Integrating genomic alterations in diffuse large B-cell lymphoma identifies new relevant pathways and potential therapeutic targets (original) (raw)
- Original Article
- Open access
- Published: 14 August 2017
Lymphoma
- A Enjuanes1,3,
- I Dlouhy1,
- P Jares1,3,
- D Martin-Garcia1,3,
- F Nadeu ORCID: orcid.org/0000-0003-2910-94401,3,
- G R Ordóñez4,
- J Rovira1,
- G Clot1,3,
- C Royo1,
- A Navarro1,3,
- B Gonzalez-Farre1,3,
- A Vaghefi1,
- G Castellano1,
- C Rubio-Perez5,
- D Tamborero5,
- J Briones6,
- A Salar7,
- J M Sancho8,
- S Mercadal9,
- E Gonzalez-Barca9,
- L Escoda10,
- H Miyoshi11,
- K Ohshima11,
- K Miyawaki12,
- K Kato12,
- K Akashi12,
- A Mozos13,
- L Colomo1,7,
- M Alcoceba ORCID: orcid.org/0000-0002-3819-48463,14,
- A Valera1,
- A Carrió1,3,
- D Costa1,3,
- N Lopez-Bigas5,15,
- R Schmitz16,
- L M Staudt16,
- I Salaverria1,3,
- A López-Guillermo1,3 &
- …
- E Campo ORCID: orcid.org/0000-0001-9850-97931,3
Leukemia volume 32, pages 675–684 (2018)Cite this article
- 14k Accesses
- 134 Citations
- 9 Altmetric
- Metrics details
Subjects
Abstract
Genome studies of diffuse large B-cell lymphoma (DLBCL) have revealed a large number of somatic mutations and structural alterations. However, the clinical significance of these alterations is still not well defined. In this study, we have integrated the analysis of targeted next-generation sequencing of 106 genes and genomic copy number alterations (CNA) in 150 DLBCL. The clinically significant findings were validated in an independent cohort of 111 patients. Germinal center B-cell and activated B-cell DLBCL had a differential profile of mutations, altered pathogenic pathways and CNA. Mutations in genes of the NOTCH pathway and tumor suppressor genes (TP53/CDKN2A), but not individual genes, conferred an unfavorable prognosis, confirmed in the independent validation cohort. A gene expression profiling analysis showed that tumors with NOTCH pathway mutations had a significant modulation of downstream target genes, emphasizing the relevance of this pathway in DLBCL. An in silico drug discovery analysis recognized 69 (46%) cases carrying at least one genomic alteration considered a potential target of drug response according to early clinical trials or preclinical assays in DLBCL or other lymphomas. In conclusion, this study identifies relevant pathways and mutated genes in DLBCL and recognizes potential targets for new intervention strategies.
Similar content being viewed by others
Introduction
Diffuse large B-cell lymphoma (DLBCL) is a highly heterogeneous neoplasm.1 Although current therapies have improved the clinical outcome, 30–40% of the patients are still not cured.2 Understanding the molecular basis of this heterogeneity may facilitate the design of alternative management strategies including specific targeted therapies. The cell of origin (COO) of these tumors, germinal center B-cell (GCB) or activated B-cell (ABC), is one of the major sources of diversity associated with different molecular alterations and clinical evolution.3, 4, 5, 6, 7, 8, 9 More recently, next-generation sequencing (NGS) studies have provided a comprehensive catalog of somatic mutations in DLBCL that may also contribute to their heterogeneous behavior.10, 11, 12, 13 However, the number of patients analyzed is still relatively small and the clinical significance of these new mutations remains unknown. On the other hand, few NGS mutational studies have compared the mutational profile of the tumors with their respective chromosomal alterations.14 Therefore, an integrative view of these two layers of genomic information may provide a better understanding of their influence on the behavior of DLBCL.
One of the major goals of large-scale genomic analyzes of tumors is to identify new targets for therapeutic intervention. However, these comprehensive studies are confronted with the challenge of identifying appropriate candidate drugs for individual patients from the increasing catalog of available drugs that could be tested in new preclinical and clinical studies. The fulfillment of this major objective of precision oncology may require the assistance of bioinformatics tools that integrate the personalized genomic profiles of the tumors with the vast information of potential available drugs.15, 16
The goal of this study was to determine the clinical relevance of recurrent genomic alterations of DLBCL and their potential value in the management of patients. We have performed an integrated analysis of genomic alterations and mutations in a large panel of genes in DLBCL and run an in silico prescription strategy that connects the individual genomic profile with druggability options.16
Patients and methods
Patients and samples
One hundred fifty patients diagnosed with de novo DLBCL, not otherwise specified (NOS),1 from 2002 to 2014, including 14 primary extranodal cases, were selected for this study. Primary mediastinal large B-cell lymphomas and other DLBCL subtypes were excluded. Cases were selected based on the availability of high quality DNA obtained from frozen tissue samples with high tumor cell content (>60%). In the same period of time, 403 patients with DLBCL-NOS were not studied due to the lack of adequate material. These patients had similar clinical features to those of the included patients (Supplementary Table 1). The tumor COO, GCB, ABC or unclassified (UC), was established using U133 Plus 2.0 arrays (Affymetrix, Santa Clara, CA, USA) and/or the Lymph2Cx assay (NanoString technologies, Seattle, WA, USA).17 The patients’ main clinical features and outcome are detailed in Supplementary Figure 1 and Table 1. Most patients (126, 85%) were treated with a median of 6 courses (range, 1–6) of R-CHOP (rituximab, cyclophosphamide, adriamycin, vincristine and prednisone) and the remainder with regimens without adriamycin mainly due to their age or previous heart disease. Only patients receiving R-CHOP were included in the prognostic analyzes.
Table 1 Initial features and outcome of patients with DLBCL of the initial and validation series
A validation series of 111 patients (54M/57 F; median age 63 years) diagnosed over the same period of time was selected from different Spanish and Japanese institutions (Table 1). Ninety patients (86%) were treated with immunochemotherapy, including adriamycin-containing regimens and only these were included in prognostic studies. Patients in the initial and validation cohorts had similar features and outcome (Table 1 and Supplementary Figure 1). This study was approved by the Institutional Review Board of Hospital Clínic (Barcelona, Spain). Informed consent was obtained from all patients in accordance with the Declaration of Helsinki.
Targeted next-generation sequencing and mutational analysis
We performed targeted NGS of 106 genes selected from previous DLBCL genome sequencing studies (Supplementary Table 2 and Supplementary Methods).10, 11, 12, 13 Libraries were generated using HaloPlex (Agilent technologies, Santa Clara, CA, USA) and sequenced in a MiSeq instrument (Illumina, San Diego, CA, USA). Sequencing data have been deposited at the European Nucleotide Archive (ENA, http://www.ebi.ac.uk/ena) under accession number ERP021212. In addition, exon1α, 1β and 2 of CDKN2A and the 3′UTR region of NOTCH1 were analyzed by Sanger sequencing (Supplementary Methods). Two different bioinformatics pipelines (DreamGenics and SureCall tools) were used for the alignment and variant calling (Supplementary Methods). Combination of the two algorithms identified 1331 variant calls (Supplementary Table 3). The accuracy of the calls was confirmed by verifying 99% (151/152) of the selected variants by Sanger sequencing (Supplementary Methods). A selection of driver mutations with potential functional effect was performed based on the criteria described in Supplementary Methods and Supplementary Figure 2. Briefly, potential driver mutations included: (1) ‘truncating mutations’ (_n_=274), (2) ‘relevant mutations’ manually curated based on previous reports in the literature and COSMIC database. This group included somatic and functional mutations and mutations clustering in known functional domains (_n_=216), and (3) missense mutations identified as ‘functional mutations’ by the Mutation Assessor (MA), OncodriveCLUST and SIFT algorithms (_n_=271).18 To test the accuracy of our ‘functional prediction’ algorithm for missense mutations, we selected 92 variants in 32 patients who had germline DNA available. We observed that 90% of the mutations classified as functional were somatic (28/31) while 89% of the germline mutations were classified as non-functional (24/27) (Supplementary Methods and Supplementary Table S15). Taking these three criteria together, we selected 761 potential driver mutations for the clinicopathological analysis (Supplementary Table 4). Virtually all identified mutations (96.3%) showed allelic frequencies ⩾10%.
Thirteen mutated genes with significant clinical impact in the initial series were selected for validation in the independent cohort of patients. Libraries of these genes were generated using the Access-Array system (Fluidigm, South San Francisco, CA, USA) and Nextera XT (Illumina), sequenced and analyzed as described (Supplementary Methods).
Copy number analysis
DNA copy number alterations (CNA) were examined in 119 cases using Cytoscan HD arrays (Affymetrix) and analyzed using Nexus CN 7.5 Discovery edition (Biodiscovery, Hawthorne, CA, USA) as described.19 Minimal common regions of gain and loss, and copy number neutral loss of heterozygosity (CNN-LOH) were defined as described in Supplementary Methods. Deletions of CDKN2A locus were examined by quantitative PCR in the validation series (Supplementary Methods). Copy number data have been deposited at GEO database under accession number GSE94705.
Statistical methods
Complete response (CR), progression-free survival (PFS) and overall survival (OS) definitions were the standard ones.20 _χ_2 method was used for categorical variables and Student’s _t_-test for continuous variables. Non-parametric tests were applied when necessary. Actuarial survival analysis was performed by the Kaplan–Meier method and differences assessed by the log-rank test. Multivariate Cox regression analysis was used to assess the independent prognostic impact of different variables in terms of PFS and OS. The _P-_values for multiple comparisons were adjusted using the Benjamini–Hochberg correction. Statistical analyzes were carried out with SPSS v.22 and R software v3.1.3.
In silico drug prescription
Genomic-guided potential therapeutic opportunities for each DLBCL patient were identified in silico by using the Cancer Genome Interpreter modified from our previous described pipeline16 (https://www.cancergenomeinterpreter.org/). The platform matches the genomic alterations of a tumor with an expert manually curated database of genomic alterations that can be used as biomarkers of drug sensitivity, resistance and severe toxicity. The biomarkers database is organized according to the level of clinical evidence supporting the genotype–phenotype association including clinical guidelines, late (phases III–IV) or early clinical trials (phases I–II), case reports and preclinical studies.21 The biomarkers are classified as: (1) ‘Biomarker and tumor match’ for those alterations reported to be targets of specific drugs in DLBCL or other lymphoid neoplasms; (2) ‘Biomarker match of different gene mutation’ for those alterations reported to confer sensitivity to a given drug in DLBCL upon other amino acid changes and (3) ‘Biomarker match and tumor repurposing’ for those genomic alterations described as biomarkers of drug response in other cancers.15
Results
Mutational profile of DLBCL
A total of 761 potential driver mutations were identified in 89 out of the 106 genes with a similar number in GCB and ABC-DLBCL (4.8±2.8 vs 4.0±2.4 mutated genes per case, respectively) (Supplementary Table 4 and Supplementary Figure 3). The most frequently mutated genes were KMT2D, MYD88, CREBBP and TP53 found in more than 15% of cases whereas 27 additional genes were mutated in more than 5% of patients (Figure 1a). As expected, some genes carried mutations with the imprint of the somatic hypermutational machinery (BCL6, IRF4, IRF8, CIITA, PIM1, MYC, SOCS1, BCL7A, BTG1 and BTG2).22, 23 MYD88, PIM1, CD79B and PRDM1 were significantly more frequently mutated in ABC-DLBCL whereas KMT2D, CREBBP, TNFRSF14, B2M, EZH2, GNA13, FOXO1, ACTB and SOCS1 mutations were more common in GCB-DLBCL (Figure 1a, Supplementary Table 5). Interestingly, MYD88 L265P mutation was almost exclusively identified in ABC-DLBCL while non-L265P mutations were also seen in GCB or UC-DLBCL (Figure 1b).24 TP53 truncating and missense mutations on the DNA binding domain (DBD) were preferentially found in ABC-DLBCL, whereas other mutations in the gene were equally distributed in both DLBCL subtypes (Figure 1b).
Figure 1
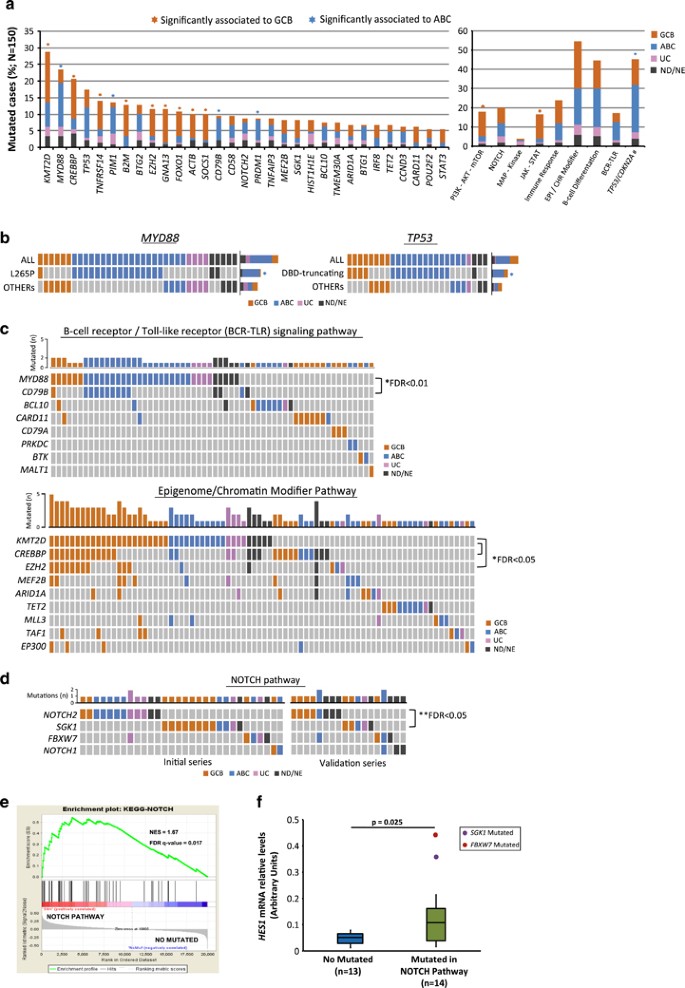
Recurrent mutated genes and pathways in 150 DLBCLs patients. (a) Bar-graphs show mutated genes in more than 5% of DLBCL patients and frequently mutated pathways. Each color bar indicates biological subtypes; GCB: germinal center B-cell type, ABC: activated B-cell type, UC: unclassified, ND/NE: not done or not evaluable. An asterisk represents mutated genes/pathways significantly enriched in one of the subtypes of COO and asterisk color denotes the enriched group. Tumor suppressor genes include mutations and deletions in TP53 and CDKN2A, respectively. (#) (b) Heat maps show the distribution of MYD88 and TP53 mutated patients in both DLBCL subtypes. TP53 mutations are divided into truncating and missense mutations located on the DNA binding domains (DBD) and ‘others’. Columns depict individual cases and rows mutated genes/mutation type. (c, d) Heat maps representing relationships among mutated genes in B-cell receptor (BCR)/Toll-like receptor signaling, Epigenome/Chromatin Modifier and NOTCH pathways. Graph-bars above show the total number of mutated cases for each gene. One black asterisk represents significant mutated gene concurrence and two asterisks significant exclusion. Significant _P_-values corrected by false discovery rate (FDR) are showed. (e) Gene-set enrichment analysis (GSEA) of NOTCH pathway mutated cases vs cases with no mutations in genes of this pathway. (f) Box plots show HES1 mRNA expression levels in NOTCH pathway mutated cases and cases with no mutations in this pathway.
To determine the possible interactions between mutated genes, we evaluated their patterns of association in the same tumors and within predefined pathogenic pathways (Figures 1c and d, Supplementary Table 6). MYD88 and CD79B mutations were significantly concurrent in the same tumors, particularly in ABC-DLBCL (FDR<0.01), whereas KMT2D mutations were associated with EZH2 and CREBBP mutations in GCB (FDR<0.05). Mutations in other epigenetic regulatory genes (MEF2B, ARID1A and EP300) were frequently seen in the same tumors but intriguingly, TET2 mutations never overlapped with mutations in other epigenetic genes. Mutations in genes of the B-cell receptor (BCR) signaling (CD79B, CARD11, BCL10, CD79A, BTK, PRKCB and MALT1) tended to occur in different cases although without statistical significance, probably due to the low number of cases for each gene (Figure 1c). Mutations in the PI3K/AKT/mTOR and JAK/STAT pathways were more frequent in GCB-DLBCL, whereas gene aberrations (mutations/deletions) in tumor suppressor genes (TP53, CDNK2A) were more represented in ABC-DLBCL (P<0.01) (Figure 1a).
We also observed mutations in genes of NOTCH pathway (NOTCH2, NOTCH1 and FBWX7) (Figure 1d). SGK1 has been suggested to be a negative regulator of NOTCH signaling enhancing NOTCH protein degradation and reducing its activation by the gamma-secretase but its potential role in lymphoid neoplasms has not been explored.25, 26 SGK1 mutations in our cases were frequently truncating and in some cases associated with loss of the wild-type allele suggesting that they may enhance NOTCH1 activity (Supplementary Table S4). On the other hand, NOTCH2 and SGK1 mutations were mutually exclusive (FDR<0.05) (Figure 1d). To evaluate whether SGK1 mutations could be considered in the NOTCH pathway in these tumors, we performed a gene-set enrichment analysis (GSEA) comparing SGK1 mutated and unmutated cases. SGK1 mutated DLBCL had a significant overexpression of genes upregulated by NOTCH activation in lymphoid cells and a concordant downregulation of gene signatures inhibited by NOTCH (Supplementary Figure 6A).27, 28 Based on these results we evaluated the relevance of NOTCH pathway in DLBCL comparing the gene expression profiling of 12 cases with NOTCH pathway mutations (5 NOTCH2, 4 SGK1, 2 NOTCH1 and 1 FBWX7) and 27 wild-type tumors (Supplementary Material). The GSEA found a significant overexpression of downstream signaling genes in cases with NOTCH pathway mutations compared with wild-type tumors (Figure 1e and Supplementary Figure 6B). In addition, a qRT-PCR analysis of HES1 expression showed significantly higher mRNA levels in cases with NOTCH pathway mutations (Figure 1f). All these findings are consistent with the downstream activation of NOTCH signaling in DLBCL with mutations in genes of this pathway.
Copy number and structural alterations
All tumors examined carried CNAs including 1226 losses, 56 homozygous deletions, 1112 gains, 96 amplifications and 270 regions of recurrent CNN-LOH (Supplementary Table 7). The profile of CNA and target genes in ABC and GCB-DLBCL were similar to those previously described (Figures 2a and b, Supplementary Methods and Supplementary Table 8). However, new alterations and potential target genes in the minimal common deleted regions were identified, including losses of TMEM30A (6q14.1) (39/119 33%, two homozygous) and EBF1 (5q33.3) (9/119, 8%, one homozygous) (Figure 2a). Additional homozygous deletions targeted CDKN2A (_n_=9), CD70 (_n_=2), PTEN (_n_=2), CD58 (_n_=1) and TNFAIP3 (_n_=1). Recurrent amplifications included REL/BCL11A (11 and 9 cases, respectively), miR17-92 cluster (_n_=7), CDK6 (_n_=6) and CDK14 (_n_=6) (Supplementary Table 8).
Figure 2
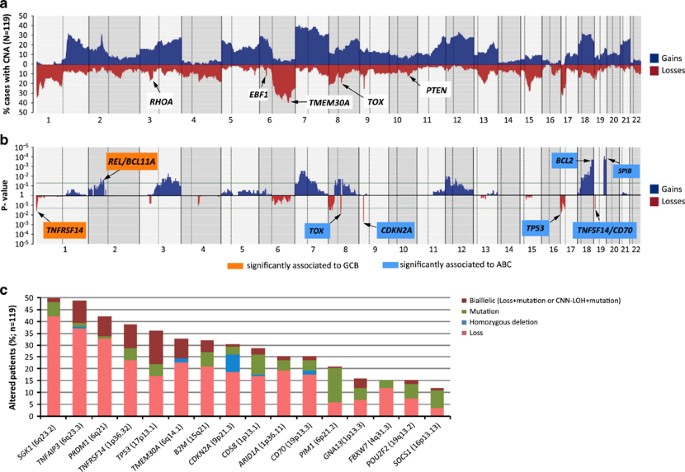
Copy number alterations (CNA) in 119 DLBCLs patients and integration with other genetic alterations. (a) Frequency of CNA of 119 DLBCL patients analyzed by Cytoscan HD assay. Each probe is aligned from chromosome 1 to 22 and p to q. Chromosomes X and Y were excluded from the analysis because sex-matched reference DNA samples were not used. The vertical axis indicates frequency of the genomic aberration among the analyzed cases. Gains are depicted in dark blue and losses are depicted in red. Genes affected by copy number alterations and not previously described in DLBCL are indicated. (b) Significant patterns of CNAs between DLBCL subtypes are depicted: ABC (light blue boxes) and GCB (orange boxes). The _X_-axis shows _P_-value among these two groups and significant threshold is marked with a green line. (c) Bar-graph represents frequency of mutations and CNAs for each gene in 119 DLBCL cases, determined by targeted NGS (CDKN2A by Sanger sequencing) and copy number analysis. Gene alterations are divided into four groups: Mutations (single-nucleotide mutations and/or small indels), homozygous deletion, loss (loss of one allele) and bialleic inactivation (Loss+mutation or CNN-LOH+mutation).
Integrative analysis of mutations and CNA identified 16 genes with biallelic inactivation including homozygous deletions or heterozygous deletions with concomitant truncating mutations (Figure 2c). As expected, 74% of TP53 mutations were associated with 17p losses or CNN-LOH. The common deleted 6q14–q23 region included PRDM1 and TNFAIP3 (Supplementary Figure 4) but we also identified SGK1 and TMEM30A as novel targets with biallelic inactivation (Figure 2c, Supplementary Figure 4). MYD88 was the only gene with known homozygous activating mutations in two cases due to CNN-LOH.
We detected chromothripsis-like patterns in 28 (24%) DLBCLs with similar distribution in ABC and GCB-DLBCL. These cases showed more TP53 aberrations (61 vs 29% _P_=0.004) and 11q23–q25 gains/amplifications, including ETS1 and FLI1, (57 vs 19%, P<0.001) than cases without chromothripsis. The most affected chromosomes were 13 (_n_=6), 2 (_n_=5) and 6 (_n_=5). Interestingly, regions targeted by chromothripsis included amplifications of miR17-92 (13q31.3) (_n_=3) and BCL11A/REL (2p16) (_n_=1) (Supplementary Figures 5A and B).
BCL2, BCL6 and MYC were rearranged in 19% (25/131), 20% (25/122) and 9% (11/124) of the cases, respectively, (Supplementary Figure 3). BCL2 and MYC translocations predominated in GCB-DLBCL whereas BCL6 translocations were equally distributed in GCB and ABC-DLBCL. Seven cases had a double hit, 6 MYC/BCL2 in 4 GCB and 2 UC, and one ABC had a MYC/BCL6.
Clinical impact
The clinical impact of mutations and CNAs present in at least five cases was evaluated. Gains of 5p15, 11q24, 12q14 and 12q15 and losses of 8q12 correlated with lower CR rate, whereas no other CNA or single-gene mutation was associated with the response to therapy. As expected, R-IPI and COO among other standard clinical variables significantly predicted PFS and OS (Supplementary Table 9). Several mutated genes and CNA also had an impact on PFS and OS (Figure 3). Interestingly, among TP53 variants, only truncating and DBD mutations were associated with shorter OS. Of note, KLHL6 and SGK1 were the only mutations associated with a worse OS independently of the IPI and COO of the tumor (Figure 3).
Figure 3
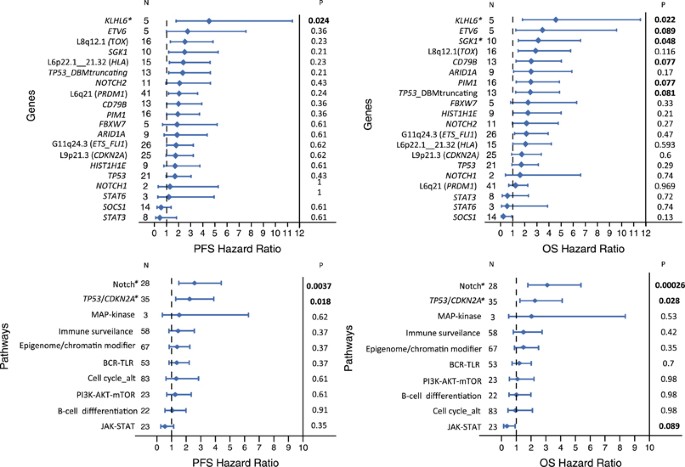
Forest plots of OS and PFS of gene alterations and pathways in the initial series. Gene alterations herein shown correspond to those with significant impact on overall (OS) or progression-free survival (PFS) in the statistical analysis before correction for multiple comparisons, as well as those drivers included in any of the three significant pathways. The _P_-values shown were corrected for multiple comparisons (Benjamini–Hochberg method). *indicates gene and pathway mutations that had prognostic value independent of the IPI and COO of the tumor in the multivariate analysis.
We then analyzed the clinical influence of genetic alterations in 10 predefined functional pathways or group of genes (Supplementary Table 6, Figure 3). The main features of the patients according to the aberrations in these pathways are listed in Table 2 and Supplementary Tables 10a and b. Alterations in NOTCH pathway (NOTCH2, NOTCH1, FBXW7 and SGK1) and in TP53/CDKN2A were associated with shorter PFS and OS, whereas patients with JAK/STAT pathway (SOCS1, STAT3 and STAT6) mutations had superior OS (Figure 4a, Table 2). Alterations in TP53/CDKN2A showed a trend for a worse response to therapy. A multivariate analysis including R-IPI (very good vs good vs poor) and COO (GCB vs ABC) along with NOTCH, TP53/CDKN2A and JAK/STAT pathways (non-altered vs altered in each case) showed in the final model with 82 cases that R-IPI (Hazard ratio (HR) 4.0; _P_=0.006), NOTCH pathway (HR 2.8; _P_=0.006) and TP53/CDKN2A (HR 2.4; _P_=0.005) maintained independent significance for OS.
Table 2 Baseline features of the patients according to NOTCH, TP53/CDKN2A and JAK/STAT pathways in the initial series
Figure 4
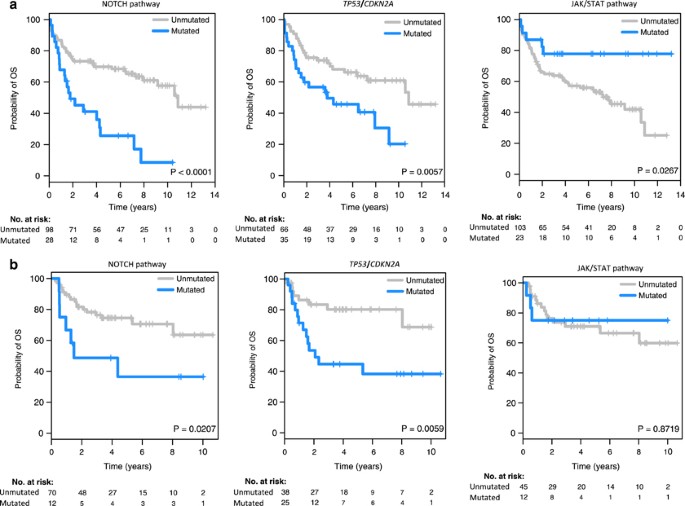
PFS and OS according to alterations in NOTCH and JAK-STAT pathways and TP53/CDKN2A (4A and 4B are for the initial and the validation series, respectively).
The prognostic impact of these three pathways was assessed in an independent cohort of patients. The distribution of the individual gene alterations was similar in both series (Supplementary Table 11). The clinical features and outcome of the patients according to the status of NOTCH, TP53/CDKN2A and JAK/STAT pathways in the validation series are listed in Supplementary Table 12. As shown in Figure 4b, the adverse prognostic impact on OS of NOTCH pathway and TP53/CDKN2A alterations was validated in this independent cohort.
Genomic-guided therapeutic opportunities
We identified 69 (46%) cases carrying at least one genomic alteration in 9 genes (CDK6, TP53, CDKN2A, PTEN, MYC, ARID1A and CD79B (with or without MYD88), EZH2 and NOTCH1) considered a biomarker of drug response as supported by the results of early clinical trials (_n_=66) or preclinical assays (_n_=3) in DLBCL or other lymphomas (Supplementary Table 13) (Figures 5a–c). The tumors of 26 additional patients (17%) showed at least one gene alteration that could be exploited by a drug repurposing strategy of two types (Figure 5a). The first one corresponds to gene alterations that are biomarkers of drug response described in other cancer types and whose effect in DLBCL has not been assessed yet. The second one were mutations observed in genes described as biomarkers of drug response mostly in preclinical assays, but had different amino acid changes in the same functional domains. These mutations are predicted to have the same oncogenic effect as the known biomarker and may therefore lead to a similar drug response.
Figure 5
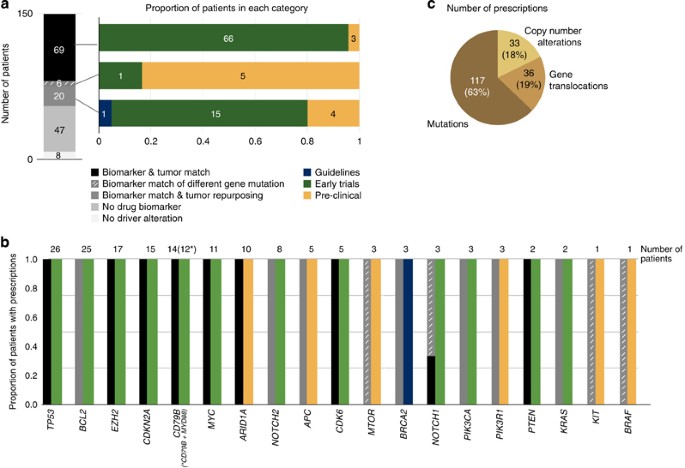
Genomic-guided therapeutic opportunities of the DLBCL cohort. Therapeutic opportunities have been classified according to the level of evidence supporting the effect of the genomic biomarker into (i) clinical guidelines (for example, FDA-approved or NCNN recommendations), (ii) late (phases III–IV) or (iii) early (phases I–II) clinical trials, (iv) case reports or (v) preclinical data. In addition to the alterations described as biomarkers of drug response in DLBCL (biomarker and tumor match), we included driver mutations in genes described as biomarkers of drug response in DLBCL upon a different amino acid change (biomarker match of different gene mutation), as well as genomic alterations described as biomarkers of drug response in other tumor types (biomarker match and tumor repurposing). (a) This panel depicts the therapeutic opportunities per patient (each patient has been counted only once according to their best therapeutic option following the above classification). (b) This panel depicts the therapeutic opportunities per gene; the numbers on top of the bars correspond to the number of patients exhibiting a biomarker of drug response in that gene (each patient has been counted only once according to their best therapeutic option given the gene alteration). Biomarkers that have been described for DLBCL and other non-Hodgkin lymphomas were also considered in the tumor match category. (c) Finally, this panel depicts the contribution of each alteration type to the overall number of in silico prescriptions per patient and altered gene.
Discussion
This study confirms the differential distribution of mutated genes, pathogenic pathways and CNA in GCB and ABC-DLBCL and also the presence of common alterations in both subtypes highlighting the molecular heterogeneity of these tumors. The larger number of cases investigated compared with previous whole-exome sequencing studies has expanded the view of the interactions among the individual mutated genes and those integrated in specific pathways. In this sense, we confirmed the association of MYD88 mutations, particularly L265P, with CD79B mutations in ABC-DLBCL whereas other MYD88 mutations occurred indistinctively in both DLBCL subtypes.24, 29 Similarly to other studies, CREBBP and KMT2D were found in both DLBCL subtypes.10, 11, 12, 13, 30 However, here they were also significantly associated between them and with EZH2 mutations in GCB-DLBCL, whereas KMT2D mutations, independent of the other two genes, were also detected in ABC-DLBCL. CREBBP and EP300 have a similar function and molecular structure.31 Mutations in these genes have been found as mutually exclusive in DLBCL associated with adverse clinical outcome.32 However, in our series most EP300 mutations occurred in tumors with CREBBP mutations. None of these mutated genes had prognostic significance (Figures 1c and 3).
In addition to individual genes, we integrated the analysis of the mutations in different components of pathogenic pathways. Mutations in genes of the PI3K-AKT-mTOR pathway were significantly more frequent in GCB-DLBCL. This finding is consistent with the activation of PI3K signaling pathway observed in these tumors frequently associated with loss of PTEN.7, 33 JAK-STAT signaling is a feature of ABC-DLBCL triggered by autocrine production of Interleukin-6 (IL-6) and Interleukin-10 (IL-10).24, 34, 35, 36 However, we found mutations in SOCS1, STAT3 and STAT6 more frequently in GCB-DLBCL, a finding that expands the previous observation of inactivating SOCS1 mutations in GCB-DLBCL.37
We also found frequent mutations in genes of the NOTCH pathway, NOTCH2 (9%), and less frequently in NOTCH1 (3%), that were confirmed in the validation cohort. All NOTCH1 and NOTCH2 mutations truncated the PEST domain. Mutations in these genes have only occasionally been detected in previous DLBCL whole-genome/exome sequencing studies, probably due to the relatively low coverage of these studies compared with ours (≈50 × vs >500 ×, respectively,) and which may have been insufficient to detect mutations in the GC-rich hot-spot region.10, 11, 12, 13, 38 We also sequenced the 3′UTR region of NOTCH1 recently described as a hot spot for activating somatic mutations in chronic lymphocytic leukemia (CLL).39 Only one case showed this type of mutation. We also found mutations in other genes of NOTCH pathway including the ubiquitin ligase FBXW7 and the kinase SGK1.25, 26, 40 To validate the role of this pathway in DLBCL, we investigated the expression of downstream genes regulated by NOTCH and observed that tumors carrying mutations in this pathway had a significant overexpression of NOTCH target genes. These findings support the role of the NOTCH pathway in a subset of DLBCL. Further studies are warranted to explore the impact of these mutations on DLBCL.
The integration of mutations and CNA has revealed new genes targeted by both types of alterations in DLBCL. In addition to biallelic alterations of known genes such as CDKN2A, TNFAIP3, PRDM1, PTEN, B2M, CD70 or CD58, we also found novel biallelic alterations of TMEM30A, SGK1, GNA13 and EBF1, among others, indicating the relevance of their inactivation in DLBCL. The frequent alterations of TMEM30A and SGK1 identify these genes as new targets of the complex 6q14–q23 deletion in DLBCL, in addition to the known PRDM1 and TNFAIP3. The role of TMEM30A in lymphomagenesis is unknown but it is interesting that its truncating mutations interfere with the function of ATP11C, a transmembrane protein involved in B-cell differentiation and BCR signaling41, 42 whereas SGK1 inactivation seems to promote NOTCH signaling.25, 26
Several studies have identified the prognostic value of individual mutated genes and CNA in DLBCL.7, 14, 43, 44, 45, 46, 47, 48 However, their significance is still controversial as very few of these alterations have been validated in independent cohorts. We confirmed the poor prognosis of CDKN2A deletions and 8p23.1 losses49, 50, 51, 52 in DLBCL. However, we could not validate the adverse effect of 3p gains, including FOXP1.7 The clinicopathological significance of CNA, especially other than CDKN2A, should be further analyzed in DLBCL.
Our study identified the prognostic impact of several individual mutated genes after correction for multiple comparisons, but only SGK1 and KLHL6 were independent of R-IPI and the COO of the tumors. The integrated analysis of alterations in pathogenic pathways has an increasing interest for strategies targeting mechanisms rather than individual genes.53, 54 This approach may also overcome the challenges of the low frequency of most mutated genes in DLBCL.53, 55 In this perspective, we found that genetic alterations in NOTCH pathway and TP53/CDKN2A genes conferred poor outcome independently of the R-IPI and COO, and these findings were confirmed in the independent cohort of DLBCL.
Recent studies have revealed the relationship between NOTCH1/2 mutations and tumor aggressiveness in different mature B-cell neoplasms including CLL,39, 56 splenic marginal zone lymphomas,57, 58 follicular lymphomas59, 60 and mantle cell lymphomas.19, 61 In our DLBCL cohort none of the individual mutated genes of the pathway had a confirmed prognostic significance in the initial and validation cohorts. On the contrary, the integrated analysis of the mutations in all the genes of the pathway conferred an adverse prognosis that was independent of the IPI and COO subtype. This finding was confirmed in the validation cohort supporting the relevance of NOTCH pathway in DLBCL and indicating that the integrated analysis of altered pathways in DLBCL may be more relevant than individual genes.
We observed a similar situation with TP53 and CDKN2A alterations. As previously observed,62 not all TP53 mutations had a prognostic impact. In the initial cohort only TP53 truncating and DBD mutations were associated with a significant shorter survival, although it was not independent of the COO subtype. TP53 mutations occurred in both ABC and GCB subtypes but truncating and mutations in DBD occurred preferentially in ABC-DLBCL. Interestingly, the combination of TP53 truncating and DBD mutations, and CDKN2A deletions was associated with adverse prognosis that was independent of the R-IPI and COO of the tumor and could be confirmed in the independent validation cohort.
We also identified a subset of tumors with mutations in JAK/STAT pathway that had a better outcome. Activation of the JAK-STAT is common in primary mediastinal large B-cell lymphoma, but we specifically excluded these tumors in our study. Interestingly, the good prognosis of DLBCL with SOCS1 inactivating mutations has been previously observed.14, 32, 37 Although the prognostic value could not be validated in the independent cohort, identifying these patients as candidates for targeted therapies may be relevant.54
The comprehensive profiling of genomic alterations in this DLBCL cohort revealed a landscape of genomic-guided therapeutic opportunities.54 Overall, 46% of the tumors exhibited biomarkers of drug response currently supported by the results of early clinical trials (phases I/II) or preclinical assays. This number is extended to 63% when drug repurposing opportunities are also taken into account. This analysis concentrated on drugs interacting directly with altered genes. Further studies considering drugs with potential effect on pathogenic altered pathways may expand the number of patients who could benefit from a personalized approach. The consideration of these therapeutic strategies may open new perspectives for patients suffering from tumors unresponsive to standard strategies.
In conclusion, we have recognized novel target genes and defined the relevance of alterations of NOTCH pathway and TP53/CDKN2A in DLBCL. Our findings suggest that the global analysis of alterations in defined pathways may be more relevant than independent genes. Using an in silico prescription pipeline we have also identified a number of candidate drugs with potential therapeutic interactions with driver oncogenic proteins. All these findings may orient future preclinical and clinical intervention strategies in DLBCL.
References
- Swerdlow S, Campo E, Harris N, Jaffe E, Pileri S, Stein H et al. World Health Organization Classification of Tumours: Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues, Fourth edition. IARC Press: Lyon, 2008.
Google Scholar - Coiffier B . Rituximab therapy in malignant lymphoma. Oncogene 2007; 26: 3603–3613.
Article CAS PubMed Google Scholar - Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000; 403: 503–511.
Article CAS PubMed Google Scholar - Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. New Engl J Med 2002; 346: 1937–1947.
Article PubMed Google Scholar - Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Delabie J, Ott G et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004; 103: 275–282.
Article CAS PubMed Google Scholar - Colomo L, Lopez-Guillermo A, Perales M, Rives S, Martinez A, Bosch F et al. Clinical impact of the differentiation profile assessed by immunophenotyping in patients with diffuse large B-cell lymphoma. Blood 2003; 101: 78–84.
Article CAS PubMed Google Scholar - Lenz G, Wright GW, Emre NC, Kohlhammer H, Dave SS, Davis RE et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci USA 2008; 105: 13520–13525.
Article CAS PubMed PubMed Central Google Scholar - Bea S, Zettl A, Wright G, Salaverria I, Jehn P, Moreno V et al. Diffuse large B-cell lymphoma subgroups have distinct genetic profiles that influence tumor biology and improve gene-expression-based survival prediction. Blood 2005; 106: 3183–3190.
Article CAS PubMed PubMed Central Google Scholar - Tagawa H, Suguro M, Tsuzuki S, Matsuo K, Karnan S, Ohshima K et al. Comparison of genome profiles for identification of distinct subgroups of diffuse large B-cell lymphoma. Blood 2005; 106: 1770–1777.
Article CAS PubMed Google Scholar - Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011; 476: 298–303.
Article CAS PubMed PubMed Central Google Scholar - Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet 2011; 43: 830–837.
Article CAS PubMed PubMed Central Google Scholar - Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci USA 2012; 109: 3879–3884.
Article CAS PubMed PubMed Central Google Scholar - Zhang J, Grubor V, Love CL, Banerjee A, Richards KL, Mieczkowski PA et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc Natl Acad Sci USA 2013; 110: 1398–1403.
Article CAS PubMed PubMed Central Google Scholar - Juskevicius D, Lorber T, Gsponer J, Perrina V, Ruiz C, Stenner-Liewen F et al. Distinct genetic evolution patterns of relapsing diffuse large B-cell lymphoma revealed by genome-wide copy number aberration and targeted sequencing analysis. Leukemia 2016; 30: 2385–2395.
Article CAS PubMed Google Scholar - Martinez-Ledesma E, de Groot JF, Verhaak RG . Seek and destroy: relating cancer drivers to therapies. Cancer Cell 2015; 27: 319–321.
Article CAS PubMed Google Scholar - Rubio-Perez C, Tamborero D, Schroeder MP, Antolin AA, Deu-Pons J, Perez-Llamas C et al. In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals targeting opportunities. Cancer Cell 2015; 27: 382–396.
Article CAS PubMed Google Scholar - Scott DW, Wright GW, Williams PM, Lih CJ, Walsh W, Jaffe ES et al. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood 2014; 123: 1214–1217.
Article CAS PubMed PubMed Central Google Scholar - Reva B, Antipin Y, Sander C . Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res 2011; 39: e118.
Article CAS PubMed PubMed Central Google Scholar - Bea S, Valdes-Mas R, Navarro A, Salaverria I, Martin-Garcia D, Jares P et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci USA 2013; 110: 18250–18255.
Article CAS PubMed PubMed Central Google Scholar - Cheson BD, Fisher RI, Barrington SF, Cavalli F, Schwartz LH, Zucca E et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol 2014; 32: 3059–3068.
Article PubMed PubMed Central Google Scholar - Dienstmann R, Jang IS, Bot B, Friend S, Guinney J . Database of genomic biomarkers for cancer drugs and clinical targetability in solid tumors. Cancer Discov 2015; 5: 118–123.
Article CAS PubMed PubMed Central Google Scholar - Mottok A, Renne C, Seifert M, Oppermann E, Bechstein W, Hansmann ML et al. Inactivating SOCS1 mutations are caused by aberrant somatic hypermutation and restricted to a subset of B-cell lymphoma entities. Blood 2009; 114: 4503–4506.
Article CAS PubMed Google Scholar - Khodabakhshi AH, Morin RD, Fejes AP, Mungall AJ, Mungall KL, Bolger-Munro M et al. Recurrent targets of aberrant somatic hypermutation in lymphoma. Oncotarget 2012; 3: 1308–1319.
Article PubMed PubMed Central Google Scholar - Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011; 470: 115–119.
Article CAS PubMed Google Scholar - Mo JS, Ann EJ, Yoon JH, Jung J, Choi YH, Kim HY et al. Serum- and glucocorticoid-inducible kinase 1 (SGK1) controls Notch1 signaling by downregulation of protein stability through Fbw7 ubiquitin ligase. J Cell Sci 2011; 124: 100–112.
Article CAS PubMed Google Scholar - Mo JS, Yoon JH, Hong JA, Kim MY, Ann EJ, Ahn JS et al. Phosphorylation of nicastrin by SGK1 leads to its degradation through lysosomal and proteasomal pathways. PloS One 2012; 7: e37111.
Article CAS PubMed PubMed Central Google Scholar - Sharma VM, Calvo JA, Draheim KM, Cunningham LA, Hermance N, Beverly L et al. Notch1 contributes to mouse T-cell leukemia by directly inducing the expression of c-myc. Mol Cell Biol 2006; 26: 8022–8031.
Article CAS PubMed PubMed Central Google Scholar - Weng AP, Millholland JM, Yashiro-Ohtani Y, Arcangeli ML, Lau A, Wai C et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev 2006; 20: 2096–2109.
Article CAS PubMed PubMed Central Google Scholar - Okosun J, Bodor C, Wang J, Araf S, Yang CY, Pan C et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet 2014; 46: 176–181.
Article CAS PubMed Google Scholar - Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet 2010; 42: 181–185.
Article CAS PubMed PubMed Central Google Scholar - Bedford DC, Brindle PK . Is histone acetylation the most important physiological function for CBP and p300? Aging 2012; 4: 247–255.
Article CAS PubMed PubMed Central Google Scholar - Juskevicius D, Jucker D, Klingbiel D, Mamot C, Dirnhofer S, Tzankov A . Mutations of CREBBP and SOCS1 are independent prognostic factors in diffuse large B cell lymphoma: mutational analysis of the SAKK 38/07 prospective clinical trial cohort. J Hematol Oncology 2017; 10: 70.
Article Google Scholar - Pfeifer M, Grau M, Lenze D, Wenzel SS, Wolf A, Wollert-Wulf B et al. PTEN loss defines a PI3K/AKT pathway-dependent germinal center subtype of diffuse large B-cell lymphoma. Proc Natl Acad Sci USA 2013; 110: 12420–12425.
Article CAS PubMed PubMed Central Google Scholar - Lam LT, Wright G, Davis RE, Lenz G, Farinha P, Dang L et al. Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-{kappa}B pathways in subtypes of diffuse large B-cell lymphoma. Blood 2008; 111: 3701–3713.
Article CAS PubMed PubMed Central Google Scholar - Rui L, Drennan AC, Ceribelli M, Zhu F, Wright GW, Huang DW et al. Epigenetic gene regulation by Janus kinase 1 in diffuse large B-cell lymphoma. Proc Natl Acad Sci USA 2016; 113: E7260–E7267.
Article CAS PubMed PubMed Central Google Scholar - Ding BB, Yu JJ, Yu RY, Mendez LM, Shaknovich R, Zhang Y et al. Constitutively activated STAT3 promotes cell proliferation and survival in the activated B-cell subtype of diffuse large B-cell lymphomas. Blood 2008; 111: 1515–1523.
Article CAS PubMed PubMed Central Google Scholar - Schif B, Lennerz JK, Kohler CW, Bentink S, Kreuz M, Melzner I et al. SOCS1 mutation subtypes predict divergent outcomes in diffuse large B-Cell lymphoma (DLBCL) patients. Oncotarget 2013; 4: 35–47.
Article PubMed Google Scholar - Morin RD, Mungall K, Pleasance E, Mungall AJ, Goya R, Huff RD et al. Mutational and structural analysis of diffuse large B-cell lymphoma using whole-genome sequencing. Blood 2013; 122: 1256–1265.
Article CAS PubMed PubMed Central Google Scholar - Puente XS, Bea S, Valdes-Mas R, Villamor N, Gutierrez-Abril J, Martin-Subero JI et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015; 526: 519–524.
Article CAS PubMed Google Scholar - O'Neil J, Look AT . Mechanisms of transcription factor deregulation in lymphoid cell transformation. Oncogene 2007; 26: 6838–6849.
Article CAS PubMed Google Scholar - Yabas M, Teh CE, Frankenreiter S, Lal D, Roots CM, Whittle B et al. ATP11C is critical for the internalization of phosphatidylserine and differentiation of B lymphocytes. Nat Immunol 2011; 12: 441–449.
Article CAS PubMed PubMed Central Google Scholar - Siggs OM, Arnold CN, Huber C, Pirie E, Xia Y, Lin P et al. The P4-type ATPase ATP11C is essential for B lymphopoiesis in adult bone marrow. Nat Immunol 2011; 12: 434–440.
Article CAS PubMed PubMed Central Google Scholar - Chan FC, Telenius A, Healy S, Ben-Neriah S, Mottok A, Lim R et al. An RCOR1 loss-associated gene expression signature identifies a prognostically significant DLBCL subgroup. Blood 2015; 125: 959–966.
Article CAS PubMed Google Scholar - Trinh DL, Scott DW, Morin RD, Mendez-Lago M, An J, Jones SJ et al. Analysis of FOXO1 mutations in diffuse large B-cell lymphoma. Blood 2013; 121: 3666–3674.
Article CAS PubMed PubMed Central Google Scholar - Novak AJ, Asmann YW, Maurer MJ, Wang C, Slager SL, Hodge LS et al. Whole-exome analysis reveals novel somatic genomic alterations associated with outcome in immunochemotherapy-treated diffuse large B-cell lymphoma. Blood Cancer J 2015; 5: e346.
Article CAS PubMed PubMed Central Google Scholar - Dubois S, Viailly PJ, Mareschal S, Bohers E, Bertrand P, Ruminy P et al. Next-generation sequencing in diffuse large B-cell lymphoma highlights molecular divergence and therapeutic opportunities: a LYSA study. Clin Cancer Res 2016; 22: 2919–2928.
Article CAS PubMed Google Scholar - Cao Y, Zhu T, Zhang P, Xiao M, Yi S, Yang Y et al. Mutations or copy number losses of CD58 and TP53 genes in diffuse large B cell lymphoma are independent unfavorable prognostic factors. Oncotarget 2016; 7: 83294–83307.
PubMed PubMed Central Google Scholar - Xia Y, Xu-Monette ZY, Tzankov A, Li X, Manyam GC, Murty V et al. Loss of PRDM1/BLIMP-1 function contributes to poor prognosis of activated B-cell-like diffuse large B-cell lymphoma. Leukemia 2017; 31: 625–636.
Article CAS PubMed Google Scholar - Jardin F, Jais JP, Molina TJ, Parmentier F, Picquenot JM, Ruminy P et al. Diffuse large B-cell lymphomas with CDKN2A deletion have a distinct gene expression signature and a poor prognosis under R-CHOP treatment: a GELA study. Blood 2010; 116: 1092–1104.
Article CAS PubMed Google Scholar - Pinyol M, Cobo F, Bea S, Jares P, Nayach I, Fernandez PL et al. p16(INK4a) gene inactivation by deletions, mutations, and hypermethylation is associated with transformed and aggressive variants of non-Hodgkin's lymphomas. Blood 1998; 91: 2977–2984.
CAS PubMed Google Scholar - Scandurra M, Mian M, Greiner TC, Rancoita PM, De Campos CP, Chan WC et al. Genomic lesions associated with a different clinical outcome in diffuse large B-Cell lymphoma treated with R-CHOP-21. Br J Haematology 2010; 151: 221–231.
Article Google Scholar - Monti S, Chapuy B, Takeyama K, Rodig SJ, Hao Y, Yeda KT et al. Integrative analysis reveals an outcome-associated and targetable pattern of p53 and cell cycle deregulation in diffuse large B cell lymphoma. Cancer Cell 2012; 22: 359–372.
Article CAS PubMed PubMed Central Google Scholar - Intlekofer AM, Younes A . Precision therapy for lymphoma—current state and future directions. Nat Rev Clin Oncol 2014; 11: 585–596.
Article CAS PubMed Google Scholar - Younes A, Ansell S, Fowler N, Wilson W, de Vos S, Seymour J et al. The landscape of new drugs in lymphoma. Nat Rev Clin Oncol 2016; 14: 335–346.
Article PubMed PubMed Central Google Scholar - Vermaat JS, Pals ST, Younes A, Dreyling M, Federico M, Aurer I et al. Precision medicine in diffuse large B-cell lymphoma: hitting the target. Haematologica 2015; 100: 989–993.
PubMed PubMed Central Google Scholar - Puente XS, Pinyol M, Quesada V, Conde L, Ordonez GR, Villamor N et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011; 475: 101–105.
Article CAS PubMed PubMed Central Google Scholar - Rossi D, Trifonov V, Fangazio M, Bruscaggin A, Rasi S, Spina V et al. The coding genome of splenic marginal zone lymphoma: activation of NOTCH2 and other pathways regulating marginal zone development. J Exp Med 2012; 209: 1537–1551.
Article CAS PubMed PubMed Central Google Scholar - Kiel MJ, Velusamy T, Betz BL, Zhao L, Weigelin HG, Chiang MY et al. Whole-genome sequencing identifies recurrent somatic NOTCH2 mutations in splenic marginal zone lymphoma. J Exp Med 2012; 209: 1553–1565.
Article CAS PubMed PubMed Central Google Scholar - Karube K, Martinez D, Royo C, Navarro A, Pinyol M, Cazorla M et al. Recurrent mutations of NOTCH genes in follicular lymphoma identify a distinctive subset of tumours. J Pathol 2014; 234: 423–430.
Article CAS PubMed Google Scholar - Krysiak K, Gomez F, White BS, Matlock M, Miller CA, Trani L et al. Recurrent somatic mutations affecting B-cell receptor signaling pathway genes in follicular lymphoma. Blood 2017; 129: 473–483.
Article CAS PubMed PubMed Central Google Scholar - Kridel R, Meissner B, Rogic S, Boyle M, Telenius A, Woolcock B et al. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood 2012; 119: 1963–1971.
Article CAS PubMed Google Scholar - Xu-Monette ZY, Wu L, Visco C, Tai YC, Tzankov A, Liu WM et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood 2012; 120: 3986–3996.
Article CAS PubMed PubMed Central Google Scholar
Acknowledgements
This study was supported by the Ministerio de Economía y Competitividad, Grant No. SAF2015-64885- R (to EC), Generalitat de Catalunya Suport Grups de Recerca AGAUR 2014-SGR-795 (to EC), Instituto de Salud Carlos III, Spanish Ministry of Health, PI12/01536 (to AL-G) and PI16/00420 (to AL-G), the Red Temática de Investigación Cooperativa en Cáncer (RTICC) grant RD12/0036/0036 (to EC), RD12/0036/0023 (to AL-G), RD12/0036/0069 (to MA), BIO/SA78/15 (to MA) and the European Regional Development Fund ‘Una manera de fer Europa’, CERCA Programme/Generalitat de Catalunya. EC is an Academia Researcher of the ‘Institució Catalana de Recerca i Estudis Avançats’ (ICREA) of the Generalitat de Catalunya. KK has received a research fellowship from the Uehara Memorial Foundation (Japan). DT is supported by the People Programme (Marie Curie Actions) of the Seventh Framework Programme of the European Union (FP7/2007–2013) under REA grant agreement number 600388 and by the Agency of Competitiveness for Companies of the Government of Catalonia, ACCIÓ and SAF2015-74072-JIN. CR-P is supported by an FPI fellowship. ID is supported by ‘Josep Font’ grant from Hospital Clinic. This work was also supported by La Fundació la Marató de TV3 and EU H2020 Programme 2014–2020 under grant agreements no. 634143 (MedBioinformatics), and the European Research Council (consolidator grant 682398) (to NL-B). This work was mainly developed at the Centre Esther Koplowitz (CEK), Barcelona, Spain. We are indebted to the Genomics core facility of the Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS) for their technical help. We are grateful to N Villahoz and MC Muro, S Guijarro, C. Capdevila, L Pla, L Gelabert, R Bermudo and M Sánchez for their excellent technical assistance. We also grateful to R Seki and K Nagafuji for their support.
Author information
Authors and Affiliations
- Institut d’Investigacions Biomèdiques August Pi i Sunyer, Hospital Clínic, Universitat de Barcelona, Barcelona, Spain
K Karube, A Enjuanes, I Dlouhy, P Jares, D Martin-Garcia, F Nadeu, J Rovira, G Clot, C Royo, A Navarro, B Gonzalez-Farre, A Vaghefi, G Castellano, L Colomo, A Valera, A Carrió, D Costa, I Salaverria, A López-Guillermo & E Campo - Department of Pathology and Cell Biology, Graduate School of Medicine and Faculty of Medicine, University of the Ryukyus, Nishihara, Japan
K Karube - CIBERONC, Madrid, Spain,
A Enjuanes, P Jares, D Martin-Garcia, F Nadeu, G Clot, A Navarro, B Gonzalez-Farre, M Alcoceba, A Carrió, D Costa, I Salaverria, A López-Guillermo & E Campo - DREAMgenics, S.L., Asturias, Spain
G R Ordóñez - Department of Experimental and Health Sciences, Institute for Research in Biomedicine (IRB Barcelona), The Barcelona Institute of Science and Technology, Research Unit on Biomedical Informatics, Universitat Pompeu Fabra, Barcelona, Spain
C Rubio-Perez, D Tamborero & N Lopez-Bigas - Servei de Patologia, Hospital de Sant Pau, Barcelona, Spain
J Briones - Department of Pathology, Hospital del Mar, Universitat Pompeu Fabra, Barcelona, Spain
A Salar & L Colomo - ICO-Hospital Germans Trias i Pujol, Barcelona, Spain
J M Sancho - ICO-Hospital Duran i Reynals, L'Hospitalet, Barcelona, Spain
S Mercadal & E Gonzalez-Barca - Department of Hematology, Hospital Universitari Joan XXIII, Tarragona, Spain
L Escoda - Department of Pathology, Kurume University School of Medicine, Kurume, Japan
H Miyoshi & K Ohshima - Department of Medicine and Biosystemic Science, Graduate School of Medical Science, Kyushu University, Fukuoka, Japan
K Miyawaki, K Kato & K Akashi - Servei de Patologia, Hospital de Sant Pau, Barcelona, Spain
A Mozos - Unidad de Biología Molecular/Histocompatibilidad, Servicio de Hematología, Hospital Universitario de Salamanca, Salamanca, Spain
M Alcoceba - Institució Catalana de Recerca i Estudis Avançats (ICREA), Barcelona, Spain
N Lopez-Bigas - Lymphoid Malignancies Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA
R Schmitz & L M Staudt
Authors
- K Karube
You can also search for this author inPubMed Google Scholar - A Enjuanes
You can also search for this author inPubMed Google Scholar - I Dlouhy
You can also search for this author inPubMed Google Scholar - P Jares
You can also search for this author inPubMed Google Scholar - D Martin-Garcia
You can also search for this author inPubMed Google Scholar - F Nadeu
You can also search for this author inPubMed Google Scholar - G R Ordóñez
You can also search for this author inPubMed Google Scholar - J Rovira
You can also search for this author inPubMed Google Scholar - G Clot
You can also search for this author inPubMed Google Scholar - C Royo
You can also search for this author inPubMed Google Scholar - A Navarro
You can also search for this author inPubMed Google Scholar - B Gonzalez-Farre
You can also search for this author inPubMed Google Scholar - A Vaghefi
You can also search for this author inPubMed Google Scholar - G Castellano
You can also search for this author inPubMed Google Scholar - C Rubio-Perez
You can also search for this author inPubMed Google Scholar - D Tamborero
You can also search for this author inPubMed Google Scholar - J Briones
You can also search for this author inPubMed Google Scholar - A Salar
You can also search for this author inPubMed Google Scholar - J M Sancho
You can also search for this author inPubMed Google Scholar - S Mercadal
You can also search for this author inPubMed Google Scholar - E Gonzalez-Barca
You can also search for this author inPubMed Google Scholar - L Escoda
You can also search for this author inPubMed Google Scholar - H Miyoshi
You can also search for this author inPubMed Google Scholar - K Ohshima
You can also search for this author inPubMed Google Scholar - K Miyawaki
You can also search for this author inPubMed Google Scholar - K Kato
You can also search for this author inPubMed Google Scholar - K Akashi
You can also search for this author inPubMed Google Scholar - A Mozos
You can also search for this author inPubMed Google Scholar - L Colomo
You can also search for this author inPubMed Google Scholar - M Alcoceba
You can also search for this author inPubMed Google Scholar - A Valera
You can also search for this author inPubMed Google Scholar - A Carrió
You can also search for this author inPubMed Google Scholar - D Costa
You can also search for this author inPubMed Google Scholar - N Lopez-Bigas
You can also search for this author inPubMed Google Scholar - R Schmitz
You can also search for this author inPubMed Google Scholar - L M Staudt
You can also search for this author inPubMed Google Scholar - I Salaverria
You can also search for this author inPubMed Google Scholar - A López-Guillermo
You can also search for this author inPubMed Google Scholar - E Campo
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toE Campo.
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Leukemia website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article
Cite this article
Karube, K., Enjuanes, A., Dlouhy, I. et al. Integrating genomic alterations in diffuse large B-cell lymphoma identifies new relevant pathways and potential therapeutic targets.Leukemia 32, 675–684 (2018). https://doi.org/10.1038/leu.2017.251
- Received: 19 April 2017
- Revised: 24 July 2017
- Accepted: 27 July 2017
- Published: 14 August 2017
- Issue Date: March 2018
- DOI: https://doi.org/10.1038/leu.2017.251