-based vaccine strategies (see Fig. 1).
Fig. 1
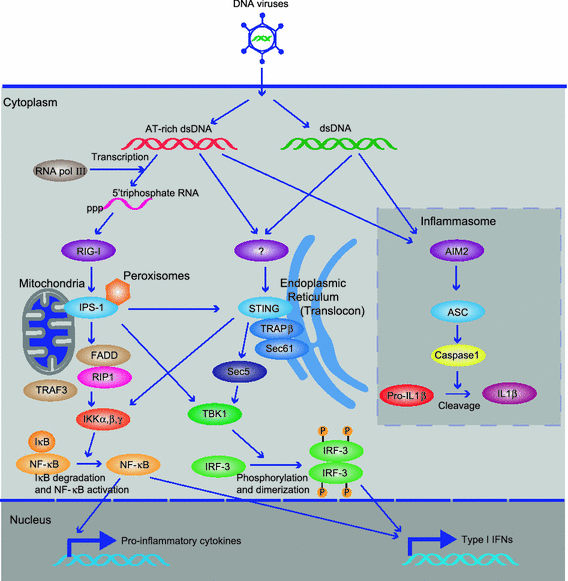
DNA-mediated activation of innate immune signaling. a TLR9-dependent innate immune signaling. TLR9 localizes in the ER and interacts with UNC-93B, which mediates TLR9 translocation. Upon stimulation of DNA-containing CpG motifs (CpG DNA) including viral genome, TLR9 traffics from ER to endosome to contact CpG DNA, and recruits a signaling complex consisting of MyD88, IRAK4, and TRAF6. TRAF6 in turn activates TAK1, and TAK1 subsequently activates MAPK and the IKK complex (IKKα, IKKβ, and IKKγ) to activate NF-κB. NF-κB and MAPK regulates inflammatory cytokines expression. IRAK4 activates TRAF3 and IRAK1, which catalyze IRF7 phosphorylation and induce type I IFNs expression. b Cytosolic DNA-mediated innate immune signaling. Cytosolic AT-rich dsDNA is recognized by RNA PolIII and transcribed into 5′ triphosphate RNA, which activates RIG-I/IPS-1 signaling. IPS1 interacts with FADD/RIP1, which activate NF-κB. IPS-1 also activates TBK1 which phosphorylates IRF-3 and induces type I IFNs expression. AT-rich dsDNA or non-AT-rich dsDNA is recognized by unknown receptor, and activate STING-dependent signaling. STING localizes in the ER with translocon complex (TRAPβ, Sec61), and activate Sec5/TBK1 to induce type I IFNs. STING also activates NF-κB to induce pro-inflammatory cytokines. dsDNA also activates AIM2/ASC/Caspase-1 inflammasome pathway to activate IL-1β
References
- Iwasaki A, Medzhitov R (2010) Regulation of adaptive immunity by the innate immune system. Science 327:291–295
Google Scholar - Blasius AL, Beutler B (2010) Intracellular Toll-like receptors. Immunity 32:305–315
Google Scholar - Kawai T, Akira S (2009) The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol 21:317–337
Article PubMed CAS Google Scholar - Nagata S, Hanayama R, Kawane K (2010) Autoimmunity and the clearance of dead cells. Cell 140:619–630
Google Scholar - Rouse BT, Sehrawat S (2010) Immunity and immunopathology to viruses: what decides the outcome? Nat Rev Immunol 10:514–526
Google Scholar - Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T (2004) The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5:730–737
Article PubMed CAS Google Scholar - Yoneyama M, Fujita T (2009) RNA recognition and signal transduction by RIG-I-like receptors. Immunol Rev 227:54–65
Article PubMed CAS Google Scholar - Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S (2006) Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105
Article PubMed CAS Google Scholar - Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T (2005) IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434:772–777
Article PubMed CAS Google Scholar - Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S (2005) IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 6:981–988
Article PubMed CAS Google Scholar - Seth RB, Sun L, Ea CK, Chen ZJ (2005) Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122:669–682
Article PubMed CAS Google Scholar - Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J (2005) Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167–1172
Article PubMed CAS Google Scholar - Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB (2005) VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell 19:727–740
Article PubMed CAS Google Scholar - Randall RE, Goodbourn S (2008) Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol 89:1–47
Article PubMed CAS Google Scholar - Muruve DA, Petrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, Parks RJ, Tschopp J (2008) The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 452:103–107
Article PubMed CAS Google Scholar - Ishikawa H, Barber GN (2008) STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455:674–678
Article PubMed CAS Google Scholar - Ishikawa H, Ma Z, Barber GN (2009) STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461:788–792
Article PubMed CAS Google Scholar - Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S (2000) A Toll-like receptor recognizes bacterial DNA. Nature 408:740–745
Article PubMed CAS Google Scholar - Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A (2003) Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med 198:513–520
Article PubMed CAS Google Scholar - Hochrein H, Schlatter B, O’Keeffe M, Wagner C, Schmitz F, Schiemann M, Bauer S, Suter M, Wagner H (2004) Herpes simplex virus type-1 induces IFN-alpha production via Toll-like receptor 9-dependent and -independent pathways. Proc Natl Acad Sci USA 101:11416–11421
Article PubMed CAS Google Scholar - Gilliet M, Cao W, Liu YJ (2008) Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol 8:594–606
Article PubMed CAS Google Scholar - Spies B, Hochrein H, Vabulas M, Huster K, Busch DH, Schmitz F, Heit A, Wagner H (2003) Vaccination with plasmid DNA activates dendritic cells via Toll-like receptor 9 (TLR9) but functions in TLR9-deficient mice. J Immunol 171:5908–5912
PubMed CAS Google Scholar - Heit A, Maurer T, Hochrein H, Bauer S, Huster KM, Busch DH, Wagner H (2003) Cutting edge: Toll-like receptor 9 expression is not required for CpG DNA-aided cross-presentation of DNA-conjugated antigens but essential for cross-priming of CD8 T cells. J Immunol 170:2802–2805
PubMed CAS Google Scholar - Ewald SE, Lee BL, Lau L, Wickliffe KE, Shi GP, Chapman HA, Barton GM (2008) The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature 456:658–662
Article PubMed CAS Google Scholar - Tabeta K, Hoebe K, Janssen EM, Du X, Georgel P, Crozat K, Mudd S, Mann N, Sovath S, Goode J, Shamel L, Herskovits AA, Portnoy DA, Cooke M, Tarantino LM, Wiltshire T, Steinberg BE, Grinstein S, Beutler B (2006) The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol 7:156–164
Article PubMed CAS Google Scholar - Kim YM, Brinkmann MM, Paquet ME, Ploegh HL (2008) UNC93B1 delivers nucleotide-sensing Toll-like receptors to endolysosomes. Nature 452:234–238
Article PubMed CAS Google Scholar - Cao W, Manicassamy S, Tang H, Kasturi SP, Pirani A, Murthy N, Pulendran B (2008) Toll-like receptor-mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin-sensitive PI(3)K-mTOR-p70S6K pathway. Nat Immunol 9:1157–1164
Article PubMed CAS Google Scholar - Koyama S, Coban C, Aoshi T, Horii T, Akira S, Ishii KJ (2009) Innate immune control of nucleic acid-based vaccine immunogenicity. Expert Rev Vaccines 8:1099–1107
Article PubMed CAS Google Scholar - Wilson HL, Dar A, Napper SK, Marianela LopezA, Babiuk LA, Mutwiri GK (2006) Immune mechanisms and therapeutic potential of CpG oligodeoxynucleotides. Int Rev Immunol 25:183–213
Article PubMed CAS Google Scholar - Ehlers M, Fukuyama H, McGaha TL, Aderem A, Ravetch JV (2006) TLR9/MyD88 signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE. J Exp Med 203:553–561
Article PubMed CAS Google Scholar - Okabe Y, Kawane K, Akira S, Taniguchi T, Nagata S (2005) Toll-like receptor-independent gene induction program activated by mammalian DNA escaped from apoptotic DNA degradation. J Exp Med 202:1333–1339
Article PubMed CAS Google Scholar - Yoshida H, Okabe Y, Kawane K, Fukuyama H, Nagata S (2005) Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat Immunol 6:49–56
Article PubMed CAS Google Scholar - Stetson DB, Medzhitov R (2006) Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24:93–103
Article PubMed CAS Google Scholar - Ishii KJ, Coban C, Kato H, Takahashi K, Torii Y, Takeshita F, Ludwig H, Sutter G, Suzuki K, Hemmi H, Sato S, Yamamoto M, Uematsu S, Kawai T, Takeuchi O, Akira S (2006) A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat Immunol 7:40–48
Article PubMed CAS Google Scholar - Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, Shu HB (2008) The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29:538–550
Article PubMed CAS Google Scholar - Sun W, Li Y, Chen L, Chen H, You F, Zhou X, Zhou Y, Zhai Z, Chen D, Jiang Z (2009) ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc Natl Acad Sci USA 106:8653–8658
Article PubMed CAS Google Scholar - Jin L, Waterman PM, Jonscher KR, Short CM, Reisdorph NA, Cambier JC (2008) MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Mol Cell Biol 28:5014–5026
Article PubMed CAS Google Scholar - Dixit E, Boulant S, Zhang Y, Lee AS, Odendall C, Shum B, Hacohen N, Chen ZJ, Whelan SP, Fransen M, Nibert ML, Superti-Furga G, Kagan JC (2010) Peroxisomes are signaling platforms for antiviral innate immunity. Cell 141:668–681
Google Scholar - Hartmann E, Gorlich D, Kostka S, Otto A, Kraft R, Knespel S, Burger E, Rapoport TA, Prehn S (1993) A tetrameric complex of membrane proteins in the endoplasmic reticulum. Eur J Biochem 214:375–381
Article PubMed CAS Google Scholar - Menetret JF, Hegde RS, Heinrich SU, Chandramouli P, Ludtke SJ, Rapoport TA, Akey CW (2005) Architecture of the ribosome-channel complex derived from native membranes. J Mol Biol 348:445–457
Article PubMed CAS Google Scholar - Menetret JF, Hegde RS, Aguiar M, Gygi SP, Park E, Rapoport TA, Akey CW (2008) Single copies of Sec61 and TRAP associate with a nontranslating mammalian ribosome. Structure 16:1126–1137
Article PubMed CAS Google Scholar - Guo W, Novick P (2004) The exocyst meets the translocon: a regulatory circuit for secretion and protein synthesis? Trends Cell Biol 14:61–63
Article PubMed CAS Google Scholar - Lipschutz JH, Lingappa VR, Mostov KE (2003) The exocyst affects protein synthesis by acting on the translocation machinery of the endoplasmic reticulum. J Biol Chem 278:20954–20960
Article PubMed CAS Google Scholar - Chien Y, Kim S, Bumeister R, Loo YM, Kwon SW, Johnson CL, Balakireva MG, Romeo Y, Kopelovich L, Gale M Jr, Yeaman C, Camonis JH, Zhao Y, White MA (2006) RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell 127:157–170
Article PubMed CAS Google Scholar - Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, Matsunaga K, Kageyama S, Omori H, Noda T, Yamamoto N, Kawai T, Ishii K, Takeuchi O, Yoshimori T, Akira S (2009) Atg9a controls dsDNA-driven dynamic translocation of STING and the innate immune response. Proc Natl Acad Sci USA 106:20842–20846
Google Scholar - Ishii KJ, Kawagoe T, Koyama S, Matsui K, Kumar H, Kawai T, Uematsu S, Takeuchi O, Takeshita F, Coban C, Akira S (2008) TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 451:725–729
Article PubMed CAS Google Scholar - Miyahira AK, Shahangian A, Hwang S, Sun R, Cheng G (2009) TANK-binding kinase-1 plays an important role during in vitro and in vivo type I IFN responses to DNA virus infections. J Immunol 182:2248–2257
Article PubMed CAS Google Scholar - Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, Ohba Y, Taniguchi T (2007) DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448:501–505
Article PubMed CAS Google Scholar - Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, Fitzgerald KA, Paludan SR, Bowie AG (2010) IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol 11:997–1004
Google Scholar - Yanai H, Ban T, Wang Z, Choi MK, Kawamura T, Negishi H, Nakasato M, Lu Y, Hangai S, Koshiba R, Savitsky D, Ronfani L, Akira S, Bianchi ME, Honda K, Tamura T, Kodama T, Taniguchi T (2009) HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature 462:99–103
Article PubMed CAS Google Scholar - Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V (2009) RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat Immunol 10:1065–1072
Article PubMed CAS Google Scholar - Chiu YH, Macmillan JB, Chen ZJ (2009) RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138:576–591
Article PubMed CAS Google Scholar - Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C (2006) RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science 314:997–1001
Article PubMed CAS Google Scholar - Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, Endres S, Hartmann G (2006) 5′-Triphosphate RNA is the ligand for RIG-I. Science 314:994–997
Article PubMed Google Scholar - Balachandran S, Thomas E, Barber GN (2004) A FADD-dependent innate immune mechanism in mammalian cells. Nature 432:401–405
Article PubMed CAS Google Scholar - Burckstummer T, Baumann C, Bluml S, Dixit E, Durnberger G, Jahn H, Planyavsky M, Bilban M, Colinge J, Bennett KL, Superti-Furga G (2009) An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol 10:266–272
Article PubMed Google Scholar - Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA (2009) AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458:514–518
Article PubMed CAS Google Scholar - Roberts TL, Idris A, Dunn JA, Kelly GM, Burnton CM, Hodgson S, Hardy LL, Garceau V, Sweet MJ, Ross IL, Hume DA, Stacey KJ (2009) HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 323:1057–1060
Article PubMed CAS Google Scholar - Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES (2009) AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458:509–513
Article PubMed CAS Google Scholar - Baccala R, Hoebe K, Kono DH, Beutler B, Theofilopoulos AN (2007) TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat Med 13:543–551
Article PubMed CAS Google Scholar - Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Moroy T (2000) Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet 25:177–181
Article PubMed CAS Google Scholar - Yasutomo K, Horiuchi T, Kagami S, Tsukamoto H, Hashimura C, Urushihara M, Kuroda Y (2001) Mutation of DNASE1 in people with systemic lupus erythematosus. Nat Genet 28:313–314
Article PubMed CAS Google Scholar - Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, Yoshikawa H, Nagata S (2006) Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature 443:998–1002
Article PubMed CAS Google Scholar - Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, Corry PC, Cowan FM, Frints SG, Klepper J, Livingston JH, Lynch SA, Massey RF, Meritet JF, Michaud JL, Ponsot G, Voit T, Lebon P, Bonthron DT, Jackson AP, Barnes DE, Lindahl T (2006) Mutations in the gene encoding the 3′–5′ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet 38:917–920
Article PubMed CAS Google Scholar - Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, de Silva U, Bailey SL, Witte T, Vyse TJ, Kere J, Pfeiffer C, Harvey S, Wong A, Koskenmies S, Hummel O, Rohde K, Schmidt RE, Dominiczak AF, Gahr M, Hollis T, Perrino FW, Lieberman J, Hubner N (2007) Mutations in the gene encoding the 3′–5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet 39:1065–1067
Article PubMed CAS Google Scholar - Stetson DB, Ko JS, Heidmann T, Medzhitov R (2008) Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134:587–598
Article PubMed CAS Google Scholar - Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD (2005) Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest 115:407–417
PubMed CAS Google Scholar - Barton GM, Kagan JC, Medzhitov R (2006) Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat Immunol 7:49–56
Article PubMed CAS Google Scholar
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
- Department of Medicine and Sylvester Comprehensive Cancer Center, University of Miami Miller School of Medicine, Rm 511 Papanicolaou Building [M700], 1550 NW 10th Ave, Miami, FL, 33136, USA
Hiroki Ishikawa & Glen N. Barber
Authors
- Hiroki Ishikawa
You can also search for this author inPubMed Google Scholar - Glen N. Barber
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toGlen N. Barber.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Ishikawa, H., Barber, G.N. The STING pathway and regulation of innate immune signaling in response to DNA pathogens.Cell. Mol. Life Sci. 68, 1157–1165 (2011). https://doi.org/10.1007/s00018-010-0605-2
- Received: 05 August 2010
- Revised: 19 November 2010
- Accepted: 25 November 2010
- Published: 15 December 2010
- Issue Date: April 2011
- DOI: https://doi.org/10.1007/s00018-010-0605-2