Non-Smad pathways in TGF-β signaling (original) (raw)
Introduction
Transforming growth factor-β (TGF-β) is the prototype of a family of secreted polypeptide growth factors. To date, up to 33 TGF-β-related genes have been identified in mammalian genomes as the result of genome sequencing projects; these include bone morphogenic proteins (BMPs), activin/inhibin, growth and differentiation factors, nodal, and anti-Müllerian hormone 1. These cytokines play very important roles during development, as well as in normal physiological and disease processes, by regulating a wide array of cellular processes, such as cell growth, differentiation, migration, apoptosis, and extracellular matrix production 2, 3. For instance, TGF-β is a potent anti-tumor agent because it strongly inhibits the growth of epithelial cells. However, in a different cellular context, TGF-β can also promote tumor growth because it is able to induce changes in transcriptional activities that re-program epithelial cells into mesenchymal cells, thereby facilitating tumor metastasis and invasion 3, 4. Over a decade ago, genetic studies in worms and fruitflies uncovered a group of genes, later dubbed as Smads, which appear to play a crucial role in mediating the intracellular responses to TGF-β and/or its related factors 5. Subsequent biochemical characterization demonstrated that Smads are transcription factors that constantly shuttle between the cytoplasm and the nucleus 6, 7, 8. When activated, the TGF-β receptors undergo conformational changes that allow direct binding of Smads and their phosphorylation by the kinase activities of the cytoplasmic domains of the type I receptors. This results in the accumulation of Smads in the nucleus to regulate target gene transcription. The identification of Smads elated the field of TGF-β signaling, but it also instigated a perplexing dilemma in terms of reconciling the diverse functions of the TGF-β family with the simplicity of the Smad signaling model. Now mounting evidence has revealed that the diversity of TGF-β signaling responses is determined by the combinatorial usage of core pathway components including ligands, receptors, Smads, and Smad-interacting transcription factors, by 'cross-talks' with other signaling pathways, and by the ability of TGF-β receptors to activate other signaling modules, in addition to Smads, to reinforce, attenuate, or otherwise modulate downstream cellular responses 9, 10. The cross-talks between TGF-β/Smads and other cell signaling pathways will be discussed in detail in another review in this special issue. This review is solely devoted to the non-Smad pathways that are activated by the TGF-β receptors through either phosphorylation or direct interaction, with an emphasis on recent advances that revealed the underlying biochemical mechanisms. These non-Smad pathways include various branches of MAP kinase (MAPK) pathways, Rho-like GTPase signaling pathways, and phosphatidylinositol-3-kinase (PI3K)/AKT pathways.
TGF-β-induced Erk activation and tyrosine phosphorylation
The initial indication that TGF-β can activate the Erk MAPK pathway came from observations showing a rapid activation of p21 (Ras) by TGF-β in rat intestine or mink lung epithelial cells 11, 12. The rapid GTP loading of Ras in response to TGF-β in the above epithelial cells may cause recruitment of Raf, a MAP kinase kinase kinase (MAP3K), to the plasma membrane and lead to activation of Erk through MEK1. Subsequently, rapid activation of Erk by TGF-β was observed in epithelial cells 13, breast cancer cells 14, and fibroblasts 15. The kinetics of Erk phosphorylation induced by TGF-β varies with cell types and culture conditions. In some cell lines, a delayed response of Erk to TGF-β was reported, typically with the peak of Erk phosphorylation occurring hours after ligand stimulation, suggesting an indirect response requiring protein translation 16. In contrast, in other types of cells, activation can occur rapidly within 5-10 min of TGF-β stimulation, which is comparable to the time course of Erk activation by mitogenic factors such as EGF 17.
Although Smad-dependent transcriptional mechanism can account, at least in part, for the delayed activation of Erk by TGF-β, it does not explain the rapid activation of Erk by TGF-β. In the receptor tyrosine kinase (RTK)/Ras/Erk signaling pathway, binding of growth factors to their RTKs induces dimerization and activation of the RTK 18, 19. This results in auto- and trans-phosphorylation of multiple tyrosine residues in the cytoplasmic domain of RTK. Once phosphorylated, these tyrosine residues serve as docking sites for numerous signaling molecules with either Src homology 2 (SH2) or phosphotyrosine binding (PTB) domains, such as Src and growth factor receptor binding protein 2 (Grb2). Grb2 is an adaptor protein that is bound to Sos in the cytoplasm in the absence of ligand stimulation. Upon RTK activation, Grb2/Sos complex is recruited to the RTK, which brings Sos to the plasma membrane, where it activates Ras by catalyzing the exchange of GDP for GTP. In its GTP-bound state, Ras can bind Raf and activate a MAPK cascade that includes MEK and Erk. Grb2/Sos complex can bind to phosphotyrosine residues of RTK through the SH2 domain of Grb2 or through another adaptor protein, Shc (Src homology domain 2 containing) 20. Shc binds to RTK and serves as a substrate of RTK. Tyrosine phosphorylation of Shc enables it to associate with Grb2 and recruit Grb2/Sos complex to activate Ras and the downstream MAPK cascade.
Although type I and type II TGF-β receptors are well-defined serine-threonine kinases, TβRII undergoes autophosphorylation on three tyrosine residues: Y259, Y336, and Y424, albeit at a much lower level than autophosphorylation on serine and threonine residues 21. TβRII can also be phosphorylated by Src, a non-RTK, on Y284, which can serve as a docking site for the recruitment of Grb2 and Shc, thereby bridging TβRII to MAPK activation 22. Moreover, TβRI can also be tyrosine phosphorylated after TGF-β stimulation 23. As TβRI is activated by TβRII upon ligand binding and forms a tetrameric receptor heterocomplex with TβRII, it is not clear whether the tyrosine phosphorylation of TβRI stems from TβRI autophosphorylation or from transphosphorylation by TβRII. Nevertheless, activated TβRI can recruit and directly phosphorylate ShcA on tyrosine and serine residues, thus promoting the formation of a ShcA/Grb2/Sos complex 23. The ShcA/Grb2/Sos complex is then capable of activating Ras at the plasma membrane, leading to sequential activation of c-Raf, MEK, and Erk. The kinase activities of both types of TGF-β receptors are required for Shc phosphorylation 23. Although ShcA can be phosphorylated by either TβRI or TβRII in vitro, ShcA interacts with, and is phosphorylated by, TβRI more efficiently than TβRII. Overexpression of a ShcA mutant lacking either the PTB or the SH2 domain attenuates TGF-β-induced ShcA tyrosine phosphorylation, abolishing the interaction between Grb2 and ShcA, and Raf phosphorylation, which leads to decreased Erk activation by TGF-β 23. Transfection of siRNA that silences Shc also diminishes TGF-β-induced Erk activation. Thus, Shc plays an essential role in TGF-β-induced Erk activation, and recruitment of tyrosine kinase signaling pathways is one of the molecular mechanisms for TGF-β to activate non-Smad signaling (Figure 1).
Figure 1
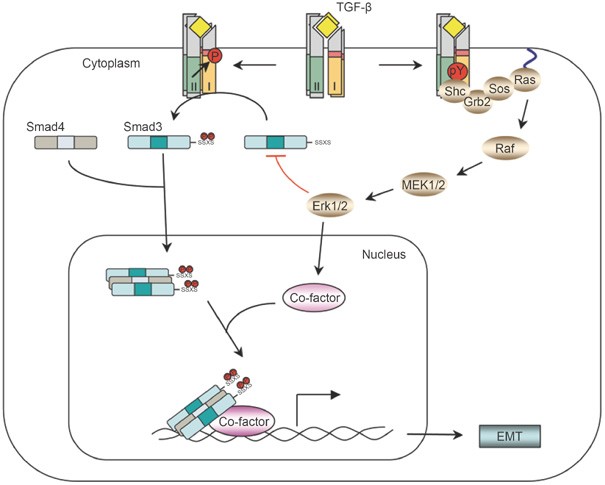
The Erk non-Smad pathway. TGF-β can induce phosphorylation of tyrosine residues on both type I and type II receptors and/or on Shc. The phosphorylated tyrosines are capable of recruiting Grb2/Sos to activate Erk through Ras, Raf, and their downstream MAPK cascades. Erk then regulates target gene transcription through its downstream transcription factors in conjunction with Smads to control EMT. Erk can also inhibit R-Smad activities through phosphorylation of R-Smads.
Erk activation is important for epithelial to mesenchymal transition (EMT), which is one of the major biological functions of TGF-β. EMT is a normal physiological process necessary for embryonic development, and is a pathological feature associated with tumor metastasis and fibrosis 24, 25. During EMT, cells lose epithelial characteristics and acquire the properties of mesenchyme, including downregulation of adherens junctions and their affiliated proteins, e.g. E-cadherin, increased MMP activity, induction of actin stress fibers, and acquisition of motile and invasive properties. In late stages of tumorigenesis, TGF-β promotes tumor growth by inducing EMT through a combination of Smad-dependent and Smad-independent effects 3, 4. Erk activation is one of the non-Smad pathways necessary for TGF-β-mediated EMT 26, 27. Erk is required for disassembly of cell adherens junctions and induction of cell motility by TGF-β. In a transcriptomic screen of genetic programs for TGF-β-induced EMT, TGF-β-stimulated Erk signal regulates a subset of target genes, which are enriched for genes with defined roles in cell-matrix interaction, cell motility, and endocytosis 26. These genes are known to function in the remodeling of integrin-based cell-matrix adhesion and promote cell motility. Consistent with the role of Erk in TGF-β-mediated EMT, reduction of ShcA or Grb2 expression using siRNA renders mammary epithelial or mammary tumor cells unresponsive to TGF-β-induced EMT, migration, and invasion 28, 29. In addition, a dominant-negative form of ShcA, lacking its three known tyrosine phosphorylation sites, completely abrogates the TGF-β-induced motility and invasion of breast cancer cells 29. Therefore, the Shc-Grb2-Erk pathway is a key component of pro-oncogenic activity of TGF-β that mediates tumor invasiveness and metastasis. Moreover, Erk can also phosphorylate receptor-activated Smads, including Smad1, Smad2 and Smad3, to regulate their activities 30, 31, 32, 33. In cell culture systems, phosphorylation of Smads by Erk inhibits Smad activity, which has been invoked to explain how oncogenic Ras overrides TGF-β-mediated growth arrest in cancer cells 31. Finally, Erk substrates, such as AP-1 family members, can interact and function in conjunction with Smads to regulate gene expression 27, 34, 35.
TGF-β-induced JNK/p38 activation
Perhaps the best-characterized non-Smad pathway is the JNK and p38 MAPK signaling cascades (Figure 2). Like Erk, JNK and p38 are at the tertiary layer of MAPK cascades, in which they are activated by the MAP kinase kinases (MKKs), specifically MKK4 and MKK3/6, respectively 36. TGF-β can rapidly activate JNK through MKK4 37, 38, 39 and p38 MAPK through MKK3/6 in various cell lines 40, 41, 42, 43. Experiments with a dominant-negative form of Smad3 or using Smad3- or Smad4-deficient cells showed that Smads are dispensable for the TGF-β-induced activation of JNK 38, 39, suggesting that the MAPK pathway is activated by TGF-β independently of Smads. A direct demonstration of Smad independence was based on the utilization of a mutant TβRI receptor with an altered L45 loop, which renders the receptor defective in Smad binding and activation, but still retaining an intact kinase activity. This mutant type I receptor is still capable of mediating TGF-β-induced activation of JNK and p38 MAPK 43, 44.
Figure 2
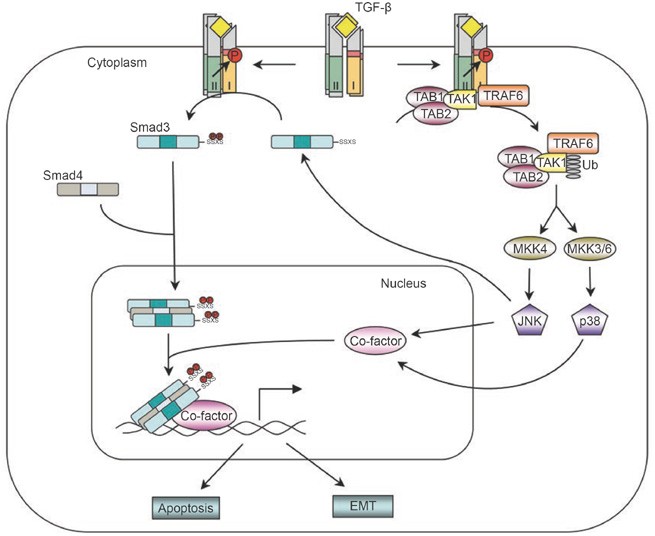
The JNK/p38 non-Smad pathway. TGF-β receptors interact with TRAF6 and induce the formation of K63-linked poly-ubiquitin chains on TRAF6. Poly-ubiquitinated TRAF6 recruits TAK1 to activate JNK/p38. Activated JNK/p38 act in conjunction with Smads to regulate apoptosis and EMT by controlling the activities of downstream transcription factors. JNK can also regulate R-Smad activity directly by phosphorylation.
Further upstream, MKKs are activated by the MAP3Ks; in the case of MKK3/6 and MKK4, the TGF-β-activated kinase 1 (TAK1) is one of the activating MAP3Ks. Vertebrate TAK1 was identified based on the ability of a mouse cDNA to substitute for a latent MAP3K of Saccharomyces cerevisiae in the yeast mating pheromone response, and the cloned kinase was found to activate and be activated by TGF-β signaling 45. During Xenopus embryonic development, TAK1 is involved in mesoderm induction and patterning mediated by BMP 46. The physiological significance of TAK1 in TGF-β signaling was later revealed in mouse genetic studies, in which the TAK1 gene was inactivated through homologous recombination 47, 48. TAK1-deficient embryos exhibit defects in the vasculature of the embryo proper and yolk sac, a phenotype with striking similarities to that exhibited by loss-of-function mutations in the genes encoding the type I receptor ALK1 or the type III receptor endoglin 48. It was also demonstrated that TAK1 is absolutely required for TGF-β-induced JNK and NFκB activation by using TAK1-deficient mouse embryonic fibroblasts 47. Subsequently, a direct physical interaction between TGF-β type II receptor and TAK1 was reported 49. However, how TGF-β activates TAK1 was not known until very recently.
TRAF6, which plays an important role in the activation of TAK1 in interleukin-1 receptor (IL-1R)- and Toll-like receptors (TLRs)-mediated signaling pathways, was found to be crucial for TGF-β-induced activation of the TAK1-JNK/p38 pathways 50, 51. Like most TRAF proteins, TRAF6 is composed of a highly conserved carboxyl-terminal TRAF domain and a more variable amino-terminal domain, which contains an E3 ligase RING finger domain and several Zn fingers 52. TRAF6 associates with members of interleukin-1β receptor/Toll-like receptors (IL-1R/TLRs) through its C-terminal TRAF domain upon ligand stimulation, which leads to activation of the RING finger E3 ligase and subsequent lysine-63 (K63)-linked polyubiquitination of TRAF6 itself. Unlike lysine-48 (K48)-linked polyubiquitination, which normally targets proteins for degradation, K63-linked polyubiquitin chains act as scaffolds to assemble protein kinase complexes and mediate their activation 53. Polyubiquitinated TRAF6 then recruits TAK1 and triggers its activation, thus allowing TAK1 to activate downstream JNK/p38 pathways 54. Similarly, TRAF6 can also associate with TGF-β type II and type I receptors through its C-terminal TRAF domain 51. The fact that TRAF6 can be pulled down by type II receptors in TβRI-deficient cells but not by type I receptors in TβRII-deficient cells initially suggests that TRAF6 interacts directly with TβRII, while its interaction with TβRI is mediated through TβRII. However, since TβRII can become activated in a homocomplex, whereas TβRI can only be activated by TβRII, an alternative explanation is that TRAF6 interacts with an activated receptor complex consisting of either a homodimer of TβRII or a heterocomplex of two types of receptors 51. The role of TβRI in the binding of TRAF6 was further supported by the identification of a conserved consensus TRAF6-binding motif in TβRI 50. Binding of TRAF6 to activated TGF-β-receptor complex induces K63-linked polyubiquitination of TRAF6, and promotes the association between TRAF6 and TAK1, as well as TAK1 polyubiquitination and activation 50, 51. Interestingly, although the kinase activity of TβRI is not required for interaction with TRAF6, it is required for polyubiquitination of TRAF6 and activation of downstream kinase pathways induced by TGF-β 51. In addition to TAK1, two other MAPKKKs, MEKK1 and MLK3, have also been proposed to function upstream of TGF-β-mediated activation of JNK or p38 MAPK via MKK4 or MKK3/MKK6 respectively 55, 56, 57, 58. It is not known whether TRAF6 or other TRAFs are involved in their activation by TGF-β.
Although TGF-β induces JNK/p38 activation independently of Smad activation, the TRAF6-TAK1-JNK/p38 cascade functions in conjunction with the Smad-dependent pathway to regulate downstream cellular responses to TGF-β. For instance, induction of apoptosis is a well-recognized mechanism by which TGF-β exerts its tumor-suppression activity. Besides Smad-dependent pathways, the TRAF6-TAK1-JNK/p38 pathway is essential for TGF-β-induced apoptosis. Overexpression of TAK1 caused cells or Xenopus embryo to undergo apoptosis, whereas cells expressing the kinase-inactive TAK1 were protected from TGF-β- or BMP-induced apoptosis 46, 59, 60. Moreover, knockdown of TRAF6 using siRNA or treating cells with a chemical inhibitor of p38 efficiently blocked TGF-β-mediated apoptosis 43, 50, 51, 61. These results indicate that the TRAF6-TAK1-JNK/p38 pathway functions in a cooperative manner with the Smad pathway in TGF-β/BMP-induced apoptosis.
Similar to its role in TGF-β-induced apoptosis, the TRAF6-TAK1-JNK/p38 pathway also plays a very important role in TGF-β-induced EMT. Inhibiting p38 activity using p38 inhibitor or dominant-negative forms of MKK3 or p38 impairs TGF-β-mediated changes in cell shape and reorganization of the actin cytoskeleton 43, 62. Knocking down TRAF6 expression also inhibits TGF-β-mediated EMT 51. Thus, activation of the TRAF6-TAK1-p38 pathway is another obligatory requirement for TGF-β-induced EMT.
Rho-like GTPases in TGF-β-mediated EMT
The Rho-like GTPases, including RhoA, Rac and Cdc42, play important roles in controlling dynamic cytoskeletal organization, cell motility, and gene expression through a variety of effectors 63. Like MAPK pathways, RhoA is also a key player in TGF-β-induced EMT (Figure 3). TGF-β rapidly activates RhoA-dependent signaling pathways to induce stress fiber formation and mesenchymal characteristics in epithelial cells and primary keratinocytes 64, 65. The rapid activation of RhoA induced by TGF-β is likely to be independent of Smad2 and/or Smad3, as suggested by the rapid onset and the inability of a dominant-negative Smad3 mutant to block RhoA activities in epithelial cells 64. It is noteworthy that a delayed peak of RhoA activation, depending upon new protein synthesis, was observed in certain cells 65; induction of this peak of RhoA activity may involve TGF-β-induced expression of NET1, a RhoA-specific guanine exchange factor that mediates RhoA activation by Smad-mediated transcription 66.
Figure 3
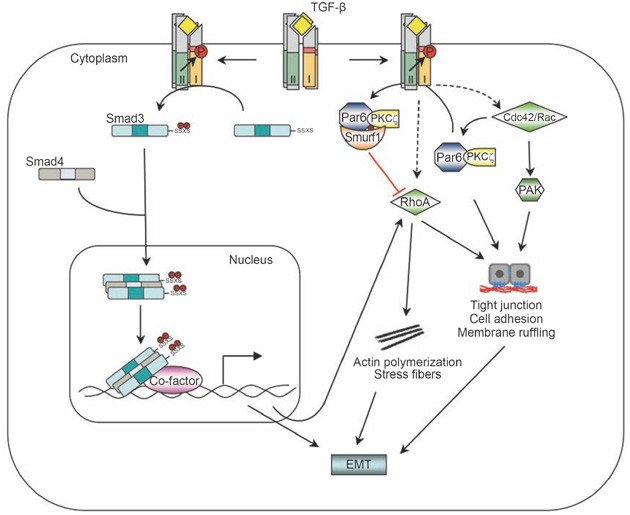
The Small GTPase non-Smad pathway. RhoA can be activated by TGF-β via either Smad-dependent or independent routes to induce actin stress fiber formation during EMT. TGF-β also induces the dissolution of tight junctions during EMT by recruiting Cdc42 to the receptor complex, and by triggering degradation of RhoA at cellular protrusions. TGF-β-induced RhoA degradation requires phosphorylation of Par6 by TβRII and the Smurf1 E3 ligase.
However, downregulation of RhoA protein levels at tight junctions by TGF-β has also been reported 67. Par6, a scaffold protein regulating epithelial cell polarity 68, interacts with TβRI at tight junctions 67. TGF-β stimulation induces the assembly and accumulation of TβRI-TβRII complexes at tight junctions, where TβRII phosphorylates Par6 at serine residue 345 67. After phosphorylation, Par6 recruits Smurf1 to the activated receptor complex at tight junctions in polarized epithelial cell sheets. The Par6-Smurf1 complex then mediates localized ubiquitination and turnover of RhoA at cellular protrusions, which enables TGF-β-dependent dissolution of tight junctions, a prerequisite for EMT 67. Mutation of serine 345 of Par6 to alanine blocks Smurf1-mediated degradation of RhoA in response to TGF-β. The Smurf1-mediated degradation of RhoA is a localized event and requires the presence of Smurf1 at lamellipodial- and filopodial-like protrusions. PKCζ, an effector of the Cdc42/Rac1-Par6 polarity complex, can bind directly to Smurf1 to regulate Smurf1 localization and RhoA degradation at cellular protrusions 67, 69, indicating that the activity of Smurf1 toward RhoA is locally restricted to sites of active protrusions. In support of this notion, silencing Smurf1 expression by siRNA-mediated RNA interference did not lead to marked changes in total RhoA protein levels; instead, RhoA and associated F-actin accumulated in cellular protrusions 69. Sites of RhoA ubiquitination by Smurf1 have been mapped to two residues, lysines 6 and 7. Mutation of these two lysines inhibited dissolution of tight junctions and EMT in response to TGF-β 67. Thus, through regulating RhoA degradation, TGF-β promotes dissolution of tight junctions and rearrangement of the actin cytoskeleton. It is possible that TGF-β regulates RhoA activity in two different modes: it induces a rapid activation of RhoA during the early phase of stimulation, and then downregulates the level of RhoA protein at later stages; both of these modes of regulation appear to be essential for TGF-β-induced EMT.
Besides RhoA, TGF-β can also induce activation of the Cdc42 GTPase 70 (Figure 3). Activation of Cdc42 by TGF-β appears to be independent of Smads, because blocking either Smad2 or Smad3 phosphorylation, or both simultaneously, does not affect activation of the p21-activated kinase (PAK) 2, which acts downstream of Cdc42 70. Physical interaction between Cdc42 and cell surface TGF-β receptor complexes has been demonstrated 71, and a cluster of proteins involved in the Cdc42 and PAK network are found in the receptor-associated protein complex. This complex includes the PAK-interacting Cdc42 GTPase, the Rac1 exchange factors α-PIX and β-PIX, PAK1 itself, a PAK1-interacting partner, oxidative stress-responsive kinase-1, and occludin (OCLN), a tight-junction accessory protein 71. One of the key steps in TGF-β-induced EMT is dissolution of tight junctions. The interaction of OCLN with TβRI regulates the localization of TβRI to tight junctions in polarized epithelial cells. A mutant OCLN without the TβRI interaction domain causes mislocalization of TβRI across the surface of the cells and inhibits TGF-β-induced dissolution of tight junctions and EMT 71. In BMP signaling, LIM kinase 1 (LIMK1), a downstream effector of the PAK network, can also associate with the BMPRII receptor and this interaction synergizes with Cdc42 to activate the catalytic activity of LIMK1, thus increasing the phospho-cofilin level and causing changes in the actin cytoskeleton 72, 73. Smad7 appears to be required for TGF-β-mediated Cdc42 activation 74, but whether Smad7 also associates with the same protein complex at the tight junction is not known.
PI3K/Akt pathway in TGF-β/Smad-mediated responses
Several findings suggest a role of PI3K in TGF-β signaling (Figure 4). First, TGF-β can rapidly activate PI3K, as indicated by the phosphorylation of its downstream effector Akt 75, 76, 77, 78, 79. This activation appears to be independent of Smad2/3 activation 78. Second, in immunoprecipitation experiments, the TβRII receptor was found to be constitutively associated with p85, the regulatory subunit of PI3K, while association of the TβRI receptor with p85 requires TGF-β stimulation 80. The association between TGF-β receptors and p85 was not direct, but the kinase activities of the TGF-β receptors were required for TGF-β-induced PI3K activation. Inhibition of TβRI activity using a chemical inhibitor prevents TGF-β-induced activation of Akt by PI3K 75, 79. In addition, TGF-β may also induce activation of PI3K indirectly through TGF-β-induced TGF-α expression and consequent activation of EGF receptor signaling 77. On the other hand, TGF-β was shown to down-regulate PI3K/Akt signaling activity through Smad-dependent expression of the lipid phosphatase SHIP 81, which may account for the transient nature of TGF-β-induced phosphorylation of Akt.
Figure 4
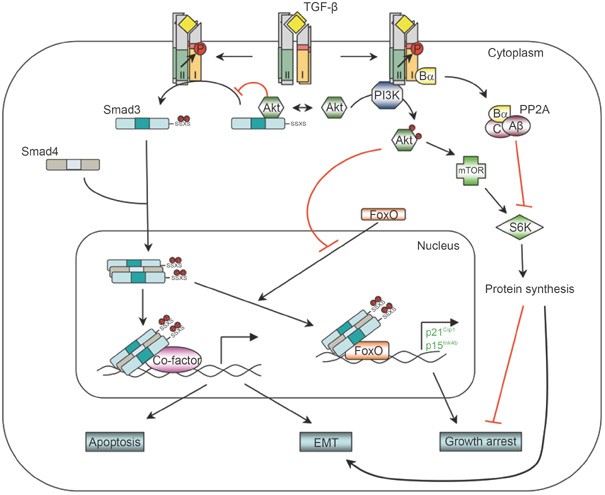
The PI3K/Akt non-Smad pathway. TGF-β can activate PI3K and Akt, possibly by inducing a physical interaction between the p85 subunit of PI3K and the receptors. The activated PI3K/Akt pathway then controls translational responses through mTOR/S6K, which collaborates with Smad-mediated transcriptional responses during EMT. In the growth arrest response, TGF-β may inhibit S6K by inducing an interaction between the Bα subunit of PP2A and the receptors. Akt is also capable of antagonizing TGF-β-induced apoptosis and growth arrest by sequestering Smad3 in the cytoplasm and by inhibiting the activity of FoxO transcription factor.
The PI3K/Akt pathway is another non-Smad pathway contributing to TGF-β-induced EMT. PI3K is implicated in TGF-β-mediated actin filament reorganization and cell migration by studies using chemical inhibitors. Studies using chemical inhibitors of PI3K should be interpreted cautiously because pharmacological inhibition of PI3K at the dosage used often reduces the phosphorylation of Smad2 by receptors 74, 75, due to interference with the SARA-dependent presentation of Smad2 and/or Smad3 to membrane-bound TGF-β receptors 82, 83. The requirement of the PI3K/Akt pathway in TGF-β-mediated EMT could be mediated, at least in part, by a downstream effector of Akt, mammalian target of rapamycin (mTOR), which is a key regulator of protein synthesis via phosphorylation of S6 kinase (S6K) and eukaryotic initiation factor 4E-binding protein 1 (4E-BP1). TGF-β-induced activation of mTOR, S6K, and 4E-BP1 has been observed in several different cell types, including murine NMUMG mammary gland epithelial cells and human HaCAT keratinocytes 79, both of which undergo EMT in response to TGF-β. A chemical inhibitor of PI3K or a dominant-negative mutant of Akt blocks TGF-β-induced mTOR activation. Consistent with the notion that phosphorylation of S6K and 4E-BP1 by mTOR enhances translational capacity, increases in protein synthesis were also observed during TGF-β-induced EMT in these cells 79. Treating these cells with either a TβRI inhibitor or rapamycin, an inhibitor of mTOR, inhibits TGF-β-induced increase in protein content 79. Although rapamycin does not affect TGF-β-induced morphological changes, it inhibits the enhanced migration and invasive behavior associated with TGF-β-induced EMT 79. Therefore, the TGF-β-induced translation pathway through PI3K/AKT/mTOR may complement the transcription pathway mediated by Smads in TGF-β-induced EMT.
Besides EMT, PI3K plays a role in TGF-β-mediated fibroblast proliferation and morphological transformation 78. Tyrosine kinase c-Abl acts downstream of PI3K and accounts for at least part of the TGF-β-mediated fibroblast response. Inhibition or loss of the c-Abl kinase prevents TGF-β-mediated morphological alterations, expression of extracellular matrix genes, and cell proliferation in fibroblasts 84, 85.
However, in many other TGF-β-induced responses, the PI3K/Akt pathway antagonizes Smad-mediated effects. For example, activation of PI3K or Akt protects cells from TGF-β-induced apoptosis and growth inhibition 76, 86, 87. This protection has been suggested to result from a physical interaction between Akt and Smad3 88, 89. Akt, which relays signals downstream of PI3K, can directly associate with Smad3. The interaction between Smad3 and Akt prevents TβRI-mediated phosphorylation and nuclear localization of Smad3, thereby resulting in inhibition of Smad3-mediated transcription 88, 89. Besides Akt, the forkhead transcription factor, FoxO, also plays a role in the antagonizing effect of PI3K/Akt on Smad-mediated transcription. TGF-β is a well-known potent growth inhibitor. Key to this function of TGF-β is its ability to suppress proto-oncogene c-Myc transcription, and to induce transcription of p15Ink4b and/or p21Cip1, which are inhibitors of G1 phase cyclin-dependent kinases 4. In the case of induction of p15Ink4b and p21Cip1 expression, a transactivation complex containing Smad3, Smad4, and FoxO family of transcription factors is required 90, 91. Since FoxO proteins are targets of the PI3K/Akt pathway, Akt can inhibit nuclear localization of FoxO proteins by phosphorylating them, and thus barring them from their target genes. This offers another mechanistic account for the antagonizing interaction between the TGF-β pathway and the PI3K/Akt pathway in TGF-β-mediated transcription response and growth arrest.
In addition, mTOR and S6K were also implicated in antagonizing TGF-β-mediated responses. Rapamycin reverses the inhibitory effect of Akt on TGF-β-mediated Smad3 phosphorylation and transcription response 87, while S6K was reported as a rate-limiting factor for TGF-β-induced growth arrest in EpH4 polarized mammary epithelial cells 92. In the latter scenario, TGF-β inhibits S6K activity by promoting its dephosphorylation. The inhibition of S6K by TGF-β requires expression of the Bα regulatory subunit of protein phosphatase 2A since S6K is not inhibited in cells with undetectable Bα expression 92. The Bα subunit interacts directly with activated TβRI 93. Since Bα suppresses the association of the catalytic C and regulatory A subunits of protein phosphatase 2A 94, the Bα interaction with the receptor is expected to result in enhanced protein phosphatase 2A activity. Although these observations strongly suggest a role for mTOR and S6K in antagonizing the response to TGF-β, their precise role and associated mechanisms need further exploration.
Future perspective and conclusion
In the past few years, considerable progress has been made toward understanding the signaling networks and downstream pathways of TGF-β receptors. It is now clear that the co-operation between Smad and non-Smad signaling pathways determines the final outcome of cellular response to TGF-β. It will not be surprising if more non-Smad signaling pathways are discovered in the future. In an automated high-throughput interaction mapping, more than 100 proteins associated with the TGF-β receptor complex were identified 71. Many of these proteins are either themselves signaling molecules or are signaling adaptors. Interestingly, many of these interactions are dynamically dependent on the activation state of the receptor complex. Further validation and characterization of these receptor-interacting proteins will undoubtedly reveal novel molecular mechanisms that contribute to both Smad-dependent and -independent TGF-β signaling and advance our understanding of the ability of TGF-β to induce a plethora of diverse biological responses. In addition, a major challenge now is to determine how addition of TGF-β to a given cell can direct cell-surface, membrane-bound receptors to recruit and activate multiple downstream effectors, and how the specificity in downstream signaling is achieved. The combination of in vivo imaging of TGF-β receptors, their regulators and their effectors, loss-of-function analysis using RNA interference and knockout mouse models, proteomics and phospho-proteomics will be powerful tools in answering these questions. Finally, as we continue to advance our understanding of the signaling network of TGF-β and how subtle perturbation of this network can result in pathological situations, we should also consider practical and clinical approaches for pharmacological intervention of different molecular targets in this signaling network for treatment of a variety of diseases.
References
- Derynck R, Miyazono K . TGF-beta and the TGF-beta family. In: Derynck R, Miyazono K, eds. The TGF-beta Family. NewYork: Cold Spring Harbor Laboratory Press, 2008:29–43.
- Massagué J, Blain SW, Lo RS . TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 2000; 103:295–309.
Article PubMed Google Scholar - Derynck R, Akhurst RJ . Differentiation plasticity regulated by TGF-beta family proteins in development and disease. Nat Cell Biol 2007; 9:1000–1004.
Article CAS PubMed Google Scholar - Massagué J . TGFbeta in cancer. Cell 2008; 134:215–230.
Article CAS PubMed PubMed Central Google Scholar - Derynck R, Zhang Y . Intracellular signalling: the mad way to do it. Curr Biol 1996; 6:1226–1229.
Article CAS PubMed Google Scholar - Massagué J, Seoane J, Wotton D . Smad transcription factors. Genes Dev 2005; 19:2783–2810.
Article CAS PubMed Google Scholar - Feng XH, Derynck R . Specificity and versatility in TGF-beta signaling through Smads. Annu Rev Cell Dev Biol 2005; 21:659–693.
Article CAS PubMed Google Scholar - Schmierer B, Hill CS . TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol 2007; 8:970–982.
Article CAS PubMed Google Scholar - Derynck R, Zhang YE . Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003; 425:577–584.
Article CAS PubMed Google Scholar - Moustakas A, Heldin CH . Non-Smad TGF-beta signals. J Cell Sci 2005; 118:3573–3584.
Article CAS PubMed Google Scholar - Mulder KM, Morris SL . Activation of p21ras by transforming growth factor beta in epithelial cells. J Biol Chem 1992; 267:5029–5031.
CAS PubMed Google Scholar - Yan Z, Winawer S, Friedman E . Two different signal transduction pathways can be activated by transforming growth factor beta 1 in epithelial cells. J Biol Chem 1994; 269:13231–13237.
CAS PubMed Google Scholar - Hartsough MT, Mulder KM . Transforming growth factor beta activation of p44mapk in proliferating cultures of epithelial cells. J Biol Chem 1995; 270:7117–7124.
Article CAS PubMed Google Scholar - Frey RS, Mulder KM . TGFbeta regulation of mitogen-activated protein kinases in human breast cancer cells. Cancer Lett 1997; 117:41–50.
Article CAS PubMed Google Scholar - Mucsi I, Skorecki KL, Goldberg HJ . Extracellular signal-regulated kinase and the small GTP-binding protein, Rac, contribute to the effects of transforming growth factor-beta1 on gene expression. J Biol Chem 1996; 271:16567–16572.
Article CAS PubMed Google Scholar - Simeone DM, Zhang L, Graziano K, et al. Smad4 mediates activation of mitogen-activated protein kinases by TGF-beta in pancreatic acinar cells. Am J Physiol Cell Physiol 2001; 281:C311–C319.
Article CAS PubMed Google Scholar - Olsson N, Piek E, Sundstrom M, ten Dijke P, Nilsson G . Transforming growth factor-beta-mediated mast cell migration depends on mitogen-activated protein kinase activity. Cell Signal 2001; 13:483–490.
Article PubMed Google Scholar - Schlessinger J . Cell signaling by receptor tyrosine kinases. Cell 2000; 103:211–225.
Article CAS PubMed Google Scholar - McKay MM, Morrison DK . Integrating signals from RTKs to ERK/MAPK. Oncogene 2007; 26:3113–3121.
Article CAS PubMed Google Scholar - Ravichandran KS . Signaling via Shc family adapter proteins. Oncogene 2001; 20:6322–6330.
Article CAS PubMed Google Scholar - Lawler S, Feng XH, Chen RH, et al. The type II transforming growth factor-beta receptor autophosphorylates not only on serine and threonine but also on tyrosine residues. J Biol Chem 1997; 272:14850–14859.
Article CAS PubMed Google Scholar - Galliher AJ, Schiemann WP . Src phosphorylates Tyr284 in TGF-beta type II receptor and regulates TGF-beta stimulation of p38 MAPK during breast cancer cell proliferation and invasion. Cancer Res 2007; 67:3752–3758.
Article CAS PubMed Google Scholar - Lee MK, Pardoux C, Hall MC, et al. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J 2007; 26:3957–3967.
Article CAS PubMed PubMed Central Google Scholar - Thiery JP . Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol 2003; 15:740–746.
Article CAS PubMed Google Scholar - Lee JM, Dedhar S, Kalluri R, Thompson EW . The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol 2006; 172:973–981.
Article CAS PubMed PubMed Central Google Scholar - Zavadil J, Bitzer M, Liang D, et al. Genetic programs of epithelial cell plasticity directed by transforming growth factor-beta. Proc Natl Acad Sci USA 2001; 98:6686–6691.
Article CAS PubMed PubMed Central Google Scholar - Davies M, Robinson M, Smith E, et al. Induction of an epithelial to mesenchymal transition in human immortal and malignant keratinocytes by TGF-beta1 involves MAPK, Smad and AP-1 signalling pathways. J Cell Biochem 2005; 95:918–931.
Article CAS PubMed Google Scholar - Galliher-Beckley AJ, Schiemann WP . Grb2 binding to Tyr284 in TbetaR-II is essential for mammary tumor growth and metastasis stimulated by TGF-beta. Carcinogenesis 2008; 29:244–251.
Article CAS PubMed Google Scholar - Northey JJ, Chmielecki J, Ngan E, et al. Signaling through ShcA is required for transforming growth factor beta- and Neu/ErbB-2-induced breast cancer cell motility and invasion. Mol Cell Biol 2008; 28:3162–3176.
Article CAS PubMed PubMed Central Google Scholar - Kretzschmar M, Doody J, Massagué J . Opposing BMP and EGF signalling pathways converge on the TGF-beta family mediator Smad1. Nature 1997; 389:618–622.
Article CAS PubMed Google Scholar - Kretzschmar M, Doody J, Timokhina I, Massagué J . A mechanism of repression of TGFbeta/ Smad signaling by oncogenic Ras. Genes Dev 1999; 13:804–816.
Article CAS PubMed PubMed Central Google Scholar - Funaba M, Zimmerman CM, Mathews LS . Modulation of Smad2-mediated signaling by extracellular signal-regulated kinase. J Biol Chem 2002; 277:41361–41368.
Article CAS PubMed Google Scholar - Matsuura I, Wang G, He D, Liu F . Identification and characterization of ERK MAP kinase phosphorylation sites in Smad3. Biochemistry 2005; 44:12546–12553.
Article CAS PubMed Google Scholar - Zhang Y, Feng XH, Derynck R . Smad3 and Smad4 cooperate with c-Jun/c-Fos to mediate TGF-beta-induced transcription. Nature 1998; 394:909–913.
Article CAS PubMed Google Scholar - Hall MC, Young DA, Waters JG, et al. The comparative role of activator protein 1 and Smad factors in the regulation of Timp-1 and MMP-1 gene expression by transforming growth factor-beta 1. J Biol Chem 2003; 278:10304–10313.
Article CAS PubMed Google Scholar - Weston CR, Davis RJ . The JNK signal transduction pathway. Curr Opin Cell Biol 2007; 19:142–149.
Article CAS PubMed Google Scholar - Frey RS, Mulder KM . Involvement of extracellular signal-regulated kinase 2 and stress-activated protein kinase/Jun N-terminal kinase activation by transforming growth factor beta in the negative growth control of breast cancer cells. Cancer Res 1997; 57:628–633.
CAS PubMed Google Scholar - Engel ME, McDonnell MA, Law BK, Moses HL . Interdependent SMAD and JNK signaling in transforming growth factor-beta-mediated transcription. J Biol Chem 1999; 274:37413–37420.
Article CAS PubMed Google Scholar - Hocevar BA, Brown TL, Howe PH . TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J 1999; 18:1345–1356.
Article CAS PubMed PubMed Central Google Scholar - Hanafusa H, Ninomiya-Tsuji J, Masuyama N, et al. Involvement of the p38 mitogen-activated protein kinase pathway in transforming growth factor-beta-induced gene expression. J Biol Chem 1999; 274:27161–27167.
Article CAS PubMed Google Scholar - Sano Y, Harada J, Tashiro S, et al. ATF-2 is a common nuclear target of Smad and TAK1 pathways in transforming growth factor-beta signaling. J Biol Chem 1999; 274:8949–8957.
Article CAS PubMed Google Scholar - Bhowmick NA, Zent R, Ghiassi M, McDonnell M, Moses HL . Integrin beta 1 signaling is necessary for transforming growth factor-beta activation of p38MAPK and epithelial plasticity. J Biol Chem 2001; 276:46707–46713.
Article CAS PubMed Google Scholar - Yu L, Hebert MC, Zhang YE . TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J 2002; 21:3749–3759.
Article CAS PubMed PubMed Central Google Scholar - Itoh S, Thorikay M, Kowanetz M, et al. Elucidation of Smad requirement in transforming growth factor-beta type I receptor-induced responses. J Biol Chem 2003; 278:3751–3761.
Article CAS PubMed Google Scholar - Yamaguchi K, Shirakabe K, Shibuya H, et al. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science 1995; 270:2008–2011.
Article CAS PubMed Google Scholar - Shibuya H, Iwata H, Masuyama N, et al. Role of TAK1 and TAB1 in BMP signaling in early Xenopus development. EMBO J 1998; 17:1019–1028.
Article CAS PubMed PubMed Central Google Scholar - Shim JH, Xiao C, Paschal AE, et al. TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev 2005; 19:2668–2681.
Article CAS PubMed PubMed Central Google Scholar - Jadrich JL, O'Connor MB, Coucouvanis E . The TGF beta activated kinase TAK1 regulates vascular development in vivo. Development 2006; 133:1529–1541.
Article CAS PubMed Google Scholar - Watkins SJ, Jonker L, Arthur HM . A direct interaction between TGFbeta activated kinase 1 and the TGFbeta type II receptor: implications for TGFbeta signalling and cardiac hypertrophy. Cardiovasc Res 2006; 69:432–439.
Article CAS PubMed Google Scholar - Sorrentino A, Thakur N, Grimsby S, et al. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat Cell Biol 2008; 10:1199–1207.
Article CAS PubMed Google Scholar - Yamashita M, Fatyol K, Jin C, et al. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol Cell 2008; 31:918–924.
Article CAS PubMed PubMed Central Google Scholar - Bradley JR, Pober JS . Tumor necrosis factor receptor-associated factors (TRAFs). Oncogene 2001; 20:6482–6491.
Article CAS PubMed Google Scholar - Haglund K, Dikic I . Ubiquitylation and cell signaling. EMBO J 2005; 24:3353–3359.
Article CAS PubMed PubMed Central Google Scholar - Wang C, Deng L, Hong M, et al. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001; 412:346–351.
Article CAS PubMed Google Scholar - Atfi A, Djelloul S, Chastre E, Davis R, Gespach C . Evidence for a role of Rho-like GTPases and stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) in transforming growth factor beta-mediated signaling. J Biol Chem 1997; 272:1429–1432.
Article CAS PubMed Google Scholar - Brown JD, DiChiara MR, Anderson KR, Gimbrone Jr MA, Topper JN . MEKK-1, a component of the stress (stress-activated protein kinase/c-Jun N-terminal kinase) pathway, can selectively activate Smad2-mediated transcriptional activation in endothelial cells. J Biol Chem 1999; 274:8797–8805.
Article CAS PubMed Google Scholar - Zhang L, Wang W, Hayashi Y, et al. A role for MEK kinase 1 in TGF-beta/activin-induced epithelium movement and embryonic eyelid closure. EMBO J 2003; 22:4443–4454.
Article CAS PubMed PubMed Central Google Scholar - Kim KY, Kim BC, Xu Z, Kim SJ . Mixed lineage kinase 3 (MLK3)-activated p38 MAP kinase mediates transforming growth factor-beta-induced apoptosis in hepatoma cells. J Biol Chem 2004; 279:29478–29484.
Article CAS PubMed Google Scholar - Kimura N, Matsuo R, Shibuya H, Nakashima K, Taga T . BMP2-induced apoptosis is mediated by activation of the TAK1-p38 kinase pathway that is negatively regulated by Smad6. J Biol Chem 2000; 275:17647–17652.
Article CAS PubMed Google Scholar - Edlund S, Bu S, Schuster N, et al. Transforming growth factor-beta1 (TGF-beta)-induced apoptosis of prostate cancer cells involves Smad7-dependent activation of p38 by TGF-beta-activated kinase 1 and mitogen-activated protein kinase kinase 3. Mol Biol Cell 2003; 14:529–544.
Article CAS PubMed PubMed Central Google Scholar - Liao JH, Chen JS, Chai MQ, Zhao S, Song JG . The involvement of p38 MAPK in transforming growth factor beta1-induced apoptosis in murine hepatocytes. Cell Res 2001; 11:89–94.
Article CAS PubMed Google Scholar - Bakin AV, Rinehart C, Tomlinson AK, Arteaga CL . p38 mitogen-activated protein kinase is required for TGFbeta-mediated fibroblastic transdifferentiation and cell migration. J Cell Sci 2002; 115:3193–3206.
CAS PubMed Google Scholar - Jaffe AB, Hall A . Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol 2005; 21:247–269.
Article CAS PubMed Google Scholar - Bhowmick NA, Ghiassi M, Bakin A, et al. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell 2001; 12:27–36.
Article CAS PubMed PubMed Central Google Scholar - Edlund S, Landstrom M, Heldin CH, Aspenstrom P . Transforming growth factor-beta-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol Biol Cell 2002; 13:902–914.
Article CAS PubMed PubMed Central Google Scholar - Shen X, Li J, Hu PP, et al. The activity of guanine exchange factor NET1 is essential for transforming growth factor-beta-mediated stress fiber formation. J Biol Chem 2001; 276:15362–15368.
Article CAS PubMed Google Scholar - Ozdamar B, Bose R, Barrios-Rodiles M, et al. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science 2005; 307:1603–1609.
Article CAS PubMed Google Scholar - Gao L, Joberty G, Macara IG . Assembly of epithelial tight junctions is negatively regulated by Par6. Curr Biol 2002; 12:221–225.
Article CAS PubMed Google Scholar - Wang HR, Zhang Y, Ozdamar B, et al. Regulation of cell polarity and protrusion formation by targeting RhoA for degradation. Science 2003; 302:1775–1779.
Article CAS PubMed Google Scholar - Wilkes MC, Murphy SJ, Garamszegi N, Leof EB . Cell-type-specific activation of PAK2 by transforming growth factor beta independent of Smad2 and Smad3. Mol Cell Biol 2003; 23:8878–8889.
Article CAS PubMed PubMed Central Google Scholar - Barrios-Rodiles M, Brown KR, Ozdamar B, et al. High-throughput mapping of a dynamic signaling network in mammalian cells. Science 2005; 307:1621–1625.
Article CAS PubMed Google Scholar - Foletta VC, Lim MA, Soosairajah J, et al. Direct signaling by the BMP type II receptor via the cytoskeletal regulator LIMK1. J Cell Biol 2003; 162:1089–1098.
Article CAS PubMed PubMed Central Google Scholar - Lee-Hoeflich ST, Causing CG, Podkowa M, et al. Activation of LIMK1 by binding to the BMP receptor, BMPRII, regulates BMP-dependent dendritogenesis. EMBO J 2004; 23:4792–4801.
Article CAS PubMed PubMed Central Google Scholar - Edlund S, Landstrom M, Heldin CH, Aspenstrom P . Smad7 is required for TGF-beta-induced activation of the small GTPase Cdc42. J Cell Sci 2004; 117:1835–1847.
Article CAS PubMed Google Scholar - Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL . Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem 2000; 275:36803–36810.
Article CAS PubMed Google Scholar - Shin I, Bakin AV, Rodeck U, Brunet A, Arteaga CL . Transforming growth factor beta enhances epithelial cell survival via Akt-dependent regulation of FKHRL1. Mol Biol Cell 2001; 12:3328–3339.
Article CAS PubMed PubMed Central Google Scholar - Vinals F, Pouyssegur J . Transforming growth factor beta1 (TGF-beta1) promotes endothelial cell survival during in vitro angiogenesis via an autocrine mechanism implicating TGF-alpha signaling. Mol Cell Biol 2001; 21:7218–7230.
Article CAS PubMed PubMed Central Google Scholar - Wilkes MC, Mitchell H, Penheiter SG, et al. Transforming growth factor-beta activation of phosphatidylinositol 3-kinase is independent of Smad2 and Smad3 and regulates fibroblast responses via p21-activated kinase-2. Cancer Res 2005; 65:10431–10440.
Article CAS PubMed Google Scholar - Lamouille S, Derynck R . Cell size and invasion in TGF-beta-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol 2007; 178:437–451.
Article CAS PubMed PubMed Central Google Scholar - Yi JY, Shin I, Arteaga CL . Type I transforming growth factor beta receptor binds to and activates phosphatidylinositol 3-kinase. J Biol Chem 2005; 280:10870–10876.
Article CAS PubMed Google Scholar - Valderrama-Carvajal H, Cocolakis E, Lacerte A, et al. Activin/TGF-beta induce apoptosis through Smad-dependent expression of the lipid phosphatase SHIP. Nat Cell Biol 2002; 4:963–969.
Article CAS PubMed Google Scholar - Tsukazaki T, Chiang TA, Davison AF, Attisano L, Wrana JL . SARA, a FYVE domain protein that recruits Smad2 to the TGFbeta receptor. Cell 1998; 95:779–791.
Article CAS PubMed Google Scholar - Hayes S, Chawla A, Corvera S . TGF beta receptor internalization into EEA1-enriched early endosomes: role in signaling to Smad2. J Cell Biol 2002; 158:1239–1249.
Article CAS PubMed PubMed Central Google Scholar - Daniels CE, Wilkes MC, Edens M, et al. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J Clin Invest 2004; 114:1308–1316.
Article CAS PubMed PubMed Central Google Scholar - Wang S, Wilkes MC, Leof EB, Hirschberg R . Imatinib mesylate blocks a non-Smad TGF-beta pathway and reduces renal fibrogenesis in vivo. FASEB J 2005; 19:1–11.
Article CAS PubMed Google Scholar - Chen RH, Su YH, Chuang RL, Chang TY . Suppression of transforming growth factor-beta-induced apoptosis through a phosphatidylinositol 3-kinase/Akt-dependent pathway. Oncogene 1998; 17:1959–1968.
Article CAS PubMed Google Scholar - Song K, Wang H, Krebs TL, Danielpour D . Novel roles of Akt and mTOR in suppressing TGF-beta/ALK5-mediated Smad3 activation. EMBO J 2006; 25:58–69.
Article CAS PubMed Google Scholar - Conery AR, Cao Y, Thompson EA, et al. Akt interacts directly with Smad3 to regulate the sensitivity to TGF-beta induced apoptosis. Nat Cell Biol 2004; 6:366–372.
Article CAS PubMed Google Scholar - Remy I, Montmarquette A, Michnick SW . PKB/Akt modulates TGF-beta signalling through a direct interaction with Smad3. Nat Cell Biol 2004; 6:358–365.
Article CAS PubMed Google Scholar - Seoane J, Le HV, Shen L, Anderson SA, Massagué J . Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 2004; 117:211–223.
Article CAS PubMed Google Scholar - Gomis RR, Alarcon C, He W, et al. A FoxO-Smad synexpression group in human keratinocytes. Proc Natl Acad Sci USA 2006; 103:12747–12752.
Article CAS PubMed PubMed Central Google Scholar - Petritsch C, Beug H, Balmain A, Oft M . TGF-beta inhibits p70 S6 kinase via protein phosphatase 2A to induce G(1) arrest. Genes Dev 2000; 14:3093–3101.
Article CAS PubMed PubMed Central Google Scholar - Griswold-Prenner I, Kamibayashi C, Maruoka EM, Mumby MC, Derynck R . Physical and functional interactions between type I transforming growth factor beta receptors and Balpha, a WD-40 repeat subunit of phosphatase 2A. Mol Cell Biol 1998; 18:6595–6604.
Article CAS PubMed PubMed Central Google Scholar - Janssens V, Goris J . Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J 2001; 353:417–439.
Article CAS PubMed PubMed Central Google Scholar
Acknowledgements
The research in Ying E Zhang's group is supported by the Intramural Research Program of the NIH, USA, National Cancer Institute, Center for Cancer Research.
Author information
Authors and Affiliations
- Laboratory of Cellular and Molecular Biology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, 20892, MD, USA
Ying E Zhang
Corresponding author
Correspondence toYing E Zhang.
Rights and permissions
About this article
Cite this article
Zhang, Y. Non-Smad pathways in TGF-β signaling.Cell Res 19, 128–139 (2009). https://doi.org/10.1038/cr.2008.328
- Published: 30 December 2008
- Issue Date: January 2009
- DOI: https://doi.org/10.1038/cr.2008.328