C. elegans ced-13 can promote apoptosis and is induced in response to DNA damage (original) (raw)
Introduction
A major pathway leading to cell death following DNA damage involves activation of the p53 tumor suppressor protein.1, 2, 3, 4, 5 The ability of p53 to act as a transcriptional activator is thought to be important for the induction of apoptosis, since mutant p53 proteins that are defective in transcriptional activation fail to promote apoptosis of cells experiencing DNA damage.6, 7 Numerous transcriptional targets of p53 have been described. Among these are several cell death genes including Bax, Apaf-1, Pidd and two BH3 (Bcl-2 homology region 3)-only genes, PUMA and Noxa.8, 9, 10, 11, 12, 13, 14, 15 The BH3 domain is a weakly conserved motif found in a variety of proteins that regulate apoptosis.16 Structural studies suggest that this domain adopts an amphipathic alpha-helical conformation.17, 18, 19 Proteins containing a BH3 domain as their only recognizable motif (BH3-only proteins) promote apoptosis in a number of settings. For example, in mammals, stimulation of the Fas receptor by ligand can trigger cell death by a number of mechanisms. For example, Fas can activate caspase-8, which in turn can directly activate caspase-3 and promote cell death. Caspase-8 can also cleave Bid, a BH3-only protein. Cleaved Bid associates with mitochondria and can promote cell death.20, 21 Bim, another BH3-only protein, is required for cytokine deprivation-induced apoptosis and seems to be involved in some aspect of negative selection in the immune system.22, 23 Mice lacking a functional PUMA gene are severely impaired in apoptosis of hematopoietic cells, fibroblasts and neurons, following exposure to ionizing radiation (IR), suggesting that PUMA is an important cell death target of p53.24, 25 Mice harboring a deletion of Noxa may also be deficient in IR-induced apoptosis, but to a much lesser degree than PUMA mutants.25, 26 Thus, in mammals, two BH3-only genes, PUMA and Noxa, are transcriptionally induced by p53 to promote cell death in response to DNA damage.
Apoptosis in Caenorhabditis elegans normally occurs during the development of somatic tissues and the maturation of female germ cells, as well as in the female germ line in response to DNA damage.27, 28, 29 The C. elegans BH3-only protein EGL-1 is required for many, if not all, somatic programmed cell deaths and for DNA-damage-induced germ-cell death.28, 30 The egl-1 gene acts upstream of the genes ced-9, ced-4 and ced-3, which encode a Bcl-2-like cell death inhibitor, an Apaf-1-like adaptor protein and a caspase, respectively. Furthermore, EGL-1 protein physically interacts with the antiapoptotic CED-9 protein. These observations suggest that EGL-1 inhibits CED-9 activity, allowing CED-4 to activate the CED-3 caspase, thus promoting apoptosis.27, 30, 31 Here we describe a second C. elegans BH3-only protein, CED-13. ced-13 mRNA accumulates following IR exposure in a _cep-1/p53_-dependent manner. We show that CED-13 can interact with CED-9 and promote apoptosis. These results are consistent with a conserved pathway in which p53 activates the transcription of two apoptosis-promoting BH3-only proteins in response to DNA damage.
Results
ced-13 encodes a conserved BH3-only protein
To identify regulators of programmed cell death in C. elegans, we searched available translated genomic sequences using the BLAST program32 for peptides with similarities to BH3 domains. We identified three such peptides: one corresponded to the EGL-1 protein, a second corresponded to the C. elegans Bnip3 protein,33 and a third was encoded by a genomic fragment of cosmid R09F10 not previously annotated as an open reading frame (ORF) (Figure 1a). To determine whether the region of R09F10 identified was part of a protein-coding gene, we inspected the genomic sequence manually and assembled a putative gene structure. Using the polymerase chain reaction (PCR) and primers corresponding to the predicted gene, we were able to amplify a cDNA from a mixed-stage cDNA library we constructed34 (S Shaham, unpublished data). The existence of a cDNA suggested that the region of R09F10 we identified indeed contained a transcribed gene, designated R09F10.9. Northern blots of purified mixed-stage C. elegans mRNA probed with the 32P-labelled cDNA failed to detect any mRNA, suggesting that R09F10.9 was expressed at low levels (data not shown). We deduced the structure of R09F10.9 by comparing the sequences of the cDNA and cosmid R09F10. Three features of the assembled structure suggested that R09F10.9 might play a role in apoptosis. First, the structure of the gene revealed a single intron in a 5′ noncoding region and a single intron within the protein-coding region (Figure 1b). This structure as well as the overall size of R09F10.9 resembled that of the egl-1 gene, suggesting that these genes may be related. Second, alignment of the amino-acid sequences of the EGL-1 protein and the predicted R09F10.9 protein (Figure 1c) revealed a distinct stretch of similarity (13/21 identical amino acids) surrounding the putative BH3 domain. Significantly, all residues contributing to the putative hydrophobic face of the BH3 helix were conserved. Third, the syntenic genomic region of the related nematode Caenorhabditis briggsae contains sequences that are most similar to R09F10.9 in the region surrounding the BH3 domain and similar sequences exist in the Caenorhabditis remanei genome (Figure 1d), suggesting functional importance. We thus named R09F10.9 ced-13 (ced, cell death abnormal; see below).
Figure 1
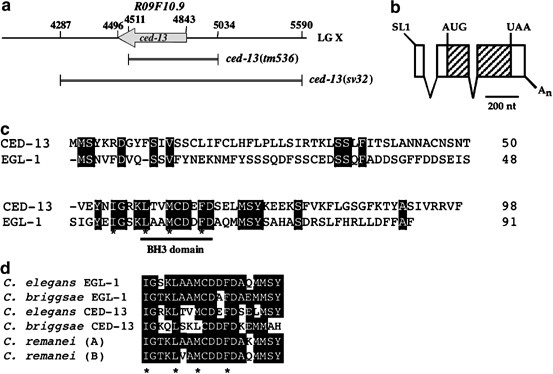
ced-13 encodes a conserved BH3-only protein. (a) A map of the region of cosmid R09F10 containing R09F10.9/ced-13. Numbers indicate sequence positions along R09F10. The arrow indicates direction of transcription. Double lines flanked by vertical bars delineate the extent and end points, respectively, of two deletion alleles of the locus: sv32 and tm536. (b) Predicted ced-13 mRNA structure as deduced from RT-PCR and cDNA sequencing experiments. Empty boxes, noncoding exon regions; hatched boxes, coding exon region; V figures, spliced introns; SL1, spliced leader sequence; A n , poly A tail. The start (AUG) and stop (UAA) codons are indicated. (c) An alignment of CED-13 and EGL-1 amino-acid sequences. Identical residues are boxed. The putative BH3 domains of EGL-1 and CED-13 are indicated by a horizontal bar under the alignment. Hydrophobic residues that are predicted to lie on one face of the BH3 domain helix are indicated by asterisks. (d) An alignment of the putative BH3 domains of EGL-1 and CED-13 proteins from C. elegans, C. briggsae and C. remanei. For C. briggsae, we could assign egl-1 and ced-13 coding regions by synteny with the C. elegans genes. For C. remanei, the genome has not yet been assembled, so the two proteins are designated A and B. Asterisks indicate hydrophobic residues that are predicted to lie on one face of the BH3 domain helix
Overexpression of ced-13 promotes apoptosis
To test whether ced-13 indeed has a role in apoptosis, we examined three independent C. elegans lines harboring stably integrated transgenes consisting of the ced-13 cDNA fused to a heat-shock-inducible promoter (PHS).35 Embryos examined 2 h after heat shock contained many more dying cells, as viewed using differential interference contrast microscopy (DIC), than embryos that were not exposed to a heat shock (Table 1 ). Heat-shocked transgenic embryos uniformly succumbed and did not hatch. These results suggest that CED-13 can promote apoptosis in C. elegans.
Table 1 Overexpression of ced-13 promotes programmed cell death
To determine the mechanism by which CED-13 causes cell death, we examined the ability of the PHS_ced-13_ transgenes to cause ectopic cell death in animals containing mutations in components of the core apoptotic machinery. Loss-of-function mutations in egl-1, ced-4 and ced-3 and a gain-of-function mutation in ced-9, block most, if not all, apoptotic cell deaths that occur in somatic tissues during C. elegans development.27 As shown in Table 1, transgenic embryos harboring loss-of-function mutations in the ced-3 or ced-4 genes, or a gain-of-function mutation in the ced-9 gene, did not contain dying cells after a heat shock. Furthermore, all such embryos hatched and developed into fertile adults. These results suggest that _ced-13_-induced cell death required ced-3 and ced-4 function and could be blocked by ced-9. Transgenic embryos containing a loss-of-function mutation in egl-1, however, consistently had slightly more dying cells after a heat shock when compared to wild-type animals. These results suggest that egl-1 function was not required for cell death induction by ced-13. Thus, ced-13 can act upstream of ced-3, ced-4 and ced-9 and in parallel to egl-1 to induce apoptosis.
CED-13 binds to CED-9
The observation that ced-9 function could block _ced-13_-induced cell death suggested that the CED-13 and CED-9 proteins may interact directly. To determine whether CED-13 could bind CED-9, we examined their interaction in a two-hybrid assay. As shown in Figure 2a, coexpression of CED-13 and CED-9 proteins fused to the Saccharomyces cerevisiae Gal4p DNA-binding domain and Gal4p activation domain, respectively, or vice versa, promoted expression of a Gal4-dependent lacZ reporter gene, as assayed by both enzymatic activity and colorimetric assays. These results suggest that CED-13 can bind CED-9. To confirm this interaction, we performed co-precipitation experiments using a bacterially produced glutathione-_S_-transferase (GST)::CED-9 fusion protein and in vitro_-translated CED-13 protein. As shown in Figure 2b, CED-13 bound the GST::CED-9 fusion protein, whereas control proteins failed to do so. Furthermore, CED-13 did not bind GST alone, supporting the hypothesis that CED-13 and CED-9 interact specifically. To test whether the BH3 domain of CED-13 was important for the interaction with CED-9, we co-precipitated GST::CED-9 with a CED-13 protein lacking nine amino acids (L59–D67) containing the BH3 domain. As shown in Figure 2c, CED-13ΔBH3 bound CED-9 about 10 times less efficiently than wild-type CED-13. Furthermore, when overexpressed using a heat-inducible promoter, CED-13ΔBH3 failed to promote cell death. Specifically, embryos of three PHS_ced-13_ΔBH3 transgenic lines contained 3±2, 2±2 and 2±1 cell corpses, whereas three lines harboring wild-type PHS_ced-13 contained 24±10, 27±15 and 18±5 corpses. These results suggest that CED-13 requires its BH3 domain to bind CED-9 and promote cell death.
Figure 2
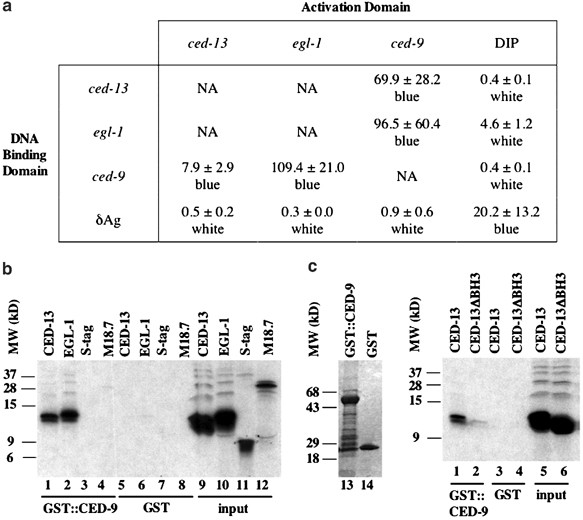
CED-13 binds CED-9. (a) Table summarizing two-hybrid interactions between CED-13 and CED-9. Each ORF was fused to DNA encoding either the DNA-binding domain or activation domain of the S. cerevisiae Gal4 protein and pairs of fusion constructs were cotransformed, as indicated, into yeast carrying a lacZ gene with a 5′ Gal4p DNA-binding site. LacZ activity was quantitated using Miller assays and numbers represent the average±standard deviation in Miller units of three separate experiments (see Materials and Methods). Each yeast strain was also used in a lacZ filter assay and the colors of the resulting patches following incubation with X-gal are indicated below the numbers. δAg (hepatitis delta virus antigen) and DIP (delta antigen interacting protein) are control proteins that are known to interact with each other in this assay.51 (b) Co-precipitation experiment of bacterially produced and purified GST::CED-9 (lanes 1–4) or GST (lanes 5–8) proteins and different _in vitro_-translated proteins (see Materials and Methods). Lanes 9–12 represent 50% of the _in vitro_-translated product used in the precipitation reaction. All _in vitro_-translated products contained a 6 kDa protein tag at their amino termini. S-tag and M18.7 are control proteins. S-tag contains the 6 kDa protein tag and additional unrelated sequences. M18.7 is a C. elegans protein. Lanes 13 and 14: GST::CED-9 and GST proteins used in the co-precipitation reactions separated on an SDS polyacrylamide gel and stained with Coomassie blue. Approximately 10 ng of either protein was used in the precipitation reactions. MW, molecular weight. (c) Similar to (b), except that CED-13 and CED-13ΔBH3 were the only in vitro translated proteins added to the precipitation reactions
Analysis of ced-13 mutants
The results presented above suggest a role for ced-13 in regulating apoptosis in C. elegans. To understand the function of ced-13 in vivo, we isolated two C. elegans mutants harboring deletions in the ced-13 locus (Figure 1a; see Materials and Methods). ced-13(sv32) animals lack the entire ced-13 coding region as well as 747 base pairs (bp) upstream of the ATG codon and 209 bp downstream of the TAA stop codon. ced-13(tm536) animals lack the entire ced-13 coding region, except for 15 bp upstream of the TAA stop codon, as well as 191 bp upstream of the ATG codon. Neither deletion allele removes coding sequences of the adjacent genes R09F10.4 and R09F10.3. sv32 and tm536, therefore, are likely to represent null alleles of the ced-13 gene. We found no defects in programmed cell death of somatic cells during development in either ced-13 mutant (Table 2 ). Furthermore, we did not observe increased cell death in double mutant combinations of ced-13(sv32) or ced-13(tm536) with the strong loss-of-function alleles egl-1(n1084n3082), ced-4(n1162), ced-3(n717), or ced-1(e1735), or the gain-of-function allele ced-9(n1950) and no reduction in cell death in double mutant combinations of ced-13(sv32) or ced-13(tm536) with the weak loss-of-function alleles egl-1(n3331), ced-4(n3312), or ced-3(n2438). Similarly, ced-13(sv32) and ced-13(tm536) did not affect the level of physiological germ-cell death, the process by which about 50% of all germ cells in the female gonad are eliminated, in an otherwise wild-type background or in a background containing the egl-1 loss-of-function mutation n3082n1084 (Figure 3a, data not shown).
Table 2 ced-13 does not control cell death in the pharynx
Figure 3
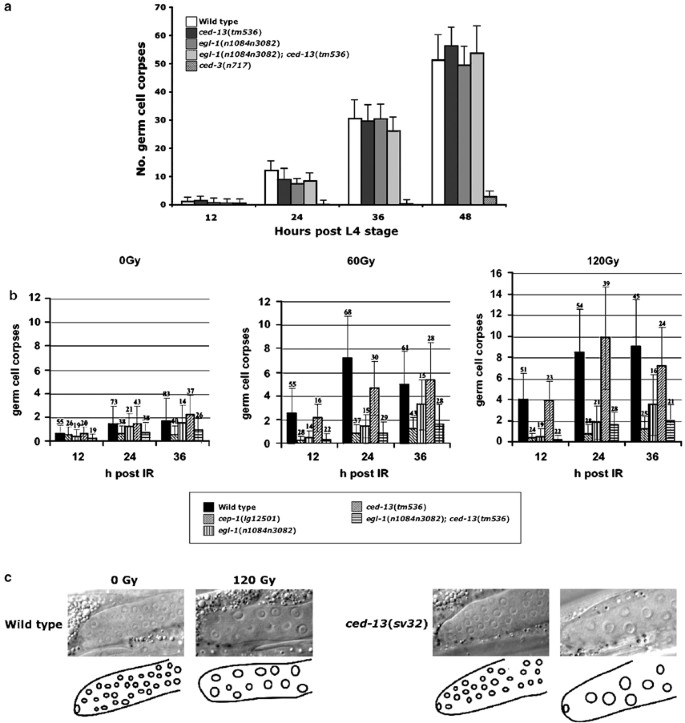
Phenotypic studies of ced-13 mutants. (a) ced-13 is not required for physiological germ-cell death. A representative histogram depicting the accumulation of apoptotic germ cells in adult C. elegans hermaphrodites in the absence of exposure to IR. All strains contained the ced-6 mutation n2095, which blocks engulfment of apoptotic cells as well as the transgene bcIs39 (P lim-7 _ced-1_-gfp), which marks apoptotic cells to allow for accurate scoring of cell corpse accumulation. Apoptotic cells were scored using DIC and epifluorescence as described in Materials and Methods. A total of 15–20 animals were observed per strain per time point. Data shown are from one representative experiment of three such experiments performed. No. germ-cell corpses, sum of apoptotic cells observed. Error bars represent the standard deviation. (b) IR-induced germ-cell death in ced-13 mutants. The histogram depicts the number of germ-cell corpses observed, following exposure to X-rays of the indicated doses, of animals of the indicated genotypes. Apoptotic cells were scored using DIC microscopy. ced-6(n2095) and bcIs39 were not present in the strains analyzed. Error bars represent the standard deviation. Numbers above bars represent the number of animals scored. (c) Cell-cycle arrest of dividing germ cells in ced-13(sv32) and wild-type animals following exposure to 120 Gy of X-rays. Images are DIC photographs of the mitotically active region of the female gonad. Note that in both strains arrested nuclei are larger in exposed animals, indicating that cell-cycle arrest has taken place
When we examined ced-13(sv32) and ced-13(tm536) hermaphrodites following exposure to IR, we obtained complex results. For example, in the experiment depicted in Figure 3b, we could detect no consistent difference in germ-cell deaths between ced-13(tm586) and wild-type animals. However, a consistent but weak difference was detected between egl-1(n1084n3082) animals and egl-1(n1084n3082); ced-13(tm586) animals across radiation doses and times postirradiation. In other experiments, we could also occasionally detect consistent weak differences between ced-13 mutants and wild-type animals (data not shown). We propose, therefore, that ced-13 might play a role in germ-cell death following IR treatment. In general, the number of dying germ cells that are observed varies dramatically in individual animals of a given genotype at a given time following IR exposure, resulting in standard deviations on the order of the mean. This variation might be due to differences in germ-cell proliferation rates as a consequence of slight temperature variations between strains, or to differences in the precise developmental time at which the animals are irradiated.36 Germ-cell death measurements can, therefore, be difficult to compare between experiments, even when many animals are scored. Thus, while large differences between strains are readily apparent, small but genuine differences between strains may not always be apparent. This contrasts with the developmental cell deaths that are often scored in the C. elegans pharynx, where the presence or absence of cells in an invariant lineage is assessed (e.g. Table 2). Thus, although loss of ced-13 may indeed result in a reduction in the number of dying germ cells, our inability to consistently reproduce this difference prevents us from concluding this with certainty.
If ced-13 does play a role in IR-induced germ-cell death, it could function upstream of or as part of the DNA damage response checkpoint (as defined by the mrt-2 gene, for example37), or downstream of this checkpoint. Two observations suggest that ced-13 may function downstream of the checkpoint. First, in response to radiation, ced-13 mutant germ cells arrested cell-cycle progression in a manner indistinguishable from wild-type germ cells (Figure 3c, data not shown). Second, mutants in the DNA damage response checkpoint produce many inviable progeny, presumably because of the accumulation of unrepaired DNA lesions in these animals. Mortality of ced-13 embryos was indistinguishable from wild-type animals over a range of radiation doses. For example, as in the wild type, about 50% of ced-13(sv32) or ced-13(tm536) embryos derived from irradiated parents survived following 120 gray (Gy) of X-irradiation, whereas 0% of mrt-2 embryos survived (data not shown).
ced-13 mRNA accumulation following IR treatment depends on cep-1/p53
The possible role of ced-13 in DNA-damage-induced apoptosis, together with the low levels of the ced-13 transcript in unirradiated animals (see above), suggested that although ced-13 was unlikely to be a checkpoint control gene, ced-13 transcription may be specifically induced in response to DNA damage. To test this hypothesis, we irradiated animals and followed ced-13 mRNA accumulation using quantitative real-time reverse transcriptase (RT)-PCR (qPCR). In unirradiated wild-type animals, we consistently detected only very low levels of ced-13 mRNA. However, when we treated wild-type animals with increasing doses of IR, ced-13 transcript was induced up to 10-fold (Figures 4a and b). The dose-dependent induction of ced-13 was comparable to egl-1 induction (Figure 4c). To examine whether ced-13 mRNA accumulation was confined to germ cells, we examined mRNA levels in glp-4(bn2) mutants following IR treatment. glp-4(bn2) mutants grown at 25°C lack germ cells, whereas those grown at 15°C possess a germ line.38 We found that at 15°C, glp-4 animals exposed to IR accumulated ced-13 transcript as in wild-type animals. However, glp-4 mutants grown at 25°C did not accumulate ced-13 mRNA following IR exposure (Supplementary Figure 1). These results suggest that ced-13 accumulation after IR treatment occurs primarily in the germ line.
Figure 4
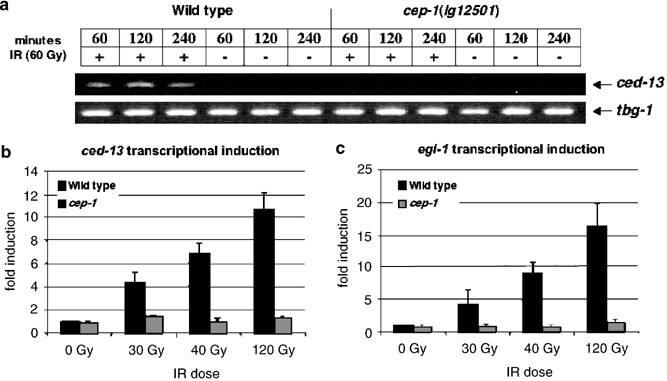
Increase in ced-13 and egl-1 mRNA levels following IR requires cep-1. (a) Wild-type or cep-1(lg12501) animals were examined for the presence of ced-13 transcript under the conditions indicated. Transcript was detected by RT-PCR followed by separation of amplified cDNAs on an agarose gel. Minutes, time following irradiation; IR, exposure (+) or no exposure (−) to X-irradiation. (b) ced-13 and (c) egl-1 quantitative real-time RT-PCR following exposure of wild-type or cep-1(lg12501) animals to increasing doses of X-rays. Average of two representative qPCR reactions is shown. Error bars indicate standard deviation. Fold induction, comparison to cDNAs amplified from age-matched unirradiated animals
The p53 tumor suppressor gene functions in mammalian cells to induce both cell-cycle arrest and apoptosis.1 Mutations in the C. elegans p53-related gene cep-1 prevent germ-cell apoptosis in response to DNA damage4, 5 but not cell-cycle arrest,4, 5, suggesting that cep-1 may function downstream of the DNA damage checkpoint. To examine whether cep-1 was required to induce ced-13 transcription following DNA damage, we examined ced-13 mRNA accumulation in response to IR in animals lacking a functional cep-1 gene. As shown in Figure 4, cep-1(lg12501) mutants failed to accumulate ced-13 (and egl-1) transcript even at high radiation doses. CEP-1 protein can function as a transcription factor and will bind to human p53 consensus binding sites.5 Interestingly, the ced-13 promoter contains several potential p53-binding sites, including two identical palindromic sites of sequence AAACATGTTT located 364 and 402 bp upstream of the ced-13 start codon. These sites match the p53 half binding-site consensus sequence RRRCWWGYYY.39 Furthermore, sequence inspection of the syntenic region in the related nematode C. briggsae revealed one copy of the identical half-site upstream of the putative C. briggsae ced-13 gene (data not shown). Taken together, these results support a model in which CEP-1 protein, activated by DNA damage, binds directly to the ced-13 promoter to activate ced-13 transcription. CED-13 protein, in turn, could bind to and inhibit CED-9, resulting in the activation of CED-4 and subsequent activation of CED-3 to promote cell death (Figure 5).
Figure 5
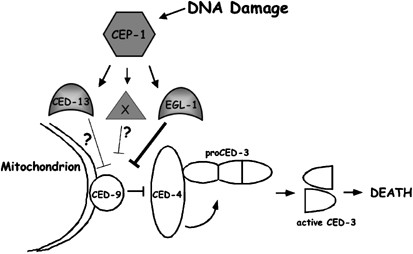
Model for DNA-damage-induced apoptosis in C. elegans female germ cells. DNA damage activates CEP-1/p53 in an unknown way, allowing for transcription of ced-13 and egl-1. CED-13 and EGL-1 inhibit CED-9/Bcl-2, allowing CED-4/Apaf-1 to activate the CED-3 caspase, leading to cell death. CED-13 activity is much lower than that of EGL-1 (fainter inhibitory arrow), either because it is inherently weaker or because it is induced in response to rare types of DNA damage
Discussion
Two mammalian BH3-only genes, Noxa and PUMA, are transcriptionally induced by p53 in response to DNA damage.9, 10, 13, 14, 15 Mice deficient in PUMA display severe defects in apoptosis of various cell types in response to DNA damage induced by IR or etoposide, suggesting that PUMA is important for DNA-damage induced apoptosis.24, 25 Cells from mice deficient in Noxa, however, are generally not resistant to DNA-damage-induced apoptosis, although fibroblasts derived from these mice may show minor defects in apoptosis following etoposide treatment.25 Neither PUMA- nor Noxa-deficient cells were as resistant to DNA damage as cells lacking p53. These observations indicate that Noxa and PUMA act downstream of p53 to promote apoptosis. Interestingly, the C. elegans BH3-only gene egl-1 has a significant role in radiation-induced germ-cell apoptosis,28 and, as shown in Figure 4c, egl-1 mRNA also accumulated in a p53-dependent fashion following irradiation (see also Hofmann et al.40). Thus, as in mammals, C. elegans possesses two BH3-only proteins that are regulated by p53. EGL-1, like PUMA, has a major role in DNA-damage-induced death and CED-13, like Noxa, may play a minor role. Furthermore, as in mammals, egl-1 or ced-13 single mutants, or egl-1; ced-13 double mutants were not as resistant to DNA-damage-induced apoptosis as cep-1/p53 mutants alone. This result further supports the notion that there may be additional cep-1/p53 responsive genes that are important for promoting DNA-damage-induced apoptosis (Figure 5).
The conservation of ced-13 sequences between C. elegans and the related nematodes C. briggsae and C. remanei (Figure 1d) suggests that there has been evolutionary pressure to conserve this gene, despite minor defects in animals lacking ced-13. Why, then, might two BH3-only proteins be involved in DNA-damage-induced apoptosis in C. elegans and in mammals? One possibility is that each protein performs a different biochemical/cell biological function during apoptosis and that these different activities are required for efficient and reproducible control of cell killing in response to DNA damage. For example, both CED-13 and EGL-1 may bind to CED-9; however, CED-13 may specifically bind to a rare conformation of CED-9. In this model, the surprisingly weak phenotype of ced-13 mutants suggests that ced-13 might serve to fine-tune the cell death process (Figure 5). Alternatively, although perhaps less likely, it is possible that both genes have the same functions, but respond to different stimuli. Thus, for example, egl-1 may become activated more generally, in response to a variety of cellular stressors, whereas ced-13 may be activated in response to a specific and rare type of DNA damage, such as persistent double-strand breaks that might occur during meiotic recombination. Our observation that ced-13 kills better in the absence of egl-1 (Table 1) hints at possible complexity in the interactions between the protein products of the two genes. The conservation of the DNA damage response and apoptotic pathways between C. elegans and vertebrates suggests that similar interactions between BH3-only proteins might also regulate the in vivo responses of vertebrate cells to DNA damage.
Materials and Methods
General methods and strains
All strains were grown at 20°C as described previously.41 The wild-type strain used was C. elegans var. Bristol (N2). Alleles used in this study are listed below and are described by Hodgkin42 except where noted otherwise: LGI: ced-1(e1735), glp-4(bn2),38 cep-1(lg12501);5 LGIII: ced-4(n1162, n3312), ced-6(n2095), ced-9(n1950); LGIV: ced-3(n717, n243843); LGV: egl-1(n1084n3082,30 n333144), bcIs39 (P lim-7 _ced-1_-gfp;45 this study); LGX: ced-13(sv32, tm536) (this study), lin-15(n765).
Isolation of ced-13 mutants and generation of transgenic animals
ced-13(sv32) and ced-13(tm536) were isolated using UV/trimethylpsoralen mutagenesis followed by identification of mutants using PCR and sib selection as previously described.46 Strains were backcrossed five times. Animals containing PHS_ced-13_ transgenes were generated by integration of extrachromosomal transgenes harboring both a PHS_ced-13_ plasmid and marker plasmid, pRF4, encoding the rol-6(su1006) allele, as previously described.31 Animals containing the P lim-7 _ced-1_-gfp transgene bcIs39 were generated by integrating an extrachromosomal transgene harboring the P lim-7 _ced-1_-gfp plasmid (pZZ63045) and plasmid pL15-EK (lin-15(+)47) in a lin-15(n765) background. Animals carrying the stably integrated DNA were backcrossed four times to N2.
Microscopic analysis of mutant animals
Overexpression in somatic cells: Following heat shock, embryos were mounted for observation using DIC, as previously described,31 and the number of cell corpses was counted. Somatic cell death: Extra cells in the anterior pharynx were counted in larvae at the L3 and L4 stages of development as previously described.48 Physiological germ-cell death: animals were picked at the L4 stage and transferred to 9-cm plates containing 100 μl Escherichia coli OP50 solution seeded the day before the experiment and incubated overnight at room temperature. Germ-cell death was measured 12, 24, 36 and 48 h after the L4 stage. Dead germ cells were counted using a GFP reporter specific for apoptotic cells (P lim-7 _ced-1_-gfp) and DIC, as not all dead germ cells were visible with either method alone. The different strains were counted double blind. IR-induced germ-cell death: Animals were picked at the L4 stage and exposed to the indicated radiation dose from a Siemens X-ray source. IR-induced germ-cell death was measured at 12, 24, 36 and 48 h post-IR using DIC.
Protein interaction studies
We used vector pAS1 for activation domain fusion constructs and vector pACT2 for DNA-binding domain fusion constructs in the two-hybrid assays.49 Miller assays: 1 ml of log-phase yeast cells was centrifuged for 5 min, washed with Z buffer (16.1 g Na2HPO4·7H2O, 5.5 g NaH2PO4·H2O, 0.75 g KCl, 0.246 g MgSO4·7H2O in 1 l water) and resuspended in Z buffer with beta-mercaptoethanol (BME; 27 μl/10 ml), 50 μl CHCl3 and 20 μl 0.1% SDS and vortexed for 15 s. 700 μl of _o_-nitrophenyl-beta-D-galactopyranoside (ONPG) substrate (1 mg/ml in Z buffer+BME) was added and the reaction was incubated for 30 min at 30°C and stopped with 1 M Na2CO3. Reaction was centrifuged and absorption of supernatant measured at 420 nm. Miller units=_A_420 × 1000/(_A_600 × time (min) × volume (ml)). LacZ filter assays: Yeast were grown in patches on plates of selective media and replica plated to a second set of plates containing a Whatman 50 circular filter. Patches were grown overnight, filter papers were then removed and frozen in liquid N2 for 10 s. Each filter was then placed on top of a second filter paper soaked in a solution of 30 μl 2% X-gal in 2.5 ml Z buffer and incubated at 37°C. GST pull-down experiments: 10 ng of glutathione Sepharose purified GST fusion protein was incubated with binding buffer (142.5 mM KCl, 10 mM HEPES (pH 7.6), 5 mM MgCl2, 1 mM EDTA, 0.25% IGEPAL CA-630/Sigma, 2.5 mg/ml BSA) and _in vitro_-translated protein (TNT system, Promega) translated from vector pCITE-4a(+) (Novagen) containing cDNA sequences of the protein of interest for 60 min at 4°C. Complexes were washed 5 × in binding buffer and analyzed by SDS–PAGE.
ced-13 and egl-1 transcriptional regulation
Approximately 2000 worms per sample were developmentally synchronized by bleaching and then treated with IR. Total RNA was extracted with 1 ml GibcoBRL Trizol according to the manufacturer's instructions. cDNA synthesis was performed using Clontech ‘Advantage RT for PCR kit’ and 1.5 μg of total RNA per reaction, according to the manufacturer's instructions. Quantitative PCR (qPCR) was performed with a Roche LightCycler and FastStart DNA Master SYBR Green 1 kit (Roche Applied Science) according to the manufacturer's instructions. Primer pairs (for egl-1 878 5′-tactcctcgtctcaggactt-3′ and 880 5′-catcgaagtcatcgcacat-3′; for ced-13 706 5′-acggtgtttgagttgcaagc-3′ and 707 5′-gtcgtacaagcgtgatggat-3′; for tbg-1 710 5′-cgtcatcagcctggtagaaca-3′ and 711 5′-tgatgactgtccacgttgga-3′) were designed to generate intron-spanning products of 110–150 bp. The generation of specific PCR products was confirmed by melting curve analysis (which measures product specificity by the decrease in the fluorescence signal when the PCR product is denatured), gel electrophoresis (using Roche Agarose MS for analyzing small PCR products) and sequencing. Each primer pair was tested with a logarithmic dilution of a cDNA mix to generate a linear standard curve (crossing point (CP) plotted versus log of template concentration), which was used to calculate the primer pair efficiency (_E_=10(−1/slope)). Gamma tubulin (tbg-1) mRNA was used as an external standard. For data analysis, the second derivative maximum method was applied and induction of egl-1 cDNA was calculated according to Pfaffl:50 (E egl-1 ΔCP(cDNA 0Gy-cDNA)egl_−_1)/(E tbg-1 ΔCP(cDNA 0Gy-cDNA)tbg_−_1).
Abbreviations
BH3:
Bcl-2 homology domain 3
IR:
ionizing radiation
ORF:
open reading frame
GST:
glutathione-_S_-transferase
Gy:
Gray
qPCR:
quantitative PCR
DIC:
differential interference contrast
References
- Michael D and Oren M (2002) The p53 and Mdm2 families in cancer. Curr. Opin. Genet. Dev. 12: 53–59
Article CAS PubMed Google Scholar - Baptiste N, Friedlander P, Chen X and Prives C (2002) The proline-rich domain of p53 is required for cooperation with anti-neoplastic agents to promote apoptosis of tumor cells. Oncogene 21: 9–21
Article CAS PubMed Google Scholar - Brodsky MH, Nordstrom W, Tsang G, Kwan E, Rubin GM and Abrams JM (2000) Drosophila p53 binds a damage response element at the reaper locus. Cell 101: 103–113
Article CAS PubMed Google Scholar - Derry WB, Putzke AP and Rothman JH (2001) Caenorhabditis elegans p53: role in apoptosis, meiosis and stress resistance. Science 294: 591–595
Article CAS PubMed Google Scholar - Schumacher B, Hofmann K, Boulton S and Gartner A (2001) The C. elegans homolog of the p53 tumor suppressor is required for DNA damage-induced apoptosis. Curr Biol 11: 1722–1727
Article CAS PubMed Google Scholar - Chao C, Saito S, Kang J, Anderson CW, Appella E and Xu Y (2000) p53 transcriptional activity is essential for p53-dependent apoptosis following DNA damage. EMBO J 19: 4967–4975
Article CAS PubMed PubMed Central Google Scholar - Jimenez GS, Nister M, Stommel JM, Beeche M, Barcarse EA, Zhang XQ, O'Gorman S and Wahl GM (2000) A transactivation-deficient mouse model provides insights into Trp53 regulation and function. Nat. Genet. 26: 37–43
Article CAS PubMed Google Scholar - Moroni MC, Hickman ES, Denchi EL, Caprara G, Colli E, Cecconi F, Muller H and Helin K (2001) Apaf-1 is a transcriptional target for E2F and p53. Nat. Cell Biol. 3: 552–558
Article CAS PubMed Google Scholar - Yu J, Wang Z, Kinzler KW, Vogelstein B and Zhang L (2003) PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc. Natl. Acad. Sci. USA 100: 1931–1936
Article CAS PubMed PubMed Central Google Scholar - Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T and Tanaka N (2000) Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288: 1053–1058
Article CAS PubMed Google Scholar - Lin Y, Ma W and Benchimol S (2000) Pidd, a new death-domain-containing protein, is induced by p53 and promotes apoptosis. Nat. Genet. 26: 122–127
Article CAS PubMed Google Scholar - Miyashita T and Reed JC (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80: 293–299
Article CAS PubMed Google Scholar - Yu J, Zhang L, Hwang PM, Kinzler KW and Vogelstein B (2001) PUMA induces the rapid apoptosis of colorectal cancer cells. Mol. Cell 7: 673–682
Article CAS PubMed Google Scholar - Nakano K and Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 7: 683–694
Article CAS PubMed Google Scholar - Han J, Flemington C, Houghton AB, Gu Z, Zambetti GP, Lutz RJ, Zhu L and Chittenden T (2001) Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc. Natl. Acad. Sci. USA 98: 11318–11323
Article CAS PubMed PubMed Central Google Scholar - Chittenden T (2002) BH3 domains: intracellular death-ligands critical for initiating apoptosis. Cancer Cell 2: 165–166
Article CAS PubMed Google Scholar - Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS, Nettesheim D, Chang BS, Thompson CB, Wong SL, Ng SL and Fesik SW (1996) X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature 381: 335–341
Article CAS PubMed Google Scholar - McDonnell JM, Fushman D, Milliman CL, Korsmeyer SJ and Cowburn D (1999) Solution structure of the proapoptotic molecule BID: a structural basis for apoptotic agonists and antagonists. Cell 96: 625–634
Article CAS PubMed Google Scholar - Chou JJ, Li H, Salvesen GS, Yuan J and Wagner G (1999) Solution structure of BID, an intracellular amplifier of apoptotic signaling. Cell 96: 615–624
Article CAS PubMed Google Scholar - Li H, Zhu H, Xu CJ and Yuan J (1998) Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94: 491–501
Article CAS PubMed Google Scholar - Luo X, Budihardjo I, Zou H, Slaughter C and Wang X (1998) Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94: 481–490
Article CAS PubMed Google Scholar - Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, Adams JM and Strasser A (1999) Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis and to preclude autoimmunity. Science 286: 1735–1738
Article CAS PubMed Google Scholar - Bouillet P, Purton JF, Godfrey DI, Zhang LC, Coultas L, Puthalakath H, Pellegrini M, Cory S, Adams JM and Strasser A (2002) BH3-only Bcl-2 family member Bim is required for apoptosis of autoreactive thymocytes. Nature 415: 922–926
Article CAS PubMed Google Scholar - Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, McKinnon PJ, Cleveland JL and Zambetti GP (2003) Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 4: 321–328
Article CAS PubMed Google Scholar - Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM and Strasser A (2003) p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 302: 1036–1038
Article CAS PubMed Google Scholar - Shibue T, Takeda K, Oda E, Tanaka H, Murasawa H, Takaoka A, Morishita Y, Akira S, Taniguchi T and Tanaka N (2003) Integral role of Noxa in p53-mediated apoptotic response. Genes Dev. 17: 2233–2238
Article CAS PubMed PubMed Central Google Scholar - Metzstein MM, Stanfield GM and Horvitz HR (1998) Genetics of programmed cell death in C. elegans: past, present and future. Trends Genet. 14: 410–416
Article CAS PubMed Google Scholar - Gartner A, Milstein S, Ahmed S, Hodgkin J and Hengartner MO (2000) A conserved checkpoint pathway mediates DNA damage-induced apoptosis and cell cycle arrest in C. elegans. Mol. Cell 5: 435–443
Article CAS PubMed Google Scholar - Gumienny TL, Lambie E, Hartwieg E, Horvitz HR and Hengartner MO (1999) Genetic control of programmed cell death in the Caenorhabditis elegans hermaphrodite germline. Development 126: 1011–1022
CAS PubMed Google Scholar - Conradt B and Horvitz HR (1998) The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9. Cell 93: 519–529
Article CAS PubMed Google Scholar - Shaham S and Horvitz HR (1996) Developing Caenorhabditis elegans neurons may contain both cell-death protective and killer activities. Genes Dev. 10: 578–591
Article CAS PubMed Google Scholar - Altschul SF, Gish W, Miller W, Myers EW and Lipman DJ (1990) Basic local alignment search tool. J. Mol. Biol. 215: 403–410
Article CAS PubMed Google Scholar - Yasuda M, D'Sa-Eipper C, Gong XL and Chinnadurai G (1998) Regulation of apoptosis by a Caenorhabditis elegans BNIP3 homolog. Oncogene 17: 2525–2530
Article CAS PubMed Google Scholar - Shaham S and Bargmann CI (2002) Control of neuronal subtype identity by the C. elegans ARID protein CFI-1. Genes Dev. 16: 972–983
Article CAS PubMed PubMed Central Google Scholar - Stringham EG, Dixon DK, Jones D and Candido EP (1992) Temporal and spatial expression patterns of the small heat shock (hsp16) genes in transgenic Caenorhabditis elegans. Mol. Biol. Cell 3: 221–233
Article CAS PubMed PubMed Central Google Scholar - Gartner A, MacQueen AJ and Villeneuve AM (2004) Methods for analyzing checkpoint responses in Caenorhabditis elegans. Methods Mol. Biol. 280: 257–274
CAS PubMed Google Scholar - Ahmed S and Hodgkin J (2000) MRT-2 checkpoint protein is required for germline immortality and telomere replication in C. elegans. Nature 403: 159–164
Article CAS PubMed Google Scholar - Beanan MJ and Strome S (1992) Characterization of a germ-line proliferation mutation in C. elegans. Development 116: 755–766
CAS PubMed Google Scholar - el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW and Vogelstein B (1992) Definition of a consensus binding site for p53. Nat. Genet. 1: 45–49
Article CAS PubMed Google Scholar - Hofmann ER, Milstein S, Boulton SJ, Ye M, Hofmann JJ, Stergiou L, Gartner A, Vidal M and Hengartner MO (2002) Caenorhabditis elegans HUS-1 is a DNA damage checkpoint protein required for genome stability and EGL-1-mediated apoptosis. Curr. Biol. 12: 1908–1918
Article CAS PubMed Google Scholar - Brenner S (1974) The genetics of Caenorhabditis elegans. Genetics 77: 71–94
CAS PubMed PubMed Central Google Scholar - Hodgkin J (1997) Genetics. In C. elegans II, Riddle D, Blumenthal T, Meyer BJ and Priess J, eds Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press pp. 898–1048
- Hengartner MO and Horvitz HR (1994) Activation of C. elegans cell death protein CED-9 by an amino-acid substitution in a domain conserved in Bcl-2. Nature 369: 318–320
Article CAS PubMed Google Scholar - Reddien PW, Cameron S and Horvitz HR (2001) Phagocytosis promotes programmed cell death in C. elegans. Nature 412: 198–202
Article CAS PubMed Google Scholar - Zhou Z, Hartwieg E and Horvitz HR (2001) CED-1 is a transmembrane receptor that mediates cell corpse engulfment in C. elegans. Cell 104: 43–56
Article CAS PubMed Google Scholar - Jansen G, Hazendonk E, Thijssen KL and Plasterk RH (1997) Reverse genetics by chemical mutagenesis in Caenorhabditis elegans. Nat. Genet. 17: 119–121
Article CAS PubMed Google Scholar - Clark SG, Lu X and Horvitz HR (1994) The Caenorhabditis elegans locus lin-15, a negative regulator of a tyrosine kinase signaling pathway, encodes two different proteins. Genetics 137: 987–997
CAS PubMed PubMed Central Google Scholar - Ellis RE and Horvitz HR (1991) Two C. elegans genes control the programmed deaths of specific cells in the pharynx. Development 112: 591–603
CAS PubMed Google Scholar - Bai C and Elledge SJ (1997) Gene identification using the yeast two-hybrid system. Methods Enzymol. 283: 141–156
Article CAS PubMed Google Scholar - Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29: e45
Article CAS PubMed PubMed Central Google Scholar - Brazas R and Ganem D (1996) A cellular homolog of hepatitis delta antigen: implications for viral replication and evolution. Science 274: 90–94
Article CAS PubMed Google Scholar
Acknowledgements
SS would like to acknowledge the late Ira Herskowitz for advice and support during the initial stages of this study. We thank Mary Abraham and Carine Waase for comments on the manuscript and Sebastian Greiss for assistance with the glp-4 studies. ST was supported by grants from Cancerfonden and The Wallenberg Foundation. BC was supported by the Max-Planck Society, the European Molecular Biology Organization (EMBO Young Investigator Award) and the Howard Hughes Medical Institute (HHMI award #76200-560801 to Dartmouth Medical School under the Biomedical Research Support Program for Medical Schools). AG was supported by the Max-Planck Society (Erich Nigg) and by the German DFG Grants GA703-1 to GA703-3. SS was supported by grants from the Helen Hay Whitney Foundation, the Kimmel Foundation and NIH Grant R01HD042680.
Author information
Author notes
- Edited by J Abrams
Authors and Affiliations
- Max-Planck-Institute of Biochemistry, Am Klopferspitz 18A, 82152, Martinsried, Germany
B Schumacher & A Gartner - Department of Genetics, Dartmouth Medical School, 7400 Remsen, Hanover, NH, 03755-3837, USA
C Schertel & B Conradt - Max-Planck-Institute of Neurobiology, Am Klopferspitz 18A, 82152, Martinsried, Germany
C Schertel, N Wittenburg & B Conradt - Umeå Center for Molecular Pathogenesis, Umeå University, SE-901 87, Umeå, Sweden
S Tuck - Department of Physiology, Tokyo Women's Medical University School of Medicine, 8-1, Kawada-cho, Shinjuku-ku, Tokyo, 162-8666, Japan
S Mitani - Laboratory of Developmental Genetics, The Rockefeller University, 1230 York Avenue, New York, NY, 10021, USA
S Shaham
Authors
- B Schumacher
You can also search for this author inPubMed Google Scholar - C Schertel
You can also search for this author inPubMed Google Scholar - N Wittenburg
You can also search for this author inPubMed Google Scholar - S Tuck
You can also search for this author inPubMed Google Scholar - S Mitani
You can also search for this author inPubMed Google Scholar - A Gartner
You can also search for this author inPubMed Google Scholar - B Conradt
You can also search for this author inPubMed Google Scholar - S Shaham
You can also search for this author inPubMed Google Scholar
Additional information
These authors contributed equally
Electronic supplementary material
Rights and permissions
About this article
Cite this article
Schumacher, B., Schertel, C., Wittenburg, N. et al. C. elegans ced-13 can promote apoptosis and is induced in response to DNA damage.Cell Death Differ 12, 153–161 (2005). https://doi.org/10.1038/sj.cdd.4401539
- Received: 20 July 2004
- Revised: 11 October 2004
- Accepted: 25 October 2004
- Published: 17 December 2004
- Issue Date: 01 February 2005
- DOI: https://doi.org/10.1038/sj.cdd.4401539