Draft genome of the living fossil Ginkgo biloba (original) (raw)
Abstract
Background
Ginkgo biloba L. (Ginkgoaceae) is one of the most distinctive plants. It possesses a suite of fascinating characteristics including a large genome, outstanding resistance/tolerance to abiotic and biotic stresses, and dioecious reproduction, making it an ideal model species for biological studies. However, the lack of a high-quality genome sequence has been an impediment to our understanding of its biology and evolution.
Findings
The 10.61 Gb genome sequence containing 41,840 annotated genes was assembled in the present study. Repetitive sequences account for 76.58% of the assembled sequence, and long terminal repeat retrotransposons (LTR-RTs) are particularly prevalent. The diversity and abundance of LTR-RTs is due to their gradual accumulation and a remarkable amplification between 16 and 24 million years ago, and they contribute to the long introns and large genome. Whole genome duplication (WGD) may have occurred twice, with an ancient WGD consistent with that shown to occur in other seed plants, and a more recent event specific to ginkgo. Abundant gene clusters from tandem duplication were also evident, and enrichment of expanded gene families indicates a remarkable array of chemical and antibacterial defense pathways.
Conclusions
The ginkgo genome consists mainly of LTR-RTs resulting from ancient gradual accumulation and two WGD events. The multiple defense mechanisms underlying the characteristic resilience of ginkgo are fostered by a remarkable enrichment in ancient duplicated and ginkgo-specific gene clusters. The present study sheds light on sequencing large genomes, and opens an avenue for further genetic and evolutionary research.
View this article's peer review reports
Similar content being viewed by others
Background
Ginkgo biloba L. is one of the best-known and most distinctive trees, and represents one of the four extant gymnosperm lineages (cycads, ginkgo, conifers and gnetophytes) and has no living relatives. Cycads are widely accepted to be the most closely related lineage [1–4] but the debate continues [5].
Gingko is a living fossil that has remained essentially unchanged in terms of gross morphology for more than 200 million years [6, 7]. It has survived glaciations as a relic in China [8, [9](/article/10.1186/s13742-016-0154-1#ref-CR9 "Fan J, Shi H, Xiao H, Zhao G. Double-Layer 3D Macro-Mesoporous Metal Oxide Modified Boron-Doped Diamond with Enhanced Photoelectrochemical Performance. ACS Appl Mater Interfaces. 2016. doi: 10.1021/acsami.6b01929
.")\] followed by a human-aided global redistribution \[[10](/article/10.1186/s13742-016-0154-1#ref-CR10 "Zhao Y, Paule J, Fu C, Koch MA. Out of China: Distribution history of Ginkgo biloba L. Taxon. 2010;59(2):495–504.")\], and thus provides an inspiring example of how humans can help a species survive and renew. The infamous resilience of ginkgo has enabled it to become widespread and popular across the world. This resilience includes an outstanding resistance or tolerance to both herbivores and pathogens, accounting in part for the longevity of individual trees, and also, in turn, for the longevity of the species \[[11](/article/10.1186/s13742-016-0154-1#ref-CR11 "Major RT. The Ginkgo, the Most Ancient Living Tree. Science. 1967;157(3794):1270–3.")\]. At least three separate defense systems act in ginkgo in response to herbivore attacks: i) repellence and antifeedance due to the presence of flavonoids and terpenic trilactones such as ginkgolides and bilobalides \[[12](/article/10.1186/s13742-016-0154-1#ref-CR12 "Pan L, Ren L, Chen F, Feng Y, Luo Y. Antifeedant Activity of Ginkgo biloba Secondary Metabolites against Hyphantria cunea Larvae: Mechanisms and Applications. PLoS One. 2016;11(5):e0155682.")\]; ii) activation of direct defense genes and the production of plant secondary metabolites including glycosylated flavonoids; iii) emission of specific volatile organic compounds (VOCs) mainly built from terpenes (e.g., sesquiterpenes α-copaene and β-caryophyllene) that potentially activate indirect defenses by attracting predators of browsing insects \[[13](/article/10.1186/s13742-016-0154-1#ref-CR13 "Mohanta TK, Occhipinti A, Atsbaha Zebelo S, Foti M, Fliegmann J, Bossi S, Maffei ME, Bertea CM. Ginkgo biloba responds to herbivory by activating early signaling and direct defenses. PLoS One. 2012;7(3):e32822.")\]. Ginkgolides and bilobalides have been linked to resistance to fungi \[[12](/article/10.1186/s13742-016-0154-1#ref-CR12 "Pan L, Ren L, Chen F, Feng Y, Luo Y. Antifeedant Activity of Ginkgo biloba Secondary Metabolites against Hyphantria cunea Larvae: Mechanisms and Applications. PLoS One. 2016;11(5):e0155682.")\] and possibly endophytic bacteria \[[14](/article/10.1186/s13742-016-0154-1#ref-CR14 "Yang R, Fan X, Cai X, Hu F. The inhibitory mechanisms by mixtures of two endophytic bacterial strains isolated from Ginkgo biloba against pepper phytophthora blight. Biol Control. 2015;85:59–67.")\], although the mechanism of resistance to bacterial pathogens remains unclear.Although numerous studies have focused on the aforementioned unique features of this fascinating species, and despite the increasing availability of genome-derived resources for ginkgo [15–18], our comprehensive and in-depth understanding has been impeded by the lack of a well-resolved and fully annotated genome that would also facilitate studies on the evolution of land plants, in particular gymnosperms. Furthermore, a complete ginkgo genome will facilitate the assembly and annotation of the published genome drafts of Picea abies [19], Picea glauca [20, 21] and Pinus taeda [22–[26](/article/10.1186/s13742-016-0154-1#ref-CR26 "Wegrzyn J, Lin B, Zieve J, Dougherty W, Martínez-García P, Koriabine M, Holtz-Morris A, de Jong P, Crepeau M, Langley C et al: Insights into the loblolly pine genome: characterization of BAC and fosmid sequences. PloS One 2013; 8, DOI: 10.1371/journal.pone.0072439
.")\], along with other newly-reported gymnosperm genomes, as well as the transcriptomes of _Cycas revoluta_, _Ephedra trifurca_ and _Pinus canariensis_ \[[27](/article/10.1186/s13742-016-0154-1#ref-CR27 "Huang Y, Kendall T, Forsythe ES, Dorantes-Acosta A, Li S, Caballero-Pérez J, Chen X, Arteaga-Vázquez M, Beilstein MA, Mosher RA. Ancient Origin and Recent Innovations of RNA Polymerase IV and V. Mol Biol Evol. 2015;32(7):1788–99.")\]. Despite the rapid advance in the publication of complete genome sequences for diverse plant species, the belated publication of the ginkgo genome may in part result from its large size, which was estimated to be 11.75 Gb \[[28](/article/10.1186/s13742-016-0154-1#ref-CR28 "Zonneveld BJM. Conifer genome sizes of 172 species, covering 64 of 67 genera, range from 8 to 72 picogram. Nord J Bot. 2012;30(4):490–502.")\]. The vast amount of repeated sequences in gymnosperm genomes, as illustrated in the genome draft of _P. abies_ \[[19](/article/10.1186/s13742-016-0154-1#ref-CR19 "Nystedt B, Street NR, Wetterbom A, Zuccolo A, Lin YC, Scofield DG, Vezzi F, Delhomme N, Giacomello S, Alexeyenko A, et al. The Norway spruce genome sequence and conifer genome evolution. Nature. 2013;497(7451):579–84.")\], further hampers ambitions to address this issue.In this study, we present the ginkgo genome sequence based on the high-quality assembly, annotation and analysis of genomic structures and evolution, and provide new insight into the evolution of large genomes and multiple defense mechanisms.
Data description
Genomic DNA was extracted from endosperm tissue from ginkgo seeds that develop directly from female gametophytes without fertilization and thus contain haploid genomes without undergoing genetic recombination. All paired-end libraries and one mate-pair library (2 kb) were constructed using DNA extracted from a single seed. The additional five mate-pair libraries (2 − 40 Kb) were constructed for scaffolding using DNA from other seeds from the same sampled tree (see details in Additional file 1: Table S1). These eight libraries were used only for scaffolding procedures to avoid the possible introduction of heterozygosity. Data were generated using a Hiseq 2000/4000 platform from 1253.09 Gb clean data (Additional file 1: Table S2). RNA was isolated from female and male reproductive organs and a 2-year seedling, respectively. Preparation of the cDNA library and sequencing was performed as described previously [[29](/article/10.1186/s13742-016-0154-1#ref-CR29 "Divi UK, Zhou X-R, Wang P, Butlin J, Zhang D-M, Liu Q, Vanhercke T, Petrie JR, Talbot M, White RG. Deep sequencing of the fruit transcriptome and lipid accumulation in a non-seed tissue of Chinese tallow, a potential biofuel crop. Plant Cell Physiol. 2016;57(1):125-37. doi: 10.1093/pcp/pcv181
.")\]. A total of 6.30 Gb, 6.40 Gb and 6.40 Gb of raw data were obtained from female, male and seedling samples, respectively, using an Illumina Hiseq 2000 (Additional file [1](/article/10.1186/s13742-016-0154-1#MOESM1): Table S3). Further details about sample collection, DNA/RNA extraction, library construction and sequencing can be found in the Methods section. All genome data have been uploaded to _Giga_DB \[[30](/article/10.1186/s13742-016-0154-1#ref-CR30 "Guan R, Zhao Y, Zhang H, Fan G, Liu X, Zhou W, Shi C, Wang J, Liu W, Liang X, Fu Y, Ma K, Zhao L, Zhang F, Lu Z, Lee SM, Xu X, Wang J, Yang H, Fu C, Ge S, Chen1 W. De novo sequencing of Ginkgo biloba. GigaScience Database 2016,
http://dx.doi.org/10.5524/100209
.")\].Analyses
Genome sequencing and assembly
Whole genome sequencing of ginkgo yielded 189.84× coverage raw sequence data. After filtering, 120.79× high-quality reads, comprising ~100× from paired-end libraries and ~20× from mate-pair libraries, remained for genome assembly (Additional file 1: Table S2). The ginkgo genome was estimated to be 10.00 Gb in size (Additional file 1: Table S4) with a high proportion of repeat elements (Additional file 2: Figure S1). All clean data were used to generate a draft genome assembly, followed by gap filling. The assembled draft genome is 10.61 Gb in length (including 493 Mb N’s) with N50 values of 48.2 kb for contigs and 1.36 Mb for scaffolds, respectively (Table 1).
Table 1 Assembly and annotation of the ginkgo genome
To evaluate the sequencing randomness and assembly quality, a fraction of reads (insert size of 500 bp and sequencing depth of 10.5-fold, Additional file 1: Table S2) was mapped to the genic region of the assembled genome, and the sequencing depth for each base exhibited a Poisson-like distribution (Additional file 2: Figure S2), suggesting no obvious bias for sequencing and assembly. Subsequent alignment of 17,902 expressed sequence tags (ESTs) to the assembled genome, along with 42,243 and 32,088 transcripts assembled from RNA-seq data from ginkgo seeds and leaves, resulted in 94.45, 92.68 and 92.57% being properly mapped, respectively (identity cutoff = 0.90, coverage ratio cutoff = 0.90).
Genome annotation
Using a combination of de novo and homology-based methods, up to 76.58% of the assembled sequences were found to be repeated sequences comprising transposable elements and tandem repeats, of which 79.20% were of the long terminal repeat (LTR) type (Additional file 1: Table S5 and Additional file 2: Figure S3; see further details below). Application of the k-mer frequency-based approach [31] resulted in an even higher proportion of repeat content (~84.43%), highlighting the challenges involved in assembling the repetitive elements in the ginkgo genome from limited short-read sequencing data.
Gene models were predicted using protein sequences from five land plants (Selaginella moellendorffii, Picea abies, Pinus taeda, Arabidopsis thaliana and Oryza sativa) coupled with transcriptomes assembled from RNA-seq data and EST data downloaded from NCBI (Additional file 1: Table S6). The length distributions of four categories of annotated genes (gene, coding sequence (CDS), exon and intron) were compared for the five species (Additional file 2: Figure S4). Calculation if the completeness of gene sets using BUSCO [32] (V1.1b) resulted in the identification of 707 complete single-copy BUSCOs (73.95% of the 956 known BUSCO groups), of which 308 were duplicated. Function annotation via mapping to functional databases identified 68.12% protein sequences with known homologous genes (Additional file 1: Table S7). The presence of 31.88% of proteins with no known function indicates a large proportion of highly diverged, species-specific genes in the ginkgo genome.
Of the 41,840 predicted ginkgo genes, 30,209 were assigned with high confidence based on supporting expression data, which is a slightly higher proportion than P. abies (28,354) [19] and far more than P. taeda (15,653; Additional file 1: Table S8) [23, [24](/article/10.1186/s13742-016-0154-1#ref-CR24 "Wegrzyn JL, Liechty JD, Stevens KA, Wu LS, Loopstra CA, Vasquez-Gross HA, Dougherty WM, Lin BY, Zieve JJ, Martinez-Garcia PJ et al: Unique features of the loblolly pine (Pinus taeda L.) megagenome revealed through sequence annotation. Genetics. 2014; 196. DOI: 10.1534/genetics.113.159996
.")\]. The average length of _G. biloba_ mRNA or CDS sequences was longer than that of _P. abies_ (Additional file [2](/article/10.1186/s13742-016-0154-1#MOESM2): Figure S4). Following comparison with the genomes of _S. moellendorffii_, _P. canariensis, Cycas revoluta_, _A. thaliana_ and _O. sativa_ (Additional file [1](/article/10.1186/s13742-016-0154-1#MOESM1): Table S9), ginkgo genes were clustered into 12,303 families (Additional file [2](/article/10.1186/s13742-016-0154-1#MOESM2): Figure S5). Phylogenetic reconstruction based on 920 single copy orthologous genes indicates that ginkgo is more closely related to cycads than is _P. canariensis_ (Fig. [1a](/article/10.1186/s13742-016-0154-1#Fig1)), further supporting the prevailing hypothesis of gymnosperm phylogeny \[[1](/article/10.1186/s13742-016-0154-1#ref-CR1 "Lee EK, Cibrian-Jaramillo A, Kolokotronis SO, Katari MS, Stamatakis A, Ott M, Chiu JC, Little DP, Stevenson DW, McCombie WR, et al. A functional phylogenomic view of the seed plants. PLoS Genet. 2011;7(12):e1002411.")–[4](/article/10.1186/s13742-016-0154-1#ref-CR4 "Ruhfel BR, Gitzendanner MA, Soltis PS, Soltis DE, Burleigh JG. From algae to angiosperms-inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes. BMC Evol Biol. 2014;14:23.")\].Fig. 1
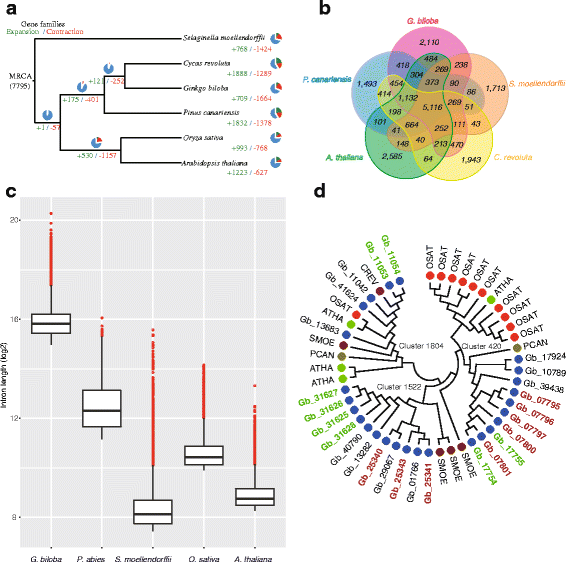
Phylogenetic relationships and comparative genomic analyses. a Phylogenetic relationships and number of gene families displaying expansion and contraction. b Comparison of the number of gene families in the five land plants Ginkgo biloba, Cycacs revoluta, Pinus canariensis, Selaginella moellendorffii and Arabidopsis thaliana. c Comparison of the longest 10% of introns in the five land plants. d Phylogenetic tree of three orthologous gene families indicating gene duplication and tandem distribution. The colors of solid circles represent species. Gene IDs of ginkgo start with ‘Gb’, and red and green text represents tandem distribution in different scaffolds. CREV, C. revoluta, PCAN, P. canariensis, SMOE, S. moellendorffii, OSAT, O. sativa, ATHA, A. thaliana
We identified 2110 gene families unique to ginkgo, and 5116 orthologous gene families shared by the five land plants were analyzed (Fig. 1b). The ginkgo-specific gene families (including 11,105 genes, of which 4247 were functionally annotated in the KEGG database) are remarkably enriched in eight specific pathways/functions, including ATP-binding cassette (ABC) transporters, monoterpenoid biosynthesis and phenylpropanoid biosynthesis (Additional file 1: Table S10). Signals characteristic of expansion and contraction were detected in 709 and 1664 gene families, respectively (Fig. 1a). GO enrichment analysis of a subset of 223 gene families of significantly altered size (P < 0.05) revealed that pathways associated with the response to biotic stimuli, such as the defense response (Cluster 368: 21 genes in ginkgo vs. 1 gene in S. moellendorffii, O. sativa and P. canariensis) and plant-type cell wall organization (16 genes encoding malate dehydrogenase in both Cluster 719 and Cluster 1793) were particularly enriched. Highly represented molecular functions included protein kinase activity (FLS2 and EFR gene families in Cluster 38 that have a key role in plant-pathogen interactions), transferase activity, transferring hexosyl groups (involved in conjugation of the growth hormone indole-3-acetic acid [33] and glucosinolate biosynthesis), and terpene synthase activity (functioning in the defense system mainly via alpha-bisabolene synthase [34].
Evolution of LTR-RTs
The ginkgo genome is mainly composed of repeated sequences, mostly LTR-RTs (58.34% of assembled sequences), of which two superfamilies, Ty3/Gypsy and Ty1/Copia, account for 45.63 and 12.71% of all assembled sequences, respectively. Two rounds of BLAST similarity searches identified 24,090 vs. 17,564, 28,352 vs. 10,915, 2416 vs. 1790, 728 vs. 2727, and 365 vs. 494 Ty1/Copia and Ty3/Gypsy domains in the genomes of G. biloba, Zea mays, P. abies, Physcomitrella patens and Populus trichocarpa, respectively.
To investigate the evolution of transposable elements (TEs) in land plants, phylogenetic trees of domains in reverse transcriptase genes were constructed for both Ty1/Copia and Ty3/Gypsy elements. In the phylogenetic tree of the Ty3/Gypsy superfamily, LTR-RTs from ginkgo were clustered in five (1, 2, 3, 4 and 7) of the seven major clades. Clade 1 was composed of elements from all five species (Fig. 2a), indicating conservation among land plants, and the origins of the other clades postdating species divergence. Notable ginkgo-specific expansions were observed in clades 4 and 7, representing two subfamilies, and in clade 2, which included a minority of components from P. abies. Despite a closer phylogenetic relationship and shared clades 2 and 3, ginkgo displayed substantially higher diversity and abundance within the Ty3/Gypsy superfamily than the other analyzed gymnosperm (P. abies), possibly indicating ancient origins for these diverse clades (subfamilies) followed by a gradual and/or rapid diversification. The two clades revealed for Z. mays (5 and 6) both diverged more recently than the other major clades, and while clade 6 shared by maize and poplar was narrow, clade 5, which is specific to maize, was far more diverse and abundant, indicating within-clade divergence intermediately followed by recent expansion of the Ty3/Gypsy family in maize.
Fig. 2
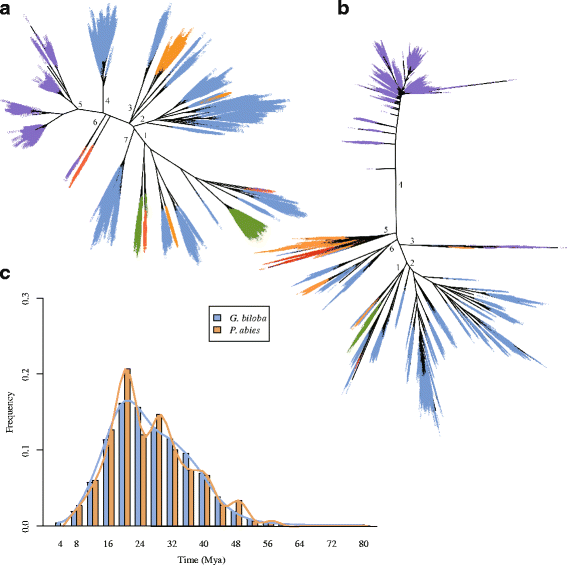
Evolution of LTR-RTs in ginkgo. a Phylogenetic relationships within the Ty3/Gypsy superfamily in the five land plants. Blue: Ginkgo biloba; green, Physcomitrella patens (moss); orange, Picea abies; red, Populus trichocarpa; purple, Zea mays. b Phylogenetic relationships within the Ty1/Copia superfamily in the five land plants. c Comparison of the timing of LTR-RTs insertions between G. biloba and P. abies
The Ty1/Copia superfamily exhibited a slightly different pattern (Fig. 2b), with four of the six major clades (1, 2, 5 and 6) present in ginkgo. Clade 1, shared by all five land plants, is presumably the most conserved. All four clades were less scattered in the phylogenetic tree, and clustered more closely to clade 1, than their counterparts in the Ty3/Gypsy tree, consistent with a more conserved evolution, as described previously [35–38]. Clade 2, the sole ginkgo-specific clade, was both highly diverse and abundant, and was a sister clade to clade 1, the most conserved clade, indicating an earlier split from the ancestral gene than occurred in the Ty3/Gypsy superfamily. Relative to clade 2, clade 6 is less well conserved in ginkgo, and clade 5 even less so. Even so, this superfamily appears to be more diverse and abundant in ginkgo than in P. abies, which was expanded mainly in clade 5. The majority of elements from maize similarly clustered in the species-specific clade 4, connected by a long branch, while a few members were clustered with spruce-derived elements in the narrow clade 3.
Furthermore, to estimate the activity of LTR-RTs at the molecular level, 47,342 putative complete LTR-RTs were identified in the ginkgo genome, and the insertion time for each pair of LTR-RTs [39] had a mutation rate of 2.2 × 10-9 substitutions per base per year [19]. This result suggests that the amplification of LTR-RTs occurred largely between 16 and 24 million years ago (mya; Fig. 2c), consistent with the timescale proposed for P. abies [19].
TE insertions in introns
A comparison of gene models for the five land plants revealed that the average length of the longest 10% of introns (13,607 for G. biloba, 5069 for P. abies, 10,034 for S. moellendorffii, 12,152 for O. sativa and 11,275 for A. thaliana) in ginkgo was significantly longer (P <2.0 × 10-6, Welch Two Sample _t_-test) than in the other four land plants (Fig. 1c). Similar results were also reported previously for P. glauca and P. taeda [21, [24](/article/10.1186/s13742-016-0154-1#ref-CR24 "Wegrzyn JL, Liechty JD, Stevens KA, Wu LS, Loopstra CA, Vasquez-Gross HA, Dougherty WM, Lin BY, Zieve JJ, Martinez-Garcia PJ et al: Unique features of the loblolly pine (Pinus taeda L.) megagenome revealed through sequence annotation. Genetics. 2014; 196. DOI: 10.1534/genetics.113.159996
."), [40](/article/10.1186/s13742-016-0154-1#ref-CR40 "Stival Sena J, Giguere I, Boyle B, Rigault P, Birol I, Zuccolo A, Ritland K, Ritland C, Bohlmann J, Jones S, et al. Evolution of gene structure in the conifer Picea glauca: a comparative analysis of the impact of intron size. BMC Plant Biol. 2014;14:95.")\]. Repeat elements occurred in 26.56% of introns in ginkgo, whereas they accounted for 65.48% of the total sequence length of introns. In contrast, repetitive elements only accounted for 2.94% of intron sequences in _P. glauca_, even though these elements occur in 32.4% of genes \[[40](/article/10.1186/s13742-016-0154-1#ref-CR40 "Stival Sena J, Giguere I, Boyle B, Rigault P, Birol I, Zuccolo A, Ritland K, Ritland C, Bohlmann J, Jones S, et al. Evolution of gene structure in the conifer Picea glauca: a comparative analysis of the impact of intron size. BMC Plant Biol. 2014;14:95.")\]. Furthermore, comparison of gene structures (Additional file [2](/article/10.1186/s13742-016-0154-1#MOESM2): Figure S6) as well as average intron length and genome size (Additional file [2](/article/10.1186/s13742-016-0154-1#MOESM2): Figure S7) revealed that the length and number of exons in ginkgo are conserved, while introns vary substantially, consistent with these elements in conifers \[[40](/article/10.1186/s13742-016-0154-1#ref-CR40 "Stival Sena J, Giguere I, Boyle B, Rigault P, Birol I, Zuccolo A, Ritland K, Ritland C, Bohlmann J, Jones S, et al. Evolution of gene structure in the conifer Picea glauca: a comparative analysis of the impact of intron size. BMC Plant Biol. 2014;14:95.")\].Further comparison of the distribution of repeats revealed a high percentage of repeats in both introns and intergenic regions, reaching a maximum of 69%. For regions with a repeat ratio less than 44%, the frequency of introns was higher than that of intergenic regions. Conversely, for regions with a repeat ratio of 44% or greater, the frequency of repeats in introns was lower than intergenic regions (Additional file 2: Figure S8). Transposon insertions tend to accumulate in genes that are expressed at lower levels [41], which may be driven by negative selection due to increased transcriptional cost for longer transcripts [42].
Gene duplications
Whole genome duplication (WGD) and tandem duplication resulting from unequal crossover represent two dominant mechanisms for generating gene copies. WGDs are usually identified from the _K_s (a measure of the substitutions per synonymous site) distribution of paralogs, or from gene collinearity data. We identified as few as 40 syntenic gene pairs within the entire gene set of the ginkgo genome. Thus, calculation of the _K_s value of paralogs of gingko [43] was based on the gene cluster results of OrthoMCL (release 5) [44]. WGD may lead to peaks of gene duplication, whereas accumulation of tandem duplicate genes (TDGs) may affect the distribution of _K_s values. Two peaks were observed in the corrected _K_s distribution (after removing TDGs) of gingko (i.e., _K_s = 0.1 − 0.2 (peak 1) and _K_s = 0.7 − 1.0 (peak 2; Fig. 3). The timing of the two WGDs at peaks 1 and 2, estimated from the mutation rates (per base per year) of 0.68 × 10-9 [[45](/article/10.1186/s13742-016-0154-1#ref-CR45 "Buschiazzo E, Ritland C, Bohlmann J, Ritland K. Slow but not low: genomic comparisons reveal slower evolutionary rate and higher dN/dS in conifers compared to angiosperms. BMC Evol Biol. 2012;12:8. doi: 10.1186/1471-2148-12-8
.")\], was found to be between 74 and 147 mya, and between 515 and 735 mya, respectively. The latter of these time periods is consistent with that previously reported for seed plants \[[46](/article/10.1186/s13742-016-0154-1#ref-CR46 "Li Z, Baniaga AE, Sessa EB, Scascitelli M, Graham SW, Rieseberg LH, Barker MS: Early genome duplications in conifers and other seed plants. Sci Adv. 2015;1(10):e1501084. doi:
10.1126/sciadv.1501084
.")–[48](/article/10.1186/s13742-016-0154-1#ref-CR48 "Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, Tomsho LP, Hu Y, Liang H, Soltis PS, et al. Ancestral polyploidy in seed plants and angiosperms. Nature. 2011;473(7345):97–100.")\], while the former time period is far later than when _Gingko_ and conifers diverged, indicating a potential independent WGD event postdating the origin of _Ginkgo_ by at least 170 mya \[[49](/article/10.1186/s13742-016-0154-1#ref-CR49 "Zhou Z-Y. An overview of fossil Ginkgoales. Palaeoworld. 2009;18(1-22):206.")\].Fig. 3

_K_s distribution of clustered genes in ginkgo. _K_s distributions were plotted for three sets of genes; all clustered genes (red), clusters of tandem duplicate genes (TDGs; black) and clustered genes excluding TDGs (green)
We detected 2061 gene clusters containing 5201 TDGs (Additional file 1: Table S11) and an unambiguous peak in the _K_s distribution of these TDGs in ginkgo that corresponded to the predicted recent WGD (Fig. 3). Two sets of TDGs (Gb_25430, Gb_25431 and Gb_25343 on scaffold4299, and Gb_31625, Gb_31626, Gb_31627 and Gb_31628 on scaffold5904; the clade in Cluster 1522 in Fig. 1d) were identified that belong to the same shikimate O-hydroxycinnamoyl transferase family, which forms a kernel in the biosynthesis of plant secondary metabolites including stilbenoid, diarylheptanoid and gingerol. A tandem distribution was also observed for gene families related to the gibberellin (GA) receptor GID1 (Fig. 1d, the clade in Cluster 420) involved in transducing GA signaling and inducing a wide range of plant growth responses. Cluster 1804, comprising Gb_11042, Gb_11053, Gb_11054, Gb_13683 and Gb_41624 encoding pathogenesis-related protein 1 that functions in plant hormone signal transduction and plant-pathogen interactions, was highly diverged between ginkgo and other land plants, and also within ginkgo (Fig. 1d).
Resistant genes
Considerable expansion was also uncovered for diverse gene families related to multiple defense mechanisms in ginkgo. The glucosinolate biosynthesis gene family comprises 29 genes involved in core pathways of both pungent components and flavonoids (flavone and flavonol) biosynthesis. The α-bisabolene synthase gene family, absent in both A. thaliana and O. sativa, is composed of 28 genes participating in terpene synthase activity, carbon-oxygen lyase activity and magnesium ion binding. Meanwhile, we observed a high incidence of gene duplication in an array of genes in two pathways of defensive metabolites (Fig. 4a), one involving the six ginkgolide/bilobalide biosynthesis genes DXS (1-deoxy-D-xylulose-5-phosphate synthase, K01662), HDS (4-hydroxy-3-methylbut-2-en-1-yl diphosphate synthase, K03562), GPPS (geranyl-diphosphate synthase, K00787), FPPS (farnesyl-diphosphate synthase, K00795), HMGS (HMG-CoA synthase, K01641), and AACT (acetyl-CoA acetyl- transferase, K00626) [16]. We also compared the copy number of each gene in this pathway between ginkgo and all other species in the KEGG database (release-76), and observed a remarkable expansion in K00938, K003526, K01641, K00021 and K10960 (Fig. 4b, shadowed in purple) in ginkgo. The other high incidence of gene duplication occurred in four sets of genes involved in flavonoid biosynthesis (ko00360 in KEGG) comprising CHS (chalcone synthase), F3’H (flavonoid 3′-hydroxylase), FLS (flavonol synthase) and DFR (dihydromyricetin reductase).
Fig. 4
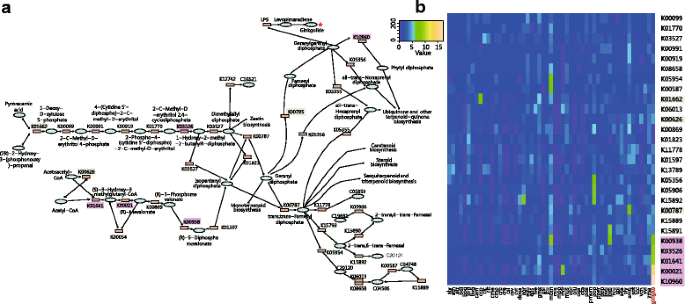
Gene expansion of terpenoid backbone biosynthesis in ginkgo. a Backbone pathways of terpenoid biosynthesis and enriched genes in ginkgo. Genes names shaded in green apparently underwent expansion in ginkgo based on comparison with S. moellendorffii, P. canariensis, C. revoluta, A. thaliana and O. sativa. Gene names shaded in purple apparently underwent expansion in ginkgo based on comparison with all released plant genomes in the KEGG database. Expansion in both K03526 and K01641 gene families was suggested by both comparisons. b The number of each gene in the pathways of all released plant genomes in the KEGG database. Genes in purple and green shadow correspond to expansions as described in (a). Full names for species can be found at http://www.genome.jp/kegg/catalog/org_list.html
The most remarkable duplication was detected in a gene family involved in plant-pathogen interactions and related to leucine rich repeat (LRR) receptor-like serine/threonine-protein kinase flagellin-sensitive 2 (FLS2) and EF-Tu receptor (EFR). _FLS2_-coded pattern recognition receptors (_PRR_s) recognize the highly conserved N-terminal epitope (flg22) that is a feature of the main bacterial flagella protein flagellin [50]. The _EFR_-coded _PRR_s for bacterial EF-Tu function similarly [[51](/article/10.1186/s13742-016-0154-1#ref-CR51 "Zipfel C, Kunze G, Chinchilla D, Caniard A, Jones JD, Boller T, Felix G. Perception of the bacterial PAMP EF-Tu by the receptor EFR restricts Agrobacterium-mediated transformation. Cell. 2006;125(4):749–60. doi: 10.1016/j.cell.2006.03.037
.")\]. In gingko, the family that includes both _FLS2_ and _EFR_ contains as many as 62 genes, far more than that in _A. thaliana_ (one copy of _FLS2_ and one copy of _EFR_). Thus, _FLS2_ could be considered an important element of the immune system in ginkgo. Furthermore, five additional duplicate genes (_RIN4, HSP90, CEBiP, CaMCML_ and _PR1_) were also enriched in this pathway (map04626 in KEGG).Discussion
Sequencing strategies and genome features
In the present study we sequenced and assembled the large ginkgo genome using high quality data. The genome (contig N50 of 48.2 kb and scaffold N50 of 1.36 Mb) is about 80 times larger than the A. thaliana genome, and is even larger than those of the angiosperm plants sorghum, maize and orchid that are renowned for their large genomes. The contiguity of the ginkgo genome is far better than previously published gymnosperm genomes, and may have benefited from several factors. Firstly, ginkgo DNA was extracted from the female gametophytic endosperm of a single seed based on the strategy described previously for the loblolly pine (P. glauca) genome [23], but P. glauca DNA was derived from apical shoots [[52](/article/10.1186/s13742-016-0154-1#ref-CR52 "Birol I, Raymond A, Jackman SD, Pleasance S, Coope R, Taylor GA, Saint Yuen MM, Keeling CI, Brand D, Vandervalk BP, et al. Assembling the 20 Gb white spruce (Picea glauca) genome from whole-genome shotgun sequencing data. Bioinformatics. 2013;29(12):1492-7. doi: 10.1093/bioinformatics/btt178
.")\], which may have introduced considerable heterozygosity. Secondly, the amount of the sequencing data generated for ginkgo was much greater than that of other gymnosperms, resulting in a deeper coverage. We generated a total of \~1969 Gb raw data for ginkgo with a sequencing depth of 196.95, compared with 970 Gb raw data and 43.32-fold depth for _P. glauca_ WS77111 \[[21](/article/10.1186/s13742-016-0154-1#ref-CR21 "Warren RL, Keeling CI, Yuen MM, Raymond A, Taylor GA, Vandervalk BP, Mohamadi H, Paulino D, Chiu R, Jackman SD, et al. Improved white spruce (Picea glauca) genome assemblies and annotation of large gene families of conifer terpenoid and phenolic defense metabolism. Plant J. 2015;83(2):189–212.")\]. Thirdly, we constructed three paired-end and five mate-pair libraries with sizes ranging from 250 to 40 kb, which facilitated sequence assembly, whereas for _P. glauca_ WS77111 only three libraries were constructed, with fragment lengths of 400 bp, 600 bp and 15 kb. Moreover, the genomes of _P. glauca_ and _P. taeda_ are approximately twice the size of the ginkgo genome, and the repeat sequences in _P. taeda_ amount to 79% of the total genome \[[21](/article/10.1186/s13742-016-0154-1#ref-CR21 "Warren RL, Keeling CI, Yuen MM, Raymond A, Taylor GA, Vandervalk BP, Mohamadi H, Paulino D, Chiu R, Jackman SD, et al. Improved white spruce (Picea glauca) genome assemblies and annotation of large gene families of conifer terpenoid and phenolic defense metabolism. Plant J. 2015;83(2):189–212."), [25](/article/10.1186/s13742-016-0154-1#ref-CR25 "Zimin A, Stevens K, Crepeau M, Holtz-Morris A, Korabline M, Marçais G, Puiu D, Roberts M, Wegrzyn J, de Jong P et al: Sequencing and assembly of the 22-Gb loblolly pine genome. Genetics. 2014; 196. DOI:
10.1534/genetics.113.159715
")\]. Thus, we were able to generate a high quality assembly of the gymnosperm genome, which has long been presumed a huge challenge due to the genome size \[[53](/article/10.1186/s13742-016-0154-1#ref-CR53 "Ohri D, Khoshoo TN. Genome size in gymnosperms. Plant Syst Evol. 1986;153(1):119–32. doi:
10.1007/BF00989421
.")\] and high proportion of repeat elements \[[54](/article/10.1186/s13742-016-0154-1#ref-CR54 "De La Torre A, Birol I, Bousquet J, Ingvarsson P, Jansson S, Jones SJ, Keeling CI, MacKay J, Nilsson O, Ritland K, et al. Insights into Conifer Giga-Genomes. Plant Physiol. 2014;166(4):1724–32.")\].The ginkgo genome is mainly constituted by repetitive DNA sequences, with TEs accounting for at least 75%. This proportion is greater than reported for other sequenced plant genomes such as 70% in Norway spruce [19], 61% in orchid [55], 58% in sorghum [56], 49% in grape [57] and 35% in rice [[58](/article/10.1186/s13742-016-0154-1#ref-CR58 "International Rice Genome Sequencing P. The map-based sequence of the rice genome. Nature. 2005;436(7052):793–800. doi: 10.1038/nature03895
.")\], but is still slightly smaller than the 79% in loblolly pine \[[23](/article/10.1186/s13742-016-0154-1#ref-CR23 "Neale DB, Wegrzyn JL, Stevens KA, Zimin AV, Puiu D, Crepeau MW, Cardeno C, Koriabine M, Holtz-Morris AE, Liechty JD, et al. Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol. 2014;15(3):1–13.")\] (Additional file [1](/article/10.1186/s13742-016-0154-1#MOESM1): Table S8). Based on our more complete and contiguous ginkgo genome, we constructed a repeat library that will likely prove more valuable for gymnosperm genome research than other currently available conifer genomes.Our approach demonstrated the sequencing, assembly and annotation of a large genome, and provides insight into the de novo sequencing of other challenging genomes such as other gymnosperms and ferns [[59](/article/10.1186/s13742-016-0154-1#ref-CR59 "Wolf PG, Sessa EB, Marchant DB, Li F-W, Rothfels CJ, Sigel EM, Gitzendanner MA, Visger CJ, Banks JA, Soltis DE, et al. An exploration into fern genome space. Genome Biol Evol. 2015;7(9):2533-44. doi: 10.1093/gbe/evv163
.")\]. Our approach may also facilitate improvements in the quality of gymnosperm genome data more generally.Evolution of gene structures and large genomes
Compared with the fern S. moellendorffii, the conifer P. abies, and the angiosperms A. thaliana and O. sativa, ginkgo genes are clearly much longer due to the insertion of TE. The length and number of exons among species is conserved, whereas the intron length varies dramatically among land plants. Like genes, introns in ginkgo are also much longer than in other land plants (Fig. 1c, Additional file 2: Figures S6 and S7). Across all gingko genes, 16,939 introns were longer than 20 kb, accounting for 12.45% of all introns, a significantly higher percentage than reported for conifers [40]. The average intron length was not correlated with genome size in conifers [40], and this was also supported by the ginkgo genome data in the present study. Although introns in Ginkgo are longer on average, the genome is only half the size of the P. abies genome (Additional file 2: Figure S7). These findings indicate a remarkable expansion of introns and a complex evolution of gene structure in the ginkgo genome.
The present results showed that the genome of ginkgo comprises a high proportion of repetitive sequences (at least 76.58%) with long introns with TE insertions. LTR elements appeared to have accumulated gradually over time, but a significant amplification and accumulation was evident between 16 and 24 mya (Fig. 2c), consistent with their proposed gradual accumulation and an overall gradual increase in genome size [19]. Compared with Norway spruce, the LTR-RTs superfamilies Ty3/Gypsy and Ty1/Copia substantially more diverse and abundant in ginkgo, indicating greater expansion and divergence in the wider ginkgo genome. It should be noted that this conclusion might be biased to some extent due to disparities in the volumes of ginkgo and spruce datasets (24,090 vs. 2416; 17,564 vs. 1790) as well as the continuity of assembled sequences. Ginkgo has a much larger genome and a comparable proportion of LTR-RTs, whereas maize appears to have undergone a recent massive expansion in a limited number of genes in both Ty3/Gypsy and Ty1/Copia superfamilies, but LTR-RTs likely expanded in multiple early-to-late-divergence events across many subfamilies in ginkgo (Fig. 2a and b).
Our evidence supports two WGDs in ginkgo, with an ancient WGD occurring 515 to 735 mya, consistent with previous reports [[46](/article/10.1186/s13742-016-0154-1#ref-CR46 "Li Z, Baniaga AE, Sessa EB, Scascitelli M, Graham SW, Rieseberg LH, Barker MS: Early genome duplications in conifers and other seed plants. Sci Adv. 2015;1(10):e1501084. doi: 10.1126/sciadv.1501084
.")–[48](/article/10.1186/s13742-016-0154-1#ref-CR48 "Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, Tomsho LP, Hu Y, Liang H, Soltis PS, et al. Ancestral polyploidy in seed plants and angiosperms. Nature. 2011;473(7345):97–100."), [60](/article/10.1186/s13742-016-0154-1#ref-CR60 "Scott AD, Stenz NW, Ingvarsson PK, Baum DA. Whole genome duplication in coast redwood (Sequoia sempervirens) and its implications for explaining the rarity of polyploidy in conifers. New Phytol. 2016;211(1):186–93."), [61](/article/10.1186/s13742-016-0154-1#ref-CR61 "Ahuja MR. Polyploidy in gymnosperms: revisited. Silvae Genetica. 2005;54(2):59–69.")\], and a more recent event reported for the first time in this study and not yet observed in other seed plants \[[46](/article/10.1186/s13742-016-0154-1#ref-CR46 "Li Z, Baniaga AE, Sessa EB, Scascitelli M, Graham SW, Rieseberg LH, Barker MS: Early genome duplications in conifers and other seed plants. Sci Adv. 2015;1(10):e1501084. doi:
10.1126/sciadv.1501084
.")\]. This recent WGD may have uniquely contributed to the morphological and biological diversity of the gingko lineage. In summary, both LTR-RT insertions and two WGD events may have contributed to the large genome of ginkgo.The recent proliferation in the maize genome may have occurred ~3 mya or even more recently [62, 63], and therefore postdated the recent WGD event that occurred between 5 and 12 mya [62]. In the maize genome, chromosomal breakages and fusions, as well as uneven gene loss, may have occurred when the ancestral cell returned to a genetically diploid state after a WGD event [62]. This active mechanism might prevent a continuous growth in genome size in angiosperms. Thus, the lack of an active mechanism for removing transposable elements such as unequal recombination [64] might have led to the enormous genome size in gymnosperms [19].
Multiple defense mechanisms underlie resilience in gingko
Ginkgo is renowned for its wide spectrum of resistance and tolerance of insects and pathogens, due to multiple chemical defense mechanisms including repellence and anti-feedance towards insect herbivores exerted by terpenic lactones (ginkgolides and bilobalides) [12], chemical toxicity towards insects and pathogens via flavonoids and/or lactones, and attraction towards predators of herbivorous insects through terpene-composed VOCs [12, 13]. Interestingly, this impressive arsenal is shared by a number of plants, which begs the question of why these defense systems result in significantly less herbivore-mediated disease in ginkgo.
One explanation for the outstanding resistance displayed by gingko is a remarkable duplication of resistance genes and enrichment of relevant pathways. Gene duplication could increase the efficiency of resistance-related reactions through dosage effects, and might also provide a platform for adaptive evolution of duplicated copies [55]. We revealed most notable gene duplications in at least two gene families responsible for biosynthesis of flavonoids and terpenes in ginkgo, respectively. Specifically, glucosinolate biosynthesis and α-bisabolene synthase gene families, comprising 29 and 28 genes, respectively, were highly duplicated in the gingko genome. The latter gene family, which is absent in both A. thaliana and O. sativa, plays an important role in terpene-mediated defense in conifers [34]. Meanwhile, biosynthesis pathways of flavonoids and terpenoids were enriched across multiple duplicate gene families (four for flavonoids and six for terpenoids). Furthermore, by comparing with all current species in the KEGG database, five terpenoid biosynthesis genes appear to be notably expanded in ginkgo, and this enrichment is likely to further increase the efficiency of biosynthesis.
Another explanation is the enrichment of ginkgo-specific gene families and genes involved in the biosynthesis of defense metabolites. Gene families unique to ginkgo are concentrated in pathways for monoterpenoid and phenylpropanoid biosynthesis (Additional file 1: Table S10) and in pathways involving biotic stimuli responses, defense responses, and terpene synthase activity (Fig. 4a). These species-specific gene families might add extra dosage effects that enhance resistance and possibly also adaptability.
The induction of direct defense metabolites such as flavonoids and the release of VOCs have been reported for many angiosperms [13]. Given the long-standing interaction between plants and insects, these shared strategies may suggest an ancient origin for these defense mechanisms. This hypothesis is further supported by the observation that the fern Pterisvittata also responds to herbivory by reactive oxygen species production and the emission of VOCs [65].
In contrast to well-studied chemical defense mechanisms, very little is known about bacterial resistance and the genes responsible in ginkgo. In the present study, we identified one gene family (containing _FLS_2 and EFR) involved in recognizing bacterial infection in gingko [50]. A striking incidence of genes duplication in this family has resulted in 62 members, far exceeding the number in A. thaliana. Furthermore, in gingko, this pathway (map04626) is enriched with an additional five gene families (RIN4, _HSP_90, CEBiP, CaMCML, and _PR_1) in addition to _FLS_2 and EFR.
Methods
Sampling and extraction of DNA and RNA
Multiple seeds at visually different developmental stages were collected from five separate large ginkgo trees at one of the ginkgo refuge populations located on Tianmu Mountain [8, [9](/article/10.1186/s13742-016-0154-1#ref-CR9 "Fan J, Shi H, Xiao H, Zhao G. Double-Layer 3D Macro-Mesoporous Metal Oxide Modified Boron-Doped Diamond with Enhanced Photoelectrochemical Performance. ACS Appl Mater Interfaces. 2016. doi: 10.1021/acsami.6b01929
.")\], Zhejiang Province, China on 19 July 2015\. Voucher specimens were deposited at the Herbarium of Zhejiang University (Zhou WB & Zhao LJ, HZU13445, HZU13446, HZU13448, HZU13449, HZU13453, HZU13459). Endosperm tissues, which directly develop from female gametophytes without fertilization and thus contain haploid genomes, were dissected from the seeds. Genomic DNA was extracted using a standard cetyltrimethylammonium bromide (CTAB) protocol \[[66](/article/10.1186/s13742-016-0154-1#ref-CR66 "Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1987;19:11–5.")\]. The DNA yield of each seed was quantified (Additional file [1](/article/10.1186/s13742-016-0154-1#MOESM1): Table S4) to select the candidate sample(s) for _de novo_ sequencing. DNA extracted from a single seed of one sampled tree (TM011) was determined for the subsequent construction of all paired-end libraries. Other seeds from the same tree were used to construct long-insert libraries.For RNA isolation, ovulate (female) and staminate (male) strobili and seedlings were collected from Tianmu Mountain in April 2015. Female and male samples from three to four ginkgo trees were pooled as mixed female and male samples (GinkgoF vs. GinkgoM), respectively. All aerial parts from three 2-year seedlings were also pooled as a mixed sample (GinkgoL1). Mixed samples were immediately frozen in liquid nitrogen and stored at -80 °C. Total RNA was isolated using a modified CTAB protocol [67].
Library construction and sequencing
Due to the large size of the gingko genome (approximately 10 Gb), we constructed libraries with a wide range of insert sizes, aiming to alleviate the technical challenge of genome assembly. Eight libraries were built with insert sizes between 250 and 40 kb according to the manufacturer’s instructions. A total of 1969.48 Gb of raw data was generated using Illumina Hiseq 2000/4000 (see details in Additional file 1: Table S1) and clean data totaled 1253.09 Gb (Additional file 1: Table S2).
For transcriptome sequencing, 20 μg RNA from each tissue was used for cDNA library preparation and sequencing as described previously [[29](/article/10.1186/s13742-016-0154-1#ref-CR29 "Divi UK, Zhou X-R, Wang P, Butlin J, Zhang D-M, Liu Q, Vanhercke T, Petrie JR, Talbot M, White RG. Deep sequencing of the fruit transcriptome and lipid accumulation in a non-seed tissue of Chinese tallow, a potential biofuel crop. Plant Cell Physiol. 2016;57(1):125-37. doi: 10.1093/pcp/pcv181
.")\]. RNA integrity was assessed using capillary electrophoresis on an Agilent BioAnalyzer 2100 (Agilent Technologies, Palo Alto, California, USA). Polyadenylated RNA isolated using oligo (dT)-attached beads was fragmented and reverse transcribed to cDNA. Paired-end libraries generated from each tissue were sequenced separately on an Illumina Hiseq 2000 platform. A total of 19.10 Gb raw data were obtained for three samples, specifically 6.40 Gb, 6.30 Gb and 6.40 Gb for male, female and seedling samples, respectively (Additional file [1](/article/10.1186/s13742-016-0154-1#MOESM1): Table S3).Genome assembly
Raw genomic sequencing data were filtered using SOAP_filter_ (V2.2). Low-quality reads including those with adapters, with a high N percentage, and low quality bases (more than 10% of bases with a Q-phred value lower than 7 and 15 for Hiseq 2000 and Hiseq 4000 platform, respectively), and PCR duplicated reads were removed. Genome size and complexity were estimated using k-mer analysis on approximate 50-fold sequencing reads from small insert size libraries. Filtered reads were processed to generate the assembled genome using SOAP_denovo2_ [68] (V2.0.4) with key parameters of -R -K 89 -D 3 -d 3. Gap filling was performed based on the assembly results utilizing KGF [[69](/article/10.1186/s13742-016-0154-1#ref-CR69 "Short Oligonucleotide Analysis Package homepage: http://soap.genomics.org.cn/
. Accessed 16 June 2016.")\] (V1.06) and Gapcloser \[[70](/article/10.1186/s13742-016-0154-1#ref-CR70 "SOAPdenovo2 GitHub:
https://github.com/gigascience/paper-luo2012
. Accessed 16 June 2016.")\] (V1.10).RNA-seq data from seeds and leaves were downloaded from NCBI (SRR2163235 and SRR1722455, respectively) and assembled using Trinity (V2012-10-05) [71], resulting in the assembly of 42,243 and 32,088 transcripts, respectively. Gingko ESTs were retrieved from the NCBI EST database. Assembled transcripts and retrieved ESTs were mapped to the ginkgo genome using blat [72] (V34) with default parameters and the sim4 output format. The blat results were filtered with a cutoff of 0.90 for identity and coverage.
Repeat annotation
TEs were found to account for a large proportion of DNA in conifer genomes [54]. TEs and other repeat elements in the ginkgo genome were identified using a combination of homology-based and de novo approaches. Specifically, TEs at DNA and protein levels were determined based on repeat searching in the known repeat database Repbase [73] using RepeatMasker (V4.0.5) and RepeatProteinMask (V4.0.5). De novo repeat libraries were constructed based on the genome using the de novo prediction programs RepeatModeler (V1.0.8) and LTR-FINDER (V1.0.6), followed by the removal of contamination and multi-copy genes from libraries. Using these libraries as a database, repeats in the genome were identified and classified using RepeatMasker. Tandem repeats including microsatellites (SSRs) with unit sizes ranging from 1 to 2000 bases were identified using the program Tandem Repeats Finder (TRF, V4.07). Finally, non-redundant repeat content was calculated and summarized using an in-house Perl script (included in GigaDB [[30](/article/10.1186/s13742-016-0154-1#ref-CR30 "Guan R, Zhao Y, Zhang H, Fan G, Liu X, Zhou W, Shi C, Wang J, Liu W, Liang X, Fu Y, Ma K, Zhao L, Zhang F, Lu Z, Lee SM, Xu X, Wang J, Yang H, Fu C, Ge S, Chen1 W. De novo sequencing of Ginkgo biloba. GigaScience Database 2016, http://dx.doi.org/10.5524/100209
.")\]).Intact LTR elements were detected using LTR_STRUC with default settings. Since loose default settings could lead to false positive results, results should be interpreted with caution. Pairs of LTR sequences were extracted and aligned using MUSCLE [74] (V3.8.31). The nucleotide distance K between one pair of LTRs was calculated using the Kimura two-parameter model in the distmat program within the EMBOSS package. Finally, the insert time T was calculated based on the formula T = K / 2_r_ with a mutation rate (r) of 2.2 × 10-9 substitutions per base per year [[45](/article/10.1186/s13742-016-0154-1#ref-CR45 "Buschiazzo E, Ritland C, Bohlmann J, Ritland K. Slow but not low: genomic comparisons reveal slower evolutionary rate and higher dN/dS in conifers compared to angiosperms. BMC Evol Biol. 2012;12:8. doi: 10.1186/1471-2148-12-8
.")\].Identification of LTR-RT elements
Two-round searches to identify Ty1/Copia and Ty3/Gypsy superfamilies in the genome of ginkgo and the other four land plants (P. patens, P. abies, P. trichocarpa and Z. mays) were performed based on the reported Ty3/Gypsy and Ty1/Copia reverse transcriptase domains with sequences EAYLDDLASRSRKRKDHPTHLRLIFERCRYFRIRLNPNKCSFCVTSGRLLGFIVSTTGIMVDPLKVGAIVQLPPPRTIVQLQSLQGKANFLRRFIANYAE and WKVYQMDVKSAFLNGYLEEEVYVQQPPRYEVRGQEDKVYRLKKALNGLKQAPRAWYSKIDSYMIKNEFIRSTSEPTLYTKVNEQGQILIVCLYVDDLIY, respectively. Genome sequences of Norway spruce were downloaded from database [[75](/article/10.1186/s13742-016-0154-1#ref-CR75 "Norway spruce genome sequence: ftp://plantgenie.org/Data/ConGenIE/Picea_abies
. Accessed 16 June 2016.")\]. The method was as follows:- (1)
We searched the Ty1- and _Ty3-_specific domain amino acid sequences using tBLASTn (V2.2.26) against whole genome sequences for gingko and the other four species using parameters -p tBLASTn -e 1e-5 -F F -m 8. Target hits were obtained using a strict filter criteria of identity ≥0.50 and coverage ratio ≥0.95”. - (2)
Sequence hits obtained from round one searches were used as queries for the second round of tBLASTn searches for all genome sequences using an enhanced filter criteria of identity ≥0.80 and coverage ratio ≥0.95. The resulting nucleotide sequences were re-aligned against the amino acid sequences of the two domains using tBLASTn with parameter -m 0, leading to the eventual identification of amino acid sequences for all Ty1 and Ty3 elements.
To construct Ty1 and Ty3 phylogenetic trees for the five species (G. biloba, P. patens, P. abies, P. trichocarpa and Z. mays), the resultant amino acid sequences were aligned using MUSCLE [74] (V3.8.31) with default parameters. Phylogenetic trees were inferred based on multiple sequence alignment using FastTree [76] (V2.1.9) with parameters of amino acid distances: BLOSUM45, joins: balanced, support: SH-like 1000, search: normal + NNI + SPR (2 rounds range 10) + ML-NNI, opt-each = 1, tophits: 1.00*sqrtN, close = default refresh = 0.80, ML model: Jones-Taylor-Thorton, and CAT approximation with 20 rate categories.
Gene model annotation
Protein sequences of S. moellendorffii, P. abies, P. taeda, A. thaliana and O. sativa were aligned against the ginkgo genome using tBLASTn (V2.2.26). Gene structure was predicted using GeneWise [77] (V2.2.0). Transcripts from seeds and leaves assembled from gingko RNA-seq and EST data separated from EST data in NCBI were mapped to the genome. The program PASA [78] was applied to assemble the spliced alignment results and annotate the candidate genes. Genes were predicted using the hidden Markov model in Augustus (V3.0.3) through a de novo approach. Results derived from different methods were integrated, and only those candidate genes consistently supported were retained in the final gene set. For function annotation, protein sequences were mapped to the Gene Ontology [79], KEGG [[80](/article/10.1186/s13742-016-0154-1#ref-CR80 "KEGG homepage: http://www.genome.jp/kegg/
. Accessed 16 June 2016.")\], InterPro \[[81](/article/10.1186/s13742-016-0154-1#ref-CR81 "Mitchell A, Chang HY, Daugherty L, Fraser M, Hunter S, Lopez R, McAnulla C, McMenamin C, Nuka G, Pesseat S, et al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 2015;43(Database issue):D213–21.")\], UniProt \[[82](/article/10.1186/s13742-016-0154-1#ref-CR82 "UniProt homepage:
http://www.ebi.ac.uk/uniprot
. Accessed 24 Apr 2016.")\] and Non-redundant protein NCBI databases.Detection of tandem duplicate genes
Pairwise self-alignment of protein sequences was conducted using BLASTp (V2.2.26), and protein sequences were sorted by coordinates in the assembled sequences. Genes with a distance greater than 2 were filtered, where the gene distance represents the number of genes between two focal genes. TDGs were calculated for G. biloba, Z. mays, A. thaliana, P. trichocarpa and G. max.
Analysis of phylogenetic reconstruction and evolution
CDSs of S. moellendorffii, G. biloba, A. thaliana and O. sativa were extracted based on genome annotation. Transcriptome sequencing data for P. canatriensis and C. revoluta were downloaded from NCBI (SRR1531151 and SRR1525778), and RNA-seq data were assembled using Trinity [71], resulting in 78,805 and 68,553 transcripts, respectively. The EMBOSS sixpack package [[83](/article/10.1186/s13742-016-0154-1#ref-CR83 "Emboss Sixpack package homepage: http://www.ebi.ac.uk/Tools/st/emboss_sixpack/
. Accessed 24 Apr 2016.")\] was employed to translate all possible proteins and to filter candidates by mapping to the NCBI plant protein database. A total of 28,925 and 32,512 proteins were annotated based on the transcriptomes of _P. canatriensis_ and _C. revoluta_, respectively_._ OrthoMCL (version 5) \[[44](/article/10.1186/s13742-016-0154-1#ref-CR44 "Li L, Stoeckert Jr CJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13(9):2178–89.")\] was used to cluster CDS from all six species and to identify the gene families. In total, 920 single copy genes were identified, and their phase1 sites were used to reconstruct phylogenetic trees using PhyML (V3.0) \[[84](/article/10.1186/s13742-016-0154-1#ref-CR84 "Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–21.")\]. To investigate the expansion and contraction of gene families, changes were identified by comparing 7795 gene families filtered from all 24,271 gene families using CAFÉ \[[85](/article/10.1186/s13742-016-0154-1#ref-CR85 "De Bie T, Cristianini N, Demuth JP, Hahn MW. CAFE: a computational tool for the study of gene family evolution. Bioinformatics. 2006;22(10):1269–71.")\].Identification of WGD events
All gingko amino acid sequences were self-aligned using BLASTp with filtering criteria identity ≥0.40, e value ≤1.0 × 10-5, more than 100 amino acids matched. To obtain paralogous gene families, we performed gene cluster analyses based on the CDS alignment. _K_s values were calculated for each paralogous family using yn00 in the PAML package. The _K_s of a given family was represented by the median value, and the distribution of corrected _K_s values was plotted by masking TDGs.
Abbreviations
CDS:
Coding sequence
EFR:
EF-Tu receptor
ESTs:
Expressed sequence tags
FLS:
Flavonol synthase
FLS2:
Flagellin-sensitive 2
LRR:
Leucine rich repeat
LTR-RTs:
Long terminal repeat retrotransposons
mya:
Million years ago
TDG:
Tandem duplicate gene
TE:
Transposable element
VOC:
Volatile organic compound
WGD:
Whole genome duplication
References
- Lee EK, Cibrian-Jaramillo A, Kolokotronis SO, Katari MS, Stamatakis A, Ott M, Chiu JC, Little DP, Stevenson DW, McCombie WR, et al. A functional phylogenomic view of the seed plants. PLoS Genet. 2011;7(12):e1002411.
Article CAS PubMed PubMed Central Google Scholar - Wu CS, Chaw SM, Huang YY. Chloroplast phylogenomics indicates that Ginkgo biloba is sister to cycads. Genome Biol Evol. 2013;5(1):243–54.
Article PubMed PubMed Central Google Scholar - Xi Z, Rest JS, Davis CC. Phylogenomics and coalescent analyses resolve extant seed plant relationships. PLoS One. 2013;8(11):e80870.
Article PubMed PubMed Central Google Scholar - Ruhfel BR, Gitzendanner MA, Soltis PS, Soltis DE, Burleigh JG. From algae to angiosperms-inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes. BMC Evol Biol. 2014;14:23.
Article PubMed PubMed Central Google Scholar - Lu Y, Ran JH, Guo DM, Yang ZY, Wang XQ. Phylogeny and divergence times of gymnosperms inferred from single-copy nuclear genes. PLoS One. 2014;9(9):e107679.
Article PubMed PubMed Central Google Scholar - Crane PR. Ginkgo : the tree that time forgot. New Haven: Yale University Press; 2013.
Google Scholar - Zhou Z-Y. An overview of fossil Ginkgoales. Palaeoworld. 2009;18(1):1–22.
Article CAS Google Scholar - Gong W, Chen C, Dobes C, Fu CX, Koch MA. Phylogeography of a living fossil: pleistocene glaciations forced Ginkgo biloba L. (Ginkgoaceae) into two refuge areas in China with limited subsequent postglacial expansion. Mol Phylogenet Evol. 2008;48(3):1094–105.
Article PubMed Google Scholar - Fan J, Shi H, Xiao H, Zhao G. Double-Layer 3D Macro-Mesoporous Metal Oxide Modified Boron-Doped Diamond with Enhanced Photoelectrochemical Performance. ACS Appl Mater Interfaces. 2016. doi:10.1021/acsami.6b01929.
Google Scholar - Zhao Y, Paule J, Fu C, Koch MA. Out of China: Distribution history of Ginkgo biloba L. Taxon. 2010;59(2):495–504.
Google Scholar - Major RT. The Ginkgo, the Most Ancient Living Tree. Science. 1967;157(3794):1270–3.
Article CAS PubMed Google Scholar - Pan L, Ren L, Chen F, Feng Y, Luo Y. Antifeedant Activity of Ginkgo biloba Secondary Metabolites against Hyphantria cunea Larvae: Mechanisms and Applications. PLoS One. 2016;11(5):e0155682.
Article PubMed PubMed Central Google Scholar - Mohanta TK, Occhipinti A, Atsbaha Zebelo S, Foti M, Fliegmann J, Bossi S, Maffei ME, Bertea CM. Ginkgo biloba responds to herbivory by activating early signaling and direct defenses. PLoS One. 2012;7(3):e32822.
Article CAS PubMed PubMed Central Google Scholar - Yang R, Fan X, Cai X, Hu F. The inhibitory mechanisms by mixtures of two endophytic bacterial strains isolated from Ginkgo biloba against pepper phytophthora blight. Biol Control. 2015;85:59–67.
Article Google Scholar - Brenner ED, Katari MS, Stevenson DW, Rudd SA, Douglas AW, Moss WN, Twigg RW, Runko SJ, Stellari GM, McCombie WR, et al. EST analysis in Ginkgo biloba: an assessment of conserved developmental regulators and gymnosperm specific genes. BMC Genomics. 2005;6:143.
Article PubMed PubMed Central Google Scholar - Lin X, Zhang J, Li Y, Luo H, Wu Q, Sun C, Song J, Li X, Wei J, Lu A, et al. Functional genomics of a living fossil tree, Ginkgo, based on next-generation sequencing technology. Physiol Plant. 2011;143(3):207–18.
Article CAS PubMed Google Scholar - He B, Gu Y, Xu M, Wang J, Cao F, Xu LA. Transcriptome analysis of Ginkgo biloba kernels. Front Plant Sci. 2015;6:819.
PubMed PubMed Central Google Scholar - Wang Y-Q, Shen J-K, Berglund T, Ohlsson AB, Tang X-F, Zhou Z-K, Wu R-Y, Zhou X-H, Chen J-N. Analysis of expressed sequence tags from Ginkgo mature foliage in China. Tree Genet Genomes. 2010;6(3):357–65.
Article Google Scholar - Nystedt B, Street NR, Wetterbom A, Zuccolo A, Lin YC, Scofield DG, Vezzi F, Delhomme N, Giacomello S, Alexeyenko A, et al. The Norway spruce genome sequence and conifer genome evolution. Nature. 2013;497(7451):579–84.
Article CAS PubMed Google Scholar - Birol I, Raymond A, Jackman SD, Pleasance S, Coope R, Taylor GA, Yuen MM, Keeling CI, Brand D, Vandervalk BP, et al. Assembling the 20 Gb white spruce (Picea glauca) genome from whole-genome shotgun sequencing data. Bioinformatics. 2013;29(12):1492–7.
Article CAS PubMed PubMed Central Google Scholar - Warren RL, Keeling CI, Yuen MM, Raymond A, Taylor GA, Vandervalk BP, Mohamadi H, Paulino D, Chiu R, Jackman SD, et al. Improved white spruce (Picea glauca) genome assemblies and annotation of large gene families of conifer terpenoid and phenolic defense metabolism. Plant J. 2015;83(2):189–212.
Article CAS PubMed Google Scholar - Kovach A, Wegrzyn JL, Parra G, Holt C, Bruening GE, Loopstra CA, Hartigan J, Yandell M, Langley CH, Korf I, et al. The Pinus taeda genome is characterized by diverse and highly diverged repetitive sequences. BMC Genomics. 2010;11:420.
Article PubMed PubMed Central Google Scholar - Neale DB, Wegrzyn JL, Stevens KA, Zimin AV, Puiu D, Crepeau MW, Cardeno C, Koriabine M, Holtz-Morris AE, Liechty JD, et al. Decoding the massive genome of loblolly pine using haploid DNA and novel assembly strategies. Genome Biol. 2014;15(3):1–13.
Article Google Scholar - Wegrzyn JL, Liechty JD, Stevens KA, Wu LS, Loopstra CA, Vasquez-Gross HA, Dougherty WM, Lin BY, Zieve JJ, Martinez-Garcia PJ et al: Unique features of the loblolly pine (Pinus taeda L.) megagenome revealed through sequence annotation. Genetics. 2014; 196. DOI: 10.1534/genetics.113.159996.
- Zimin A, Stevens K, Crepeau M, Holtz-Morris A, Korabline M, Marçais G, Puiu D, Roberts M, Wegrzyn J, de Jong P et al: Sequencing and assembly of the 22-Gb loblolly pine genome. Genetics. 2014; 196. DOI: 10.1534/genetics.113.159715
- Wegrzyn J, Lin B, Zieve J, Dougherty W, Martínez-García P, Koriabine M, Holtz-Morris A, de Jong P, Crepeau M, Langley C et al: Insights into the loblolly pine genome: characterization of BAC and fosmid sequences. PloS One 2013; 8, DOI: 10.1371/journal.pone.0072439.
- Huang Y, Kendall T, Forsythe ES, Dorantes-Acosta A, Li S, Caballero-Pérez J, Chen X, Arteaga-Vázquez M, Beilstein MA, Mosher RA. Ancient Origin and Recent Innovations of RNA Polymerase IV and V. Mol Biol Evol. 2015;32(7):1788–99.
Article CAS PubMed PubMed Central Google Scholar - Zonneveld BJM. Conifer genome sizes of 172 species, covering 64 of 67 genera, range from 8 to 72 picogram. Nord J Bot. 2012;30(4):490–502.
Article Google Scholar - Divi UK, Zhou X-R, Wang P, Butlin J, Zhang D-M, Liu Q, Vanhercke T, Petrie JR, Talbot M, White RG. Deep sequencing of the fruit transcriptome and lipid accumulation in a non-seed tissue of Chinese tallow, a potential biofuel crop. Plant Cell Physiol. 2016;57(1):125-37. doi: 10.1093/pcp/pcv181.
- Guan R, Zhao Y, Zhang H, Fan G, Liu X, Zhou W, Shi C, Wang J, Liu W, Liang X, Fu Y, Ma K, Zhao L, Zhang F, Lu Z, Lee SM, Xu X, Wang J, Yang H, Fu C, Ge S, Chen1 W. De novo sequencing of Ginkgo biloba. GigaScience Database 2016, http://dx.doi.org/10.5524/100209.
- Liu B, Yuan J, Hu X, Zhang H, Li N, Li Z, Chen Y, Mu D, Fan W: Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. 2013.
- Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–2.
Article CAS PubMed Google Scholar - Szerszen JB, Szczyglowski K, Bandurski RS. iaglu, a gene from Zea mays involved in conjugation of growth hormone indole-3-acetic acid. Science. 1994;265(5179):1699–701.
Article CAS PubMed Google Scholar - Bohlmann J, Crock J, Jetter R, Croteau R. Terpenoid-based defenses in conifers: cDNA cloning, characterization, and functional expression of wound-inducible (E)-alpha-bisabolene synthase from grand fir (Abies grandis). Proc Natl Acad Sci U S A. 1998;95(12):6756–61.
Article CAS PubMed PubMed Central Google Scholar - Wicker T, Keller B. Genome-wide comparative analysis of copia retrotransposons in Triticeae, rice, and Arabidopsis reveals conserved ancient evolutionary lineages and distinct dynamics of individual copia families. Genome Res. 2007;17(7):1072–81.
Article CAS PubMed PubMed Central Google Scholar - Smykal P, Kalendar R, Ford R, Macas J, Griga M. Evolutionary conserved lineage of Angela-family retrotransposons as a genome-wide microsatellite repeat dispersal agent. Heredity. 2009;103(2):157–67.
Article CAS PubMed Google Scholar - Moisy C, Schulman AH, Kalendar R, Buchmann JP, Pelsy F. The Tvv1 retrotransposon family is conserved between plant genomes separated by over 100 million years. Theor Appl Genet. 2014;127(5):1223–35.
Article CAS PubMed Google Scholar - Zuccolo A, Scofield DG, De Paoli E, Morgante M. The Ty1-copia LTR retroelement family PARTC is highly conserved in conifers over 200 MY of evolution. Gene. 2015;568(1):89–99.
Article CAS PubMed Google Scholar - SanMiguel P, Gaut BS, Tikhonov A, Nakajima Y, Bennetzen JL. The paleontology of intergene retrotransposons of maize. Nat Genet. 1998;20(1):43–5.
Article CAS PubMed Google Scholar - Stival Sena J, Giguere I, Boyle B, Rigault P, Birol I, Zuccolo A, Ritland K, Ritland C, Bohlmann J, Jones S, et al. Evolution of gene structure in the conifer Picea glauca: a comparative analysis of the impact of intron size. BMC Plant Biol. 2014;14:95.
Article PubMed PubMed Central Google Scholar - Schnable JC, Springer NM, Freeling M. Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc Natl Acad Sci U S A. 2011;108(10):4069–74.
Article CAS PubMed PubMed Central Google Scholar - Castillo-Davis CI, Mekhedov SL, Hartl DL, Koonin EV, Kondrashov FA. Selection for short introns in highly expressed genes. Nat Genet. 2002;31(4):415–8.
CAS PubMed Google Scholar - Blanc G, Wolfe KH. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell. 2004;16(7):1667–78.
Article CAS PubMed PubMed Central Google Scholar - Li L, Stoeckert Jr CJ, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13(9):2178–89.
Article CAS PubMed PubMed Central Google Scholar - Buschiazzo E, Ritland C, Bohlmann J, Ritland K. Slow but not low: genomic comparisons reveal slower evolutionary rate and higher dN/dS in conifers compared to angiosperms. BMC Evol Biol. 2012;12:8. doi:10.1186/1471-2148-12-8.
Article PubMed PubMed Central Google Scholar - Li Z, Baniaga AE, Sessa EB, Scascitelli M, Graham SW, Rieseberg LH, Barker MS: Early genome duplications in conifers and other seed plants. Sci Adv. 2015;1(10):e1501084. doi:10.1126/sciadv.1501084.
- Cui L, Wall PK, Leebens-Mack JH, Lindsay BG, Soltis DE, Doyle JJ, Soltis PS, Carlson JE, Arumuganathan K, Barakat A, et al. Widespread genome duplications throughout the history of flowering plants. Genome Res. 2006;16(6):738–49.
Article CAS PubMed PubMed Central Google Scholar - Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, Tomsho LP, Hu Y, Liang H, Soltis PS, et al. Ancestral polyploidy in seed plants and angiosperms. Nature. 2011;473(7345):97–100.
Article CAS PubMed Google Scholar - Zhou Z-Y. An overview of fossil Ginkgoales. Palaeoworld. 2009;18(1-22):206.
Google Scholar - Gomez-Gomez L, Boller T. FLS2: an LRR receptor-like kinase involved in the perception of the bacterial elicitor flagellin in Arabidopsis. Mol Cell. 2000;5(6):1003–11.
Article CAS PubMed Google Scholar - Zipfel C, Kunze G, Chinchilla D, Caniard A, Jones JD, Boller T, Felix G. Perception of the bacterial PAMP EF-Tu by the receptor EFR restricts Agrobacterium-mediated transformation. Cell. 2006;125(4):749–60. doi:10.1016/j.cell.2006.03.037.
Article CAS PubMed Google Scholar - Birol I, Raymond A, Jackman SD, Pleasance S, Coope R, Taylor GA, Saint Yuen MM, Keeling CI, Brand D, Vandervalk BP, et al. Assembling the 20 Gb white spruce (Picea glauca) genome from whole-genome shotgun sequencing data. Bioinformatics. 2013;29(12):1492-7. doi:10.1093/bioinformatics/btt178.
- Ohri D, Khoshoo TN. Genome size in gymnosperms. Plant Syst Evol. 1986;153(1):119–32. doi:10.1007/BF00989421.
Article Google Scholar - De La Torre A, Birol I, Bousquet J, Ingvarsson P, Jansson S, Jones SJ, Keeling CI, MacKay J, Nilsson O, Ritland K, et al. Insights into Conifer Giga-Genomes. Plant Physiol. 2014;166(4):1724–32.
Article Google Scholar - Cai J, Liu X, Vanneste K, Proost S, Tsai WC, Liu KW, Chen LJ, He Y, Xu Q, Bian C, et al. The genome sequence of the orchid Phalaenopsis equestris. Nat Genet. 2015;47(1):65–72.
Article CAS PubMed Google Scholar - Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A, et al. The Sorghum bicolor genome and the diversification of grasses. Nature. 2009;457(7229):551–6.
Article CAS PubMed Google Scholar - Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449(7161):463–7.
Article CAS PubMed Google Scholar - International Rice Genome Sequencing P. The map-based sequence of the rice genome. Nature. 2005;436(7052):793–800. doi:10.1038/nature03895.
Article Google Scholar - Wolf PG, Sessa EB, Marchant DB, Li F-W, Rothfels CJ, Sigel EM, Gitzendanner MA, Visger CJ, Banks JA, Soltis DE, et al. An exploration into fern genome space. Genome Biol Evol. 2015;7(9):2533-44. doi:10.1093/gbe/evv163.
- Scott AD, Stenz NW, Ingvarsson PK, Baum DA. Whole genome duplication in coast redwood (Sequoia sempervirens) and its implications for explaining the rarity of polyploidy in conifers. New Phytol. 2016;211(1):186–93.
Article CAS PubMed Google Scholar - Ahuja MR. Polyploidy in gymnosperms: revisited. Silvae Genetica. 2005;54(2):59–69.
Google Scholar - Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326(5956):1112–5.
Article CAS PubMed Google Scholar - Baucom RS, Estill JC, Chaparro C, Upshaw N, Jogi A, Deragon JM, Westerman RP, Sanmiguel PJ, Bennetzen JL. Exceptional diversity, non-random distribution, and rapid evolution of retroelements in the B73 maize genome. PLoS Genet. 2009;5(11):e1000732.
Article PubMed PubMed Central Google Scholar - Bennetzen JL, Ma J, Devos KM. Mechanisms of recent genome size variation in flowering plants. Ann Bot. 2005;95(1):127–32.
Article CAS PubMed PubMed Central Google Scholar - Imbiscuso G, Trotta A, Maffei M, Bossi S. Herbivory induces a ROS burst and the release of volatile organic compounds in the fern Pteris vittata L. J Plant Interact. 2009;4(1):15–22.
Article CAS Google Scholar - Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull. 1987;19:11–5.
Google Scholar - Tel-Zur N, Abbo S, Myslabodski D, Mizrahi Y. Modified CTAB procedure for DNA isolation from epiphytic cacti of the genera Hylocereus and Selenicereus (Cactaceae). Plant Mol Biol Report. 1999;17(3):249–54.
Article CAS Google Scholar - Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 2012;1(1):18.
Article PubMed PubMed Central Google Scholar - Short Oligonucleotide Analysis Package homepage: http://soap.genomics.org.cn/. Accessed 16 June 2016.
- SOAPdenovo2 GitHub: https://github.com/gigascience/paper-luo2012. Accessed 16 June 2016.
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–52.
Article CAS PubMed PubMed Central Google Scholar - Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12(4):656–64.
Article CAS PubMed PubMed Central Google Scholar - Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110(1-4):462–7.
Article CAS PubMed Google Scholar - Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7.
Article CAS PubMed PubMed Central Google Scholar - Norway spruce genome sequence: ftp://plantgenie.org/Data/ConGenIE/Picea_abies. Accessed 16 June 2016.
- Price MN, Dehal PS, Arkin AP. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5(3):e9490.
Article PubMed PubMed Central Google Scholar - Birney E, Clamp M, Durbin R. GeneWise and Genomewise. Genome Res. 2004;14(5):988–95.
Article CAS PubMed PubMed Central Google Scholar - Haas BJ, Delcher AL, Mount SM, Wortman JR, Smith Jr RK, Hannick LI, Maiti R, Ronning CM, Rusch DB, Town CD, et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003;31(19):5654–66.
Article CAS PubMed PubMed Central Google Scholar - Gene Ontology C. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43(Database issue):D1049–56.
Article Google Scholar - KEGG homepage: http://www.genome.jp/kegg/. Accessed 16 June 2016.
- Mitchell A, Chang HY, Daugherty L, Fraser M, Hunter S, Lopez R, McAnulla C, McMenamin C, Nuka G, Pesseat S, et al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 2015;43(Database issue):D213–21.
Article PubMed Google Scholar - UniProt homepage: http://www.ebi.ac.uk/uniprot. Accessed 24 Apr 2016.
- Emboss Sixpack package homepage: http://www.ebi.ac.uk/Tools/st/emboss_sixpack/. Accessed 24 Apr 2016.
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–21.
Article CAS PubMed Google Scholar - De Bie T, Cristianini N, Demuth JP, Hahn MW. CAFE: a computational tool for the study of gene family evolution. Bioinformatics. 2006;22(10):1269–71.
Article PubMed Google Scholar
Funding
This work was financially supported by Technology Innovation Program Support from the Shenzhen Municipal Government (NO.JSGG20130918102805062), Basic Research Program Support from the Shenzhen Municipal Government (NO.JCYJ20120618172523025), the Shenzhen Municipal Government of China (NO.JCYJ20150529150505656), the Public Technology Research Project of Zhejiang Province (2014C32107), the National Natural Science Foundation of China (31000102) and the CAS/SAFEA International Partnership Program for Creative Research Teams.
Availability of supporting data and materials
Raw and transcriptomic data are available from NCBI bioproject PRJNA307642. Supporting genome assemblies, annotations, supplemental data and custom scripts are hosted in the GigaScience GigaDB repository [[30](/article/10.1186/s13742-016-0154-1#ref-CR30 "Guan R, Zhao Y, Zhang H, Fan G, Liu X, Zhou W, Shi C, Wang J, Liu W, Liang X, Fu Y, Ma K, Zhao L, Zhang F, Lu Z, Lee SM, Xu X, Wang J, Yang H, Fu C, Ge S, Chen1 W. De novo sequencing of Ginkgo biloba. GigaScience Database 2016, http://dx.doi.org/10.5524/100209
.")\].Authors’ contributions
RG participated in the acquisition of sequencing data, gene set annotation, evolution analysis, gene duplication analysis and manuscript co-drafting. YZ designed sample collection and transcriptome analysis, coordinated scientific questions, and co-drafted and integrated the manuscript. HZ performed data input and processing, genome assembly, repeat sequence annotation, and sample management, and co-drafted the manuscript. GF participated in the LTR-RT evolution analysis, transcriptome assembly, project management and manuscript co-drafting. XX and X Liu participated in study design, data output and project management. CS, WL and JW participated in the LTR-RT insertion analysis. WZ collected samples, performed the transcriptome analysis and co-drafted the manuscript. YF, KM and X Liang helped with data processing. LZ participated in transcriptome analysis. Fumin Zhang contributed to the study design. ZL and SMYL participated in manuscript revision. WC, SG and CF conceived the overall study and revised the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Author notes
Authors and Affiliations
- BGI-Shenzhen, Shenzhen, 518083, China
Rui Guan, He Zhang, Guangyi Fan, Xin Liu, Chengcheng Shi, Jiahao Wang, Xinming Liang, Yuanyuan Fu, Kailong Ma, Xun Xu, Jian Wang, Huanming Yang & Wenbin Chen - The Key Laboratory of Conservation Biology for Endangered Wildlife of the Ministry of Education, College of Life Sciences, Zhejiang University, Hangzhou, 310058, China
Yunpeng Zhao, Wenbin Zhou, Lijun Zhao & Chengxin Fu - State Key Laboratory of Systematic and Evolutionary Botany, Institute of Botany, Chinese Academy of Sciences, Beijing, 100093, China
Fumin Zhang & Song Ge - BGI-Qingdao, Qingdao, 266555, China
Rui Guan, He Zhang, Guangyi Fan & Wenbin Chen - State Key Laboratory of Bioelectronics, School of Biological Science and Medical Engineering, Southeast University, Nanjing, 210096, China
Rui Guan, Yuanyuan Fu & Zuhong Lu - Laboratory of Systematic & Evolutionary Botany and Biodiversity, Institute of Ecology and Conservation Center for Gene Resources of Endangered Wildlife, Zhejiang University, Hangzhou, 310058, China
Yunpeng Zhao, Wenbin Zhou, Lijun Zhao & Chengxin Fu - State Key Laboratory of Quality Research in Chinese Medicine and Institute of Chinese Medical Sciences, Macao, China
Guangyi Fan & Simon Ming-Yuen Lee - Stanley Ho Centre for Emerging Infectious Diseases, Faculty of Medicine, The Chinese University of Hong Kong, Shatin, Hong Kong
He Zhang - BGI-Wuhan, BGI-Shenzhen, Wuhan, 430074, China
Weiqing Liu - James D. Watson Institute of Genome Sciences, Hangzhou, 310058, China
Jian Wang & Huanming Yang
Authors
- Rui Guan
You can also search for this author inPubMed Google Scholar - Yunpeng Zhao
You can also search for this author inPubMed Google Scholar - He Zhang
You can also search for this author inPubMed Google Scholar - Guangyi Fan
You can also search for this author inPubMed Google Scholar - Xin Liu
You can also search for this author inPubMed Google Scholar - Wenbin Zhou
You can also search for this author inPubMed Google Scholar - Chengcheng Shi
You can also search for this author inPubMed Google Scholar - Jiahao Wang
You can also search for this author inPubMed Google Scholar - Weiqing Liu
You can also search for this author inPubMed Google Scholar - Xinming Liang
You can also search for this author inPubMed Google Scholar - Yuanyuan Fu
You can also search for this author inPubMed Google Scholar - Kailong Ma
You can also search for this author inPubMed Google Scholar - Lijun Zhao
You can also search for this author inPubMed Google Scholar - Fumin Zhang
You can also search for this author inPubMed Google Scholar - Zuhong Lu
You can also search for this author inPubMed Google Scholar - Simon Ming-Yuen Lee
You can also search for this author inPubMed Google Scholar - Xun Xu
You can also search for this author inPubMed Google Scholar - Jian Wang
You can also search for this author inPubMed Google Scholar - Huanming Yang
You can also search for this author inPubMed Google Scholar - Chengxin Fu
You can also search for this author inPubMed Google Scholar - Song Ge
You can also search for this author inPubMed Google Scholar - Wenbin Chen
You can also search for this author inPubMed Google Scholar
Corresponding authors
Correspondence toChengxin Fu, Song Ge or Wenbin Chen.
Additional files
Additional file 1: Table S1.
Statistics for gingko genome raw sequencing data. Table S2. Statistics for ginkgo genome clean sequencing data. Table S3. Statistics for RNA-seq data from male and female ginkgo samples. Table S4. Comparisons of DNA yield and genome survey analysis. Table S5. Transposable elements in the ginkgo genome. Table S6. Gene model annotation. Table S7. Gene functional annotation. Table S8. Comparison of gene metrics for the genomes of ginkgo and seven other land plants. Table S9. Comparison of gene family clusters between ginkgo and five other land plants. Table S10. Functional enrichment of specific genes in ginkgo. Table S11. Comparison of TDGs in the genomes of ginkgo and seven other land plants. (DOCX 41 kb)
Additional file 2: Figure S1.
17-mer frequency distribution of ginkgo genome sequencing. Figure S2. Distribution of sequence depth for the ginkgo genome. The x-axis represents the depth and the y-axis represents the proportion of the corresponding DNA bases. Clean reads were aligned against genic regions and the percentage of bases with different sequencing depth was calculated. Figure S3. The distribution of sequence divergence rates of TEs in the ginkgo genome. (a) Based on the Repbase-comparison approach; (b) Based on the de novo approach. Figure S4. Comparison of the length distribution of gene sets in ginkgo and four other land plants. Figure S5. Comparison of orthologous genes in ginkgo and five other land plants. Figure S6. Comparison of gene structure in G. biloba and four other land plants. Five land plants are included in each gene cluster: A. thalina in orange, G. biloba in yellow, O. sativa in green, P. abies in pink and S. moellendorffii in blue. Figure S7. Comparison of genome size and average intron length between ginkgo and four other land plants. Note that the intron size in P. abies might be underestimated due to the assembly quality of its genome. Figure S8. Comparison of the distribution of repeat elements between intergenic and intron regions in ginkgo. (DOCX 641 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article
Cite this article
Guan, R., Zhao, Y., Zhang, H. et al. Draft genome of the living fossil Ginkgo biloba.GigaSci 5, 49 (2016). https://doi.org/10.1186/s13742-016-0154-1
- Received: 17 August 2016
- Accepted: 01 November 2016
- Published: 21 November 2016
- DOI: https://doi.org/10.1186/s13742-016-0154-1