The Big Bang of picorna-like virus evolution antedates the radiation of eukaryotic supergroups (original) (raw)
Key Points
- RNA viruses, which are most common in eukaryotes, are among the simplest forms of life.
- Genomic and metagenomic studies have highlighted remarkable diversity of a major class of RNA viruses, the extended picornavirus-like superfamily.
- Phylogenetic analysis reveals close evolutionary relationships between RNA viruses infecting unicellular eukaryotes and distinct families of picorna-like viruses of plants and animals.
- This suggests that diversification of picorna-like viruses antedated radiation of the eukaryotes and probably occurred in a 'Big Bang' concomitant with the key events of eukaryogenesis.
- The origins of the conserved genes of picorna-like viruses can be traced to specific prokaryotic ancestors.
- The Big Bang of picorna-like virus evolution might have been triggered by chance assembly of these ancestral genes at the earliest stages of eukaryogenesis.
Abstract
The recent discovery of RNA viruses in diverse unicellular eukaryotes and developments in evolutionary genomics have provided the means for addressing the origin of eukaryotic RNA viruses. The phylogenetic analyses of RNA polymerases and helicases presented in this Analysis article reveal close evolutionary relationships between RNA viruses infecting hosts from the Chromalveolate and Excavate supergroups and distinct families of picorna-like viruses of plants and animals. Thus, diversification of picorna-like viruses probably occurred in a 'Big Bang' concomitant with key events of eukaryogenesis. The origins of the conserved genes of picorna-like viruses are traced to likely ancestors including bacterial group II retroelements, the family of HtrA proteases and DNA bacteriophages.
Similar content being viewed by others
Main
In the past few years the importance of virology for understanding fundamental aspects of biological evolution has grown. In particular, RNA viruses might hold clues to the origin of genetic systems, being, possibly, the living relics of the ancient RNA world that is widely believed to predate the extant DNA-based genetic cycle of cellular organisms1,2. On a more practical level, knowledge of RNA virus evolution is indispensable for unravelling the origins of devastating emergent diseases such as AIDS, severe acute respiratory syndrome and haemorrhagic Ebola fever3. In addition, metagenomic research has revealed an enormous diversity of DNA and RNA viruses in the environment and has shown that, at least in marine habitats, viruses are the most abundant biological entities, with as many as 10 virus particles per cell4,5,6,7. Because most marine viruses kill host cells, they substantially contribute to the global carbon cycle7.
Several complementary developments have led to a dramatic expansion of the explored part of the 'virosphere'. The most conspicuous discoveries include many unusual archaeal viruses8,9,10, large phycodnaviruses infecting green algae and stramenopiles11,12, insect polydnaviruses13 and the giant mimivirus14,15. In addition, bacteriophage genomics has uncovered enormous, unanticipated diversity of this part of the viral world16,17,18,19,20,21. Evolutionary genomic analysis of the rapidly growing collection of viral genomes has revealed both deep unity, as exemplified by the demonstration of the common ancestry of diverse families of large DNA viruses of eukaryotes22, and the enormous variability of genome content, for example, in the case of archaeal viruses for which common origins are typically not traceable8,10.
In parallel, there has been a resurgence of interest in viruses and virus-like selfish genetic elements as major players in the origin and evolution of cellular life23,24,25,26,27,28,29. Two concepts of ancient origin and early evolution of viruses have been proposed, both emphasizing the tight connections between the evolution of viruses and cells25,28. One concept expounds the 'three RNA cells' scenario, according to which RNA viruses 'invented' DNA and introduced it, complete with the replication machinery, into putative primordial RNA cells that are envisaged to have been ancestors of each of the three domains of extant life25. The second, 'virus world' concept, based primarily on the mounting evidence from comparative genomics, posits that both RNA and DNA viruses evolved from primordial genetic systems that existed before the emergence of fully fledged cells, and that the large DNA genomes of the first cellular life forms evolved by accretion of virus-like and plasmid-like DNA replicons28. The virus world model also suggests that the major classes of viruses of eukaryotes evolved through mixing and matching the genes that were derived from prokaryotic viruses, plasmids and chromosomes at the time of eukaryogenesis. The collective result of these developments is a new landscape of data, models and ideas that calls for rewriting the fundamentals of virology27,28,30.
A long-standing enigma in virology is the non-uniform distribution of the major classes of RNA, DNA and retroid viruses among the branches of host organisms28. For instance, vertebrates can be infected by all classes of viruses, whereas green plants do not seem to be infected with retroid RNA viruses or true (non-pararetro) double-stranded (ds) DNA viruses31,32. Even more intriguing are the disparities between the abundance and diversity of positive-strand RNA viruses in plants and animals33, the extreme paucity of such viruses in bacteria34,35, and their apparent absence in archaea8,10 (M. Young, personal communication). These striking but largely unexplained patterns of virus distribution suggest that tight connections exist between major evolutionary transitions in the history of life and the global ecology of viruses. Understanding these connections is essential for the development of a general picture of the evolution of viruses and cells.
The current view of evolution of viruses and their host ranges derives primarily from studies on a few model organisms, such as mammals, birds, green plants (mostly cultured), and, to a lesser extent, insects, fungi and several groups of well-characterized bacteria. Until recently, there has been almost no research on viruses that infect the diverse groups of unicellular eukaryotes. However, this has changed as viruses have recently been isolated from a variety of marine eukaryotes such as algae and dinoflagellates7. These studies have resulted in the identification and sequencing of many positive-strand RNA viruses, which has dramatically increased the size and diversity of this virus class36,37,38,39 (see Supplementary information S1 (table)). In addition, several RNA viruses have been identified and sequenced as a result of metagenomic studies40,41,42,43.
In this Analysis article, we exploit the growing collection of diverse viral genome sequences that infect a wide range of eukaryotes to carry out a genomic comparison and phylogenetic analysis of a major division of eukaryotic positive-strand RNA viruses, the picorna-like superfamily, in an attempt to shed light on the early stages of its evolution. We conclude that the diverse groups of picorna-like viruses probably evolved in a Big Bang that antedated the radiation of the five supergroups of eukaryotes. Our analysis provides independent evidence in support of the concept of the major transitions in the history of life as explosive, non-linear events44 and suggests that the Big Bangs of host organism evolution trigger concomitant bursts of viral evolution.
The extended picornavirus-like superfamily
There seems to be an inherent paradox about the evolution of RNA viruses in general and picornaviruses in particular. RNA replication is extremely error-prone, especially in picornaviruses, with a mutation rate that is high enough to maintain a broad quasispecies distribution of RNA sequences and push the viruses to the brink of a mutational meltdown or error catastrophe45,46,47,48. Moreover, it has been shown that the distribution of variants in a quasispecies is not a biologically irrelevant consequence of error-prone replication but rather a crucial factor of viral evolution. The interaction of variants within a quasispecies ensures the adaptability of viruses in changing environments and, in particular, substantially contributes to viral pathogenesis48,49,50. Nevertheless, there is readily detectable conservation of protein domain sequences among viruses that infect diverse hosts and have widely different structures and reproduction strategies. As pointed out by Biebricher and Eigen, RNA viruses “operate close to the error threshold that allows maximum exploration of sequence space while conserving the information content of the genotype”51. However, it seems that the functional constraints on the viral proteins that have key functions in reproduction are strong enough to maintain the alignment of the sequences of the respective domains over a broad range of viral groups, in spite of the mutational pressure. This allows deep phylogenetic analyses52.
In early comparative genomic analyses, positive-strand RNA viruses of eukaryotes were classified into three superfamilies: picorna-like, alpha-like and flavi-like33,53,54. These three superfamilies include most known positive-strand RNA viruses, although the classification of nidoviruses and RNA bacteriophages remained uncertain. The superfamilies were delineated through a combination of phylogenetic analysis of conserved protein sequences, primarily those of RNA-dependent RNA polymerases (RdRps)55, and comparison of diagnostic features of genome organization that are linked to replication and expression strategies. Phylogenetic analysis of RNA viruses at the level of superfamilies is difficult owing to their deep divergence and the high rate of sequence evolution, so it has been argued that the phylogenetic signal contained in the RdRp sequences might be insufficient to define the superfamilies56. Nevertheless, the core subsets of each superfamily were readily identified by straightforward sequence comparison and phylogenetic analyses, and the existence of signature arrangements of conserved genes clinches the case for the objective existence of the superfamilies57,58.
The picornavirus-like superfamily, in particular, is characterized by a partially conserved set of genes that consists of the RdRp, a chymotrypsin-like protease (3CPro, named after the picornavirus 3C protease), a superfamily 3 helicase (S3H) and a genome-linked protein (viral protein, genome-linked, VPg) (Fig. 1; Supplementary information S1 (table)). This set of four genes can be considered to be a signature of the picorna-like superfamily because these genes are not found in other characterized RNA viruses (with the exception of the distinct 3CPro-like proteases of nidoviruses59). Furthermore, most of the viruses in the picorna-like superfamily have icosahedral virions that are composed of capsid proteins with the characteristic jelly-roll fold (jelly-roll capsid protein, JRC). It has to be emphasized that the presence of all four signature genes is not an absolute requirement for classifying a virus as a member of the picorna-like superfamily. In some of the viruses included in the superfamily this genomic layout (bauplan) is incomplete or substantially altered (Fig. 1) but there is additional, strong evidence of their evolutionary relationship to picorna-like viruses. For example, astroviruses have no helicase, whereas nodaviruses lack the helicase, the protease and the VPg (Fig. 1). However, even in the case of the nodaviruses, a connection to the picornavirus superfamily seems convincing thanks to the presence of characteristic motifs and the overall sequence conservation of the RdRp33,55,60.
Figure 1: The genome layouts in the main evolutionary lineages (clades) of picorna-like viruses.
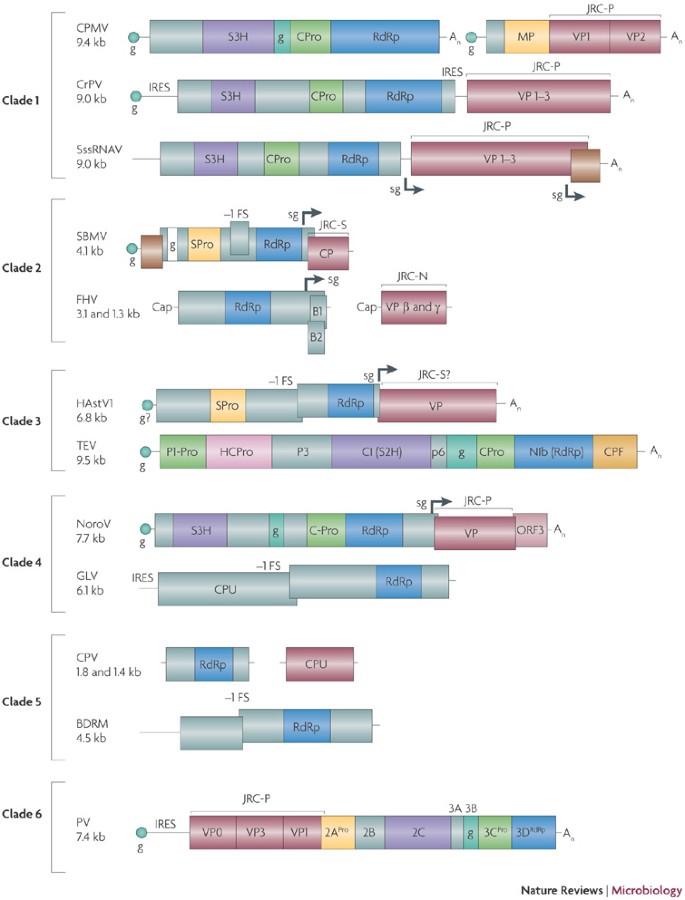
The boxes and lines represent open reading frames (ORFs) and non-coding sequences, respectively, roughly to scale. The signature proteins of the picorna-like superfamily are RdRp (RNA-dependent RNA polymerase, S3H (superfamily 3 helicase), CPro or SPro (chymotrypsin-like cysteine or serine proteases), JRC (jelly-roll capsid protein, three structural subsets of which are P for picornavirus-like (P)68, sobemovirus-like (S)119 and nodavirus-like (N)120), and VPg (viral protein, genome-linked; denoted g). −1FS, minus one frameshift; 2APro, 2A protease; 3CPro, 3C protease; 3DRdRp, 3D RdRp; An, poly(A) sequence; BDRM, Bryopsis cinicola dsRNA replicon from mitochondria; CI, cylindrical inclusion protein; CPF, capsid protein, filamentous capsid; CPMV, cowpea mosaic virus; CPU, capsid protein, unknown evolutionary origin; CPV, Cryptosporidium parvum virus; CrPV, cricket paralysis virus; FHV, flock house virus; GLV, Giardia lamblia virus; HAstV1, human astrovirus 1; HCPro, helper component proteinase; IRES, internal ribosome entry site; MP, movement protein; NIb, nuclear inclusion b protein; NoroV, norovirus; ORF, open reading frame; P1Pro, protein 1 proteinase; P3, protein 3; p6, 6 kDa protein; PV, poliovirus; S2H, superfamily 2 helicase; sg, subgenomic RNA promoter; SBMV, southern bean mosaic virus; SssRNAV, Schizochytrium single-stranded RNA virus; TEV, tobacco etch virus; VP, virion protein.
We carried out additional sequence analysis in order to validate and update the roster of viruses in the picorna-like superfamily. To this end, we defined the core of the superfamily to include all viruses that contain the 'picorna-like' RdRp and one (3CPro) or two (3CPro and S3H) of the additional signature genes. The amino acid sequence alignment of the RdRps of the viruses that comprise this core was used to generate a position-specific scoring matrix (PSSM), which was screened against the National Center for Biotechnology Information's RefSeq database in order to identify potential additional members of the picorna-like superfamily. This analysis confirmed that the RdRps of nodaviruses had highly significant and specific similarity to those of the picorna-like viruses (Supplementary information S1, S2 (table,figure); the original outputs of the PSSM searches are available on request). Notably, and in accord with the previous conclusions on the multiple originations of dsRNA viruses from positive-strand RNA viruses61,62,63,64, we found that the RdRps of two distinct families of dsRNA viruses, Partitiviridae and Totiviridae, also seemed to be related to the picorna-like superfamily (Fig. 1; Supplementary information S1 (table)).
Genome analysis of the recently isolated positive-strand RNA viruses of unicellular eukaryotes yielded an unexpected result. All four of these viruses, which infect taxonomically diverse hosts, belong to the picorna-like superfamily according to the criteria outlined above, namely, the (partial) conservation of the picorna-type set of signature genes and specific sequence conservation of at least some of the proteins encoded by these signature genes36,37,38,39, for example, Schizochytrium ssRNA virus (Fig. 1). Metagenomic analyses also revealed an apparent prevalence of picorna-like viruses among marine RNA viruses (hosts are unknown)41,42. The current sampling of the diversity of eukaryotic viruses is not sufficient to conclude whether this is a true reflection of the host ranges of the superfamilies of eukaryotic ssRNA viruses or an unrecognized bias in sequencing studies. This uncertainty notwithstanding, identification of RNA viruses in unicellular eukaryotes has led to a notable expansion of the picorna-like superfamily. Remarkably, this superfamily is now represented in four of the five supergroups of eukaryotes65,66, namely Unikonta (including animals, fungi and Amoebozoa), Plantae (land plants, and green and red algae), Chromalveolata (for example, apicomplexa, dinoflagellates, diatoms and oomycetes) and Excavata (for example, kinetoplastids, trichomonads and diplomonads such as Giardia lamblia) (Fig. 2). By contrast, the alpha-like and flavi-like superfamilies of positive-strand RNA viruses have so far only been detected in unikonts (primarily, animals) and plants, with only two known exceptions41,67.
Figure 2: The host ranges of picorna-like viruses.
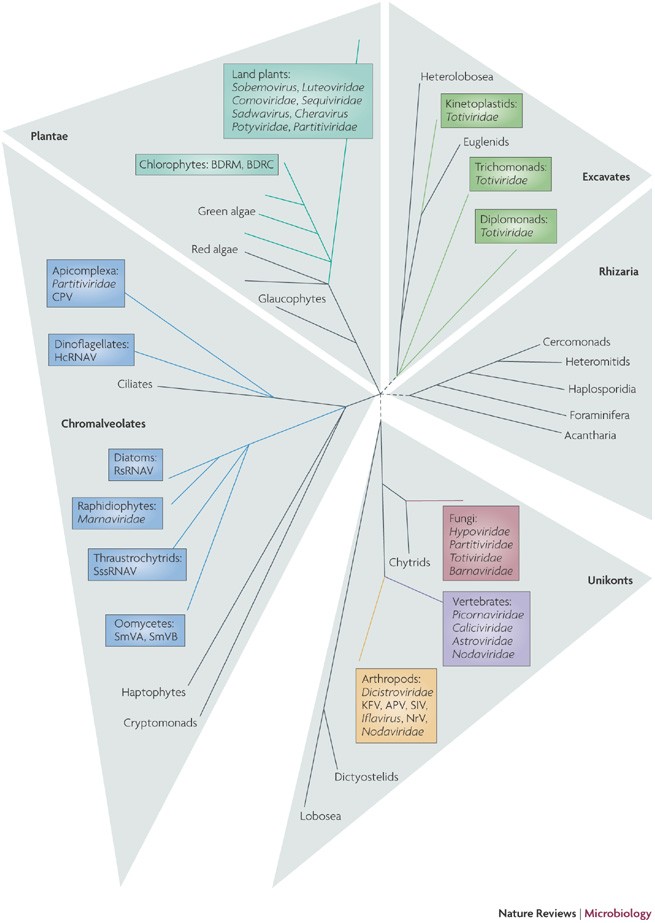
This simplified evolutionary tree of eukaryotes represents five supergroups, the relationships between which remain unresolved65,66. Black lines and names correspond to evolutionary lineages for which no picorna-like viruses have been described so far, whereas coloured lines correspond to lineages known to be infected by picorna-like viruses (named in adjacent coloured boxes). APV, Acyrthosiphon pisum virus; CPV, Cryptosporidium parvum virus; HcRNAV, Heterocapsa circularisquama RNA virus; KFV, kelp fly virus; NrV, Nora virus; RsRNA, Rhizosolenia setigera RNA virus; SIV, Solenopsis invicta virus-2; SmVA, Sclerophtora macrospora virus A; SmVB, Sclerophtora macrospora virus B; SssRNAV, Schizochytrium single-stranded RNA virus.
The extended picorna-like superfamily of positive-strand RNA viruses identified here includes the recently proposed order Picornavirales68, which has five families and three floating genera, along with an additional nine families, one genus and 15 unclassified viruses. It includes extremely diverse viruses and virus-like elements, many of which do not closely resemble picornaviruses. As discussed previously28, the notion of monophyly has limited applicability when broad groups of viruses are considered, given the important roles of gene sampling and recombination in the evolution of viruses (as captured, in particular, in the concept of reticulate evolution of bacteriophages69). Nevertheless, we believe that the picorna-like superfamily as described here is a valid group based on current sequence resources, although changes, especially expansion, will undoubtedly result from future analyses. New developments in the taxonomy of the picorna-like viruses should also be expected (see International Committee on Taxonomy of Viruses).
Here we refrain from further discussion of taxonomy and focus on the evolution of picorna-like viruses, with the aim of clarifying the phylogenetic positions of new viruses of unicellular eukaryotes, superimposing the evolutionary trees of viruses and hosts and, hopefully, gaining new insights into the original diversification of viruses of eukaryotes.
Phylogenies of RdRps and helicases
Only two proteins that are encoded in most picorna-like viruses show sequence conservation that is sufficient to obtain resolved phylogenetic trees: the RdRp and the S3H. Multiple alignments of these proteins (Supplementary information S2, S3 (figures) for RdRp and S3H, respectively) were used for maximum-likelihood phylogenetic analysis (Fig. 3). The RdRp tree consists of six strongly or moderately supported major clades that form a star phylogeny with short, apparently unresolvable internal branches (Fig. 3). The clades are as follows, roughly in the order of the decreasing diversity of viruses and hosts.
Figure 3: The phylogenetic tree of the RNA-dependent RNA polymerases of picorna-like viruses.
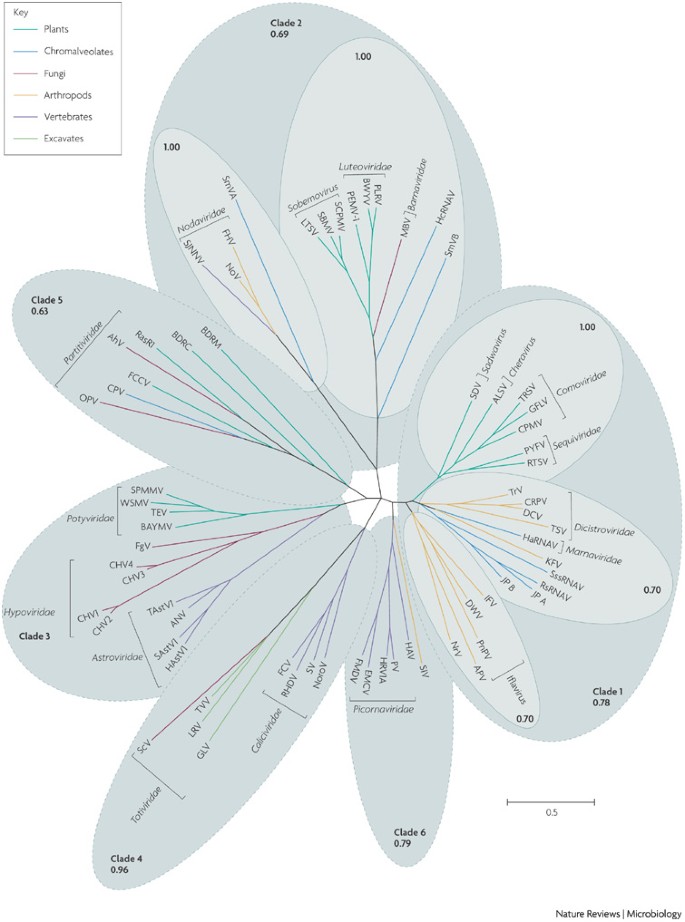
All the sequences of viral genomes and encoded proteins were from GenBank. Viral lineages are colour-coded to reflect their host range. Multiple alignments of protein sequences were constructed using the MUSCLE program121, with subsequent manual adjustment using the corresponding crystal structures. Maximum likelihood trees were constructed using the TREEFINDER program122, with the Whelan And Goldman (WAG123) evolutionary model with γ-distributed site rates. The obtained trees were then used to initialize Monte Carlo Markov Chain (MCMC) computations using the MrBayes program124. For two runs of four MCMCs, 1.1 × 106 generations were retained, with the first 105 generations discarded as burn-in. We pooled together 1,000 samples taken every 1,000 generations from each of the runs, and constructed consensus trees. Support values (fraction of sampled trees with the given tree bipartition present) are indicated for selected clades. The extremely short, apparently unresolvable internal branches of both trees indicative of star phylogeny are best compatible with the rapid diversification of picorna-like viruses in a Big Bang-type event44. AhV, Atkinsonella hypoxylon virus; ALSV, apple latent spherical virus; ANV, avian nephritis virus; APV, Acyrthosiphon pisum virus; BAYMV, barley yellow mosaic virus; BDRC, Bryopsis cinicola dsRNA replicon from chloroplasts; BDRM, Bryopsis cinicola dsRNA replicon from mitochondria; BWYV, beet western yellows virus; CHV, cryphonectria parasitica hypovirus; CPMV, cowpea mosaic virus; CPV, Cryptosporidium parvum virus; CrPV, cricket paralysis virus; DCV, Drosophila C virus; DWV, deformed wing virus; EMCV, Encephalomyocarditis virus; FCCV, Fragaria chiloensis cryptic virus; FCV, feline calicivirus; FgV, Fusarium graminearum virus; FHV, flock house virus; FMDV, foot-and-mouth disease virus; GFLV, grapevine fanleaf virus; GLV, Giardia lamblia virus; HaRNAV, Heterosigma akashiwo RNA virus; HAstV1, human astrovirus 1; HAV, hepatitis A virus; HcRNAV, Heterocapsa circularisquama RNA virus; HRV1A, human rhinovurus 1A; IFV, infectious flacherie virus; JP, Jericho pier; KFV, kelp fly virus; LRV, leishmania RNA virus 1-1; LTSV, lucerne transient streak virus; MBV, mushroom bacilliform virus; NoroV, norovirus; NoV, Nodamura virus; NrV, Nora virus; OPV, Ophiostoma himal-ulmi virus 1; PEMV-1, pea enation mosaic virus-1; PLRV, potato leafroll virus; PnPV, Perina nuda picorna-like virus; PV, poliovirus; PYFV, parsnip yellow fleck virus; RasR1, Raphanus sativus dsRNA 1; RHDV, rabbit haemorrhagic disease virus; RsRNA, Rhizosolenia setigera RNA virus; RTSV, rice tungro spherical virus; SAstV1, sheep astrovirus-1; SBMV, southern bean mosaic virus; SCPMV, southern cowpea mosaic virus; ScV, Saccharomyces cerevisiae virus L-A; SDV, satsuma dwarf virus; SIV, Solenopsis invicta virus-2; SJNNV, striped jack nervous necrosis virus; SmVA, Sclerophtora macrospora virus A; SmVB, Sclerophtora macrospora virus B; SPMMV, sweet potato mild mottle virus; SssRNAV, Schizochytrium single-stranded RNA virus; SV, Sapporo virus; TAstV1, turkey astrovirus-1; TEV, tobacco etch virus; TRSV, tobacco ringspot virus; TrV, Triatoma virus; TSV, Taura syndrome virus; TVV, Trichomonas vaginalis virus; WSMV, wheat streak mosaic virus.
Comovirus and dicistrovirus clade (clade 1 in Fig. 3 ). This group has the greatest diversity and includes viruses that infect host organisms of three eukaryotic supergroups: Plantae, Unikonta and Chromalveolata. There are three distinct subclades: the comovirus lineage, which encompasses a variety of plant viruses; the dicistrovirus and marnavirus lineage, which is an assemblage of insect viruses70, recently isolated viruses infecting marine chromalveolates36,38,39, and closely related marine viruses with unknown hosts42; and the third lineage, consisting of iflaviruses and other insect viruses70,71,72.
Sobemovirus and nodavirus clade (clade 2). This clade is only moderately supported. However, it consists of two definitively supported subclades, each of which combines viruses infecting hosts from three (sobemovirus lineage: Plantae, Fungi73 and Chromalveolata37,74) or two (nodavirus lineage: opisthokonts60 and Chromalveolata75) eukaryotic supergroups.
Astrovirus and potyvirus clade (clade 3). This strongly supported clade unites animal astroviruses76, plant potyviruses and dsRNA hypoviruses. dsRNA hypoviruses infect fungal pathogens of plants and have been proposed to have evolved from potyviruses77,78,79,80. Although specific sequence similarities between astrovirus and potyvirus RdRps have been noticed previously81, the recent expansion in the number of relevant sequenced viruses allows confident validation of this clade.
Calicivirus and totivirus clade (clade 4). This is an unexpected but strongly supported unification of a distinct family of animal viruses, the caliciviruses82, with the dsRNA totiviruses, which have been isolated from several diverse excavates and fungi83,84.
Partitivirus clade (clade 5). This clade contains dsRNA viruses of plants, fungi83 and an apicomplexan (which is a chromalveolate)85. Some of the partitivirus-related genetic RNA elements do not have capsids and replicate in the mitochondria or chloroplasts of green algae86.
Picornavirus clade (clade 6). This is the only monotypic group that consists entirely of the Picornaviridae family of vertebrate viruses68 allied with a solitary insect virus87.
Phylogenetic analysis
Strikingly, five of the six major clades of picorna-like virus RdRps include viruses whose hosts belong to two or three eukaryotic supergroups. Evolution of viruses cannot be reduced to the evolution of their RdRps. However, RdRp is the only universal protein in the picorna-like superfamily, so in this Analysis we use the RdRp tree as a standard against which to compare trees and distributions of other genes.
Phylogenetic analysis of RNA helicases (S3H) of picorna-like viruses is more limited in scope than the RdRp analysis because viruses in three of the six RdRp clades do not encode this protein (Figs 1, 3). The S3H tree consists of four well-supported clades (Fig. 4). The largest and most diverse clade mainly corresponds to RdRp clade 1. However, there are notable exceptions: dicistroviruses fall outside the clade and form a lineage of their own; the S3Hs of two insect viruses (kelp fly virus and Acyrthosiphon pisum virus) belong to the calicivirus clade; and the S3H of another insect virus (nora virus) belongs to the picornavirus clade (Fig. 4). Although artefacts of tree topology cannot be ruled out, the respective clades are well supported, so these limited discrepancies between the phylogenies of the RdRp and the S3H of picorna-like viruses suggest the possibility of multiple recombination events during viral evolution.
Figure 4: The phylogenetic tree of superfamily 3 helicases of picorna-like viruses.
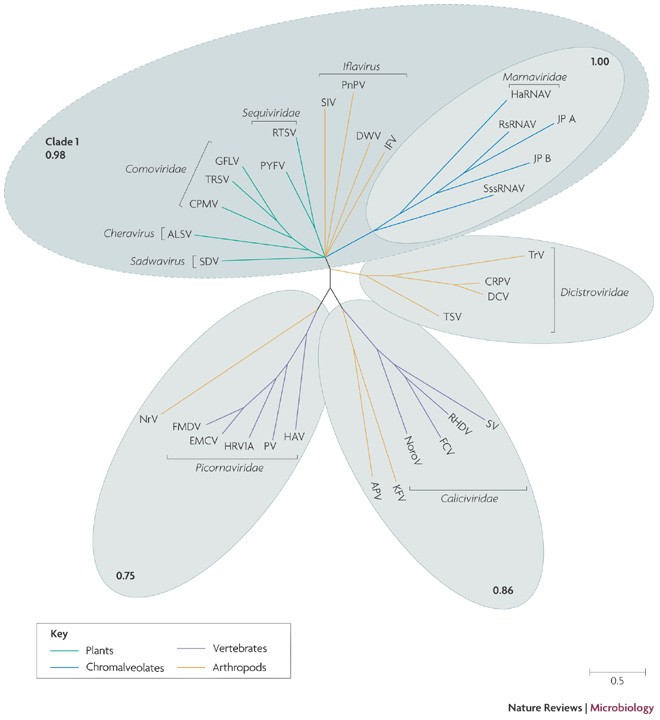
Note that only a subset of viruses in the picorna-like superfamily encode a superfamily 3 helicase. All the sequences of viral genomes and encoded proteins were from GenBank. Viral lineages are colour-coded to reflect their host range. Multiple alignments of protein sequences were constructed using the MUSCLE program121, with subsequent manual adjustment using the corresponding crystal structures. Maximum likelihood trees were constructed using the TREEFINDER program122, with the Whelan And Goldman (WAG123) evolutionary model with γ-distributed site rates. The obtained trees were then used to initialize Monte Carlo Markov Chain (MCMC) computations using the MrBayes program124. For two runs of four MCMCs, 1.1 × 106 generations were retained, with the first 105 generations discarded as burn-in. We pooled together 1,000 samples taken every 1,000 generations from each of the runs, and constructed consensus trees. Support values (fractions of sampled trees with the given tree bipartition present) are indicated for selected clades. The short, apparently unresolvable internal branches of both trees that are indicative of star phylogeny are best compatible with the rapid diversification of picorna-like viruses in a Big Bang-type event. ALSV, apple latent spherical virus; APV, Acyrthosiphon pisum virus; CPMV, cowpea mosaic virus; CRPV, cricket paralysis virus; DCV, Drosophila C virus; DWV, deformed wing virus; EMCV, Encephalomyocarditis virus; FCV, feline calicivirus; FMDV, foot-and-mouth disease virus; GFLV, grapevine fanleaf virus; HaRNAV, Heterosigma akashiwo RNA virus; HAV, hepatitis A virus; HRV1A, human rhinovurus 1A; IFV, infectious flacherie virus; JP, Jericho pier; KFV, kelp fly virus; NoroV, norovirus; NrV, Nora virus; PnPV, Perina nuda picorna-like virus; PV, poliovirus; PYFV, parsnip yellow fleck virus; RHDV, rabbit haemorrhagic disease virus; RsRNA, Rhizosolenia setigera RNA virus; RTSV, rice tungro spherical virus; SDV, satsuma dwarf virus; SIV, Solenopsis invicta virus-2; SssRNAV, Schizochytrium single-stranded RNA virus; SV, Sapporo virus; TRSV, tobacco ringspot virus; TrV, Triatoma virus; TSV, Taura syndrome virus.
The third conserved protein of picorna-like viruses, 3CPro, is more common than the S3H and is present in families from all RdRp clades apart from the partitivirus clade (Fig. 1). Most viral proteases have a catalytic cysteine that replaces the active serine residue that is characteristic of the rest of trypsin-like proteases88. However, at least two groups of viruses — the sobemovirus lineage of the RdRp clade 2 and astroviruses — possess serine proteases (Fig. 1). A reliable tree of virus proteases could not be obtained owing to the relatively low information content of the multiple alignment (Supplementary information S4 (figure)). However, it is noteworthy that viral serine proteases were polyphyletic, that is, the serine proteases of astroviruses formed a strongly supported clade with the cysteine proteases of potyviruses, whereas the serine proteases of sobemoviruses, luteoviruses and related viruses of fungi and chromalveolates comprised a distinct clade (data not shown).
The Big Bang of picorna-like virus evolution
The phylogenetic analyses presented in this article show that five of the six clades in the RdRp tree encompass picorna-like viruses that infect hosts from two or three eukaryotic supergroups. Early and, presumably, rapid diversification of picorna-like viruses, antedating the divergence of eukaryotic supergroups, seems to be the most parsimonious evolutionary scenario. However, the contribution of subsequent horizontal virus transfer (HVT) could be substantial as well, in accord with the concept of the reticulate evolution of viruses69. In particular, transmission of viruses between plants and fungi seems possible given the close associations between plants and their fungal pathogens. HVT might have been particularly important in the evolution of the Partitiviridae family, in which plant and fungal viruses are intermixed in phylogenetic trees89 (Fig. 3), and is also likely to account for the evolution of the Hypoviridae77 (Fig. 3).
However, it seems that HVT only confounded the results of a Big Bang of virus diversification, a scenario that conforms to the recently proposed general model of major evolutionary transitions44. In the Big Bang scenario, major branches of picorna-like viruses had already emerged by the time the eukaryotic supergroups radiated from their common ancestor and, then, viruses from this ancestral pool explored the evolving hosts and infected those that were susceptible. One prediction of the Big Bang model is that picorna-like viruses will eventually be identified that infect hosts from all the major lineages of eukaryotic organisms, although viruses of this superfamily so far have not been isolated from Amoebozoa, red algae and Rhizaria (which are generally poorly studied organisms).
The alternative hypothesis — namely, emergence of the ancestors of the six major clades of picorna-like viruses in one of the eukaryotic supergroups, with subsequent HVT to hosts from other supergroups — seems to be substantially less parsimonious, considering that this scenario would require numerous HVT events between organisms with widely different global ecologies and lifestyles. Furthermore, none of the supergroups of eukaryotes are known to host picorna-like viruses from all of the six clades that are present in an RdRp tree, a distribution that seems to be most compatible with viruses from a pre-existing ancestral pool infecting the emerging eukaryotic supergroups (Fig. 3).
How does this scenario of picorna-like virus evolution relate to the existing notions on the evolution of their cellular hosts? The Big Bang of picorna-like viruses is consistent with the probably rapid and tumultuous nature of eukaryogenesis that, under the symbiogenetic scenarios, was initiated by the archaeo-bacterial symbiosis90,91,92,93. Under this model, eukaryogenesis would involve extensive recombination between the symbiont and host genomes and, apparently, infestation of the host genes by group II retroelements that came from the symbiont and gave rise to the spliceosomal introns91. Explosive evolution of eukaryotic viruses in general, and the Big Bang of picorna-like virus evolution in particular, would be inherent to this turbulent era28. As discussed in detail elsewhere, symbiogenesis appears to be the most parsimonious scenario for the emergence of the eukaryotic cell, considering the presence of mitochondria or related organelles in all extensively characterized modern eukaryotes and the explanatory power of this model with respect to the origin of the nucleus and other eukaryotic organelles. However, the alternative scenario, namely the origin of an amitochondrial ancestor of eukaryotes as one of the three primary domains of life, has also been strongly defended in recent theoretical studies94,95. Adopting this scenario would not affect our conclusion on the Big Bang of picorna-like virus evolution but would push this event to an early, primordial stage of the evolution of life. This stage is believed to have involved rampant recombination between diverse genetic elements, a state that would be conducive to the explosive diversification of viruses24,28.
The origins of picorna-like viruses
The picorna-like superfamily is defined by the presence of a partially conserved set of genes that includes those encoding RdRp, the S3H, the 3CPro, VPg and JRC (Fig. 1). Among sequenced genomes of viruses infecting bacteria and archaea, none contain any pair of genes from this set. Barring the unlikely possibility that such viruses of prokaryotes remain to be discovered, it follows that the ancestor(s) of the picorna-like viral superfamily was assembled from individual genes during eukaryogenesis. Can we trace the sources of these genes? Despite the rapidity of the evolutionary processes during a Big Bang and the high rate of evolution of RNA virus genes, database searches seem to provide tangible clues.
We derived PSSMs for the RdRps, S3H and 3CPro of the picorna-like superfamily and compared them with the non-redundant protein database using PSI-BLAST (position-specific iterative basic local alignment search tool)96 to identify the closest homologues outside the picorna-like superfamily that could be the ancestors of these signature genes. The RdRp PSSM produced highly significant hits to the RdRps of the other two superfamilies of eukaryotic positive-strand RNA viruses and, notably, the reverse transcriptases (RTs) of bacterial group II retroelements (Table 1 and Supplementary information S2 (figure)). The similarity between the RdRps of picorna-like viruses and the RdRps of RNA bacteriophages was substantially lower (Table 1). The conservation of several sequence motifs and the structural similarity between RdRps of positive-strand RNA viruses and RTs have been described previously97,98,99,100, and the relationship between the two classes of polymerases is complemented by biochemical evidence, for example, the ability of RdRps to efficiently use dNTPs as substrates in the presence of Mn2+ cations101,102,103.
Table 1 Homologues of the signature genes of picorna-like superfamily viruses*
Considering the symbiotic scenario of eukaryogenesis, it is notable that the RdRps of picorna-like viruses are most similar to RTs from prokaryotic retroelements, as opposed to those from eukaryotic retroid viruses or retroelements. Given these findings and the wide spread of group II retroelements in bacteria, in a sharp contrast to the scarcity of RNA bacteriophages, it appears plausible that the RdRps of eukaryotic positive-strand RNA viruses evolved from prokaryotic RTs. Group II retroelements are widely believed to be the progenitors of eukaryotic spliceosomal introns104,105,106, as well as ancestors of the eukaryotic telomerase and retroid viruses107,108,109. So this hypothesis places the origin of the picorna-like superfamily and other eukaryotic positive-strand RNA viruses in the middle of the turbulent process of eukaryogenesis.
The roots of the 3CPros of picorna-like viruses appear even clearer. Most of the statistically significant hits observed with the 3CPro PSSM are members of a distinct family of bacterial and mitochondrial serine proteases typified by the Escherichia coli periplasmic protease HtrA110 (Table 1). This relationship is supported by the analysis of structural neighbours, in which the mitochondrial protease HTRA2 (also known as OMI)111 comes up as the closest non-viral neighbour of 3CPro (data not shown). The similarity between the serine proteases of the HtrA family and the cysteine proteases of picornaviruses has been noticed previously88 but, at the time, the sequence information was insufficient to infer the nature of the evolutionary relationship between these protein families. With the current genomic data and considering the bacterial provenance, mitochondrial localization and function of the HtrA family of proteases in eukaryotes, it can be concluded that the 3CPro descends from an HtrA-family protease, and that this protease in turn is most probably derived from the mitochondrial endosymbiont.
The case of the SF3 helicase of picorna-like viruses is more complex. The PSSM-initiated sequence searches reveal that the highest similarity is to the helicases of circoviruses, followed by bacterial AAA+ ATPases; the available bacteriophage S3H sequences are much less similar to the picorna-like virus helicases (Table 1). However, the S3Hs have several sequence and structure features pointing to their monophyly112,113,114, which suggests that the S3Hs of eukaryotic viruses evolved from their bacteriophage homologues. In this scenario, the observed hierarchy of sequence similarity could be explained by the slower evolution of AAA+ ATPases of cellular organisms compared with the related viral S3H, or by the absence in the current database of the phage group that provided the putative ancestral helicase. Conceivably, the circoviruses are derivatives of this putative phage family.
The JRCs of picorna-like viruses, similarly, might have derived from capsid proteins of DNA-containing viruses of bacteria or archaea18. It should be noted, however, that the known icosahedral capsid proteins of prokaryotic DNA viruses, such as bacteriophages PRD1 or phi29 or Sulfolobus turret icosahedral virus115, have double JRC domains, whereas the capsid proteins of picorna-like viruses contain single JRC domains116. The similarity between the picorna-like virus JRCs and the capsid proteins of bacterial and archaeal viruses can be traced only through structural comparisons and is limited in extent (Ref. 18 and E.V.K., unpublished data), attesting to a substantial modification and, possibly, partial degradation of the JRC fold that was required to encapsidate small RNAs of picorna-like viruses. Alternatively, the picorna-like viral version of the JRC might have been derived from an unknown small prokaryotic virus.
Thus, the available evidence points to the assembly of the ancestral picorna-like viruses from diverse building blocks during eukaryogenesis and before the radiation of the eukaryotic supergroups (Fig. 5). The emergence of these ancestral viruses is probably best depicted as a Big-Bang-type event, so the order of emergence of the individual clades and the specific relationships between them could be undecipherable. In accordance with the concept of reticulate evolution of viruses, it is even conceivable that a common viral ancestor of picorna-like viruses never existed, that is, that the major clades of picorna-like viruses obtained their signature genes from different prokaryotic viruses and genetic elements. However, given the consistent presence of the five signature genes in the majority of picorna-like viruses, this possibility appears to be non-parsimonious. It is more likely that the Big Bang of picorna-like virus evolution was precipitated by accidental assembly of the signature genes in an ancestral virus (Fig. 5).
Figure 5: The proposed evolutionary scenario for the picorna-like superfamily of positive-strand RNA viruses of eukaryotes.
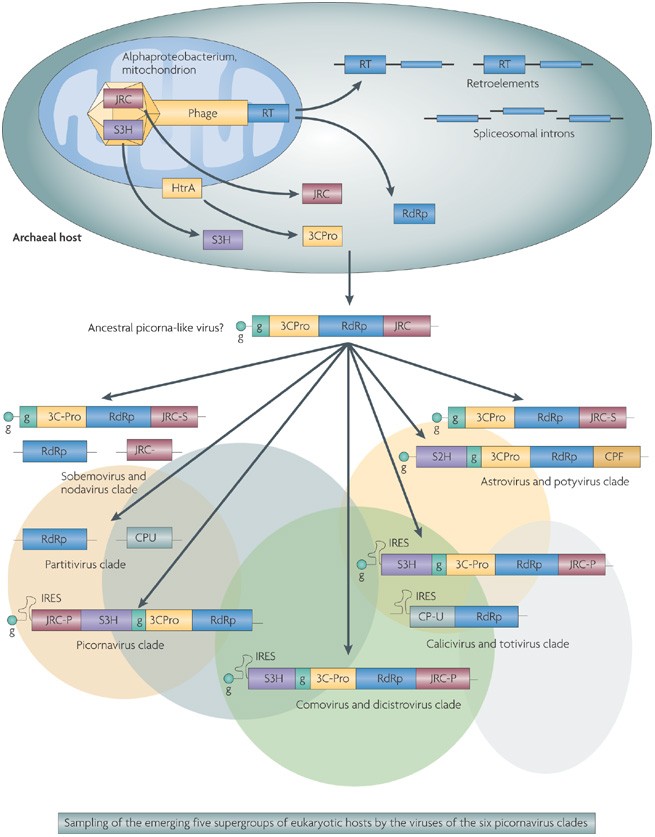
This scenario is based on the symbiogenetic model of the origin of eukaryotes, according to which eukaryogenesis was initiated by the engulfment of an α-proteobacterium (the future mitochondrion) by an archaeon90,91,92,93. As discussed in the text, reverse transcriptase (RT)-encoding group II retroelements originating from the bacterial symbiont are the likely ancestors of both eukaryotic retroelements and spliceosomal introns, and the RT of these elements might have given rise to the RNA-dependent RNA polymerase (RdRp) of picorna-like viruses. Under this scenario, the superfamily 3 helicase (S3H) and the jelly-roll capsid protein (JRC) of the ancestral picorna-like virus are tentatively derived from a bacteriophage of the symbiont, and the 3C-like proteinase (3CPro) is derived from a symbiont's membrane protease (see text for details). Coloured ovals and the arrows at the bottom of the diagram symbolize burst-like emergence of the five supergroups of eukaryotes and of the six clades of the picorna-like viruses, respectively. CPF, capsid protein, filamentous capsid; CPU, capsid protein, unknown evolutionary origin; g, viral protein, genome-linked; IRES, internal ribosome entry site; JRC-N, nodavirus -like JRC; JRC-P, picornavirus -like JRC; JRC-S, sobemovirus-like JRC; S2H, superfamily 2 helicase.
The evolutionary scenario schematically depicted in Fig. 5 is predicated on the symbiogenetic model of eukaryogenesis. At least one piece of evidence, the distinct bacterial origin of 3CPro, seems to be best compatible with this model. In general, however, the scenario of picorna-like virus evolution is robust with respect to the concepts of eukaryogenesis and would fit the three-domain model as well. The main difference would be pushing the assembly of the ancestral virus back to the pre-cellular stage of virus evolution24,28. Moreover, this scenario seems to be better compatible with the current data on the diversity of the JRC18 because, in this case, the JRC of picorna-like viruses could be considered the primitive form of this fold.
Subsequent evolution of picorna-like viruses seems to have involved a variety of substantial modifications of the viral genome layout, which often occurred in parallel in different clades (Fig. 5). The apparent replacement of the S3H by a superfamily 2 helicase in potyviruses is a case in point, as is the replacement of the JRC gene with a gene for an unrelated capsid protein that forms filamentous capsids117 in the same viral family. In this case, the changes to the viral bauplan can be linked to a specific host range that would facilitate recombination between viruses: in plants — the host organisms of potyviruses — viruses of the alpha-like supergroup, which typically have a superfamily 2 helicase and a filamentous capsid, are extremely abundant and were the likely source of the respective genes acquired by potyviruses. The hypoviruses, which are probable derivatives of potyviruses (although this is not obvious from the RdRp tree), have apparently lost both the capsid protein and the 3CPro. In this case, the loss of the capsid is linked to the predominantly vertical transmission of viruses in fungi. The nodaviruses (and, apparently, Sclerophtora macrospora virus A, the related virus from a chromalveolate) present perhaps the most dramatic case of gene loss and bauplan modification in the picorna-like superfamily, with both the 3C-like protease and VPg lost. A parallel loss of 3CPro is seen in the totiviruses and, apparently, in the entire partitivirus clade.
The Big Bang model implies that the early stages of the evolution of picorna-like viruses did not involve virus–host co-evolution inasmuch as different major clades of picorna-like viruses invaded the same eukaryotic supergroups. Of course, co-evolution is common at later, less turbulent phases of evolution that involve extensive virus–host co-adaptation as has been amply documented, for example, for mammalian herpesviruses118.
Conclusions
The results of phylogenetic analysis presented here suggest that diversification of the picorna-like superfamily of eukaryotic positive-strand RNA viruses occurred in a Big Bang at an early stage of eukaryogenesis, before the divergence of the supergroups of eukaryotes. This scenario implies that viruses from the ancestral pool invaded the emerging supergroups of eukaryotes. Thus, at least at this early stage in the evolution of RNA viruses of eukaryotes, there seems to have been no virus–host co-evolution in the sense of concomitant evolution of the host and viral lineages. However, evolution of picorna-like viruses was tightly intertwined with the pivotal events of eukaryogenesis such as the emergence of mitochondria and spliceosomal introns.
References
- Joyce, G. F. The antiquity of RNA-based evolution. Nature 418, 214–221 (2002). In-depth analysis of the RNA world concept of the primordial genetic systems.
Article CAS PubMed Google Scholar - Koonin, E. V. & Martin, W. On the origin of genomes and cells within inorganic compartments. Trends Genet. 21, 647–654 (2005). A conceptual framework for the origin of life within microscopic mineral compartments at hydrothermal vents through Darwinian selection of self-replicating, recombining RNA molecules that gradually evolved into complex molecular ensembles.
Article CAS PubMed PubMed Central Google Scholar - Holmes, E. C. & Drummond, A. J. The evolutionary genetics of viral emergence. Curr. Top. Microbiol. Immunol. 315, 51–66 (2007).
CAS PubMed PubMed Central Google Scholar - Suttle, C. A. Viruses in the sea. Nature 437, 356–361 (2005).
Article CAS PubMed Google Scholar - Edwards, R. A. & Rohwer, F. Viral metagenomics. Nature Rev. Microbiol. 3, 504–510 (2005).
Article CAS Google Scholar - Angly, F. E. et al. The marine viromes of four oceanic regions. PLoS Biol. 4, e368 (2006).
Article CAS PubMed PubMed Central Google Scholar - Suttle, C. A. Marine viruses — major players in the global ecosystem. Nature Rev. Microbiol. 5, 801–812 (2007). This incisive review provides a broad prospective on the abundance, diversity and role of the marine viruses in the biosphere.
Article CAS Google Scholar - Prangishvili, D., Garrett, R. A. & Koonin, E. V. Evolutionary genomics of archaeal viruses: unique viral genomes in the third domain of life. Virus Res. 117, 52–67 (2006).
Article CAS PubMed Google Scholar - Khayat, R. et al. Structure of an archaeal virus capsid protein reveals a common ancestry to eukaryotic and bacterial viruses. Proc. Natl Acad. Sci. USA 102, 18944–18949 (2005).
Article CAS PubMed PubMed Central Google Scholar - Ortmann, A. C., Wiedenheft, B., Douglas, T. & Young, M. Hot crenarchaeal viruses reveal deep evolutionary connections. Nature Rev. Microbiol. 4, 520–528 (2006).
Article CAS Google Scholar - Nandhagopal, N. et al. The structure and evolution of the major capsid protein of a large, lipid-containing DNA virus. Proc. Natl Acad. Sci. USA 99, 14758–14763 (2002).
Article CAS PubMed PubMed Central Google Scholar - Dunigan, D. D., Fitzgerald, L. A. & Van Etten, J. L. Phycodnaviruses: a peek at genetic diversity. Virus Res. 117, 119–132 (2006).
Article CAS PubMed Google Scholar - Dupuy, C., Huguet, E. & Drezen, J. M. Unfolding the evolutionary story of polydnaviruses. Virus Res. 117, 81–89 (2006).
Article CAS PubMed Google Scholar - Raoult, D. et al. The 1.2-megabase genome sequence of Mimivirus. Science 306, 1344–1350 (2004).
Article CAS PubMed Google Scholar - Claverie, J. M. et al. Mimivirus and the emerging concept of “giant” virus. Virus Res. 117, 133–144 (2006).
Article CAS PubMed Google Scholar - Hendrix, R. W. Bacteriophage genomics. Curr. Opin. Microbiol. 6, 506–511 (2003).
Article CAS PubMed Google Scholar - Casjens, S. R. Comparative genomics and evolution of the tailed-bacteriophages. Curr. Opin. Microbiol. 8, 451–458 (2005).
Article CAS PubMed Google Scholar - Bamford, D. H., Grimes, J. M. & Stuart, D. I. What does structure tell us about virus evolution? Curr. Opin. Struct. Biol. 15, 655–663 (2005). Homologous capsid proteins are seen in a wide variety of superficially unrelated icosahedral viruses that infect diverse hosts, in a striking demonstration of far-reaching evolutionary connections between viruses.
Article CAS PubMed Google Scholar - Liu, J., Glazko, G. & Mushegian, A. Protein repertoire of double-stranded DNA bacteriophages. Virus Res. 117, 68–80 (2006).
Article CAS PubMed Google Scholar - Pedulla, M. L. et al. Origins of highly mosaic mycobacteriophage genomes. Cell 113, 171–182 (2003).
Article CAS PubMed Google Scholar - Sullivan, M. B. et al. Prevalence and evolution of core photosystem II genes in marine cyanobacterial viruses and their hosts. PLoS Biol. 4, e234 (2006).
Article CAS PubMed PubMed Central Google Scholar - Iyer, L. M., Balaji, S., Koonin, E. V. & Aravind, L. Evolutionary genomics of nucleo-cytoplasmic large DNA viruses. Virus Res. 117, 156–184 (2006).
Article CAS PubMed Google Scholar - Claverie, J. M. Viruses take center stage in cellular evolution. Genome Biol. 7, 110 (2006).
Article CAS PubMed PubMed Central Google Scholar - Forterre, P. The origin of viruses and their possible roles in major evolutionary transitions. Virus Res. 117, 5–16 (2006).
Article CAS PubMed Google Scholar - Forterre, P. Three RNA cells for ribosomal lineages and three DNA viruses to replicate their genomes: a hypothesis for the origin of cellular domain. Proc. Natl Acad. Sci. USA 103, 3669–3674 (2006). A hypothesis that implicates viruses in the independent origins of the DNA replication machineries of the three domains of cellular life.
Article CAS PubMed PubMed Central Google Scholar - Gorinsek, B., Gubensek, F. & Kordis, D. Phylogenomic analysis of chromoviruses. Cytogenet. Genome Res. 110, 543–552 (2005).
Article CAS PubMed Google Scholar - Koonin, E. V. & Dolja, V. V. Evolution of complexity in the viral world: the dawn of a new vision. Virus Res. 117, 1–4 (2006).
Article CAS PubMed Google Scholar - Koonin, E. V., Senkevich, T. G. & Dolja, V. V. The ancient virus world and evolution of cells. Biol. Direct 1, 29 (2006). This article developed the concept of 'viral hallmark genes' — genes that are present in a variety of viruses but not in cellular life forms — and proposed that these genes comprise an uninterrupted flow of genetic information from pre-cellular stages of evolution to this day.
Article CAS PubMed PubMed Central Google Scholar - Pritham, E. J., Putliwala, T. & Feschotte, C. Mavericks, a novel class of giant transposable elements widespread in eukaryotes and related to DNA viruses. Gene 390, 3–17 (2007).
Article CAS PubMed Google Scholar - Raoult, D. & Forterre, P. Redefining viruses: lessons from Mimivirus. Nature Rev. Microbiol. 6, 315–319 (2008). A new definition of viruses capitalizes on the sharp distinction between viruses as capsid-encoding organisms and cellular life forms as ribosome-encoding organisms.
Article CAS Google Scholar - Hull, R. Matthews' Plant Virology (Academic Press, San Diego, 2001).
Google Scholar - Knipe, D. M. & Howley, P. M. Fields Virology (Lippincott Williams & Wilkins, Philadelphia, 2001).
Google Scholar - Koonin, E. V. & Dolja, V. V. Evolution and taxonomy of positive-strand RNA viruses: implications of comparative analysis of amino acid sequences. Crit. Rev. Biochem. Mol. Biol. 28, 375–430 (1993). A conceptual synthesis on the early studies in comparative genomics and evolution of positive-strand RNA viruses; advances the concept of the three major superfamilies of the positive-strand RNA viruses.
Article CAS PubMed Google Scholar - Bollback, J. P. & Huelsenbeck, J. P. Phylogeny, genome evolution, and host specificity of single-stranded RNA bacteriophage (family Leviviridae). J. Mol. Evol. 52, 117–128 (2001).
Article CAS PubMed Google Scholar - Ruokoranta, T. M., Grahn, A. M., Ravantti, J. J., Poranen, M. M. & Bamford, D. H. Complete genome sequence of the broad host range single-stranded RNA phage PRR1 places it in the Levivirus genus with characteristics shared with Alloleviviruses. J. Virol. 80, 9326–9330 (2006).
Article CAS PubMed PubMed Central Google Scholar - Lang, A. S., Culley, A. I. & Suttle, C. A. Genome sequence and characterization of a virus (HaRNAV) related to picorna-like viruses that infects the marine toxic bloom-forming alga Heterosigma akashiwo. Virology 320, 206–217 (2004).
Article CAS PubMed Google Scholar - Nagasaki, K. et al. Comparison of genome sequences of single-stranded RNA viruses infecting the bivalve-killing dinoflagellate Heterocapsa circularisquama. Appl. Environ. Microbiol 71, 8888–8894 (2005).
Article CAS PubMed PubMed Central Google Scholar - Takao, Y., Mise, K., Nagasaki, K., Okuno, T. & Honda, D. Complete nucleotide sequence and genome organization of a single-stranded RNA virus infecting the marine fungoid protist Schizochytrium sp. J. Gen. Virol. 87, 723–733 (2006).
Article CAS PubMed Google Scholar - Shirai, Y. et al. Genomic and phylogenetic analysis of a single-stranded RNA virus infecting Rhizosolenia setigera (Stramenopiles: Baccilariophyceae). J. Mar. Biol. Ass. UK 86, 475–483 (2006).
Article CAS Google Scholar - Culley, A. I., Lang, A. S. & Suttle, C. A. High diversity of unknown picorna-like viruses in the sea. Nature 424, 1054–1057 (2003).
Article CAS PubMed Google Scholar - Culley, A. I., Lang, A. S. & Suttle, C. A. Metagenomic analysis of coastal RNA virus communities. Science 312, 1795–1798 (2006). This article uses the power of metagenomics to address diversity and evolutionary affinities of uncultured marine RNA viruses.
Article CAS PubMed Google Scholar - Culley, A. I., Lang, A. S. & Suttle, C. A. The complete genomes of three viruses assembled from shotgun libraries of marine RNA virus communities. Virol. J. 4 (2007).
- Culley, A. I. & Steward, G. F. New genera of RNA viruses in subtropical seawater, inferred from polymerase gene sequences. Appl. Environ. Microbiol. 73, 5937–5944 (2007).
Article CAS PubMed PubMed Central Google Scholar - Koonin, E. V. The Biological Big Bang model for the major transitions in evolution. Biol. Direct 2, 21 (2007). A unifying concept of the major transitions in evolution as episodes of explosive diversification powered by rampant gene exchange and recombination.
Article CAS PubMed PubMed Central Google Scholar - Domingo, E., Escarmis, C., Mendez-Arias, L. & Holland, J. J. in Origin and Evolution of Viruses (eds Domingo, E., Webster, R. & Holland, J.) 141–161 (Academic Press, San Diego, 1999).
Book Google Scholar - Gromeier, M., Wimmer, E. & Gorbalenya, A. E. in Origin and Evolution of Viruses (eds Domingo, E., Webster, R. & Holland, J.) 287–344 (Academic Press, San Diego, 1999).
Book Google Scholar - Crotty, S., Cameron, C. E. & Andino, R. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc. Natl Acad. Sci. USA 98, 6895–6900 (2001).
Article CAS PubMed PubMed Central Google Scholar - Domingo, E. et al. Viruses as quasispecies: biological implications. Curr. Top. Microbiol. Immunol. 299, 51–82 (2006). A recent review that emphasizes the significance of quasispecies for the adaptability and pathogenesis of RNA viruses and the ongoing evolution of the viral populations.
CAS PubMed PubMed Central Google Scholar - Vignuzzi, M., Stone, J. K., Arnold, J. J., Cameron, C. E. & Andino, R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 439, 344–348 (2006).
Article CAS PubMed Google Scholar - Domingo, E., Martin, V., Perales, C. & Escarmis, C. Coxsackieviruses and quasispecies theory: evolution of enteroviruses. Curr. Top. Microbiol. Immunol. 323, 3–32 (2008).
CAS PubMed Google Scholar - Biebricher, C. K. & Eigen, M. What is a quasispecies? Curr. Top. Microbiol. Immunol. 299, 1–31 (2006). A broad analysis of the quasispecies concept and its application to the rapidly evolving RNA viruses.
CAS PubMed Google Scholar - Koonin, E. V. & Gorbalenya, A. E. Evolution of RNA genomes: does the high mutation rate necessitate high rate of evolution of viral proteins? J. Mol. Evol. 28, 524–527 (1989).
Article CAS PubMed Google Scholar - Goldbach, R. Genome similarities between plant and animal RNA viruses. Microbiol. Sci. 4, 197–202 (1987). The beginnings of the concept of superfamilies of positive-strand RNA viruses that span wide ranges of hosts.
CAS PubMed Google Scholar - Goldbach, R. & Wellink, J. Evolution of plus-strand RNA viruses. Intervirology 29, 260–267 (1988).
Article CAS PubMed Google Scholar - Koonin, E. V. The phylogeny of RNA-dependent RNA polymerases of positive-strand RNA viruses. J. Gen. Virol. 72 (Pt 9), 2197–2206 (1991).
Article PubMed Google Scholar - Zanotto, P. M., Gibbs, M. J., Gould, E. A. & Holmes, E. C. A reevaluation of the higher taxonomy of viruses based on RNA polymerases. J. Virol. 70, 6083–6096 (1996).
CAS PubMed PubMed Central Google Scholar - Strauss, E. G., Strauss, J. H. & Levine, A. J. in Fields Virology (eds Fields, B. N., Knipe, D. M. & Howley, P. M.) 153–171 (Lippincott-Raven, Philadelphia, 1996).
Google Scholar - Gibbs, M. J., Koga, R., Moriyama, H., Pfeiffer, P. & Fukuhara, T. Phylogenetic analysis of some large double-stranded RNA replicons from plants suggests they evolved from a defective single-stranded RNA virus. J. Gen. Virol. 81, 227–233 (2000).
Article CAS PubMed Google Scholar - Gorbalenya, A. E., Enjuanes, L., Ziebuhr, J. & Snijder, E. J. Nidovirales: evolving the largest RNA virus genome. Virus Res. 117, 17–37 (2006).
Article CAS PubMed PubMed Central Google Scholar - Johnson, K. N., Johnson, K. L., Dasgupta, R., Gratsch, T. & Ball, A. L. Comparisons among the larger genome segments of six nodaviruses and their encoded RNA replicases. J. Gen. Virol. 82, 1855–1866 (2001).
Article CAS PubMed Google Scholar - Koonin, E. V. Evolution of double-stranded RNA viruses: a case for polyphyletic origin from different groups of positive-stranded RNA viruses. Semin. Virol. 3, 327–339 (1992).
CAS Google Scholar - Koonin, E. V., Gorbalenya, A. E. & Chumakov, K. M. Tentative identification of RNA-dependent RNA polymerases of dsRNA viruses and their relationship to positive strand RNA viral polymerases. FEBS Lett. 252, 42–46 (1989).
Article CAS PubMed Google Scholar - Gorbalenya, A. E. et al. The palm subdomain-based active site is internally permuted in viral RNA-dependent RNA polymerases of an ancient lineage. J. Mol. Biol. 324, 47–62 (2002).
Article CAS PubMed PubMed Central Google Scholar - Ahlquist, P. Parallels among positive-strand RNA viruses, reverse-transcribing viruses and double-stranded RNA viruses. Nature Rev. Microbiol. 4, 371–382 (2006). A recent perspective on structural, functional and mechanistic similarities in replication of the diverse viruses that have RNA genomes.
Article CAS Google Scholar - Keeling, P. J. et al. The tree of eukaryotes. Trends Ecol. Evol. 20, 670–676 (2005). A conceptually important overview of eukaryotic evolution that introduces five supergroups, the exact relationships between which are difficult to determine.
Article PubMed Google Scholar - Keeling, P. J. Genomics. Deep questions in the tree of life. Science 317, 1875–1876 (2007).
Article CAS PubMed Google Scholar - Hacker, C. V., Brasier, C. M. & Buck, K. W. A double-stranded RNA from a Phytophtora species is related to the plant endornaviruses and contains a putative UDP glycosyltransferase gene. J. Gen. Virol. 86, 1561–1570 (2005).
Article CAS PubMed Google Scholar - Le Gall, O. et al. Picornavirales, a proposed order of positive-sense single-stranded RNA viruses with a pseudo-T = 3 virion architecture. Arch. Virol. 153, 715–727 (2008). A formal description of the proposed order Picornavirales that comprises the core of the picorna-like virus superfamily.
Article CAS PubMed Google Scholar - Lima-Mendez, G., Van Helden, J., Toussaint, A. & Leplae, R. Reticulate representation of evolutionary and functional relationships between phage genomes. Mol. Biol. Evol. 25, 762–777 (2008).
Article CAS PubMed Google Scholar - Gordon, K. H. J. & Waterhouse, P. M. Small RNA viruses of insects: expression in plants and RNA silencing. Adv. Virus Res. 68, 459–502 (2006).
Article CAS PubMed Google Scholar - Van der Wilk, F., Dullemans, A. M., Verbeek, M. & Van der Heuvel, J. F. J. M. Nucleotide sequence and genomic organization of Acyrthosiphon pisum virus. Virology 238, 353–362 (1997).
Article CAS PubMed Google Scholar - Habayeb, M. S., Ekengren, S. K. & Hultmark, D. Nora virus, a persistent virus in Drosophila, defines a new picorna-like family. J. Gen. Virol. 87, 3045–3051 (2006).
Article CAS PubMed Google Scholar - Revill, P. A., Davidson, A. D. & Wright, P. J. The nucleotide sequence and genome organization of mushroom bacilliform virus. Virology 202, 904–911 (1994).
Article CAS PubMed Google Scholar - Yokoi, T., Takemoto, Y., Suzuki, M., Yamashita, S. & Hibi, T. The nucleotide sequence and genome organization of Sclerophtora macrospora virus B. Virology 264, 344–349 (1999).
Article CAS PubMed Google Scholar - Yokoi, T., Yamashita, S. & Hibi, T. The nucleotide sequence and genome organization of Sclerophtora macrospora virus A. Virology 311, 394–399 (2003).
Article CAS PubMed Google Scholar - Matsui, S. M. & Greenberg, H. B. in Fields Virology (eds Knipe, D. M. & Howley, P. M.) 875–893 (Lippncott Williams & Wilkins, Philadelphia, 2001).
Google Scholar - Koonin, E. V., Choi, G. H., Nuss, D. L., Shapira, R. & Carrington, J. C. Evidence for common ancestry of a chestnut blight hypovirulence-associated double-stranded RNA and a group of positive-strand RNA plant viruses. Proc. Natl Acad. Sci. USA 88, 10647–10651 (1991).
Article CAS PubMed PubMed Central Google Scholar - Nuss, D. L. Hypovirulence: mycoviruses at the fungal-plant interface. Nature Rev. Microbiol. 3, 632–642 (2005). This article provides conceptual analysis of the interactions between viruses and their plant pathogenic fungal hosts.
Article CAS Google Scholar - Linder-Basso, D., Dynek, J. N. & Hillman, B. I. Genome analysis of Cryphonectria hypovirus 4, the most common hypovirus species in North America. Virology 337, 192–203 (2005).
Article CAS PubMed Google Scholar - Chu, Y. M. et al. Double-stranded RNA mycovirus from Fusarium graminearum. Appl. Environ. Microbiol. 68, 2529–2534 (2002).
Article CAS PubMed PubMed Central Google Scholar - Jiang, B., Monroe, S. S., Koonin, E. V., Stine, S. E. & Glass, R. I. RNA sequence of astrovirus: distinctive genomic organization and a putative retrovirus-like ribosomal frameshifting signal that directs the viral replicase synthesis. Proc. Natl. Acad. Sci. USA 90, 10539–10543 (1993).
Article CAS PubMed PubMed Central Google Scholar - Green, K. Y., Chanock, R. M. & Kapikian, A. Z. in Fields Virology (eds. Knipe, D. M. & Howley, P. M.) 841–874 (Lippincott Williams & Wilkins, Philadelphia, 2001).
Google Scholar - Ghabrial, S. A. Origin, adaptation and evolutionary pathways of fungal viruses. Virus Genes 16, 119–131 (1998).
Article CAS PubMed PubMed Central Google Scholar - Caston, J. R. et al. Three-dimentional structure and stoichometry of Helmintosporium victroriae 190S totivirus. Virology 347, 323–332 (2006).
Article CAS PubMed Google Scholar - Khramtsov, N. V. & Upton, S. J. Association of RNA polymerase complexes of the parasitic protozoan Cryptosporidium parvum with virus-like particles: heterogeneous system. J. Virol. 74, 5788–5795 (2000).
Article CAS PubMed PubMed Central Google Scholar - Koga, R., Horiuchi, H. & Fukuhara, T. Double-stranded RNA replicons associated with chloroplasts of a green alga, Bryopsis cinicola. Plant Mol. Biol. 51, 991–999 (2003).
Article CAS PubMed Google Scholar - Valles, S. M., Strong, C. A. & Hashimoto, Y. A new positive-strand RNA virus with unique genome characteristics from the red imported fire ant, Solenopsis invicta. Virology 365, 457–463 (2007).
Article CAS PubMed Google Scholar - Gorbalenya, A. E., Donchenko, A. P., Blinov, V. M. & Koonin, E. V. Cysteine proteases of positive strand RNA viruses and chymotrypsin-like serine proteases. A distinct protein superfamily with a common structural fold. FEBS Lett. 243, 103–114 (1989). The first demonstration of a highly significant sequence similarity between picornaviral 3CPros and the HtrA family of bacterial proteases.
Article CAS PubMed Google Scholar - Crawford, L. J. et al. Molecular characterization of a partitivirus from Ophiostoma himal-ulmi. Virus Genes 33, 33–39 (2006).
Article CAS PubMed Google Scholar - Embley, T. M. & Martin, W. Eukaryotic evolution, changes and challenges. Nature 440, 623–630 (2006). A comprehensive review of the current concepts of the origin of the eukaryotic cell that makes the sharp distinction between symbiotic and archezoan scenarios.
Article CAS PubMed Google Scholar - Martin, W. & Koonin, E. V. Introns and the origin of nucleus–cytosol compartmentation. Nature 440, 41–45 (2006). A hypothesis of the major role of the invasion of group II introns as the principal driving force behind the emergence of the nucleus during eukaryogenesis.
Article CAS PubMed Google Scholar - Martin, W. & Muller, M. The hydrogen hypothesis for the first eukaryote. Nature 392, 37–41 (1998).
Article CAS PubMed Google Scholar - Rivera, M. C. & Lake, J. A. The ring of life provides evidence for a genome fusion origin of eukaryotes. Nature 431, 152–155 (2004). An original method of phylogenetic analysis provides evidence in support of the origin of eukaryotic cell through fusion of prokaryotic genomes.
Article CAS PubMed Google Scholar - Kurland, C. G., Collins, L. J. & Penny, D. Genomics and the irreducible nature of eukaryote cells. Science 312, 1011–1014 (2006).
Article CAS PubMed Google Scholar - Poole, A. & Penny, D. Eukaryote evolution: engulfed by speculation. Nature 447 913 (2007).
Article CAS PubMed Google Scholar - Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Article CAS PubMed PubMed Central Google Scholar - Poch, O., Sauvaget, I., Delarue, M. & Tordo, N. Identification of four conserved motifs among the RNA-dependent polymerase encoding elements. EMBO J. 8, 3867–3874 (1989). The first clear demonstration of structural and evolutionary relationships between viral RdRps and reverse transcriptases.
Article CAS PubMed PubMed Central Google Scholar - Ago, H. et al. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Structure 7, 1417–1426 (1999).
Article CAS PubMed Google Scholar - Hansen, J. L., Long, A. M. & Schultz, S. C. Structure of the RNA-dependent RNA polymerase of poliovirus. Structure 5, 1109–1122 (1997).
Article CAS PubMed Google Scholar - Ng, K. K., Arnold, J. J. & Cameron, C. E. Structure-function relationships among RNA-dependent RNA polymerases. Curr. Top. Microbiol. Immunol. 320, 137–156 (2008).
CAS PubMed PubMed Central Google Scholar - Arnold, J. J., Ghosh, S. K. & Cameron, C. E. Poliovirus RNA-dependent RNA polymerase (3Dpol). Divalent cation modulation of primer, template, and nucleotide selection. J. Biol. Chem. 274, 37060–37069 (1999).
Article CAS PubMed Google Scholar - Arnold, J. J., Gohara, D. W. & Cameron, C. E. Poliovirus RNA-dependent RNA polymerase (3Dpol): pre-steady-state kinetic analysis of ribonucleotide incorporation in the presence of Mn2+. Biochemistry 43, 5138–5148 (2004).
Article CAS PubMed Google Scholar - Hung., M., Gibbs, C. S. & Tsiang, M. Biochemical characterization of rhinovirus RNA-dependent RNA polymerase. Antiviral Res. 56, 99–114 (2002).
Article CAS PubMed Google Scholar - Lambowitz, A. M. & Zimmerly, S. Mobile group II introns. Annu. Rev. Genet. 38, 1–35 (2004). This article reviews the mechanistic and evolutionary aspects of group II introns that were implicated in the origin of spliceosomal introns.
Article CAS PubMed Google Scholar - Robart, A. R. & Zimmerly, S. Group II intron retroelements: function and diversity. Cytogenet. Genome Res. 110, 589–597 (2005).
Article CAS PubMed Google Scholar - Toor, N., Keating, K. S., Taylor, S. D. & Pyle, A. M. Crystal structure of a self-spliced group II intron. Science 320, 77–82 (2008). This article reviews the mechanistic and evolutionary aspects of group II introns that were implicated in the origin of spliceosomal introns.
Article CAS PubMed PubMed Central Google Scholar - Eickbush, T. H. & Jamburunthugoda, V. K. The diversity of retrotransposons and the properties of their reverse transcriptases. Virus Res. 134, 221–234 (2008).
Article CAS PubMed PubMed Central Google Scholar - Arkhipova, I. R., Pyatkov, K. I., Meselson, M. & Evgen'ev, M. B. Retroelements containing introns in diverse invertebrate taxa. Nature Genet. 33, 123–124 (2003).
Article CAS PubMed Google Scholar - Gladyshev, E. A. & Arkhipova, I. R. Telomere-associated endonuclease-deficient Penelope-like retroelements in diverse eukaryotes. Proc. Natl. Acad. Sci. USA 104, 9352–9357 (2007).
Article CAS PubMed PubMed Central Google Scholar - Clausen, T., Southan, C. & Ehrmann, M. The HtrA family of proteases: implications for protein composition and cell fate. Mol. Cell 10, 443–455 (2002).
Article CAS PubMed Google Scholar - Li, W. et al. Structural insights into the pro-apoptotic function of mitochondrial serine protease HtrA2/Omi. Nature Struct. Biol. 9, 436–441 (2002).
Article CAS PubMed Google Scholar - Gorbalenya, A. E., Koonin, E. V. & Wolf, Y. I. A new superfamily of putative NTP-binding domains encoded by genomes of small DNA and RNA viruses. FEBS Lett. 262, 145–148 (1990).
Article CAS PubMed Google Scholar - Neuwald, A. F., Aravind, L., Spouge, J. L. & Koonin, E. V. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 9, 27–43 (1999).
CAS PubMed Google Scholar - Iyer, L. M., Leipe, D. D., Koonin, E. V. & Aravind, L. Evolutionary history and higher order classification of AAA+ ATPases. J. Struct. Biol. 146, 11–31 (2004). An evolutionary classification of the vast class of the cellular and viral ATPases in the context of the origins of primordial genetic systems, last universal common ancestor, bacteria, archaea and eukaryotes. It describes S3Hs as a distinct branch within the AAA+ class of ATPases.
Article CAS PubMed Google Scholar - Maaty, W. S. et al. Characterization of the archaeal thermophile Sulfolobus turreted icosahedral virus validates an evolutionary link among double-stranded DNA viruses from all domains of life. J. Virol. 80, 7625–7635 (2006).
Article CAS PubMed PubMed Central Google Scholar - Benson, S. D., Bamford, J. K., Bamford, D. H. & Burnett, R. M. Does common architecture reveal a viral lineage spanning all three domains of life? Mol. Cell 16, 673–685 (2004).
Article CAS PubMed Google Scholar - Dolja, V. V., Boyko, V. P., Agranovsky, A. A. & Koonin, E. V. Phylogeny of capsid proteins of rod-shaped and filamentous plant viruses: two families with distinct patterns of sequence and probably structure conservation. Virology 184, 79–86 (1991).
Article CAS PubMed Google Scholar - McGeoch, D. J., Rixon, F. J. & Davison, A. J. Topics in herpesvirus genomics and evolution. Virus Res. 117, 90–104 (2006).
Article CAS PubMed Google Scholar - Dolja, V. V. & Koonin, E. V. Phylogeny of capsid proteins of small icosahedral RNA plant viruses. J. Gen. Virol. 72 1481–1486 (1991).
Article PubMed Google Scholar - Schneemann, A., Reddy, V. & Johnson, J. E. The structure and function of nodavirus particles: a paradigm for understanding chemical biology. Adv. Virus Res. 50, 381–466 (1998).
Article CAS PubMed Google Scholar - Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Article CAS PubMed PubMed Central Google Scholar - Jobb, G., von Haeseler, A. & Strimmer, K. TREEFINDER: a powerful graphical analysis environment for molecular phylogenetics. BMC Evol. Biol. 4, 18 (2004).
Article PubMed PubMed Central Google Scholar - Whelan, S. & Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 18, 691–699 (2001).
Article CAS PubMed Google Scholar - Ronquist, F. & Huelsenbeck, J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003).
Article CAS PubMed Google Scholar
Acknowledgements
This paper is dedicated to Professor Vadim I. Agol. We thank V. Agol and T. Senkevich for critical reading of the manuscript and useful comments. E.V.K. and Y.I.W. are supported by the Department of Health and Human Services (National Library of Medicine, National Institutes for Health) intramural research funds. The research in V.V.D.'s laboratory is partially supported by National Institutes for Health grant GM053190 and BARD award no. IS-3,784-05.
Author information
Authors and Affiliations
- National Center for Biotechnology Information, National Institutes of Health, Bethesda, 20894, Maryland, USA
Eugene V. Koonin & Yuri I. Wolf - National Research Institute of Fisheries and Environment of Inland Sea, Fisheries Research Agency, 2-17-5 Maruishi, Hiroshima, 739-0452, Japan
Keizo Nagasaki - Department of Botany and Plant Pathology and Center for Genome Research and Biocomputing, Oregon State University, Corvallis, 97331, Oregon, USA
Valerian V. Dolja
Authors
- Eugene V. Koonin
You can also search for this author inPubMed Google Scholar - Yuri I. Wolf
You can also search for this author inPubMed Google Scholar - Keizo Nagasaki
You can also search for this author inPubMed Google Scholar - Valerian V. Dolja
You can also search for this author inPubMed Google Scholar
Corresponding authors
Correspondence toEugene V. Koonin or Valerian V. Dolja.
Supplementary information
Related links
Glossary
Virosphere
Also termed virus world, the virosphere is the entirety of viruses and virus-like agents comprising a genetic pool that is continuous in space and time and encompasses, in particular, hallmark viral genes that encode essential functions of many diverse viruses but are not found in genomes of cellular life forms.
Superfamily
In this context, a superfamily is a large group of viral families that are thought to have evolved from a common ancestor.
Picornaviruses
Narrowly defined, picornaviruses are a family of small, positive-strand RNA viruses that infect animals including humans (for example, poliovirus and foot-and-mouth disease virus). Broadly defined, the superfamily of picorna-like viruses consists of many families of RNA viruses that infect animals, plants and diverse unicellular eukaryotes, and appear to be evolutionarily related to picornaviruses.
Jelly-roll fold
The jelly-roll fold is a characteristic structural fold of the capsid proteins that comprise the icosahedral capsids of a variety of viruses including most of the picorna-like viruses.
Maximum likelihood
Generally, maximum likelihood is the statistical methodology used to fit a mathematical model of a process to the available data. In the context of phylogenetic analysis, maximum-likelihood methods use evolution models of various degrees of complexity to infer probability distributions for all possible topologies of a phylogenetic tree and, accordingly, assign likelihood values to particular topologies.
Clade
A clade is a taxonomic group that consists of a single common ancestor and all its descendants; in a phylogenetic tree, a clade is always either a terminal branch or a compact subtree.
Horizontal virus transfer
(HVT). Cross-species virus transmission and adaptation to a new host.
Retroelements
Diverse genetic elements that encode a reverse transcriptase and, accordingly, replicate through a genetic cycle that includes a step of DNA synthesis on a RNA template.
Rights and permissions
About this article
Cite this article
Koonin, E., Wolf, Y., Nagasaki, K. et al. The Big Bang of picorna-like virus evolution antedates the radiation of eukaryotic supergroups.Nat Rev Microbiol 6, 925–939 (2008). https://doi.org/10.1038/nrmicro2030
- Published: 10 November 2008
- Issue Date: December 2008
- DOI: https://doi.org/10.1038/nrmicro2030