Hypoxia promotes stem cell phenotypes and poor prognosis through epigenetic regulation of DICER (original) (raw)
Introduction
Cancer mortality is largely attributable to distant metastasis. The mechanisms underlying the metastatic process are complex and are part of a series of events that ultimately result in the formation of macroscopic metastasis in distant organs from cells with tumour initiating or ‘stem cell’ properties1. Acquisition of stem cell and metastatic traits that enable this process, and the conditions in tumours that stimulate it, are poorly understood. However, many recent studies indicate that some tumour cells are able to transition from an epithelial to mesenchymal (EMT) phenotype through a process similar to that which occurs in development. Acquisition of the mesenchymal phenotype is associated with both increased tumour-initiating properties and the ability to form metastases in experimental models. However, in tumours this process requires some degree of plasticity, as formation of a tumour at a secondary metastatic site requires transition back to the epithelial cell state (mesenchymal to epithelial transition).
Micro RNAs (miRNAs) are small regulatory RNAs that play an important role in normal development and in disease by regulating the expression of a vast number of target messenger RNAs2. miRNA biogenesis begins with transcription of long primary miRNAs (pri-miRNA) containing one or more hairpin structures that are processed by the nuclear endonuclease DROSHA, generating a 70-nucleotide stem loop known as the precursor miRNA (pre-miRNA). The pre-miRNA is exported to the cytoplasm by XPO5, and cleaved by DICER in a complex with TRBP2 to generate a ~22-nucleotide mature miRNA duplex. One strand is loaded into the RNA-induced silencing complex, which controls gene expression through sequence-specific interactions with target mRNAs causing their degradation or translational repression3. Several members of this miRNA biogenesis pathway have been identified as haplo-insufficient tumour suppressors, including DICER itself, XPO5 and TRBP2 (refs 4, 5, 6, 7, 8, 9, 10). Using a variety of mouse models, these studies indicate that partial suppression of miRNA biogenesis is sufficient to accelerate tumour development. Loss of one DICER allele in mouse models results in a reduction in overall levels of mature miRNA and increased lung and soft tissue sarcomas4,5. These studies extend earlier clinical findings, demonstrating that miRNA levels are frequently reduced in tumours6. It is not clear how a reduction in miRNA biogenesis promotes cancer and whether loss of one or more specific miRNAs underlies this effect. However, miRNA has been hypothesized to confer ‘robustness’ to biological processes including stabilizing differentiated cell states11. In patients, low levels of DICER in breast, ovarian and other cancers are associated with aggressive, invasive disease, distant recurrence and poor overall survival12,13,14. In several model systems, DICER repression has also been shown to stimulate metastasis7,10.
In addition to monoallelic loss in cancer5, several mechanisms have been described as potential regulators of DICER including the transcription factors MITF15 and Tap63 (ref. 10) and miR-103/107 (ref. 7). DICER expression has also been reported to be inhibited by hypoxia through an unknown mechanism16. Hypoxia is a common feature of tumours strongly associated with poor prognosis in multiple sites, including breast cancer17,18,19. Clinical studies show a strong association between hypoxia and distant metastasis or relapse19,20,21,22,23,24. Laboratory data support a direct role for hypoxia in driving metastasis, including in vivo studies with cell line-derived25,26, and more recently, patient-derived xenografts grown in the orthotopic site. Hypoxia has been suggested to promote stemness in both normal tissues and tumours27,28,29,30,31,32. However, the mechanisms driving this aggressive phenotype are poorly understood.
In this study we have identified a new mechanism linking hypoxia, reduced miRNA biogenesis and acquisition of phenotypes associated with poor outcome. We show that tumour hypoxia is associated with reduced DICER expression in large cohorts of breast cancer patients and identify an epigenetic mechanism that suppresses DICER transcription through inhibition of oxygen-dependent H3K27me3 demethylases KDM6A/B. In breast cancer, reduced expression of DICER leads to a selective decrease in processing of the miR-200 family and consequently to derepression of ZEB1 and activation of the EMT and associated stem cell phenotypes.
Results
Reduced DICER expression in hypoxic human breast cancers
Both experimental and clinical data have demonstrated a strong correlation between hypoxia and more aggressive disease, including phenotypes recently linked to DICER suppression, such as stemness33 and metastasis18,25. We therefore examined the association between DICER expression, DICER copy number and hypoxia in breast cancer. We stratified breast cancer patients from two data sets (METABRIC34 and The Cancer Genome Atlas (TCGA)35) having normal DICER copy number by the amount of hypoxia as determined using the validated Winter hypoxia signature36 (Fig. 1a and Supplementary Fig. 1a). The median RNA expression of 99 hypoxia associated genes in the Winter signature is an independent prognostic factor in head and neck squamous cell carcinoma and breast cancer series. In both data sets, patients with the largest hypoxic fraction exhibited the lowest DICER mRNA expression (Fig. 1a and Supplementary Fig. 1a). A significant inverse correlation between hypoxia and DICER expression was found for the TCGA, METABRIC, Harris and 14 out of 18 smaller breast cancer gene expression studies (Fig. 1b and Supplementary Table 1). A pooled data set, consisting of 19 studies with long-term clinical follow-up, also demonstrated a highly significant (_P_=5.80 × 10−13) inverse correlation between DICER and hypoxia. Interestingly, in both the pooled and METABRIC data sets, low levels of DICER and high levels of hypoxia were associated with poor outcome (Fig. 1c and Supplementary Figs 2 and 3). Notably, the reduction in DICER expression in the most hypoxic quartile was reduced to levels similar to that in the ~5% of tumours that had monoallelic loss of DICER. These data suggest that hypoxia is a key contributor to DICER expression and is responsible for reduced DICER levels in significantly more patients than genetic loss. We confirmed that hypoxia suppresses DICER at both the mRNA and protein level in a panel of breast cancer (MCF7, MDA-MB-468, MDA-MB-231, SUM149 and HCC1954), normal (MCF10A) or transformed (HMLER) mammary epithelial cell lines after exposure to oxygen levels commonly found in human tumours (<0.02 to 1.0% O2) (Fig. 1d–f and Supplementary Fig. 4). Hypoxic suppression varied from 26% to 74% at the mRNA level and 16% to 84% at the protein level over a period of 24–48 h of hypoxia. DICER repression in hypoxia was not associated with any particular breast cancer subtype in either the cell lines or in the breast cancer clinical data sets (Supplementary Figs 2 and 3, and Supplementary Table 1).
Figure 1: Impaired DICER expression in hypoxic human breast cancers.
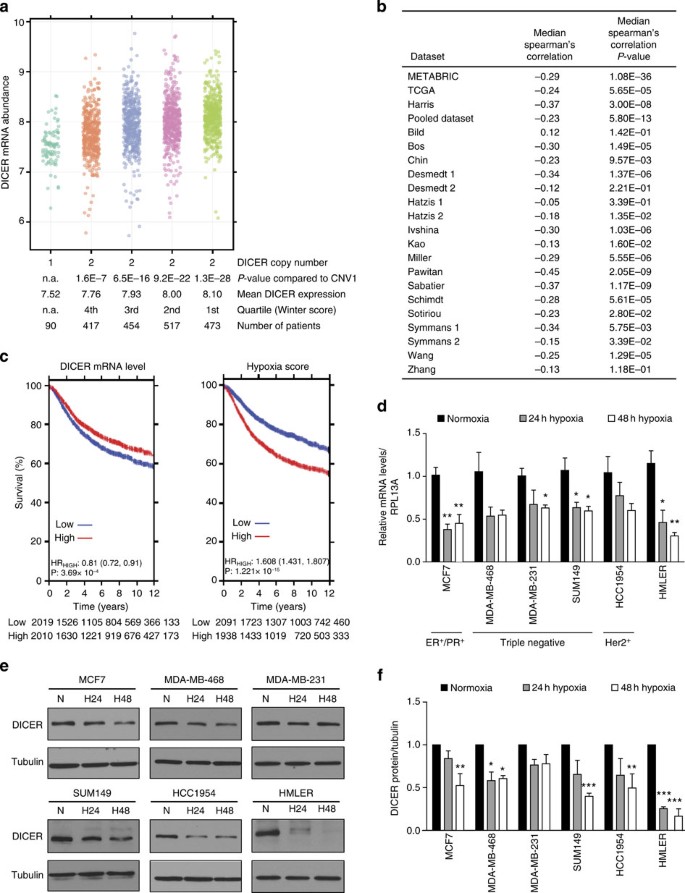
(a) DICER mRNA abundance in breast cancer patients from METABRIC stratified by DICER copy number and hypoxic fraction as determined with the Winter hypoxia metagene. _P_-values obtained with Wilcoxon rank sum test. (b) Inverse correlation between DICER mRNA abundance and hypoxia score. _P_-values obtained with Spearman’s correlation. (c) Kaplan–Meier survival curves for DICER and hypoxia in pooled breast cancer data set consisting of 19 studies. (d) RNA was extracted from indicated breast cancer cell lines exposed to 0.2% O2 for 24 or 48 h and subjected to quantitative reverse transcriptase–PCR analysis of DICER with RPL13A as control (_n_≥3). Data represent mean±s.e.m. _P_-values obtained with one-way analysis of variance (ANOVA), Bonferroni’s post-hoc test. (e) Representative western blottings were performed with antibody against DICER and anti-tubulin as control. (f) Densitometric analysis of western blottings in e where the intensity of the DICER bands were normalized for tubulin signal (_n_=3). Error bars represent s.e.m. _P_-values obtained with one-way ANOVA, Bonferroni’s post-hoc test. *P<0.05, **P<0.01, ***P<0.001.
In addition to monoallelic loss in cancer, several mechanisms have been implicated in DICER regulation. These include the transcription factors MITF15 and Tap63 (ref. 10), which induce DICER, miR-103/107, which repress DICER7, and a Von Hippel-Lindau (VHL)-dependent mechanism affecting DICER protein stability16. We examined each of these and found that none could explain suppression of DICER by hypoxia in breast cancer. Reporter constructs containing 2.5 kb of the DICER promoter (with or without mutations in the MITF E-box elements) showed no regulation by hypoxia (Supplementary Fig. 5a,b). Similarly, no increase in miR-103/107 and no decrease in DICER transcript or protein stability were observed during hypoxia (Supplementary Fig. 5c–f). We also observed no significant difference in DICER repression in cell lines isogenic for VHL (Supplementary Fig. 5g). Importantly, MITF, Tap63 and miR-103/107 also showed weak or no correlation with DICER expression in breast cancer patients in the TCGA and METABRIC data sets (Supplementary Fig. 1b–h).
DICER is epigenetically regulated during hypoxia
The lack of regulation of the DICER reporter construct (Supplementary Fig. 5a,b) during hypoxia was surprising. To directly test whether transcription of the endogenous DICER locus was affected, we measured changes in de novo transcription during hypoxia by pulse-labelling RNA. These experiments demonstrated that DICER transcription decreased 40% to 50% during hypoxia (Fig. 2a), whereas the HIF1 target gene carbonic anhydrase-9 (CA9) increased 20-fold (Supplementary Fig. 7a). Transcriptional suppression of DICER was not dependent on known hypoxia response pathways, including either the HIF (hypoxia-inducible factor) pathway (as reported previously16) or the PERK/ATF4 arm of the unfolded protein response (Supplementary Fig. 6a–e). DICER repression occurred normally in both HIF1α knockout cells and in multiple lines where HIF1α was depleted by RNA interference (Supplementary Fig. 6a–c). In addition, hypoxic downregulation of DICER did not affect HIF regulation in breast cancer cell lines, which has been previously reported in other cell types16 (Supplementary Fig. 6f).
Figure 2: Epigenetic silencing of DICER promoter in response to hypoxia.
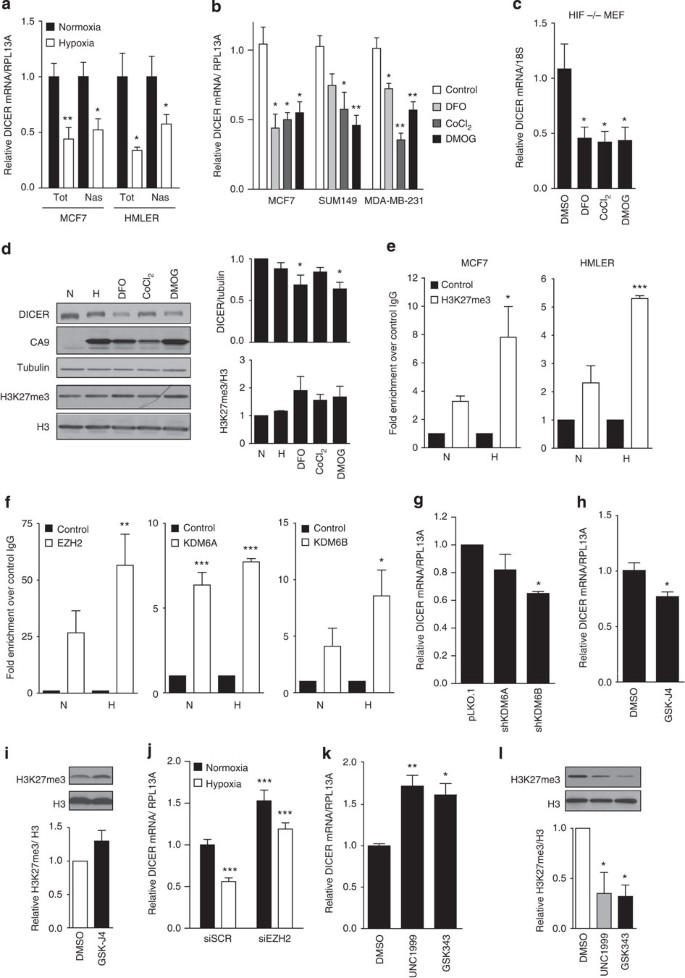
(a) Total (Tot) and nascent (Nas) DICER mRNA expression during hypoxia in MCF7 and HMLER cell lines was determined using quantitative reverse transcriptase–PCR (qRT–PCR) with RPL13A as control (_n_=4). (b) Indicated breast cancer cell lines (_n_=3) or (c) HIF1α-null mouse embryonic fibrobasts (MEFs) (_n_=4) were exposed to 500 μM iron chelator DFO, 250 μM antagonist CoCl2 or 500 μM DMOG for 24 h. DICER mRNA expression was determined using qRT–PCR with RPL13A or 18S as control. (d) Western blot analysis of MCF7 cell extracts prepared after 24 h exposure to 0.2% O2, 500 μM DFO, 250 μM CoCl2 or 500 μM DMOG with antibodies specific for DICER, CA9 and H3K27me3. Tubulin and H3 serve as respective loading controls. Representative blot (right) and densitometric analysis of western blottings from four independent experiments (left). (e) Validation of the enrichment in H3K27me at the DICER promoter was done using ChIP analysis of H3K27me3 mark in combination with qRT–PCR analysis in MCF7 and HMLER cells during normoxia (N) or hypoxia (H). Fold-enrichment shown over control IgG (_n_=3). (f) Enrichment of EZH2, KDM6A or KDM6B at the DICER promoter was done using ChIP analysis of each enzyme in combination with qRT–PCR analysis in MCF7 cells during normoxia (N) and hypoxia (H) (_n_=3). (g,h) qRT–PCR analysis of DICER expression with RPL13A as control in MCF7 cells (g) bearing shRNAs against KDM6A/B (_n_=3) or (h) treated with 10 μM KDM6A/B inhibitor GSK-J4 24 h (_n_=3). (i) Representative blot (top) and quantification of global H3K27me3 (bottom) after GSK-J4 treatment in h. DICER mRNA expression in MCF7 cells (j) transiently transfected with siRNA directed against EZH2 during aerobic and hypoxic conditions (_n_=4) or (k) treated with 5 μM EZH2 inhibitor UNC1999 or GSK343 for 48 h (_n_=3). (l) Representative western blotting (top) and quantification of global H3K27me3 levels (bottom) after EZH2 inhibition in k. Error bars represent s.e.m. _P_-values obtained with Student’s _t_-test or one-way analysis of variance, Bonferroni’s post-hoc test. *P<0.05, **P<0.01, ***P<0.001.
Although HIF1 was not required for DICER suppression during hypoxia, we found that agents that stabilize HIF by inhibiting the HIF prolyl-hydroxylases (EGLN1/2/3) did cause DICER suppression. Deferoxamine (DFO), cobalt chloride (CoCl2) and dimethyloxalylglycine (DMOG) at concentrations sufficient to activate transcription of HIF1 target genes (for example, _CA9_—Supplementary Fig. 7b) all resulted in a significant reduction in DICER at the mRNA and protein level (Fig. 2b,d). DFO and CoCl2 stabilize HIF by chelating or competing with iron [Fe(II)], whereas DMOG does so by competitive inhibition of 2-oxogluterate, which in addition to oxygen, are required co-factors for the HIF prolyl-hydroxylases that mediate HIF stability. However, similar to hypoxia, treatment with DFO, CoCl2 and DMOG also caused DICER repression in HIF1α knockout cells (Fig. 2c).
As hypoxia, DFO, CoCl2 and DMOG all influenced DICER expression in a HIF1α-independent manner, we hypothesized that DICER was regulated through inhibition of alternative iron, oxygen and 2-oxogluterate-dependent enzymes, such as the Jumonji-domain containing hydroxylases KDM6A and KDM6B37 that regulate epigenetic silencing through removal of repressive histone 3 lysine 27 trimethylation (H3K27me3) marks38. Indeed, inhibition of all three required cofactors of the KDM6A/B enzymes by hypoxia, DFO, CoCl2 and DMOG resulted in an increase in total H3K27me3 in multiple cell types (Fig. 2d and Supplementary Fig. 7c). More importantly, H3K27me3 chromatin immunoprecipitation followed by sequencing (ChIP-seq) demonstrated that hypoxic exposure for as short as 8 h led to an increase in repressive H3K27me3 marks in the DICER promoter region (Supplementary Fig. 7d). Enrichment in H3K27me3 at the DICER promoter during hypoxia was confirmed using conventional ChIP–quantitative PCR (qPCR) in MCF7 (eightfold enrichment versus hypoxic IgG control) and HMLER (fivefold enrichment versus hypoxic IgG control) cell lines (Fig. 2e). In both cell lines, this translated into an approximate doubling of H3K27me3 as compared with levels under normoxia. Furthermore, ChIP–qPCR analysis using specific antibodies against the H3K27 methyltransferase EZH2 (writer) and the oxygen-dependent H3K27me3 demethylases KDM6A and KDM6B (erasers) revealed significant enrichment over IgG controls at the DICER promoter for each respective enzyme, with no significant difference in enrichment between normoxic and hypoxic conditions (Fig. 2f). Consistent with a role for epigenetic regulation of DICER, knockdown of KDM6A or KDM6B resulted in reduced DICER expression (Fig. 2g and Supplementary Fig. 7e–g). Similarly, inhibition of KDM6A/B with the inhibitor GSK-J4 at concentrations that increased overall levels of H3K27me3 by 1.3-fold caused a 23% and 50% decrease in DICER expression in MCF7 and HMLER cells, respectively (Fig. 2h,i and Supplementary Fig. 7h,i) without affecting HIF activity (Supplementary Fig. 7h). Conversely, knockdown of EZH2 resulted in a significant increase in DICER expression and was able to largely prevent DICER repression during hypoxia (Fig. 2j and Supplementary Fig. 7j). Similarly, inhibition of EZH2 using UNC1999 and GSK343 at levels that caused decreases in global H3K27me3 levels by 50%–70% (Fig. 2l and Supplementary Fig. 7k) also increased DICER to levels comparable with EZH2 knockdown (Fig. 2k and Supplementary Fig. 7l). Together, these data indicate that basal expression of DICER is regulated by dynamic and opposing activities of KDM6A/B and EZH2, which are both constitutively present at the DICER locus, and that suppression of KDM6A/B activity under hypoxia is sufficient to increase H3K27me3 in an EZH2-dependent manner and suppress DICER transcription.
Hypoxia causes an miRNA processing defect
To examine the consequences of DICER suppression, we created MCF7 and HMLER breast cancer cell lines with stable knockdown of DICER and assessed changes in levels of mature miRNA. Suppression of DICER to ~30% resulted in a widespread reduction in mature miRNA for the majority of miRNA species (Fig. 3a,b and Supplementary Fig. 8a). Hypoxia also led to an analogous impairment in the processing of exogenously introduced short hairpin (shRNA) (Supplementary Fig. 8b). Short interfering RNA (siRNA) and shRNA targeting HIF1α showed similar efficiency under aerobic conditions, but under hypoxia HIF1α shRNA, which requires DICER processing, was significantly less efficient. Under hypoxia, HIF1α siRNA reduced the HIF target gene CA9 by ~50%, whereas HIF1α shRNA failed to prevent CA9 induction.
Figure 3: Impaired miRNA processing in hypoxic cells.
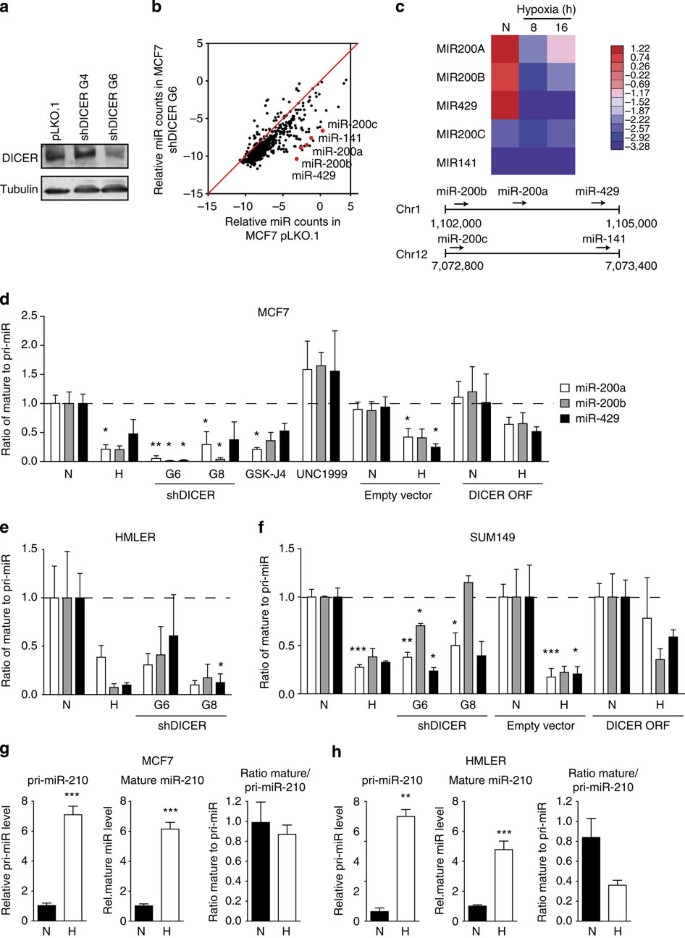
(a) MCF7 cells were transduced with lentiviral shRNA constructs directed at DICER. Cell extracts from MCF7 cells bearing empty vector pLKO.1, non-functional shDICER_G4 or functional shDICER_G6 were subjected to western blot analysis using a DICER-specific antibody. Tubulin served as loading control. (b) Total RNA was extracted from MCF7 pLKO.1 or shDICER_G6. miRNA levels were determined by Nanostring technology (_n_=3). (c) MCF7 cells exposed to hypoxia (<0.02% O2) for 16 h were used for total RNA isolation and subsequently subjected to deep-sequence analysis of miRNA content. Heat map of the miR-200 family in MCF7 during hypoxia is shown. Bottom panel shows schematic representation of the genomic organization of the miR-200 family. (d) The ratio of mature miRNA to pri-miRNA for miR-200a, b and 429 in MCF7 cells after hypoxia, DICER knockdown (shRNA G6 and G8), KDM6 inhibitor GSK-J4 (10 μM), EZH2 inhibitor UNC1999 (5 μM) and DICER overexpression. Mature and pri-miRNA levels were determined by quantitative reverse transcriptase–PCR (qRT–PCR). Similar experiments were performed in (e) HMLER and (f) SUM149 cells. (g,h) Processing of hypoxia-inducible miR-210 was assessed as described in d for (g) MCF7 and (h) HMLER cells. Expression levels of mature and pri-miRNAs for the miR-210 family was determined by qRT–PCR. N, normoxia; H, hypoxia. Data in d–h represent mean ratios (_n_=3)±s.e.m. _P_-values obtained with Student’s _t_-test. *P<0.05, **P<0.01, ***P<0.001.
Sensitivity to DICER knockdown varied among different miRNA and the five members of the miR-200 family (miR-200a, miR-200b, miR-429, miR-200c and miR-141) were among the most strongly repressed in both lines (Fig. 3b and Supplementary Fig. 8a). Similarly, the miR-200a/b/429 cluster was among the most strongly repressed miRNAs in response to hypoxia (Fig. 3c). We confirmed that both hypoxia and DICER knockdown led to a substantial defect in the processing of miR-200 family precursors (pri-miRNAs) into mature miRNAs. The ratio of mature to pri-miRNA, an indicator of DICER activity, dropped substantially in response to both DICER knockdown and hypoxia in MCF7, HMLER and SUM149 cells (Fig. 3d–f and Supplementary Fig. 8c). Furthermore, the reduction in DICER caused by inhibition of KDM6A/B using GSK-J4 also resulted in a decrease in miR-200a/b/429 processing similar to that during hypoxia or DICER knockdown (Fig. 3d). Conversely, inhibition of EZH2 using UNC1999, which increased DICER expression, increased miR-200a/b/429 processing (Fig. 3d). Importantly, transient overexpression of DICER during hypoxia increased miR-200a/b/429 processing to near basal levels, demonstrating that these effects on miRNA processing under hypoxia are due to its effects on DICER (Fig. 3d,f and Supplementary Fig. 8d).
Despite the defect in miRNA processing caused by DICER repression, some mature miRNAs increased during hypoxia, including the widely reported hypoxia-inducible miR-210. However, in this case the increase is due entirely to a transcriptional effect (Supplementary Fig. 8e,f and Fig. 3g,h). The processing of miR-210 mediated by DICER is reduced under hypoxia, but this effect is smaller than the overall transcriptional increase, resulting in increased mature levels of the miRNA.
Hypoxia stimulates EMT and CSC associated properties
The miR-200 family is implicated in regulation of the ZEB1 and ZEB2 transcription factors, which repress E-cadherin and stimulate the EMT39. DICER knockdown has previously been implicated in promoting the EMT and metastasis through the miR-200 family7, and miR-200b repression and ZEB1 induction during hypoxia has been reported40,41. We hypothesized that DICER suppression during hypoxia may similarly regulate the EMT and perhaps underlie the known association of tumour hypoxia with metastasis, stemness and aggressive disease. Indeed, DICER repression in response to hypoxia or knockdown resulted in enhanced expression of ZEB1, loss of epithelial marker expression (E-cadherin) and increased expression of mesenchymal markers (N-cadherin and Vimentin) (Fig. 4a–c). The transcription factor TWIST, previously implicated in hypoxia-induced EMT and metastasis42, remained unchanged, as did the transcription factor SNAIL (Supplementary Fig. 9a,b). The hypoxia- and DICER knockdown-induced EMT is mechanistically distinct from the classical EMT inducer transforming growth factor-β1 (TGFβ1), which was associated with increased expression of TWIST and SNAIL, but caused no change in DICER expression or miRNA biogenesis (Supplementary Fig. 9c–e).
Figure 4: DICER repression promotes cancer stem cell-associated phenotypes by reduced miR-200 expression during hypoxia.
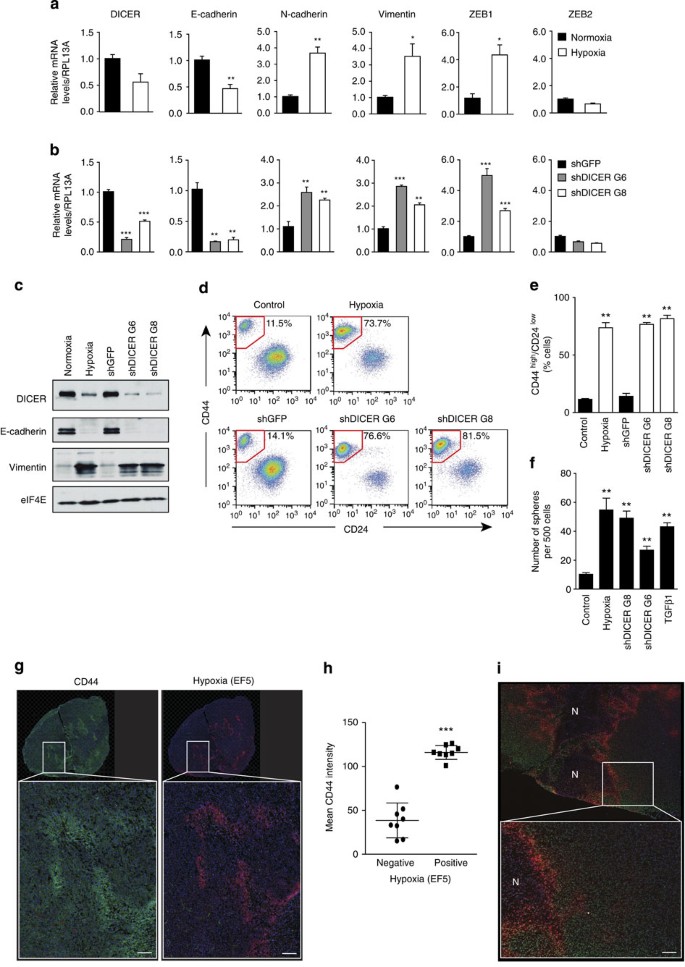
(a) HMLER cells were grown for 7 days under 1% O2. RNA was extracted and used to determine DICER mRNA levels by quantitative reverse transcriptase–PCR analysis. Expression of epithelial and mesenchymal-associated genes was assessed simultaneously. RPL13A served as control (_n_=3). (b) Similar analysis as in a was performed on HMLER cells transduced with two independent shRNA constructs targeting DICER (_n_=3). (c) HMLER protein extracts were subjected to western blot analysis of DICER, E-cadherin, Vimentin and eIF4E as loading control. (d) Representative FACS analysis of HMLER cells using antibodies specific for CD44 and CD24. (e) Quantification of the percentage of cells with CD44highCD24low for three independent experiments. (f) Sphere formation assay, number of spheres formed per 500 cells plotted (_n_=3). (g) Representative image of whole tumour (top) and 25 × region (bottom) of CD44 (green), EF5 (red) and DAPI (blue) staining in HMLER orthotopic xenograft. (h) Mean CD44 intensity in hypoxic (EF5 negative) versus non-hypoxic (EF5 positive) tumour regions of HMLER xenografts (_n_=8 mice). (i) Representative image of whole tumour (top) and 25 × region (bottom) of H3K27me3 (green), EF5 (red) and DAPI (blue) staining in HMLER orthotopic xenograft. N indicates regions of tumour necrosis. Scale bar, 100 μm. Error bars represent s.e.m. _P_-values obtained with Student’s _t_-test or one-way analysis of variance, Bonferroni’s post-hoc test. *P<0.05, **P<0.01, ***P<0.001.
In breast cancer, the EMT has been linked to acquisition of stem cell phenotypes, including expression of cell surface antigens associated with human breast stem cells (CD44high CD24low), increased mammosphere formation and increased tumour initiation capacity43,44,45. In HMLER cells, hypoxic exposure led to an increase in the frequency of CD44high CD24low cells from 11.5% to 73.7% (Fig. 4d,e), comparable to levels observed following exposure to TGFβ1 (Supplementary Fig. 9e) or reported following forced exposure of SNAIL or TWIST43. A similar increase in the fraction of CD44highCD24low cells occurred following DICER knockdown alone (76.6% and 81.5% versus 14.1%) (Fig. 4d,e). Both hypoxic exposure and DICER knockdown resulted in a greater than fivefold increase in mammosphere formation similar to that observed following TGFβ1 exposure (Fig. 4f). Importantly, hypoxia is similarly able to influence H3K27me3, DICER, EMT and stem cell phenotypes in vivo. We established HMLER xenografts and examined the spatial relationship between hypoxia, H3K27me3 and CD44 using multi-fluorescence immunohistochemistry. As shown in Fig. 4g,h, tumour hypoxia in vivo (as assessed by EF5) is strongly associated with increased expression of the stem cell marker CD44 (Fig. 4g,h). Hypoxic tumour areas also show increased overall levels of H3K27me3, with discernable gradients in expression away from hypoxic areas (Fig. 4i). Furthermore, we assessed DICER, ZEB1 and E-cadherin expression in vivo using a panel of breast cancer xenografts and found that DICER and E-cadherin were inversely correlated with the endogenous hypoxia marker CA9, whereas ZEB1 showed a positive correlation with CA9 (Supplementary Fig. 9f).
Finally, we tested directly if hypoxia- and DICER knockdown-induced EMT and acquisition of stem cell phenotypes is dependent on loss of mature miR-200 levels. DICER overexpression during hypoxia, which rescued the defect in miR-200 processing (Fig. 3d), also prevented increased expression of ZEB1 (Supplementary Fig. 9g). Furthermore, overexpression of miR-200b alone prevented increased expression of ZEB1 and loss of E-cadherin during hypoxia, without affecting DICER repression and the defect in miRNA biogenesis (Fig. 5a,b). miR-200b overexpression during hypoxia also prevented the increase in the CD44highCD24low population and mammosphere formation (Fig. 5c–e). Together, these demonstrate that hypoxic suppression of DICER causes an EMT-driven acquisition of stem cell properties in breast cancer through a reduction in the biogenesis of mature miR-200 family members (Fig. 5f).
Figure 5: Hypoxia stimulates cancer stem cell-associated phenotypes in a miR-200b-dependent manner.
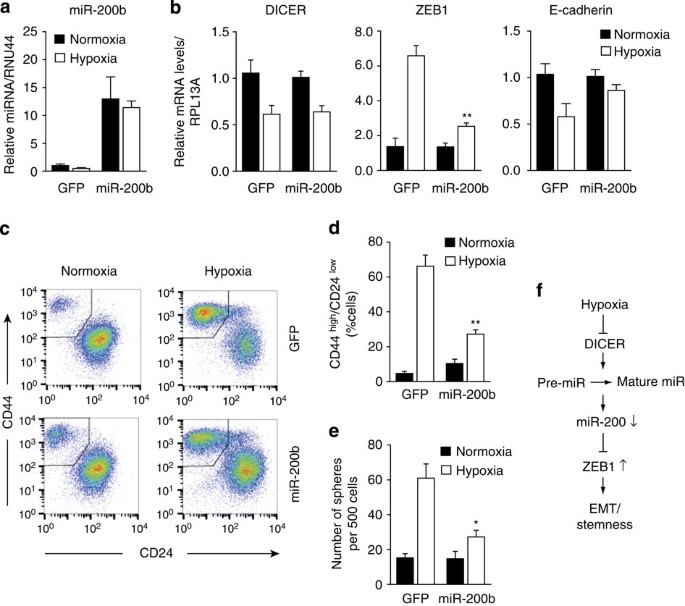
HMLER cells overexpressing miR-200b or green fluorescent protein (GFP) (_n_=3) grown for 7 days under 1% O2 or control conditions. (a,b) quantitative reverse transcriptase–PCR analysis was performed to measure (a) mature miR-200b levels and (b) DICER, ZEB1 and E-cadherin mRNA levels where RPL13A served as control (_n_=3). (c) Representative FACS analysis using antibodies specific for CD44 and CD24. (d) Quantification of the percentage of cells with CD44 highCD24low for three independent experiments. (e) Sphere formation assay, number spheres formed per 500 cells plotted (_n_=3). (f) Model of hypoxia-mediated EMT and stemness. Error bars represent s.e.m. _P_-values obtained with Student’s _t_-test. *P<0.05, **P<0.01.
Discussion
Our study demonstrates that hypoxia in the tumour microenvironment is a contributor to DICER expression and repression of miRNA biogenesis. In large independent cohorts of breast cancer patients, the association between hypoxia and DICER is stronger than any previously reported regulator of DICER, and the number of patients affected by this mechanism exceeds those harbouring monoallelic loss. This analysis also indicates that DICER suppression may underlie part of the known association of hypoxia with metastasis and poor outcome, although hypoxia remains a better discriminator than DICER in breast cancer patients. This is perhaps not surprising given that hypoxia influences other important biological processes, including angiogenesis, altered metabolism and chromosomal instability. The changes in histone methylation and DICER expression are relatively small (approximately twofold) in response to hypoxia or inhibition of EZH2 and KDM6A/B. Previous studies unambiguously identified DICER as a haploinsufficient tumour suppressor and an important driver of tumorigenesis under conditions of <50% repression4,5. Our study is consistent with these earlier reports and demonstrates that relatively small changes in DICER expression during hypoxia underlie functional changes in miRNA biogenesis, EMT and properties associated with stemness. Interestingly, Rupaimoole et al. have demonstrated that hypoxia causes additional suppression of miRNA biogenesis through silencing of DROSHA46 and that the combined defect in miRNA biogenesis resulting from DICER and DROSHA suppression leads to increased metastasis in ovarian cancer. The coordinated suppression of these two key enzymes required for miRNA biogenesis as well as hypoxia-dependent suppression of AGO2 (ref. 47) suggests a particularly important and broad role for oxygen in the regulation of miRNA levels. The consequences of such regulation are likely to be different in specific tissues and cancer types, depending on the expression of different miRNAs. In breast cancer, we show that selective sensitivity of the miR-200 family to loss of DICER plays a dominant role in regulation of EMT and cancer stem cell phenotypes, which may underlie the known association of hypoxia with aggressive disease in breast and other cancer types12,13,14,17,18.
Previous studies have reported multiple mechanisms of DICER regulation, which can occur at the level of transcription through MITF15 and Tap63 (ref. 10), mRNA stability through miR-103/107, as well as at the protein level in a VHL-dependent manner16. Our study reveals that DICER transcription is also regulated dynamically by acquisition or loss of the repressive H3K27me3 polycomb mark that is typically associated with gene silencing38. This mark is also found on promoters of so-called ‘poised’ genes expressed at low levels in embryonic stem cells where it is often associated with co-occurrence of the activating H3K4 mark48. Several previous studies have indicated that the balance between writing (EZH2) and erasing (KDM6A/B) H3K27me3 marks within the genome can play an important role in cancer49,50. This is supported by the finding that EZH2 is frequently overexpressed or activated in cancer51,52,53, and associated with increased metastasis54. Conversely, the H3K27me3 demethylase KDM6A is frequently mutated in human cancers55,56 and KDM6B expression is decreased in subsets of human cancers57,58. Our study identifies DICER as an important integrator of EZH2 methyltransferase and KDM6A/B demethylase activity. We showed that EZH2 and KDM6A/B are present at the DICER locus, and that inhibition of these enzymes can cause acute changes in the levels of H3K27me3, DICER expression and DICER activity. The hydroxylase activity of KDM6A/B requires molecular oxygen, 2-oxogluterate and Fe(II) to demethylate H3K27me3. Correspondingly, we found that hypoxia, or depletion/competition of the other co-factors with DFO, CoCl2 or DMOG, all suppress DICER. To our knowledge, this is the first such example of a gene whose expression is dynamically regulated at the epigenetic level by the availability of metabolic enzymatic co-factors. As the basal levels of H3K27me3 are influenced by both EZH2 and KDM6A/B activities, we expect that additional metabolic co-factors that regulate their activity including methyl donation (EZH2) or 2-oxogluterate (KDM6A/B) will similarly influence DICER H3K27me3 levels and its expression.
In contrast to genetic alteration of EZH2 and/or KDM6A/B, hypoxia-driven changes in the epigenetic state of DICER enables transient changes in cell phenotype in response to the local tumour microenvironment. In breast cancer, our findings demonstrate that suppression of DICER during hypoxia is sufficient to reduce miR-200 levels and enable cells to undergo an EMT and acquire stem cell properties. The fact that this occurs at the epigenetic level in an EZH2-dependent manner is consistent with a recent report that implicates EZH2 in controlling the EMT through epigenetic reprogramming59, but provides a potential mechanism for cells to retain plasticity and restore DICER and miR-200 levels when oxygen becomes available. Cancer cells need to undergo the reverse process, mesenchymal to epithelial transition, to ensure successful colonization and metastatic outgrowth60,61. We speculate that the clinical importance of hypoxia in driving metastasis and poor outcome is linked to this ability to transiently influence cell phenotype via DICER expression and miRNA biogenesis.
Finally, our results provide new potential therapeutic opportunities for targeting hypoxia and its influence on poor outcome in cancer. Hypoxia has been demonstrated to influence patient outcome, both through its ability to promote metastatic growth of disseminated stem cells and for regrowth of tumours following treatment. Hypoxic cells are intrinsically resistant to radiation and other forms of chemotherapy and small numbers of these cells can ‘re-seed’ the tumour, enabling regrowth following therapy. Hypoxic tumour cells also arise as a consequence of treatment with anti-angiogenic agents and can contribute to tumour regrowth post therapy62. Consequently, the ability of hypoxic cells to stimulate tumour initiation—either in naive metastases or following therapy of primary tumours—is considered to play a large role in treatment outcome. It may be possible to interfere with the epigenetic regulation of DICER during hypoxia and/or its influence on the expression of key miRNAs that promote these adverse clinical effects. Recent potent, orally available inhibitors of EZH2 have been reported63,64 and our results suggest that treatment with these agents induce DICER expression. Alternatively, an siRNA and/or miRNA strategy that does not rely on Drosha/Dicer processing and is not compromised within the tumour microenvironment could be applied. Effective delivery of siRNA and/or miRNA in vivo has improved remarkably and pre-clinical studies have demonstrated the potential of miR-200b delivery in breast, ovarian and other orthotopic models65.
Methods
Data set analyses
Retrieval and processing of breast cancer data sets. Preprocessing of raw mRNA abundance data sets was performed in R statistical environment (v2.14.1). Raw affymetrix-based data (PMIDs: 16273092, 17545524, 16141321, 16280042, 16478745, 18498629, 17157792, 19421193, 20098429, 20064235, 20490655, 20697068, 18821012, 18593943, 15721472, 21501481, 17079448, 21422418 and 21558518) were normalized using RMA (robust multi-array average) algorithm66 (R package: affy v1.32.1). ProbeSet annotation to Entrez Gene IDs was performed using custom CDFs67 (R packages: hgu133ahsentrezgcdf v15.0.0, hgu133bhsentrezgcdf v15.0.0, hgu133plus2hsentrezgcdf v15.0.0, hthgu133ahsentrezgcdf v15.0.0 and hgu95av2hsentrezgcdf v15.0.0 for affymetrix-based breast cancer data sets). The METABRIC breast cancer data set was preprocessed, summarized and quantile-normalized from the raw expression files generated by Illumina BeadStudio (R packages: beadarray v2.4.2 and illuminaHuman v3.db_1.12.2). Raw data files were downloaded from European genome-phenome archive (EGA) (Study ID: EGAS00000000083). Data files of one sample were not available at the time of this analysis and were therefore excluded. All data sets were normalized independently. Preprocessed segmented genome copy number aberration (CNA) data from the METABRIC cohort was downloaded from EGA. Preprocessed mRNA abundance data from TCGA were downloaded from cBioPortal for Cancer Genomics of Memorial Sloan-Kettering Cancer Center http://www.cbioportal.org/public-portal/ on 6 November 2012. Level 4 processed CNA data from TCGA were downloaded from GISTIC2 from the Broad Institute at http://gdac.broadinstitute.org/runs/analyses__2012_09_13/reports/cancer/BRCA/copynumber/gistic2/nozzle.html Discrete amplification and deletion calls for each sample based on the obtained CNA data were tabulated. For both mRNA abundance and CNA data, patient clinical information were obtained online from the original publication35.
Calculation of Winter signature scores. For each gene in the Winter signature, each patient in a given cohort was assigned an initial score of either +1 or −1 as follows: a patient was assigned +1 if her expression of that gene exceeded the median expression of that gene in that complete cohort. Otherwise, the patient was assigned −1 for that gene. For each patient in a given cohort, the +1’s and −1’s obtained as described above over all genes in the Winter signature were summed and the resulting sum was the Winter signature score for that patient in that cohort.
mRNA abundance correlation of Dicer with MITF or TP63. For each of the Metabric (Training and Validation datasets combined) and TCGA breast cancer mRNA abundance data sets, a scatter plot was generated of the Dicer mRNA abundance against that of MITF. Pearson’s product–moment correlation coefficient of the mRNA abundance of the two genes was computed and its _P_-value, based on the two-sided _t_-test for the Pearson’s product–moment correlation, was computed. This analysis was repeated for the correlation of Dicer mRNA abundance and TP63 mRNA abundance.
Comparison of Dicer mRNA abundance and Winter signature. For each of the Metabric (Training and Validation datasets combined) and TCGA breast cancer data sets, only patients with no Dicer copy number variation and those with monoallelic Dicer loss were retained. These retained patients were then divided into five groups as follows: those with monoallelic Dicer loss formed one group, and those with no Dicer copy number variation were ordered by their Winter signature scores and divided into four quarters, with the first quarter comprising patients with the lowest Winter signature scores, while the fourth represents those with highest Winter signature scores. A strip plot was generated to visualize the differences in Dicer mRNA abundance levels among these five groups. The Dicer mRNA abundance of patients in each of the four quarters of the group with no Dicer copy number variation was compared against the group with monoallelic Dicer loss, by computing the _P_-value based on the Wilcoxon rank sum test.
Comparison of miR-103/107 expression and Winter signature. Analyses were carried out in R statistical environment (version 3.0.1) (http:///www.r-project.org/). All tests were two-sided and considered statistically significant at the 0.05 level. Clinical and miRNA expression were downloaded from the Cancer Genome Atlas Project (TCGA) available through the associated files of the paper: ‘Comprehensive molecular portraits of human breast tumors, Nature, 27 September 2012’ (https://tcga-data.nci.nih.gov/docs/publications/brca_2012/). We also downloaded Level 3 (RNASeqV2) genes profiles for Breast Invasive Carcinoma from TCGA Data Portal: https://tcga-data.nci.nih.gov/tcga/. A hypoxia score was assigned to each TCGA sample based on the Winter hypoxia metagene signature and following the previously reported methodology68. The Shapiro–Wilk test was applied to verify whether the data follows a normal distribution. Accordingly, an analysis of variance test with Tukey post-hoc test was applied to assess the relationship between miR-103 and Winter hypoxia score, whereas a non-parametric test Kruskal–Wallis test with Nemenyi post-hoc test was applied to assess the relationship between miR-107 expression and Winter hypoxia score.
Cell culture and treatment
The following cell lines were obtained from ATCC and grown according to provided subculturing instructions: MCF7, MDA-MB-468, MDA-MB-231, T47D, U373, HCT116, Hela, ME180 and SiHa. HCC1954 and SUM149 cell lines were provided as a gift by Dr Benjamin Neel (Princess Margaret Cancer Centre). HCC1954 cells were grown in RPMI medium supplemented with 10% fetal bovine serum (FBS) and SUM149 cells were grown in Ham’s F12 medium supplemented with 5% FBS, insulin (5 μg ml−1), hydrocortisone (1 μg ml−1) and 10 mM Hepes (pH 7.4). HMLER cells were a gift from Dr Robert Weinberg (MIT) and were grown as previously described69. MCF10A cells were provided by Dr Senthil Muthuswamy (Princess Margaret Cancer Centre) and were grown in DMEM/F12 (Gibco BRL) supplemented with 5% donor horse serum, 20 ng ml−1 epidermal growth factor, 10 μg ml−1 insulin, 1 ng ml−1 cholera toxin, 100 μg ml−1 hydrocortisone, 50 μg ml−1 penicillin and 50 μg ml−1 streptomycin. Wild type and HIF1α−/− mouse embryonic fibrobasts, RCC4 and RCC4+pVHL were grown in DMEM medium supplemented with 10% FBS. All cell lines were routinely tested to confirm the absence of Mycoplasma. For hypoxic exposure, cells were transferred into a HypOxygen H35 workstation. The atmosphere in the chamber consisted of 5% H2, 5% CO2, the desired % O2 and residual N2. For protein stability experiments, MCF7 cells were exposed to 100 μg ml−1 cycloheximide (Sigma) for 0, 2, 4 or 8 h of normoxia or 0.2% O2 hypoxia. For exposure to stress-inducing agents, indicated cell lines were grown for 24 h in 250 μM CoCl2, 500 μM DFO or 500 μM DMOG. For inhibition of KDM6A/B activity, MCF7 and HMLER cells were grown for 24 h in GSK-J4 at indicated concentrations. For inhibition of EZH2 activity, MCF7 and HMLER cells were grown for 48–72 h in UNC1999 or GSK343 at indicated concentrations. For exposure to TGFβ1, HMLER cells were cultured in standard growth medium with the addition of 5% FBS and treated with 2.5 ng ml−1 TGFβ1 for 12 days.
RNA extraction and quantitative reverse–transcription PCR
RNA was isolated using TRI reagent (Sigma) and samples were reverse transcribed using qScript cDNA SuperMix (Quantas). Quantitative real-time PCR was performed on an Eppendorf Realplex2 mastercycler using SYBR green (Quantas). Specific primers used are listed in Supplementary Table 2. For determination of DICER mRNA half-life, cells were treated with 5 μg ml−1 actinomycin D (Sigma) for the indicated times. De novo RNA synthesis was measured using the Click-iT Nascent RNA Capture Kit (Molecular Probes). Briefly, MCF7 and HMLER cells were pulse labelled with 0.2 mM 5-ethynyl Uridine for 1 h during aerobic conditions or 24 h hypoxia (0.2% O2) after which RNA was isolated as described above. Nascent RNA was captured using magnetic streptavidin beads, reverse transcribed and analysed by quantitative real-time PCR. For determination of pri-miRNA and mature miRNA levels, the following assays from Applied Biosystems were used: pri-miR200a (Hs03303376_pri), pri-miR200b (Hs03303027_pri), pri-miR429 (Hs03303727_pri), pri-miR200c (Hs03303157_pri), pri-miR141 (Hs03303157_pri), pri-miR210 (Hs03302948_pri), hsa-miR200a (000502), hsa-miR200b (002251), hsa-miR429 (001024), hsa-miR200c (002300), miR141 (002145), hsa-miR103 (000439), hsa-miR107 (000443), hsa-miR210 (000512), RNU44 (001094) and RNU48 (001006).
Western blot analysis
Cells were washed twice with cold PBS and scraped in 50 mM Tris HCl pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate and 0.1% SDS supplemented with protease and phosphatase inhibitors (Roche). After centrifugation at 10,000 g, supernatants were boiled in Laemmli buffer for 10 min and proteins were resolved by SDS–PAGE. Proteins were subsequently transferred onto polyvinylidene difluoride membrane and blocked for 1 h in TBS containing 0.05% Tween 20 (TBS-T) supplemented with 5% skim milk powder. Membranes were probed overnight at 4 °C with antibodies directed against: DICER (1:200, H-212; Santa Cruz Biotechnology), CA9 (1:1,000, M75; gift from Dr Silvia Pastorekova), H3K27me3 (1:20,000, 07-449; Upstate), H3 (1:5,000, D1H2; Cell Signaling Technology), E-cadherin (1:1,000, Cell Signaling Technology), Vimentin (1:500, clone V9; Sigma), β-tubulin (1:20,000, Abcam), β-actin (1:20,000, Sigma) or eIF4E (1:1,000, BD Transduction Laboratories). Bound antibodies were visualized using horseradish peroxidase-linked secondary antibodies (GE Healthcare) and ECL luminescence (Pierce).
Luciferase reporter assay
A 2.5-kb fragment from the 5′-flanking region of the DICER1 gene was previously described15. MCF7 cells were transiently co-transfected with the DICER or CA9 promoter70 constructs and pcDNALacZ using Lipofectamine (Invitrogen). Transfected cells were subcultured 16 h post transfection, exposed to hypoxia and finally harvested 48 h after transfection. Luciferase and β-galactosidase activity was measured using a commercial kit (Applied Biosystems) and measured on the Fluorstar Optima plate reader (BMG Labtech).
Plasmids and viral infections
Knockdown was achieved using lentiviral shRNA constructs directed against DICER: TRCN0000004386, TRCN000000439; HIF-1α: TRCN0000003810; KDM6A: TRCN0000107760; KDM6B: TRCN0000236677; green fluorescent protein as control: TRCN0000072181. DICER open reading frame was cloned into pLenti CMV DEST using Gateway LR Clonase II (Invitrogen). Pri-miR200b was cloned into pLJM1 as _Age_1/_Eco_R1 fragment. Lentiviral particles were generated by co-transfection of 293T cells with packaging plasmids pCMVdR8.74psPAX2 and pMD2.G, together with shRNA vector pLKO.1 using Lipofectamine 2000. Virus supernatant was harvested 48 and 72 h post transfection. Cell lines were transduced with lentiviral supernatant in the presence of 8 μg ml−1 polybrene. Infected cells were selected for 48 h in 2 μg ml−1 puromycin-containing media or 7 days in 5 μg ml−1 blasticidin-containing media. Validated siRNA duplexes directed against EZH2 (ref. 52) 5′- AAGACTCTGAATGCAGTTGCT -3′ and HIF1α 5′- CUGAUGACCAGCAACUUGA -3′ were ordered from Sigma. Stealth RNA interference-negative control was ordered from Invitrogen (12935-300). For siRNA experiments, cells were transfected 72 h before analysis with 2 nM siRNA duplex using Lipofectamine (Invitrogen).
Chromatin immunoprecipitation
MCF7 and HMLER cells were fixed in 1% formaldehyde. Cross-linking was allowed to proceed for 10 min at room temperature and stopped by addition of glycine at a final concentration of 0.125 M, followed by an additional incubation for 5 min. Fixed cells were washed twice with PBS and harvested in SDS buffer (50 mM Tris at pH 8.1, 0.5% SDS, 100 mM NaCl, 5 mM EDTA), supplemented with protease inhibitors (Aprotinin, Antipain and Leupeptin all at 5 μg ml−1 and 1 mM phenylmethylsulfonyl fluoride). Cells were pelleted by centrifugation and suspended in IP Buffer (100 mM Tris at pH 8.6, 100 mM NaCl, 0.3% SDS, 1.7% Triton X-100 and 5 mM EDTA), containing protease inhibitors. Cells were disrupted by sonication, yielding genomic DNA fragments with a bulk size of 200–500 bp. For each immunoprecipitation, 1 ml of lysate was precleared by addition of 35 μl of blocked protein A beads (50% slurry protein A-Sepharose (Amersham); 0.5 mg ml−1 fatty acid-free BSA (Sigma); and 0.2 mg ml−1 herring sperm DNA in TE), followed by clarification by centrifugation. Ten-microlitre aliquots of precleared suspension were reserved as input DNA and kept at 4 °C. Samples were immunoprecipitated overnight at 4 °C using 1 μg of antibodies for either HA as a negative control (sc-805; Santa Cruz), H3K27me3 (07-449; Upstate), EZH2 (AC22, Millipore), KDM6A (ab36938, Abcam) or KDM6B (ab85392, Abcam). Immune complexes were recovered by adding 40 μl of blocked protein A beads and incubated for 4 h at 4 °C. Beads were washed three times in 1 ml of Mixed Micelle Buffer (20 mM Tris at pH 8.1, 150 mM NaCl, 5 mM EDTA, 5% w/v sucrose, 1% Triton X-100 and 0.2% SDS), twice in 1 ml of Buffer 500 (50 mM HEPES at pH 7.5, 0.1% w/v deoxycholic acid, 1% Triton X-100 and 1 mM EDTA), twice in 1 ml of LiCl Detergent Wash Buffer (10 mM Tris at pH 8.0, 0.5% deoxycholic acid, 0.5% NP-40, 250 mM LiCl and 1 mM EDTA) and once in 1 ml of TE. Immunocomplexes were eluted from beads in 250 μl elution buffer (1% SDS and 0.1 M NaHCO3) for 2 h at 65 °C with continuous shaking at 1,000 r.p.m., and after centrifugation supernatants were collected. Two hundred and fifty microlitres elution buffer was added to input DNA samples and these were processed in parallel with eluted samples. Cross-links were reversed overnight at 65 °C followed by a 2-h digestion with RNAseA at 37 °C and 2 h proteinase K (0.2 μg ml−1) at 55 °C. DNA fragments were recovered using QIAquick PCR purification columns, according to manufacturers’ instructions. Samples were eluted in 75 μl EB buffer and then further 1/5 diluted in TE buffer. The immunoprecipitated DNA was quantified by real-time qPCR using SYBR Green I (Applied Biosystems) and following forward and reverse primers: DICER: F-5′- CGGTGGGCGTTAAATAAGTG -3′ and R-5′- CCCCCATACTGAGATGCTGT -3′. After PCR, melting curves were acquired to ensure that a single product was amplified in the reaction.
Global miRNA profiling
Total RNA was isolated from cells bearing shDICER G6 and pLKO.1 as control or cells exposed to hypoxia, and samples were subjected to Nanostring analysis using the Nanostring Human v2 miRNA code set at the UHN Microarray Centre. miRNAs with counts equal or lower than the negative controls were discarded from the analysis. miRNA counts were normalized to housekeeping gene RPLP0 and averaged for three independent experiments.
Fluorescence-activated cell sorting
FACS analysis was performed on a BD FACS Calibur (Becton Dickinson) using PE-conjugated anti-CD24 antibody (clone ML5) and APC-conjugated anti-CD44 (clone G44-26) (BD Bioscience).
Mammosphere culture
Mammosphere culture was performed as previously described71, with slight modifications. The mammospheres were cultured for 7–10 days in MammoCult media (StemCell Technologies) supplemented with 4 μg ml−1 heparin (Sigma) and 0.5 μg ml−1 hydrocortisone (Sigma). For sphere formation assays, mammospheres were dissociated to single cells with trypsin and 500 dissociated cells were plated in a 96-well plate and cultured for 10 days. Mammospheres with diameter >75 μm were counted.
In vivo models
All animal experiments were performed under protocols approved by the Ontario Cancer Institute’s Animal Care Committee, according the regulations of the Canadian Council on Animal Care. Female NOD-SCID mice at 6–8 weeks old were used to inject HMLER cells into the inguinal mammary fat pad (1 × 106 cells in a 1:1 mixture of BD Matrigel and media) following anaesthetization with isoflurane. One week before the injection of cells, a 60-day release pellet containing 2 mg 17β-estradiol and 20 mg progesterone (Innovative Research of America) was implanted subcutaneously into each mouse. At end point, mice were sacrificed, tumours harvested and optical coherence tomography embedded for immunohistochemical staining and analysis.
Immunohistochemical staining and image analysis
The expression of CD44 and EF5 in orthotopic xenografts was investigated as follows: Flash-frozen tissue samples were embedded in optical coherence tomography and stored at −80 °C until sectioned. Sections were thawed at room temperature before fixation in 2% paraformaldehyde for 20 min. After washing sections in PBS, sections were permeabilized in PBS containing 0.5% Triton X-100 for 15 min, wash 3 × 5 min in PBS-T and then incubated in a primary antibody cocktail of mouse anti-human CD44 (1:75, BD Pharmingen) and rat anti-mouse CD31 (1:300, BD Pharmingen) or anti-human H3K27me3 (1:500, C36B11; Cell Signalling) overnight at room temperature. Sections were subsequently washed 3 × 5 min in PBS-T followed by incubation in a secondary antibody cocktail of goat anti-mouse Alexafluor 488 (1:200, Life Technologies) and goat anti-rat Alexafluor 555 (1:200, Life technologies) or goat anti-mouse Alexafluor 488 (1:200, Life Technologies) for 1 h at room temperature. After washing in PBS-T, sections were incubated in EF5-Cy5 (1:50, provided by Dr Cameron Koch) for 3 h at room temperature. After washing, sections were finally incubated in a working solution of DAPI (4',6-diamidino-2-phenylindole) for 5 min, washed, dried and imaged on a laser scanning microscope (Huron Technologies). Images were analysed using Definiens Tissue Studio software, which allows for semi-automatic histology image analysis. Briefly, the software was trained to identify viable tumour areas, necrotic areas, tumour stroma and empty areas within the scanned section. A threshold was determined by mean +2 s.d. intensity in the EF5 and CD44 channels. The average CD44 staining intensity within the tumour area was measured in both EF5-negative and -positive areas. EF5-positive area above a background threshold was obtained from the average intensity of all tumour sections.
Statistical analyses
Unless otherwise stated, a Student’s _t_-test or one-way analysis of variance with Bonferroni’s post-hoc test were used to test significance between populations. A significance threshold of P<0.05 was applied. Points and error bars plotted in graphs represent the mean±s.e.m for three or more independent experiments.
Additional information
How to cite this article: van den Beucken, T. et al. Hypoxia promotes stem cell phenotypes and poor prognosis through epigenetic regulation of DICER. Nat. Commun. 5:5203 doi: 10.1038/ncomms6203 (2014).
Accessions codes: miRNA profiling and ChIP-Seq data have been deposited in the NCBI Gene Expression Omnibus database under accession codes GSE61722 and GSE61740, respectively.
Accession codes
Accessions
Gene Expression Omnibus
References
- Valastyan, S. & Weinberg, R. A. Tumor metastasis: molecular insights and evolving paradigms. Cell 147, 275–292 (2011).
Article CAS Google Scholar - Bartel, D. P. MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233 (2009).
Article CAS Google Scholar - Filipowicz, W., Bhattacharyya, S. N. & Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat. Rev. Genet. 9, 102–114 (2008).
Article CAS Google Scholar - Kumar, M. S., Lu, J., Mercer, K. L., Golub, T. R. & Jacks, T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat. Genet. 39, 673–677 (2007).
Article CAS Google Scholar - Kumar, M. S. et al. Dicer1 functions as a haploinsufficient tumor suppressor. Genes Dev. 23, 2700–2704 (2009).
Article CAS Google Scholar - Lu, J. et al. MicroRNA expression profiles classify human cancers. Nature 435, 834–838 (2005).
Article CAS ADS Google Scholar - Martello, G. et al. A microRNA targeting dicer for metastasis control. Cell 141, 1195–1207 (2010).
Article CAS Google Scholar - Melo, S. A. et al. A genetic defect in exportin-5 traps precursor microRNAs in the nucleus of cancer cells. Cancer Cell 18, 303–315 (2010).
Article CAS Google Scholar - Melo, S. A. et al. A TARBP2 mutation in human cancer impairs microRNA processing and DICER1 function. Nat. Genet. 41, 365–370 (2009).
Article CAS Google Scholar - Su, X. et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature 467, 986–990 (2010).
Article CAS ADS Google Scholar - Ebert, M. S. & Sharp, P. A. Roles for microRNAs in conferring robustness to biological processes. Cell 149, 515–524 (2012).
Article CAS Google Scholar - Grelier, G. et al. Prognostic value of Dicer expression in human breast cancers and association with the mesenchymal phenotype. Br. J. Cancer 101, 673–683 (2009).
Article CAS Google Scholar - Karube, Y. et al. Reduced expression of Dicer associated with poor prognosis in lung cancer patients. Cancer Sci. 96, 111–115 (2005).
Article CAS Google Scholar - Merritt, W. M. et al. Dicer, Drosha, and outcomes in patients with ovarian cancer. New Engl. J. Med. 359, 2641–2650 (2008).
Article CAS Google Scholar - Levy, C. et al. Lineage-specific transcriptional regulation of DICER by MITF in melanocytes. Cell 141, 994–1005 (2010).
Article CAS Google Scholar - Ho, J. J. et al. Functional importance of Dicer protein in the adaptive cellular response to hypoxia. J. Biol. Chem. 287, 29003–29020 (2012).
Article CAS Google Scholar - Vaupel, P. Prognostic potential of the pre-therapeutic tumor oxygenation status. Adv. Exp. Med. Biol. 645, 241–246 (2009).
Article Google Scholar - Fyles, A. et al. Tumor hypoxia has independent predictor impact only in patients with node-negative cervix cancer. J. Clin. Oncol. 20, 680–687 (2002).
Article CAS Google Scholar - Schindl, M. et al. Overexpression of hypoxia-inducible factor 1alpha is associated with an unfavorable prognosis in lymph node-positive breast cancer. Clin. Cancer Res. 8, 1831–1837 (2002).
CAS PubMed Google Scholar - Hussain, S. A. et al. Hypoxia-regulated carbonic anhydrase IX expression is associated with poor survival in patients with invasive breast cancer. Br. J. Cancer 96, 104–109 (2007).
Article CAS Google Scholar - Yan, M., Rayoo, M., Takano, E. A. & Fox, S. B. BRCA1 tumours correlate with a HIF-1alpha phenotype and have a poor prognosis through modulation of hydroxylase enzyme profile expression. Br. J. Cancer 101, 1168–1174 (2009).
Article CAS Google Scholar - Hiraga, T., Kizaka-Kondoh, S., Hirota, K., Hiraoka, M. & Yoneda, T. Hypoxia and hypoxia-inducible factor-1 expression enhance osteolytic bone metastases of breast cancer. Cancer Res. 67, 4157–4163 (2007).
Article CAS Google Scholar - Brennan, D. J. et al. CA IX is an independent prognostic marker in premenopausal breast cancer patients with one to three positive lymph nodes and a putative marker of radiation resistance. Clin. Cancer Res. 12, 6421–6431 (2006).
Article CAS Google Scholar - Vaupel, P., Briest, S. & Hockel, M. Hypoxia in breast cancer: pathogenesis, characterization and biological/therapeutic implications. Wien. Med. Wochenschr. 152, 334–342 (2002).
Article CAS Google Scholar - Cairns, R. A. & Hill, R. P. Acute hypoxia enhances spontaneous lymph node metastasis in an orthotopic murine model of human cervical carcinoma. Cancer Res. 64, 2054–2061 (2004).
Article CAS Google Scholar - Cairns, R. A., Kalliomaki, T. & Hill, R. P. Acute (cyclic) hypoxia enhances spontaneous metastasis of KHT murine tumors. Cancer Res. 61, 8903–8908 (2001).
CAS PubMed Google Scholar - Tamara Marie-Egyptienne, D., Lohse, I. & Hill, R. P. Cancer stem cells, the epithelial to mesenchymal transition (EMT) and radioresistance: potential role of hypoxia. Cancer Lett. 341, 63–72 (2012).
Article Google Scholar - Hill, R. P., Marie-Egyptienne, D. T. & Hedley, D. W. Cancer stem cells, hypoxia and metastasis. Semin. Radiat. Oncol. 19, 106–111 (2009).
Article Google Scholar - Das, B. et al. Hypoxia enhances tumor stemness by increasing the invasive and tumorigenic side population fraction. Stem Cells 26, 1818–1830 (2008).
Article Google Scholar - Li, Z. et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 15, 501–513 (2009).
Article CAS Google Scholar - Keith, B. & Simon, M. C. Hypoxia-inducible factors, stem cells, and cancer. Cell 129, 465–472 (2007).
Article CAS Google Scholar - Chaturvedi, P. et al. Hypoxia-inducible factor-dependent breast cancer-mesenchymal stem cell bidirectional signaling promotes metastasis. J. Clin. Invest. 123, 189–205 (2013).
Article CAS Google Scholar - Lock, F. E. et al. Targeting carbonic anhydrase IX depletes breast cancer stem cells within the hypoxic niche. Oncogene 32, 5210–5219 (2012).
Article Google Scholar - Curtis, C. et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352 (2012).
Article CAS Google Scholar - Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012).
- Winter, S. C. et al. Relation of a hypoxia metagene derived from head and neck cancer to prognosis of multiple cancers. Cancer Res. 67, 3441–3449 (2007).
Article CAS Google Scholar - Pollard, P. J. et al. Regulation of Jumonji-domain-containing histone demethylases by hypoxia-inducible factor (HIF)-1alpha. Biochem. J. 416, 387–394 (2008).
Article CAS Google Scholar - Bernstein, B. E., Meissner, A. & Lander, E. S. The mammalian epigenome. Cell 128, 669–681 (2007).
Article CAS Google Scholar - Gregory, P. A. et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 10, 593–601 (2008).
Article CAS Google Scholar - Okajima, M. et al. Anoxia/reoxygenation induces epithelial-mesenchymal transition in human colon cancer cell lines. Oncol. Rep. 29, 2311–2317 (2013).
Article CAS Google Scholar - Chan, Y. C., Khanna, S., Roy, S. & Sen, C. K. miR-200b targets Ets-1 and is down-regulated by hypoxia to induce angiogenic response of endothelial cells. J. Biol. Chem. 286, 2047–2056 (2011).
Article CAS Google Scholar - Yang, M. H. et al. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell Biol. 10, 295–305 (2008).
Article CAS Google Scholar - Mani, S. A. et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715 (2008).
Article CAS Google Scholar - Shimono, Y. et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 138, 592–603 (2009).
Article CAS Google Scholar - Blick, T. et al. Epithelial mesenchymal transition traits in human breast cancer cell lines parallel the CD44(hi/)CD24 (lo/-) stem cell phenotype in human breast cancer. J. Mammary Gland Biol. Neoplasia 15, 235–252 (2010).
Article Google Scholar - Rupaimoole, R. et al. Hypoxia-mediated downregulation of miRNA biogenesis promotes tumour progression. Nat. Commun. 5:5202 doi:10.1038/ncomms6202 (2014).
- Shen, J. et al. EGFR modulates microRNA maturation in response to hypoxia through phosphorylation of AGO2. Nature 497, 383–387 (2013).
Article CAS ADS Google Scholar - Bernstein, B. E. et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326 (2006).
Article CAS Google Scholar - Yap, D. B. et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 117, 2451–2459 (2011).
Article CAS Google Scholar - Morin, R. D. et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 42, 181–185 (2010).
Article CAS Google Scholar - Varambally, S. et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 322, 1695–1699 (2008).
Article CAS ADS Google Scholar - Bracken, A. P. et al. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 22, 5323–5335 (2003).
Article CAS Google Scholar - Banerjee, R. et al. The tumor suppressor gene rap1GAP is silenced by miR-101-mediated EZH2 overexpression in invasive squamous cell carcinoma. Oncogene 30, 4339–4349 (2011).
Article CAS Google Scholar - Crea, F., Paolicchi, E., Marquez, V. E. & Danesi, R. Polycomb genes and cancer: time for clinical application? Crit. Rev. Oncol. Hematol. 83, 184–193 (2012).
Article Google Scholar - Dalgliesh, G. L. et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 463, 360–363 (2010).
Article CAS ADS Google Scholar - van Haaften, G. et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat. Genet. 41, 521–523 (2009).
Article CAS Google Scholar - Agger, K. et al. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene- and stress-induced senescence. Genes Dev. 23, 1171–1176 (2009).
Article CAS Google Scholar - Barradas, M. et al. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 23, 1177–1182 (2009).
Article CAS Google Scholar - Tiwari, N. et al. Sox4 is a master regulator of epithelial-mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer Cell 23, 768–783 (2013).
Article CAS Google Scholar - Ocana, O. H. et al. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 22, 709–724 (2012).
Article CAS Google Scholar - Tsai, J. H., Donaher, J. L., Murphy, D. A., Chau, S. & Yang, J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 22, 725–736 (2012).
Article CAS Google Scholar - Mehta, S. et al. Assessing early therapeutic response to bevacizumab in primary breast cancer using magnetic resonance imaging and gene expression profiles. J. Natl Cancer Inst. Monogr. 2011, 71–74 (2011).
Article Google Scholar - Konze, K. D. et al. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS. Chem. Biol. 8, 1324–1334 (2013).
Article CAS Google Scholar - Amatangelo, M. D. et al. Three-dimensional culture sensitizes epithelial ovarian cancer cells to EZH2 methyltransferase inhibition. Cell Cycle 12, 2113–2119 (2013).
Article CAS Google Scholar - Pecot, C. V., Calin, G. A., Coleman, R. L., Lopez-Berestein, G. & Sood, A. K. RNA interference in the clinic: challenges and future directions. Nat. Rev. Cancer 11, 59–67 (2011).
Article CAS Google Scholar - Irizarry, R. A. et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264 (2003).
Article Google Scholar - Dai, M. et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 33, e175 (2005).
Article Google Scholar - Starmans, M. H. et al. The prognostic value of temporal in vitro and in vivo derived hypoxia gene-expression signatures in breast cancer. Radiother. Oncol. 102, 436–443.
Article CAS Google Scholar - Elenbaas, B. et al. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 15, 50–65 (2001).
Article CAS Google Scholar - van den Beucken, T. et al. Hypoxia-induced expression of carbonic anhydrase 9 is dependent on the unfolded protein response. J. Biol. Chem. 284, 24204–24212 (2009).
Article CAS Google Scholar - Dontu, G. et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 17, 1253–1270 (2003).
Article CAS Google Scholar
Acknowledgements
We thank R. Weinberg for providing the HMLER cells. We also thank C. Arrowsmith for providing the SGC inhibitors. We thank Alison Casey, Milan Ganguly and Trevor Do for advice and technical assistance on the immunohistochemical staining in these and other tissue samples. This work was financially supported by the Dutch Cancer Society (KWF grant UM 2008-4068 to B.W.), the Ontario Ministry of Health and Long Term Care (OMOHLTC), the Terry Fox New Frontiers Research Program PPG-1036 to B.G.W. and M.K.), the Ontario Institute for Cancer Research and Terry Fox Research Institute (Stem Cell Program to B.G.W.), the Canadian Institute for Health Research (CIHR grant 201592 to B.G.W. and M.K.) and the EU 7th framework programme (METOXIA project 222741 to B.G.W. and M.K.). The views expressed do not necessarily reflect those of the OMOHLTC. This study was conducted with the support of the Ontario Institute for Cancer Research to P.C.B. through funding provided by the Government of Ontario. E.K. and C.Q.Y. were supported by fellowships from the Canadian Breast Cancer Foundation (CBCF). A.L.H. was supported by funding from Cancer Research UK (CRUK_A11359) and the Breast Cancer Research Foundation (BCRF). A.K.S. was supported by funding from the National Institute of Health (U54 151668, P50 CA083639, and UH2 TR000943). M.I. was supported by funding from the National Institute of Health (R01 CA155332-01). This study makes use of data generated by the Molecular Taxonomy of Breast Cancer International Consortium34, which was funded by Cancer Research UK and the British Columbia Cancer Agency Branch.
Author information
Author notes
- Twan van den Beucken and Elizabeth Koch: These authors contributed equally to this work
Authors and Affiliations
- Princess Margaret Cancer Centre and Campbell Family Institute for Cancer Research, University Health Network, 101 College Street, Room 12-310, Toronto Medical Discovery Tower, Toronto, M5G 1L7, Ontario, Canada
Twan van den Beucken, Elizabeth Koch, Marianne Koritzinsky & Bradly G. Wouters - Department of Radiation Oncology, GROW School for Oncology and Developmental Biology, Maastricht University Medical Center, Maastricht, 6200 MD, The Netherlands
Twan van den Beucken, Maud H. W. Starmans & Bradly G. Wouters - Department of Toxicogenomics, GROW School for Oncology and Developmental Biology, Maastricht University Medical Center, Maastricht, 6200 MD, The Netherlands
Twan van den Beucken - Department of Medical Biophysics, University of Toronto, Toronto, M5G 1L7, Ontario, Canada
Elizabeth Koch, Cindy Q. Yao, Paul C. Boutros & Bradly G. Wouters - Informatics and Bio-computing Program, Ontario Institute for Cancer Research, Toronto, M5G 1L7, Ontario, Canada
Kenneth Chu, Syed Haider, Maud H. W. Starmans, Cindy Q. Yao & Paul C. Boutros - Departments of Gynecologic Oncology and Cancer Biology, The University of Texas MD Anderson Cancer Center, Houston, 77030, Texas, USA
Rajesha Rupaimoole & Anil K. Sood - Department of Molecular Genetics, GROW School for Oncology and Developmental Biology, Maastricht University Medical Center, Maastricht, 6200 MD, The Netherlands
Peggy Prickaerts & Jan Willem Voncken - Department of Experimental Cardiology, Heart Failure Research Centre, Academic Medical Center, 1100 DD Amsterdam, The Netherlands
Michiel Adriaens - Department of Bioinformatics—BiGCaT, Maastricht University, 6200 MD Maastricht, The Netherlands
Michiel Adriaens - Cancer Research UK Department of Oncology, The Weatherall Institute of Molecular Medicine, John Radcliffe Hospital, Oxford, OX3 9DS, UK
Adrian L. Harris & Francesca M. Buffa - Department of Medicine, The Indiana University Melvin and Bren Simon Cancer Center, Indianapolis, 46202, Indiana, USA
Mircea Ivan - Center for RNA Interference and Non-Coding RNAs, The University of Texas MD Anderson Cancer Center, Houston, 77030, Texas, USA
Cristina Ivan & Anil K. Sood - Department of Thoracic, Head and Neck Oncology, The University of Texas MD Anderson Cancer Center, Houston, 77030, Texas, USA
Chad V. Pecot - Department of Pharmacology and Toxicology, University of Toronto, Toronto, M5S 1A8, Ontario, Canada
Paul C. Boutros - Institute of Medical Science, University of Toronto, Toronto, M5S 1A8, Ontario, Canada
Marianne Koritzinsky - Department of Radiation Oncology, University of Toronto, Toronto, M5T 1P5, Ontario, Canada
Marianne Koritzinsky & Bradly G. Wouters - Selective Therapies Program, Ontario Institute for Cancer Research, Toronto, M5G 1L7, Ontario, Canada
Bradly G. Wouters
Authors
- Twan van den Beucken
You can also search for this author inPubMed Google Scholar - Elizabeth Koch
You can also search for this author inPubMed Google Scholar - Kenneth Chu
You can also search for this author inPubMed Google Scholar - Rajesha Rupaimoole
You can also search for this author inPubMed Google Scholar - Peggy Prickaerts
You can also search for this author inPubMed Google Scholar - Michiel Adriaens
You can also search for this author inPubMed Google Scholar - Jan Willem Voncken
You can also search for this author inPubMed Google Scholar - Adrian L. Harris
You can also search for this author inPubMed Google Scholar - Francesca M. Buffa
You can also search for this author inPubMed Google Scholar - Syed Haider
You can also search for this author inPubMed Google Scholar - Maud H. W. Starmans
You can also search for this author inPubMed Google Scholar - Cindy Q. Yao
You can also search for this author inPubMed Google Scholar - Mircea Ivan
You can also search for this author inPubMed Google Scholar - Cristina Ivan
You can also search for this author inPubMed Google Scholar - Chad V. Pecot
You can also search for this author inPubMed Google Scholar - Paul C. Boutros
You can also search for this author inPubMed Google Scholar - Anil K. Sood
You can also search for this author inPubMed Google Scholar - Marianne Koritzinsky
You can also search for this author inPubMed Google Scholar - Bradly G. Wouters
You can also search for this author inPubMed Google Scholar
Contributions
T.v.d.B., E.K., M.K. and B.G.W. designed and conceived the study; T.v.d.B., E.K. and B.G.W. wrote the manuscript; P.P., M.A., J.W.V. and B.G.W. performed the ChIP-seq study; K.C., P.C.B., S.H., C.Q.Y., M.H.W.S., A.L.H, F.M.B., M.I. and C.I. performed analysis of breast cancer data sets. E.K., R.R., C.V.P. and A.K.S. performed the in vivo study. All authors edited and approved the final manuscript.
Corresponding author
Correspondence toBradly G. Wouters.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
About this article
Cite this article
van den Beucken, T., Koch, E., Chu, K. et al. Hypoxia promotes stem cell phenotypes and poor prognosis through epigenetic regulation of DICER.Nat Commun 5, 5203 (2014). https://doi.org/10.1038/ncomms6203
- Received: 20 January 2014
- Accepted: 09 September 2014
- Published: 29 October 2014
- DOI: https://doi.org/10.1038/ncomms6203