Notch1 antiapoptotic activity is abrogated by caspase cleavage in dying T lymphocytes (original) (raw)
Introduction
Proteins belonging to the Notch family are evolutionarily conserved surface receptors associated with a broad spectrum of cellular processes including differentiation, proliferation and apoptosis, from Drosophila melanogaster to Homo sapiens.1 Notch proteolysis plays a critical role in the signaling of these receptors, which represents a new paradigm of signal transduction, the regulated intramembrane proteolysis (RIP).2 Precursors of Notch receptors are constitutively cleaved by a furin class serine protease in the _trans_-Golgi compartment before migrating to the plasma membrane.3 Upon binding to a member of the Delta/Serrate/Lag-2 (DSL) family of protein, intramembrane cleavage of Notch1 produces a fragment, the Notch1 intracellular domain (NICD).4 The NICD then translocates to the nucleus to regulate the expression of specific genes. Notch1 main cofactor in the nucleus is the repressor CBF-1/Suppressor of Hairless/Lag-1 (CSL), which upon association with the NICD is converted to a transcriptional activator.5
Several groups have demonstrated the importance of the Notch pathway during T-cell development.6, 7 Murine models of genetic interference have shown that Notch1 activity is required for the commitment of common lymphoid precursors to the T-cell lineage8, 9, 10 and for αβ pre-T-cell differentiation. 11, 12 Considering the role of the Notch pathway in lymphopoiesis, it is not surprising that the human ortholog of D. melanogaster Notch1 was identified in three cases of human T-cell leukemia, where a translocation of the NICD led to constitutive Notch1 signaling,13 suggesting that this receptor was endowed with an oncogenic effect in T lymphocytes. This hypothesis was confirmed by retroviral transduction of the NICD in murine bone marrow cells, leading to the appearance of clonal leukemic cells of an immature T-cell phenotype all expressing the truncated Notch1.14 A recent study shows that activating mutations can be found in the Notch1 gene in more than 50% of human primary T-ALL samples.15 Both the ankyrin repeats and a C-terminal transactivation domain (TAD) are required for leukemogenesis;16 however, the precise mechanism leading to leukemia remains elusive.
The role of Notch1 in oncogenesis may be related to its ability to regulate the apoptotic process. Indeed, the constitutively active intracellular fragment of murine Notch1 is capable of rendering thymocytes resistant to glucocorticoid-induced apoptosis.17 Furthermore, overexpression of the human NICD blocks activation-induced cell death (AICD) in a T-cell hybridoma18 or death receptor-induced apoptosis in the Jurkat leukemic cell line.19 Several mechanisms have been proposed for the inhibition of apoptosis by Notch1. Abrogation of T-cell hybridoma apoptosis by the NICD was first associated with inhibition of Nur77 activity.18 The activity of this orphan nuclear receptor from the steroid receptor family is required for AICD.20, 21, 22 More recently, Notch1 was also shown to trigger the p56lck-dependent activation of the antiapoptotic kinase AKT and to increase the levels of the antiapoptotic proteins c-FLIP, IAP-2 and Bcl-XL.19 The contribution of each pathway to Notch1-mediated inhibition of apoptosis is not yet clear.
Many antiapoptotic proteins are cleaved by cysteine proteases of the caspase family during apoptosis, which results in the abrogation of their inhibitory function. The human genome encodes 11 caspases among which caspase-9, -8 and -3 play a central role during apoptosis in many cell types.23 Their function is mediated through proteolysis of cellular proteins such as the DNA fragmentation factor, leading to internucleosomal cleavage of DNA24 or cleavage of proteins able to antagonize the apoptotic process, including Bcl-2, XIAP or the AKT/PKB kinase.25, 26, 27, 28, 29 The inhibitory effect of Notch1 on T-cell death prompted us to investigate whether Notch1 was also a caspase substrate during T-cell apoptosis. We show here that the NICD is completely fragmented by caspases during T-lymphocyte apoptosis and that the cleavage products are no longer able to inhibit AICD in a T-cell hybridoma. Finally, caspase-mediated fragments are unable to antagonize the activity of the Nur77 transcription factor, supporting a role for this protein in Notch1 antiapoptotic activity.
Results
Notch1 is cleaved by caspases in apoptotic Jurkat cells
The intracellular domain of Notch1 contains several caspase consensus sites (DxxD or VxxD), which are evolutionarily conserved from Xenopus to human (Figure 1). These sites are distributed throughout the cytoplasmic portion of the receptor and can be found within the NF-_κ_B binding domain (NBD), the ankyrin repeats (ANK) and the C-terminal transactivation domain (TAD) involved in the control of gene expression by Notch1.30 The potential cleavage of Notch1 by caspases during apoptosis was initially investigated in the Jurkat T-cell line. Treatment of Jurkat cells with an anti-Fas antibody induced the cleavage of the well-known caspase-3 substrate PARP,31, 32 caspase-3 and caspase-6 activation and the exposure of phosphatidylserine at the cell surface in 85% of the cells (Figure 2a). Concomitant with caspase activation, the 115 kDa intracellular domain of Notch1 disappeared and six new bands of 82, 78, 77, 70, 48 and 45 kDa were observed in the apoptotic cell lysates, as revealed by a monoclonal antibody (Figure 2a) or a polyclonal antibody (Figure 2b) raised against the C-terminal region of the NICD.3 Inhibition of Notch1 fragmentation by the caspase inhibitors DEVD-fmk and VEID-fmk but not FA-fmk (Figure 2b) suggested that caspase-3- and caspase-6-like activities were required for this cleavage. Proteins such as p56lck and actin were not cleaved in apoptotic cells (Figure 2b), demonstrating that nonspecific protein degradation could not account for Notch1 and PARP cleavage. Similar experiments were performed using the Jurkat clone JIB2, where Bcl-2 expression was induced by the addition of doxycyclin (Dox), thereby inhibiting Fas-mediated mitochondrial cytochrome c release, caspase activation and apoptosis.33, 34, 35 In the presence of Bcl-2, the disappearance of the full-length NICD and the generation of Notch1 fragments were abrogated (Figure 2c), showing that Notch1 cleavage was dependent on cytochrome c release from the mitochondria and activation of caspases following Fas crosslinking. To confirm that the protein processed in Jurkat cells undergoing apoptosis was indeed Notch1, lysates of untreated or apoptotic Jurkat cells were incubated in the presence of a radiolabeled, recombinant NICD produced by using an in vitro coupled transcription/translation system. This recombinant NICD lacking Notch1 extracellular and transmembrane domains is not subjected to post-translational modifications and only migrates at 77 kDa, versus 115 kDa for the endogenous NICD. In three independent experiments, the recombinant NICD was cleaved into fragments of 60, 53, 28 and 24 kDa by apoptotic Jurkat cell lysates, but not by proteins from untreated Jurkat cells (Figure 2d). Adding the caspase inhibitor DEVD-CHO but not YVAD-CHO prevented digestion of the recombinant NICD by the apoptotic cell lysate, further supporting the role of caspase-3-like proteases in Notch1 cleavage. Due to a difference in size between the endogenous and the recombinant NICD, the fragments detected by Western blot or in vitro in the presence of apoptotic Jurkat proteins were different in size. However, the cleavage pattern obtained following digestion of the recombinant NICD with apoptotic Jurkat cell extracts was similar to the one observed in Western blot analysis of apoptotic Jurkat cells, with major fragments of 60 and 53 kDa (reminiscent of the 77–82 kDa major fragments detected in Western blot) and minor fragments at 24–28 kDa (corresponding to the 45–48 kDa fragments). Taken together, these results demonstrate that Notch1 is cleaved by caspases in Jurkat cells during Fas-mediated apoptosis.
Figure 1

Caspase consensus sites in Notch1 are evolutionary conserved. The sequence of human (Genebank accession number: AAG33848), mouse (A46019), rat (Q07008) and Xenopus (AAB02039) Notch1 starting at the transmembrane domain were aligned using the Bioedit software. The consensus caspase cleavage sites are shown in boxes. Aspartic acid residues are highlighted in bold. The transmembrane domain (TM), CSL binding domain (RAM), NF-_κ_B binding domain (NBD), ankyrin repeats (ANK) and transactivation domain (TAD) are underlined
Figure 2
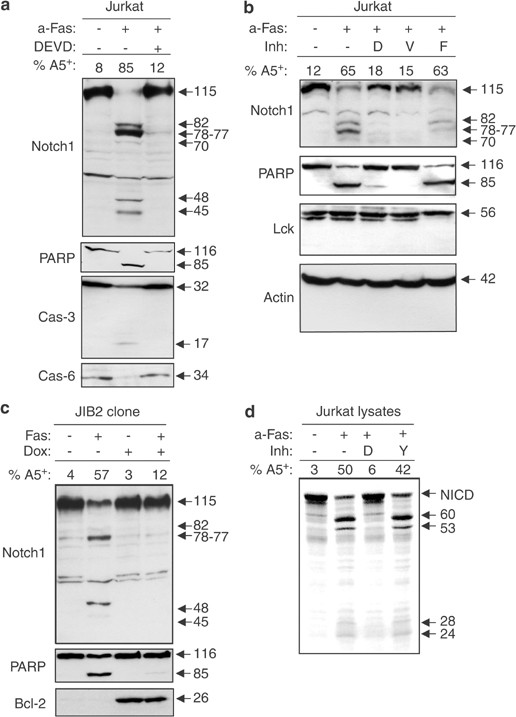
Notch1 is cleaved by caspases during Fas-mediated apoptosis. (a) Jurkat T cells were cultured in the presence or absence of anti-Fas antibody (CH11), with or without the caspase-3-like protease inhibitor DEVD-fmk. After 5 h at 37°C, an aliquot was kept for the apoptosis assay using Annexin-V staining. The remaining cells were lysed and proteins were subjected to Western blot analysis using antibodies against Notch1 (btan20), PARP, caspase-3 and caspase-6. The molecular weights of all the bands are indicated in the left of the autoradiography. (b) Jurkat cells were incubated in the presence of CH11, with or without 50 _μ_M of DEVD-fmk (D), VEID-fmk (V) or the negative control FA-fmk (F). Cell lysates were subjected to Western blot analysis using polyclonal anti-Notch1, -PARP, -lck or -_β_-actin antibodies. (c) The Bcl-2-inducible Jurkat clone JIB2 was preincubated in the presence or absence of Dox during 36 h. JIB2 cells were then cultured with or without anti-Fas antibody for 5 h and treated as in (a). (d) Jurkat lysates used for in vitro cleavage assays were prepared by incubating Jurkat cells in medium alone or with CH11 for 5 h and by performing four freeze–thaw cycles in caspase buffer. Apoptosis was assessed by flow cytometry and the percentage of Annexin-V-positive cells is indicated. Radiolabeled NICD was incubated for 1 h at 37°C with 50 _μ_g of cellular proteins in the presence or absence of 1 _μ_M of DEVD-CHO (D) or YVAD-CHO (Y) and reactions were analyzed by SDS–PAGE and autoradiography. Results shown are representative of two (panels a–c) and four (panel d) independent experiments
Caspase-3 and caspase-6 are the major proteases involved in Notch1 cleavage
We then sought to identify the caspases involved in the cleavage of Notch1. To determine whether Notch1 was a caspase-3 substrate, the radiolabeled NICD was incubated with recombinant caspase-3. Recombinant PARP was also produced in vitro and used as a positive control for caspase-3 activity. The NICD was fragmented into six peptides following 30 min of incubation at 37°C with recombinant caspase-3 (Figure 3a). This processing was completely inhibited by adding 1 _μ_M DEVD-CHO. Several fragments appeared simultaneously after 15 min of incubation with the protease and bands of 66, 55, 45, 40, 28 and 26 kDa could clearly be seen after 1 h of digestion, at a time when PARP was completely cleaved to its 85 kDa fragment (Figure 3a). The main caspase-3 consensus sites in the NICD sequence are DQTD1840H and DITD2092H (Figure 1). To determine the role of the other caspase-3 consensus sites, NICD deletion mutants starting at the two major sites were generated by PCR (Figure 3b) and produced in vitro. Digestion of the NΔ1 deletion starting at residue D1853 located downstream of the RAM domain led to a pattern very similar to the full-length NICD (Figure 3b). In contrast, digestion of the Notch1 portion located downstream of the ankyrin repeats (N3 deletion) generated only one new band (identified by an asterisk in Figure 3b). These results suggested that most caspase-3 cleavage sites were located between the ends of the RAM domain and the ankyrin repeats, with the exception of one cleavage site located downstream of the ankyrin repeats.
Figure 3
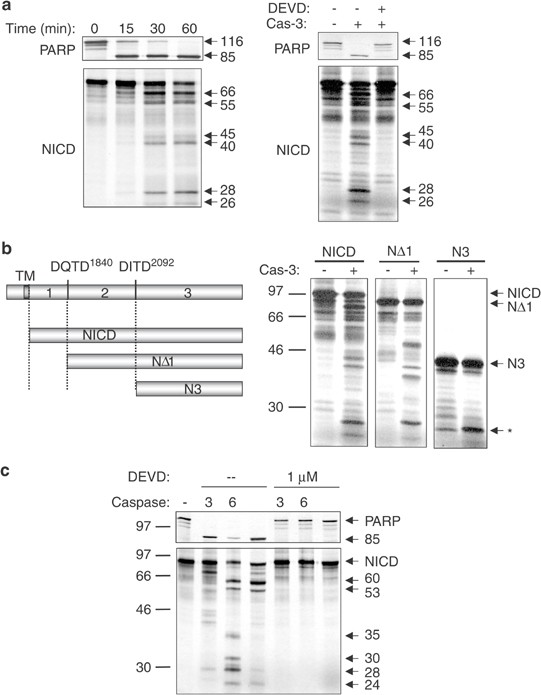
Notch1 is cleaved by recombinant caspase-3 and caspase-6. (a) Left panel: radiolabeled recombinant NICD and PARP were generated by in vitro transcription/translation. Both proteins were incubated with 10 U of caspase-3 for 15–60 min at 37°C. Proteins were separated by SDS–PAGE and analyzed by autoradiography. Right panel: radiolabeled recombinant proteins were incubated for 1 h at 37°C in caspase buffer in the presence of 10 U of recombinant caspase-3, with or without 1 _μ_M of DEVD-CHO. Reactions were analyzed as in the left panel. (b) Deletion mutants corresponding to two main caspase-3 sites DQTD1840 and DITD2092 were amplified by PCR. The NΔ1 deletion starts at residue D1853 and excludes the RAM domain and most of the NBD. The N3 deletion starts at residue R2096 located downstream of the ankyrin repeats. The full-length NICD or deletion mutants were incubated with recombinant caspase-3 and the digestions were subjected to SDS–PAGE and autoradiography as described for (a). Positions of the molecular weight standards are indicated on the left of the autoradiography. The band indicated by the asterisk represents the only fragment generated by caspase-3 digestion of the N3 deletion. (c) Radiolabeled PARP or NICD was incubated with 10 U of caspase-3 (3) or caspase-6 (6), or with 50 _μ_g of proteins from an apoptotic Jurkat cell lysate (Jf) in the presence or absence of 1 _μ_M DEVD-CHO. After 1 h of digestion at 37°C, reactions were stopped by the addition of 2 × loading buffer and were loaded on the same 12% polyacrylamide gel. Cleavage patterns were revealed by autoradiography. Molecular weight of caspase-6-mediated fragments is indicated on the right and the position of molecular weight markers on the left of the autoradiography. All digestions were repeated at least three times with similar results
Consistent with the inhibition of Notch-1 cleavage by the caspase-6 inhibitor VEID-fmk in vivo, recombinant caspase-6 was also able to cleave the NICD, albeit in a different pattern than caspase-3 (Figure 3c). Incubation of the NICD with recombinant caspase-6 consistently generated fragments of 60, 53, 35, 30, 28 and 24 kDa in five independent experiments. None of these fragments were observed in the presence of 1 _μ_M of DEVD-CHO (Figure 3c). Digestion of the recombinant NICD by caspase-7 resulted in a similar pattern as caspase-3, although the cleavage was less efficient (data not shown). Incubation of the NICD with recombinant caspase-3, caspase-6 or with apoptotic Jurkat cell lysates was performed in an attempt to identify the contribution of each caspase in Notch1 cleavage in Jurkat cells. Notch1 fragments produced by apoptotic Jurkat lysates were very similar to the ones generated by caspase-6, although fragments corresponding to caspase-3 activity were also weakly detected (Figure 3c). Therefore, caspase-6 was the main protease involved in Notch1 cleavage in apoptotic Jurkat cells. These results correlate with the inhibition of Notch1 cleavage by the caspase inhibitors DEVD-fmk and VEID-fmk and demonstrate that caspase-6 and caspase-3 are the major proteases involved in Notch1 cleavage in Jurkat cells undergoing Fas-mediated apoptosis.
Caspase cleavage sites are mainly located in the NBD and the ankyrin repeats
A mutation analysis was performed by PCR to localize precisely caspase-3 and caspase-6 cleavage sites in the NICD. Aspartic acid to glutamic acid mutations at each putative site was confirmed by DNA sequencing. NICD mutants were produced by in vitro transcription/translation and their digestion by caspase-3 or caspase-6 was analyzed by SDS–PAGE (Figure 4a). Results demonstrate that caspase-3 uses at least four sites including DQTD1840, DADCMD1874, DITDHMD2095 and CLLD2192, whereas caspase-6 cleaves Notch1 at the DADCMD1874, EEED1906, DITDHMD2095 and CLLD2192 sites (Figure 4b). Mutation of other putative cleavage sites did not affect the cleavage pattern of Notch1 by either caspase. To further confirm the cleavage sites identified by mutagenesis, a GST-NICD fusion protein was produced in bacteria and incubated for 1 h at 37°C with each caspase. The fragments obtained were purified by SDS–PAGE and subjected to N-terminal sequencing. Digestion of the GST-NICD fusion with caspase-6 and caspase-3 produced six and five fragments, respectively (data not shown). As illustrated in Figure 4b, the amino-terminus sequence of these fragments confirmed Notch1 cleavage by caspase-3 at the DQTD1840H site. Sequencing of the fragments purified from caspase-6 digestion of GST-Notch1 indicated the VDADCMD1874, EEED1906, VSAD1973, ANRD2089ITDHMD and CLLD2193 sites. However, mutation of the VSAD1973 site did not affect Notch1 cleavage by caspase-6 (Figure 4a, lane 8), suggesting that this site is either weakly cleavable by caspases or not used at all. In summary, caspase-6 and caspase-3 cleave the NICD at five different sites, located in the NBD, the ankyrin repeats, which are involved in the antiapoptotic function of Notch1,18 and the TAD domain, the latter being associated with the transforming activity of Notch1.16
Figure 4

The caspase cleavage sites are located within the NBD, ankyrin repeats and TAD domain. (a) Point mutations were performed by PCR to change the aspartic acid residues to glutamic residues in each putative caspase cleavage site of the NICD. The mutants are identified on the right of the figure (lanes 3–13). These mutants were incubated with recombinant caspase-3 or caspase-6 and cleavage was analyzed as in Figure 3. Small dots indicate the missing fragments for each mutant as compared to the wild-type molecule (lane 2). The molecular weight of the fragments is indicated on the right. (b) Illustration of the identified caspase cleavage sites. The NICD is represented with all its functional domains, as well as the putative caspase cleavage sites (black lines). The sites identified by mutagenesis are indicated by vertical arrows. Some of these sites were confirmed by N-terminal sequencing of Notch1 fragments as follows: a GST-NICD fusion molecule was produced in bacteria and purified using glutathione-coupled beads. The beads coated with GST-NICD were incubated with caspase-3 or caspase-6 and the reactions were stopped by addition of 2 × loading buffer. Proteins were separated on a 12% acrylamide gel, transferred on a PVDF membrane, revealed by Coomassie staining and cut from the membrane to perform N-terminal sequencing. The sequence obtained allowed to confirm the DQTD site for caspase-3 and the DADCMD, EEED, DITDHMD and CLLD sites for caspase-6
Notch1 is cleaved by caspases in peripheral blood T cells during AICD
Notch1 expression and activity have been described in stimulated peripheral CD4+ T cells.36, 37, 38 The Notch ligand Delta1 is able to increase the expression of HES1, which is controlled by Notch signaling, in peripheral CD4 and CD8 lymphocytes.39 However, the role of Notch receptors in peripheral T lymphocytes remains unclear. Notch1-mediated inhibition of apoptosis in the Jurkat T-cell line19 suggested that this receptor could antagonize apoptosis in peripheral T cells. Therefore, we hypothesized that the NICD should be cleaved during AICD, in order to allow cell death to occur. To verify this hypothesis, AICD was induced by stimulating peripheral blood mononuclear cells (PBMCs) during 4 days with anti-CD3 and IL-2, followed by restimulation on anti-CD3-coated plates. Following 16 h of restimulation, the percentage of Annexin-V-positive cells rose to 59% and new Notch1 fragments could be observed at 70, 45, 40, 32, 28 and 22 kDa by Western blot analysis (Figure 5a). Concomitantly, the processing of caspase-3, caspase-6 and PARP was observed in the same lysates containing Notch1 fragments (Figure 5a). To confirm that the bands observed by Western blot resulted from the cleavage of Notch1 by caspases, the recombinant NICD was incubated with lysates from untreated (−) or apoptotic (+) lymphocytes. Proteins from apoptotic but not resting peripheral lymphocytes could cleave Notch1 in vitro (Figure 5b), confirming that Notch1 was a caspase substrate during AICD in peripheral blood T cells. Addition of DEVD-CHO but not YVAD-CHO prevented the cleavage of the recombinant NICD by proteins from apoptotic peripheral T cells (Figure 5b), further supporting a role for caspase-3 and caspase-6 in this response.
Figure 5

Notch1 is cleaved by caspases during AICD in peripheral T cells. (a) Purified PBMCs were left untreated (−) or stimulated by addition of anti-CD3 and IL-2 and left in culture for 4 days before transfer to anti-CD3-coated plates to induce AICD (+/+). After 16 h of restimulation, cells were harvested and apoptosis was assessed using the Annexin-V assay. Proteins from untreated or restimulated cells were analyzed by Western blot using the monoclonal anti-Notch1 antibody btan20, or the anti-caspase-3, -caspase-6 and -PARP antibodies. (b) The radiolabeled, recombinant NICD was incubated for 1 h at 37°C with 5 _μ_g of proteins extracted from untreated (−) or apoptotic (+) PBMCs in the presence or absence of DEVD-CHO (D) or YVAD-CHO (Y). Reactions were analyzed as in Figure 3. Results are representative of three independent experiments giving similar results. The molecular weights of the bands are indicated on the right. (c) PBMCs were left untreated or stimulated with 1 _μ_g/ml of anti-CD3 antibody for 6 days. Activated T cells were then incubated with or without anti-Fas antibody and apoptosis was assessed by Annexin-V staining. Cell lysates were subjected to Western blot analysis using anti-HES1 or _β_-actin antibodies and a densitometric analysis was performed. Results are expressed as the ratio between HES1 and _β_-actin band intensities for each condition
To determine whether Notch1 transcriptional activity was abrogated following Notch1 cleavage in dying cells, the expression of the protein HES1 was assessed by Western blot in activated or apoptotic T cells. Peripheral T lymphocytes were left untreated, or activated with anti-CD3 and anti-CD28 antibodies and cell death was triggered 6 days later by Fas crosslinking. As a control for HES1 expression, Jurkat T cells were incubated for 8 or 24 h with the Notch1 ligand Delta1 as previously described.40 A four fold increase in HES1 levels was observed following T-cell activation and concomitant with Notch1 cleavage, HES1 levels dropped to the level of untreated cells following apoptosis induction (Figure 5c). This result suggests that Notch1 cleavage during apoptosis abrogates its transcriptional activity.
Notch1 fragments cannot antagonize AICD
To demonstrate the role of a caspase substrate in the apoptotic process, it is a standard strategy to overexpress an uncleavable substrate and verify whether it counteracts apoptosis. However overexpression of an antiapoptotic protein such as Notch1 already protects against cell death19 and an uncleavable mutant would exhibit the same activity, first through its antiapoptotic activity and second by sequestration of active caspases, in the same way as caspase inhibitors. For instance, Deveraux et al.27 have shown that an uncleavable XIAP mutant inhibited anti-Fas-induced apoptosis as well as the wild-type XIAP. However, as for many antiapoptotic proteins including XIAP, Bcl-2 or AKT, cleavage of Notch1 by caspases should abrogate its inhibitory effect on cell death. Therefore, the ability of caspase-3- or caspase-6-mediated fragments to inhibit cell death was analyzed. The full-length NICD or the deletion mutants NΔ1 and N3, corresponding to the main fragments generated by caspases cleavage at the DADCMD1874 and DITDHMD2095 sites (Figure 4b), were subcloned in the retroviral vector AP2, upstream of an IRES sequence and the GFP cDNA. The NICD was also cloned in the antisense direction (NICDAS) as a control. Viral supernatants from the BOSC23 packaging cell line transfected with these constructs were used to transduce the different mutants in the DO11.10 T-cell hybridoma where the antiapoptotic function of Notch1 was first demonstrated.18, 19 GFP-positive infected DO11.10 cells were then sorted by flow cytometry and expression of all Notch1 forms was confirmed by Western blot analysis using the anti-Notch1 monoclonal antibody btan20 (data not shown). Then, AICD was triggered by incubating these cell lines in anti-CD3-coated plates for 6 h and apoptosis levels were assessed by staining for chromatin condensation using the fluorescent dye Hoechst 33342. In four independent experiments, only the expression of the NICD could significantly inhibit AICD (Figure 6), confirming that once cleaved by caspases, Notch1 cannot antagonize cell death anymore.
Figure 6
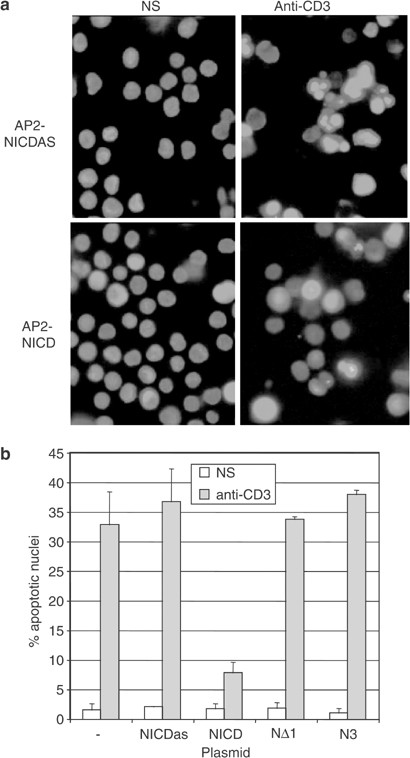
Notch1 fragments are unable to inhibit AICD. (a) The NICD, NΔ1 and N3 cDNAs were cloned in the AP2 retroviral vector upstream of an IRES and GFP cDNA. The NICD was also cloned in the antisense direction as a control (NICDAS). These constructs or the empty AP2 vector were used to transduce the DO11.10 T cell hybridoma. Infected cells were sorted for GFP expression, verified by Western blot using an anti-Notch antibody and stimulated during 8 h with anti-CD3 antibodies. Stimulated cells were stained with the fluorescent dye Hoechst 33342, which revealed chromatin condensation during apoptosis. (b) The percentage of apoptotic nuclei was assessed for each cell line stimulated in duplicate. Results are expressed as the mean±s.d. of duplicates from one representative experiment among four
Notch1 fragments exhibit a reduced capacity to inhibit Nur77 activity
Both caspase-3 and caspase-6 cleave Notch1 within the ankyrin repeats, a domain involved in the association between the NICD and Nur77,18 which suggests that Notch1 cleavage during apoptosis may abrogate its inhibitory effect on Nur77 activity. To determine the influence of Notch1 cleavage on Nur77 inhibition, the full-length NICD, the NΔ1 and the N3 deletion mutant were expressed in 293T cells along with the human Nur77 (hNur77) and a Nur77 reporter construct. The transcriptional activity of hNur77 was inhibited in a dose-dependent manner in the presence of the full-length NICD (Figure 7a), demonstrating that human Notch1 can also block transactivation by the human ortholog of the murine Nur77. In contrast, the NΔ1 deletion did not affect hNur77 activity (Figure 7b). Interestingly, this deletion mutant corresponds to a fragment that is produced by both caspase-3 and caspase-6 following cleavage at the DADCMD1874 site (Figure 4b). The N3 deletion only had a small but reproducible inhibitory effect on Nur77 transactivation (20% inhibition versus 60% for the full-length NICD). The experiments were carried out under saturating amounts of the NICD or deletion mutants, and using higher amounts of NΔ1 or N3 vectors (up to four times) did not increase their capacity to inhibit Nur77 activity (data not shown). Finally, the influence of Notch1 fragments on the inhibitory activity of the full-length NICD was assessed by cotransfection of the NICD with each fragment, along with hNur77. Neither deletion interfered with the inhibition of hNur77 activity by the full-length NICD in cotransfection experiments (Figure 7c), excluding a potential dominant-negative effect of these fragments on Notch1 inhibitory activity (_n_=2). In conclusion, Notch1 deletion mutants corresponding to the main caspase-mediated fragments have a reduced capacity to inhibit hNur77 activity, supporting a role for this factor in Notch1 antiapoptotic activity.
Figure 7

Notch1 cleavage abrogates its inhibitory effect on hNur77 activity. (a) 293T cells were transfected in triplicate with hNur77 with or without the NICD, with a Nur77 reporter construct containing the luciferase gene under the control of Nur77 and with beta-galactosidase. Following 48 h of culture at 37°C, cells were lysed and the beta-galactosidase and luciferase activities were quantified. The results are expressed as percentage of Nur77 activity, calculated as follows: the mean ratio of the luciferase and beta-galactosidase activities obtained in the presence of the NICD or deletion mutants was divided by the mean ratio obtained with hNur77 alone. (b) Cotransfection of 293T cells with hNur77 and either the full-length NICD, the NΔ1 or the N3 deletions was performed and after 48 h of culture, luciferase and beta-galactosidase activities were quantified. Results were expressed as the percentage of Nur77 activity in the presence of hNur77 alone and are representative of four independent experiments performed in triplicate, giving similar results. (c) 293T cells were transfected with hNur77, with or without the NICD, and cotransfected with either the GST (as a control) or the NΔ1 or N3 deletions. Analysis of Nur77 activity was performed as described for (a). Results are from one out of two experiments performed in triplicate and giving identical results
Discussion
The Notch1 receptor is an oncogenic molecule in T lymphocytes13, 14 and has been shown to inhibit apoptosis in T-cell lines.18, 19 Antiapoptotic molecules such as Bcl-2, XIAP or AKT are subjected to caspase-mediated cleavage during apoptosis,25, 26, 27, 28, 29 resulting in the abrogation of their antiapoptotic activity. Results presented in this study demonstrate that Notch1 antiapoptotic activity is also regulated by caspase-mediated cleavage. Indeed, the NICD is completely fragmented in Jurkat cells undergoing apoptosis and is cleaved in vitro by recombinant caspase-3 and caspase-6. The cleavage sites used by both caspases are localized in the NBD, the ankyrin repeats and the C-terminal transactivation domain. Importantly, Notch1 deletions corresponding to fragments generated by caspases are no longer able to antagonize apoptosis in DO11.10 cells and exhibit a reduced capacity to inhibit the transcription factor Nur77, an essential factor during AICD.20, 21, 22
The maturation and signaling of Notch receptors is known to depend on the activity of several types of proteases. Following synthesis as long precursors, these receptors are constitutively cleaved by a Furin-like protease in the _trans_-Golgi network.41 Ligand binding to the extracellular portion of the receptor first triggers cleavage of the Notch N-terminal moiety on the cell surface by the TACE metalloprotease. This step is required for an intramembrane proteolytic event mediated by a Presenilin-dependent complex, which releases the NICD from the plasma membrane and allows this fragment to migrate to the nucleus. Finally, the active form of Notch1 is degraded by the proteasome following phosphorylation and association with SEL-10 through a C-terminal PEST domain.42 Our study demonstrates that another family of proteases, the proapoptotic caspases, modulates Notch1 activity following apoptosis induction in T lymphocytes. Notch1 cleavage by caspase-3 and caspase-6 results in the production of fragments unable to inhibit hNur77 activity and antagonize AICD.
Our results suggest that caspase-6 is the main protease involved in Notch1 cleavage during apoptosis, since the main NICD fragments observed in the presence of apoptotic Jurkat cell lysates corresponded to the fragments generated by caspase-6 cleavage (Figure 3c). The role of this caspase in the apoptotic process has been associated so far to cytoskeleton alterations through cleavage of structural proteins such as lamin A and C,43, 44, 45 cytokeratin 1846 and vimentin.47 However, the identification of Notch1 as a caspase-6 substrate suggests another function for this caspase during apoptosis. Through its association with CSL, Notch1 directly regulates chromatin structure and gene transcription by recruiting histone acetyl transferases (HATs) to DNA regions containing the CSL response element 5′-TGGGAAA-3′.48, 49 Cleavage of the NICD by caspase-6 results in the removal of the RAM domain involved in its binding to CSL, disruption of the ankyrin repeats necessary for Mastermind association and cleavage of the TAD domain that is required for Notch1-mediated transcription.50 Therefore, caspase-6 could affect gene expression through Notch1 fragmentation. Consistent with this hypothesis, caspase-6 also cleaves special AT-rich sequence-binding protein 1 (SATB1) during T-cell apoptosis.51 SATB1 coordinates gene expression during T-cell development, through binding to matrix attachment regions involved in the loop domain organization of chromatin.52 Cleavage of SATB1, exclusively by caspase-6, during T-cell apoptosis induces its dissociation from the chromatin and is likely to affect its ability to coordinate gene expression.51
Notch1 antiapoptotic activity in DO11.10 cells has first been associated with inhibition of the transcription factor Nur77.18 Nur77 activity is required for AICD since a Nur77 dominant-negative form or Nur77 antisense oligonucleotides inhibits AICD in a T-cell hybridoma or in thymocytes.20, 22, 53 Notch1 fragmentation by caspases following stimulation of AICD should disrupt its association with Nur77 and release its proapoptotic activity. Results presented here confirm that cleavage of Notch1 at the DADCMD1874 site leads to the production of a fragment of approximately 80 kDa unable to inhibit Nur77 activity or AICD as shown by using the NΔ1 deletion mutant. The fragment generated by cleavage at the DITDHMD2095 site and corresponding to the N3 deletion mutant is only endowed with a weak inhibitory capacity (20% inhibition versus 60% for the full-length NICD) and it is further cleaved by caspase-3 and caspase-6 (Figures 3b and 4b). The weak inhibition of Nur77 activity by the N3 deletion may not be sufficient to affect the apoptotic response since DO11.10 transduced with this deletion are not resistant to cell death (Figure 6).
The NICD was reported to bind to Nur77 through Notch1 ankyrin repeats.18 However, the domains of Notch1 required for this inhibitory effect are not known. The NΔ1 deletion containing the ankyrin repeats but lacking the RAM and NBD domain did not affect Nur77 activity in the cotransfection assay, which suggests that the N-terminal portion of the NICD may be required for Nur77 inhibition. Since the RAM domain is involved in Notch1 binding to CSL, this factor may be essential for Notch1 inhibitory effect. Alternatively, removal of the N-terminal portion of Notch1 may affect the overall protein conformation and compromise Notch1 association with Nur77. Since the N3 deletion lacks the ankyrin repeats, it probably does not bind to Nur77 and possesses a weak capacity to block Nur77 activity. This residual inhibitory effect could be due to the capacity of N3 to interact with factors necessary for Nur77 function, such as CPB/p300.54, 55
Although these results support a role for Nur77 inhibition in Notch1 antiapoptotic effect, other potential mechanisms for Notch1-mediated inhibition of apoptosis have been proposed. Deftos et al.17 have shown that a tryptophan residue in the RAM domain is involved in the protective effect of Notch1 against dexamethasone-induced cell death in a murine T-cell hybridoma. The lack of inhibitory effect of fragments similar to the NΔ1 deletion mutant could be explained by the removal of the RAM domain by caspases through cleavage at the DADCMD1874 site. The NICD can also block NF-_κ_B activity by direct binding and inhibition of NF-_κ_B p50 subunit by Notch1,56 stimulate the p56lck-dependent activation of the antiapoptotic kinase AKT or trigger an increase in Bcl-XL, IAP-2 and c-FLIP expression.19 Since Notch1 is completely fragmented during T-cell apoptosis, its transcriptional activity as well as its interaction with Nur77, p56lck or NF-_κ_B p50 may all be affected by caspase activity. Therefore, cleavage of the NICD by caspases is likely to release several factors required during AICD, namely NF-_κ_B and Nur77, while simultaneously disrupting the activation of the AKT survival pathway by Notch1. In conclusion, Notch1 can now be added to a list of antiapoptotic proteins inactivated by caspases during apoptosis, which include Bcl-2, AKT and XIAP.
Materials and Methods
Reagents and cell lines
Recombinant caspase-3 and caspase-6 from the MultiCasPak-2 kit were purchased from BIOMOL (Plymouth Meeting, PA, USA), the caspase inhibitors DEVD-fmk and VEID-fmk from Enzyme Systems products (Dublin, CA, USA) and R&D Systems (Minneapolis, MN, USA) and the DEVD-CHO and YVAD-CHO inhibitors from Peptides International Inc. Recombinant IL-2 was obtained from the NIH AIDS Research and Reference Reagent Program. FITC-conjugated Annexin V was purchased from Biosource (Nivelles, Belgium) and propidium iodide and Dox were from Sigma (Mississauga, Ontario, Canada). The anti-Fas antibody (CH11, Beckman/Immunotech) was used to induce apoptosis in Jurkat cells; the following antibodies were used in Western blot analyses: anti-PARP (Biomol), anti-Bcl-2 (DAKO), anti-caspase-3, anti-caspase-6 (R&D Systems), anti-HES1 (Chemicon), anti-Notch1 (Santa Cruz Biotech) and anti-_β_-actin (Sigma). The anti-Notch1 and anti-CD3 monoclonal antibodies were purified by protein G-Sepharose chromatography from supernatants of the btan20 hybridoma (produced by Dr. S Artavanis-Tsakonas, distributed by the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by the University of Iowa, Department of Biological sciences, Iowa City, IA 52242) and the OKT3 hybridoma (American Type Culture Collection), respectively. The Jurkat and 293T cell lines were obtained from the American Type Culture Collection and maintained in RPMI 1640 (Gibco BRL, Burlington, Canada) or DMEM (Gibco BRL) supplemented with 10% fetal calf serum (FCS). The DO11.10 and the BOSC23 cell lines were cultured in DMEM containing 10% FCS. The JIB2 clone allowing inducible Bcl-2 expression was generated as previously described.57
DNA constructs
NICD and deletion mutants (N3, NΔ1) were amplified by PCR from the pcDNA1-Tan1 vector (provided by Dr. S Artavanis-Tsakonas, Massachusetts General Hospital Cancer Center, Charlestown, MA). The following primers were used to clone the NICD and its deletion mutants into the _Eco_RI/_Xho_I sites of the pcDNA3.1 His-A vector (Invitrogen): NICD starting at residue L1768, primer A: 5′-TGC GGG GGA ATT CTG TCC CGC AAG CGC CGG-3′ and F: 5′-GCG GCG CTC GAG CTA CAC GTC TGC CTG GCT CGG CTC-3′; N3, primer E: 5′-GCG GCG GGA ATT CTG CCG CGC GAC ATC GCA CAG GAG-3′ and F; NΔ1, primer C: 5′-CAC CTG GAT GGA ATT CTG CGC ATG TCT GCC ATG GCC-3′ and F. All the constructs were sequenced to confirm the absence of mutations. The plasmid encoding the GST-NICD fusion was generated as follows: using the primers 5′-ACT GAA TTC GCT GCT GTC CCG CAA G-3′ and 5′-CCG CTC GAG TCG GCT CTC CAC TCA GGA AG-3′, the NICD was amplified from the pcDNA1-Tan1 vector and cloned into pCRII-TOPO (Invitrogen). The NICD was then subcloned into the _Eco_RI and _Xho_I restriction sites of the pGEX-4T3 vector. The full-length cDNA encoding NR4A1, the human ortholog of the murine Nur77 (hNur77), was cloned from activated primary T cells by RT-PCR into the pCMVTAG5a vector using the restriction sites present in the primers 5′-TAT GGA TCC ATG ATG CCC TGT ATC CAA GCC CAA TA-3′ and 5′-TAT TCT CGA GGA AGG GCA GCG TGT CCA TGA AGA-3′. The Nur77 reporter plasmid (NurRE3-Luc) as well as the control vector (POMC-Luc) provided by Dr. J Drouin (Clinical Research Institute of Montreal, Québec, Canada) are described elsewhere.58 The _Sac_I/_Xba_I fragment of the PARP cDNA (obtained from Dr. G Poirier, Laval University, Québec, Canada) was inserted into the pBS-KS+ vector to produce the pBS-PARP construct.
Retroviral production and cell transduction
The various forms of Notch1 were subcloned by PCR from the pCDNA3.1 vector into the AP2 retroviral construct using the primers 5′-TAC CAG ATC TTA ATA CGA CTC ACT ATA GGG-3′ and 5′-TCA TAG ATC TTA GAA GGC ACA GTC GAG G-3′ containing the _Bgl_II site. Using the calcium phosphate precipitation method, BOSC23 cells were transfected with the AP2 vectors containing the NICD, NΔ1 and N3 cDNA or the NICD cloned in the antisense direction (NICDAS) as a control. After 48 h of culture, cell supernatants were harvested, supplemented with 1 _μ_g/ml of polybrene and added to DO11.10 cells before a spin-infection for 90 min at 2200 r.p.m. GFP-positive cells were sorted 48 h after infection and the expression of Notch1 forms was confirmed by Western blot using the anti-Notch1 antibody btan20.
Cell stimulation and apoptosis assay
Jurkat cells were suspended at 106 cells/ml in the absence or presence of 250 ng/ml of anti-Fas antibody (CH11). After 5 h of incubation at 37°C, an aliquot was used for Annexin V staining (as described previously59) and the remaining cells were washed in PBS and lysed in SDS–PAGE sample buffer. The JIB2 cells were treated similarly after incubation for 36 h in the presence or absence of 1 _μ_g/ml of Dox to induce Bcl-2 expression. Human PBMCs were purified from whole blood by Ficoll gradient and were stimulated by the addition of 1 _μ_g/ml of OKT3 and 20 U/ml of IL-2. Following 24 h of stimulation, cells were resuspended at 106 cells/ml in RPMI containing 10% FCS and 200 U/ml of IL-2. After 3 days, they were placed for 16 h in plates coated with 1 _μ_g/ml of OKT3 to induce AICD, or with 250 ng/ml of anti-Fas antibody (CH11) to trigger directly apoptosis, which was quantified by the Annexin V assay. AICD was induced in DO11.10 cell lines expressing either the full-length NICD or the deletion mutants by plating 106 cells per well in duplicate in 24-well plates coated with 1 _μ_g/ml of the anti-CD3 antibody 145-2C11 followed by incubation for 8 h at 37°C. Stimulated cells were then washed with PBS, fixed in 5% formaldehyde and stained in PBS containing 1 _μ_g/ml of Hoechst 33342 (Molecular Probes) for 10 min in the dark. After washing with PBS, stained cells were analyzed using an Axioplan 2 microscope (Zeiss) under UV light. The percentage of apoptotic nuclei exhibiting chromatin condensation was calculated after counting 100–200 cells in each duplicate sample.
Cotransfection assay
Using the calcium phosphate precipitation method, 293T cells were transfected in triplicate, with 2 _μ_g of the pCMV-hNur77 vector, 0.1 _μ_g of the reporter plasmid NurRE3-Luc and 2 _μ_g of pcDNA3.1 vector containing either the wild-type NICD, the various deletions or the GST as a control. The vector pUT535 constitutively expressing beta-galactosidase was also cotransfected to standardize the samples for transfection efficiency and a carrier vector (pBS-PARP) was used to transfect the same amount of total DNA under different conditions. After 48 h of culture, the cells were harvested, lysed in 300 _μ_l of Reporter Lysis Buffer (Promega) and beta-galactosidase and luciferase activities were determined.
Western blot analysis
Following stimulation, cells were lysed directly in Laemli sample buffer and the lysates were boiled for 5 min. Proteins were then separated using 12% SDS–PAGE and transferred to PVDF membranes. After a blocking step in PBS containing 5% nonfat dry milk and 0.05% Tween -20, membranes were incubated for 1 h with primary antibodies in blocking buffer. After three washes and incubation with HRP-coupled secondary antibodies, the proteins were detected using the enhanced chemiluminescence (ECL) kit (Amersham, Oakville, Canada).
In vitro transcription/translation and cleavage assay
The coupled transcription/translation of the cDNAs encoding PARP and NICD were carried out with the TNT Coupled Reticulocyte Lysate System (Promega, Madison, WI, USA) as previously described.60 For in vitro cleavage by recombinant caspases, 2 _μ_l of 35S-labeled proteins was incubated with 10 U of caspase-3 or -6 in caspase buffer (25 mM Hepes pH 7.5, 5 mM EDTA, 2 mM DTT, 0.1% CHAPS, 10% sucrose). Cleavage products were fractionated on 12% SDS–PAGE and analyzed by autoradiography. Cell lysates used for in vitro cleavage assays were prepared by four freeze–thaw cycles of 5 × 106 cells in 100 _μ_l of caspase buffer with 2% sucrose and protease inhibitors. 35S-labeled proteins were incubated for 1 h at 37°C with 50 or 5 _μ_g of proteins obtained from apoptotic Jurkat cells or apoptotic PBMCs, respectively.
Amino-terminal sequence analysis of Notch1 fragments
The expression of the GST-NICD fusion was induced in Escherichia coli cultures (BL21 strain) by adding 0.1 _μ_M of isopropyl-_β_-D-thiogalactopyranoside (IPTG) for 1 h at 30°C. After centrifugation, the bacteria were lysed by sonication in ice-cold PBS containing protease inhibitors (Roche). The supernatants of this lysate were incubated for 1 h at 4°C in the presence of glutathione-Sepharose 4b beads (Pharmacia). After centrifugation, the beads were washed in ice-cold PBS and kept at 4°C. For digestion by caspases, the beads were incubated in a 50 _μ_l volume with 50–100 U of caspase-3, -6 or -7 for 1 h at 37°C. The reactions were stopped by adding 50 _μ_l of 2 × Laemli buffer and by 5 min incubation at 100°C. After separation of the fragments on a 12% acrylamide gel, they were transferred to a PVDF membrane, stained with Coomassie blue and cut from the membranes for N-terminal sequence analysis using a Procise 494 cLC sequencer (Applied Biosystems) operated following the manufacturer's protocol.
Abbreviations
AICD:
activation-induced cell death
CSL:
CBF-1/Suppressor of Hairless/Lag-1
DSL:
Delta/Serrate/Lag-2
FasL:
Fas-ligand
NBD:
NF-_κ_B binding domain
NICD:
Notch1 intracellular domain
PBMCs:
peripheral blood mononuclear cells
RIP:
regulated intramembrane proteolysis
TACE:
TNF_α_-converting enzyme
TAD:
transactivation domain
SATB1:
special AT-rich binding protein 1
References
- Artavanis-Tsakonas S, Rand MD and Lake RJ (1999) Notch signaling: cell fate control and signal integration in development. Science 284: 770–776
Article CAS PubMed Google Scholar - Ebinu JO and Yankner BA (2002) A RIP tide in neuronal signal transduction. Neuron 34: 499–502
Article CAS PubMed Google Scholar - Blaumueller CM, Qi H, Zagouras P and Artavanis-Tsakonas S (1997) Intracellular cleavage of Notch leads to a heterodimeric receptor on the plasma membrane. Cell 90: 281–291
Article CAS PubMed Google Scholar - Schroeter EH, Kisslinger JA and Kopan R (1998) Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 393: 382–386
Article CAS PubMed Google Scholar - Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R and Israel A (1995) Signalling downstream of activated mammalian Notch. Nature 377: 355–358
Article CAS PubMed Google Scholar - Brenner M (2000) To be or notch to be. Nat. Med. 6: 1210–1211
Article CAS PubMed Google Scholar - Robson MacDonald H, Wilson A and Radtke F (2001) Notch1 and T-cell development: insights from conditional knockout mice. Trends Immunol. 22: 155–160
Article Google Scholar - Pui JC, Allman D, Xu L, DeRocco S, Karnell FG, Bakkour S, Lee JY, Kadesch T, Hardy RR, Aster JC and Pear WS (1999) Notch1 expression in early lymphopoiesis influences B versus T lineage determination. Immunity 11: 299–308
Article CAS PubMed Google Scholar - Radtke F, Wilson A, Stark G, Bauer M, van Meerwijk J, MacDonald HR and Aguet M (1999) Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity 10: 547–558
Article CAS PubMed Google Scholar - Schmitt TM and Zuniga-Pflucker JC (2002) Induction of T cell development from hematopoietic progenitor cells by delta-like-1 in vitro. Immunity 17: 749–756
Article CAS PubMed Google Scholar - Washburn T, Schweighoffer E, Gridley T, Chang D, Fowlkes BJ, Cado D and Robey E (1997) Notch activity influences the alphabeta versus gammadelta T cell lineage decision. Cell 88: 833–843
Article CAS PubMed Google Scholar - Ciofani M, Schmitt TM, Ciofani A, Michie AM, Cuburu N, Aublin A, Maryanski JL and Zuniga-Pflucker JC (2004) Obligatory role for cooperative signaling by pre-TCR and Notch during thymocyte differentiation. J. Immunol. 172: 5230–5239
Article CAS PubMed Google Scholar - Ellisen LW, Bird J, West DC, Soreng AL, Reynolds TC, Smith SD and Sklar J (1991) TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 66: 649–661
Article CAS PubMed Google Scholar - Pear WS, Aster JC, Scott ML, Hasserjian RP, Soffer B, Sklar J and Baltimore D (1996) Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. J. Exp. Med. 183: 2283–2291
Article CAS PubMed Google Scholar - Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT and Aster JC (2004) Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306: 269–271
Article CAS PubMed Google Scholar - Aster JC, Xu L, Karnell FG, Patriub V, Pui JC and Pear WS (2000) Essential roles for ankyrin repeat and transactivation domains in induction of T-cell leukemia by notch1. Mol. Cell. Biol. 20: 7505–7515
Article CAS PubMed PubMed Central Google Scholar - Deftos ML, He YW, Ojala EW and Bevan MJ (1998) Correlating notch signaling with thymocyte maturation. Immunity 9: 777–786
Article CAS PubMed PubMed Central Google Scholar - Jehn BM, Bielke W, Pear WS and Osborne BA (1999) Cutting edge: protective effects of notch-1 on TCR-induced apoptosis. J. Immunol. 162: 635–638
CAS PubMed Google Scholar - Sade H, Krishna S and Sarin A (2004) The anti-apoptotic effect of Notch-1 requires p56lck-dependent, Akt/PKB-mediated signaling in T cells. J. Biol. Chem. 279: 2937–2944
Article CAS PubMed Google Scholar - Woronicz JD, Calnan B, Ngo V and Winoto A (1994) Requirement for the orphan steroid receptor Nur77 in apoptosis of T-cell hybridomas. Nature 367: 277–281
Article CAS PubMed Google Scholar - Liu ZG, Smith SW, McLaughlin KA, Schwartz LM and Osborne BA (1994) Apoptotic signals delivered through the T-cell receptor of a T-cell hybrid require the immediate-early gene nur77. Nature 367: 281–284
Article CAS PubMed Google Scholar - Calnan BJ, Szychowski S, Chan FK, Cado D and Winoto A (1995) A role for the orphan steroid receptor Nur77 in apoptosis accompanying antigen-induced negative selection. Immunity 3: 273–282
Article CAS PubMed Google Scholar - Salvesen GS and Abrams JM (2004) Caspase activation – stepping on the gas or releasing the brakes?. Lessons from humans and flies. Oncogene 23: 2774–2784
Article CAS PubMed Google Scholar - Liu X, Zou H, Slaughter C and Wang X (1997) DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell 89: 175–184
Article CAS PubMed Google Scholar - Fadeel B, Hassan Z, Hellstrom-Lindberg E, Henter JI, Orrenius S and Zhivotovsky B (1999) Cleavage of Bcl-2 is an early event in chemotherapy-induced apoptosis of human myeloid leukemia cells. Leukemia 13: 719–728
Article CAS PubMed Google Scholar - Kirsch DG, Doseff A, Chau BN, Lim DS, de Souza-Pinto NC, Hansford R, Kastan MB, Lazebnik YA and Hardwick JM (1999) Caspase-3-dependent cleavage of Bcl-2 promotes release of cytochrome c. J. Biol. Chem. 274: 21155–21161
Article CAS PubMed Google Scholar - Deveraux QL, Leo E, Stennicke HR, Welsh K, Salvesen GS and Reed JC (1999) Cleavage of human inhibitor of apoptosis protein XIAP results in fragments with distinct specificities for caspases. EMBO J. 18: 5242–5251
Article CAS PubMed PubMed Central Google Scholar - Widmann C, Gibson S and Johnson GL (1998) Caspase-dependent cleavage of signaling proteins during apoptosis. A turn-off mechanism for anti-apoptotic signals. J. Biol. Chem. 273: 7141–7147
Article CAS PubMed Google Scholar - Rokudai S, Fujita N, Hashimoto Y and Tsuruo T (2000) Cleavage and inactivation of antiapoptotic Akt/PKB by caspases during apoptosis. J. Cell. Physiol. 182: 290–296
Article CAS PubMed Google Scholar - Kurooka H, Kuroda K and Honjo T (1998) Roles of the ankyrin repeats and C-terminal region of the mouse notch1 intracellular region. Nucleic Acids Res. 26: 5448–5455
Article CAS PubMed PubMed Central Google Scholar - Tewari M, Quan LT, O'Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS and Dixit VM (1995) Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell 81: 801–809
Article CAS PubMed Google Scholar - Schlegel J, Peters I, Orrenius S, Miller DK, Thornberry NA, Yamin TT and Nicholson DW (1996) CPP32/apopain is a key interleukin 1 beta converting enzyme-like protease involved in Fas-mediated apoptosis. J. Biol. Chem. 271: 1841–1844
Article CAS PubMed Google Scholar - Armstrong RC, Aja T, Xiang J, Gaur S, Krebs JF, Hoang K, Bai X, Korsmeyer SJ, Karanewsky DS, Fritz LC and Tomaselli KJ (1996) Fas-induced activation of the cell death-related protease CPP32 Is inhibited by Bcl-2 and by ICE family protease inhibitors. J. Biol. Chem. 271: 16850–16855
Article CAS PubMed Google Scholar - Sun XM, Bratton SB, Butterworth M, MacFarlane M and Cohen GM (2002) Bcl-2 and Bcl-xL inhibit CD95-mediated apoptosis by preventing mitochondrial release of Smac/DIABLO and subsequent inactivation of X-linked inhibitor-of-apoptosis protein. J. Biol. Chem. 277: 11345–11351
Article CAS PubMed Google Scholar - Bossy-Wetzel E and Green DR (1999) Caspases induce cytochrome c release from mitochondria by activating cytosolic factors. J. Biol. Chem. 274: 17484–17490
Article CAS PubMed Google Scholar - Adler SH, Chiffoleau E, Xu L, Dalton NM, Burg JM, Wells AD, Wolfe MS, Turka LA and Pear WS (2003) Notch signaling augments T cell responsiveness by enhancing CD25 expression. J. Immunol. 171: 2896–2903
Article CAS PubMed Google Scholar - Hoyne GF, Dallman MJ and Lamb JR (2000) T-cell regulation of peripheral tolerance and immunity: the potential role for Notch signalling. Immunology 100: 281–288
Article CAS PubMed PubMed Central Google Scholar - Palaga T, Miele L, Golde TE and Osborne BA (2003) TCR-mediated Notch signaling regulates proliferation and IFN-gamma production in peripheral T cells. J. Immunol. 171: 3019–3024
Article CAS PubMed Google Scholar - Wong KK, Carpenter MJ, Young LL, Walker SJ, McKenzie G, Rust AJ, Ward G, Packwood L, Wahl K, Delriviere L, Hoyne G, Gibbs P, Champion BR, Lamb JR and Dallman MJ (2003) Notch ligation by Delta1 inhibits peripheral immune responses to transplantation antigens by a CD8+ cell-dependent mechanism. J. Clin. Invest. 112: 1741–1750
Article CAS PubMed PubMed Central Google Scholar - Varnum-Finney B, Xu L, Brashem-Stein C, Nourigat C, Flowers D, Bakkour S, Pear WS and Bernstein ID (2000) Pluripotent, cytokine-dependent, hematopoietic stem cells are immortalized by constitutive Notch1 signaling. Nat. Med. 6: 1278–1281
Article CAS PubMed Google Scholar - Schweisguth F (2004) Notch signaling activity. Curr. Biol. 14: R129–R138
Article CAS PubMed Google Scholar - Gupta-Rossi N, Le Bail O, Gonen H, Brou C, Logeat F, Six E, Ciechanover A and Israel A (2001) Functional interaction between SEL-10, an F-box protein, and the nuclear form of activated Notch1 receptor. J. Biol. Chem. 276: 34371–34378
Article CAS PubMed Google Scholar - Takahashi A, Alnemri ES, Lazebnik YA, Fernandes-Alnemri T, Litwack G, Moir RD, Goldman RD, Poirier GG, Kaufmann SH and Earnshaw WC (1996) Cleavage of lamin A by Mch2 alpha but not CPP32: multiple interleukin 1 beta-converting enzyme-related proteases with distinct substrate recognition properties are active in apoptosis. Proc. Natl. Acad. Sci. USA 93: 8395–8400
Article CAS PubMed PubMed Central Google Scholar - Slee EA, Adrain C and Martin SJ (2001) Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J. Biol. Chem. 276: 7320–7326
Article CAS PubMed Google Scholar - Ruchaud S, Korfali N, Villa P, Kottke TJ, Dingwall C, Kaufmann SH and Earnshaw WC (2002) Caspase-6 gene disruption reveals a requirement for lamin A cleavage in apoptotic chromatin condensation. EMBO J. 21: 1967–1977
Article CAS PubMed PubMed Central Google Scholar - Caulin C, Salvesen GS and Oshima RG (1997) Caspase cleavage of keratin 18 and reorganization of intermediate filaments during epithelial cell apoptosis. J. Cell Biol. 138: 1379–1394
Article CAS PubMed PubMed Central Google Scholar - Byun Y, Chen F, Chang R, Trivedi M, Green KJ and Cryns VL (2001) Caspase cleavage of vimentin disrupts intermediate filaments and promotes apoptosis. Cell Death Differ. 8: 443–450
Article CAS PubMed Google Scholar - Dou S, Zeng X, Cortes P, Erdjument-Bromage H, Tempst P, Honjo T and Vales LD (1994) The recombination signal sequence-binding protein RBP-2N functions as a transcriptional repressor. Mol. Cell. Biol. 14: 3310–3319
Article CAS PubMed PubMed Central Google Scholar - Tun T, Hamaguchi Y, Matsunami N, Furukawa T, Honjo T and Kawaichi M (1994) Recognition sequence of a highly conserved DNA binding protein RBP-J kappa. Nucleic Acids Res. 22: 965–971
Article CAS PubMed PubMed Central Google Scholar - Fryer CJ, Lamar E, Turbachova I, Kintner C and Jones KA (2002) Mastermind mediates chromatin-specific transcription and turnover of the Notch enhancer complex. Genes Dev. 16: 1397–1411
Article CAS PubMed PubMed Central Google Scholar - Galande S, Dickinson LA, Mian IS, Sikorska M and Kohwi-Shigematsu T (2001) SATB1 cleavage by caspase 6 disrupts PDZ domain-mediated dimerization, causing detachment from chromatin early in T-cell apoptosis. Mol. Cell. Biol. 21: 5591–5604
Article CAS PubMed PubMed Central Google Scholar - Alvarez JD, Yasui DH, Niida H, Joh T, Loh DY and Kohwi-Shigematsu T (2000) The MAR-binding protein SATB1 orchestrates temporal and spatial expression of multiple genes during T-cell development. Genes Dev. 14: 521–535
CAS PubMed PubMed Central Google Scholar - Garcia I, Pipaon C, Alemany S and Perez-Castillo A (1994) Induction of NGFI-B gene expression during T cell activation. Role of protein phosphatases. J. Immunol. 153: 3417–3425
CAS PubMed Google Scholar - Wallberg AE, Pedersen K, Lendahl U and Roeder RG (2002) p300 and PCAF act cooperatively to mediate transcriptional activation from chromatin templates by notch intracellular domains in vitro. Mol. Cell. Biol. 22: 7812–7819
Article CAS PubMed PubMed Central Google Scholar - Wansa KD, Harris JM and Muscat GE (2002) The activation function-1 domain of Nur77/NR4A1 mediates trans-activation, cell specificity, and coactivator recruitment. J. Biol. Chem. 277: 33001–33011
Article CAS PubMed Google Scholar - Wang J, Shelly L, Miele L, Boykins R, Norcross MA and Guan E (2001) Human Notch-1 inhibits NF-kappa B activity in the nucleus through a direct interaction involving a novel domain. J. Immunol. 167: 289–295
Article CAS PubMed Google Scholar - Aouad SM, Cohen LY, Sharif-Askari E, Haddad EK, Alam A and Sekaly RP (2004) Caspase-3 is a component of Fas death-inducing signaling complex in lipid rafts and its activity is required for complete caspase-8 activation during Fas-mediated cell death. J. Immunol. 172: 2316–2323
Article CAS PubMed Google Scholar - Maira M, Martens C, Philips A and Drouin J (1999) Heterodimerization between members of the Nur subfamily of orphan nuclear receptors as a novel mechanism for gene activation. Mol. Cell. Biol. 19: 7549–7557
Article CAS PubMed PubMed Central Google Scholar - Alam A, Cohen LY, Aouad S and Sekaly RP (1999) Early activation of caspases during T lymphocyte stimulation results in selective substrate cleavage in nonapoptotic cells. J. Exp. Med. 190: 1879–1890
Article CAS PubMed PubMed Central Google Scholar - Rheaume E, Cohen LY, Uhlmann F, Lazure C, Alam A, Hurwitz J, Sekaly RP and Denis F (1997) The large subunit of replication factor C is a substrate for caspase-3 in vitro and is cleaved by a caspase-3-like protease during Fas-mediated apoptosis. EMBO J. 16: 6346–6354
Article CAS PubMed PubMed Central Google Scholar
Acknowledgements
We thank Dr. S Artavanis-Tsakonas (MGH Cancer Center, Charlestown, MA) for providing the human Notch1 cDNA and Dr. J Drouin (IRCM, Montreal) for the Nur77 reporter construct and the POMC-Luciferase vector. We are grateful to Dr. G Poirier for the PARP cDNA, to Dr. John Hiscott (Lady Davis Institute, Montreal) for kindly providing the neo-CMVt inducible system and to Dr. Irwin D Bernstein (Fred Hutchinson Cancer Research Center) for the Delta1-Myc fusion protein. We are most thankful to Ms. Dany Gauthier (IRCM) for excellent technical assistance. This work was supported by the CIHR (grant # MOP38105). The protein sequencer was purchased with a grant from the Canadian Foundation for Innovation. MB was supported by a fellowship from the MRC of Canada and the Alzheimer's Society of Canada. LS was supported by the FRSQ-FCAR-Santé doctoral research bursary and the Doctoral Research Award from the CIHR. RPS is a recipient of a Canada research Chair in human Immunology and a senior scientist of the CIHR.
Author information
Authors and Affiliations
- Laboratoire d'Immunologie, CR-CHUM, campus St-Luc, Pavillon Edouard-Asselin, 264 Bd. René Lévesque E., Montréal, Québec, Canada, H2X 1P1
L Y Cohen, M Bourbonnière, L Sabbagh, A Bouchard, T Chew, P Jeannequin & R-P Sékaly - Department of Microbiology and Immunology, McGill University, Montreal, Québec, Canada, H3A 2B4
L Sabbagh & R-P Sékaly - Laboratoire de Structure et métabolisme des neuropeptides, Institut de Recherches Cliniques de Montréal, 110 Av. des Pins O, Montréal, Québec, Canada, H2W 1R7
C Lazure
Authors
- L Y Cohen
You can also search for this author inPubMed Google Scholar - M Bourbonnière
You can also search for this author inPubMed Google Scholar - L Sabbagh
You can also search for this author inPubMed Google Scholar - A Bouchard
You can also search for this author inPubMed Google Scholar - T Chew
You can also search for this author inPubMed Google Scholar - P Jeannequin
You can also search for this author inPubMed Google Scholar - C Lazure
You can also search for this author inPubMed Google Scholar - R-P Sékaly
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toL Y Cohen.
Additional information
Edited by BA Osborne
Rights and permissions
About this article
Cite this article
Cohen, L., Bourbonnière, M., Sabbagh, L. et al. Notch1 antiapoptotic activity is abrogated by caspase cleavage in dying T lymphocytes.Cell Death Differ 12, 243–254 (2005). https://doi.org/10.1038/sj.cdd.4401568
- Received: 05 July 2004
- Revised: 20 October 2004
- Accepted: 29 October 2004
- Published: 14 January 2005
- Issue Date: 01 March 2005
- DOI: https://doi.org/10.1038/sj.cdd.4401568