Comprehensive molecular profiling of lung adenocarcinoma (original) (raw)
Main
Lung cancer is the most common cause of global cancer-related mortality, leading to over a million deaths each year and adenocarcinoma is its most common histological type. Smoking is the major cause of lung adenocarcinoma but, as smoking rates decrease, proportionally more cases occur in never-smokers (defined as less than 100 cigarettes in a lifetime). Recently, molecularly targeted therapies have dramatically improved treatment for patients whose tumours harbour somatically activated oncogenes such as mutant EGFR1 or translocated ALK, RET, or ROS1 (refs 2, 3, 4). Mutant BRAF and ERBB2 (ref. 5) are also investigational targets. However, most lung adenocarcinomas either lack an identifiable driver oncogene, or harbour mutations in KRAS and are therefore still treated with conventional chemotherapy. Tumour suppressor gene abnormalities, such as those in TP53 (ref. 6), STK11 (ref. 7), CDKN2A8, KEAP1 (ref. 9), and SMARCA4 (ref. 10) are also common but are not currently clinically actionable. Finally, lung adenocarcinoma shows high rates of somatic mutation and genomic rearrangement, challenging identification of all but the most frequent driver gene alterations because of a large burden of passenger events per tumour genome11,12,13. Our efforts focused on comprehensive, multiplatform analysis of lung adenocarcinoma, with attention towards pathobiology and clinically actionable events.
Clinical samples and histopathologic data
We analysed tumour and matched normal material from 230 previously untreated lung adenocarcinoma patients who provided informed consent (Supplementary Table 1). All major histologic types of lung adenocarcinoma were represented: 5% lepidic, 33% acinar, 9% papillary, 14% micropapillary, 25% solid, 4% invasive mucinous, 0.4% colloid and 8% unclassifiable adenocarcinoma (Supplementary Fig. 1)14. Median follow-up was 19 months, and 163 patients were alive at the time of last follow-up. Eighty-one percent of patients reported past or present smoking. Supplementary Table 2 summarizes demographics. DNA, RNA and protein were extracted from specimens and quality-control assessments were performed as described previously15. Supplementary Table 3 summarizes molecular estimates of tumour cellularity16.
Somatically acquired DNA alterations
We performed whole-exome sequencing (WES) on tumour and germline DNA, with a mean coverage of 97.6× and 95.8×, respectively, as performed previously17. The mean somatic mutation rate across the TCGA cohort was 8.87 mutations per megabase (Mb) of DNA (range: 0.5–48, median: 5.78). The non-synonymous mutation rate was 6.86 per Mb. MutSig2CV18 identified significantly mutated genes among our 230 cases along with 182 similarly-sequenced, previously reported lung adenocarcinomas12. Analysis of these 412 tumour/normal pairs highlighted 18 statistically significant mutated genes (Fig. 1a shows co-mutation plot of TCGA samples (n = 230), Supplementary Fig. 2 shows co-mutation plot of all samples used in the statistical analysis (n = 412) and Supplementary Table 4 contains complete MutSig2CV results, which also appear on the TCGA Data Portal along with many associated data files (https://tcga-data.nci.nih.gov/docs/publications/luad_2014/). TP53 was commonly mutated (46%). Mutations in KRAS (33%) were mutually exclusive with those in EGFR (14%). BRAF was also commonly mutated (10%), as were PIK3CA (7%), MET (7%) and the small GTPase gene, RIT1 (2%). Mutations in tumour suppressor genes including STK11 (17%), KEAP1 (17%), NF1 (11%), RB1 (4%) and CDKN2A (4%) were observed. Mutations in chromatin modifying genes SETD2 (9%), ARID1A (7%) and SMARCA4 (6%) and the RNA splicing genes RBM10 (8%) and U2AF1 (3%) were also common. Recurrent mutations in the MGA gene (which encodes a Max-interacting protein on the MYC pathway19) occurred in 8% of samples. Loss-of-function (frameshift and nonsense) mutations in MGA were mutually exclusive with focal MYC amplification (Fisher’s exact test P = 0.04), suggesting a hitherto unappreciated potential mechanism of MYC pathway activation. Coding single nucleotide variants and indel variants were verified by resequencing at a rate of 99% and 100%, respectively (Supplementary Fig. 3a, Supplementary Table 5). Tumour purity was not associated with the presence of false negatives identified in the validation data (P = 0.31; Supplementary Fig. 3b).
Figure 1: Somatic mutations in lung adenocarcinoma.
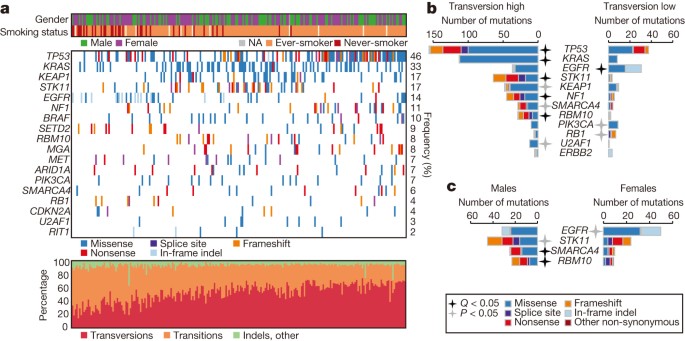
a, Co-mutation plot from whole exome sequencing of 230 lung adenocarcinomas. Data from TCGA samples were combined with previously published data12 for statistical analysis. Co-mutation plot for all samples used in the statistical analysis (n = 412) can be found in Supplementary Fig. 2. Significant genes with a corrected P value less than 0.025 were identified using the MutSig2CV algorithm and are ranked in order of decreasing prevalence. b, c, The differential patterns of mutation between samples classified as transversion high and transversion low samples (b) or male and female patients (c) are shown for all samples used in the statistical analysis (n = 412). Stars indicate statistical significance using the Fisher’s exact test (black stars: q < 0.05, grey stars: P < 0.05) and are adjacent to the sample set with the higher percentage of mutated samples.
Past or present smoking associated with cytosine to adenine (C >A) nucleotide transversions as previously described both in individual genes and genome-wide12,13. C > A nucleotide transversion fraction showed two peaks; this fraction correlated with total mutation count (_R_2 = 0.30) and inversely correlated with cytosine to thymine (C > T) transition frequency (_R_2 = 0.75) (Supplementary Fig. 4). We classified each sample (Supplementary Methods) into one of two groups named transversion-high (TH, n = 269), and transversion-low (TL, n = 144). The transversion-high group was strongly associated with past or present smoking (P < 2.2 × 10−16), consistent with previous reports13. The transversion-high and transversion-low patient cohorts harboured different gene mutations. Whereas KRAS mutations were significantly enriched in the transversion-high cohort (P = 2.1 × 10−13), EGFR mutations were significantly enriched in the transversion-low group (P = 3.3 × 10−6). PIK3CA and RB1 mutations were likewise enriched in transversion-low tumours (P < 0.05). Additionally, the transversion-low tumours were specifically enriched for in-frame insertions in EGFR and ERBB2 (ref. 5) and for frameshift indels in RB1 (Fig. 1b). RB1 is commonly mutated in small-cell lung carcinoma (SCLC). We found RB1 mutations in transversion-low adenocarcinomas were enriched for frameshift indels versus single nucleotide substitutions compared to SCLC (P < 0.05)20,21 suggesting a mutational mechanism in transversion-low adenocarcinoma that is probably distinct from smoking in SCLC.
Gender is correlated with mutation patterns in lung adenocarcinoma22. Only a fraction of significantly mutated genes from the complete set reported in this study (Fig. 1a) were enriched in men or women (Fig. 1c). EGFR mutations were enriched in tumours from the female cohort (P = 0.03) whereas loss-of-function mutations within RBM10, an RNA-binding protein located on the X chromosome23 were enriched in tumours from men (P = 0.002). When examining the transversion-high group, 16 out of 21 RBM10 mutations were observed in males (P = 0.003, Fisher’s exact test).
Somatic copy number alterations were very similar to those previously reported for lung adenocarcinoma24 (Supplementary Fig. 5, Supplementary Table 6). Significant amplifications included NKX2-1, TERT, MDM2, KRAS, EGFR, MET, CCNE1, CCND1, TERC and MECOM (Supplementary Table 6), as previously described24, 8q24 near MYC, and a novel peak containing CCND3 (Supplementary Table 6). The CDKN2A locus was the most significant deletion (Supplementary Table 6). Supplementary Table 7 summarizes molecular and clinical characteristics by sample. Low-pass whole-genome sequencing on a subset (n = 93) of the samples revealed an average of 36 gene–gene and gene–inter-gene rearrangements per tumour. Chromothripsis25 occurred in six of the 93 samples (6%) (Supplementary Fig. 6, Supplementary Table 8). Low-pass whole genome sequencing-detected rearrangements appear in Supplementary Table 9.
Description of aberrant RNA transcripts
Gene fusions, splice site mutations or mutations in genes encoding splicing factors promote or sustain the malignant phenotype by generating aberrant RNA transcripts. Combining DNA with mRNA sequencing enabled us to catalogue aberrant RNA transcripts and, in many cases, to identify the DNA-encoded mechanism for the aberration. Seventy-five per cent of somatic mutations identified by WES were present in the RNA transcriptome when the locus in question was expressed (minimum 5×) (Supplementary Fig. 7a) similar to prior analyses15. Previously identified fusions involving ALK (3/230 cases), ROS1 (4/230) and RET (2/230) (Fig. 2a, Supplementary Table 10), all occurred in transversion-low tumours (P = 1.85 × 10−4, Fisher’s exact test).
Figure 2: Aberrant RNA transcripts in lung adenocarcinoma associated with somatic DNA translocation or mutation.
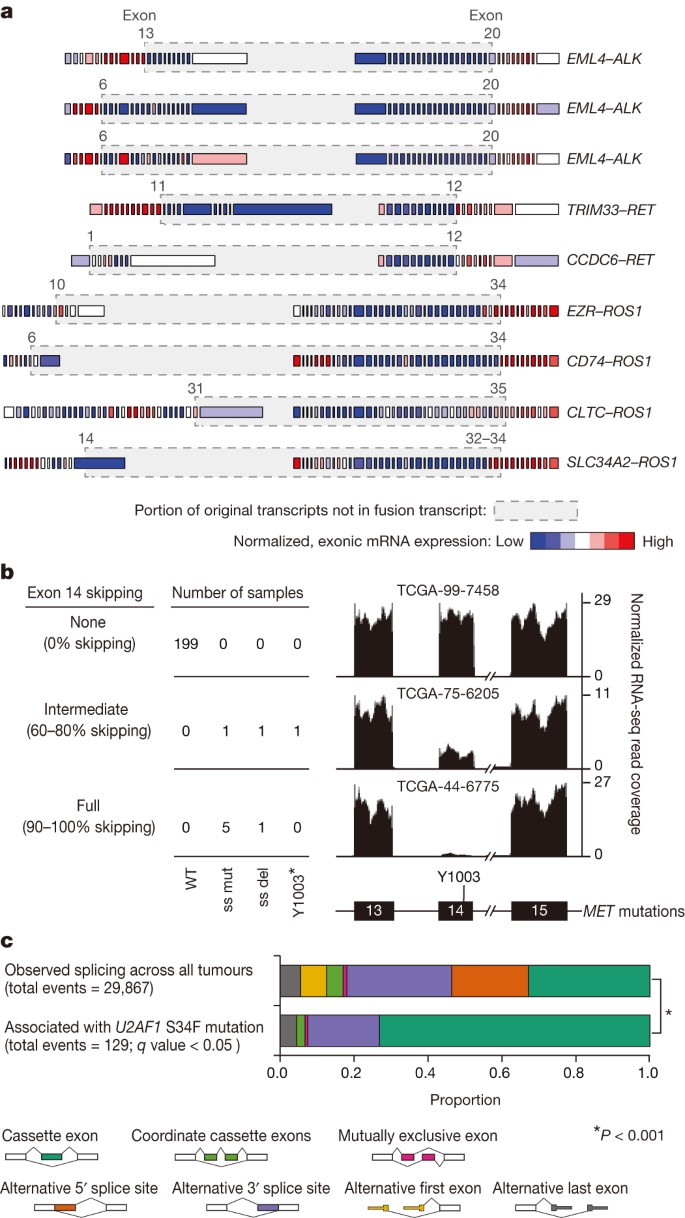
a, Normalized exon level RNA expression across fusion gene partners. Grey boxes around genes mark the regions that are removed as a consequence of the fusion. Junction points of the fusion events are also listed in Supplementary Table 9. Exon numbers refer to reference transcripts listed in Supplementary Table 9. b, MET exon 14 skipping observed in the presence of exon 14 splice site mutation (ss mut), splice site deletion (ss del) or a Y1003* mutation. A total of 22 samples had insufficient coverage around exon 14 for quantification. The percentage skipping is (total expression minus exon 14 expression)/total expression. c, Significant differences in the frequency of 129 alternative splicing events in mRNA from tumours with U2AF1 S34F tumours compared to U2AF1 WT tumours (q value <0.05). Consistent with the function of U2AF1 in 3′ splice site recognition, most splicing differences involved cassette exon and alternative 3′ splice site events (chi-squared test, P < 0.001).
MET activation can occur by exon 14 skipping, which results in a stabilized protein26. Ten tumours had somatic MET DNA alterations with MET exon 14 skipping in RNA. In nine of these samples, a 5′ or 3′ splice site mutation or deletion was identified27. MET exon 14 skipping was also found in the setting of a MET Y1003* stop codon mutation (Fig. 2b, Supplementary Fig. 8a). The codon affected by the Y1003* mutation is predicted to disrupt multiple splicing enhancer sequences, but the mechanism of skipping remains unknown in this case.
S34F mutations in U2AF1 have recently been reported in lung adenocarcinoma12 but their contribution to oncogenesis remains unknown. Eight samples harboured _U2AF1_S34F. We identified 129 splicing events strongly associated with _U2AF1_S34F mutation, consistent with the role of U2AF1 in 3′-splice site selection28. Cassette exons and alternative 3′ splice sites were most commonly affected (Fig. 2c, Supplementary Table 11)29. Among these events, alternative splicing of the CTNNB1 proto-oncogene was strongly associated with U2AF1 mutations (Supplementary Fig. 8b). Thus, concurrent analysis of DNA and RNA enabled delineation of both cis and trans mechanisms governing RNA processing in lung adenocarcinoma.
Candidate driver genes
The receptor tyrosine kinase (RTK)/RAS/RAF pathway is frequently mutated in lung adenocarcinoma. Striking therapeutic responses are often achieved when mutant pathway components are successfully inhibited. Sixty-two per cent (143/230) of tumours harboured known activating mutations in known driver oncogenes, as defined by others30. Cancer-associated mutations in KRAS (32%, n = 74), EGFR (11%, n = 26) and BRAF (7%, n = 16) were common. Additional, previously uncharacterized KRAS, EGFR and BRAF mutations were observed, but were not classified as driver oncogenes for the purposes of our analyses (see Supplementary Fig. 9a for depiction of all mutations of known and unknown significance); explaining the differing mutation frequencies in each gene between this analysis and the overall mutational analysis described above. We also identified known activating ERBB2 in-frame insertion and point mutations (n = 5)6, as well as mutations in MAP2K1 (n = 2), NRAS and HRAS (n = 1 each). RNA sequencing revealed the aforementioned MET exon 14 skipping (n = 10) and fusions involving ROS1 (n = 4), ALK (n = 3) and RET (n = 2). We considered these tumours collectively as oncogene-positive, as they harboured a known activating RTK/RAS/RAF pathway somatic event. DNA amplification events were not considered to be driver events before the comparisons described below.
We sought to nominate previously unrecognized genomic events that might activate this critical pathway in the 38% of samples without a RTK/RAS/RAF oncogene mutation. Tumour cellularity did not differ between oncogene-negative and oncogene-positive samples (Supplementary Fig. 9b). Analysis of copy number alterations using GISTIC31 identified unique focal ERBB2 and MET amplifications in the oncogene-negative subset (Fig. 3a, Supplementary Table 6); amplifications in other wild-type proto-oncogenes, including KRAS and EGFR, were not significantly different between the two groups.
Figure 3: Identification of novel candidate driver genes.
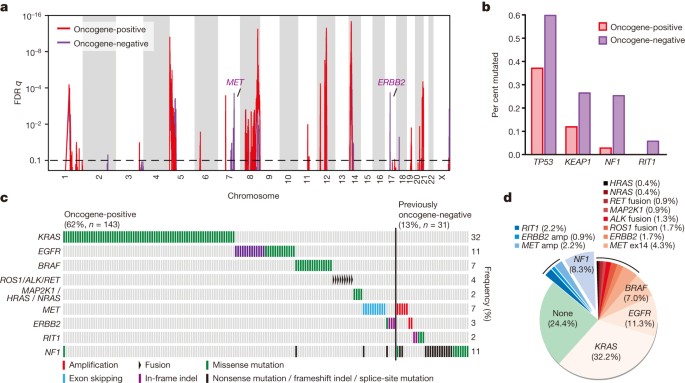
a, GISTIC analysis of focal amplifications in oncogene-negative (n = 87) and oncogene-positive (n = 143) TCGA samples identifies focal gains of MET and ERBB2 that are specific to the oncogene-negative set (purple). b, TP53, KEAP1, NF1 and RIT1 mutations are significantly enriched in samples otherwise lacking oncogene mutations (adjusted P < 0.05 by Fisher’s exact test). c, Co-mutation plot of variants of known significance within the RTK/RAS/RAF pathway in lung adenocarcinoma. Not shown are the 63 tumours lacking an identifiable driver lesion. Only canonical driver events, as defined in Supplementary Fig. 9, and proposed driver events, are shown; hence not every alteration found is displayed. d, New candidate driver oncogenes (blue: 13% of cases) and known somatically activated drivers events (red: 63%) that activate the RTK/RAS/RAF pathway can be found in the majority of the 230 lung adenocarcinomas.
We next analysed WES data independently in the oncogene-negative and oncogene-positive subsets. We found that TP53, KEAP1, NF1 and RIT1 mutations were significantly enriched in oncogene-negative tumours (P < 0.01; Fig. 3b, Supplementary Table 12). NF1 mutations have previously been reported in lung adenocarcinoma11, but this is the first study, to our knowledge, capable of identifying all classes of loss-of-function NF1 defects and to statistically demonstrate that NF1 mutations, as well as KEAP1 and TP53 mutations are enriched in the oncogene-negative subset of lung adenocarcinomas (Fig. 3c). All RIT1 mutations occurred in the oncogene-negative subset and clustered around residue Q79 (homologous to Q61 in the switch II region of RAS genes). These mutations transform NIH3T3 cells and activate MAPK and PI(3)K signalling[32](/articles/nature13385#ref-CR32 "Berger, A. H. et al. Oncogenic RIT1 mutations in lung adenocarcinoma. Oncogene http://dx.doi.org/10.1038/onc.2013.581
(2014)"), supporting a driver role for mutant _RIT1_ in 2% of lung adenocarcinomas. This analysis increases the rate at which putative somatic lung adenocarcinoma driver events can be identified within the RTK/RAS/RAF pathway to 76% ([Fig. 3d](/articles/nature13385#Fig3)).Recurrent alterations in key pathways
Recurrent aberrations in multiple key pathways and processes characterize lung adenocarcinoma (Fig. 4a). Among these were RTK/RAS/RAF pathway activation (76% of cases), PI(3)K-mTOR pathway activation (25%), p53 pathway alteration (63%), alteration of cell cycle regulators (64%, Supplementary Fig. 10), alteration of oxidative stress pathways (22%, Supplementary Fig. 11), and mutation of various chromatin and RNA splicing factors (49%).
Figure 4: Pathway alterations in lung adenocarcinoma.
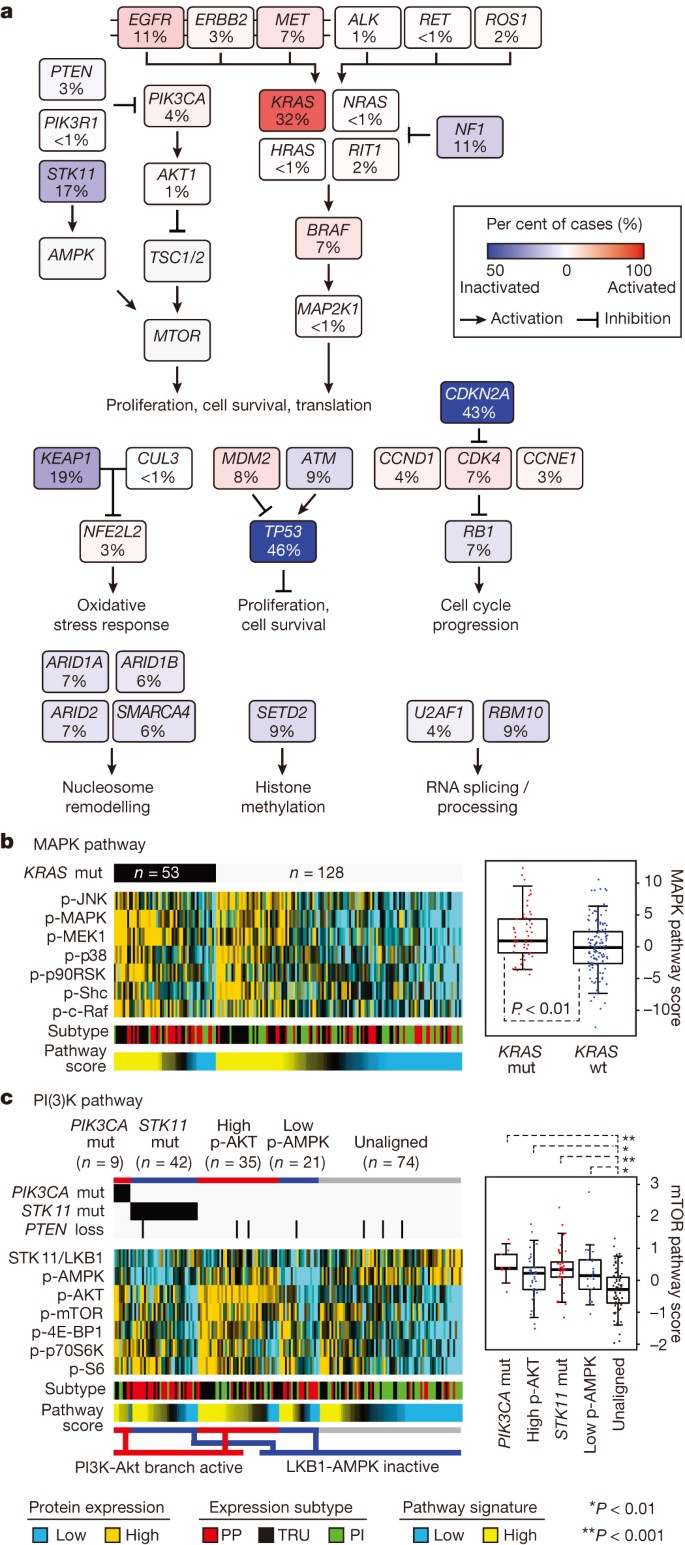
a, Somatic alterations involving key pathway components for RTK signalling, mTOR signalling, oxidative stress response, proliferation and cell cycle progression, nucleosome remodelling, histone methylation, and RNA splicing/processing. b, c, Proteomic analysis by RPPA (n = 181) P values by two-sided _t_-test. Box plots represent 5%, 25%, 75%, median, and 95%. PP, proximal proliferative; TRU, terminal respiratory unit; PI, proximal inflammatory. c, mTOR signalling may be activated, by either Akt (for example, via PI(3)K) or inactivation of AMPK (for example, via STK11 loss). Tumours were separated into three main groups: those with PI(3)K-AKT activation, through either PIK3CA activating mutation or unknown mechanism (high p-AKT); those with LKB1-AMPK inactivation, through either STK11 mutation or unknown mechanism with low levels of LKB1 and p-AMPK; and those showing none of the above features.
We then examined the phenotypic sequelae of some key genomic events in the tumours in which they occurred. Reverse-phase protein arrays provided proteomic and phosphoproteomic phenotypic evidence of pathway activity. Antibodies on this platform are listed in Supplementary Table 13. This analysis suggested that DNA sequencing did not identify all samples with phosphoprotein evidence of activation of a given signalling pathway. For example, whereas _KRAS_-mutant lung adenocarcinomas had higher levels of phosphorylated MAPK than KRAS wild-type tumours had on average, many KRAS wild-type tumours displayed significant MAPK pathway activation (Fig. 4b, Supplementary Fig. 10). The multiple mechanisms by which lung adenocarcinomas achieve MAPK activation suggest additional, still undetected RTK/RAS/RAF pathway alterations. Similarly, we found significant activation of mTOR and its effectors (p70S6kinase, S6, 4E-BP1) in a substantial fraction of the tumours (Fig. 4c). Analysis of mutations in PIK3CA and STK11, STK11 protein levels, and AMPK and AKT phosphorylation33 led to the identification of three major mTOR patterns in lung adenocarcinoma: (1) tumours with minimal or basal mTOR pathway activation, (2) tumours showing higher mTOR activity accompanied by either _STK11_-inactivating mutation or combined low STK11 expression and low AMPK activation and (3) tumours showing high mTOR activity accompanied by either phosphorylated AKT activation, PIK3CA mutation, or both. As with MAPK, many tumours lack an obvious underlying genomic alteration to explain their apparent mTOR activation.
Molecular subtypes of lung adenocarcinoma
Broad transcriptional and epigenetic profiling can reveal downstream consequences of driver mutations, provide clinically relevant classification and offer insight into tumours lacking clear drivers. Prior unsupervised analyses of lung adenocarcinoma gene expression have used varying nomenclature for transcriptional subtypes of the disease34,35,36,37. To coordinate naming of the transcriptional subtypes with the histopathological38, anatomic and mutational classifications of lung adenocarcinoma, we propose an updated nomenclature: the terminal respiratory unit (TRU, formerly bronchioid), the proximal-inflammatory (PI, formerly squamoid), and the proximal-proliferative (PP, formerly magnoid)39 transcriptional subtypes (Fig. 5a). Previously reported associations of expression signatures with pathways and clinical outcomes34,36,39 were observed (Supplementary Fig. 7b) and integration with multi-analyte data revealed statistically significant genomic alterations associated with these transcriptional subtypes. The PP subtype was enriched for mutation of KRAS, along with inactivation of the STK11 tumour suppressor gene by chromosomal loss, inactivating mutation, and reduced gene expression. In contrast, the PI subtype was characterized by solid histopathology and co-mutation of NF1 and TP53. Finally, the TRU subtype harboured the majority of the _EGFR_-mutated tumours as well as the kinase fusion expressing tumours. TRU subtype membership was prognostically favourable, as seen previously34 (Supplementary Fig. 7c). Finally, the subtypes exhibited different mutation rates, transition frequencies, genomic ploidy profiles, patterns of large-scale aberration, and differed in their association with smoking history (Fig. 5a). Unsupervised clustering of miRNA sequencing-derived or reverse phase protein array (RPPA)-derived data also revealed significant heterogeneity, partially overlapping with the mRNA-based subtypes, as demonstrated in Supplementary Figs 12 and 13.
Figure 5: Integrative analysis.
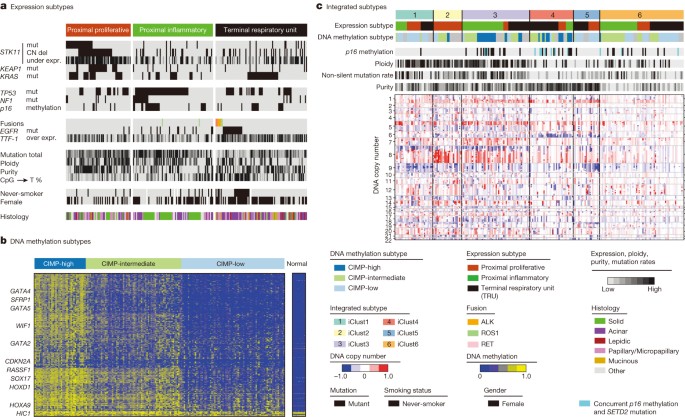
a–c, Integrating unsupervised analyses of 230 lung adenocarcinomas reveals significant interactions between molecular subtypes. Tumours are displayed as columns, grouped by mRNA expression subtypes (a), DNA methylation subtypes (b), and integrated subtypes by iCluster analysis (c). All displayed features are significantly associated with subtypes depicted. The CIMP phenotype is defined by the most variable CpG island and promoter probes.
Mutations in chromatin-modifying genes (for example, SMARCA4, ARID1A and SETD2) suggest a major role for chromatin maintenance in lung adenocarcinoma. To examine chromatin states in an unbiased manner, we selected the most variable DNA methylation-specific probes in CpG island promoter regions and clustered them by methylation intensity (Supplementary Table 14). This analysis divided samples into two distinct subsets: a significantly altered CpG island methylator phenotype-high (CIMP-H(igh)) cluster and a more normal-like CIMP-L(ow) group, with a third set of samples occupying an intermediate level of methylation at CIMP sites (Fig. 5b). Our results confirm a prior report40 and provide additional insights into this epigenetic program. CIMP-H tumours often showed DNA hypermethylation of several key genes: CDKN2A, GATA2, GATA4, GATA5, HIC1, HOXA9, HOXD13, RASSF1, SFRP1, SOX17 and WIF1 among others (Supplementary Fig. 14). WNT pathway genes are significantly over-represented in this list (P value = 0.0015) suggesting that this is a key pathway with an important driving role within this subtype. MYC overexpression was significantly associated with the CIMP-H phenotype as well (P = 0.003).
Although we did not find significant correlations between global DNA methylation patterns and individual mutations in chromatin remodelling genes, there was an intriguing association between SETD2 mutation and CDKN2A methylation. Tumours with low CDKN2A expression due to methylation (rather than due to mutation or deletion) had lower ploidy, fewer overall mutations (Fig. 5c) and were significantly enriched for SETD2 mutation, suggesting an important role for this chromatin-modifying gene in the development of certain tumours.
Integrative clustering41 of copy number, DNA methylation and mRNA expression data found six clusters (Fig. 5c). Tumour ploidy and mutation rate are higher in clusters 1–3 than in clusters 4–6. Clusters 1–3 frequently harbour TP53 mutations and are enriched for the two proximal transcriptional subtypes. Fisher’s combined probability tests revealed significant copy number associated gene expression changes on 3q in cluster one, 8q in cluster two, and chromosome 7 and 15q in cluster three (Supplementary Fig. 15). The low ploidy and low mutation rate clusters four and five contain many TRU samples, whereas tumours in cluster 6 have comparatively lower tumour cellularity, and few other distinguishing molecular features. Significant copy number-associated gene expression changes are observed on 6q in cluster four and 19p in cluster five. The CIMP-H tumours divided into a high ploidy, high mutation rate, proximal-inflammatory CIMP-H group (cluster 3) and a low ploidy, low mutation rate, TRU-associated CIMP-H group (cluster 4), suggesting that the CIMP phenotype in lung adenocarcinoma can occur in markedly different genomic and transcriptional contexts. Furthermore, cluster four is enriched for CDKN2A methylation and SETD2 mutations, suggesting an interaction between somatic mutation of SETD2 and deregulated chromatin maintenance in this subtype. Finally, cluster membership was significantly associated with mutations in TP53, EGFR and STK11 (Supplementary Fig. 15, Supplementary Table 6).
Conclusions
We assessed the mutation profiles, structural rearrangements, copy number alterations, DNA methylation, mRNA, miRNA and protein expression of 230 lung adenocarcinomas. In recent years, the treatment of lung adenocarcinoma has been advanced by the development of multiple therapies targeted against alterations in the RTK/RAS/RAF pathway. We nominate amplifications in MET and ERBB2 as well as mutations of NF1 and RIT1 as driver events specifically in otherwise oncogene-negative lung adenocarcinomas. This analysis increases the fraction of lung adenocarcinoma cases with somatic evidence of RTK/RAS/RAF activation from 62% to 76%. While all lung adenocarcinomas may activate this pathway by some mechanism, only a subset show tonic pathway activation at the protein level, suggesting both diversity between tumours with seemingly similar activating events and as yet undescribed mechanisms of pathway activation. Therefore, the current study expands the range of possible targetable alterations within the RTK/RAS/RAF pathway in general and suggests increased implementation of MET and ERBB2/HER2 inhibitors in particular. Our discovery of inactivating mutations of MGA further underscores the importance of the MYC pathway in lung adenocarcinoma.
This study further implicates both chromatin modifications and splicing alterations in lung adenocarcinoma through the integration of DNA, transcriptome and methylome analysis. We identified alternative splicing due to both splicing factor mutations in trans and mutation of splice sites in cis, the latter leading to activation of the MET gene by exon 14 skipping. Cluster analysis separated tumours based on single-gene driver events as well as large-scale aberrations, emphasizing lung adenocarcinoma’s molecular heterogeneity and combinatorial alterations, including the identification of coincident SETD2 mutations and CDKN2A methylation in a subset of CIMP-H tumours, providing evidence of a somatic event associated with a genome-wide methylation phenotype. These studies provide new knowledge by illuminating modes of genomic alteration, highlighting previously unappreciated altered genes, and enabling further refinement in sub-classification for the improved personalization of treatment for this deadly disease.
Methods Summary
All specimens were obtained from patients with appropriate consent from the relevant institutional review board. DNA and RNA were collected from samples using the Allprep kit (Qiagen). We used standard approaches for capture and sequencing of exomes from tumour DNA and normal DNA15 and whole-genome shotgun sequencing. Significantly mutated genes were identified by comparing them with expectation models based on the exact measured rates of specific sequence lesions42. GISTIC analysis of the circular-binary-segmented Affymetrix SNP 6.0 copy number data was used to identify recurrent amplification and deletion peaks31. Consensus clustering approaches were used to analyse mRNA, miRNA and methylation subtypes using previous approaches15. The publication web page is (https://tcga-data.nci.nih.gov/docs/publications/luad_2014/). Sequence files are in CGHub (https://cghub.ucsc.edu/).
Change history
20 June 2018
In Supplementary Table 7 of this Article, the iCLUSTER output column contained incorrect cluster labels. This table has been corrected online. Corrected online 08 October 2014 - The surname of author Kristen Rodgers was misspelled 'Rogers'. This has been corrected online
References
- Paez, J. G. et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304, 1497–1500 (2004)
Article ADS CAS PubMed Google Scholar - Kwak, E. L. et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 363, 1693–1703 (2010)
Article CAS PubMed PubMed Central Google Scholar - Bergethon, K. et al. ROS1 rearrangements define a unique molecular class of lung cancers. J. Clin Oncol. 30, 863–870 (2012)
Article CAS PubMed PubMed Central Google Scholar - Drilon, A. et al. Response to cabozantinib in patients with RET fusion-positive lung adenocarcinomas. Cancer Discov. 3, 630–635 (2013)
Article CAS PubMed PubMed Central Google Scholar - Stephens, P. et al. Lung cancer: intragenic ERBB2 kinase mutations in tumours. Nature 431, 525–526 (2004)
Article ADS CAS PubMed Google Scholar - Takahashi, T. et al. p53: a frequent target for genetic abnormalities in lung cancer. Science 246, 491–494 (1989)
Article ADS CAS PubMed Google Scholar - Sanchez-Cespedes, M. et al. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 62, 3659–3662 (2002)
CAS PubMed Google Scholar - Shapiro, G. I. et al. Reciprocal Rb inactivation and p16INK4 expression in primary lung cancers and cell lines. Cancer Res. 55, 505–509 (1995)
CAS PubMed Google Scholar - Singh, A. et al. Dysfunctional KEAP1–NRF2 interaction in non-small-cell lung cancer. PLoS Med. 3, e420 (2006)
Article PubMed PubMed Central CAS Google Scholar - Medina, P. P. et al. Frequent _BRG1/SMARCA4_-inactivating mutations in human lung cancer cell lines. Hum. Mutat. 29, 617–622 (2008)
Article CAS PubMed Google Scholar - Ding, L. et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455, 1069–1075 (2008)
Article ADS CAS PubMed PubMed Central Google Scholar - Imielinski, M. et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150, 1107–1120 (2012)
Article CAS PubMed PubMed Central Google Scholar - Govindan, R. et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 150, 1121–1134 (2012)
Article CAS PubMed PubMed Central Google Scholar - Travis, W. D., Brambilla, E. & Riely, G. J. New pathologic classification of lung cancer: relevance for clinical practice and clinical trials. J. Clin. Oncol. 31, 992–1001 (2013)
Article CAS PubMed Google Scholar - The Cancer Genome Atlas Research Network Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525 (2012)
Article ADS PubMed Central CAS Google Scholar - Carter, S. L. et al. Absolute quantification of somatic DNA alterations in human cancer. Nature Biotechnol. 30, 413–421 (2012)
Article CAS Google Scholar - Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature Biotechnol. 31, 213–219 (2013)
Article CAS Google Scholar - Lawrence, M. S. et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501 (2014)
Article ADS CAS PubMed PubMed Central Google Scholar - Hurlin, P. J., Steingrimsson, E., Copeland, N. G., Jenkins, N. A. & Eisenman, R. N. Mga, a dual-specificity transcription factor that interacts with Max and contains a T-domain DNA-binding motif. EMBO J. 18, 7019–7028 (1999)
Article CAS PubMed PubMed Central Google Scholar - Peifer, M. et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nature Genet. 44, 1104–1110 (2012)
Article CAS PubMed Google Scholar - Rudin, C. M. et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nature Genet. 44, 1111–1116 (2012)
Article CAS PubMed Google Scholar - Tokumo, M. et al. The relationship between epidermal growth factor receptor mutations and clinicopathologic features in non-small cell lung cancers. Clin. Cancer Res. 11, 1167–1173 (2005)
CAS PubMed Google Scholar - Coleman, M. P. et al. A novel gene, DXS8237E, lies within 20 kb upstream of UBE1 in Xp11.23 and has a different X inactivation status. Genomics 31, 135–138 (1996)
Article CAS PubMed Google Scholar - Weir, B. A. et al. Characterizing the cancer genome in lung adenocarcinoma. Nature 450, 893–898 (2007)
Article ADS CAS PubMed PubMed Central Google Scholar - Stephens, P. J. et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40 (2011)
Article MathSciNet CAS PubMed PubMed Central Google Scholar - Kong-Beltran, M. et al. Somatic mutations lead to an oncogenic deletion of Met in lung cancer. Cancer Res. 66, 283–289 (2006)
Article CAS PubMed Google Scholar - Seo, J. S. et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 22, 2109–2119 (2012)
Article CAS PubMed PubMed Central Google Scholar - Wu, S., Romfo, C. M., Nilsen, T. W. & Green, M. R. Functional recognition of the 3′ splice site AG by the splicing factor U2AF35. Nature 402, 832–835 (1999)
Article ADS CAS PubMed Google Scholar - Brooks, A. N. et al. A pan-cancer analysis of transcriptome changes associated with somatic mutations in U2AF1 reveals commonly altered splicing events. PLoS ONE 9, e87361 (2014)
Article ADS PubMed PubMed Central CAS Google Scholar - Pao, W. & Hutchinson, K. E. Chipping away at the lung cancer genome. Nature Med. 18, 349–351 (2012)
Article CAS PubMed Google Scholar - Beroukhim, R. et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc. Natl Acad. Sci. USA 104, 20007–20012 (2007)
Article ADS CAS PubMed PubMed Central Google Scholar - Berger, A. H. et al. Oncogenic RIT1 mutations in lung adenocarcinoma. Oncogene http://dx.doi.org/10.1038/onc.2013.581 (2014)
- Creighton, C. J. et al. Proteomic and transcriptomic profiling reveals a link between the PI3K pathway and lower estrogen-receptor (ER) levels and activity in ER+ breast cancer. Breast Cancer Res. 12, R40 (2010)
Article PubMed PubMed Central CAS Google Scholar - Wilkerson, M. D. et al. Differential pathogenesis of lung adenocarcinoma subtypes involving sequence mutations, copy number, chromosomal instability, and methylation. PLoS ONE 7, e36530 (2012)
Article ADS CAS PubMed PubMed Central Google Scholar - Beer, D. G. et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nature Med. 8, 816–824 (2002)
Article CAS PubMed Google Scholar - Hayes, D. N. et al. Gene expression profiling reveals reproducible human lung adenocarcinoma subtypes in multiple independent patient cohorts. J. Clin. Oncol. 24, 5079–5090 (2006)
Article CAS PubMed Google Scholar - Bhattacharjee, A. et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc. Natl Acad. Sci. USA 98, 13790–13795 (2001)
Article ADS CAS PubMed PubMed Central Google Scholar - Travis, W. D. et al. International association for the study of lung cancer/American Thoracic Society/European Respiratory Society international multidisciplinary classification of lung adenocarcinoma. J. Thoracic Oncol. 6, 244–285 (2011)
Article Google Scholar - Yatabe, Y., Mitsudomi, T. & Takahashi, T. TTF-1 expression in pulmonary adenocarcinomas. Am. J. Surg. Pathol. 26, 767–773 (2002)
Article PubMed Google Scholar - Shinjo, K. et al. Integrated analysis of genetic and epigenetic alterations reveals CpG island methylator phenotype associated with distinct clinical characters of lung adenocarcinoma. Carcinogenesis 33, 1277–1285 (2012)
Article CAS PubMed Google Scholar - Mo, Q. et al. Pattern discovery and cancer gene identification in integrated cancer genomic data. Proc. Natl Acad. Sci. USA 110, 4245–4250 (2013)
Article ADS CAS PubMed PubMed Central Google Scholar - Lawrence, M. S. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 (2013)
Article ADS CAS PubMed PubMed Central Google Scholar
Acknowledgements
This study was supported by NIH grants: U24 CA126561, U24 CA126551, U24 CA126554, U24 CA126543, U24 CA126546, U24 CA137153, U24 CA126563, U24 CA126544, U24 CA143845, U24 CA143858, U24 CA144025, U24 CA143882, U24 CA143866, U24 CA143867, U24 CA143848, U24 CA143840, U24 CA143835, U24 CA143799, U24 CA143883, U24 CA143843, U54 HG003067, U54 HG003079 and U54 HG003273. We thank K. Guebert and L. Gaffney for assistance and C. Gunter for review.
Author information
Authors and Affiliations
- University of California San Francisco, San Francisco, 94158, California, USA
Eric A. Collisson & Barry S. Taylor - The Eli and Edythe L. Broad Institute, Cambridge, 02142, Massachusetts, USA
Joshua D. Campbell, Angela N. Brooks, Alice H. Berger, Juliann Chmielecki, Gad Getz, Peter S. Hammerman, Bryan Hernandez, Carrie Sougnez, Andrew D. Cherniack, Mara Rosenberg, Matthew Meyerson, Mara Rosenberg, Stacey B. Gabriel, Kristian Cibulskis, Carrie Sougnez, Jaegil Kim, Chip Stewart, Lee Lichtenstein, Eric S. Lander, Michael S. Lawrence, Gad Getz, Joshua D. Campbell, Angela N. Brooks, Juliann Chmielecki, Marcin Imielinski, Robert C. Onofrio, Travis Zack, Carrie Sougnez, Elena Helman, Chandra Sekhar Pedamallu, Jill Mesirov, Andrew D. Cherniack, Gordon Saksena, Steven E. Schumacher, Scott L. Carter, Bryan Hernandez, Levi Garraway, Rameen Beroukhim, Stacey B. Gabriel, Gad Getz, Matthew Meyerson, Mara Rosenberg, Juok Cho, Daniel DiCara, David Heiman, Pei Lin, William Mallard, Douglas Voet, Hailei Zhang, Lihua Zou, Michael S. Noble, Michael S. Lawrence, Gordon Saksena, Nils Gehlenborg, Helga Thorvaldsdottir, Jill Mesirov, Marc-Danie Nazaire, Jim Robinson & Gad Getz - Dana Farber Cancer Institute, Boston, 02115, Massachusetts, USA
Angela N. Brooks, Matthew Meyerson, Angela N. Brooks, Levi Garraway, Rameen Beroukhim & Matthew Meyerson - Memorial Sloan-Kettering Cancer Center, New York, 10065, New York, USA
William Lee, Marc Ladanyi, Nikolaus Schultz, Ronglai Shen, William D. Travis, William Lee, B. Arman Aksoy, Giovanni Ciriello, Gideon Dresdner, Jianjiong Gao, Benjamin Gross, Venkatraman E. Seshan, Marc Ladanyi, Boris Reva, Rileen Sinha, S. Onur Sumer, Nils Weinhold, Nikolaus Schultz, Ronglai Shen, Chris Sander, William D. Travis, Natasha Rekhtman, Maureen Zakowski, Valerie W. Rusch, William D. Travis & Natasha Rekhtman - University of Michigan, Ann Arbor, Michigan 48109, USA.,
David G. Beer - Johns Hopkins University, Baltimore, 21287, Maryland, USA
Leslie Cope, Ludmila Danilova, James G. Herman, Stephen B. Baylin, Leslie Cope, Ludmila Danilova, Stephen B. Baylin, Peter Illei, Edward Gabrielson, James Shin, Beverly Lee, Kristen Rodgers, Dante Trusty, Malcolm V. Brock & Peter Illei - Baylor College of Medicine, Houston, 77030, Texas, USA
Chad J. Creighton, Chad J. Creighton & David Wheeler - Washington University, St. Louis, 63108, Missouri, USA
Li Ding, Ramaswamy Govindan, Cyriac Kandoth, Robert Fulton, Lucinda L. Fulton, Michael D. McLellan, Richard K. Wilson, Kai Ye, Catrina C. Fronick, Christopher A. Maher, Christopher A. Miller, Michael C. Wendl, Christopher Cabanski, Li Ding, Elaine Mardis, Ramaswamy Govindan, Mark A. Watson, Sandra McDonald & Bryan Meyers - Harvard Medical School, Boston, 02115, Massachusetts, USA
Gad Getz, Raju Kucherlapati, Angela Hadjipanayis, Gad Getz, Marcin Imielinski, Eran Hodis, Levi Garraway, Rameen Beroukhim, Gad Getz, Matthew Meyerson, Angela Hadjipanayis, Semin Lee, Angeliki Pantazi, Xiaojia Ren, Lixing Yang, Peng-Chieh Chen, Michael Parfenov, Andrew Wei Xu, Netty Santoso, Peter J. Park, Raju Kucherlapati & Gad Getz - Massachusetts General Hospital, Boston, 02114, Massachusetts, USA
Gad Getz, Gad Getz, Marcin Imielinski, Gad Getz & Gad Getz - University of North Carolina at Chapel Hill, Chapel Hill, 27599, North Carolina, USA
D. Neil Hayes, Matthew D. Wilkerson, Katherine A. Hoadley, J. Todd Auman, Shaowu Meng, Yan Shi, Elizabeth Buda, Scot Waring, Umadevi Veluvolu, Donghui Tan, Piotr A. Mieczkowski, Corbin D. Jones, Janae V. Simons, Matthew G. Soloway, Tom Bodenheimer, Stuart R. Jefferys, Jeffrey Roach, Alan P. Hoyle, Junyuan Wu, Saianand Balu, Darshan Singh, Jan F. Prins, J.S. Marron, Joel S. Parker, D. Neil Hayes, Charles M. Perou, Matthew D. Wilkerson, Joel S. Parker, Katherine A. Hoadley, Patrick K. Kimes, D. Neil Hayes, Charles M. Perou, Mei Huang, Leigh B. Thorne, Lori Boice, Ashley Hill Salazar, William K. Funkhouser, W. Kimryn Rathmell & William K. Funkhouser - University of Texas MD Anderson Cancer Center, Houston, 77054, Texas, USA
John V. Heymach, Rileen Sinha, John N. Weinstein, Lauren Averett Byers, Robert A. Holt, Harshad S. Mahadeshwar, Alexei Protopopov, Sahil Seth, Xingzhi Song, Jiabin Tang, Jianhua Zhang, Lynda Chin, Lynda Chin, Jianhua Zhang, Bradley M. Broom, Jing Wang, Yiling Lu, Patrick Kwok Shing Ng, Lixia Diao, Lauren Averett Byers, Wenbin Liu, John V. Heymach, Christopher I. Amos, John N. Weinstein, Rehan Akbani, Gordon B. Mills & Richard Hajek - Princess Margaret Cancer Centre, Toronto, Ontario M5G 2M9, Canada.,
Igor Jurisica, Ming-Sound Tsao & Ming-Sound Tsao - Brigham and Women’s Hospital Boston, 02115, Massachusetts, USA
David Kwiatkowski, Angela Hadjipanayis, Semin Lee, Angeliki Pantazi, Michael Parfenov, Andrew Wei Xu, Netty Santoso, Peter J. Park, Raju Kucherlapati & William G. Richards - BC Cancer Agency, Vancouver, British Columbia V5Z 4S6, Canada.,
Gordon Robertson, Andy Chu, Miruna Balasundaram, Yaron S. N. Butterfield, Rebecca Carlsen, Andy Chu, Eric Chuah, Noreen Dhalla, Ranabir Guin, Carrie Hirst, Darlene Lee, Haiyan I. Li, Michael Mayo, Richard A. Moore, Andrew J. Mungall, Jacqueline E. Schein, Payal Sipahimalani, Angela Tam, Richard Varhol, A. Gordon Robertson, Natasja Wye, Nina Thiessen, Steven J. M. Jones & Marco A. Marra - Mayo Clinic, Rochester, 55905, Minnesota, USA
Dennis A. Wigle, Dennis A. Wigle, Michael K. Asiedu & Farhad Kosari - University of Southern California, Los Angeles, 90033, California, USA
Daniel J. Weisenberger, Peter W. Laird, Daniel J. Weisenberger, Dennis T. Maglinte, Philip H. Lai, Moiz S. Bootwalla, David J. Van Den Berg, Timothy Triche Jr & Peter W. Laird - University of California Santa Cruz, Santa Cruz, 95064, California, USA
Amie Radenbaugh, Singer Ma, Joshua M. Stuart, Sam Ng, Singer Ma, Jingchun Zhu, Amie Radenbaugh, Joshua M. Stuart & David Haussler - Massachusetts Institute of Technology, Cambridge, 02142, Massachusetts, USA
Eric S. Lander - University of Kentucky, Lexington, 40515, Kentucky, USA
Jinze Liu - Buck Institute for Age Research, Novato, 94945, California, USA
Christopher C. Benz & Christina Yau - Howard Hughes Medical Institute, University of California Santa Cruz, Santa Cruz, 95064, California, USA
David Haussler - Oregon Health and Science University, Portland, 97239, Oregon, USA
Paul T. Spellman - International Genomics Consortium, Phoenix, 85004, Arizona, USA
Erin Curley, Joseph Paulauskis, Kevin Lau, Scott Morris, Troy Shelton, David Mallery, Johanna Gardner & Robert Penny - Analytical Biological Services, Inc., Wilmington, 19801, Delaware, USA
Charles Saller & Katherine Tarvin - University of Alabama at Birmingham, Birmingham, 35294, Alabama, USA
Robert Cerfolio & Ayesha Bryant - Cleveland Clinic, Cleveland, 44195, Ohio, USA
Daniel P. Raymond, Nathan A. Pennell, Carol Farver & Carol Farver - Christiana Care, Newark, 19713, Delaware, USA
Christine Czerwinski, Lori Huelsenbeck-Dill, Mary Iacocca, Nicholas Petrelli, Brenda Rabeno, Jennifer Brown & Thomas Bauer - Cureline, Inc., South San Francisco, 94080, California, USA
Oleg Dolzhanskiy, Olga Potapova, Daniil Rotin, Olga Voronina, Elena Nemirovich-Danchenko & Konstantin V. Fedosenko - Emory University, Atlanta, 30322, Georgia, USA
Anthony Gal, Madhusmita Behera, Suresh S. Ramalingam, Gabriel Sica & Gabriel Sica - Fox Chase Cancer Center, Philadelphia, 19111, Philadelphia, USA
Douglas Flieder, Jeff Boyd & JoEllen Weaver - ILSbio, Chestertown, 21620, Maryland, USA
Bernard Kohl & Dang Huy Quoc Thinh - Indiana University School of Medicine, Indianapolis, 46202, Indiana, USA
George Sandusky - Individumed, Silver Spring, 20910, Maryland, USA
Hartmut Juhl - The Prince Charles Hospital and the University of Queensland Thoracic Research Center, Brisbane, 4032, Australia.,
Edwina Duhig, Belinda Clarke, Ian A. Yang, Kwun M. Fong, Lindy Hunter, Morgan Windsor, Rayleen V. Bowman, Belinda Clarke & Edwina Duhig - Sullivan Nicolaides Pathology & John Flynn Hospital, Tugun 4680, Australia.,
Edwina Duhig & Edwina Duhig - Lahey Hospital and Medical Center, Burlington, 01805, Massachusetts, USA
Christina Williamson, Eric Burks, Kimberly Rieger-Christ, Antonia Holway & Travis Sullivan - NYU Langone Medical Center, New York, 10016, New York, USA
Paul Zippile, James Suh, Harvey Pass, Chandra Goparaju, Yvonne Owusu-Sarpong & James Suh - Ontario Tumour Bank, Ontario Institute for Cancer Research, Toronto, Ontario M5G 0A3, Canada.,
John M. S. Bartlett, Sugy Kodeeswaran, Jeremy Parfitt, Harmanjatinder Sekhon & Monique Albert - Penrose St. Francis Health Services, Colorado Springs, 80907, Colorado, USA
John Eckman & Jerome B. Myers - Roswell Park Cancer Center, Buffalo, 14263, New York, USA
Richard Cheney, Carl Morrison, Carmelo Gaudioso & Richard Cheney - Rush University Medical Center, Chicago, 60612, Illinois, USA
Jeffrey A. Borgia, Philip Bonomi, Mark Pool & Michael J. Liptay - St. Petersburg Academic University, St Petersburg 199034, Russia.,
Fedor Moiseenko & Irina Zaytseva - Thoraxklinik am Universitätsklinikum Heidelberg, 69126 Heidelberg, Germany.,
Hendrik Dienemann, Michael Meister & Thomas R. Muley - University Heidelberg, 69120 Heidelberg, Germany.,
Philipp A. Schnabel - University of Cologne, 50931 Cologne, Germany.,
Martin Peifer - University of Miami, Sylvester Comprehensive Cancer Center, Miami, 33136, Florida, USA
Carmen Gomez-Fernandez, Lynn Herbert & Sophie Egea - University of Pittsburgh, Pittsburgh, 15213, Pennsylvania, USA
Rajiv Dhir, Samuel A. Yousem, Sanja Dacic, Frank Schneider, Jill M. Siegfried & Sanja Dacic - Center Hospitalier Universitaire Vaudois, Lausanne and European Thoracic Oncology Platform, CH-1011 Lausanne, Switzerland.,
Solange Peters & Igor Letovanec - Ziauddin University Hospital, Karachi, 75300, Pakistan.,
Khurram Z. Khan - SRA International, Inc., Fairfax, 22033, Virginia, USA
Mark A. Jensen, Eric E. Snyder, Deepak Srinivasan, Ari B. Kahn, Julien Baboud & David A. Pot - National Cancer Institute, National Institutes of Health, Bethesda, 20892, Maryland, USA
Kenna R. Mills Shaw, Margi Sheth, Tanja Davidsen, John A. Demchok, Liming Yang, Zhining Wang, Roy Tarnuzzer & Jean Claude Zenklusen - National Human Genome Research Institute, National Institutes of Health, Bethesda, 20892, Maryland, USA
Bradley A. Ozenberger & Heidi J. Sofia
Consortia
The Cancer Genome Atlas Research Network
Disease analysis working group
- Eric A. Collisson
- , Joshua D. Campbell
- , Angela N. Brooks
- , Alice H. Berger
- , William Lee
- , Juliann Chmielecki
- , David G. Beer
- , Leslie Cope
- , Chad J. Creighton
- , Ludmila Danilova
- , Li Ding
- , Gad Getz
- , Peter S. Hammerman
- , D. Neil Hayes
- , Bryan Hernandez
- , James G. Herman
- , John V. Heymach
- , Igor Jurisica
- , Raju Kucherlapati
- , David Kwiatkowski
- , Marc Ladanyi
- , Gordon Robertson
- , Nikolaus Schultz
- , Ronglai Shen
- , Rileen Sinha
- , Carrie Sougnez
- , Ming-Sound Tsao
- , William D. Travis
- , John N. Weinstein
- , Dennis A. Wigle
- , Matthew D. Wilkerson
- , Andy Chu
- , Andrew D. Cherniack
- , Angela Hadjipanayis
- , Mara Rosenberg
- , Daniel J. Weisenberger
- , Peter W. Laird
- , Amie Radenbaugh
- , Singer Ma
- , Joshua M. Stuart
- , Lauren Averett Byers
- , Stephen B. Baylin
- , Ramaswamy Govindan
- & Matthew Meyerson
Genome sequencing centres: The Eli & Edythe L. Broad Institute
- Mara Rosenberg
- , Stacey B. Gabriel
- , Kristian Cibulskis
- , Carrie Sougnez
- , Jaegil Kim
- , Chip Stewart
- , Lee Lichtenstein
- , Eric S. Lander
- , Michael S. Lawrence
- & Gad Getz
Washington University in St. Louis
- Cyriac Kandoth
- , Robert Fulton
- , Lucinda L. Fulton
- , Michael D. McLellan
- , Richard K. Wilson
- , Kai Ye
- , Catrina C. Fronick
- , Christopher A. Maher
- , Christopher A. Miller
- , Michael C. Wendl
- , Christopher Cabanski
- , Li Ding
- , Elaine Mardis
- & Ramaswamy Govindan
Baylor College of Medicine
- Chad J. Creighton
- & David Wheeler
Genome characterization centres: Canada’s Michael Smith Genome Sciences Centre, British Columbia Cancer Agency
- Miruna Balasundaram
- , Yaron S. N. Butterfield
- , Rebecca Carlsen
- , Andy Chu
- , Eric Chuah
- , Noreen Dhalla
- , Ranabir Guin
- , Carrie Hirst
- , Darlene Lee
- , Haiyan I. Li
- , Michael Mayo
- , Richard A. Moore
- , Andrew J. Mungall
- , Jacqueline E. Schein
- , Payal Sipahimalani
- , Angela Tam
- , Richard Varhol
- , A. Gordon Robertson
- , Natasja Wye
- , Nina Thiessen
- , Robert A. Holt
- , Steven J. M. Jones
- & Marco A. Marra
The Eli & Edythe L. Broad Institute
- Joshua D. Campbell
- , Angela N. Brooks
- , Juliann Chmielecki
- , Marcin Imielinski
- , Robert C. Onofrio
- , Eran Hodis
- , Travis Zack
- , Carrie Sougnez
- , Elena Helman
- , Chandra Sekhar Pedamallu
- , Jill Mesirov
- , Andrew D. Cherniack
- , Gordon Saksena
- , Steven E. Schumacher
- , Scott L. Carter
- , Bryan Hernandez
- , Levi Garraway
- , Rameen Beroukhim
- , Stacey B. Gabriel
- , Gad Getz
- & Matthew Meyerson
Harvard Medical School/Brigham & Women’s Hospital/MD Anderson Cancer Center
- Angela Hadjipanayis
- , Semin Lee
- , Harshad S. Mahadeshwar
- , Angeliki Pantazi
- , Alexei Protopopov
- , Xiaojia Ren
- , Sahil Seth
- , Xingzhi Song
- , Jiabin Tang
- , Lixing Yang
- , Jianhua Zhang
- , Peng-Chieh Chen
- , Michael Parfenov
- , Andrew Wei Xu
- , Netty Santoso
- , Lynda Chin
- , Peter J. Park
- & Raju Kucherlapati
University of North Carolina, Chapel Hill
- Katherine A. Hoadley
- , J. Todd Auman
- , Shaowu Meng
- , Yan Shi
- , Elizabeth Buda
- , Scot Waring
- , Umadevi Veluvolu
- , Donghui Tan
- , Piotr A. Mieczkowski
- , Corbin D. Jones
- , Janae V. Simons
- , Matthew G. Soloway
- , Tom Bodenheimer
- , Stuart R. Jefferys
- , Jeffrey Roach
- , Alan P. Hoyle
- , Junyuan Wu
- , Saianand Balu
- , Darshan Singh
- , Jan F. Prins
- , J.S. Marron
- , Joel S. Parker
- , D. Neil Hayes
- & Charles M. Perou
University of Kentucky
- Jinze Liu
The USC/JHU Epigenome Characterization Center
- Leslie Cope
- , Ludmila Danilova
- , Daniel J. Weisenberger
- , Dennis T. Maglinte
- , Philip H. Lai
- , Moiz S. Bootwalla
- , David J. Van Den Berg
- , Timothy Triche Jr
- , Stephen B. Baylin
- & Peter W. Laird
Genome data analysis centres: The Eli & Edythe L. Broad Institute
- Mara Rosenberg
- , Lynda Chin
- , Jianhua Zhang
- , Juok Cho
- , Daniel DiCara
- , David Heiman
- , Pei Lin
- , William Mallard
- , Douglas Voet
- , Hailei Zhang
- , Lihua Zou
- , Michael S. Noble
- , Michael S. Lawrence
- , Gordon Saksena
- , Nils Gehlenborg
- , Helga Thorvaldsdottir
- , Jill Mesirov
- , Marc-Danie Nazaire
- , Jim Robinson
- & Gad Getz
Memorial Sloan-Kettering Cancer Center
- William Lee
- , B. Arman Aksoy
- , Giovanni Ciriello
- , Barry S. Taylor
- , Gideon Dresdner
- , Jianjiong Gao
- , Benjamin Gross
- , Venkatraman E. Seshan
- , Marc Ladanyi
- , Boris Reva
- , Rileen Sinha
- , S. Onur Sumer
- , Nils Weinhold
- , Nikolaus Schultz
- , Ronglai Shen
- & Chris Sander
University of California, Santa Cruz/Buck Institute
- Sam Ng
- , Singer Ma
- , Jingchun Zhu
- , Amie Radenbaugh
- , Joshua M. Stuart
- , Christopher C. Benz
- , Christina Yau
- & David Haussler
Oregon Health & Sciences University
- Paul T. Spellman
University of North Carolina, Chapel Hill
- Matthew D. Wilkerson
- , Joel S. Parker
- , Katherine A. Hoadley
- , Patrick K. Kimes
- , D. Neil Hayes
- & Charles M. Perou
The University of Texas MD Anderson Cancer Center
- Bradley M. Broom
- , Jing Wang
- , Yiling Lu
- , Patrick Kwok Shing Ng
- , Lixia Diao
- , Lauren Averett Byers
- , Wenbin Liu
- , John V. Heymach
- , Christopher I. Amos
- , John N. Weinstein
- , Rehan Akbani
- & Gordon B. Mills
Biospecimen core resource: International Genomics Consortium
- Erin Curley
- , Joseph Paulauskis
- , Kevin Lau
- , Scott Morris
- , Troy Shelton
- , David Mallery
- , Johanna Gardner
- & Robert Penny
Tissue source sites: Analytical Biological Service, Inc.
- Charles Saller
- & Katherine Tarvin
Brigham & Women’s Hospital
- William G. Richards
University of Alabama at Birmingham
- Robert Cerfolio
- & Ayesha Bryant
Cleveland Clinic
- Daniel P. Raymond
- , Nathan A. Pennell
- & Carol Farver
Christiana Care
- Christine Czerwinski
- , Lori Huelsenbeck-Dill
- , Mary Iacocca
- , Nicholas Petrelli
- , Brenda Rabeno
- , Jennifer Brown
- & Thomas Bauer
Cureline
- Oleg Dolzhanskiy
- , Olga Potapova
- , Daniil Rotin
- , Olga Voronina
- , Elena Nemirovich-Danchenko
- & Konstantin V. Fedosenko
Emory University
- Anthony Gal
- , Madhusmita Behera
- , Suresh S. Ramalingam
- & Gabriel Sica
Fox Chase Cancer Center
- Douglas Flieder
- , Jeff Boyd
- & JoEllen Weaver
ILSbio
- Bernard Kohl
- & Dang Huy Quoc Thinh
Indiana University
- George Sandusky
Indivumed
- Hartmut Juhl
John Flynn Hospital
- Edwina Duhig
Johns Hopkins University
- Peter Illei
- , Edward Gabrielson
- , James Shin
- , Beverly Lee
- , Kristen Rodgers
- , Dante Trusty
- & Malcolm V. Brock
Lahey Hospital & Medical Center
- Christina Williamson
- , Eric Burks
- , Kimberly Rieger-Christ
- , Antonia Holway
- & Travis Sullivan
Mayo Clinic
- Dennis A. Wigle
- , Michael K. Asiedu
- & Farhad Kosari
Memorial Sloan-Kettering Cancer Center
- William D. Travis
- , Natasha Rekhtman
- , Maureen Zakowski
- & Valerie W. Rusch
NYU Langone Medical Center
- Paul Zippile
- , James Suh
- , Harvey Pass
- , Chandra Goparaju
- & Yvonne Owusu-Sarpong
Ontario Tumour Bank
- John M. S. Bartlett
- , Sugy Kodeeswaran
- , Jeremy Parfitt
- , Harmanjatinder Sekhon
- & Monique Albert
Penrose St. Francis Health Services
- John Eckman
- & Jerome B. Myers
Roswell Park Cancer Institute
- Richard Cheney
- , Carl Morrison
- & Carmelo Gaudioso
Rush University Medical Center
- Jeffrey A. Borgia
- , Philip Bonomi
- , Mark Pool
- & Michael J. Liptay
St. Petersburg Academic University
- Fedor Moiseenko
- & Irina Zaytseva
Thoraxklinik am Universitätsklinikum Heidelberg, Member of Biomaterial Bank Heidelberg (BMBH) & Biobank Platform of the German Centre for Lung Research (DZL)
- Hendrik Dienemann
- , Michael Meister
- , Philipp A. Schnabel
- & Thomas R. Muley
University of Cologne
- Martin Peifer
University of Miami
- Carmen Gomez-Fernandez
- , Lynn Herbert
- & Sophie Egea
University of North Carolina
- Mei Huang
- , Leigh B. Thorne
- , Lori Boice
- , Ashley Hill Salazar
- , William K. Funkhouser
- & W. Kimryn Rathmell
University of Pittsburgh
- Rajiv Dhir
- , Samuel A. Yousem
- , Sanja Dacic
- , Frank Schneider
- & Jill M. Siegfried
The University of Texas MD Anderson Cancer Center
- Richard Hajek
Washington University School of Medicine
- Mark A. Watson
- , Sandra McDonald
- & Bryan Meyers
Queensland Thoracic Research Center
- Belinda Clarke
- , Ian A. Yang
- , Kwun M. Fong
- , Lindy Hunter
- , Morgan Windsor
- & Rayleen V. Bowman
Center Hospitalier Universitaire Vaudois
- Solange Peters
- & Igor Letovanec
Ziauddin University Hospital
- Khurram Z. Khan
Data Coordination Centre
- Mark A. Jensen
- , Eric E. Snyder
- , Deepak Srinivasan
- , Ari B. Kahn
- , Julien Baboud
- & David A. Pot
Project team: National Cancer Institute
- Kenna R. Mills Shaw
- , Margi Sheth
- , Tanja Davidsen
- , John A. Demchok
- , Liming Yang
- , Zhining Wang
- , Roy Tarnuzzer
- & Jean Claude Zenklusen
National Human Genome Research Institute
- Bradley A. Ozenberger
- & Heidi J. Sofia
Expert pathology panel
- William D. Travis
- , Richard Cheney
- , Belinda Clarke
- , Sanja Dacic
- , Edwina Duhig
- , William K. Funkhouser
- , Peter Illei
- , Carol Farver
- , Natasha Rekhtman
- , Gabriel Sica
- , James Suh
- & Ming-Sound Tsao
Contributions
The Cancer Genome Atlas Research Network contributed collectively to this study. Biospecimens were provided by the tissue source sites and processed by the biospecimen core resource. Data generation and analyses were performed by the genome sequencing centres, cancer genome characterization centres and genome data analysis centres. All data were released through the data coordinating centre. The National Cancer Institute and National Human Genome Research Institute project teams coordinated project activities. We also acknowledge the following TCGA investigators who made substantial contributions to the project: E. A. Collisson (manuscript coordinator); J. D. Campbell, J. Chmielecki, (analysis coordinators); C. Sougnez (data coordinator); J. D. Campbell, M. Rosenberg, W. Lee, J. Chmielecki, M. Ladanyi, and G. Getz (DNA sequence analysis); M. D. Wilkerson, A. N. Brooks, and D. N. Hayes (mRNA sequence analysis); L. Danilova and L. Cope (DNA methylation analysis); A. D. Cherniack (copy number analysis); M. D. Wilkerson and A. Hadjipanayis (translocations); N. Schultz, W. Lee, E. A. Collisson, A. H. Berger, J. Chmielecki, C. J. Creighton, L. A. Byers and M. Ladanyi (pathway analysis); A. Chu and A. G. Robertson (miRNA sequence analysis); W. Travis and D. A. Wigle (pathology and clinical expertise); L. A. Byers and G. B. Mills (reverse phase protein arrays); S. B. Baylin, R. Govindan and M. Meyerson (project chairs).
Corresponding authors
Correspondence toMatthew Meyerson or Matthew Meyerson.
Ethics declarations
Competing interests
The author declare no competing financial interests.
Additional information
The primary and processed data used to generate the analyses presented here can be downloaded by registered users from The Cancer Genome Atlas at (https://tcga-data.nci.nih.gov/tcga/tcgaDownload.jsp). All of the primary sequence files are deposited in cgHub and all other data are deposited at the Data Coordinating Center (DCC) for public access (http://cancergenome.nih.gov/), (https://cghub.ucsc.edu/) and (https://tcga-data.nci.nih.gov/docs/publications/luad_2014/).
Supplementary information
Supplementary Information
This file contains Supplementary Methods and Results, Supplementary Figures 1-15 and additional references – see separate excel for Supplementary Tables 1-14). (PDF 12490 kb)
Supplementary Tables
This file contains Supplementary Tables 1-14 (XLSX 3072 kb)
PowerPoint slides
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported licence. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons licence, users will need to obtain permission from the licence holder to reproduce the material. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-sa/3.0/.
About this article
Cite this article
The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma.Nature 511, 543–550 (2014). https://doi.org/10.1038/nature13385
- Received: 11 June 2013
- Accepted: 22 April 2014
- Published: 09 July 2014
- Issue Date: 31 July 2014
- DOI: https://doi.org/10.1038/nature13385