Protein and lipid kinase inhibitors as targeted anticancer agents of the Ras/Raf/MEK and PI3K/PKB pathways (original) (raw)
Introduction
The growth factor-stimulated Ras/Raf/MEK (Fig. 1) and PI3K/PKB (Fig. 2) signal transduction pathways have been demonstrated to play central roles in the biology of human cancers. Whatever the mechanism, aberrant activation of these signaling cascades is a frequent event in human tumors due to their involvement in key biological processes like cancer cell survival, growth, proliferation, and invasion. Drug discovery activities to control the irregular activation of these pathways have led to the development of novel target-based agents for cancer treatment. To this end, several approaches have been adopted to inhibit or decrease pathway activity. Much of the original therapeutic efforts was directed to block receptor tyrosine kinases (RTKs), which are upstream activators of these pathways (Figs. 1 and 2). The clinical efficacy of antibodies that inhibit ligand-mediated activation of RTKs—e.g., Herceptin™, Erbitux™, or Avastin™—or low-molecular mass inhibitors that target the ATP-binding cleft of these enzymes—e.g., Iressa™, Tarceva™, or Tykerb™—have provided proof of concept of the therapeutic validity of this strategy. Parallel to the development of RTK inhibitors, pharmaceutical efforts have also been directed to target some of the cytosolic molecules of the Ras/Raf/MEK and PI3K//PKB pathways, in particular protein and lipid kinases. In this review, I will briefly summarize recent salient medicinal chemistry achievements in the identification and development of kinase inhibitors of these cytoplasmic pathways for the treatment of cancer. Several articles on individual topics are available and the reader is referred to the most recent publications in the text.
Fig. 1
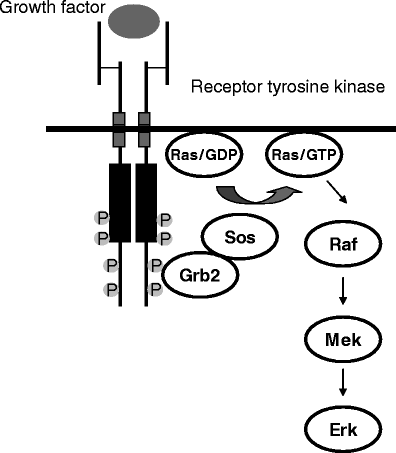
Schematic representation of the main cytosolic components of the Ras/Raf/MEK signal transduction pathway
Fig. 2
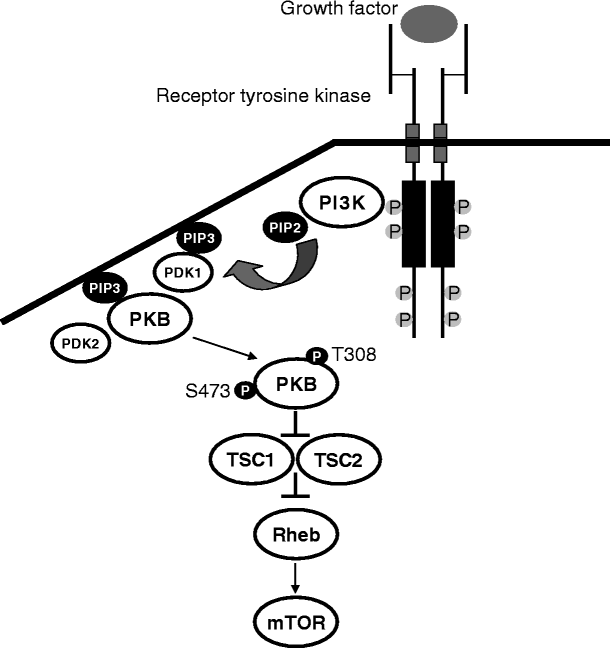
Schematic representation of the main cytosolic components of the PI3K/PKB pathway
Modulators of the Ras/Raf/MEK signaling cascade
The Ras/Raf/MEK pathway (Fig. 1) is activated through the initial extracellular binding and stimulation of RTKs by their respective cognate ligands. Upon autophosphorylation of specific tyrosine residues in the cytosolic domain of RTKs, the Grb2-Sos complex translocates to the plasma membrane and converts the inactive Ras guanosine diphosphate (GDP) to active Ras guanosine triphosphate (GTP). The interaction between Grb2 and the activated kinases or the phosphorylated receptor-associated proteins is mediated by the SH2 domain of the signaling protein that recognizes specific phosphotyrosine sequences. Ras undergoes conformational changes upon GTP binding and causes the recruitment of Raf-1 to the cytoplasmic membrane where it is phosphorylated by several kinases and simultaneously diphosphorylated at key residues by protein phosphatase 2B. Activated Raf phosphorylates MEK on two serine residues in the activation loop, which results in the activation of this protein kinase. MEK then phosphorylates and activates ERK, allowing its translocation to the nucleus where it phosphorylates transcriptional factors permitting the expression of a variety of genes. As shown in this section, several components of the Raf/MEK/ERK cascade represent unique opportunities for cancer treatment [1, 2].
Inhibitors of the kinase activity of Raf
Raf kinases exist as three isoforms A-, B-, and C-Raf. As previously mentioned, activated Raf phosphorylates and activates MEK and downstream efforts [3]. B-Raf is one of the most frequently mutated genes in human cancers, with an overall frequency of 7–8% [4]. B-Raf mutations have been found in 27–70% of melanomas, approximately 30% of papillary thyroid cancers, and 10–15% of colorectal and ovarian cancers. Among the reported mutations, the most prominent one results in a change of the Val residue in the activation loop to Glu [5], leading to a constitutive active kinase [3]. Accumulating preclinical and clinical studies have indicated that tumors harboring B-Raf mutations may exhibit an oncogenic addiction and that this phenotype presents an unique opportunity for therapeutic interference by blocking the kinase activity of Raf [6].
Sorafenib (Nexavar™, BAY 43–9006, compound 1, Fig. 3) is the only Raf kinase modulator to receive marketing approval—December 2005—for treatment of advanced renal cell carcinoma (RCC). This biaryl urea derivative inhibits in in vitro biochemical assays not only Raf kinase (e.g., IC50 = 6 nM, 22 nM, and 40 nM for C-Raf, B-Raf, and B-RafV600E, respectively), but also shows potent activity for other protein kinases, including the proangiogenic receptor tyrosine kinases PDFGR-β (IC50 = 57 nM), VEGFR-2 (IC50 = 90 nM), and FGFR-1 (IC50 = 580 nM) [7]. The in vivo antitumor activity reported for this compound is most likely driven by its multikinase inhibitory profile. In this context, the relative broad-spectrum in vivo antitumor activity exhibited by sorafenib in preclinical animal models usually did not correlate with reduced ERK activation. Thus, tumor stasis was observed when the compound was administered to mice bearing the B-RafV600E mutant Colo205 tumor xenograft, but no significant effect on ERK phosphorylation was detected at the end of this efficacy study [7].
Fig. 3
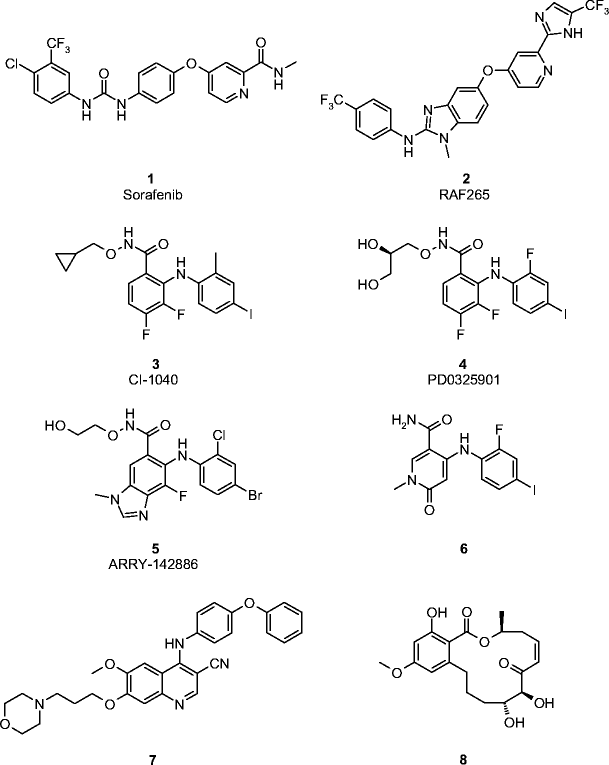
Representative examples of Raf and MEK kinase inhibitors
The resolution of the X-ray structure of the kinase domain of B-Raf in complex with sorafenib helped to understand some of the structural features of this kinase and provided the basis for the rational design of other Raf kinase modulators [3]. The inhibitor interacts with the ATP-binding cleft of the enzyme and stabilizes the protein in a conformation that prevents substrate binding and phosphorylation. In its binding mode, the distal pyridyl group occupies the ATP-binding pocket and the opposite trifluoromethyl-containing phenyl moiety inserts into a hydrophobic pocket. The interactions mediated by these groups are complemented by two critical hydrogen bonds with the urea moiety: one via its amide nitrogen atom to the carboxylate side chain of the catalytic Glu500 residue and the second via the carbonyl moiety to the main chain nitrogen of Asp593 of the DFG moiety. Similar types of hydrogen bonds have been reported between c-Abl and p38 MAP kinase and their respective inhibitors Gleevec™ and BIRB-796, probing the importance of these type of interactions in the unique mechanism of action of these kinase inhibitors [8–10].
RAF265 (previously known as CHIR-265, compound 2, Fig. 3) is another orally bioavailable Raf modulator currently being investigated in phase I clinical trials. The compound inhibits in biochemical assays C-Raf, B-Raf, B-RafV600E mutant, VEGFR-2, and c-Kit with IC50 values in the low nM range and has shown potent downregulation of the ERK pathway and tumorigenesis in relevant in vivo models [11]. PLX4032 (structure not available), which was identified following a scaffold-based drug discovery strategy, has also recently started phase I clinical trials. The compound significantly inhibits V600E B-Raf (IC50 = 31 nM) compared with a panel of over 70 other protein kinases. Its kinase profile translates into selective antiproliferative activity against Raf- but not Ras-driven melanoma and colorectal cancer cell lines (IC50 = 0.04–0.4 vs 6.5–16 μM against cell lines bearing mutant Ras). Statistically significant antitumor activity was observed in mice bearing human colorectal cancer Colo205 xenografts when given orally at 20 mg/kg per day, with no effect on body weight and strong inhibition of pERK as determined by ex vivo analyses of tumor tissue at the end of the efficacy study. XL281 (structure not disclosed) is another potent B-Raf, V600E-B-Raf, and C-Raf inhibitor (IC50 = 4.5, 6, and 2.5 nM, respectively) that has initiated phase I clinical trials in 2007. Significant inhibition of pERK levels was observed in B-Raf-driven tumor xenograft models with ED50 values ranging from 60 to 90 mg/kg.
Inhibitors of the kinase activity of MEK
The dual–specificity MEK1 and MEK2 kinases are capable of phosphorylating both serine/threonine and tyrosine residues of their physiological substrates ERK1 and ERK2. The observation that ERK is the only known MEK substrate and that ERK is commonly activated in tumor cells has triggered substantial interest in the identification and development of inhibitors of MEK as a means to block ERK activation in human cancers [1].
A common feature of the available MEK kinase modulators is their allosteric non-ATP-competitive inhibitory mechanism. The X-ray structure of the kinase domain of MEK in complex with one of the early MEK inhibitors—a derivative of compound 3, Fig. 3—has shown that the compound did not perturb ATP binding [12]. Instead, the molecule is bound to a unique, probably exclusive, pocket adjacent to the Mg/ATP-binding site locking the enzyme in a catalytically inactive conformation. The recognition of the amino acids involved in this pocket—Met143, Ile141, Leu118, and Phe209—and the conformation adopted by the enzyme upon compound binding may account for the exclusive target selectivity reported for the current non-ATP-competitive MEK kinase inhibitors.
CI-1040 (PD184352, compound 3, Fig. 3) was the first orally active, potent, and selective MEK1/2 inhibitor to enter clinical trials in cancer patients [13, 14]. Unfortunately, the compound was determined to have suboptimal human pharmacokinetic properties—low oral bioavailability and metabolic instability—and clinical investigations were discontinued after negative phase II clinical studies in cancer patients with solid tumors [15]. Two second-generation MEK1- and 2-specific inhibitors—PD0325901 (compound 4, Fig. 3) and ARRY-142886/AZD6244 (compound 5, Fig. 3)—believed to have addressed some of the pharmacological deficiencies of CI-1040 have been developed and are currently undergoing clinical trials [16].
PD0325901 is a structurally related analog of CI-1040. The modifications introduced in the scaffold resulted in a significant increase in potency (> 50-fold, IC50 = 1 nM), improved oral bioavailability, and better in vivo target coverage compared to CI-1040. ARRY-142886 is also an orally bioavailable, potent MEK1/2 inhibitor (IC50 = 12 nM) that exploits a similar binding mode as CI-1040 and PD0325901. Preclinical evaluation of these compounds showed statistically significant antitumor activity in several tumor human xenograft models at tolerated doses [2]. The observed antitumor activity was found to correlate with suppression of pERK as determined by ex vivo analyses of tumor tissue. PD0325901 and ARRY-142886/AZD6244 are currently been evaluated in phase II clinical trials in patients with solid tumors [1].
The available X-ray structural information has also been used to design new, non-ATP-competitive MEK inhibitors. This approach has been nicely illustrated in a recent publication [17]. The pyridone derivative (compound 6, Fig. 3) was found to be more potent than CI-1040 (IC50 = 4 vs 46 nM, colon26 pERK cellular assay) and more efficacious in in vivo settings as measured by response rate as well as tumor growth delay.
In addition to the non-ATP-competitive compounds described previously, a limited number of chemotypes have been described as classic ATP-MEK1/2-competitive inhibitors. Representative examples of this class of inhibitors are cyanoquinoline (compound 7, Fig. 3) [18] and resorcyclic acid (compound 8, Fig. 3) [19] derivatives. In addition to MEK1/2 modulation (e.g., IC50 = 4 nM for L-783,277) [19], resorcyclic-based compounds have shown inhibitory activity against Flt-3, FLT-1, KDR, PDGFR, TRKB, ERK, and Src. The key pharmacophore in these broad-spectrum kinase modulators is the enone functionality, which is involved in the formation of a covalent adduct with Cys-166. The inherent chemical reactivity of the enone group to Michael addition poses a major liability to develop these derivatives as potential new therapeutic agents. For other examples of competitive and noncompetitive ATP MEK inhibitors, see [1].
Modulators of the PI3K/PKB signaling cascade
A substantial number of epidemiological and experimental studies support an important role of the PI3K/PKB signaling cascade in the biology of human cancer. Thus, the activation of this pathway (Fig. 2) has been clearly validated as an essential step for the initiation and maintenance of the tumorigenic phenotype. Upregulation can be the result of: (1) constitutively active mutants or overexpression of some receptor tyrosine kinases (e.g., EGFR or ErbB2) leading to constitutive recruitment and activation of PI3K; (2) amplification and/or overexpression of the p110α catalytic subunit; (3) presence of activating mutations in the PIK3CA gene encoding the p110α catalytic subunit; (4) overexpression of the downstream kinase PKB (also known as Akt); (5) loss or inactivating mutation of the tumor suppressor gene phosphatase and tensin homolog deleted on chromosome 10 (PTEN), a natural negative regulator of the PI3K pathway; or (6) constitutive recruitment and activation by mutant forms of the Ras oncogene. The preceding alterations trigger a cascade of biological events, from cell growth and proliferation to survival and migration, that drive tumor progression, survival, angiogenesis, and metastasis. In addition to its role in the biology of human tumors, this pathway also plays a major part in the resistance of tumor cells to conventional anticancer therapies. Blocking the PI3K/PKB pathway could therefore simultaneously inhibit the proliferation and growth of tumor cells and sensitize them toward programmed cell death. To this end, drug discovery activities have been directed to identify agents that effectively and specifically disrupt this pathway in tumor cells. Herein, the progress in the development of compounds designed to target PI3K, one of the main components of the pathway, is briefly reviewed [20, 21].
Inhibitors of the lipid kinase activity of PI3K
3-Phosphorylated inositol lipids are phosphorylated by members of a family of closely related enzymes known as PI3Ks. These lipid kinases are ubiquitously expressed in cells and function as signal transducers downstream of cell surface receptors. The products of PI3K-catalyzed reactions, phosphatidylinositol 3,4,5-trisphosphate (PtdIns(3,4,5)P3), phosphoinositidylinositol 3,4-bisphosphate (PtdIns(3,4)P2), and phosphoinositidylinositol 3-phosphate (PtdIns(3)P), are second messengers and have central roles in a number of cellular processes, including cell growth, differentiation, mobility, proliferation, and survival.
The eight members of the PI3K family have been classified into three groups based on their primary sequence, in vitro substrate preference, domain structure, and mode of regulation. The subject of this section is class I PI3Ks, which is composed of two subgroups, IA and IB [22]. The class IA PI3K subgroup consists of there catalytic subunits, p110α, p110β, and p110δ, that form heterodimers with one of five regulatory domains: p85α, p85β, p85γ, p50α, and p55α. As shown in Fig. 2, these PI3Ks are activated by cell surface receptor tyrosine kinases. The class IB PI3K consists of one member, a heterodimer of p110γ and a regulatory p101 domain, that is activated by G proteins βγ subunits following the stimulation of G protein-coupled receptors. PI3Ks IA and IB catalyze the formation of PtdIns(3,4,5)P3, a process that is reversed by the action of a phosphatase, PTEN.
Two well-known and isoform nonselective PI3K inhibitors are the fungal metabolite wortmannin (compound 9, Fig. 4) [23] and LY294002 (compound 10, Fig. 4) [24]. Although these two compounds bind to the ATP-binding cleft of the lipid kinase, they block the enzymatic activity of PI3K by different mechanisms. Thus, wortmannin is an irreversible inhibitor (IC50≈ 2 nM) that forms a covalent bond with a conserved lysine residue in the ATP-binding cleft of PI3K [25], while LY294002 is a classic ATP-competitive PI3K inhibitor (IC50 = 1.40 μM) [24, 26]. In spite of the crossover inhibition of other lipid and protein kinases and their unfavorable pharmaceutical properties, these two compounds have served as powerful research tools for more than a decade to elucidate the role of PI3Ks in human tumorigenesis and evaluate the potential utility of PI3K modulators as cancer therapeutics.
Fig. 4
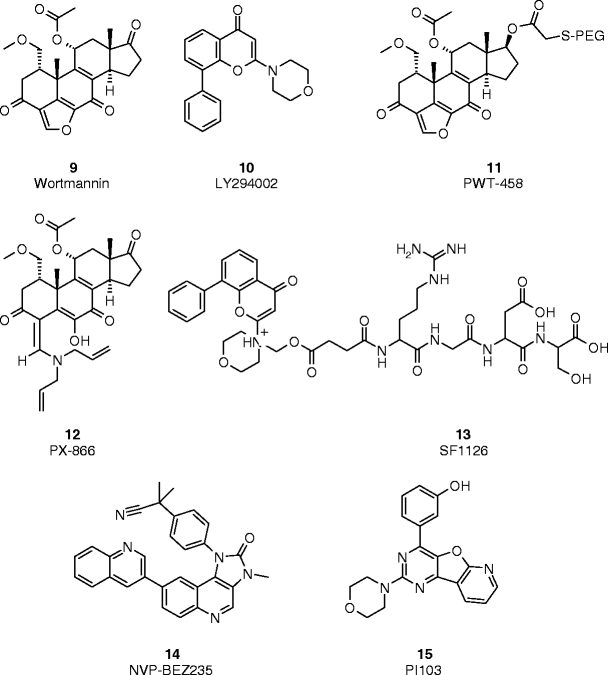
Representative examples of PI3K kinase inhibitors
The scaffolds of wortmannin and LY294002 have been the subject of intense lead optimization efforts to overcome the pharmaceutical limitations of the parent compounds. Thus, using wortmannin as a starting point, broad-spectrum PI3K inhibitors with improved pharmaceutical properties and therapeutic indexes like PWT-458 (compound 11, Fig. 4) [27] and PX-866 (compound 12, Fig. 4) [28, 29] have been reported. PWT-458 is a PEGylated derivative of wortmannin that has shown to have a higher therapeutic index over its parent compound. Upon i.v. administration, the polyethylene-glycol moiety is cleaved and 17-hydroxywortmannin is released. In vivo antitumor activity has been reported in several human xenograft models (e.g., U87MG, A549, and A498). PX-866, which is a semisynthetic viridin derivative, potently and irreversibly inhibits PI3Kα, γ, and δ (IC50 = 5.5, 9.0, and 2.7 nM, respectively), but unlike wortmannin shows a certain level of selectivity over PI3Kβ (IC50 > 300 nM) and higher biological stability. The compound has also shown antitumor activity against several human solid tumor models (e.g., OvCar-3, HT-29, or A-549) when used alone (2 mg/kg, p.o.) or in combination with chemotherapeutic or targeted anticancer agents (e.g., paclitaxel or gefitinib). In addition to a decrease in body weight and leukocytosis, a major toxicity of PX-866 administration to SCID mice was hyperglycemia with decreased glucose tolerance. These changes were dose dependent and reversible upon discontinuation of the drug. PX-866 is expected to enter phase I clinical trials in 2008.
Similar lead optimization approaches have been used for LY29002 and derivatives thereof. SF1126 (compound 13, Fig. 4) [30] is a water-soluble prodrug of LY294002. The compound, which is designed to target specific integrins, inhibits all paralogs of PI3K (IC50 = 0.5–5.7 μM) and other members of the PI3K family, including DNA-PK and mTOR. It blocks the phosphorylation and activation of PKB/Akt in cellular settings with IC50 values in the low μM range (e.g., IC50 = 2.4 μM, MM.1S). As in the case of the wortmannin derivatives, SF1126 has demonstrated significant antitumor activity in several xenograft models of brain, neuroblastoma, non-small cell lung, prostate, myeloma, renal, and colon carcinoma. In addition to its direct activity in cancer cells, microvessel density analysis of tumor tissue demonstrated that the compound also had significant antiangiogenic activity in vivo [30]. SF1126 is currently in phase I clinical trials.
In addition to the efforts made to overcome the pharmaceutical hurdles encountered with wortmannin and LY294002, high-throughput screening and medicinal chemistry activities have been utilized to discover pan- or paralog-selective PI3K inhibitors. Overall, the new generation of PI3K inhibitors have better pharmacological characteristics than the early compounds as well as improved PI3K selectivity profile. Some of these compounds are briefly reviewed in the following paragraphs.
NVP-BEZ235 (compound 14, Fig. 4) is a dual pan-PI3K/mTOR inhibitor: IC50 = 4, 76, 7, 5, and 21 nM against p110α, β, γ, δ, and mTOR, respectively [31, 32]. The compound exhibits potent antiproliferative activity against a broad panel of tumor cell lines (e.g., IC50 = 10 nM, U87MG cells) by specifically blocking the biological function of PI3K signaling components (e.g., IC50 = 10 ± 1 nM, Ser473-PKB/Akt inhibition in U87MG cells). The antiproliferative activity of this imidazoquinoline derivative in cellular settings translates well in in vivo models of human cancer (e.g., U87MG, PC3M, and A549). Thus, the compound displayed disease stasis or tumor regression when administered orally—25–50 mg/kg per day—and enhanced the efficacy of other anticancer agents when used in in vivo combination studies (e.g., docetaxel or temozolomide). Ex vivo PK/PD analyses of tumor tissues upon acute dose or after termination of in vivo efficacy studies showed a time-dependent correlation between compound concentration and inhibition of PKB/Akt phosphorylation. NVP-BEZ235 was well tolerated at the efficacious doses when compared with vehicle-treated animals, with no significant difference seen in the body weight. Unlike other modulators of the PI3K/PKB pathway, no elevated blood glucose levels were observed in the animals treated with NVP-BEZ235 after in vivo efficacy experiments in mice or rats. Moreover, no effect on insulin or glucose levels was reported for NVP-BEZ235 at efficacious doses in a 13-week rat toxicity study [32]. Phase I trials with this dual PI3K/mTOR inhibitor were initiated in patients with solid tumors at the end of 2006.
XL147 and XL765 (structures not disclosed) are also low-molecular mass PI3K inhibitors in phase I clinical trials. XL147 is active against class I PI3Ks (IC50 = 39, 383, 23, and 36 nM for p110α, β, γ, and δ, respectively) without inhibiting the kinase activity of Vps34 (IC50 = 6,975 nM), DNA-PK, or mTOR (IC50 > 15,000 nM) [33, 34]. The compound inhibits PI3K signaling in cultured tumor cells (IC50 = 0.40–1 μM, p-Thr308PKB) and blocks VEGF-induced tubule formation in cellular setting (IC50 = 529 nM, HMVEC-L). Oral dosing slows tumor growth or induces shrinkage in preclinical models of breast, lung, ovarian, prostate, and glioma tumors. Increased tumor growth inhibition over the corresponding single agents was obtained by combining this PI3K inhibitor with cytotoxic or targeted anticancer agents (e.g., carboplatin, paclitaxel, or erlotinib). XL765 has similar activity towards class I PI3K (IC50 = 39, 113, 9, and 43 nM for p110α, β, γ, and δ, respectively) as XL147, but, in addition, it also inhibits DNA-PK (IC50 = 150 nM) and mTOR (IC50 = 157 nM) [35, 36]. As in the case of XL147, tumor growth is delayed or halted in in vivo efficacy experiments using suitable xenograft tumor models. Interim analyses of ongoing phase I studies with these compounds have shown interpatient variability with terminal half-life values that ranged from 2.5 to 8 days for XL147 and 2 to 15 h for XL765. Preliminary signs of exposure-dependent pharmacodynamic modulation (changes in plasma insulin levels) were observed at 30 mg/60 mg for XL147 and 15 mg/30 mg b.i.d. for XL765. Analyses of other surrogate tissues, including PBMC lysates and hair follicles, for target modulation are also under investigation.
Imidazopyrimidines and pyridofuropyrimidines with class I PI3K activity have been disclosed in several publications. For examples, PI103 (compound 15, Fig. 4) is a pan-PI3K inhibitor with enzymatic IC50 values in the 2 nM (PI3Kα) to 15 nM range (PI3Kγ) [37, 38]. Despite its fast in vivo metabolism, the compound, which is rapidly distributed to tumor tissue, delays the growth of different subcutaneous human cancer xenografts. Recently, it has been shown that PI-103 as other pan-PI3K modulators also inhibits both complexes of mTOR: the rapamycin-sensitive mTORC1 (IC50 = 0.02 μM) and the rapamycin-insensitive mTORC2 (IC50 = 0.083 μM). The mTOR biochemical activity was further confirmed in cells, where the compound blocks downstream markers of mTOR signaling (e.g., phosphorylation of p70 S6 kinase IC50 < 0.1 μM). PI-103 has shown a significant antitumor activity in human glioma xenografts with a significant therapeutic window.
The identification and optimization of the preceding PI3K modulators has certainly benefitted by having early access to structural information. The resolution of the X-ray structure of the p110 subunit of PI3Kγ [39, 40], and more recently of the human p110α/p85α complex [[41](/article/10.1007/s11302-008-9111-5#ref-CR41 "Huang CH, Mandelker D, Schmidt-Kittler O et al (2007) The structure of a human p110α/p85
<span class="katex"><span class="katex-mathml"><math xmlns="http://www.w3.org/1998/Math/MathML"><semantics><mrow></mrow><annotation encoding="application/x-tex"></annotation></semantics></math></span><span class="katex-html" aria-hidden="true"></span></span>{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle\cdot}$}}{\alpha } }<span class="katex-display"><span class="katex"><span class="katex-mathml"><math xmlns="http://www.w3.org/1998/Math/MathML" display="block"><semantics><mrow><mi>c</mi><mi>o</mi><mi>m</mi><mi>p</mi><mi>l</mi><mi>e</mi><mi>x</mi><mi>e</mi><mi>l</mi><mi>u</mi><mi>c</mi><mi>i</mi><mi>d</mi><mi>a</mi><mi>t</mi><mi>e</mi><mi>s</mi><mi>t</mi><mi>h</mi><mi>e</mi><mi>e</mi><mi>f</mi><mi>f</mi><mi>e</mi><mi>c</mi><mi>t</mi><mi>s</mi><mi>o</mi><mi>f</mi><mi>o</mi><mi>n</mi><mi>c</mi><mi>o</mi><mi>g</mi><mi>e</mi><mi>n</mi><mi>i</mi><mi>c</mi><mi>P</mi><mi>I</mi><mn>3</mn><mi>K</mi></mrow><annotation encoding="application/x-tex">complex elucidates the effects of oncogenic PI3K</annotation></semantics></math></span><span class="katex-html" aria-hidden="true"><span class="base"><span class="strut" style="height:0.8889em;vertical-align:-0.1944em;"></span><span class="mord mathnormal">co</span><span class="mord mathnormal">m</span><span class="mord mathnormal" style="margin-right:0.01968em;">pl</span><span class="mord mathnormal">e</span><span class="mord mathnormal">x</span><span class="mord mathnormal">e</span><span class="mord mathnormal" style="margin-right:0.01968em;">l</span><span class="mord mathnormal">u</span><span class="mord mathnormal">c</span><span class="mord mathnormal">i</span><span class="mord mathnormal">d</span><span class="mord mathnormal">a</span><span class="mord mathnormal">t</span><span class="mord mathnormal">es</span><span class="mord mathnormal">t</span><span class="mord mathnormal">h</span><span class="mord mathnormal">ee</span><span class="mord mathnormal" style="margin-right:0.10764em;">ff</span><span class="mord mathnormal">ec</span><span class="mord mathnormal">t</span><span class="mord mathnormal">so</span><span class="mord mathnormal" style="margin-right:0.10764em;">f</span><span class="mord mathnormal">o</span><span class="mord mathnormal">n</span><span class="mord mathnormal">co</span><span class="mord mathnormal" style="margin-right:0.03588em;">g</span><span class="mord mathnormal">e</span><span class="mord mathnormal">ni</span><span class="mord mathnormal">c</span><span class="mord mathnormal" style="margin-right:0.13889em;">P</span><span class="mord mathnormal" style="margin-right:0.07847em;">I</span><span class="mord">3</span><span class="mord mathnormal" style="margin-right:0.07153em;">K</span></span></span></span></span>{\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle\cdot}$}}{\alpha } }$$
mutations. Science 318:1744–1748")\], has been fundamental in providing a molecular map of the ATP-binding cleft of this special family of kinases.Parallel to the drug discovery efforts to develop pan-PI3K modulators, there is an increase in interest to identify inhibitors with some level of selectivity against the different p110 enzymes. Sequence alignment and homology models have revealed that some amino acid differences exist between PI3K members, in particular, at the entrance of the ATP-binding pocket. Modulating and fine-tuning the interactions of the modified molecular template with these amino acids may provide paralog- or mutation-selective PI3K inhibitors for cancer treatment [42, 43]. To this end, selectivity trends among compound classes that target PI3K members have already been identified by extensive biochemical profiling against all the members of this lipid kinase family [44]. According to these results, inhibitors of p110β often tend to inhibit also p110δ, whereas inhibitors of p110α seems to inhibit preferentially p110γ. Interestingly, and in spite of a clear sequence homology in the kinase domain, modulators of class I PI3K tend to inhibit also DNA-PK (e.g., SF1126, XL765). The in vivo exploration—efficacy and toxicity studies—of compounds with different selectivity profiles is likely to provide important insights into the optimal kinase activity profile to exploit for drug discovery. However, as with any other drug discovery project, the ultimate proof of the “most desirable lipid kinase activity profile” will come from clinical trials.
Conclusions and outlook
Rapid progress has occurred in the last few years in the development of kinase modulators that target different components of the Ras/Raf/MEK and PI3K/PKB pathways. As shown in the preceding sections, aberrant activation of these signaling cascades is a frequent event in human tumors providing unique opportunities for therapeutic intervention. Equally important as the epidemiological data, a better understanding of the different components and their roles in the Ras/Raf/MEK and PI3K/PKB pathways has allowed the identification and development of clinical candidates that control the irregular activation of these pathways. Medicinal chemists and molecular modelers have faced the challenge of identifying structural features around the ATP-binding site of the targeted enzyme to design specific kinase inhibitors. Despite the lack of complete specificity, with the exception of the MEK inhibitors that are among the most selective inhibitors reported to date, the compounds that have entered clinical trials have shown in preclinical models significant in vivo antitumor activities in the absence of limiting toxicities. It is still unclear if the potential therapeutic benefit and tolerability that direct inhibition of components of the Ras/Raf/MEK and PI3K/PKB pathways and potential concomitant inhibition of other off-targets will be beneficial in the treatment of cancer. We can only hope that novel and safe targeted anticancer agents will come from these drug discovery efforts.
References
- Wang JW, Wilcoxen KM, Nomoto K, Wu S (2007) Recent advances of MEK inhibitors and their clinical progress. Curr Top Med Chem 7:1364–1378
Article PubMed CAS Google Scholar - Roberts PJ, Der CJ (2007) Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26:3291–3310
Article PubMed CAS Google Scholar - Wan PT, Garnett MJ, Roe SM et al (2004) Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116:855–867
Article PubMed CAS Google Scholar - Garnett MJ, Marais R (2004) Guilty as charged: B-RAF is a human oncogene. Cancer Cell 6:313–319
Article PubMed CAS Google Scholar - Davies H, Bignell GR, Cox C et al (2002) Mutations of the BRAF gene is human cancer. Nature 417:949–954
Article PubMed CAS Google Scholar - Li N, Batt D, Wartmuth M (2007) B-Raf kinase inhibitors for cancer treatment. Curr Opin Investig Drugs 8:452–456
PubMed CAS Google Scholar - Wilhelm SM, Carter C, Tang L et al (2004) BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 64:7099–7109
Article PubMed CAS Google Scholar - Nagar B, Bormmann WG, Pellicena P et al (2002) Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res 62:4236–4243
PubMed CAS Google Scholar - Schindler T, Bornmann W, Pellicena P et al (2000) Structural mechanism for STI-571 inhibition of Abelson tyrosine kinase. Science 289(5486):1938–1942
Article PubMed CAS Google Scholar - Pargellis C, Tong L, Churchill L et al (2002) Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat Struct Biol 9:268–272
Article PubMed CAS Google Scholar - Amiri P, Aikawa ME, Dove J et al (2006) CHIR-265 is a potent selective inhibitor of c-Raf/B-Raf/mutB-Raf that effectively inhibits proliferation and survival of cancer cell lines with Ras/Raf pathway mutations (abstract 4855). Proc Am Assoc Cancer Res 47
- Ohren JF, Chen H, Pavlovsky A et al (2004) Structures of human MAP kinase I (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat Struct Mol Biol 11:1192–1197
Article CAS Google Scholar - Sebolt-Leopold JS, Dudley DT, Herrera R et al (1999) Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat Med 5(7):810–816
Article PubMed CAS Google Scholar - Herrera R, Sebolt-Leopold JS (2002) Unraveling the complexities of the Raf/MAP kinase pathway for pharmacological intervention. Trends Mol Med 8(4):S27–S31
Article PubMed CAS Google Scholar - Rinehart J, Adjei AA, Lorusso PM et al (2004) Multicenter phase II study of the oral MEK inhibitor, CI-1040 in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol 22:4456–4462
Article PubMed CAS Google Scholar - Sebolt-Leopold JS, Herrera R (2004) Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat Rev Cancer 4:937–947
Article PubMed CAS Google Scholar - Spicer JA, Rewcastle GW, Kaufman MD et al (2007) 4-anilino-5-caboxamide-2-pyridone derivatives as noncompetitive inhibitors of mitogen-activated protein kinase kinase. J Med Chem 50:5090–5102
Article PubMed CAS Google Scholar - Zhang N, Wu B, Eudy N et al (2001) MEK (MAPKK) inhibitors. Part 2: structure-activity relationships of 4-anilino-3-cyano-6,7-dialkoxyquinolines. Bioorg Med Chem Lett 11:1407–1410
Article PubMed CAS Google Scholar - Zhao A, Lee SH, Mojena M et al (1999) Resorcyclic acid lactones: naturally occurring potent and selective inhibitors of MEK. J Antioiot (Tokyo) 52:1086–1094
CAS Google Scholar - Garcia-Echeverria C (2007) Survival signaling. Top Med Chem 1:169–206
Article Google Scholar - Stauffer F, Holzer P, Garcia-Echeverria C (2005) Blocking the PI3K/PKB pathway in tumor cells. Curr Med Chem Anticancer Agents 5:449–462
Article PubMed CAS Google Scholar - Maira SM, Voliva C, Garcia-Echeverria C (2008) Class IA phosphatidylinositol 3-kinase: from their biologic implication in human cancers to drug discovery. Expert Opin Ther Targets 12:223-238
Article PubMed CAS Google Scholar - Brian PW, Hemming HG, Norris GLF (1957) Wortmannin, an antibiotic produced by Penicillium wortmanni. Br Mycol Soc Trans 40:365–368
Article CAS Google Scholar - Vlahos CJ, Matter WF, Hui KY, Brown RF (1994) A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol Chem 269:5241–5248
PubMed CAS Google Scholar - Wymann MP, Bulgarelli-Leva G, Zvelebil MJ et al (1996) Wortmannin inactivates phosphoinositide 3-kinase by covalent modification of Lys-802, a residue involved in the phosphate transfer reaction. Mol Cell Biol 16:1722–1733
PubMed CAS Google Scholar - Davies SP, Reddy H, Caivano M, Cohen P (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351:95–105
Article PubMed CAS Google Scholar - Yu K, Lucas J, Zhu T et al (2005) PWT-458, a novel pegylated-17-hydroxywortmannin, inhibits phosphatidylinositol 3-kinase signaling and suppresses growth of solid tumors. Cancer Biol Ther 4:538–545
Article PubMed CAS Google Scholar - Ihle NT, Paine-Murrieta G, Berggren MI et al (2005) The phosphatidylinositol-3-kinase inhibitor PX-866 overcomes resistance to the epidermal growth factor receptor inhibitor gefitinib in A-549 human non-small cell lung cancer xenografts. Mol Cancer Ther 4:1349–1357
Article PubMed CAS Google Scholar - Ihle NT, Williams R, Chow S et al (2004) Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol Cancer Ther 3:763–772
PubMed CAS Google Scholar - Garlich JR, De P, Dey N et al (2008) A vascular targeted pan phosphoinositide 3-kinase inhibitor prodrug, SF1126, with antitumor and antiangiogenic activity. Cancer Res 68:206–215
Article PubMed CAS Google Scholar - Stauffer F, Garcia-Echeverria C, Furet P et al (2007) Medicinal chemistry efforts towards the identification and development of inhibitors of phosphatidylinositol 3-kinase (PI3Ks) and related kinases for cancer treatment (abstract 3953). Proc Am Assoc Cancer Res
- Maira SM (2007) Biochemical, cellular and in vivo profiling of a new PI3K inhibitor from the imidazoquinoline series (oral presentation). Proc Am Assoc Cancer Res
- Foster PG (2007) Potentiating the antitumor effects of chemotherapy with the selective PI3K inhibitor XL147 (abstract C199). EORTC Meeting
- Shapiro GI, Edelman G, Calvo E et al (2007) Targeting aberrant PI3K pathway signaling with XL147, a potent, selective, and orally bioavailable PI3K inhibitor (abstract C205). EORTC Meeting
- Laird D (2007) XL765 targets tumor growth, survival, and angiogenesis in preclinical models by dual inhibition of PI3K and mTOR (abstract B250). EORTC Meeting
- Patnaik A, Lorusso PM, Tabernero J et al (2007) Biomarker development for XL765, a potent and selective oral dual inhibitor of PI3K and mTOR currently being administered to patients in a phase I clinical trial (abstract B265). EORTC Meeting
- Raynaud FI, Eccles S, Clarke PA et al (2007) Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res 67:5840–5850
Article PubMed CAS Google Scholar - Fan QW, Cheng CK, Nicolaodes TP et al (2007) A dual phosphoinositide-3-kinase alpha/mTOR inhibitor cooperates with blockade of epidermal growth factor receptor in PTEN-mutant glioma. Cancer Res 67:7960–7965
Article PubMed CAS Google Scholar - Walker EH, Pacold ME, Perisic O et al (2000) Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol Cell 6:909–919
Article PubMed CAS Google Scholar - Walker EH, Perisic O, Ried C et al (2000) Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature 402:313–320
Article Google Scholar - Huang CH, Mandelker D, Schmidt-Kittler O et al (2007) The structure of a human p110α/p85\({\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle\cdot}$}}{\alpha } }\) complex elucidates the effects of oncogenic PI3K\({\underset{\raise0.3em\hbox{$\smash{\scriptscriptstyle\cdot}$}}{\alpha } }\) mutations. Science 318:1744–1748
Article PubMed CAS Google Scholar - Knight ZA, Chiang GG, Alaimo PJ et al (2004) Isoform-specific phosphoinositide 3-kinase inhibitors from an arylmorpholine scaffold. Bioorg Med Chem 12:4749–4759
Article PubMed CAS Google Scholar - Redaelli A, Lee JM, Stephens JM, Pashos CL (2003) Epidemiology and clinical burden of acute myeloid leukemia. Expert Rev Anticancer Ther 3:695–710
Article PubMed CAS Google Scholar - Knight ZA, Gonzalez B, Feldman ME et al (2006) Pharmacological map of the PI3K family defines a role for p110α in insulin signaling. Cell 125:733–747
Article PubMed CAS Google Scholar