Effector and memory T-cell differentiation: implications for vaccine development (original) (raw)
Key Points
- Long-term T-cell memory can be attributed to the following characteristics of memory T-cell populations compared with naive T-cell populations: an increased frequency of antigen-specific precursors (∼1000-fold); changes in gene-expression profile that allow faster responses; re-distribution near the site of microbial entry; and homeostatic proliferation leading to longevity.
- Compared with CD4+ T cells, CD8+ T cells seem to be less dependent on co-stimulation for their initial activation. Two models have been proposed to explain this: either CD8+ T cells have lower activation thresholds or CD8+ T cells accumulate activation signals faster than CD4+ T cells.
- Both CD4+ and CD8+ T cells readily commit to a proliferative programme after a relatively brief period of stimulation with antigen.
- CD8+ T cells divide faster and show a greater level of clonal expansion than CD4+ T cells.
- For CD8+ T cells, commitment to proliferation is tightly coupled with commitment to effector- and memory-cell differentiation. This developmental programme might ensure that a large enough pool of memory T cells is formed despite decreasing levels of antigen at later stages of the response.
- Variations in the duration of signals (T-cell receptor or cytokines) might lead to the development of effector T-cell subsets with distinct effector and migratory patterns.
- The lineage of memory T cells is not fully understood, but available evidence suggests that memory cells can be derived from effector cells or alternatively, in some cases without passing through an effector stage.
- The extent of cell death determines the size of the memory T-cell pool; this might be regulated by interleukin-2 family cytokines, molecules of the tumour-necrosis factor receptor family, and effector molecules such as perforin and interferon-γ.
- Vaccine efficacy might be improved by strategies that are designed to modulate the expansion, contraction or memory phases of the T-cell response.
Abstract
Recent work shows that after stimulation with antigen, CD4+ and CD8+ T cells embark on a programme of proliferation that is closely linked with the acquisition of effector functions and leads ultimately to memory-cell formation. Here, we discuss the signals required for commitment to this programme of development and the factors that might influence its progression. Models of the pathways of effector and memory T-cell differentiation are discussed, and we highlight the implications of this new understanding for the optimization of vaccine strategies.
Similar content being viewed by others
Main
Vaccination has had an important role in reducing the mortality and morbidity that is caused by infectious diseases. The ultimate goal of a vaccine is to develop long-lived immunological protection, whereby the first encounter with a pathogen is 'remembered', which leads to enhanced memory responses that either completely prevent reinfection or greatly reduce the severity of disease.
Specialized cells known as memory T and B cells, and long-lived effector B cells (plasma cells), which constitutively secrete high-affinity 'neutralizing' antibodies, are the basis of immunological memory. The memory T-cell compartment consists of both CD4+ and CD8+ T-cells that can rapidly acquire effector functions to kill infected cells and/or secrete inflammatory cytokines that inhibit replication of the pathogen. Effector CD4+ T cells also help B-cell responses and enhance CD8+ T-cell development, through the activation of antigen-presenting cells (APCs) or secretion of cytokines, such as interleukin-2 (IL-2), IL-4 and IL-5. In some situations, protective immunity can be mediated by just one of the branches of the immune system — such as by antibodies or CD8+ T cells — but for optimal control of pathogens, both the humoural and cellular immune responses need to be mobilized.
Although there might be interesting parallels between memory T- and B-cell development, this review will focus on the formation of effector and memory T cells, and in particular, we will examine some of the recent models that have been proposed for the development of these cells. We will discuss primarily the formation of memory CD8+ T cells, rather than CD4+ T cells, as this process has been better characterized in vivo, but this will be discussed in conjunction with memory CD4+ T-cell development, because of the interesting similarities and differences that are being identified.
Tenets of T-cell immunological protection
It is well established that the anamnestic response that is mediated by memory CD4+ and CD8+ T cells is more rapid and aggressive than the primary response. This faster T-cell response, in association with antibody responses, can control secondary infections quickly and fully eliminate the pathogen. Comparisons between naive and memory T cells have begun to reveal the physiological basis for the heightened recall responses of memory T cells.
First, as a consequence of clonal expansion during the primary infection, experiments in mice have shown that there can be a substantial increase (∼1000-fold) in the precursor frequency of antigen-specific T cells in immune animals compared with naive animals1,2,3,4,5.
Second, as naive T cells differentiate into memory cells, their gene-expression profile is reprogrammed by changes in chromatin structure and the profile of active transcription factors6. For example, the genes that encode interferon-γ (IFN-γ) and cytotoxic molecules, such as perforin and granzyme B, are not expressed in naive CD8+ T cells, but are constitutively expressed in effector and memory CD8+ T cells7,8,9,10,11. Although the synthesis of these proteins occurs in an 'on–off' fashion that is regulated by antigen contact, elevated levels of the messenger RNA transcripts endow memory CD8+ T cells with the capacity to produce larger quantities of these proteins more rapidly than naive T cells2,8,9,12,13,14.
Third, memory CD8+ T cells express a different pattern of surface proteins that are involved in cell adhesion and chemotaxis from naive T cells, which allows memory T cells to extravasate into non-lymphoid tissues and mucosal sites (for a review, see Ref. 15). This enables memory T cells to survey peripheral tissues where microbial infections are generally initiated. Recently, memory CD8+ T cells that reside in these peripheral tissues have been termed 'effector' memory T cells, whereas those that are found in lymphoid organs are termed 'central' memory T cells (Box 1)16,17,18,19.
Fourth, memory T-cell populations are maintained for a long time due to homeostatic cell proliferation, which occurs at a slow, yet steady, pace (Box 2). Interestingly, the rate of this homeostatic cell division must equal the rate of cell death, because the number of memory CD8+ T cells remains relatively constant over time2,20. To determine the factors that regulate memory CD8+ T-cell proliferation is an important goal; cytokines such as IL-2, IL-15 and IL-7 might make important contributions, but their roles have yet to be fully defined21,22,23. In contrast to CD8+ T-cell memory, a recent study indicated that virus-specific CD4+ T-cell memory decreases slowly over time20(Fig. 1). Cytokines that regulate the number of memory CD4+ T cells have not been identified, but it is interesting to speculate that the differential stability of CD4+ and CD8+ T-cell memory could stem from the distinct effects of IL-15 on the respective populations — memory CD8+ T cells proliferate in response to IL-15, whereas memory CD4+ T cells do not21.
Figure 1: Antiviral CD8+ and CD4+ T-cell responses.
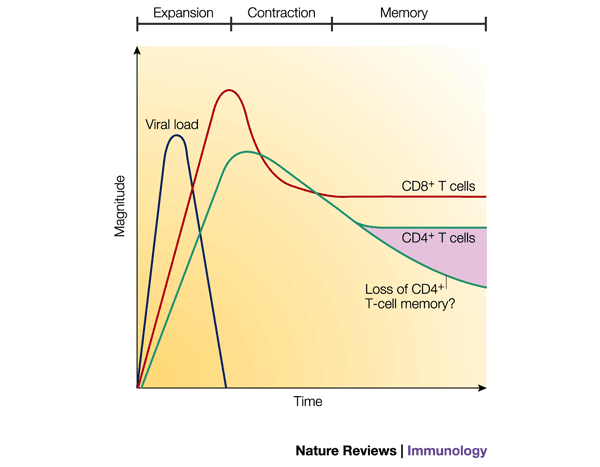
The three phases of the T-cell immune response (expansion, contraction and memory) are indicated. Antigen-specific T cells clonally expand during the first phase in the presence of antigen. Soon after the virus is cleared, the contraction phase ensues and the number of antigen-specific T cells decreases due to apoptosis. After the contraction phase, the number of virus-specific T cells stabilizes and can be maintained for great lengths of time (the memory phase). Note that, typically, the magnitude of the CD4+ T-cell response is lower than that of the CD8+ T-cell response, and the contraction phase can be less pronounced than that of CD8+ T cells. The number of memory CD4+ T cells might decline slowly over time, as reported recently20.
So, it is the increased number of antigen-specific T cells, and their faster responses, anatomical location (that is, near the sites of microbial entry) and longevity that collectively explain how memory T cells confer long-term protective immunity.
Stages of T-cell responses
The path towards memory T-cell development continues to be delineated, but there are clearly three stages that T cells pass through as they differentiate into memory cells24 (Fig. 1). The first stage, the 'expansion' phase, is initiated in the lymphoid tissues, where encounter with antigen induces naive T cells to clonally expand and differentiate into effector T cells — known as T helper (TH) cells or cytotoxic T lymphocytes (CTLs) for CD4+ and CD8+ T cells, respectively. Through the combined abilities of CD4+ and CD8+ effector T cells to secrete inflammatory cytokines and kill infected cells, a typical acute viral infection can be resolved within days. Over the weeks that follow pathogen clearance, the majority (>90%) of effector T cells die, and this second stage is often referred to as the 'death' phase or contraction period. The surviving T cells enter the third stage — the 'memory' phase — in which the number of memory T cells stabilizes, and these cells are maintained for long periods of time. The decisive factors that determine which T cells live or die are still unclear and will be discussed in greater detail below.
Thresholds of CD4 + and CD8 + T-cell activation
The context in which the T cell recognizes antigen, the abundance of antigen and the duration of antigen exposure are important parameters that can affect the speed and nature of the T-cell response. The most effective activators of T cells are mature dendritic cells (DCs) that have been activated — by either inflammatory stimuli, such as type I interferons (type I IFN), which are produced by the innate immune response, or cognate CD4+ TH cells through CD40–CD40-ligand (CD154) interactions. This increases their expression of MHC and co-stimulatory molecules, which are required for maximal T-cell stimulation. Interactions between co-stimulatory molecules and their ligands, such as CD28–CD80, CD40–CD154, 4-1BB–4-1BBL, OX40–OX40L and CD27–CD70 interactions, can affect the level of T-cell activation by enhancing T-cell receptor (TCR) signalling and/or providing additional signals that increase the expansion of T-cell populations and their responses25,26,27,28,29,30,31. Furthermore, co-stimulatory molecules might act early to augment TCR-mediated signals (for example, CD28 and CD40) or later to sustain T-cell responses (for example, OX40 and 4-1BB)28,32,33. However, several analyses of viral or bacterial infections in mice that are deficient in co-stimulatory molecules, such as CD28, CD154, OX40 and 4-1BBL, have shown that the effects of these accessory molecules on CD4+ and CD8+ T-cell responses can vary (Table 1). For example, in the absence of CD28, CD154 and OX40, CD4+ T-cell responses are severely impaired in all infectious model systems that have been tested, whereas CD8+ T-cell responses are affected moderately or not at all25,27,34,35,36,37,38,39. By contrast, CD4+ T-cell responses seem to be normal in 4-1BBL−/− animals, whereas the numbers of CTLs are slightly reduced26. The differential requirements for co-stimulatory molecules in CD4+ and CD8+ T-cell responses indicate that distinct mechanisms are involved in developing effector CD8+ and CD4+ T cells, as described recently by Szabo et al.40. Moreover, this suggests that different thresholds exist for CD4+ and CD8+ T-cell activation. It is possible that the threshold is inherently lower in CD8+ T cells or that extrinsic factors cause CD8+ T cells to accumulate TCR signals faster than CD4+ T cells, which reduces the requirement for co-stimulation (Fig. 2). Indeed, the stringent requirement for CD28 in CD4+ T-cell activation can be overcome by higher concentrations of antigen or longer durations of exposure to antigen35,41. Perhaps, an important contributing factor is that the expression of MHC class I molecules is nearly ubiquitous, whereas MHC class II molecules are expressed on a more limited set of cells. This difference might provide CD8+ T cells with more opportunities to encounter antigen than CD4+ T cells, which causes CD8+ T cells to reach their threshold of activation faster than CD4+ T cells.
Table 1 T-cell responses in the abscence of co-stimulatory molecules
Figure 2: Differences in the activation thresholds of CD4+ and CD8+ T cells.
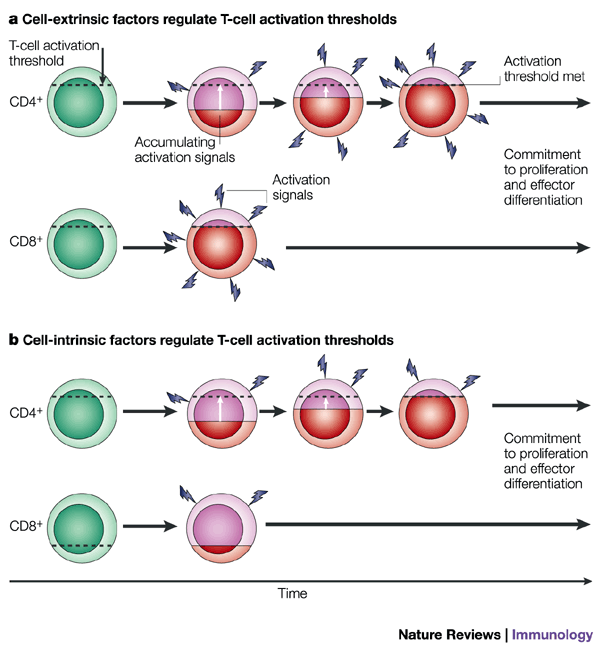
a | Factors that are extrinsic to the T cell might differentially mediate CD4+ and CD8+ T-cell activation. In this model, CD4+ and CD8+ T cells have similar activation thresholds, but CD8+ T cells 'see' different levels (and/or types) of activation signals than do CD4+ T cells, and CD8+ T cells commit more rapidly to proliferation and differentiation. Red shading indicates increasing activation status relative to the activation threshold. b | An intrinsic model in which the activation thresholds are different for CD4+ and CD8+ T cells. CD8+ T cells have a lower activation requirement, which permits the threshold for activation to be passed more rapidly than for CD4+ T cells.
Commitment to T-cell clonal expansion
As naive T cells receive appropriate antigenic and co-stimulatory signals, they become committed to clonal expansion and differentiation into effector cells. Understanding how this commitment is regulated could provide important therapeutic opportunities to augment or abrogate immune responses. Recent studies have focused on the duration of antigenic stimulation that is required for T cells to commit to proliferation by removing T cells from antigen at different time points. In the presence of professional APCs, naive CD4+ T cells required a minimum of six hours of antigenic stimulation, but in conditions that lacked co-stimulation, more than 24 hours of exposure to antigen was required to induce differentation41,42,43. These data indicate that specific activation thresholds must be reached for naive CD4+ T cells to commit to responding to antigen and that co-stimulatory signals facilitate this process.
In similar types of experiments, CD8+ T cells stimulated for brief periods (2–24 hours) could commit to at least 7–10 cell divisions14,44,45,46. It is important to emphasize that, in these experiments, only the 'parental' naive CD8+ T cells saw antigen, but the initial encounter was sufficient to sustain proliferation in the daughter cells in the absence of further antigenic stimulation. This indicated that once CD8+ T cells reach a certain threshold of activation, a programmed proliferative response ensues (Fig. 3). More investigations are required to determine clearly whether CD8+ T cells can commit faster than CD4+ T cells (two versus six hours), but if so, this might support the different activation/co-stimulatory requirements of naive CD4+ and CD8+ T cells that are discussed above (Fig. 2).
Figure 3: Programmed development of effector and memory CD8+ T cells.
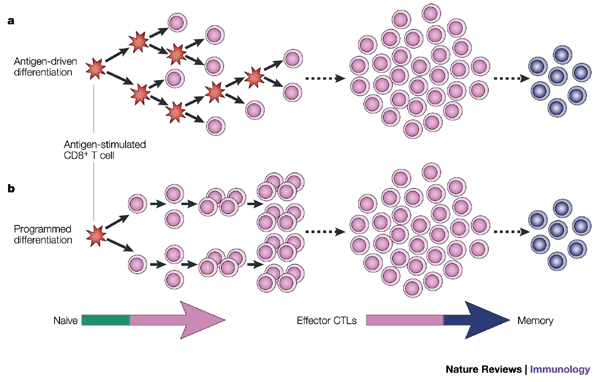
a | CD8+ T-cell proliferation is dependent on repeated encounters with antigen. Each cell that is stimulated by antigen (red) divides and progressively differentiates into effector cytotoxic T lymphocytes (CTLs) then memory CD8+ T cells with each successive cell division. According to this model, it is essential that each daughter cell be stimulated with antigen; CD8+ T-cell division, and possibly differentiation, would be halted on antigen removal. b | CD8+ T cells are developmentally programmed to divide at least 7–10 times and to differentiate into effector CTLs and long-lived, functional memory CD8+ T cells. The initial antigenic stimulus triggers this developmental programme, such that the CD8+ T cells become committed to proliferation and differentiation. Further antigenic stimulation of the daughter cells might increase the number of times the activated CD8+ T cells divide, but it is not necessary to complete this developmental programme.
T-cell proliferation rates and burst sizes
Although CD4+ and CD8+ T cells both commit to proliferation after relatively short periods of antigenic stimulation, CD4+ T cells have a slower rate of cell division in vitro and in vivo compared with CD8+ T cells. During the first 24 hours of stimulation, CD4+ and CD8+ T cells prepare for clonal expansion and increase in size, but no cell division is observed. Soon after, CD8+ T-cell division commences at a rapid rate (∼6–8 hours per cell division), whereas CD4+ T-cell division is typically delayed for another 12–24 hours (36–48 hours after the initial stimulus) and then occurs at a slightly slower rate (∼10 hours per cell division)9,14,20,42,46,47,48. These proliferative differences between CD4+ and CD8+ T cells were shown clearly in vivo in a recent study by Foulds et al.49 The slower rate of CD4+ T-cell proliferation might explain partially why the CD4+ T-cell response typically peaks 1–2 days after the CD8+ T-cell response during viral infection20,27.
In addition to faster rates of cell division, the increase in the number of antigen-specific CD8+ T cells is substantially greater than that of CD4+ T cells during viral and bacterial infections in mice, which suggests that CD8+ T cells have a higher proliferative potential than CD4+ T cells (Fig. 1)20,27,49,50,51. It has been estimated that CD8+ T cells divide ∼15–20 times during an acute infection with lymphocytic choriomeningitis virus (LCMV), whereas CD4+ T cells divide approximately nine times2,20. However, the number of times that CD4+ and CD8+ T cells divide in vivo might also be a consequence of the differential expression patterns of MHC class I and class II molecules that are referred to earlier. Also, it might not be necessary for CD4+ T-cell expansion to be as great as CD8+ T-cell expansion, because a single CD4+ T cell can provide help for multiple CTLs and B cells.
The mechanisms that control the rate and extent of cell division of CD4+ and CD8+ T cells are unknown. What is clear is that the extent of T-cell proliferation is governed by the amount of antigen available in vivo. By infecting mice with recombinant vaccinia strains that produced either high or low quantities of an ovalbumin (OVA) epitope, one group showed that the magnitude of the responding CTL population was proportional to epitope abundance52. Other experiments that involved the transfer of equal numbers of TCR-transgenic T cells into mice and then the titration of different doses of stimuli — such as peptide, peptide-loaded DCs, or epitope-expressing recombinant Listeria — have shown also that the greater the antigen load, the larger the number of effector cells that are produced14,45,53,54. Taken together with the model of programmed proliferation of T cells, these data show that, with minimal antigen contact, activated CD8+ T cells can divide at least 7–10 times, but if antigen persists, even greater expansion can be achieved. However, effector-cell populations do not continue to expand exponentially during chronic viral infection, which indicates that antigen-driven CD8+ T-cell proliferation can not be sustained indefinitely. In the case of chronic LCMV infection in mice, the rate of proliferation of effector CD8+ T cells slows, the cells lose function (EXHAUSTION) and some are physically deleted55,56,57,58. So, the number and function of effector CD8+ T cells is reduced when antigen levels remain high for extended periods of time.
Commitment to effector T-cell differentiation
CD8+ T-cell differentiation. The development of effector T-cell responses is tightly coupled to clonal expansion, but are the factors that guide commitment to proliferation and effector-cell differentiation the same? Recent studies of CD8+ T cells have shown that the link between the commitment to clonal expansion and effector-cell differentiation is remarkably tight; the same duration of antigenic stimulation (2–24 hours) that drove naive CD8+ T cells to proliferate was sufficient for them to commit to differentiate into effector cells that could secrete IFN-γ, tumour-necrosis factor (TNF) and IL-2, and kill infected cells14,44,45. These data indicate that naive CD8+ T cells are developmentally programmed to clonally expand and differentiate into CTLs after brief encounter with antigen (Fig. 3). Even though CTL effector properties were acquired after as little as 2–24 hours of stimulation, it remains to be determined whether the quality of effector properties is affected by the duration of antigenic stimulation in vivo. It seems that T cells that are activated under different conditions, such as with heat-killed bacteria or in the presence of high concentrations of IL-2 or IL-15, might develop suboptimal and/or altered effector CD8+ T-cell functions59,60.
The programmed development of CD8+ T cells has several advantages. First, it alleviates the need for prolonged confinement of CTLs to the lymphoid organs, which allows their migration to peripheral sites of infection and/or inflammation to remove infected cells. Second, it might also considerably affect the number of memory CD8+ T cells that are generated, because the size of the memory T-cell pool is directly correlated to that of the effector-cell population1,2,3,61. In several models of acute viral and bacterial infection, the number of effector CD8+ T cells peaks 2–3 days after the infectious pathogen is cleared. If each CD8+ T-cell division was regulated strictly by antigen contact, the number of effector CTLs would peak earlier and reach a lower maximum, and consequently, fewer memory CD8+ T cells would be generated (Fig. 4).
Figure 4: Comparison of the effect of programmed versus antigen-dependent T-cell proliferation on the number of memory T cells formed in antigen-limiting conditions.
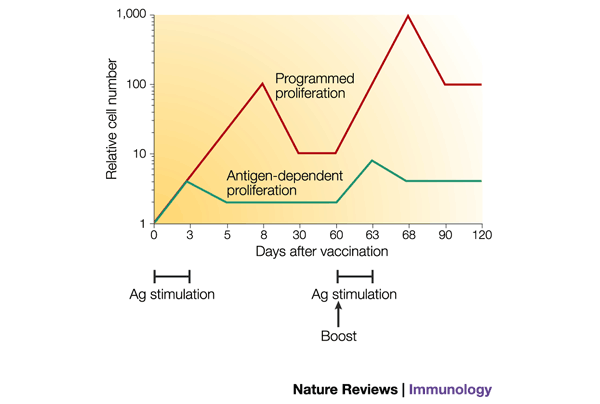
If T-cell proliferation were strictly regulated by antigen (Ag) contact (green line), then effector cells would stop dividing sooner in antigen-limiting conditions and the effector-cell burst size would be smaller than if the T cells were programmed to continue proliferating in the absence of antigen (red line). Consequently, a larger number of memory T cells would be generated with programmed T-cell development. If the memory cells re-encountered antigen — for example, due to a vaccine booster — then the effects of programmed development would be amplified, because the secondary expansion would again be greater than that of antigen-dependent proliferation. So, protective immunity might be established sooner.
CD4+ T-cell differentiation. A similar type of developmental programme might also drive the differentiation of activated CD4+ T cells, but the formation of effector CD4+ T cells might be influenced to a greater extent than for CD8+ T cells by extrinsic factors, such as the duration of antigen exposure and the types of cytokines that are present35,41,42,62,63. Unlike naive CD8+ T cells, which commit to effector and memory T-cell development within 24 hours of stimulation, naive CD4+ T cells required more than 48 hours of continual antigenic stimulation to commit to the formation of polarized TH1 or TH2 effector phenotypes in vitro (that is, the secretion of IFN-γ or IL-4, respectively). Even after 48 hours, a large proportion of the CD4+ T cells did not develop effector properties (that is, they were nonpolarized)62. Nonpolarized CD4+ T cells produced IL-2, but little or no IFN-γ or IL-4 (Refs 63,64). A more detailed analysis showed that CD4+ T cells became increasingly 'locked' into the TH1 or TH2 phenotypes with each successive cell division; cells that had divided more than four times in TH1-polarizing conditions could not revert to a TH2 phenotype, but cells that had divided fewer than four times could revert65,66,67. However, it was reported recently that CD4+ T-cell proliferation and effector differentiation can be uncoupled68. CD4+ T cells that were exposed briefly to antigen could divide approximately seven times, but optimal TH1 effector properties did not develop unless the TCR was re-engaged and the cells were exposed continuously to TH1-polarizing cytokines68. This agrees with previous findings that the addition of polarizing cytokines significantly decreases the duration of antigenic stimulation that is required for CD4+ T-cell commitment to differentiation. In the presence of IL-12, TH1 effector functions (secretion of IFN-γ) were present after 24 hours of stimulation, whereas ∼72 hours of stimulation was necessary to induce effector function in cultures that lacked this cytokine62. A similar effect was observed for TH2-polarizing cytokines, except that a longer duration of stimulation was required for the development of a TH2 effector phenotype. In addition, IL-2 can affect the progression of programmed CD8+ and CD4+ T-cell division; proliferation was increased or decreased by the presence or absence of IL-2, respectively14,42,46. So, it seems that naive CD8+ T cells commit to effector-cell differentiation more readily than CD4+ T cells. As described above, this might result from the apparent differences between CD4+ and CD8+ T cells in their activation requirements or the rates at which their thresholds for activation are reached (Fig. 2). Lastly, the impact of extrinsic signals, such as cytokines, indicates that both cell-intrinsic and cell-extrinsic mechanisms are involved in effector-cell differentiation.
Commitment to effector migration patterns. As naive T cells differentiate into effector cells, their migration patterns are altered and we are beginning to understand how this change is regulated. Effector T cells have a reduced potential for homing to lymph nodes — owing to decreased expression of lymph-node-homing receptors, such as CC-chemokine receptor 7 (CCR7) and L-selectin (CD62L) — and a greater capacity to migrate to inflamed tissues — owing to increased expression of chemokine receptors such as CCR5 and CCR2. The expression pattern of CD62L on activated T cells is triphasic and seems to be regulated by the duration of antigenic stimulation. Initially, TCR stimulation induces the rapid shedding of CD62L from the T-cell surface by proteolytic cleavage, but within 24–48 hours, CD62L is re-expressed69. However, if TCR stimulation continues, the locus that encodes CD62L becomes transcriptionally silenced and surface expression of CD62L becomes fixed at a low level for an extended period of time10,69,70 (E.J.W, V. Teichgräber, L. Harrington, T. Becker, S.M.K., R. Antia and R.A., unpublished observations). So, activated T cells might remain CD62Lhi after brief exposure to antigen, whereas longer antigenic stimulations might generate CD62Llo effector T cells (Fig. 5). After ∼24 hours of antigenic stimulation, the levels of CD62L and CCR7 remained high on activated CD4+ T cells, and these cells retained lymph-node-homing properties, whereas migration to peripheral sites, such as the peritoneum and lungs, was inefficient (Box 1)64. However, if the exposure to antigen was sustained over several days, the T cells lost surface expression of these receptors and trafficking to the lymph nodes was markedly reduced18,63,64. Cytokines, such as IL-2 and IL-15, might also modulate T-cell migration patterns by decreasing or increasing the level of expression of CCR7 and CD62L, respectively19,60. It should be emphasized that, although the expression of CD62L and CCR7 will selectively bias T-cell migration towards lymph nodes, it does not necessarily impede the migration of T cells into peripheral tissues19,64 (E.J.W., V. Teichgräber and R.A., unpublished observations).
Figure 5: Models of memory T-cell differentiation.
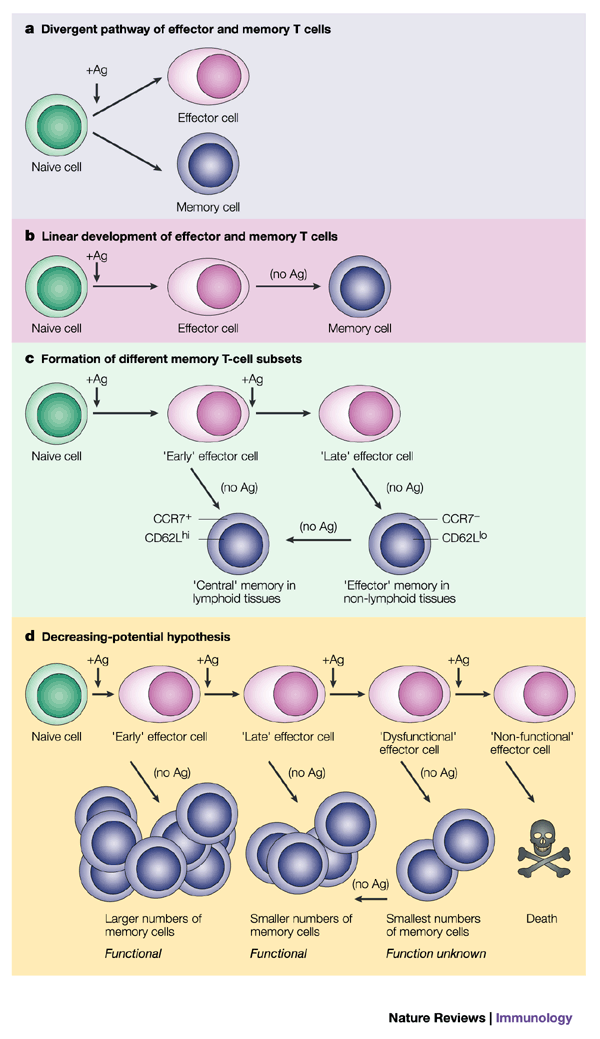
a | Model 1 represents a divergent pathway, whereby a naive T cell can give rise to daughter cells that develop into either effector or memory T cells, a decision that could be passive or instructive. In this model, naive T cells can bypass an effector-cell stage and develop directly into memory T cells. b | Model 2 represents a linear-differentiation pathway, whereby memory T cells are direct descendants of effector cells. This model indicates that memory T-cell development does not occur until antigen (Ag) is removed or greatly decreased in concentration. c | In model 3, which is a variation of model 2, a short duration of antigenic stimulation favours the development of central memory T cells, whereas a longer duration of stimulation favours the differentiation of effector memory T cells. d | Model 4 represents the decreasing-potential hypothesis, which suggests that effector T-cell functions steadily decrease as a consequence of persisting antigen (as observed in chronic infections). In addition, accumulative encounters with antigen lead to increased susceptibility of effector cells to apoptosis, and reduced numbers of memory T cells are formed. As suggested in model 2, the development of memory T cells occurs following antigen clearance. It is not known whether dysfunctional effector T cells can give rise to functional memory T cells, but this model suggests that T cells might regain function over time following the removal of antigen. CCR7, CC-chemokine receptor 7.
Overall, it appears that the manifestation of CD4+ and CD8+ T-cell responses is programmed to some degree, but that variations in the duration of TCR stimulation and the cytokine milieu might individually shape each T-cell response (that is, in terms of proliferation, cytokine secretion, cell-killing, B-cell help and migration) to create subsets of effector T cells with distinct effector functions and migratory patterns18,19,63,64,71,72.
Lineage of memory T cells
The lineage of memory T-cell development is still not fully understood. Are memory cells direct descendants of effector cells or do they arise from a second lineage (Fig. 5)? The use of a CRE/LOXP system in transgenic mice to 'mark' virus-specific effector T cells showed that 'marked' cells were maintained in the memory T-cell pool, which indicated that these cells were direct descendants of effector cells73. A second approach that used the adoptive transfer of effector T cells showed that memory T cells arise directly from this population74,75 (S.M.K. and R.A., unpublished observations). When BROMODEOXYURIDINE - and CFSE-labelling techniques were used to determine whether any T cells within the effector population proliferated during the contraction phase, minimal cell division was observed75 (S.M.K. and R.A., unpublished observations). These data suggest that the memory T-cell population is not generated from a subset of effector cells that 'divide-out' during the contraction phase, but rather, is formed directly from the effector cells themselves. Other studies have shown that activated CD8+ T cells seem to be programmed to develop into memory T cells, because CD8+ T cells that were stimulated briefly (∼24 hours), proliferated and differentiated into CTLs without further antigenic stimulation, as described above, but surprisingly, these cells continued to develop into long-lived, protective memory CD8+ T cells14,45. Therefore, the instructive programme that guides effector CD8+ T-cell development is sufficient to guide the formation of memory CD8+ T cells. However, recent studies suggest that memory T cells might also develop without passing through an effector-cell stage, and these memory cells are referred to as 'central' memory T cells18,59,60,63,64. It has been proposed that central memory T cells do not adopt effector-cell properties during the primary T-cell response, but they persist and form a protective reservoir that can give rise to secondary effector T cells if antigen is re-encountered (Box 1). Therefore, it is important to consider that memory T-cell development might occur in a non-linear fashion and that it can result in qualitatively different memory T-cell subsets (Fig. 5a and c)18,71,76. Different priming conditions (for example, the duration of antigenic stimulation and the type of cytokines present) might affect the formation of these subsets.
Effector T-cell contraction phase
What is the nature of the signal(s) that directs whether an effector T cell lives and differentiates into a long-lived memory T cell or dies? The contraction phase most probably functions as a safeguard to prevent excessive immunopathology by limiting the duration of T-cell responses. However, the extent of cell death directly determines the size of the memory T-cell pool. Therefore, it is crucial to identify the factors that positively and negatively regulate this stage of the T-cell response.
IL-2-family cytokines. One popular model is that as infection wanes, the level of cytokines that support clonal expansion and T-cell survival also declines, which triggers the apoptosis of activated T cells owing to growth-factor withdrawal. Candidate factors include type I IFNs and members of the IL-2 family (that is, IL-2, IL-4, IL-7 and IL-15), because these cytokines can reduce the rate of cell death in vitro, and in some cases in vivo, by the direct inhibition of apoptosis and/or increased effector T-cell proliferation (Table 2)21,47,77,78,79,80. So, could the administration of these factors result in the production of greater numbers of memory T cells during vaccinations? The exogenous administration of IL-2 has been shown to increase the total number of CD4+ T cells, and in combination with anti-viral therapy, to reduce viral load in patients and non-human primates infected with HIV and simian immunodeficiency virus (SIV), respectively81,82. In addition, IL-2 treatment can boost antigen-specific T-cell responses and delay the death of superantigen- and LCMV-reactive effector CD8+ T cells in mice83,84,85,86 (J. Blattman, K.A. Smith and R.A., unpublished observations). Increased levels of IL-4, IL-7 and IL-15 in vivo can increase the numbers of antigen-specific CD4+ and CD8+ T cells, and IL-15 can enhance protective immunity77,87,88,89. Whether these cytokines can have lasting effects on the generation or maintenance of antigen-specific memory T cells needs to be investigated further. It is possible that in isolation, these cytokines might not be sufficient, but in combination with other signals, greater effects could be achieved. However, driving greater expansion of the effector T-cell pool (in an antigen-independent manner) might not necessarily lead to an increase in the number of memory T cells that are formed. This was evident when mice were infected with a wild-type strain of Listeria and then, six days later, with a mutant strain that did not express the listeriolysin (LL0)91–99 epitope90. The secondary infection induced substantial proliferation of the LLO91–99-specific CD8+ T cells that were generated during the primary infection (presumably due to the production of supplementary cytokines), but a correlative increase in LLO91–99-specific memory CD8+ T cells was not observed90.
Table 2 In vivo effects of exogenous cytokines on mouse T-cell responses
TNF-family molecules. Examination of mouse knockouts has revealed that members of the TNF receptor (TNFR) family and their ligands, such as CD27 and CD154 (CD40L), might have interesting roles in the formation of memory T-cell populations. In CD154-deficient mice, the death of effector CD8+ T cells is enhanced and approximately tenfold fewer memory T cells are formed after LCMV infection, but interestingly, the lack of CD154 has no effect on CD8+ T-cell clonal expansion, as discussed previously30,91 (J. Whitmire and R.A., unpublished observations). Therefore, CD40–CD154 interactions can regulate memory T-cell setpoints by interfering with the contraction phase. Surprisingly, mice deficient in Fas (CD95), TNFR1 or both show minimal effects on effector-cell death and memory-cell setpoints, which suggests that other pathways mediate apoptosis of effector T cells92,93,94. An alternative pathway of apoptosis might involve phosphatase and tensin homologue (PTEN), a negative regulator of phosphatidylinositol 3-kinase and AKT kinase, because PTEN−/− T cells had increased AKT kinase activity and were resistant to superantigen-induced deletion in vivo95. Most probably, various mechanisms, that possibly overlap, contribute to effector-cell death, because disruption of a single mechansim has not been found to inhibit T-cell death.
Effector-cell molecules: perforin and IFN-γ. In addition to the striking cytolytic defects in perforin-knockout mice, the down-sizing of the effector CD8+ T-cell population (contraction) does not occur in the normal manner58,96,97,98. This might be due partially to the delayed or impaired pathogen clearance that is observed commonly in these mice; however, this effect is also evident when infections are resolved96. A separate study showed that the more target cells a CTL kills, the less likely it is to develop into a memory T cell99. Whether the role of perforin in CD8+ T-cell death is direct (in that perforin ultimately kills the cells that secrete it) or indirect (decreased killing of APCs that results in prolonged antigen exposure) is unclear. IFN-γ also seems to have a role in the down-modulation of the effector-cell population after infection96. So, perforin and IFN-γ are important not only for controlling the infection, but also for regulating effector-cell numbers. Because T-cell memory is determined by the magnitude of expansion and the extent of effector-cell death, strategies that interfere with cell death mediated by these molecules might enhance T-cell memory in response to vaccination.
Decreasing-potential hypothesis. A model that deserves attention is the 'decreasing-potential' hypothesis, which states that the primary factor that distinguishes effector T cells that die from those that survive and differentiate into memory T cells is the duration and level of antigenic stimulation to which the T cells are exposed (Fig. 5d)24. One prediction of this model is that effector-cell death would be reduced if T cells encountered antigen for shorter periods of time, but as discussed above, a potential caveat is that the effector-cell burst size would be smaller. The decreasing-potential hypothesis is strongly supported by the fate of antigen-specific CD4+ and CD8+ T cells during periods of chronic antigen exposure. It has been observed that antigen-specific T cells either disappear (deletion) or persist but become dysfunctional (exhaustion) in several mouse models — such as during chronic LCMV infection or in transgenic mice that express both antigen and specific TCR — and in patients with HIV or hepatitis C virus infection, or melanoma55,56,100,101,102,103,104. The degree to which the CTLs become defective seems to correlate with antigen load and can range from complete lack of function (no cytolytic activity or production of IFN-γ, TNF and IL-2) to partial loss of function (modest production of cytokines)104,105,106,107. If excessive antigen exposure induces effector T-cell dysfunction, is this effect reversible or are the cells permanently impaired? Moreover, it is not known if memory T cells can ever develop under these conditions and, if so, whether the degree of effector-cell dysfunction determines the functional state of the memory T cells that are formed (Fig. 5d). This point is particularly relevant for the design of current vaccine strategies that are aimed at treating patients with existing infections and/or tumours. Most probably, the conventional methods of vaccination — delivery of antigen by some type of vector — will not be successful, because the high levels of pre-existing antigen already impair T-cell responses. Therefore, novel vaccine strategies that help T-cells to regain their responsiveness will need to be developed, and this most certainly will be aided by determining the nature of effector T-cell non-responsiveness at the biochemical level.
T-cell development and vaccine design
Long-term immunological protection depends on both the quantity and quality of the memory T cells that are formed. Depending on the dose and virulence of the pathogen, different threshold numbers of memory CD8+ T cells might be needed to protect against disease (Fig. 6). Because the number of memory T cells formed is determined primarily by the burst size, it is essential that vaccines induce as large an effector T-cell population as possible. This poses a challenge for vaccines, because for vaccine vectors that can not replicate the widespread distribution of antigen can be a limitation. The effector-cell burst size is a function of both the number of naive T cells that are recruited into the immune response and the number of times these cells divide. A recent study showed that the recruitment of antigen-specific CD8+ T cells was reduced in antigen-limiting conditions and, even though the recruited cells divided at least 7–10 times (due to programmed proliferation), the effector-cell burst size was still ∼20 times smaller than when recruitment was complete14. Fortunately, the programmed model of memory CD8+ T-cell development predicts that even if the number of APCs is small, functional memory CD8+ T cells can develop. However, given that the quality and quantity of memory T cells that are produced might be modulated by the degree of T-cell stimulation and the types of co-stimulation and cytokines present, it is important to consider how different forms of vaccination might affect these parameters. For example, the level of antigen that can be provided by non-replicative vaccines, such as killed pathogens or recombinant protein, is limited by the innoculum dose and therefore, might be short-lived. These vaccines might induce recruited T cells to divide multiple times, but the effector-cell burst size might be smaller than if antigen were more prevalent. Furthermore, a reduced number of antigen encounters might result in the development of 'central' memory T cells rather than 'effector' memory T cells. Whether protective immunity would be enhanced or adversely affected by this is not known, because it has not been determined whether central and effector memory T cells confer different levels of protection (Box 1). Vaccination with DNA plasmids that encode antigens is gaining popularity because of the inherent stability of the vaccines, the cheap cost of producing and administering the DNA, and their ability to elicit protective immunity in animal models108,109. But, the details of T-cell priming with DNA vaccination are not fully understood, because it is not clear whether dendritic cells express antigens directly (through plasmid transfection), indirectly (through cross-presentation) or both. Also, the duration of antigen expression with DNA vaccination or the types of effector and memory T cells that are formed has not been delineated.
Figure 6: Modulation of the three phases of T-cell responses and the quantitative effects on memory T-cell formation.
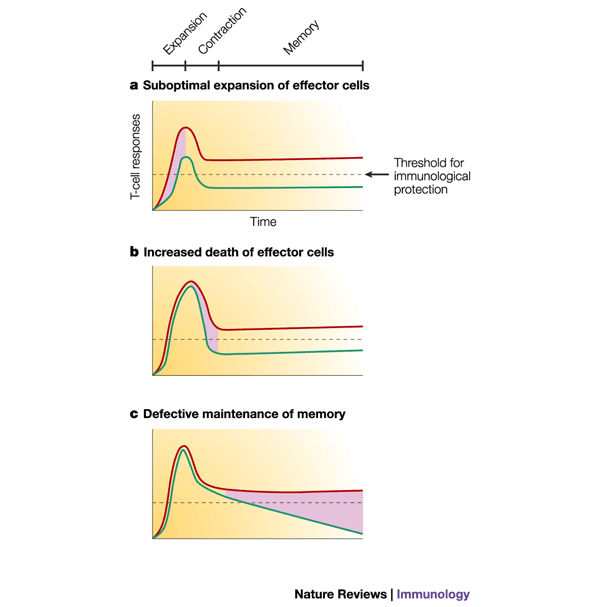
Alteration of any phase of a T-cell response — expansion, contraction or memory — can affect the number of memory T cells that is formed and whether protective immunity is established. The dashed line denotes the minimum number of antigen-specific memory T cells required to confer protection against disease. If all three phases occur optimally, then the number of memory T cells formed is above the threshold for protective immunity (red line), but if a | priming and expansion are suboptimal; b | cell death is increased; or c | memory T cells are not maintained, then the number of memory T cells that is formed will be insufficient for long-term immunological protection (green line). Pink shading indicates the suboptimal phase of the response. Therapeutic intervention at any of these three steps might lead to greater numbers of memory T cells and protective immunity.
Vaccine boosters most probably enhance immunological protection by affecting the quality and quantity of memory T cells. Higher-affinity T-cell clones can out-compete lower-affinity T cells for antigen, which suggests that repeated vaccine boosters might skew the memory T-cell population towards higher-affinity clones110,111,112,113.
There might be several ways to intervene during the different stages of a T-cell response to enhance vaccine efficacy (Fig. 6). For example, in cases where antigen distribution by a vaccine is limited or short-lived, it might be possible to increase the expansion of effector T-cell populations by modulating the factors that regulate antigen-independent cell division. Alternatively, it might be possible to intervene during the contraction phase to reduce effector-cell death, thereby increasing the number of memory T cells that are formed. Finally, the number of memory T cells might be increased by interfering with homeostasis and offsetting the balance between proliferation and attrition. Although several candidate approaches are currently under trial and show promise, these strategies remain a challenge because of a lack of understanding of the mechanisms and signals that regulate these stages of T-cell differentiation (for a review, see Ref. 114).
Concluding remarks
This review focuses on the mechanisms that regulate the development of effector and memory T cells. Although much has been learned, we still do not understand immunological memory at the molecular level. Only a handful of molecules have been identified that have important roles in this process and our understanding of how these molecules act biochemically is incomplete. For example, how does a T cell 'count' TCR and co-stimulatory signals and how do these signals collectively meet T-cell activation requirements? How are these signals translated into discrete T-cell responses, and can these responses be augmented to modulate the types of effector and memory T cells that are formed? How are changes to the T-cell phenotype made permanent as naive cells differentiate into effector and memory T cells, and what maintains this altered state in the absence of antigen? What controls the proliferative capacity of T cells in the presence and absence of antigen? Why does excessive antigenic stimulation decrease T-cell function and survival, and what are the molecular changes associated with this process? Because the cellular processes that are implicated in memory T-cell development are vast and might be essential to survival or used earlier in development, conventional knockout systems are of limited use. Insight will hopefully be gained by the new opportunities that genome-wide analysis will provide, and hypotheses should be testable using inducible-systems for disrupting genes in mice (conditional knockouts). Understanding the molecular makeup of T cells during acute and chronic immune responses will lead to more-effective vaccines for preventing disease, as well as for fighting chronic infections or tumours.
References
- Hou, S., Hyland, L., Ryan, K. W., Portner, A. & Doherty, P. C. Virus-specific CD8+ T-cell memory determined by clonal burst size. Nature 369, 652–654 (1994).
CAS PubMed Google Scholar - Murali-Krishna, K. et al. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity 8, 177–187 (1998).
Article CAS PubMed Google Scholar - Busch, D. H., Pilip, I. M., Vijh, S. & Pamer, E. G. Coordinate regulation of complex T-cell populations responding to bacterial infection. Immunity 8, 353–362 (1998).
CAS PubMed Google Scholar - Bousso, P., Levraud, J. P., Kourilsky, P. & Abastado, J. P. The composition of a primary T-cell response is largely determined by the timing of recruitment of individual T-cell clones. J. Exp. Med. 189, 1591–1600 (1999).
CAS PubMed PubMed Central Google Scholar - Whitmire, J. K., Asano, M. S., Murali-Krishna, K., Suresh, M. & Ahmed, R. Long-term CD4 Th1 and Th2 memory following acute lymphocytic choriomeningitis virus infection. J. Virol. 72, 8281–8288 (1998).
CAS PubMed PubMed Central Google Scholar - Agarwal, S. & Rao, A. Modulation of chromatin structure regulates cytokine gene expression during T-cell differentiation. Immunity 9, 765–775 (1998).
CAS PubMed Google Scholar - Grayson, J. M., Murali-Krishna, K., Altman, J. D. & Ahmed, R. Gene expression in antigen-specific CD8+ T cells during viral infection. J. Immunol. 166, 795–799 (2001).
CAS PubMed Google Scholar - Bachmann, M. F., Barner, M., Viola, A. & Kopf, M. Distinct kinetics of cytokine production and cytolysis in effector and memory T cells after viral infection. Eur. J. Immunol. 29, 291–299 (1999).This report shows that effector and memory CD8+ T cells can commit to cell division and secrete cytokines faster than naive cells, and that effector CD8+ T cells have immediate cytotoxic activity, whereas memory T cells require 12–24 hours of antigenic stimulation for this activity to be reacquired.
CAS PubMed Google Scholar - Veiga-Fernandes, H., Walter, U., Bourgeois, C., McLean, A. & Rocha, B. Response of naive and memory CD8+ T cells to antigen stimulation in vivo. Nature Immunol. 1, 47–53 (2000).This report shows that naive and memory CD8+ T cells have distinct functional properties and that when stimulated with antigen, memory T cells can proliferate and express IFN-γ, IL-2 and perforin faster than naive T cells.
CAS Google Scholar - Teague, T. K. et al. Activation changes the spectrum but not the diversity of genes expressed by T cells. Proc. Natl Acad. Sci. USA 96, 12691–12696 (1999).
CAS PubMed PubMed Central Google Scholar - Yang, Y., Chang, J. F., Parnes, J. R. & Fathman, C. G. T-cell receptor (TCR) engagement leads to activation-induced splicing of tumor necrosis factor (TNF) nuclear pre-mRNA. J. Exp. Med. 188, 247–254 (1998).
CAS PubMed PubMed Central Google Scholar - Badovinac, V. P., Corbin, G. A. & Harty, J. T. Cutting edge: OFF cycling of TNF production by antigen-specific CD8+ T cells is antigen independent. J. Immunol. 165, 5387–5391 (2000).
CAS PubMed Google Scholar - Slifka, M. K., Rodriguez, F. & Whitton, J. L. Rapid on/off cycling of cytokine production by virus-specific CD8+ T cells. Nature 401, 76–79 (1999).
CAS PubMed Google Scholar - Kaech, S. M. & Ahmed, R. Memory CD8+ T-cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nature Immunol. 2, 415–422 (2001).Together with references 44–46 , this study showed that naive CD8+ T cells could commit to proliferation and differentiation into effector and memory T cells after a brief period of antigenic stimulation (24 hours). The daughter cells of the initially activated parental CD8+ T cells continued to divide and differentiate in the absence of further antigenic stimulation, which suggested that activated T cells are developmentally programmed to clonally expand and differentiate into effector cells.
CAS Google Scholar - Moser, B. & Loetscher, P. Lymphocyte traffic control by chemokines. Nature Immunol. 2, 123–128 (2001).
CAS Google Scholar - Masopust, D., Vezys, V., Marzo, A. L. & Lefrancois, L. Preferential localization of effector memory cells in nonlymphoid tissue. Science 291, 2413–2417 (2001).
Article CAS PubMed Google Scholar - Reinhardt, R. L., Khoruts, A., Merica, R., Zell, T. & Jenkins, M. K. Visualizing the generation of memory CD4 T cells in the whole body. Nature 410, 101–105 (2001).
CAS PubMed Google Scholar - Sallusto, F., Lenig, D., Forster, R., Lipp, M. & Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401, 708–712 (1999).This study shows that different subsets of memory T cells exist and can be distinguished on the basis of their expression of the lymph-node-homing molecules CCR7 and L-selectin.
Article CAS PubMed Google Scholar - Weninger, W., Crowley, M. A., Manjunath, N. & von Andrian, U. H. Migratory properties of naive, effector, and memory CD8(+) T cells. J. Exp. Med. 194, 953–966 (2001).
CAS PubMed PubMed Central Google Scholar - Homann, D., Teyton, L. & Oldstone, M. B. Differential regulation of antiviral T-cell immunity results in stable CD8+ but declining CD4+ T-cell memory. Nature Med. 7, 913–919 (2001).
CAS PubMed Google Scholar - Zhang, X., Sun, S., Hwang, I., Tough, D. F. & Sprent, J. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity 8, 591–599 (1998).
CAS PubMed Google Scholar - Ku, C. C., Murakami, M., Sakamoto, A., Kappler, J. & Marrack, P. Control of homeostasis of CD8+ memory T cells by opposing cytokines. Science 288, 675–678 (2000).
CAS PubMed Google Scholar - Schluns, K. S., Kieper, W. C., Jameson, S. C. & Lefrancois, L. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nature Immunol. 1, 426–432 (2000).
CAS Google Scholar - Ahmed, R. & Gray, D. Immunological memory and protective immunity: understanding their relation. Science 272, 54–60 (1996).
CAS PubMed Google Scholar - Shahinian, A. et al. Differential T-cell costimulatory requirements in CD28-deficient mice. Science 261, 609–612 (1993).
CAS PubMed Google Scholar - Tan, J. T., Whitmire, J. K., Ahmed, R., Pearson, T. C. & Larsen, C. P. 4-1BB ligand, a member of the TNF family, is important for the generation of antiviral CD8 T-cell responses. J. Immunol. 163, 4859–4868 (1999).In this report, CD8+ T-cell responses to LCMV infection were found to be slightly lower in 4-1BBL −/− mice compared with normal mice, but antiviral CD4+ T-cell responses were unaffected.
CAS PubMed Google Scholar - Whitmire, J. K. et al. CD40–CD40 ligand costimulation is required for generating antiviral CD4 T-cell responses but is dispensable for CD8 T-cell responses. J. Immunol. 163, 3194–3201 (1999).This report shows that CD4+ and CD8+ T cells differentially depend on CD40–CD154 interactions. During an acute LCMV infection, the virus-specific CD8+ T-cell response was normal, but the CD4+ T-cell response was severely reduced.
CAS PubMed Google Scholar - Kopf, M. et al. OX40-deficient mice are defective in Th-cell proliferation but are competent in generating B-cell and CTL responses after virus infection. Immunity 11, 699–708 (1999).This study showed that in OX40 −/− mice, CD8+ T-cell responses were minimally affected during infection with influenza virus and LCMV, whereas CD4+ T-cell responses were greatly reduced.
CAS PubMed Google Scholar - Gravestein, L. A., Nieland, J. D., Kruisbeek, A. M. & Borst, J. Novel mAbs reveal potent co-stimulatory activity of murine CD27. Int. Immunol. 7, 551–557 (1995).
CAS PubMed Google Scholar - Borrow, P. et al. CD40L-deficient mice show deficits in antiviral immunity and have an impaired memory CD8+ CTL response. J. Exp. Med. 183, 2129–2142 (1996).
CAS PubMed Google Scholar - Chen, A. I. et al. OX40-ligand has a critical costimulatory role in dendritic cell:T-cell interactions. Immunity 11, 689–698 (1999).
CAS PubMed Google Scholar - Gramaglia, I., Weinberg, A. D., Lemon, M. & Croft, M. OX40 ligand: a potent costimulatory molecule for sustaining primary CD4 T-cell responses. J. Immunol. 161, 6510–6517 (1998).
CAS PubMed Google Scholar - Tuosto, L. & Acuto, O. CD28 affects the earliest signaling events generated by TCR engagement. Eur. J. Immunol. 28, 2131–2142 (1998).
CAS PubMed Google Scholar - Liu, Y., Wenger, R. H., Zhao, M. & Nielsen, P. J. Distinct costimulatory molecules are required for the induction of effector and memory cytotoxic T lymphocytes. J. Exp. Med. 185, 251–262 (1997).
CAS PubMed PubMed Central Google Scholar - Kundig, T. M. et al. Duration of TCR stimulation determines costimulatory requirement of T cells. Immunity 5, 41–52 (1996).
CAS PubMed Google Scholar - Andreasen, S. O., Christensen, J. E., Marker, O. & Thomsen, A. R. Role of CD40 ligand and CD28 in induction and maintenance of antiviral CD8+ effector T-cell responses. J. Immunol. 164, 3689–3697 (2000).
CAS PubMed Google Scholar - Suresh, M. et al. Role of CD28–B7 interactions in generation and maintenance of CD8 T-cell memory. J. Immunol. 167, 5565–5573 (2001).
CAS PubMed Google Scholar - Sepulveda, H., Cerwenka, A., Morgan, T. & Dutton, R. W. CD28, IL-2-independent costimulatory pathways for CD8 T-lymphocyte activation. J. Immunol. 163, 1133–1142 (1999).
CAS PubMed Google Scholar - Pardigon, N. et al. Role of co-stimulation in CD8+ T-cell activation. Int. Immunol. 10, 619–630 (1998).
CAS PubMed Google Scholar - Szabo, S. J. et al. Distinct effects of T-bet in TH1 lineage commitment and IFN-γ production in CD4 and CD8 T cells. Science 295, 338–342 (2002).
CAS PubMed Google Scholar - Iezzi, G., Karjalainen, K. & Lanzavecchia, A. The duration of antigenic stimulation determines the fate of naive and effector T cells. Immunity 8, 89–95 (1998).This study showed that naive CD4+ T cells required at least six hours of antigenic stimulation by professional APCs to commit to proliferation in vitro . In the absence of co-stimulation, >20 hours were required. Effector cells could commit to proliferation after only one hour of stimulation, and if stimulation was prolonged, the effector cells would die.
CAS PubMed Google Scholar - Jelley-Gibbs, D. M., Lepak, N. M., Yen, M. & Swain, S. L. Two distinct stages in the transition from naive CD4 T cells to effectors, early antigen-dependent and late cytokine-driven expansion and differentiation. J. Immunol. 165, 5017–5026 (2000).
CAS PubMed Google Scholar - Gett, A. V. & Hodgkin, P. D. A cellular calculus for signal integration by T cells. Nature Immunol. 1, 239–244 (2000).
CAS Google Scholar - van Stipdonk, M. J., Lemmens, E. E. & Schoenberger, S. P. Naive CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nature Immunol. 2, 423–429 (2001).This report showed that naive CD8+ T cells could commit to dividing at least 7–10 times and differentiating into CTLs after only two hours of stimulation in vitro.
CAS Google Scholar - Mercado, R. et al. Early programming of T-cell populations responding to bacterial infection. J. Immunol. 165, 6833–6839 (2000).These authors showed that a brief duration of exposure to antigen in vivo could elicit effector CD8+ T-cell development. Twenty-four hours after infection with L. monocytogenes , mice were treated with antibiotics to rapidly eliminate the bacteria and functional effector CD8+ T cells were still generated.
CAS PubMed Google Scholar - Wong, P. & Pamer, E. G. Cutting edge: antigen-independent CD8 T-cell proliferation. J. Immunol. 166, 5864–5868 (2001).
CAS PubMed Google Scholar - Tan, J. T. et al. IL-7 is critical for homeostatic proliferation and survival of naive T cells. Proc. Natl Acad. Sci. USA 98, 8732–8737 (2001).
CAS PubMed PubMed Central Google Scholar - Murali-Krishna, K. & Ahmed, R. Cutting edge: naive T cells masquerading as memory cells. J. Immunol. 165, 1733–1737 (2000).
CAS PubMed Google Scholar - Foulds, K. E. et al. CD4 and CD8 T cells are intrinsically different in their proliferative responses. J. Immunol. 168, 1528–1532 (2002).A direct comparison of the proliferation rates of activated CD4+ and CD8+ T cells in vivo.
CAS PubMed Google Scholar - Whitmire, J. K., Slifka, M. K., Grewal, I. S., Flavell, R. A. & Ahmed, R. CD40-ligand-deficient mice generate a normal primary cytotoxic T-lymphocyte response but a defective humoral response to a viral infection. J. Virol. 70, 8375–8381 (1996).
CAS PubMed PubMed Central Google Scholar - Busch, D. H. & Pamer, E. G. T-lymphocyte dynamics during Listeria monocytogenes infection. Immunol. Lett. 65, 93–98 (1999).
CAS PubMed Google Scholar - Wherry, E. J., Puorro, K. A., Porgador, A. & Eisenlohr, L. C. The induction of virus-specific CTL as a function of increasing epitope expression: responses rise steadily until excessively high levels of epitope are attained. J. Immunol. 163, 3735–3745 (1999).
CAS PubMed Google Scholar - Kedl, R. M. et al. T cells compete for access to antigen-bearing antigen-presenting cells. J. Exp. Med. 192, 1105–1113 (2000).
CAS PubMed PubMed Central Google Scholar - Ludewig, B. et al. Dendritic cells efficiently induce protective antiviral immunity. J. Virol. 72, 3812–3818 (1998).
CAS PubMed PubMed Central Google Scholar - Matloubian, M., Concepcion, R. J. & Ahmed, R. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J. Virol. 68, 8056–8063 (1994).
CAS PubMed PubMed Central Google Scholar - Zajac, A. J. et al. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 188, 2205–2213 (1998).This study shows that during chronic LCMV infection, antigen-specific CD8+ T cells can either be deleted or persist in a non-functional state. CD4+ T cells are important for maintaining effector CD8+ T-cell responses.
CAS PubMed PubMed Central Google Scholar - Moskophidis, D., Lechner, F., Pircher, H. & Zinkernagel, R. M. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 362, 758–761 (1993).
CAS PubMed Google Scholar - Zhou, S., Ou, R., Huang, L. & Moskophidis, D. Critical role for perforin-, Fas/FasL- and TNFR1-mediated cytotoxic pathways in down-regulation of antigen-specific T cells during persistent viral infection. J. Virol. 76, 829–840 (2002).
CAS PubMed PubMed Central Google Scholar - Lauvau, G. et al. Priming of memory but not effector CD8 T cells by a killed bacterial vaccine. Science 294, 1735–1739 (2001).
CAS PubMed Google Scholar - Manjunath, N. et al. Effector differentiation is not prerequisite for generation of memory cytotoxic T lymphocytes. J. Clin. Invest. 108, 871–878 (2001).
CAS PubMed PubMed Central Google Scholar - Vijh, S. & Pamer, E. G. Immunodominant and subdominant CTL responses to Listeria monocytogenes infection. J. Immunol. 158, 3366–3371 (1997).
CAS PubMed Google Scholar - Iezzi, G., Scotet, E., Scheidegger, D. & Lanzavecchia, A. The interplay between the duration of TCR and cytokine signaling determines T-cell polarization. Eur. J. Immunol. 29, 4092–4101 (1999).
CAS PubMed Google Scholar - Langenkamp, A., Messi, M., Lanzavecchia, A. & Sallusto, F. Kinetics of dendritic-cell activation: impact on priming of TH1, TH2 and nonpolarized T cells. Nature Immunol. 1, 311–316 (2000).
CAS Google Scholar - Iezzi, G., Scheidegger, D. & Lanzavecchia, A. Migration and function of antigen-primed nonpolarized T lymphocytes in vivo. J. Exp. Med. 193, 987–993 (2001).
CAS PubMed PubMed Central Google Scholar - Bird, J. J. et al. Helper T-cell differentiation is controlled by the cell cycle. Immunity 9, 229–237 (1998).
CAS PubMed Google Scholar - Gett, A. V. & Hodgkin, P. D. Cell division regulates the T-cell cytokine repertoire, revealing a mechanism underlying immune class regulation. Proc. Natl Acad. Sci. USA 95, 9488–9493 (1998).
CAS PubMed PubMed Central Google Scholar - Grogan, J. L. et al. Early transcription and silencing of cytokine genes underlie polarization of T-helper-cell subsets. Immunity 14, 205–215 (2001).
CAS PubMed Google Scholar - Bajenoff, M., Wurtz, O. & Guerder, S. Repeated antigen exposure is necessary for the differentiation, but not the initial proliferation, of naive CD4(+) T cells. J. Immunol. 168, 1723–1729 (2002).
CAS PubMed Google Scholar - Chao, C. C., Jensen, R. & Dailey, M. O. Mechanisms of L-selectin regulation by activated T cells. J. Immunol. 159, 1686–1694 (1997).
CAS PubMed Google Scholar - Oehen, S. & Brduscha-Riem, K. Differentiation of naive CTL to effector and memory CTL: correlation of effector function with phenotype and cell division. J. Immunol. 161, 5338–5346 (1998).
CAS PubMed Google Scholar - Campbell, D. J., Kim, C. H. & Butcher, E. C. Separable effector T-cell populations specialized for B-cell help or tissue inflammation. Nature Immunol. 2, 876–881 (2001).
CAS Google Scholar - Campbell, D. J. & Butcher, E. C. Rapid acquisition of tissue-specific homing phenotypes by CD4(+) T cells activated in cutaneous or mucosal lymphoid tissues. J. Exp. Med. 195, 135–141 (2002).
CAS PubMed PubMed Central Google Scholar - Jacob, J. & Baltimore, D. Modelling T-cell memory by genetic marking of memory T cells in vivo. Nature 399, 593–597 (1999).
CAS PubMed Google Scholar - Opferman, J. T., Ober, B. T. & Ashton-Rickardt, P. G. Linear differentiation of cytotoxic effectors into memory T lymphocytes. Science 283, 1745–1748 (1999).
CAS PubMed Google Scholar - Hu, H. et al. CD4(+) T-cell effectors can become memory cells with high efficiency and without further division. Nature Immunol. 2, 705–710 (2001).
CAS Google Scholar - Champagne, P. et al. Skewed maturation of memory HIV-specific CD8 T lymphocytes. Nature 410, 106–111 (2001).
CAS PubMed Google Scholar - Vella, A. T., Dow, S., Potter, T. A., Kappler, J. & Marrack, P. Cytokine-induced survival of activated T cells in vitro and in vivo. Proc. Natl Acad. Sci. USA 95, 3810–3815 (1998).
CAS PubMed PubMed Central Google Scholar - Marrack, P., Kappler, J. & Mitchell, T. Type I interferons keep activated T cells alive. J. Exp. Med. 189, 521–530 (1999).
CAS PubMed PubMed Central Google Scholar - Qin, J. Z. et al. Interleukin-7 and interleukin-15 regulate the expression of the bcl-2 and c-myb genes in cutaneous T-cell lymphoma cells. Blood 98, 2778–2783 (2001).
CAS PubMed Google Scholar - Sprent, J., Zhang, X., Sun, S. & Tough, D. T-cell proliferation in vivo and the role of cytokines. Philos Trans R Soc Lond B Biol Sci 355, 317–322 (2000).
CAS PubMed PubMed Central Google Scholar - Lalezari, J. P. et al. Low-dose daily subcutaneous interleukin-2 in combination with highly active antiretroviral therapy in HIV+ patients: a randomized controlled trial. HIV Clin. Trials 1, 1–15 (2000).
CAS PubMed Google Scholar - Barouch, D. H. et al. Augmentation of immune responses to HIV-1 and simian immunodeficiency virus DNA vaccines by IL-2/Ig plasmid administration in rhesus monkeys. Proc. Natl Acad. Sci. USA 97, 4192–4197 (2000).
CAS PubMed PubMed Central Google Scholar - Ahlers, J. D., Dunlop, N., Alling, D. W., Nara, P. L. & Berzofsky, J. A. Cytokine-in-adjuvant steering of the immune response phenotype to HIV-1 vaccine constructs: granulocyte–macrophage colony-stimulating factor and TNF-α synergize with IL-12 to enhance induction of cytotoxic T lymphocytes. J. Immunol. 158, 3947–3958 (1997).
CAS PubMed Google Scholar - Kuroda, K. et al. Implantation of IL-2-containing osmotic pump prolongs the survival of superantigen-reactive T cells expanded in mice injected with bacterial superantigen. J. Immunol. 157, 1422–1431 (1996).
CAS PubMed Google Scholar - Shimizu, K., Fields, R. C., Giedlin, M. & Mule, J. J. Systemic administration of interleukin-2 enhances the therapeutic efficacy of dendritic-cell-based tumor vaccines. Proc. Natl Acad. Sci. USA 96, 2268–2273 (1999).
CAS PubMed PubMed Central Google Scholar - Kang, B. Y. et al. Antigen-specific cytotoxicity and cell number of adoptively transferred T cells are efficiently maintained in vivo by re-stimulation with an antigen/interleukin-2 fusion protein. Int. J. Cancer 82, 569–573 (1999).
CAS PubMed Google Scholar - Yajima, T. et al. Overexpression of IL-15 in vivo increases antigen-driven memory CD8(+) T cells following a microbe exposure. J. Immunol. 168, 1198–1203 (2002).
CAS PubMed Google Scholar - Khan, I. A. & Casciotti, L. IL-15 prolongs the duration of CD8+ T-cell-mediated immunity in mice infected with a vaccine strain of Toxoplasma gondii. J. Immunol. 163, 4503–4509 (1999).
CAS PubMed Google Scholar - Maeurer, M. J. et al. Interleukin-7 or interleukin-15 enhances survival of _Mycobacterium tuberculosis_-infected mice. Infect. Immun. 68, 2962–2970 (2000).
CAS PubMed PubMed Central Google Scholar - Busch, D. H., Kerksiek, K. M. & Pamer, E. G. Differing roles of inflammation and antigen in T-cell proliferation and memory generation. J. Immunol. 164, 4063–4070 (2000).
CAS PubMed Google Scholar - Whitmire, J. K. & Ahmed, R. Costimulation in antiviral immunity: differential requirements for CD4(+) and CD8(+) T-cell responses. Curr. Opin. Immunol. 12, 448–455 (2000).
CAS PubMed Google Scholar - Zimmermann, C., Rawiel, M., Blaser, C., Kaufmann, M. & Pircher, H. Homeostatic regulation of CD8+ T cells after antigen challenge in the absence of Fas (CD95). Eur. J. Immunol. 26, 2903–2910 (1996).
CAS PubMed Google Scholar - Nguyen, L. T. et al. TNF receptor 1 (TNFR1) and CD95 are not required for T-cell deletion after virus infection but contribute to peptide-induced deletion under limited conditions. Eur. J. Immunol. 30, 683–688 (2000).
CAS PubMed Google Scholar - Reich, A., Korner, H., Sedgwick, J. D. & Pircher, H. Immune down-regulation and peripheral deletion of CD8 T cells does not require TNF-receptor–ligand interactions nor CD95 (Fas, APO-1). Eur. J. Immunol. 30, 678–682 (2000).
CAS PubMed Google Scholar - Suzuki, A. et al. T-cell-specific loss of PTEN leads to defects in central and peripheral tolerance. Immunity 14, 523–534 (2001).
CAS PubMed Google Scholar - Badovinac, V. P., Tvinnereim, A. R. & Harty, J. T. Regulation of antigen-specific CD8+ T-cell homeostasis by perforin and interferon-γ. Science 290, 1354–1358 (2000).
CAS PubMed Google Scholar - Kagi, D., Odermatt, B. & Mak, T. W. Homeostatic regulation of CD8+ T cells by perforin. Eur. J. Immunol. 29, 3262–3272 (1999).
CAS PubMed Google Scholar - Matloubian, M. et al. A role for perforin in downregulating T-cell responses during chronic viral infection. J. Virol. 73, 2527–2536 (1999).
CAS PubMed PubMed Central Google Scholar - Opferman, J. T., Ober, B. T., Narayanan, R. & Ashton-Rickardt, P. G. Suicide induced by cytolytic activity controls the differentiation of memory CD8(+) T lymphocytes. Int. Immunol. 13, 411–419 (2001).
CAS PubMed Google Scholar - Lee, P. P. et al. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nature Med. 5, 677–685 (1999).
CAS PubMed Google Scholar - Appay, V. et al. HIV-specific CD8(+) T cells produce antiviral cytokines but are impaired in cytolytic function. J. Exp. Med. 192, 63–75 (2000).
CAS PubMed PubMed Central Google Scholar - Kostense, S. et al. High viral burden in the presence of major HIV-specific CD8(+) T-cell expansions: evidence for impaired CTL effector function. Eur. J. Immunol. 31, 677–686 (2001).
CAS PubMed Google Scholar - Lechner, F. et al. Analysis of successful immune responses in persons infected with hepatitis C virus. J. Exp. Med. 191, 1499–1512 (2000).
CAS PubMed PubMed Central Google Scholar - Tanchot, C., Barber, D. L., Chiodetti, L. & Schwartz, R. H. Adaptive tolerance of CD4+ T cells in vivo: multiple thresholds in response to a constant level of antigen presentation. J. Immunol. 167, 2030–2039 (2001).
CAS PubMed Google Scholar - McKay, P. F. et al. Vaccine protection against functional CTL abnormalities in simian human immunodeficiency virus-infected rhesus monkeys. J. Immunol. 168, 332–337 (2002).
CAS PubMed Google Scholar - Edwards, B. H. et al. Magnitude of functional CD8(+) T-cell responses to the gag protein of human immunodeficiency virus type 1 correlates inversely with viral load in plasma. J. Virol. 76, 2298–2305 (2002).
CAS PubMed PubMed Central Google Scholar - Betts, M. R. et al. Analysis of total human immunodeficiency virus (HIV)-specific CD4(+) and CD8(+) T-cell responses: relationship to viral load in untreated HIV infection. J. Virol. 75, 11983–11991 (2001).
CAS PubMed PubMed Central Google Scholar - Herrmann, J. E. et al. Protection against rotavirus infections by DNA vaccination. J. Infect. Dis. 174, S93–S97 (1996).
CAS PubMed Google Scholar - Amara, R. R. et al. Control of a mucosal challenge and prevention of AIDS by a multiprotein DNA/MVA vaccine. Science 292, 69–74 (2001).
CAS PubMed Google Scholar - Savage, P. A., Boniface, J. J. & Davis, M. M. A kinetic basis for T-cell receptor repertoire selection during an immune response. Immunity 10, 485–492 (1999).
CAS PubMed Google Scholar - Kedl, R. M., Schaefer, B. C., Kappler, J. W. & Marrack, P. T cells down-modulate peptide–MHC complexes on APCs in vivo. Nature Immunol. 3, 27–32 (2002).
CAS Google Scholar - Butz, E. A. & Bevan, M. J. Massive expansion of antigen-specific CD8+ T cells during an acute virus infection. Immunity 8, 167–175 (1998).
CAS PubMed PubMed Central Google Scholar - Busch, D. H. & Pamer, E. G. T-cell affinity maturation by selective expansion during infection. J. Exp. Med. 189, 701–710 (1999).
CAS PubMed PubMed Central Google Scholar - Berzofsky, J., Ahlers, J. & Belyakov, I. Strategies for designing and optimizing new generation vaccines. Nature Rev. Immunol. 1, 209–219 (2001).
CAS Google Scholar - Doherty, P. C., Topham, D. J. & Tripp, R. A. Establishment and persistence of virus-specific CD4+ and CD8+ T-cell memory. Immunol. Rev. 150, 23–44 (1996).
CAS PubMed Google Scholar - Arbones, M. L. et al. Lymphocyte homing and leukocyte rolling and migration are impaired in L-selectin-deficient mice. Immunity 1, 247–260 (1994).
CAS PubMed Google Scholar - Warnock, R. A., Askari, S., Butcher, E. C. & von Andrian, U. H. Molecular mechanisms of lymphocyte homing to peripheral lymph nodes. J. Exp. Med. 187, 205–216 (1998).
CAS PubMed PubMed Central Google Scholar - Campbell, J. J. et al. 6-C-kine (SLC), a lymphocyte adhesion-triggering chemokine expressed by high endothelium, is an agonist for the MIP-3β receptor CCR7. J. Cell. Biol. 141, 1053–1059 (1998).
CAS PubMed PubMed Central Google Scholar - Marshall, D. R. et al. Measuring the diaspora for virus-specific CD8+ T cells.. Proc Natl Acad Sci U S A 98, 6313–6318 (2001).
CAS PubMed PubMed Central Google Scholar - Watt, F. M. & Hogan, B. L. Out of Eden: stem cells and their niches. Science 287, 1427–1430 (2000).
CAS PubMed Google Scholar - Plunkett, F. J. et al. The flow cytometric analysis of telomere length in antigen-specific CD8+ T cells during acute Epstein–Barr virus infection. Blood 97, 700–707 (2001).
CAS PubMed Google Scholar - Weng, N. P., Hathcock, K. S. & Hodes, R. J. Regulation of telomere length and telomerase in T and B cells: a mechanism for maintaining replicative potential. Immunity 9, 151–157 (1998).
CAS PubMed Google Scholar - Hathcock, K. S., Weng, N. P., Merica, R., Jenkins, M. K. & Hodes, R. Cutting edge: antigen-dependent regulation of telomerase activity in murine T cells. J. Immunol. 160, 5702–5706 (1998).
CAS PubMed Google Scholar - Morrison, S. J., Prowse, K. R., Ho, P. & Weissman, I. L. Telomerase activity in hematopoietic cells is associated with self-renewal potential. Immunity 5, 207–216 (1996).
CAS PubMed Google Scholar - Voehringer, D. et al. Viral infections induce abundant numbers of senescent CD8 T cells. J. Immunol. 167, 4838–4843 (2001).
CAS PubMed Google Scholar - Deeths, M. J., Kedl, R. M. & Mescher, M. F. CD8+ T cells become nonresponsive (anergic) following activation in the presence of costimulation. J. Immunol. 163, 102–110 (1999).
CAS PubMed Google Scholar - Bikah, G., Pogue-Caley, R. R., McHeyzer-Williams, L. J. & McHeyzer-Williams, M. G. Regulating T-helper-cell immunity through antigen responsiveness and calcium entry. Nature Immunol. 1, 402–412 (2000).
CAS Google Scholar - Sigal, L. J., Reiser, H. & Rock, K. L. The role of B7-1 and B7-2 costimulation for the generation of CTL responses in vivo. J. Immunol. 161, 2740–2745 (1998).
CAS PubMed Google Scholar - Lumsden, J. M., Roberts, J. M., Harris, N. L., Peach, R. J. & Ronchese, F. Differential requirement for CD80 and CD80/CD86-dependent costimulation in the lung immune response to an influenza virus infection. J. Immunol. 164, 79–85 (2000).
CAS PubMed Google Scholar - Schoenberger, S. P., Toes, R. E., van der Voort, E. I., Offringa, R. & Melief, C. J. T-cell help for cytotoxic T lymphocytes is mediated by CD40–CD40L interactions. Nature 393, 480–483 (1998).
CAS PubMed Google Scholar - Wu, Y. & Liu, Y. Viral induction of co-stimulatory activity on antigen-presenting cells bypasses the need for CD4+ T-cell help in CD8+ T-cell responses. Curr. Biol. 4, 499–505 (1994).
CAS PubMed Google Scholar - Ridge, J. P., Di Rosa, F. & Matzinger, P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature 393, 474–478 (1998).
CAS PubMed Google Scholar - Geiselhart, L. A. et al. IL-7 administration alters the CD4:CD8 ratio, increases T-cell numbers, and increases T-cell function in the absence of activation. J. Immunol. 166, 3019–3027 (2001).
CAS PubMed Google Scholar - Kennedy, M. K. et al. Reversible defects in natural killer and memory CD8 T-cell lineages in interleukin-15-deficient mice. J. Exp. Med. 191, 771–780 (2000).
CAS PubMed PubMed Central Google Scholar - Ahlers, J. D., Belyakov, I. M., Matsui, S. & Berzofsky, J. A. Mechanisms of cytokine synergy essential for vaccine protection against viral challenge. Int. Immunol. 13, 897–908 (2001).
CAS PubMed Google Scholar - Khatami, S., Brummer, E. & Stevens, D. A. Effects of granulocyte–macrophage colony-stimulating factor (GM-CSF) in vivo on cytokine production and proliferation by spleen cells. Clin. Exp. Immunol. 125, 198–201 (2001).
CAS PubMed PubMed Central Google Scholar
Author information
Authors and Affiliations
- Emory Vaccine Center and Department of Microbiology and Immunology, Emory University School of Medicine, 1510 Clifton Road, Atlanta, 30322, Georgia, USA
Susan M. Kaech, E. John Wherry & Rafi Ahmed
Authors
- Susan M. Kaech
You can also search for this author inPubMed Google Scholar - E. John Wherry
You can also search for this author inPubMed Google Scholar - Rafi Ahmed
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toRafi Ahmed.
Related links
Glossary
EXHAUSTION
An 'operational' definition that refers to the loss of antigen-specific T-cell responses in vivo after prolonged or repetitive stimulation with antigen, such as during chronic viral infection. The antigen-specific T cells are not deleted, but persist in a non-functional state for extended periods of time.
CRE/LOXP
A site-specific recombination system. Two short DNA sequences (LoxP sites) are engineered to flank the target DNA. Expression of Cre recombinase leads to excision of the intervening sequence. Depending on the type of promoter, Cre can be expressed at specific times during development or in specific sets of cells.
BROMODEOXYURIDINE
(BrdU). A thymidine analogue that can be incorporated into DNA during S-phase when cells are exposed to this substance. Cells that have incorporated BrdU, and presumably have divided, can be visualized using anti-BrdU antibodies by flow cytometry.
5,6-CARBOXYFLUORESCEIN DIACETATE SUCCINIMIDYL ESTER
(CFSE). A stable green-fluorescent dye that can be used to label populations of cells homogenously. When a cell divides, the fluorescent intensity dcreases by 50%, and this allows cells that have divided specific numbers of times to be visualized by flow cytometry. This reagent can successfully measure ∼7–10 cell divisions.
Rights and permissions
About this article
Cite this article
Kaech, S., Wherry, E. & Ahmed, R. Effector and memory T-cell differentiation: implications for vaccine development.Nat Rev Immunol 2, 251–262 (2002). https://doi.org/10.1038/nri778
- Issue Date: 01 April 2002
- DOI: https://doi.org/10.1038/nri778