A BAC clone fingerprinting approach to the detection of human genome rearrangements (original) (raw)
Abstract
We present a method, called fingerprint profiling (FPP), that uses restriction digest fingerprints of bacterial artificial chromosome clones to detect and classify rearrangements in the human genome. The approach uses alignment of experimental fingerprint patterns to in silico digests of the sequence assembly and is capable of detecting micro-deletions (1-5 kb) and balanced rearrangements. Our method has compelling potential for use as a whole-genome method for the identification and characterization of human genome rearrangements.
Similar content being viewed by others
Background
The phenomenon of genomic heterogeneity, and the implications of this heterogeneity to human phenotypic diversity and disease, have recently been widely recognized [1–5], energizing efforts to develop catalogues of genomic variation [6–12]. Among efforts to understand the role and effect of genomic variability, landmark studies have described changes in the genetic landscape of both normal and diseased genomes [13–15], the presence of heterogeneity at different length scales [5, 16] and variability within normal individuals of various ethnicities [17–19]. Genome rearrangements have been repeatedly linked to a variety of diseases, such as cancer [20] and mental retardation [21], and the evolution of alterations during disease progression continues to be an emphasis of current studies.
Presently, various array-based methods, such as the 32 K bacterial artificial chromosome (BAC) array and Affy 100 K SNP array [21–23], are the most common approaches to detecting and localizing copy number variants, which are one class of genomic variation. The ubiquity of arrays is largely due to the fact that array experiments are relatively inexpensive, and collect information genome-wide. The advent of high-density oligonucleotide arrays, with probes spaced approximately every 5 kb, has increased the resolution of array methods to about 20-30 kb (multiple adjacent probes must confirm an aberration to be statistically significant) [21]. Despite their advantages, commonly available array-based methods have several shortcomings. These include the inability to: detect copy number neutral variants, such as balanced rearrangements; precisely delineate breakpoints and other fine structure details of genomic rearrangements; and directly provide substrates for functional sequence-based characterization once a rearrangement has been detected.
Clone-based approaches have been developed to study genome structure, in part motivated by shortcomings of array-based methods [16, 24, 25]. In addition to their use in identifying both balanced and unbalanced rearrangements, clones have the potential to be directly used as reagents for downstream sequence characterization and cell-based functional studies [24]. Despite the advantages of clone-based methods, relatively few studies have reported their use for detecting and characterizing genomic rearrangements. End sequences from fosmid clones have been compared to the human reference genome sequence to catalogue human genome structural variation [16]. End sequence profiling (ESP) [25], which uses BAC end sequences, has been used to study genomic rearrangements in MCF7 breast cancer cells [24]. The principal drawbacks of clone-based methods are cost and speed of data acquisition. For example, in the case of end sequencing approaches that sample only the clone's termini, deeply redundant clone sampling would be required to approach coverage of the human genome. This might require millions of clones and end sequences. More tractable might be an approach capable of sampling the entire insert of a clone rather than only the ends, thereby enhancing coverage of the target genome with fewer sampled clones. Clone coverage of the human genome could then be achieved with only a small fraction of the clones required to achieve comparable genome coverage in clone end sequences.
One method for sampling clone inserts is restriction fragment clone fingerprinting, which has been used by us and others to produce redundant clone maps of whole genomes [21, 26–30]. Whole-genome clone mapping projects have shown that it is possible to achieve saturation of mammalian genome coverage with 150,000-200,000 fingerprinted BACs, with the number of BACs required inversely proportional to BAC library insert sizes. This relatively tractable number of clones suggests that whole genome surveys using BAC fingerprinting are feasible. What is not known is whether fingerprints are capable of identifying clones bearing genome rearrangements. In this study we address this question using computational simulations and fingerprint analysis of a small number of BAC clones, previously characterized by ESP. We collected restriction enzyme fingerprints from a set of 493 BACs that represented regions of the MCF7 breast cancer cell line genome. Using an alignment algorithm we developed (called fingerprint profiling (FPP)), we fingerprinted clones and aligned these fingerprints to locations on the reference genome sequence and used the alignment profiles to detect candidate genomic rearrangements. Our analysis reveals fingerprint analysis can detect small focal rearrangements and more complex events occurring within the span of a single clone. By varying the number of fingerprints collected for a clone, the sensitivity of FPP can be tuned to balance throughput with satisfactory detection performance. We also show that FPP is relatively insensitive to certain sequence repeats. Our analysis is compatible with the concept of using clone fingerprinting to profile entire genomes in screens for genome rearrangements.
Results
We explored the utility of FPP for the identification of genome rearrangements. The method involved generating one or more fingerprint patterns by digesting clones with several restriction enzymes, and comparing these patterns to in silico digests of the reference human genome sequence. Differences detected in this comparison identified the coordinates of candidate genome rearrangements.
Restriction enzyme selection
We analyzed the distribution of recognition sequences for 4,060 restriction enzyme combinations (Figure 1) on human chromosome 7 (Materials and methods). From this, we identified five restriction enzyme combinations of potential utility for FPP. All five combinations included _Hin_dIII and _Eco_RI, and one of: _Bcl_I/_Bgl_II/_Pvu_II, _Bal_I/_Bcl_I/_Bgl_II, _Nco_I/_Pvu_II/_Xba_I, Bcl/_Nco_I/_Pvu_II, or _Bgl_II/_Nco_I/_Pvu_II. Each of these combinations represented at least 99.98% of the chromosome7 sequence in restriction fragments of sizes that are generally accurately determined using our BAC clone fingerprinting method. Ultimately, we selected the combination _Hin_dIII/_Eco_RI/_Bgl_II/_Nco_I/_Pvu_II for its desirable cut site distribution, ease of use in the laboratory and our favorable experience with the high quality of fingerprints from these enzymes.
Figure 1
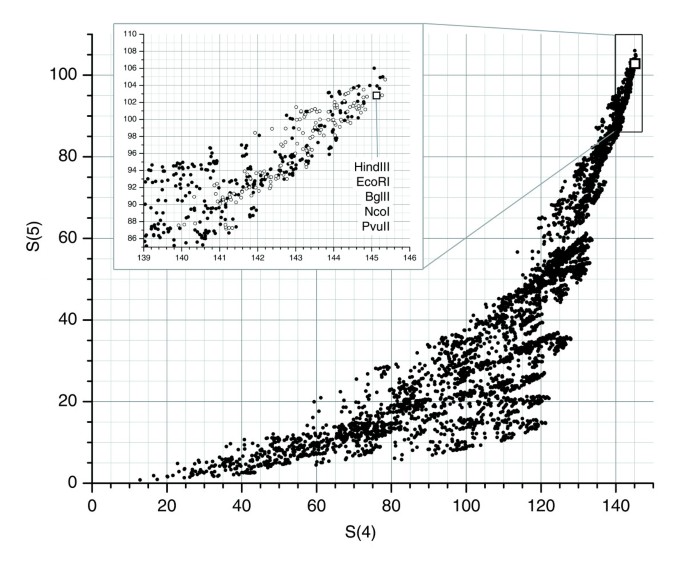
Desirability ranking of 4,060 five-enzyme combinations. We determined desirability of enzyme combinations based on S(n), defined as the fraction of the chromosome 7 that is represented by restriction fragments in the range 1-20 kb (a subset of our sizing range within which sizing accuracy is increased) for ≥n enzymes. Enzyme combinations with high values of S(n) are desirable because a large fraction of fragments in their fingerprint patterns can be accurately sized and because the number of large fragment covers found in regions represented exclusively by large fragments in all digests is minimized. Points represented by hollow glyphs correspond to enzyme combinations which achieved rank in top 10% for each of S(n = 1..5).
Theoretical sensitivity of fingerprint alignments
To demonstrate that fingerprint patterns are sufficiently complex to uniquely identify genomic intervals, we devised in silico simulations to determine specificities of fingerprint fragments and patterns and to align virtual clones with simulated rearrangement breakpoints to the reference genome sequence.
We computed the fragment specificity for a given fragment as the fraction of fragments in the genome that are experimentally indistinguishable in size (Materials and methods). Figure 2 shows the specificity for an individual _Hin_dIII fragment of a given size in the human genome (hg17), and depicts the practical specificity where experimental sizing error is used to determine whether fragment sizes can be distinguished. Our sizing error depends on fragment size (Figure 3), effectively dividing the sizing range into approximately 380 unique bins. Also depicted is the case of exact sizing, where fragments are considered indistinguishable only if their sizes are identical. Although exact sizing is not possible in the laboratory, we include the case of exact sizing here because it represents the theoretical best possible performance of FPP with the enzymes we selected, and because it helps to contrast FPP's practical performance.
Figure 2
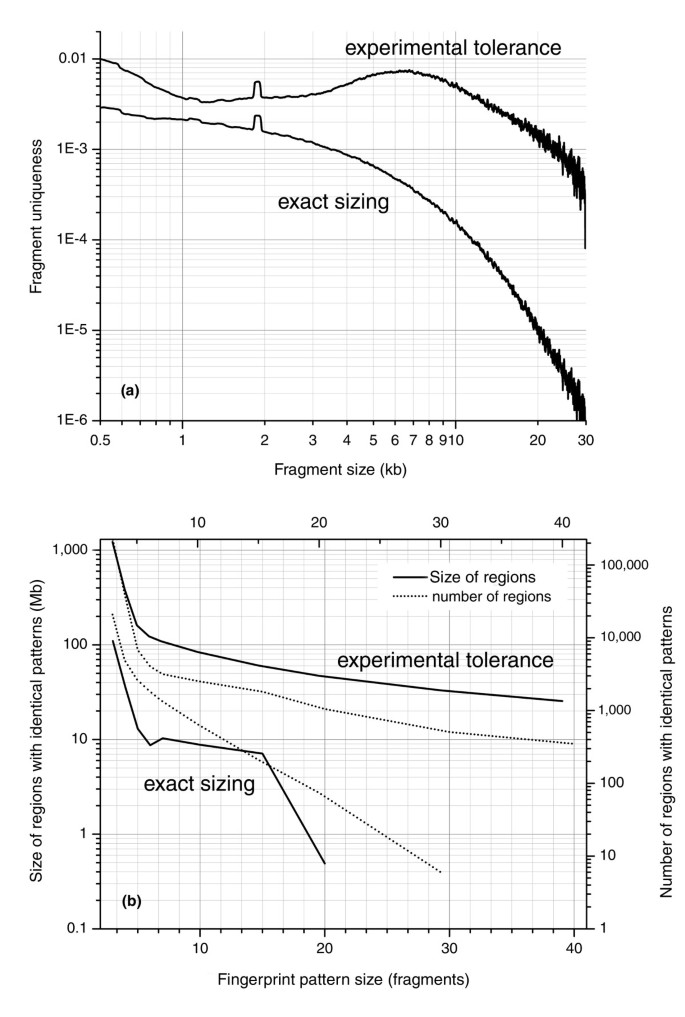
Specificity of individual restriction fragments and patterns based on exact and experimental sizing tolerance. (a) _Hin_dIII restriction fragment specificity for the human genome for fragments within the experimental size range of 500 bp to 30 kb. For a given fragment size, the vertical scale represents the fraction of fragments in the genome that are indistinguishable by size in the case of either exact sizing (fragments in common between two fingerprints must be of identical size) or within experimental tolerance (fragments in common between two fingerprints must be within experimental sizing error; Figure 3) on a fingerprinting gel. When sizing is exact, fragment specificity follows approximately the exponential distribution of fragment sizes and spans a range of 3.5 orders of magnitude. When experimental tolerance is included, the number of distinguishable fragment size bins is reduced and the range of fragment specificity drops to two orders of magnitude. (b) The specificity of a fingerprint pattern of a given size in the human genome. Fingerprint pattern size is measured in terms of number of fragments. Regions with identical patterns are those in which there is a 1:1 mapping within tolerance between all sizeable fragments. The specificity of experimental fingerprint patterns is cumulatively affected by specificity of individual fragments. The specificity of fragments is sufficiently low (that is, due to high experimental precision) so that 96.5% of the genome is uniquely represented by fragment patterns of 8 fragments or more.
Figure 3
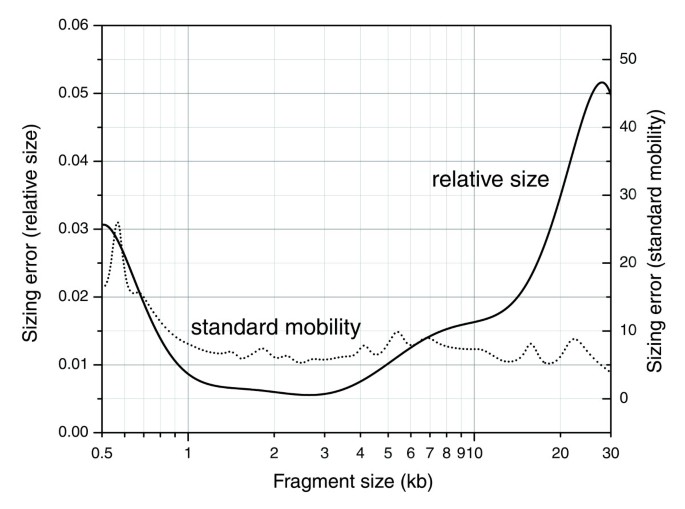
Experimental error of fragment sizing within the 0.5-30 kb sizing range of our single digest protocol. The error is expressed in relative size (left axis) and standard mobility (right axis). Standard mobility is a distance unit that takes into account inter-gel variation and is approximately linear with the distance traveled by the fragment on the gel.
This analysis revealed that _Hin_dIII fingerprints with approximately 15 fragments exhibit a high degree of specificity, as only approximately 1.5% of the genome cannot be uniquely distinguished using patterns composed of this number of fragments. This high specificity results from accurate experimental fragment sizing, and from the fact that the length of genomic repeats is generally much shorter than restriction fragments. Therefore, a specific combination of adjacent fragment sizes represents a relatively unique event in the human genome.
To evaluate the accuracy and sensitivity of actual fingerprint alignments, we performed an in silico study (Materials and methods), in which we computationally generated virtual clones containing simulated genomic rearrangement breakpoints and used these fingerprints as inputs into the alignment algorithm. Figure 4 illustrates the sensitivity and positional accuracy of the mapping of these synthetic clones as a function of the number of digests and segment size. When a single _Hin_dIII fingerprint digest is used, we successfully aligned 50% of 35 kb segments. This cutoff size can be decreased to 25 kb if two digests are used (_Hin_dIII/_Eco_RI) and to 16 kb if five digests are used (_Hin_dIII/_Eco_RI/_Bgl_II/_Nco_I/_Pst_II). The number of digests used has a large impact on the smallest alignable segment size due to the fact that the positions of cut sites of distinct enzymes are generally uncorrelated and that the individual digest patterns can be aligned independently and used together to increase sensitivity. Figure 4 suggests the number of digests that would be required to detect 90% of rearrangements of a certain size. For example, if we wish to identify a breakpoint in 90% of simulated cloned rearrangements, then the shortest rearrangements that can be detected for 1, 2, 3, 4 and 5 digests are 60, 45, 34, 28, and 25 kb, respectively. Stated differently, one can be 90% certain that when using 5 enzymes, a segment of length 25 kb within a BAC would be sufficient to identify the BAC as bearing a genome rearrangement.
Figure 4
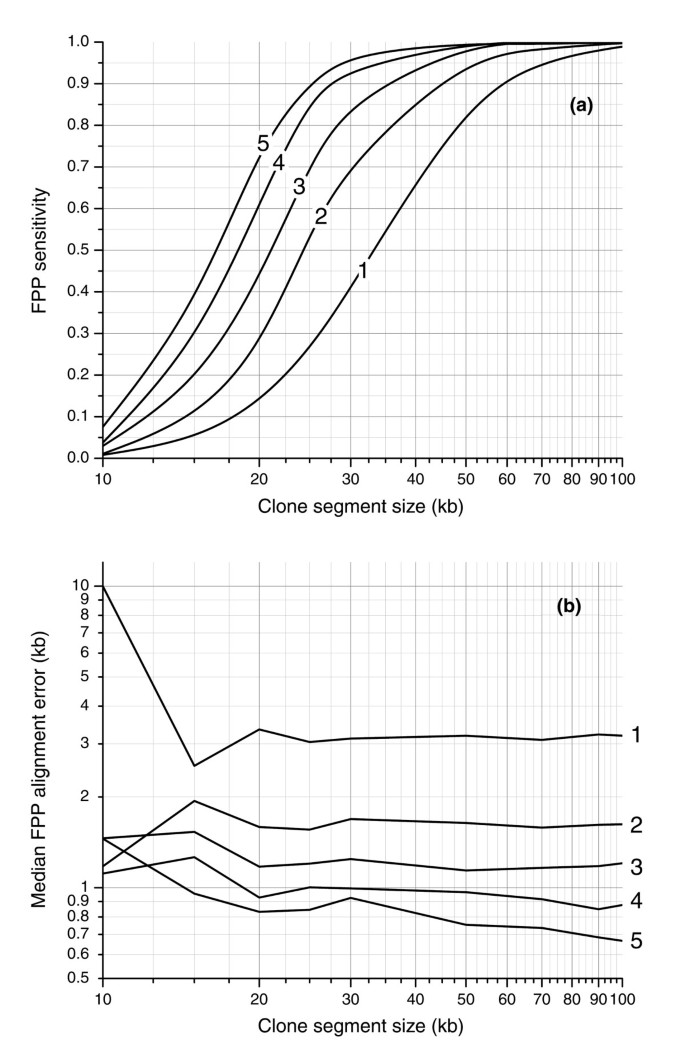
Simulation results of sensitivity and spatial error of rearrangement detection by FPP using experimental sizing tolerance. (a) Sensitivity is measured as the fraction of clone regions of a given size with successful FPP alignments and is plotted for five digests (labeled 1-5). (b) Spatial error is measured by the median distance between FPP and theoretical alignment positions. The largest improvement in both sensitivity and spatial error is realized by migrating FPP from one digest to two. With two fingerprint patterns used to align the clone, 50% of >25 kb clone regions are aligned (90% of >45 kb regions) with a spatial error of 1.7 kb.
Figure 4 shows the median distance between the left and right edges of the alignment and known segment spans for segments of varying sizes. While the values for 10 kb segments are difficult to interpret because of relatively few successful alignments, the error is otherwise constant for segment sizes and depends primarily on the number of digests. The error is 3.0 kb for an alignment based on a single digest and drops to 1.7 kb when two digests are used. When the number of digests is increased to 5, the error drops as low as 700 base-pairs (bp).
MCF7 clone fingerprint-based alignments
With knowledge gained from our simulations, we sought to apply FPP to a test set of 493 BAC clones derived from the MCF7 breast cancer cell line. Each clone was fingerprinted and aligned to the genome with FPP, and the results of the alignments were compared to alignments performed using BAC end sequences (Materials and methods, Additional data file 2). Alignments were evaluated based on their size and number, with multiple alignments indicating identification of a candidate rearrangement. We were able to obtain FPP alignments for 487/493 of the clones. On average, we were able to map 88% of a clone's fingerprint fragments to the genome, and 90% of clones had more than 72% of their fingerprint fragments mapped. Table 1 summarizes FPP and ESP rearrangement detection and Table 2 shows a detailed comparison of rearrangement detection for clones that had an FPP alignment that indicated a breakpoint. The positional accuracy of FPP alignments is shown in Table 3.
Table 1 Comparison of number of rearrangements detected by ESP and FPP in a 487 MCF7 BACs
Table 2 Profile of candidate rearrangements detected by FPP
Table 3 Positional accuracy of FPP alignments
Because ESP uses BAC end sequences that produce data for only the ends of clones, ESP has limited capacity to localize the locations of rearrangement breakpoints within clones. To investigate the precision of FPP in defining the position of breakpoints within BACs, we used clone alignments spanning regions of chromosomes 1, 3, 17 and 20 that contained known breakpoints. We selected these regions because of the enriched coverage provided by our test clone set. The breakpoint position was determined to be the average FPP alignment position with the error given by the standard deviation of the alignments. Additional data file 2 shows the layout of these breakpoints in the MCF7 genome and all FPP and ESP alignments for clones in these regions. Additional data file 3 expands several of the regions from Additional data file 2, and illustrates the relative position of FPP and ESP alignments. Additional data file 6 further increases the detail shown in Additional data file 2, depicting restriction maps and fragment matching status within each clone alignment for all five enzymes. We found 51 breakpoints in 118 unique clones (Table 4). We tested the presence of breakpoints in three clones using PCR (Table 5), and demonstrated the presence of PCR products (Figure 5) to verify fusions within the clone's insert of regions non-adjacent in the reference genome sequence.
Table 4 Location of breakpoints in the MCF7 genome in regions sampled by clones on chromosomes 1, 3, 17 and 20
Table 5 PCR primers used to validate the presence of breakpoints detected by fingerprints
Figure 5
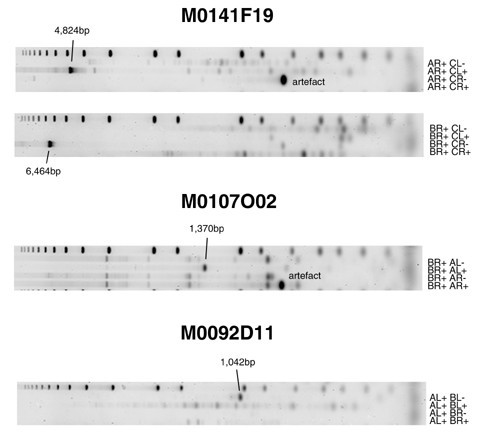
PCR reactions validating the presence of breakpoints in clones listed in Table 5. Each reaction is labeled by the primer combination (e.g. AR+ CL+) used to test order and orientation of the clone's fused regions (Materials and methods; primer combination nomenculature is described in detail in Additional data file 1). The presence of a product demonstrates the adjacency of the regions within the clone's insert.
To demonstrate that FPP can resolve complex rearrangements, we closely examined the FPP results for clone 3F5. In the original MCF7 ESP analysis, Volik et al. [25] concluded that the shotgun sequence assembly of this clone is highly rearranged and composed of five distant regions of chromosomes 3 and 20 (3p14.1, 20q13.2, 20q13, 20q13.3 and 20q13.2). Our FPP analysis generally recapitulated the shotgun sequencing results - out of the five distinct insert segments found by sequencing, we detected four (Figure 6; detailed fingerprint alignments are shown in Additional data file 4; individual restriction fragment accounting is shown in Additional data file 5). The fifth segment, sized at 4,695 bp based on alignment of the clone's sequence to the reference genome, lacked the fragment complexity to confidently identify it by FPP. This small segment includes only two entire restriction fragments (marked with asterisks in the following list of intersecting fragments) in the restriction map of our enzyme combination (_Hin_dIII, 1 fragment (7.4 kb); _Eco_RI, 3 fragments (7.2 kb, 0.9 kb*, 8.5 kb); _Bgl_II, 2 fragments (4.1 kb, 8.6 kb); _Nco_II, 3 fragments (2.0 kb, 1.9 kb*, 6.2 kb); _Pvu_II, 2 fragments (5.8 kb, 13.1 kb)).
Figure 6
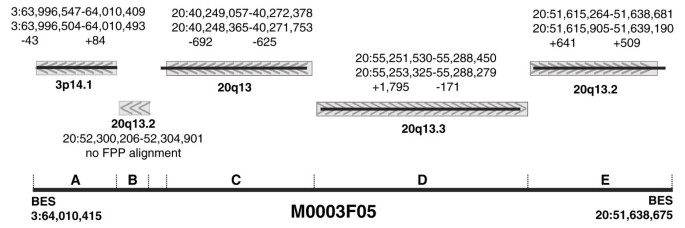
Detailed reconciliation of sequence and fingerprint alignments for clone 3F05, which contains at least four internal breakpoints. FPP is capable of dissecting complex rearrangements in a clone, as illustrated in this figure showing the internal structure of M0003F05. This BAC was sequenced [26] and found to be composed of content from at least five distinct regions (A-E). FPP detected 4/5 of these regions. BLAT (grey rectangles with alignment orientation arrows) and FPP (thin black lines) alignments of M0003F05 are shown; values underneath coordinate pairs are differences in edge positions between BLAT and FPP alignments.
Micro-rearrangements
Fingerprints provide a representation of the entire length of a clone's insert and, thus, are capable of mapping genome rearrangements internal to the clone insert that do not involve the ends of the clone. We identified 17 such small-scale candidate aberrations, and validated 4 of these using PCR (Table 6, Figure 7). PCR analysis of clone 12G17 yielded an amplicon approximately 400 bp smaller than expected, which supports the observation that experimental fragments were approximately 300 bp smaller than expected in this area. The fingerprint results are consistent with a hypothesis of a loss of a 313 bp SINE element evident in the genome sequence for this region. PCR analysis of clone 15O22 indicated an insertion of approximately 560 bp relative to the reference genome sequence. The experimental fragments nearest to the unmatched in silico fragments in this clone's fingerprints are all about 300 bp larger than expected. The results are consistent with a hypothesis of increased copy number of Alu (300 bp) or SINE (100 bp) elements evident in the genome sequence of this region.
Table 6 Location of 17 putative small-scale aberrations identified in MCF7 clones
Figure 7

PCR reactions validating small-scale aberrations listed in Table 6. Reactions are labeled A-D, corresponding to the aberrations with the same label in Table 6. In each case the observed product sizes, shown here, are different from the expected sizes based on the inter-primer distance on the reference sequence.
Discussion
Using computational simulations and restriction fingerprinting of a small number of BAC clones, we assessed the utility of clone fingerprints in detecting genomic rearrangements. We fingerprinted 493 BAC clones derived from the MCF7 breast cancer cell line genome that were previously analyzed by ESP [25]. Using the clone fingerprints, we aligned the clones to the reference genome sequence assembly (UCSC, hg17) and have mapped the candidate positions of 51 rearrangement breakpoints and 17 micro-rearrangements within clones in the set. Further, we identified other rearrangement events within the clone set that were cryptic to ESP.
The use of fingerprints to detect rearrangements provides several advantages, based on the fact that fingerprints sample essentially all of a clone's insert. First, at equivalent sampling depths, the position of a rearrangement breakpoint within a clone can be more precisely determined using FPP than with ESP. Second, fingerprint patterns can be used to locate differences internal to the insert between the clone and the reference genome. This advantage, which is not shared by ESP (Additional data files 2 and 3), can be leveraged to detect small rearrangements such as single nucleotide polymorphisms, micro-deletions, micro-insertions or other local rearrangements. There is currently no experimental method that can be applied on a whole-genome level that is sensitive to the identification of both balanced and unbalanced rearrangements on the order of 1-5 kb in size within the genome. While extremely high-density oligonucleotide arrays can, in principle, detect aberrations with a spatial frequency equal to probe spacing, confirmation of multiple adjacent probes are required to assign statistical significance to the result. Finally, a major strength of fingerprint alignments is their relative insensitivity to sequence repeats. Although approximately 50% of the human genome sequence assembly (hg17) lies in repeat regions, only 7% is found in contiguous repeat units longer than 3.9 kb, which is the average sizeable _Hin_dIII restriction fragment.
Fingerprint-based alignments confirmed a lack of rearrangement in the vast majority of clones (96%) and also confirmed the presence of rearrangements in 68% of those clones in the test set whose ESP data indicated a breakpoint. The high level of confirmation of clone integrity reflects the low incidence of false-positive alignments for clones derived from a single location. The fraction of rearrangements detected is lower than in ESP due to the inherent limitation of fingerprint-based alignments to align small regions of the genome. The use of larger BACs or greater levels of coverage redundancy (Figure 8) would be expected to address a significant portion of these apparent false-negative FPP results.
Figure 8
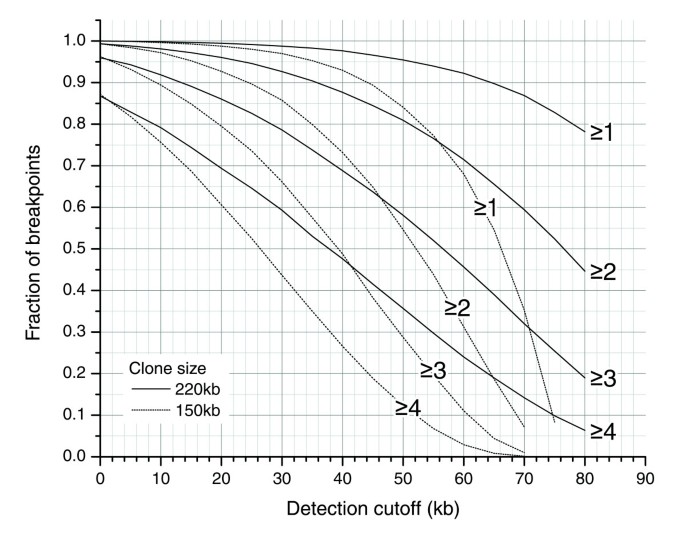
Expected fraction of breakpoints, given five-fold redundant clone coverage, captured by ≥N clones with the distance between breakpoint and clone terminus larger than detection cutoff. The plot shows detection profiles for 150 kb and 220 kb clones. The plot illustrates the benefit of redundant coverage and of using clones with larger inserts - for a given detection cutoff, a breakpoint is captured by significantly more clones on average. The detection sensitivity (Figure 4) needs to be applied to the fraction of breakpoints on this plot (for example, 80% of breakpoints found in ≥2 clones within 50 kb of the ends of the clone; assuming 2 digests, 95% of 50 kb regions can be aligned (Figure 4); therefore, 80% × 0.95 = 76% of breakpoints are expected to be detected in these conditions).
A number of studies (reviewed in [31]) have reported on the increasing prevalence of human genome structural alterations in both healthy and diseased individuals. Much of the work has been done using genome-wide microarray technologies, and the median lengths of many of the structural alterations reported are in the range of tens to hundreds of kilobases or more [32]. These lengths correspond to the resolutions possible using the microarray technologies employed for these studies. The resolving power of the FPP approach we report here improves upon the resolution possible with commonly available microarray platforms, and could easily be applied to whole genome characterization. We believe characterization of tens to hundreds of human genome samples using FPP would provide a powerful data set from which to deduce the lengths and types of genome rearrangements in human populations, as well as providing information on the sequences affected and flanking such rearrangements.
Conclusion
To explore the utility of fingerprint-based rearrangement detection, we used computational simulations and fingerprinted a set of clones derived from the MCF7 breast tumor cell line for which ESP data were available [25]. By collecting multiple fingerprints obtained with different enzymes for each clone and comparing FPP and ESP results for the same clones, we were able to conclude that FPP is well-suited for accurate study of genomic differences. Moreover, we were able to define the boundaries of differences between the reference and MCF7 genomes more precisely than with ESP, and to demonstrate complex rearrangements with FPP that otherwise required BAC shotgun sequencing to fully characterize. Using a set of 493 clones from the MCF7 BAC library sampled primarily to represent content from chromosomes 1, 3, 17 and 20, we used 5 fingerprints to identify 51 breakpoints within the regions sampled by the clones with a median positional error of 2 kb. We were able to reconcile the ESP and FPP data sets and used in silico simulations to explore the practical limitations of FPP. Based on our observations, we feel FPP has compelling potential to be used as a whole-genome method to identify and characterize human genome rearrangements.
Materials and methods
Here we describe the computational and algorithmic components of FPP. The sections broadly comprise generation of target fingerprint patterns and pattern matching, theoretical considerations in generating and using fingerprints for alignment, description of an experimental data set to characterize FPP performance and a detailed description of the FPP algorithm.
_In silico_simulations: sequence assembly digest
We performed in silico simulations to explore the theoretical limitations of using fingerprints to unambiguously identify genomic regions. We used the UCSC May 2004 (hg17) assembly of the human genome for these simulations, using in silico digests of sequence assemblies of each chromosome (1-22, X, Y). For each in silico digest the size and start/end position for all restriction fragments were calculated and stored. To generate virtual clone fingerprints, groups of adjacent restriction fragments were randomly sampled in accordance with a hypothetical clone size distribution. During the sampling process, we avoided regions of the sequence assemblies that contained undetermined base pairs.
_In silico_simulations: fingerprint comparison
We calculated similarity between fingerprint patterns using Needleman-Wunsch global alignment [33]. The similarity of two fingerprint patterns was proportional to the number of fragments that were common between fingerprints being compared. Common fragments were defined as fragments whose sizes were equal within measurement error (Additional data file 1). Such fragments have experimentally indistinguishable electrophoretic mobilities. For an estimate of experimental sizing error, we used values obtained from comparing fingerprints of sequenced BAC clones to their computationally predicted counterparts (Figure 3).
_In silico_simulations: fragment and fingerprint specificity
The degree to which a fingerprint pattern can uniquely represent a genomic region is directly proportional to the efficiency of FPP. See Additional data file 1 for a description of the method used to calculate specificity shown in Figure 2.
_In silico_simulations: enzyme selection
The choice of restriction enzymes affects the effectiveness of FPP - ideal enzymes are those which cut frequently and in a complementary manner, with cut sites of one enzyme populating regions where another enzyme lacks them. See Additional data file 1 for a description of the simulation performed to select an optimal combination of 5 enzymes.
_In silico_simulations: generation of virtual clone fingerprints to determine fingerprint-based alignment accuracy
To determine the theoretical performance of fingerprint-based alignment accuracy and the sensitivity and specificity of rearrangement breakpoint detection, we generated in silico fingerprint patterns of hypothetical clones derived from a genomic region that contained a simulated breakpoint. To simulate a clone harboring a breakpoint, a fingerprint pattern was created by combining two groups of fragments containing fragments totaling N kb and 180-N kb, derived from randomly sampling two non-overlapping regions of the genome. To simulate the restriction fragment that contained the fusion point, two edge fragments, selected randomly from each group, were combined into a single fragment. We generated 384 180-kb synthetic clones for each value of N = 5, 10, 15, 20, 30, 50, 60, 70 and 90 kb and used FPP (see below) to align the fingerprints to the sequence assembly. We quantified the accuracy and detection limits of the alignment method by comparing the alignment results with known clone locations. The positional accuracy of fingerprint-based alignments was evaluated by comparing the difference in position between the fingerprint alignments and the known span of the in silico segments of the synthetic clones.
MCF7 clone test set and end sequencing
We used a subset of BAC clones prepared from MCF7 breast tumor cell line DNA, and identified by S Volik, an author on this study. The average insert size for these clones was 141 kb [25]. We analyzed 493 clones for which paired end sequence alignments to the human sequence assembly (UCSC, hg17) were available [25]. Clone selection was performed by S Volik based on analysis of the alignments of the end sequences to the reference human genome sequence. The set of 493 clones was enriched for clones whose end sequence alignments indicated that the clones identified rearrangements on chromosomes 1, 3, 17 or 20.
MCF7 clone fingerprinting
We attempted to fingerprint each of the 493 clones as described [34]. Five fingerprints were collected for each clone using the combination of restriction enzymes that was identified as optimal: _Hin_dIII (a|agctt), _Eco_RI (g|aattc), _Bgl_II (a|gatct), _Nco_I (c|catgg) and _Pvu_II (cag|ctg). The average clone size of the test set, based on the average sum of fragments in each fingerprint, was 146 kb. This included the 7.5 kb pECBAC1 vector. We obtained a full complement of 5 fingerprints for 484 of the clones, 4 fingerprints for 6 clones, 2 fingerprints for 2 clones and no fingerprints for 1 clone.
Fingerprint profiling
The fingerprinted MCF7 clones were mapped to the reference sequence assembly (UCSC, hg17) by aligning their fingerprints against in silico fingerprints produced computationally from the assembly. The FPP algorithm is composed of four distinct steps: a global search that broadly identifies BAC-sized (or smaller) regions of the genome that yield digest patterns similar to the clone being aligned; a local search that uses a fragment accounting approach to more precisely delineate the correspondence between fragments found in both the clone fingerprint and the assembly within the boundary of each region; an edge detection algorithm that identifies the extent of the alignment; and a partitioning step that finds a minimal set of alignments that maximally account for all clone fragments on the genome.
FPP: global search
The fingerprint-based alignment was performed in two steps, first as a global search across the entire genome, followed by a local search. First, the sequence assembly was digested in silico with the recognition sequences for the same restriction enzymes used to produce the clone fingerprints. Next, 20 kb regions, spaced every 10 kb, were delineated and in silico fragments overlapping a given region (by any fraction of their length) were grouped together into bins. Each region was thus associated with five bins of in silico fragments, with each bin composed of fragments from a different enzyme. Clone fingerprints were then compared to patterns formed by binned fragments for the corresponding enzyme. Each region was assigned a similarity score (sr) that reflected the similarity between the in silico fingerprint and the experimental fingerprint. The individual 20 kb regions were rank-ordered based on their similarity score, and adjacent, possibly overlapping highly scoring regions were grouped together. Region groups were sorted by size and rank-sum.
FPP: local search and alignment edge detection
A local evidence-based search was performed in the neighborhood of 20 highest ranking grouped regions. The purpose of the local search was to identify more precisely the start and end of the region of the genome whose fingerprint pattern matched the clone's fingerprint. While the global search evaluated similarity for bins spaced every 10 kb across the genome, the local search was sensitive to specific positions of restriction enzyme recognition sequences across all digests. Each clone fingerprint was compared with the corresponding in silico fingerprint pattern derived from the area subject to the local search. Fragment covers were defined by the start and end positions of fragments across all digests. Each cover was assigned a score that reflected the extent to which the fragments forming that cover matched the clone fingerprint.
FPP: alignment edge detection
Once each cover was scored, we used a cumulative evidence model to determine the most likely start and end position of the clone fingerprint alignment. The evidence model used a running sum of cover scores (Additional data file 1) across a region. Covers having low similarity lowered the running sum and covers having high similarity increased the sum. Alignment detection was triggered when the sum grew beyond a cutoff value.
FPP: identification of minimal set of alignments
To identify the most likely combination of alignments that mapped the clone insert to the genome, we applied a partitioning model based on rules of parsimony. In addition to one or more true-positive alignments, we expected a certain number of false-positive alignments located in regions of the genome with sufficient fingerprint similarity, but distant from the actual points of origin of the clone. To identify these alignments as false-positive, we used the assumption that these alignments were coincidental and, thus, involved clone fragments independent of those involved in the true-positive alignment. To identify the best combination of alignments, we constructed and scored all possible alignment combinations of up to four alignments. For every alignment in a combination, we tabulated the number of fragments that were unique to that alignment (that is, not participating in other alignments in the combination), and fragments found in one or more alignments in the combination. Any combination for which one or more alignments failed the criteria based on unique and redundant alignment content was not considered. Each alignment was required to have at most 20 kb or 2 fragments of redundant content, which could not be more than 20% of the alignment's length. Each alignment was also required to have more than 7.5 kb and 2 fragments of unique content. Combinations composed of alignments that passed were scored on the basis of the reconstruction fraction, defined as the total size of all alignments in the combination relative to the average fingerprint size of the clone. The highest scoring combination of alignments was designated as the real alignment region.
Comparison of FPP to ESP
Clones with FPP alignments to more than one genome location were considered to harbor a rearrangement. For identifying rearrangements by ESP, we required that the end sequence alignments satisfied one of the following criteria: they had the same orientation (reverse orientation is expected in the normal case); they were separated by more than 500 kb ([25] used the criteria that the observed size be within 3 standard deviations, which was approximately sd = 35 kb, of the library's average insert size); or they aligned to different chromosomes.
ESP data were compared to the FPP alignments to explore the performance characteristics of fingerprint alignments. This comparison was designed to account for the fact that the FPP alignment is a set of one or more spans, while the clone's ESP data are a pair of end sequence alignments that are essentially defined by two points on the genome. Thus, for each of the end sequence alignments, we determined whether there existed an FPP alignment within 50 kb of the end sequence alignment and, if so, the distance between the nearest FPP alignment edge and end sequence alignment. A clone's FPP and ESP data were considered to be in agreement if both end sequence alignments were in the proximity of FPP alignments (Tables 1 and 2).
Identification of micro-aberrations
Fingerprint-based clone alignments were inspected for evidence of potential small-scale aberrations. Localized regions of incongruence between the reference and MCF7 genomes result in unmatched experimental restriction fragments. Such regions are associated with adjacent covers with a zero, or unusually low, cover score sc. The cover score quantifies the level of similarity between the fingerprint patterns of all 5 digests and the in silico pattern of a region of the genome. The cover score is described in greater detail in additional data file 1. We identified these regions by enumerating all unmatched fragments within the FPP alignment bounds for each digest and looking for non-empty intersections of unmatched fragments across all digests.
Validation of aberrations identified by FPP results
For a subset of clones whose FPP alignments indicated a translocation or a local aberration, we designed PCR primers to establish the presence and nature of the aberration. For gross aberrations, such as translocations, we designed primers to form an amplicon across the breakpoint to demonstrate its presence in the clone. For local aberrations, we designed PCR primers (Additional data file 1) spanning the affected region and sought an amplicon of a size different than suggested by primer placement on the reference genome.
Additional data files
The following additional data are available with the online version of this paper. Additional data file 1 provides additional details about the algorithms used to evaluate fingerprint similarity and fragment specificity, to select enzymes, to score fingerprint alignments, to determine alignment edges and to design PCR primers. Additional data file 2 is a figure that shows FPP and BES alignments on regions of chromosomes 1, 3, 17 and 20. Additional data file 3 is a figure that shows a more detailed view of selected regions of chromosomes 1, 3, 17 and 20 from Additional data file 2. Additional data file 4 is a figure that shows a detailed reconciliation of sequence and fingerprint alignments for regions A, C, D and E (Figure 6) of clone 3F05 which contains at least four internal breakpoints. Additional data file 5 is a figure that shows the restriction fingerprint fragment accounting for alignments of 3F05. Additional data file 6 is a figure that shows a high-resolution representation of FPP alignments shown in Additional data file 2.
Abbreviations
BAC:
bacterial artificial chromosome
bp:
base-pairs
ESP:
end sequence profiling
FPP:
fingerprint profiling.
References
- Lupski JR: Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998, 14: 417-422. 10.1016/S0168-9525(98)01555-8.
Article PubMed CAS Google Scholar - Shaw CJ, Lupski JR: Implications of human genome architecture for rearrangement-based disorders: the genomic basis of disease. Hum Mol Genet. 2004, 13 (Suppl 1): R57-64. 10.1093/hmg/ddh073.
Article PubMed CAS Google Scholar - Freeman JL, Perry GH, Feuk L, Redon R, McCarroll SA, Altshuler DM, Aburatani H, Jones KW, Tyler-Smith C, Hurles ME, et al: Copy number variation: new insights in genome diversity. Genome Res. 2006, 16: 949-961. 10.1101/gr.3677206.
Article PubMed CAS Google Scholar - Feuk L, Carson AR, Scherer SW: Structural variation in the human genome. Nat Rev Genet. 2006, 7: 85-97. 10.1038/nrg1767.
Article PubMed CAS Google Scholar - Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M, et al: Large-scale copy number polymorphism in the human genome. Science. 2004, 305: 525-528. 10.1126/science.1098918.
Article PubMed CAS Google Scholar - Smigielski EM, Sirotkin K, Ward M, Sherry ST: dbSNP: a database of single nucleotide polymorphisms. Nucleic Acids Res. 2000, 28: 352-355. 10.1093/nar/28.1.352.
Article PubMed CAS PubMed Central Google Scholar - Strausberg RL, Buetow KH, Greenhut SF, Grouse LH, Schaefer CF: The cancer genome anatomy project: online resources to reveal the molecular signatures of cancer. Cancer Invest. 2002, 20: 1038-1050. 10.1081/CNV-120005922.
Article PubMed CAS Google Scholar - Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C: Detection of large-scale variation in the human genome. Nat Genet. 2004, 36: 949-951. 10.1038/ng1416.
Article PubMed CAS Google Scholar - Thorisson GA, Smith AV, Krishnan L, Stein LD: The International HapMap Project Web site. Genome Res. 2005, 15: 1592-1593. 10.1101/gr.4413105.
Article PubMed CAS PubMed Central Google Scholar - Mills RE, Luttig CT, Larkins CE, Beauchamp A, Tsui C, Pittard WS, Devine SE: An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res. 2006, 16: 1182-1190. 10.1101/gr.4565806.
Article PubMed CAS PubMed Central Google Scholar - Crawford DC, Akey DT, Nickerson DA: The patterns of natural variation in human genes. Annu Rev Genomics Hum Genet. 2005, 6: 287-312. 10.1146/annurev.genom.6.080604.162309.
Article PubMed CAS Google Scholar - Cavalli-Sforza LL: The Human Genome Diversity Project: past, present and future. Nat Rev Genet. 2005, 6: 333-340. 10.1038/nrg1596.
PubMed CAS Google Scholar - Hezel AF, Bardeesy N, Maser RS: Telomere induced senescence: end game signaling. Curr Mol Med. 2005, 5: 145-152. 10.2174/1566524053586563.
Article PubMed CAS Google Scholar - Bisoffi M, Heaphy CM, Griffith JK: Telomeres: prognostic markers for solid tumors. Int J Cancer. 2006, 119: 2255-2260. 10.1002/ijc.22120.
Article PubMed CAS Google Scholar - Fordyce CA, Heaphy CM, Bisoffi M, Wyaco JL, Joste NE, Mangalik A, Baumgartner KB, Baumgartner RN, Hunt WC, Griffith JK: Telomere content correlates with stage and prognosis in breast cancer. Breast Cancer Res Treat. 2006, 99: 193-202. 10.1007/s10549-006-9204-1.
Article PubMed Google Scholar - Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM, Haugen E, Hayden H, Albertson D, Pinkel D, et al: Fine-scale structural variation of the human genome. Nat Genet. 2005, 37: 727-732. 10.1038/ng1562.
Article PubMed CAS Google Scholar - Conrad DF, Andrews TD, Carter NP, Hurles ME, Pritchard JK: A high-resolution survey of deletion polymorphism in the human genome. Nat Genet. 2006, 38: 75-81. 10.1038/ng1697.
Article PubMed CAS Google Scholar - Hinds DA, Kloek AP, Jen M, Chen X, Frazer KA: Common deletions and SNPs are in linkage disequilibrium in the human genome. Nat Genet. 2006, 38: 82-85. 10.1038/ng1695.
Article PubMed CAS Google Scholar - McCarroll SA, Hadnott TN, Perry GH, Sabeti PC, Zody MC, Barrett JC, Dallaire S, Gabriel SB, Lee C, Daly MJ, et al: Common deletion polymorphisms in the human genome. Nat Genet. 2006, 38: 86-92. 10.1038/ng1696.
Article PubMed CAS Google Scholar - Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR: A census of human cancer genes. Nat Rev Cancer. 2004, 4: 177-183. 10.1038/nrc1299.
Article PubMed CAS PubMed Central Google Scholar - Friedman JM, Baross A, Delaney AD, Ally A, Arbour L, Armstrong L, Asano J, Bailey DK, Barber S, Birch P, et al: Oligonucleotide microarray analysis of genomic imbalance in children with mental retardation. Am J Hum Genet. 2006, 79: 500-513. 10.1086/507471.
Article PubMed CAS PubMed Central Google Scholar - Ishkanian AS, Malloff CA, Watson SK, DeLeeuw RJ, Chi B, Coe BP, Snijders A, Albertson DG, Pinkel D, Marra MA, et al: A tiling resolution DNA microarray with complete coverage of the human genome. Nat Genet. 2004, 36: 299-303. 10.1038/ng1307.
Article PubMed CAS Google Scholar - Krzywinski M, Bosdet I, Smailus D, Chiu R, Mathewson C, Wye N, Barber S, Brown-John M, Chan S, Chand S, et al: A set of BAC clones spanning the human genome. Nucleic Acids Res. 2004, 32: 3651-3660. 10.1093/nar/gkh700.
Article PubMed CAS PubMed Central Google Scholar - Volik S, Raphael BJ, Huang G, Stratton MR, Bignel G, Murnane J, Brebner JH, Bajsarowicz K, Paris PL, Tao Q, et al: Decoding the fine-scale structure of a breast cancer genome and transcriptome. Genome Res. 2006, 16: 394-404. 10.1101/gr.4247306.
Article PubMed CAS PubMed Central Google Scholar - Volik S, Zhao S, Chin K, Brebner JH, Herndon DR, Tao Q, Kowbel D, Huang G, Lapuk A, Kuo WL, et al: End-sequence profiling: sequence-based analysis of aberrant genomes. Proc Natl Acad Sci USA. 2003, 100: 7696-7701. 10.1073/pnas.1232418100.
Article PubMed PubMed Central Google Scholar - Gregory SG, Sekhon M, Schein J, Zhao S, Osoegawa K, Scott CE, Evans RS, Burridge PW, Cox TV, Fox CA, et al: A physical map of the mouse genome. Nature. 2002, 418: 743-750. 10.1038/nature00957.
Article PubMed CAS Google Scholar - Krzywinski M, Wallis J, Gosele C, Bosdet I, Chiu R, Graves T, Hummel O, Layman D, Mathewson C, Wye N, et al: Integrated and sequence-ordered BAC- and YAC-based physical maps for the rat genome. Genome Res. 2004, 14: 766-779. 10.1101/gr.2336604.
Article PubMed CAS PubMed Central Google Scholar - Schein JE, Tangen KL, Chiu R, Shin H, Lengeler KB, MacDonald WK, Bosdet I, Heitman J, Jones SJ, Marra MA, et al: Physical maps for genome analysis of serotype A and D strains of the fungal pathogen Cryptococcus neoformans. Genome Res. 2002, 12: 1445-1453. 10.1101/gr.81002.
Article PubMed CAS PubMed Central Google Scholar - McPherson JD, Marra M, Hillier L, Waterston RH, Chinwalla A, Wallis J, Sekhon M, Wylie K, Mardis ER, Wilson RK, et al: A physical map of the human genome. Nature. 2001, 409: 934-941. 10.1038/35057157.
Article PubMed CAS Google Scholar - Marra M, Kucaba T, Sekhon M, Hillier L, Martienssen R, Chinwalla A, Crockett J, Fedele J, Grover H, Gund C, et al: A map for sequence analysis of the Arabidopsis thaliana genome. Nat Genet. 1999, 22: 265-270. 10.1038/10327.
Article PubMed CAS Google Scholar - Altshuler D, Daly M: Guilt beyond a reasonable doubt. Nat Genet. 2007, 39: 813-815. 10.1038/ng0707-813.
Article PubMed CAS Google Scholar - Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, Bailey R, Nejentsev S, Field SF, Payne F, et al: Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007, 39: 857-864. 10.1038/ng2068.
Article PubMed CAS PubMed Central Google Scholar - Needleman SB, Wunsch CD: A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970, 48: 443-453. 10.1016/0022-2836(70)90057-4.
Article PubMed CAS Google Scholar - Schein J, Kucaba T, Sekhon M, Smailus D, Waterston R, Marra M: High-throughput BAC fingerprinting. Methods Mol Biol. 2004, 255: 143-156.
PubMed CAS Google Scholar - Rozen S, Skaletsky H: Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000, 132: 365-386.
PubMed CAS Google Scholar - Schuler GD: Sequence mapping by electronic PCR. Genome Res. 1997, 7: 541-550.
PubMed CAS PubMed Central Google Scholar
Acknowledgements
We gratefully acknowledge the expert technical assistance of the BAC fingerprinting team at the BC Cancer Agency Genome Sciences Centre. This study was supported by grant 5-U01-HG002743-03 from the National Human Genome Research Institute and funding support from The National Cancer Institute of Canada, Genome BC, Genome Canada and the BC Cancer Foundation. M Marra is a senior scholar of the Michael Smith Foundation for Health Research.
Author information
Authors and Affiliations
- BC Cancer Agency Genome Sciences Centre, West 7th Avenue, Vancouver, British Columbia, V5Z 4S6, Canada
Martin Krzywinski, Ian Bosdet, Carrie Mathewson, Natasja Wye, Readman Chiu, Richard Corbett, Matthew Field, Darlene Lee, Trevor Pugh, Asim Siddiqui, Steven Jones, Jacquie Schein & Marco Marra - Cancer Research Institute, University of California at San Francisco, San Francisco, California, 94143-0808, USA
Jay Brebner, Stas Volik & Collin Collins
Authors
- Martin Krzywinski
You can also search for this author inPubMed Google Scholar - Ian Bosdet
You can also search for this author inPubMed Google Scholar - Carrie Mathewson
You can also search for this author inPubMed Google Scholar - Natasja Wye
You can also search for this author inPubMed Google Scholar - Jay Brebner
You can also search for this author inPubMed Google Scholar - Readman Chiu
You can also search for this author inPubMed Google Scholar - Richard Corbett
You can also search for this author inPubMed Google Scholar - Matthew Field
You can also search for this author inPubMed Google Scholar - Darlene Lee
You can also search for this author inPubMed Google Scholar - Trevor Pugh
You can also search for this author inPubMed Google Scholar - Stas Volik
You can also search for this author inPubMed Google Scholar - Asim Siddiqui
You can also search for this author inPubMed Google Scholar - Steven Jones
You can also search for this author inPubMed Google Scholar - Jacquie Schein
You can also search for this author inPubMed Google Scholar - Collin Collins
You can also search for this author inPubMed Google Scholar - Marco Marra
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toMarco Marra.
Additional information
Authors' contributions
MK: data analysis lead, algorithm and software development, manuscript preparation. IB, CM, NW: protocol development, laboratory fingerprint generation. JB: end sequencing and analysis. RC, RC, MF: data analysis. DL: protocol development, laboratory fingerprint generation. TP: PCR and sequencing. SV: ESP data and experiment lead and collaboration. AS, SJ: bioinformatics leads, project management. JS: laboratory lead, project management. MM, CC: principal investigator, laboratory lead, project management. All authors have read and approved the final manuscript.
Electronic supplementary material
13059_2007_1704_MOESM1_ESM.doc
Additional data file 1: Details about the algorithms used to evaluate fingerprint similarity and fragment specificity, to select enzymes, to score fingerprint alignments, to determine alignment edges and to design PCR primers. (DOC 78 KB)
13059_2007_1704_MOESM2_ESM.tiff
Additional data file 2: Regions containing clone alignments are composited horizontally. Alternating vertical white/grey bands indicate disparate regions. Distant alignments of a given clone are joined by horizontal lines. FPP alignments are either black, if validated by BES alignments, or red if no BES alignment is nearby. BES alignments are indicated by small white circles. Blunt termination of FPP alignments is indicative of a breakpoint and these boundaries are denoted by vertical red lines. The boundaries are coded by N(L, R), where N uniquely indexes the breakpoint and L, R indicates whether the breakpoint is on the left or right of the FPP alignment. The breakpoint map generated by FPP alignments is more accurate and complete. FPP alignments captured nearly all breakpoints detected by FPP to a higher degree of spatial accuracy (for example, 3R, 18L). In numerous cases, internal breakpoints were detected by FPP (for example, 7L, 8R). Regions of this figure are presented in greater detail in Figure 7. (TIFF 204 KB)
13059_2007_1704_MOESM3_ESM.tiff
Additional data file 3: Detailed view of selected regions of chromosomes 1, 3, 17 and 20 from Additional data file 2. (TIFF 517 KB)
13059_2007_1704_MOESM4_ESM.tiff
Additional data file 4: Restriction maps for each of the five enzymes (_Eco_RI (e) _Hin_dIII (h) _Bgl_II (g) _Nco_I (n) and _Pvu_II (p)) in the neighborhood of regions of M0003F05 detected by FPP. Restriction map fragments matched by experimental fragments in the corresponding fingerprint are shown in green. The FPP alignment is delineated by black vertical lines and the extent of the BLAT alignment is delineated by blue vertical lines. (TIFF 458 KB)
13059_2007_1704_MOESM5_ESM.tiff
Additional data file 5: Images of individual fingerprints of M0003F05 are accompanied by the image of the nearest marker lane. Fragment sizes are shown to the left of the fragment's band. Correspondence to FPP alignments A, C, D or E (Figure 6) for each fragment is shown. Fragments marked by an asterisk are derived from the digest of the vector. (TIFF 956 KB)
13059_2007_1704_MOESM6_ESM.tiff
Additional data file 6: The figure depicts the restriction map for each of the five digests used to fingerprint the MCF7 clones. Each clone is shown as a set of five rows, with each row corresponding to the local restriction map of _Eco_RI, _Hin_dIII, _Nco_I, _Bgl_II and _Pvu_II (top to bottom). Fragments within each map are colored green if they were experimentally detected, or red if they were not found in the clone's fingerprint. Adjacent fragments with the same match status are shown with alternating brightness to aid in distinguishing the fragments' extent. (TIFF 3 MB)
Authors’ original submitted files for images
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Krzywinski, M., Bosdet, I., Mathewson, C. et al. A BAC clone fingerprinting approach to the detection of human genome rearrangements.Genome Biol 8, R224 (2007). https://doi.org/10.1186/gb-2007-8-10-r224
- Received: 30 April 2007
- Revised: 28 August 2007
- Accepted: 22 October 2007
- Published: 22 October 2007
- DOI: https://doi.org/10.1186/gb-2007-8-10-r224