Fast and sensitive multiple alignment of large genomic sequences (original) (raw)
Abstract
Background
Genomic sequence alignment is a powerful method for genome analysis and annotation, as alignments are routinely used to identify functional sites such as genes or regulatory elements. With a growing number of partially or completely sequenced genomes, multiple alignment is playing an increasingly important role in these studies. In recent years, various tools for pair-wise and multiple genomic alignment have been proposed. Some of them are extremely fast, but often efficiency is achieved at the expense of sensitivity. One way of combining speed and sensitivity is to use an anchored-alignment approach. In a first step, a fast search program identifies a chain of strong local sequence similarities. In a second step, regions between these anchor points are aligned using a slower but more accurate method.
Results
Herein, we present CHAOS, a novel algorithm for rapid identification of chains of local pair-wise sequence similarities. Local alignments calculated by CHAOS are used as anchor points to improve the running time of DIALIGN, a slow but sensitive multiple-alignment tool. We show that this way, the running time of DIALIGN can be reduced by more than 95% for BAC-sized and longer sequences, without affecting the quality of the resulting alignments. We apply our approach to a set of five genomic sequences around the stem-cell-leukemia (SCL) gene and demonstrate that exons and small regulatory elements can be identified by our multiple-alignment procedure.
Conclusion
We conclude that the novel CHAOS local alignment tool is an effective way to significantly speed up global alignment tools such as DIALIGN without reducing the alignment quality. We likewise demonstrate that the DIALIGN/CHAOS combination is able to accurately align short regulatory sequences in distant orthologues.
Similar content being viewed by others
Background
Cross-species sequence comparison is playing an increasingly important role in genome analysis and annotation, see [1–3] for review. The functional parts of genomes are under selective pressure, and therefore evolve more slowly than non-functional parts, where random mutations can be tolerated without affecting the evolutionary fitness of the organism. Consequently, conserved sequences often correspond to functional elements. Comparative sequence analysis has been used for a variety of purposes, e.g. gene prediction [4–10], identification of regulatory elements [11–17] and identification of signature sequences to detect pathogene microorganisms [18]. One major advantage of comparative approaches is that they are based on simple measurement of sequence similarity and require little additional information about the features to be detected. While more traditional methods need large sets of training data to construct species-specific statistical models of genes or regulatory elements, comparative methods essentially depend on the availability of syntenic sequences at an appropriate evolutionary distance, making them effective for analysis of newly sequenced genomes, when little training data is available.
In recent years, a number of algorithms have been proposed for pair-wise genomic alignment; these algorithms combine local and global alignment features by returning ordered chains of local similarities. Some approaches use suffix-tree or hashing algorithms to identify pairs of _k_-mers of a certain minimum length (and, possibly, a maximum number of mismatches) [19–21]. These methods are extremely time-efficient but are most effective at aligning sequences from closely related genomes, e.g. from different strains of a bacterium [19]. A more flexible approach has been implemented in the PipMaker [22] set of tools, where a local alignment program implementing a gapped BLAST algorithm, BLASTZ [23], is used.
A sensitive and versatile tool for multiple alignment of distal sequences is DIALIGN [24]. Originally, this approach has been developed to align protein and DNA sequences of limited length, e.g. [25], but in more recent studies the program has also been applied to large genomic sequences. Göttgens et al. [14, 15] used DIALIGN to detect small regulatory sites in vertebrate genome sequences. Fitch et al. identified consensus sequences in pathogen viral genomes based on DIALIGN multiple alignments; these consensus sequences were used to identify sequence signatures for pathogen detection [18]. Unfortunately, the use of DIALIGN for analysis of genomic sequences has been limited by the long program running time: the original algorithm for pair-wise alignment required time proportional to the product of the lengths of the input sequences [26], which is too slow for long sequences.
One way of combining speed and sensitivity for genomic alignment is to use an anchored-alignment approach. In a first step, a fast search tool is used to identify a chain of high-scoring sequence similarities. These similarities are then used as anchor points for the final alignment, where a more sensitive method aligns those regions that are left over between the identified anchor points. Such an approach was initially proposed by Batzoglou et al. [6]. These authors developed GLASS, a system that aligns genomic sequences based on matching _k_-mers. Obviously, the more dense a chain of anchor points is, the higher is the reduction of the search space and gain in speed for the final procedure – on the other hand, too many anchor points could overly restrict the search space, leading to decreased alignment quality. The main challenge in the anchored-alignment approach is therefore to find a trade-off between speed and alignment quality – to locate anchor points that are as dense as possible while still leading to optimal or near-optimal alignments.
Results
In this section we first describe the CHAOS procedure for local alignment of two sequences. We then explain how pair-wise similarities identified by CHAOS can be used as anchor points for pairwise or multiple alignment. Finally, we evaluate our approach in detail, using pair-wise and multiple test data sets.
CHAOS local alignment algorithm
The CHAOS algorithm works by chaining together pairs of similar regions, one from each of the two input DNA sequences; we call such pairs of regions seeds. More precisely, a seed is a pair of words of length k with at least n identical base pairs (bp). A seed s (1) can be chained to another seed s (2) whenever (i) the indices of s (1) in both sequences are higher than the indices of s (2), and (ii) s (1) and s (2) are "near" each other, with "near" defined by both a distance and a gap criteria as illustrated in Figure 1. The final score of a chain is the total number of matching bp in it. The default parameters used by CHAOS are words of length 10, with a degeneracy of one (n = k-1), a distance and gap criteria of 20 and 5 bp respectively, and a score cutoff of 25. The detailed algorithms used for finding seeds and computing the maximal chains are specified in Methods.
Figure 1
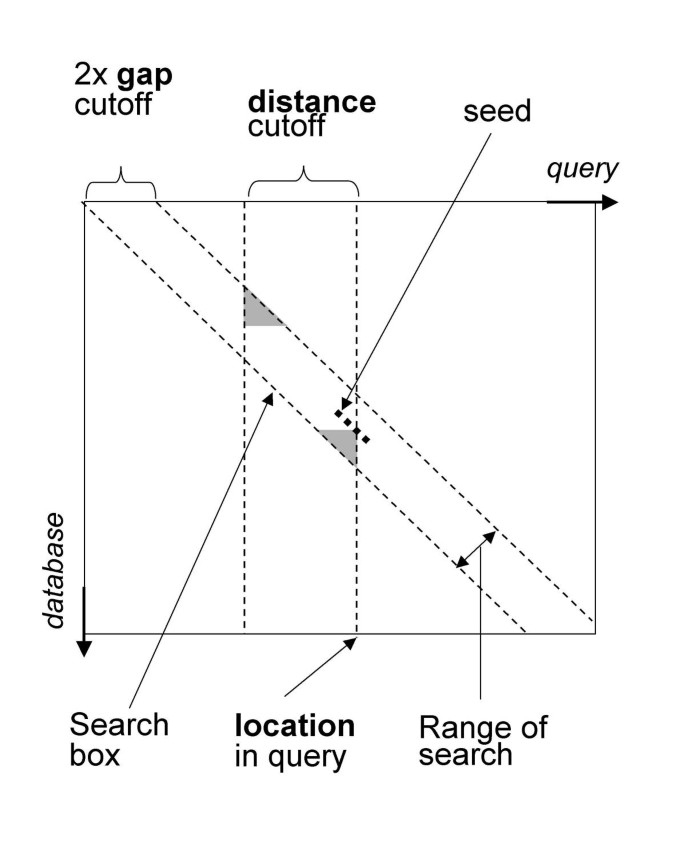
The figure shows a matrix representation of sequence alignment. The seed shown can be chained to any seed which lies inside the search box. All seeds located less then distance bp from the current location are stored in a skip list, in which we do a range query for seeds located within a gap cutoff from the diagonal on which the current seed is located. The seeds located in the grey areas are not available for chaining to make the algorithm independent of sequence order.
After computing the maximal chains, CHAOS scores each chain by using match and mismatch penalties for the letters of each seed. For two seeds seperated by x and base y pairs in the first and second sequences, a gap penalty proportional to |x - y| is incurred. CHAOS throws away chains that score below some threshold t. We augment this scoring method, by adding a rapid rescoring step: chains that score below t are immediately thrown away. Chains that score above t are rescored by performing ungapped extensions in both directions from each seed, and finding the optimal location to insert exactly one gap of size |x - y|. The matches and mismatches can be scored with an arbitrary substitution matrix. CHAOS can be used as a stand-alone program for local sequence alignment or as a pre-processing step to find anchor points for global alignment procedures.
Anchored pair-wise and multiple alignment
In the present study, we use CHAOS to identify chains of local sequence similarities that can be used as anchor points for DIALIGN. Once CHAOS has identified a collection of local alignments for a pair of input sequences, we use an algorithm based on the longest increasing subsequence problem [27] to find the highest scoring chain of local alignments in time O(N log N), where N is the number of local alignments. For pair-wise alignment, this chain is directly used to anchor the DIALIGN alignment as described in [28].
For anchored multiple alignment, we proceed as follows: in a first step, we apply CHAOS to all possible pairs of input sequences; this way we obtain a list of similarities that we consider as candidate anchor points. The problem with these similarities is that they may contradict each other, i.e. it may not be possible to include all of them simultaneously in one single multiple alignment. To solve this consistency problem, we use the same greedy algorithm that DIALIGN uses to find consistent sets of local pairwise alignments in the process of multile alignment calculation [29]. A quality score is associated with each of the identified candidate anchors and the set of all candidate anchors is sorted by these scores. Starting with the highest-scoring one, candidate anchors are accepted one-by-one as final anchor points – provided they are consistent with those candidates that have been accepted previously. Non-consistent similarities are discarded. This way, we finally obtain a consistent set of pair-wise anchor points, i.e. a set of anchor points that would fit into one single multiple alignment, see also [29, 24, 30] where our greedy procedure is explained in the context of the DIALIGN algorithm.
Program Evaluation
It is common practice to evaluate sequence alignment programs by applying them to real-world sequences with known functional sites or 3D structure. For protein alignment, several sets of benchmark sequences are available [31–33]; they are routinely used as standards of truth to evaluate and compare the performance of multiple alignment programs. For pair-wise comparison of genomic sequences, benchmark data have been compiled by Jareborg et al. [12] and Batzoglou et al. [6], these data have been used for comparative gene finding. So far, however, there are no generally accepted reference data with which to evaluate software programs for multiple genomic alignment. Herein, we first use the Jareborg benchmark data to demonstrate that our anchored-alignment procedure improves the running time of DIALIGN by up to two orders of magnitude while the resulting alignments are essentially the same as with the original non-anchored algorithm. Secondly, we apply our method to a set of five genomic sequences around the stem-cell-leukemia (SCL) gene. For all evaluations we start by masking the repeats in the sequences with RepeatMasker. We analyze the resulting multiple alignment in detail and we show that not only is the speed of DIALIGN is improved, but also important functional elements missed by the original DIALIGN can be detected by using the CHAOS anchors. Additional multiple sequence sets are used to demonstrate how the improvement in running time that we achieve depends on the length of the input sequences.
Running time for pair-wise alignment
The Jareborg data set consists of 42 annotated sequence pairs from human and mouse varying in length between less than 6 kb and more than 227 kb, with an average length of 38 kb. These sequences have been used in a paper for a systematic comparison of five different genomic alignment programs [10]. The result of this previous study was that DIALIGN was superior to other methods in terms of alignment quality, but inferior in terms of running time. Since these results have been published previously, we do not repeat the evaluation of DIALIGN for pair-wise alignment. Instead, we focus on how our anchoring procedure affects running time and alignment quality compared with the non-anchored DIALIGN.
We first applied CHAOS to our data in order to obtain chains of anchor points. Next, we aligned the sequence pairs with DIALIGN, first without anchoring and then using the anchor points identified by CHAOS, and we compared the program running time and quality of the resulting alignments. DIALIGN was run with the translation option where local similarity among DNA sequences is compared at the peptide level, see [29]. When CHAOS is run with default parameters the density of the returned anchor points was, on average, 2.1 anchor points per kb. The results in terms of alignment quality and program running time are summarized in Table 1. With a cutoff value of 20 for CHAOS, the program running time of the anchored DIALIGN could be improved by 95% compared to the non-anchored program, while the scores of the resulting alignments were reduced by about 1%. Alignment quality was measured at two distinct levels, (a) by considering the numerical score of the produced alignments and (b) by considering their biological quality. To this end, alignments were compared to annotated protein-coding exons and sensitivity and specificity were measured at the nucleotide level, i.e. a nucleotide that is part of a selected fragment is considered a true positive (TP) if it is also part an annotated exon and as false positives (FP) if it is not; true and false negatives (TN and FN) are defined accordingly. We used the usual measures for prediction accuracy, namely sensitivity = TP/(TP + FN), specificity = TP/(TP + FP), and approximate correlation = 0.5 ((TP/(TP + FN)+(TP/(TP + FP)+(TN/(TN + FP)+(TN/(TN + FN))-1.
Table 1 Total CPU time and alignment quality for DIALIGN (D) and DIALIGN anchored with CHAOS (C+D) applied to a set of 42 pairs of genomic sequences from human and mouse [12]. CHAOS was run with varying cutoff parameters. Lower cutoff values for CHAOS produced higher numbers of anchor points resulting in a decreased search space for the final DIALIGN alignment procedure thus leading to improved running time but slightly decreased alignment quality. The average number of anchor points per kilobase is shown (anc./kb). Score is the total numerical score of all produced DIALIGN alignments, i.e. the sum of the scores of the segment pairs in the alignments. As a rough measure of the biological quality of the produced alignments, we compared local sequence similarities identified by DIALIGN and CHAOS to known protein-coding regions. Here, Sn, Sp and AC are sensitivity, specificity and approximate correlation, respectively. For the D and C+D results, DIALIGN was evaluated by comparing all segment pairs contained in the alignment to annotated exons.
Multiple alignment of the stem-cell-leukemia (SCL) region
To test the combined CHAOS-DIALIGN algorithm for multiple alignment, we used a set of five genomic sequences around the stem cell leukaemia (SCL) gene. SCL is a critical regulator of haematopoiesis, with a pattern of expression that is conserved in all species studied, from mammals to teleost fish [34]. Locations of the exons and of a number of important regulatory regions have been previously experimentally determined. We took SCL sequence from immediately after the upstream gene to the end of the sequence or just after the downstream gene – whichever was longer – in five species: human, mouse, chicken, pufferfish, and zebrafish. We aligned these with DIALIGN, both with and without prior CHAOS anchoring. We then examined the alignments for regions of sequence conservation between all five species.
A total of 265,145 bases were aligned. With a new mixed-alignment option and the -o option, the combined CHAOS-DIALIGN algorithm completed the task in 1 hour and 35 minutes while the non-anchored DIALIGN took 6 hours and 6 minutes. Mixed-alignments means that local similarities are evaluated in two ways, at the nucleotide level and at the peptide level where segments are translated according to the genetic code and the resulting peptide segments are compared. This option is appropriate where genomic sequences are aligned that may contain coding as well as non-coding homologies but it is relatively time consuming. The -o option is used for reduced running time, see the DIALIGN user guide for details. By contrast, if our sequences were compared at the peptide level only, the running time was 13.8 minutes with the anchoring procedure and 49.2 minutes with the non-anchored version of DIALIGN. These test runs were carried out on a Linux PC with a 2.4 GHz Pentium 4 processor. With both program options, the running-time improvement achieved by CHAOS anchoring procedure was more than 70 percent while the numerical score of the output alignments differed by less than 1 percent ('translated' option) and less than 0.1 percent ('mixed alignment' option).
Of the four fish SCL exons, all of which have homologues in the higher species [35, 15], the three coding exons were successfully aligned across all species by both algorithms. The downstream gene, membrane associated protein-17 (MAP17), is not present in pufferfish and contains four, rather short, exons. Moreover, the chicken sequence only extends to the first of these. It is therefore perhaps not surprising that these were only aligned between human and mouse by both algorithms. Within the non-coding DNA, one further region of homology across all species was identified (see Figure 2). This region just upstream of exon 1 has promoter activity in haematopoietic cell lines and also contains a midbrain enhancer [36–38]. Within this region and in all species, CHAOS-DIALIGN perfectly aligned five motifs, each of which is essential for the appropriate pattern or level of SCL transcription [36–39]. Unanchored DIALIGN misaligned the first GATA binding site; otherwise, alignments of the SCL promoter were identical. In the immediate downstream region, within the non-coding exon 1, a further motif was identified by CHAOS-DIALIGN alone. This represents a perfect binding consensus (5 -AANATGGC-3) for the zinc finger transcription factor YY1 [40]. This motif was conserved in all five species and may act as a transcriptional enhancer for the nearby promoter. Alternatively, it may be an RNA-binding element involved in post-transcriptional processing. There is one further non-coding sequence known to be conserved in the five species, but which is not aligned by either DIALIGN algorithm – the AAUAAA polyadenylation sequence [15]. However, previous alignment of this region was only possible with a priori knowledge of its existence and following extraction and local alignment of the relevant sequences. Other multiple alignment algorithms (MAVID [41], LAGAN [42]) also fail to align this region. It is interesting to note that in two cases the CHAOS/DIALIGN combination produces biologically superior alignments than unanchored DIALIGN. This is likely due to the anchor points limiting the search area of DIALIGN and not allowing it to accept a numerically superior alignment that is incorrect biologically.
Figure 2
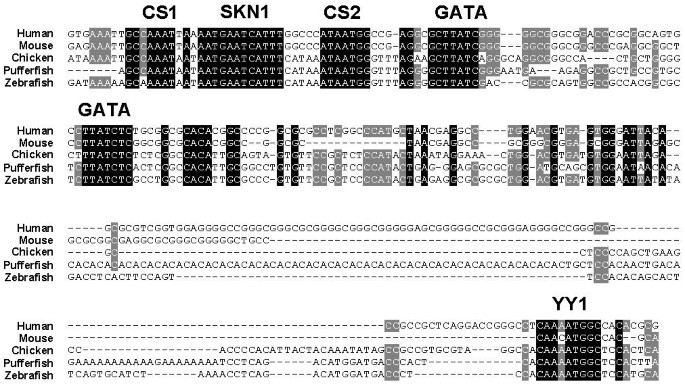
CHAOS-DIALIGN correctly aligns the SCL promoter and a conserved non-coding sequence in exon 1. The alignment was extracted from the CHAOS-DIALIGN global alignment of SCL sequences from human, mouse, chicken, zebrafish, and pufferfish. Consensus binding motifs are labelled. All except YY1 have been previously demonstrated to be essential for the appropriate pattern or level of SCL expression. The factors binding conserved sequence (CS) 1 and 2 are unknown. Shading of bases is at (grey) and (black) conservation.
Running time for longer sequences
We wanted to explore how the relative improvement in program running time that we achieved by our anchoring method depends on the length of the input sequences. The main benefit of reduced running time of DIALIGN is that this way the program becomes applicable to genomic sequences that were previously beyond its scope, so we wanted to estimate the behavior of the running time for very long sequences. It has been previously shown in [42] that given certain assumptions about the distribution of anchor points on the sequences the running time of an anchored alignment algorithm would be linear in the sequence lengths. In reality, it is difficult to predict the distribution of distances between anchor points since this depends, of course, on the sequences being compared. Nevertheless, for our data we could confirm that the relative improvement in running time for pairwise sequences was far more significant for longer sequences than for shorter ones (Figure 3).
Figure 3
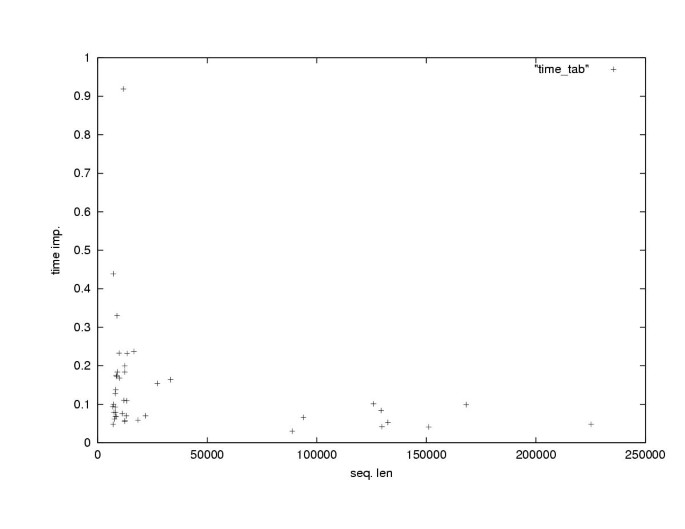
Relative improvement in program running time for 42 pairs of genomic sequences form human and mouse of different length. Each point represents one sequence pair. The _x_-axis is the medium sequence length of sequence pairs while the _y_-axis is the relative running time of the anchored-alignment procedure compared to the non-anchored procedure.
The SCL sequences that we used as a test example for multiple alignment were only 53 kb in length, so we did two additional test runs to test the performance of our approach for longer, multiple sequence sets. First, we applied the anchored and non-anchored procedures to a set of three genomic sequences from human, mouse and dog from the interleukin region [43] with an average length of 222 kb. We used the translation option together with the -o option. Without anchoring, the running time of DIALIGN was 8 h and 36 min ; with anchor points created by CHAOS, the running time was reduced to only 24 min and 40 s, so the CPU time was reduced by more than 95%. At the same time all the annotated features (all exons and known reguatory sequences) were properly aligned. The numerical score of the anchored alignment was 1.5% below the score of the non-anchored alignment. As a third example, we aligned syntenic sequences from human chromosome 20, mouse chromosome 2 and rat chromosome 3 that had an average length of more than 1 MB. The anchored program run terminated after 8 h and 17 min. We did not complete the non-anchored run but based on the first 2 days we estimated that without anchoring, the program would have terminated after 18 days, so for these sequences, the running time was reduced by around 98%.
Discussion
Multiple alignment of large genomic sequences is now a crucial tool for genome data analysis and annotation. Several studies demonstrated that DIALIGN is a highly efficient and versatile tool for this purpose. It has been used to identify biological relevant signals in raw sequence data, such as regulatory elements [14, 16, 44, 45] or protein-coding regions [10] and a new gene-prediction program called AGenDA (A lignment-based Gen e D etection A lgorithm) has been developed that relies on DIALIGN alignments as input information [9, 46]. Most recently, DIALIGN has been succesfully used to identify signature patterns for pathogen microorganisms [18]. However, DIALIGN was originally designed to align protein and short DNA sequences and its application to genomic sequences was severely limited by the long program running time. To make the program applicable to larger sequences, we implemented an anchored-alignment option where pre-defined anchor points can be used to reduce the search space and running time of the alignment procedure. To identify appropriate anchor points, we developed a fast similarity search tool called CHAOS. With the new anchoring option and anchor points created by CHAOS, DIALIGN can now be applied to data sets that were previously beyond its scope.
Most of the methods for heuristic local alignment, such as BLAST [47] and FASTA [48] were developed when the bulk of available sequence were proteins. It has been shown that such algorithms are not as efficient in aligning non-coding sequences [49]. With the new availability of genomic sequences it is appropriate to refine the algorithms used for local alignment so that they more closely reflect the fashion in which the genomic sequences are conserved. Unlike other fast algorithms for genomic alignment, CHAOS does not depend on long exact matches, does not require extensive ungapped homology, and allows mismatches in seeds, all of which are important when comparing distantly related organisms or non-coding regions, where conservation is generally much poorer than in coding areas.
Some previous algorithms for anchored global alignment have worked by first identifying very strong local similarities among the input sequences and adding weaker similarities later. The problem with this approach is that one high-scoring spurious match can lead to a wrong output alignment while many weaker but biologically important homologies may be missed. By contrast, CHAOS searches for the highest scoring chain of local alignments. This way, a numerically high-scoring but biologically wrong local alignment can be conterbalanced by a chain of several weaker local alignments – provided that the total score of these alignments exceeds the score of the one wrong alignment.
We demonstrate that the chains of local alignments returned by CHAOS can be used to anchor the DIALIGN alignment procedure, significantly improving the alignment speed, without affecting the quality of the output alignments. To compare the quality of the anchored and non-anchored alignments, we applied both versions of the program to a database of genomic sequence pairs from human and mouse. We compared the numerical scores of the resulting alignments as well as their biological quality. For multiple genomic alignment, no benchmark data are presently available to compare the perfomance of different alignment algorithms systematically. However, the first step in the DIALIGN multiple-alignment procedure is the pair-wise alignment of all possible pairs of input sequences; fragments of these pair-wise alignments are then used to assemble a multiple alignment. Thus, the results that we obtained for pair-wise alignment can be directly applied to multiple alignment.
We could confirm this in a detailed study of a set of five genomic sequences around the stem cell leukemia (SCL) gene from vertebrates ranging from fish to human. As with our test runs for pair-wise alignment, the anchoring procedure led to a considerable improvement in running time while the output alignments were virtually the same as without anchoring. The numerical scores of the anchored multiple alignments differed by less than 1 percent from the scores of the non-anchored alignments and, again, the biological quality of the anchored alignments was even improved. For the SCL sequences, the improvement in running time was less dramatic than with the human-mouse seuqence pairs used to evaluate the pair-wise alignment procedure. There are two obvious reasons for this result. (a) The SCL sequences are shorter than the sequences used for pair-wise alignment and, as discussed above, the relative improvement in running time increases with sequence length. (b) The SCL sequences are more distantly related than the human-mouse sequence pairs. Thus, the density of anchoring points identified by CHAOS is lower than in the previous examples.
In the SCL example, we demonstrated that our method is able to identify small regulatory elements. It should be mentioned that there are a number of limitations associated with distal species comparisons for the identification of putative regulatory regions. In the SCL locus, many known mammalian enhancers cannot be identified in chicken or fish species [15, 14]. This may be because sequence divergence is so extensive as to mask short regulatory motifs. In support of this is the observation that some functional regions (e.g. exon 1 and the polyA site) could be aligned only with a priori knowledge of their location, extraction of the surrounding sequence, and subsequent local alignment [15]. Alternatively, it may be because regulatory mechanisms differ. An example of this is provided by the enhancer of the IgH locus in catfish, which is capable of activity in mammalian transgenics, but which differs both in its location and critical regulatory motifs between fish and mammals [50]. Where non-coding homology in distal comparisons exists, it is usually a powerful indicator of the presence of a regulatory region. The CHAOS-DIALIGN algorithm was capable of detecting the SCL promoter in a five-way alignment of sequences from human, mouse, chicken, pufferfish, and zebrafish. Furthermore, it correctly aligned all the critical motifs within this region, and a further YY1 motif in exon 1. As discussed above, homology in all five species for this latter motif has only previously been demonstrated following extraction and local alignment of the relevant sequences using DIALIGN [15]. Other multiple alignment algorithms (MAVID [41], LAGAN [42]) fail to align this motif. Therefore, with the SCL dataset, the quality of the CHAOS-DIALIGN output in terms of biological relevance is superior to that of other multiple global alignment tools. It is also better than that of unanchored DIALIGN and, at the same time, the anchored program is between one and two orders of magnitude faster.
Finally, we want to emphasize the need for further work in the general area of multiple alignments. Perhaps the most pressing problem right now is the inability of researchers to evaluate the alignment programs except by looking at examples which have been annotated by biologists. At the same time the methods that simulate evolution of DNA sequences, such as ROSE [51], are unable to create biologically realistic sequences. Thus it is necessary to create some measure of alignment quality that is based on real sequences without biological annotation.
Conclusion
In this paper, we present a fast local pair-wise alignment tool called CHAOS (CHAins Of Seeds); we use this program to speed up the DIALIGN program. For a pair of input sequences, CHAOS returns a chain of local sequence alignments that can be used as anchor points to reduce the search space and running time of any sensitive global alignment procedure: it has also been used for anchoring in the LAGAN [42] alignment tool. We extend the anchoring approach to the problem of multiple alignment of large genomic sequences. Multiple alignments are likely to contain much more information about functional sites than pair-wise alignments, and with the increasing amount of genome sequence data, the development of methods for multiple alignment is a high priority.
Systematic test runs with pair-wise alignments demonstrate that this way the running time of DIALIGN can be reduced by one to two orders of magnitude while the quality of the resulting alignments is only minimally affected. Moreover, the relative improvement in speed increases with the length of the input sequences, making our approach particularly effective for alignment of large genomic sequences.
We also applied CHAOS/DIALIGN to a set of five genomic sequences from human, mouse, chicken, zebrafish, and pufferfish around the stem-cell-leukemia (SCL) locus. Our method correctly aligned three coding exons and five motifs involved in transcription regulation. To make our method easily available for the scientific community, we set up an internet server where CHAOS/DIALIGN can be used through a WWW interface.
Methods
In this section we describe the details of the CHAOS local alignment algorithm.
Finding the seeds
Formally, a seed is a pair of words of length k with at least n identical base pairs (bp). The seeds are located using a simplified version of the Aho-Corasick [52] algorithm. A variation on the trie data structure [53] which we call a threaded trie (T-trie) is used to store the _k_-mers of one sequence. A trie is a tree for storing strings in which there is one node for every common prefix. A node which corresponds to the word w 1...w p would have as its parent a node that corresponds to w 1...w _p_-1. A trie that contains all of the _k_-mers of some string has each leaf at depth k, and each leaf stores all of the locations where this _k_-mer occurs in the indexed sequence.
A T-trie differs from a regular trie in that a node that corresponds to the string w 1...w p will also have a back pointer to the node which corresponds to w 2...w p . We start by inserting into the T-trie all of the _k_-mers of one of the sequences, which we will call the database. Then we do a "walk" using the other, query sequence, where we start by making the root of the T-trie our current node, and for every letter of the query:
1. If the current node has a child corresponding to this letter we make this child our current node, and return any seeds stored in it,
2. Otherwise make the node pointed to by our back pointer our current node, and return to step 1.
As an illustration of why this method works well in practice, assume that all of the possible _k_-mers are present in the database (which is most likely the case). Then, finding the _k_-mers that correspond to the next letter of the query requires only two pointer operations: the first is to follow a back pointer from the k level node which is our current node, the second to follow a down pointer from the resulting node to the appropriate child. Because in practice most k-mers will be present in the database sequence this process will work quickly. To allow degeneracy we permit multiple current nodes, which correspond to the possible degenerate words. It also offers a space saving over the traditional Aho-Corasick automaton as it requires the storage of one rather than four "failure links".
Chaining the seeds
A seed s (1) can be chained to another seed s (2) whenever (i) the indices of s (1) in both sequences are higher than the indices of s (2), and (ii) s (1) and s (2) are "near" each other, with "near" defined by both a distance and a gap criteria as illustrated in Figure 1.
To find the chains of seeds we use the following algorithm. Let D be the maximum distance between two adjacent seeds. The seeds generated while examining the last D base pairs of the query sequence are stored in a skip list, a probabilistic data structure that allows for fast searches and easy in-order traversal of its elements [54]. The seeds are ordered by the difference of its indices in the two sequences (diagonal number). For each seed s found at the current location do a search in the skip list for previously stored seeds which have diagonal numbers within the permitted gap criterion of the diagonal number of s. We thus find the possible previous seeds with which s can be chained. The highest scoring chain is picked, and this chain can be further extended by future seeds. In order to enforce the distance criterion we then remove from the skip list all seeds which were generated D base pairs from the positions of the new seeds, and insert the new seeds into the skip list.
Availability and requirements
The combined CHAOS-DIALIGN software is available online at Göttingen Bioinformatics Compute Server (GoBiCS): http://dialign.gobics.de/chaos-dialign-submission
The source code for CHAOS is available at: http://www.cs.stanford.edu/~brudno/chaos/ together with a PERL script that transforms CHAOS output to the format that can be used to anchor DIALIGN. A version of DIALIGN that accepts such anchors is available at: http://bibiserv.techfak.uni-bielefeld.de/dialign/
Authors Contribution
MB developed CHAOS and drafted parts of the manuscript. MC and BG analyzed the SCL genomic sequences and drafted parts of the manuscript. SB contributed ideas to CHAOS development and rewrote portions of the manuscript. BM implemented the new version of DIALIGN and drafted parts of the manuscript. All authors read and approved the final manuscript.
References
- Miller W: Comparison of genomic DNA sequences: solved and unsolved problems. Bioinformatics 2001, 17: 391–397. 10.1093/bioinformatics/17.5.391
Article CAS PubMed Google Scholar - Frazer KA, Elnitski L, Church DM, Dubchak I, Hardison RC: Cross-species sequence comparisons: A review of methods and available resources. Genome Research 2003, 13: 1–12. 10.1101/gr.222003
Article PubMed Central CAS PubMed Google Scholar - Chain P, Kurtz S, Ohlebusch E, Slezak T: An applications-focused review of comparative genomics tools: capabilities, limitations, and future challenges. Briefings in Bioinformatics 2003, 4: 105–123.
Article CAS PubMed Google Scholar - Gelfand MS, Mironov AA, Pevzner PA: Gene recognition via spliced sequence alignment. Proc Natl Acad Sci USA 1996, 93(17):9061–9066. 10.1073/pnas.93.17.9061
Article PubMed Central CAS PubMed Google Scholar - Bafna V, Huson DH: The conserved exon method for gene finding. Bioinformatics 2000, 16: 190–202. 10.1093/bioinformatics/16.3.190
Article Google Scholar - Batzoglou S, Pachter L, Mesirov JP, Berger B, Lander ES: Human and mouse gene structure: comparative analysis and application to exon prediction. Genome Research 2000, 10(7):950–958. 10.1101/gr.10.7.950
Article PubMed Central CAS PubMed Google Scholar - Korf I, Flicek P, Duan D, Brent MR: Integrating genomic homology into gene structure prediction. Bioinformatics 2001, (17):S140-S148.
- Wiehe T, Gebauer-Jung S, Mitchell-Olds T, Guigó R: SGP-1: Prediction and validation of homologous genes based on sequence alignments. Genome Research 2001, 11: 1574–1583. 10.1101/gr.177401
Article PubMed Central CAS PubMed Google Scholar - Rinner O, Morgenstern B: AGenDA: Gene prediction by comparative sequence analysis. In Silico Biology 2002, 2: 195–205.
CAS PubMed Google Scholar - Morgenstern B, Rinner O, Abdeddaïm S, Haase D, Mayer K, Dress A, Mewes H-W: Exon discovery by genomic sequence alignment. Bioinformatics 2002, 18: 777–787. 10.1093/bioinformatics/18.6.777
Article CAS PubMed Google Scholar - Hardison R, Slightom JL, Gumucio DL, Goodman M, Stojanovic N, Miller W: Locus control regions of mammalian β-globin gene clusters: combining phylo-genetic analyses and experimental results to gain functional insights. Gene 1998, 205: 73–94. 10.1016/S0378-1119(97)00474-5
Article Google Scholar - Jareborg N, Birney E, Durbin R: Comparative analysis of noncoding regions of 77 orthologous mouse and human gene pairs. Genome Research 1999, 9: 815–824. 10.1101/gr.9.9.815
Article PubMed Central CAS PubMed Google Scholar - Loots GG, Locksley RM, Blankespoor CM, Wang ZE, Miller W, Rubin EM, Frazer KA: Identification of a coordinate regulator of interleukins 4, 13, and 5 by cross-species sequence comparisons. Science 2000, 288(5463):136–140. 10.1126/science.288.5463.136
Article CAS PubMed Google Scholar - Göttgens B, Barton LM, Gilbert JGR, Bench AJ, Sanchez MJ, Bahn S, Mistry S, Grafham D, McMurray A, Vaudin M, Amaya E, Bentley DR, Green AR: Analysis of vertebrate SCL loci identifies conserved enhancers. Nature Biotechnology 2000, 18: 181–186. 10.1038/72635
Article PubMed Google Scholar - Göttgens B, Barton L, Chapman M, Sinclair A, Knudsen B, Grafham D, Gilbert J, Rogers J, Bentley DR, Green AR: Transcriptional regulation of the stem cell leukemia gene (SCL) comparative analysis of five vertebrate SCL loci. Genome Res 2002, 12: 749–759. 10.1101/gr.45502
Article PubMed Central PubMed Google Scholar - Göttgens B, Gilbert JGR, Barton LM, Grafham D, Rogers J, Bentley DR, Green AR: Long-range comparison of human and mouse SCL loci: localized regions of sensitivity to restriction endonucleases correspond precisely with peaks of conserved noncoding sequences. Genome Res 2001, 11: 87–97. 10.1101/gr.153001
Article PubMed Central PubMed Google Scholar - Dieterich C, Wang H, Rateitschak K, Krause A, Vingron M: Annotating regulatory DNA based on man-mouse genomic comparison. Bioinformatics 2002, 18: S84-S90.
Article PubMed Google Scholar - Fitch JP, Gardner SN, Kuczmarski TA, Kurtz S, Myers R, Ott LL, Slezak TR, Vitalis EA, Zemla AT, McCready PM: Rapid Development of Nucleic Acid Diagnostics. Proceedings of the IEEE 2002, 90: 1708–1721. 10.1109/JPROC.2002.804680
Article CAS Google Scholar - Delcher LA, Kasif S, Fleischmann AD, Peterson J, White O, Salzberg SL: Alignment of whole genomes. Nucleic Acids Res 1999, 27(11):2369–2376. 10.1093/nar/27.11.2369
Article PubMed Central CAS PubMed Google Scholar - Kurtz S, Schleiermacher C: REPuter : Fast computation of maximal repeats in complete genomes. Bioinformatics 1999, 15(5):426–427. 10.1093/bioinformatics/15.5.426
Article CAS PubMed Google Scholar - Kurtz S, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R: Computation and visualization of degenerate repeats in complete genomes. In Proceedings of the 8th International Conference on Intelligent Systems for Molecular Biology Menlo Parc, CA, AAAI Press 2000, 228–238.
Google Scholar - Schwartz S, Zhang Z, Frazer KA, Smit A, Riemer C, Bouck J, Gibbs R, Hardison R, Miller W: PipMaker–a web server for aligning two genomic DNA sequences. Genome Research 2000, 10: 577–586. 10.1101/gr.10.4.577
Article PubMed Central CAS PubMed Google Scholar - Schwartz S, Kent WJ, Smit A, Zhang Z, Baertsch RHR, Haussler D, Miller W: Human-mouse alignments with BLASTZ. Genome Research 2003, 13: 103–107. 10.1101/gr.809403
Article PubMed Central CAS PubMed Google Scholar - Morgenstern B: DIALIGN 2: improvement of the segment-to-segment approach to multiple sequence alignment. Bioinformatics 1999, 15: 211–218. 10.1093/bioinformatics/15.3.211
Article CAS PubMed Google Scholar - Morgenstern B, Atchley WR: Evolution of bhlh transcription factors: modular evolution by domain shuffling? Mol Biol Evol 1999, 16: 1654–1663.
Article CAS PubMed Google Scholar - Morgenstern B: A simple and space-efficient fragment-chaining algorithm for alignment of DNA and protein sequences. Applied Mathematics Letters 2002, 15: 11–16. 10.1016/S0893-9659(01)00085-4
Article Google Scholar - Gusfield D: Algorithms on Strings, Trees, and Sequences: Computer Science and Computational Biology Cambridge, UK: Cambridge University Press 1997.
Book Google Scholar - Brudno M, Morgenstern B: Fast and sensitive alignment of large genomic sequences. In Proceedings IEEE Computer Society Bioinformatics Conference: 14 – 16 August 2002; Paolo Alto (Edited by: Vicky Markstein and Peter Markstein). IEEE Computer Society 2002, 138–147.
Chapter Google Scholar - Morgenstern B, Dress AWM, Werner T: Multiple DNA and protein sequence alignment based on segment-to-segment comparison. Proc Natl Acad Sci USA 1996, 93: 12098–12103. 10.1073/pnas.93.22.12098
Article PubMed Central CAS PubMed Google Scholar - Abdeddaïm S, Morgenstern B: Speeding up the DIALIGN multiple alignment program by using the 'greedy alignment of biological sequences library' (GABIOS-LIB). Lecture Notes in Computer Science 2001, 2066: 1–11.
Article Google Scholar - McClure MA, Vasi TK, Fitch WM: Comparative analysis of multiple protein-sequence alignment methods. Mol Biol Evol 1994, 11: 571–592.
CAS PubMed Google Scholar - Thompson JD, Plewniak F, Poch O: BAliBASE: A benchmark alignment database for the evaluation of multiple sequence alignment programs. Bioinformatics 1999, 15: 87–88. 10.1093/bioinformatics/15.1.87
Article CAS PubMed Google Scholar - Lassmann T, Sonnhammer ELL: Quality assessment of multiple alignment programs. FEBS Letters 2002, 529: 126–130. 10.1016/S0014-5793(02)03189-7
Article CAS PubMed Google Scholar - Begley CG, Green AR: The SCL gene: from case report to critical hematopoietic regulator. Blood 1999, 93: 2760–2770.
CAS PubMed Google Scholar - Barton LM, Göttgens B, Gering M, Gilbert JG, Grafham D, Rogers J, Bentley D, Patient R, Green AR: Regulation of the stem cell leukemia (SCL) gene: a tale of two fishes. Proc Natl Acad Sci USA 2001, 98: 6747–6752. 10.1073/pnas.101532998
Article PubMed Central CAS PubMed Google Scholar - Bockamp EO, McLaughlin F, Göttgens B, Murrell AM, Elefanty AG, Green AR: Distinct mechanisms direct SCL/TAL-1 expression in erythroid cells and CD34 positive primitive myeloid cells. J Biol Chem 1997, 272: 8781–8790. 10.1074/jbc.272.13.8781
Article CAS PubMed Google Scholar - Bockamp EO, McLaughlin F, Murrell AM, Göttgens B, Robb L, Begley CG, Green AR: Lineage-restricted regulation of the murine SCL/TAL-1 promoter. Blood 1995, 86: 1502–1514.
CAS PubMed Google Scholar - Sinclair AM, Göttgens B, Barton LM, Stanley ML, Pardanaud L, Klaine M, Bahn MGS, Sanchez M, Bench AJ: Distinct 5' SCL enhancers direct transcription to developing brain, spinal cord, and endothelium: neural expression is mediated by GATA factor binding sites. Dev Biol 1999, 209: 128–142. 10.1006/dbio.1999.9236
Article CAS PubMed Google Scholar - Lecointe N, Bernard O, Naert K, Joulin V, Larsen JC, Romeo PH, Mathieu-Mahul D: GATA-and SP1-binding sites are required for the full activity of the tissue-specific promoter of the TAL-1 gene. Oncogene 1994, 9: 2623–2632.
CAS PubMed Google Scholar - Hyde-DeRuyscher RP, Jennings E, Shenk T: DNA binding sites for the transcriptional activator/repressor YY1. Nuc Acids Res 1995, 23: 4457–4465.
Article CAS Google Scholar - Bray N, Pachter L: MAVID multiple alignment server. Nucleic Acids Research 2003, 31: 3525–3526. 10.1093/nar/gkg623
Article PubMed Central CAS PubMed Google Scholar - Brudno M, Do C, Cooper G, Kim MF, Davydov E, NISC Sequencing Consortium, Green ED, Sidow A, Batzoglou S: LAGAN and multi-LAGAN: Efficient tools for large-scale multiple alignment of genomic DNA. Genome Research 2003, 13: 721–731. 10.1101/gr.926603
Article PubMed Central CAS PubMed Google Scholar - Dubchak I, Brudno M, Loots GG, Pachter L, Mayor C, Rubin EM, Frazer KA: Active conservation of noncoding sequences revealed by three-way species comparisons. Genome Research 2000, 10: 1304–1306. 10.1101/gr.142200
Article PubMed Central CAS PubMed Google Scholar - Blanchette M, Schwikowski B, Tompa M: Algorithms for phylogenetic footprinting. Journal of Computational Biology 2002, 9: 211–223. 10.1089/10665270252935421
Article CAS PubMed Google Scholar - Blanchette M, Tompa M: Discovery of regulatory elements by a computational method for phylogenetic footprinting. Genome Research 2002, 12: 739–748. 10.1101/gr.6902
Article PubMed Central CAS PubMed Google Scholar - Taher L, Rinner O, Gargh ASS, Brudno M, Batzoglou S, Morgenstern B: AGenDA: Homology-based gene prediction. Bioinformatics 2003, 19: 1575–1577. 10.1093/bioinformatics/btg181
Article CAS PubMed Google Scholar - Altschul SF, Gish W, Miller W, Myers E-M, Lipman DJ: Basic local alignment search tool. J Mol Biol 1990, 215: 403–410. 10.1006/jmbi.1990.9999
Article CAS PubMed Google Scholar - Pearson WR, Lipman DJ: Improved tools for biological sequence comparison. Proc Natl Acad Sci USA 1988, 85: 2444–2448.
Article PubMed Central CAS PubMed Google Scholar - Bergman CM, Kreitman M: Analysis of conserved noncoding dna in drosophila reveals similar constraints in intergenic and intronic sequences. Genome Research 2001, 11: 1335–1345. 10.1101/gr.178701
Article CAS PubMed Google Scholar - Cioffi CC, Middleton DL, Wilson MR, Miller NW, Clem LW, Warr GW: An IgH enhancer that drives transcription through basic helix-loop-helix and Oct transcription factor binding motifs. Functional analysis of the E(mu)3' enhancer of the catfish. J Biol Chem 2001, 276: 27825–27830. 10.1074/jbc.M100110200
Article CAS PubMed Google Scholar - Stoye J, Evers D, Meyer F: Rose: Generating sequence families. Bioinformatics 1998, 14: 157–163. 10.1093/bioinformatics/14.2.157
Article CAS PubMed Google Scholar - Aho A, Corasick M: Efficient string matching: an aid to bibliographic search. Comm ACM 1975, 18: 333–340. 10.1145/360825.360855
Article Google Scholar - Fredkin E: Trie memory. Comm ACM 1960, 3: 490–500. 10.1145/367390.367400
Article Google Scholar - Pugh W: Skip lists: A probabilistic alternative to balanced trees. Comm ACM 1990, 33: 668–676. 10.1145/78973.78977
Article Google Scholar
Acknowledgements
The authors would like to thank Chuong B. Do for help with CHAOS development, Nadine Werner for assistance with the manuscript, and Inna Dubchak for valuable conversations during this study. Rasmus Steinkamp developed the WWW interface for the CHAOS/DIALIGN software at GoBiCS. MB is supported by the NSF Graduate Research Fellowship. MC and BG are supported by the Wellcome Trust and the Leukaemia Research Fund. The work is partly supported by DFG grant MO 1048/1-1.
Author information
Authors and Affiliations
- Department of Computer Science, Stanford University, Stanford, CA, 94305, USA
Michael Brudno & Serafim Batzoglou - Department of Haematology, University of Cambridge, Cambridge Institute for Medical Research, Hills Road, Cambridge, CB2 2XY, United Kingdom
Michael Chapman & Berthold Göttgens - International Graduate School in Bioinformatics and Genome Research, Universität Bielefeld, Postfach 100131, 33501, Bielefeld, Germany
Burkhard Morgenstern - University of Göttingen, Institute of Microbiology and Genetics, Goldschmidtstr. 1, 37077, Göttingen, Germany
Burkhard Morgenstern
Authors
- Michael Brudno
You can also search for this author inPubMed Google Scholar - Michael Chapman
You can also search for this author inPubMed Google Scholar - Berthold Göttgens
You can also search for this author inPubMed Google Scholar - Serafim Batzoglou
You can also search for this author inPubMed Google Scholar - Burkhard Morgenstern
You can also search for this author inPubMed Google Scholar
Corresponding authors
Correspondence toMichael Brudno or Burkhard Morgenstern.
Authors’ original submitted files for images
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Brudno, M., Chapman, M., Göttgens, B. et al. Fast and sensitive multiple alignment of large genomic sequences.BMC Bioinformatics 4, 66 (2003). https://doi.org/10.1186/1471-2105-4-66
- Received: 01 September 2003
- Accepted: 23 December 2003
- Published: 23 December 2003
- DOI: https://doi.org/10.1186/1471-2105-4-66