Gene Transfer of Tumor-Reactive TCR Confers Both High Avidity and Tumor Reactivity to Nonreactive Peripheral Blood Mononuclear Cells and Tumor-Infiltrating Lymphocytes (original) (raw)
. Author manuscript; available in PMC: 2008 Jan 4.
Abstract
Cell-based antitumor immunity is driven by CD8+ cytotoxic T cells bearing TCR that recognize specific tumor-associated peptides bound to class I MHC molecules. Of several cellular proteins involved in T cell:target-cell interaction, the TCR determines specificity of binding; however, the relative amount of its contribution to cellular avidity remains unknown. To study the relationship between TCR affinity and cellular avidity, with the intent of identifying optimal TCR for gene therapy, we derived 24 MART-1:27–35 (MART-1) melanoma Ag-reactive tumor-infiltrating lymphocyte (TIL) clones from the tumors of five patients. These MART-1-reactive clones displayed a wide variety of cellular avidities. α and β TCR genes were isolated from these clones, and TCR RNA was electroporated into the same non-MART-1-reactive allogeneic donor PBMC and TIL. TCR recipient cells gained the ability to recognize both MART-1 peptide and MART-1-expressing tumors in vitro, with avidities that closely corresponded to the original TCR clones (p = 0.018–0.0003). Clone DMF5, from a TIL infusion that mediated tumor regression clinically, showed the highest avidity against MART-1 expressing tumors in vitro, both endogenously in the TIL clone, and after RNA electroporation into donor T cells. Thus, we demonstrated that the TCR appeared to be the core determinant of MART-1 Ag-specific cellular avidity in these activated T cells and that nonreactive PBMC or TIL could be made tumor-reactive with a specific and predetermined avidity. We propose that inducing expression of this highly avid TCR in patient PBMC has the potential to induce tumor regression, as an “off-the-shelf” reagent for allogeneic melanoma patient gene therapy.
In vivo, the antitumor immune response relies on CD8+ CTL recognition of antigenic tumor peptides bound to MHC class I molecules (pMHC)3 displayed on the tumor cell surface (1–3). Currently, an effective treatment for patients with metastatic melanoma involves obtaining tumor-reactive T cells from excised tumors, expanding these T cells in vitro and reinfusing them into a lymphodepleted patient (4, 5). This treatment, commonly referred to as adoptive cell transfer (ACT), achieves a 51% objective response rate as determined by Response Evaluation Criteria in Solid Tumors (6). Patients must satisfy several requirements to receive this ACT immunotherapy: they must have surgically resectable tumors, these tumors must generate viable tumor-infiltrating lymphocytes (TIL) in the laboratory, the TIL must be reactive against tumor Ags, and these tumor-Ag reactive TIL must expand in vitro to sufficient numbers for patient treatment. Fewer than half of patients chosen for ACT therapy meet these treatment requirements. Of those patients that do receive ACT, the ability of each TIL infusion to recognize and lyse tumor can vary dramatically, likely influencing the outcome of therapy (7–9). For many patients with cancers other than melanoma, it is difficult to obtain tumor-reactive TIL. A potential solution to these problems is the transduction of genes encoding tumor-reactive TCR into patient PBL to convert them into tumor-reactive T cells (10–13). This type of experiment has recently been reported in a mouse model, showing that T cells transduced with a retrovirus encoding a TCR against a self-expressed OVA Ag can persist and function in vivo in OVA transgenic mice (14).
Several groups have shown that highly avid T cells have superior function over low-avidity cells in CD8+ CTL-mediated immunity against tumors (15–17) and viral infections (18, 19) in vivo. Recent results have linked high-affinity TCR with functional avidity in vivo in a mouse model (20), suggesting that expression of a high affinity TCR may confer high functional avidity to a T cell. Determination of TCR:pMHC affinity can be measured directly in vitro by surface plasmon resonance evaluation of binding interaction between the isolated proteins (21). In vivo, many factors contribute to the avidity of TCR-specific T cells interacting with target cells displaying cognate Ag bound to MHC. While TCR:pMHC binding is known to determine the specificity of interaction (22–24) and TCR affinity contributes to overall cellular avidity (25, 26), additional factors also affect T cell activation. The numbers and immune synapse clustering of TCR:pMHC interactions have been shown to play a role in T cell activation (27–31), and additional coreceptor:ligand interactions such as CD8/MHC H chain binding and CD28/CD80 and CD86 are also known to contribute to overall cellular avidity (8, 28, 32–35).
The substantial clinical response rate in ACT-based tumor immunotherapy demonstrates that T cells with TCR of sufficiently high avidity to eliminate tumors do exist in vivo. If these TCR were sufficient to determine cellular avidity, then the identification of an exceptionally high avidity TCR would provide an ideal candidate gene for allogeneic TCR gene therapy. Although it has been suggested that in vitro T cell binding of fluorescently labeled multimeric pMHC complexes may provide a surrogate measure of TCR affinity, a number of authors have reported a disconnect between mean fluorescence intensity of tetramer staining and TCR affinity (35–38). Several models of TCR:pMHC interaction are suggestive that naturally occurring T cell avidity may be primarily determined by TCR:pMHC affinity (25, 26); however, this hypothesis has yet to be tested empirically. Although there is no clear predictive relationship between tetramer binding intensity and functional avidity of T cells, it has recently been shown that high avidity T cells can be identified by their ability to bind pMHC tetramers independent of CD8 coreceptor binding (39).
In this study, multiple T cell clones reactive to the melanoma tumor Ag, MART-1:27–35 (MART-1) (40, 41) were isolated from TIL obtained from tumors from five different melanoma patients treated at the National Cancer Institute, Surgery Branch. MART-1-specific T cell clones from these TIL were characterized according to their ability to produce IFN-γ in response to coculture with MART-1 peptide or MART-1 expressing HLA-A*0201-positive melanoma tumor cells. Clones were then evaluated for their ability to bind MART-1:26–35(27L)/HLA-A*0201 tetramers in a CD8-dependent and independent fashion. Each clone was determined to be of high, medium, or low avidity based on their IFN-γ production in response to coculture with MART-1 peptide-pulsed targets and MART-1-expressing tumor cells, combined with an assessment of TCR affinity based on the ability to bind MART tetramer in a CD8-dependent or independent manner. The genes encoding the TCR _α_- and _β_-chains from multiple clones were then expressed in a common cell to study the degree to which the TCR affinity determines the overall avidity of effector T cells and to evaluate means to increase TCR expression and function.
Materials and Methods
Cell lines, TIL, and PBMC
All primary tissues and cells used in this study were obtained from patient samples at the Surgery Branch, National Cancer Institute.
Melanoma cell lines, mel526, mel624 (MART-1+, HLA-A*0201+), and mel888 and mel938 (HLA-A*0201−) were generated from resected tumor lesions. T2 is a lymphoblastoid cell line deficient in TAP function, whose HLA-A*0201 molecules can be readily loaded by pulsing with exogenous peptide. Jurkat T3.5 is a CD4-restricted T cell line lacking surface expression of TCR and CD3 due to a defect in the TCR _β_-chain.
PBMC used were cryopreserved leukopheresis samples of donor blood. TIL and tumor digests were generated from resected patient tumors, and provided by the TIL lab (Surgery Branch, National Cancer Institute).
Cloning of MART-1-reactive TIL
Fresh melanoma tumors from three patients were digested and cultured in vitro for 0–14 days in presence of 6000 IU (1000 Cetus units (CU) of IL-2; Chiron) in a 1:1 mix of Stemline medium (Sigma-Aldrich) (STEM):RPMI 1640 (Invitrogen Life Technologies) culture medium plus 10% human AB serum (Gemini Bioproduct), HEPES, 2-ME, 100 U/ml penicillin, and 100 _μ_g/ml Fungizone. Cells were costained with a panel of TCR VB Abs (Coulter Immunotech) and MART-1:26–35(27L)/HLA-A*0201 tetramer (Beckman-Coulter Immunomics) at day 0, 7, and 14. The widest diversity VB of MART-1-reactive T cells combined with the highest proportion of MART-1-reactive cells was observed between day 7–14 and varied between patients. TIL samples were taken from the time point at which MART-1-reactive T cell numbers and diversity were maximal and cloned by limiting dilution at two or five cells per well in 96-well plates in the presence of 400-fold excess 4000-rad irradiated allogeneic PBMC feeders, 30 ng/ml OKT-3, and 1000 CU of IL-2. Clones were sampled at day 14 for their ability to produce IFN-γ specifically in response to MART-1 peptide-pulsed target cells. These TIL clones were expanded with a second exposure to irradiated PBMC feeder cells and OKT-3 in 1000 CU of IL-2.
Evaluating cellular avidity
On day 14 of expansion, the clones were evaluated for their cellular avidity to MART-1 Ag by measuring IFN-γ secretion (ELISA paired Abs obtained from Pierce Endogen) in supernatant of 18-h cocultures with titrated MART-1 peptide on target cells or with MART-1 expressing HLA-A*0201 melanoma tumor cells. These assays were conducted with 1 × 105 effectors and 1 × 105 target cells in 0.2 ml of RPMI 1640 + 10% FBS (Invitrogen Life Technologies) in round-bottom 96-well plates at 37°C, 5% CO2. TCR CD8 independence was evaluated by determining the ability of the clones to bind to MART-1:26–35(27L)/HLA-A*0201 tetramers generated either with an intact CD8-binding region (WT), or with this binding abrogated by inclusion of a double mutation in the HLA-A*A0201 molecule (mut). These tetramers were generated by the National Institute of Allergy and Infectious Diseases tetramer facility.
TCR gene isolation
RNA was purified from each clone using Qiagen RNEasy, and 5′ RACE was performed using BD SmartRace reagents and protocol, using the universal 5′ primer, and a 3′ gene-specific primer for the TCR α constant region, or C1 or C2 β constant regions. Results were run on a gel and appropriately sized bands (800–900 bp) were excised, subcloned into pCR2.1 (Invitrogen Life Technologies) vector, and sequenced.
In vitro TCR RNA transcription and expression in PBMC
Gene-specific oligonucleotide primers were generated for the production of in vitro RNA transcription (IVT). 5′ primer design included the T7 polymerase binding sequence, followed immediately by a Kozak sequence, a start codon and the next 19–15 bp of V_α_ or V_β_ region for each TCR gene; JKF6 B28 fwd T7 gene-specific primer 5′-TAA TAC GAC TCA CTA TAG GGA GAA CCG CCA TGG GAA TCA GGC TCC TCT GTC G-3′; M5 B6.5 fwd 5′-TAA TAC GAC TCA CTA TAG GGA GAA CCG CCA TGA GCA TCG GCC TCC TGT GCT G-3′; M7 B19 fwd 5′-TAA TAC GAC TCA CTA TAG GGA GAA CCG CCA TGA GCA ACC AGG TGC TCT GCT G-3′; M9/20 B4.1 fwd 5′-TAA TAC GAC TCA CTA TAG GGA GAA CCG CCA TGG GCT GCA GGC TGC TCT GCT G-3′; #17 B11.2 fwd 5′-TAA TAC GAC TCA CTA TAG GGA GAA CCG CCA TGG GCA CCA GGC TCC TCT GCT G-3′; DMF4 B10.3 fwd 5′-TAA TAC GAC TCA CTA TAG GGA GAA CCG CCA TGG CAC AAG GTT GTT CTT C-3′; DMF5 B6.4 fwd 5′-TAA TAC GAC TCA CTA TAG GGA GAA CCG CCA TGA GAA TCA GGC TCC TGT-3′. VA regions were either 12.2 fwd 5′-TAA TAC GAC TCA CTA TAG GGA GAA CCG CCA GCA AAT CCT TGA GAG GTT TAC-3′; 12.3 fwd 5′-TAA TAC GAC TCA CTA TAG GGA GAA CCG CCA TGA TGA AAT CCT TGA GAG TTT TAC-3′; OR 35 fwd 5′-TAA TAC GAC TCA CTA TAG GGA GAA CCG CCA TGC TCC TTG AAC ATT TAT TAA TC-3′. 3′ primers included 64T and 18–25 bp of the relevant α or β constant region sequence. Reverse primers were C-α 5′-(64)T TTC AAC TGG ACC ACA GCC TCA GC-3′; C1-β 5′-(64)T TTC ATG AAT TCT TTC TTT TCA CC-3′ or C2-β 5′-(64)T TCT AGC CTC TGG AAT CCT TTC TCT TG-3′.
For IVT, a PCR product was generated using the subcloned cDNA in pCR2.1 as a template with the above oligonucleotide primer sets. Resulting bands were gel purified and used for a second round of PCR amplification. PCR product was cleaned using Zymogen DNA purification columns. One to 3 _μ_g of PCR product was used as template for IVT using Ambion T7 mMESSAGE MACHINE as per the manufacturer’s instructions, followed by RNA clean up using Qiagen RNEasy. RNA quantity was measured at OD260 by spectrophotometer, and quality was determined by running 1 _μ_g on a 2% agarose gel in denaturing loading buffer, following 5 min of incubation at 70°C.
In preparation for RNA electroporation, donor TIL or PBMC from phereses were stimulated in vitro with 50 ng/ml OKT-3, 50 CU of IL-2 in STEM:RPMI 1640 medium for 3 days, when CD8+ or CD4+ cells were positively selected using Miltenyi Biotec microbeads and magnetic columns. PBMC were then grown in vitro an additional 2–15 days in IL-2 containing medium before use. For TCR electroporation, 0.5–2.0 _μ_g of RNA from each TCR gene was used per 1 × 106 cells (at 2.5 × 107 cells/ml in Opti-MEM serum-free medium (Invitrogen Life Technologies). Cells were rested for 2–16 h without IL-2 after electroporation before use in FACS staining or coculture experiments.
Evaluating T cell function
Cocultures were set up at 1 × 105:1 × 105 (E:T) cells in complete medium (RPMI 1640 + 10% FBS (Invitrogen Life Technologies)) in 96-well plates and incubated overnight at 37°C. All samples were run in duplicate, and RNA electroporations were repeated in PBMC or TIL from three different donors. Supernatant was tested for cytokine secretion by IFN-γ ELISA (Pierce Endogen).
CTL assays coincubated decreasing ratios of effectors and 51Cr-labeled target cells (E:T) in complete medium in 96-well plates at 37°C for 4 h. Lysis was measured by 51Cr release in the medium: percent lysis = (sample release − minimum release)/(maximum release − minimum release) × 100%, average of duplicate samples.
Peptides used
Peptides used for coculture included MART-1:27–35, gp100:154–162, gp100:209–217, and gp100:280–288. Dilutions were made 1000× in DMSO before use. With the exception of MART-1, following 1 h pulsing, all target cells were washed three times before use; due to low solubility, MART-1 peptide was left in final coculture medium.
Flow cytometry
Cell surface expression of CD3 and CD8 was measured using FITC-conjugated Abs in conjunction with MART-1:26–35(27L)/HLA-A*0201 tetramers (Beckman Coulter) to measure surface MART-1 TCR on electroporated cells. Isotype controls were used to gate all samples. Cells were run on FACSCalibur or FACScan flow cytometers (BD Biosciences), with CellQuest software and analyzed using FlowJo software (Tree Star).
Results
Generating TIL clones directly from patient tumor digests reveals a diversity of MART-1-reactive T cells with varying cellular avidities
Surgically resected tumor specimens were obtained from three patients who had shown prior TIL reactivity against the HLA-A*0201 restricted melanoma tumor Ag MART-1. An array of MART-1-reactive TIL clones were generated by limiting dilution cloning after growth of the tumor digests in the presence of high-dose IL-2 in vitro for 10–14 days. A total of 576 individual clones (192 from each patient TIL) were isolated and assayed for reactivity to various melanoma tumor Ags, including MART-1, by coculturing with peptide-pulsed T2 target cells (42) and assessment of IFN-γ secretion following overnight incubation. Individual T cell clones were found that reacted with each of the melanoma Ag epitopes tested (Table I). Forty-one clones (7.1% per patient ± 0.3%) showed specific recognition of the native MART-1:27–35 Ag, 3 clones (0.5% per patient ± 0%) recognized gp100: 209–217, 2 clones (0.3% per patient ± 0.3%) recognized gp100:154–162, and 31 (5.4% per patient ± 0.6%) responded to gp100:280–288. The prevalence of these specific tumor-Ag reactive cells was remarkably similar between patients. Cells were expanded by exposure to anti-CD3 Ab (OKT-3) in the presence of high-dose IL-2, and 21 MART-1 clones proliferated and retained Ag-specific reactivity.
Table I.
Naturally occurring melanoma tumor-reactive TIL clones, prevalence by patient
| No. of Reactive Clonesa | ||||
|---|---|---|---|---|
| Patient (192 clones each) | MART-1: 27–35 | gp100: 154–162 | gp100: 209–217 | gp100: 280–288 |
| 1-D | 14; 7.3% | 1; 0.5% | 1; 0.5% | 9; 4.7% |
| 2-M | 13; 6.8% | 1; 0.5% | 1; 0.5% | 11; 5.7% |
| 3-S | 14; 7.3% | 0; 0% | 1; 0.5% | 11; 5.7% |
| Total (576 clones) | 41; 7.1% | 2; 0.3% | 3; 0.5% | 31; 5.2% |
Three additional CTL clones from two additional patients were included for analysis. A MART-1 reactive T cell clone, DMF4 (previously referred to as M1F12), was obtained from a clinically administered TIL that resulted in in vivo tumor regression (10). DMF5 was a second MART-1-reactive CTL clone from the same patient treatment TIL as DMF4. Additionally, clone JKF6, previously obtained from another patient TIL known to be highly MART-1 reactive in vitro, was also included in this study.
Functional avidity of these 24 T cell clones was evaluated by the production of IFN-γ in response to coculture with T2 target cells pulsed with diminishing amounts of MART-1 peptide and to naturally MART-1 expressing HLA-A*0201-restricted melanoma tumor cell lines. The clones DMF4, DMF5, and JKF6 were among the most highly avid CTLs, producing high amounts of IFN-γ even in response to low concentrations of MART-1 peptide or endogenous MART-1 on tumor cells (Fig. 1, A and B). Of all CTLs tested, DMF5 and JKF6 displayed the highest avidity for both tumors and exogenously supplied peptide. IFN-γ production in response to MART-1 peptide was compared with the response to MART-1-expressing tumors, and found to be strongly correlated (p = 0.006) (Fig. 1_C_), confirming that the Ag the clones recognized on tumors was MART-1, and that MART-1 peptide reactivity correlated with the ability to recognize MART-1 bearing tumors (Fig. 1_C_). In evaluating TCR for the potential use in antitumor allogeneic gene transfer, it is crucial that the receptor recognize a number of different HLA-A*0201-restricted melanoma tumors. In Fig. 1_D_, we show that the ability of any given T cell clone to recognize a MART-1-expressing melanoma tumor cell line was strongly correlated with its ability to recognize another MART-1 bearing melanoma cell line (p < 0.0005).
FIGURE 1.

MART-1-reactive TIL clones derived from several patients display a wide diversity of peptide and MART-1 tumor reactivities. A, IFN-γ production by 24 MART-1 clones derived from five patients, coincubated with titered MART-1 peptide on T2 target cells. B, IFN-γ response of MART-1 clones following coculture with MART-1/HLA-A*0201-positive (mel526+, mel624+) and HLA-A*0201-negative melanoma tumors (mel888−, mel938−). IFN-γ release was evaluated from 18-h culture supernatant by ELISA. C, Correlation of IFN-γ produced by each clone in response to coculture with T2 cells pulsed with MART-1 peptide or mel624+ MART-1-expressing tumor. D, Correlation of IFN-γ produced by each clone in response to coculture with two different MART-1/HLA-A*0201-expressing tumors. Statistical analysis by ANOVA regression analysis.
Tetramer binding of CTLs to pMHC complexes is accomplished by a combination of TCR:pMHC association and CD8 coreceptor interaction with the MHC class I H chain (27, 32). Clones were evaluated for their ability to bind pMHC complexes in a CD8-dependent or independent fashion using MART-1:26–35(27L)/HLA-A*0201 tetramers consisting of either nonmutated HLA H chains, or with an HLA H chain double-mutation eliminating CD8 binding (43). While most MART-1-reactive clones were capable of binding tetramer in the presence of CD8 coreceptor binding, only select clones (10 of 24) were able to do so in the absence of CD8 coreceptor binding (Fig. 2, Table II), indicative of a higher-affinity TCR.
FIGURE 2.
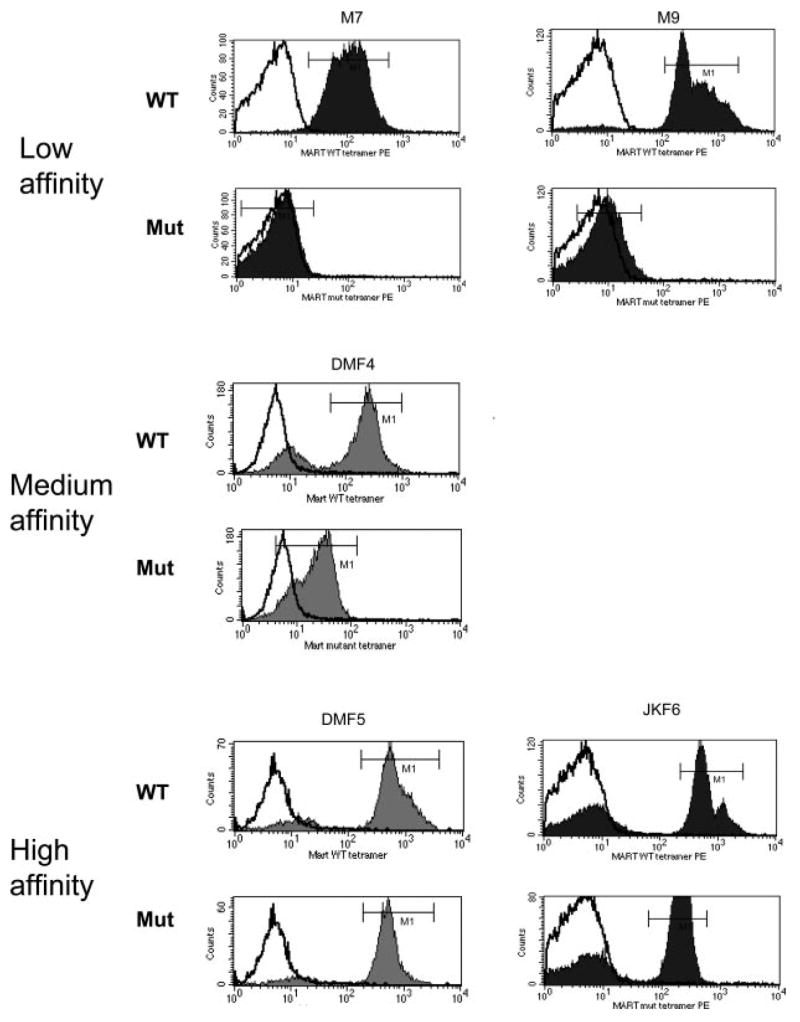
Different MART-1-reactive TIL clones vary in their ability to bind MART-1/HLA-A*0201 tetramer with CD8 independence. Each MART-1-reactive TIL clone was stained with MART-1:26–35(27L)/HLA-A*0201 (MART-1) unmodified tetramer (WT), or MART-1 tetramer with a HLA H chain double mutation abrogating CD8 binding to the HLA-A*0201 molecule (Mut). Representative graphs of each type of TCR binding to tetramer are shown: low-affinity clones were unable to bind tetramer in absence of CD8 coreceptor binding, medium-affinity clones bound CD8-independent tetramer at low levels, and high-affinity clones bound tetramer at high levels in absence of CD8 coreceptor binding. In each graph the isotype control is shown (white) as well as MART-1 tetramer binding (shaded).
Table II.
Avidity and affinity characterization of MART-1-reactive TIL clones
| Tetramer MFI | IFN-γ Produced (pg/ml) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Clone | gp100: 209 | WT MART-1 | mut MART-1 | 1.0 _μ_M MART-1 | 1.0 _μ_M gp100 | mel526+ | mel624+ | mel888− | Avidity-Affinity |
| M2 | 5 | 51 | 6 | 2,777 | 10 | 2,070 | 2,864 | 2 | Medium |
| M3 | 6 | 365 | 106 | 528 | 15 | 38 | 43 | 2 | Low/Medium |
| M5 | 5 | 621 | 98 | 5,381 | 50 | 1,554 | 3,346 | 1 | High |
| M6 | 5 | 5 | 5 | 377 | 47 | 935 | 990 | 20 | Medium |
| M7 | 5 | 109 | 6 | 35 | 23 | 102 | 135 | 4 | Low |
| M8 | 5 | 31 | 5 | 2,709 | 48 | 759 | 2,171 | 41 | Medium |
| M9 | 5 | 316 | 9 | 92 | 64 | 236 | 182 | 3 | Low |
| M12 | 5 | 246 | 5 | 1,403 | 14 | 709 | 687 | 1 | Medium |
| M13 | 6 | 372 | 6 | 1,531 | 59 | 3,207 | 6,522 | 4 | Medium |
| M14 | 5 | 5 | 5 | 20 | 24 | 13 | 8 | 11 | N/A |
| M19 | 5 | 799 | 279 | 794 | 4 | 581 | 679 | 1 | Medium/High |
| M20 | 7 | 271 | 7 | 34 | 27 | 489 | 465 | 3 | Low |
| M21 | 6 | 84 | 6 | 3,303 | 34 | 4,428 | 6,096 | 19 | Medium |
| M23 | 10 | 378 | 10 | 686 | 3 | 682 | 699 | 10 | Medium |
| M26 | 6 | 594 | 302 | 541 | 102 | 405 | 367 | 12 | Medium |
| M29 | 5 | 193 | 5 | 7,789 | 557 | 1,352 | 4,704 | 17 | Medium |
| M30 | 5 | 294 | 6 | 7,340 | 2 | 1,364 | 2,111 | 14 | Medium |
| M31 | 6 | 352 | 7 | 3,974 | 48 | 735 | 692 | 19 | Medium |
| #10 | 6 | 259 | 6 | 897 | 105 | 3,703 | 6,158 | 13 | Medium |
| #17 | 5 | 533 | 84 | 4,330 | 633 | 1,678 | 2,322 | 9 | Medium/High |
| #18 | 6 | 649 | 118 | 751 | 23 | 815 | 796 | 16 | Medium/High |
| JKF6 | 6 | 537 | 207 | 6,917 | 122 | 5,896 | 6,224 | 15 | High |
| DMF4 | 5 | 308 | 25 | 1,987 | 45 | 1,780 | 2,397 | 24 | Medium |
| DMF5 | 5 | 538 | 474 | 17,161 | 80 | 5,806 | 10,865 | 29 | High |
To determine the degree to which TCR transfer can confer cellular avidity, clones were selected that showed low avidity (low IFN-γ production plus inability to bind tetramer in the absence of CD8 binding), medium avidity (high IFN-γ production in response to target cells but unable to bind tetramer with CD8-independence, or produced low IFN-γ in response to target cells but could bind tetramer with CD8-independence), or high avidity (high IFN-γ production combined with the ability to bind tetramer independently of CD8), the results of which are tabulated in Table II. While high TCR affinity often coincided with high production of IFN-γ in response to target cells, this was not always observed. Certain clones (M3, M19, M26, #18) were able to bind tetramer independent of CD8 coreceptor binding, yet produced only modest amounts of cytokine in response to target cell stimulus. The reason for this is not clear, but differing TCR affinities for the HLA-A*0201 molecule or the native MART-1:27–35 peptide on target cells vs the altered MART-1:26–35(27L) peptide used to stabilize tetramers may be a contributing factor. Alternatively, the overall capability of these T cell clones to produce IFN-γ may have been reduced in comparison to other clones. The highest-avidity IFN-_γ_-producing clones DMF5 and JKF6 were able to bind tetramer independent of CD8, as was one other highly avid clone, M5.
Analysis of TCR variable region diversity indicates a strong Vα gene restriction for recognizing MART-1 Ag
The ability of T cells to respond functionally to MART-1 Ag in both peptide- and tumor-associated Ag form was combined with TCR affinity (estimated by mutated MART-1:26–35(27L)/HLA-A*0201 tetramer binding) to characterize T cell clones as high, medium, or low avidity-affinity (Table II). Representative clones were chosen from each category: three low (M7, M9, M20), two medium (#17, DMF4), and three high (M5, JKF6, DMF5). RNA was purified from each clone, and TCR α and β genes were isolated using 5′ RACE, subcloned, and sequenced. Of these eight MART-1-reactive clones, none exhibited similar β_-chain variable regions, with the exception of clones M9 and M20, which were derived from the same patient and upon sequencing proved to be duplicate cells. This CTL clone (M9/M20) will be referred to as M9 throughout the remainder of this study. Five of seven clones showed V_α region 12-2 restriction, and a sixth clone consisted of the closely related V_α_12-3 (Table III), confirming previous reports of a strong TCR α variable region restriction in recognition of MART-1 Ag (44–46).
Table III.
TCR variable-region restriction of MART-1-reactive clones of various avidities
| Mart Clone | TCR V_α_ | TCR V_β_ | Clone Avidity/Affinity (IFN-γ/CD8 independence) |
|---|---|---|---|
| DMF4 | 35 | 10-3 | Medium/Medium |
| DMF5 | 12-2 | 6-4 | High/High |
| JKF6 | 12-2 | 28 | High/High |
| #17 | 12-2 | 11-2 | Medium/High |
| M5 | 12-2 | 6-5 | High/High |
| M7 | 12-2 | 19 | Low/Low |
| M9a | 12-3 | 4-1 | Low/Low |
| M20a | 12-3 | 4-1 | Low/Low |
TCR transfer is sufficient to confer overall cellular avidity to donor PBMC in an Ag-specific manner
RNA was generated by in vitro transcription of each pair of α and β TCR genes isolated from the seven clones indicated above. TCR RNA was electroporated into the TCR _β_-chain-deficient Jurkat T3.5 cell line (47, 48). As shown in Fig. 3_A_, CD3 Ab staining levels were similar on the surfaces of cells electroporated with each of the seven TCR pairs. This indicated that following electroporation of the same amounts of RNA, the α and β chains of each TCR were translated into similar amounts of TCR proteins within the cells, which in turn were able to form appropriate heterodimeric structures and recruit the CD3 portion of the TCR complex to the cell surface. Appropriate MART-1-reactive tertiary αβ TCR pairing was evaluated by the ability to bind MART-1:26–35(27L)/HLA-A*0201 tetramers (Beckman Coulter), analyzed by flow cytometry (Fig. 3_B_). Intriguingly, although TCR surface pairing assessed by CD3 staining was similar for all TCRs tested, there were marked differences in the ability of each TCR to bind MART-1 tetramer, correlating with the TCR affinity. RNA encoding these TCR was then electroporated into 7-day OKT-3-stimulated PBMC and stained for MART-1 TCR formation by tetramer staining (Fig. 3_C_). The tetramer staining of TCR electroporated CD8+ PBMC was similar to that observed in Jurkat cells. Although PBMC bear the CD8 coreceptor, they also express endogenous TCR _α_- and _β_-chains to compete with the electroporated MART-1 TCR genes for heterodimer pairing. Notably, in both cell types, those TCR derived from high-avidity T cells (DMF5, JKF6, M5) were able to bind tetramer more readily than those with low avidity (M7, M9). Although RNA electroporation efficiency was observed to be >95% (by GFP controls, data not shown), only a fraction of the TCR-electroporated cells were able to bind MART-1 tetramer at all, suggesting that levels of TCR expression achieved by this method are still lower than TCR levels observed in endogenous TCR-expressing T cells.
FIGURE 3.
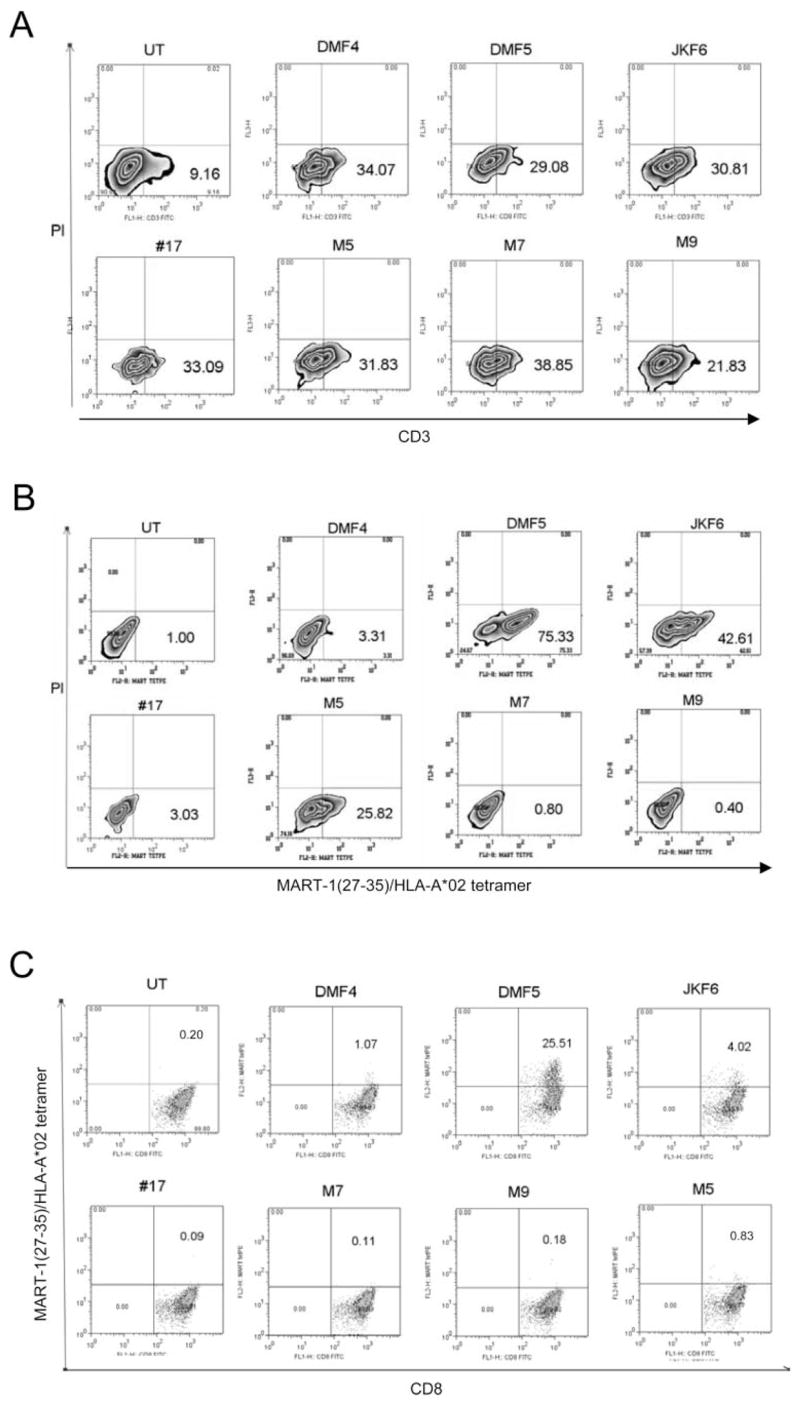
RNA electroporation of TCR derived from MART-1 clones into Jurkat and donor CD8+ PBMC resulted in appropriate αβ TCR translation and surface expression. Percentage of live cells staining positive is indicated. A and B, Two micrograms per 1 × 106 cells in vitro transcribed RNA from TCR _α_- and _β_-chains of each MART-1 TIL clone was electroporated into TCR-deficient, CD8-negative Jurkat T3.5 cells that were then stained for CD3 cell surface expression (A) or MART-1: 26–35(27L)/HLA-A*0201 tetramer (B), and evaluated by flow cytometry. C, Flow cytometric evaluation of CD8+ donor PBMC electroporated with 2 _μ_g per 1 × 106 cells TCR RNA, costained with CD8 and MART-1:26–35(27L)/HLA-A*0201 tetramer. UT = untreated cells.
Ag-specific response by the TCR-electroporated T cells was evaluated by coculture with MART-1 peptide-pulsed T2 targets, or MART-1-expressing melanoma tumor cells, and assayed for supernatant IFN-γ (Fig. 4, A and B). The TCR-electroporated cells were capable of generating an Ag-specific response, and this response varied in intensity. Three TCR (DMF5, JKF6, and M5) were able to generate high amounts of IFN-γ, two (DMF4, #17) generated mid-level responses, and two (M7, M9) generated only negligible responses to MART-1 Ag. In order of diminishing functional avidity, the response induced by each TCR was DMF5 > JKF6 > M5 > #17 > DMF4 > M7 = M9. These cells retained the same respective avidities observed for the original TIL clones, and this pattern was repeated regardless of the amount of TCR RNA electroporated (2–0.5 _μ_g RNA per million cells, data not shown). Statistical correlation (ANOVA regression analysis) between IFN-γ production of the original TIL clones and corresponding TCR RNA electroporated PBMC in response to MART-1-bearing target cells was evaluated (Fig. 4_C_) and found to be highly significant ( p = 0.018 for 1.0 _μ_M MART-1 peptide; p = 0.0003 and p = 0.009 for mel624 and mel526 tumor targets, respectively). CD8+ PBMC electroporated with MART-1 TCR were also able to induce specific target cell lysis of peptide-pulsed targets or MART-1-bearing tumors in chromium release assays (Fig. 5_A_), at levels consistent with each relative TCR avidity: DMF5 > JKF6 > M5 > DMF4 > #17 > M7 = M9.
FIGURE 4.
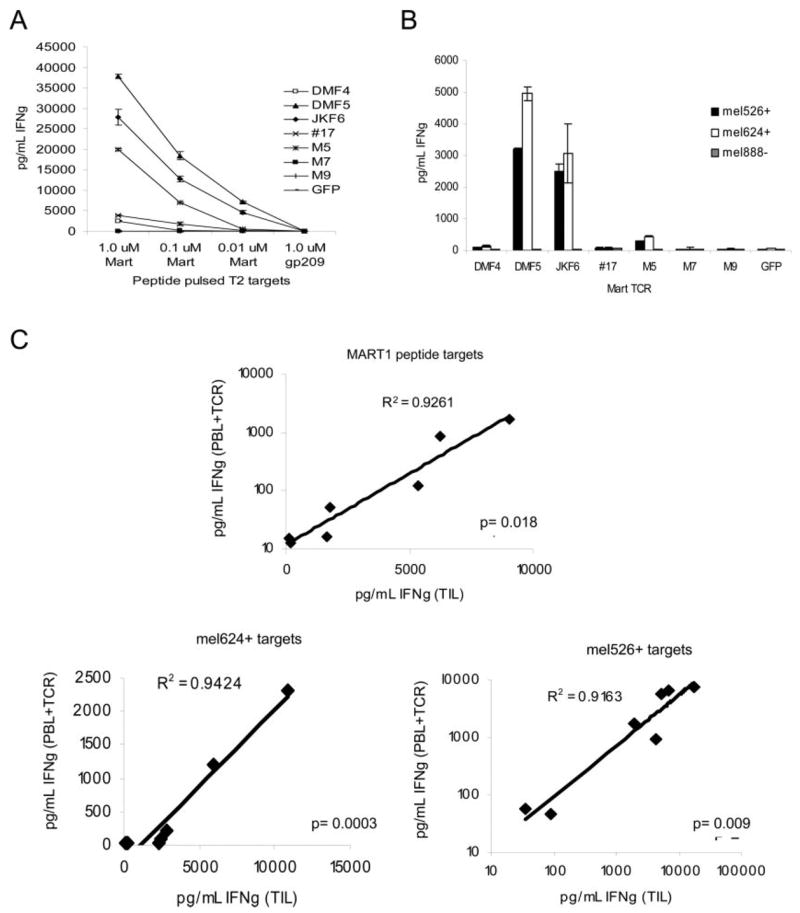
RNA electroporation of MART-1 TCR derived from clones of varying avidities into activated donor CD8+ PBMC resulted in similar Ag-specific functional avidities. A and B, Donor CD8+-enriched PBMC previously activated with OKT-3 Ab were electroporated with 2 _μ_g of RNA from each TCR chain per 1 × 106 cells, and cocultured overnight with MART-1 peptide pulsed T2 target cells (A) or MART-1/HLA-A*0201-expressing (mel526+, mel624+) or nonexpressing (mel888−) melanoma tumors (B). IFN-γ production was measured in supernatants by ELISA. Values shown are the average of duplicate samples, ±SEM. C, Correlation of IFN-γ produced by native TIL clones and donor CD8+ PBMC electroporated with matched TCR RNA. Responses to cocultures with T2 cells pulsed with MART-1 peptide, mel526+, and mel624+ MART-1-expressing melanoma tumors are shown. The p values were obtained by ANOVA regression analysis.
FIGURE 5.
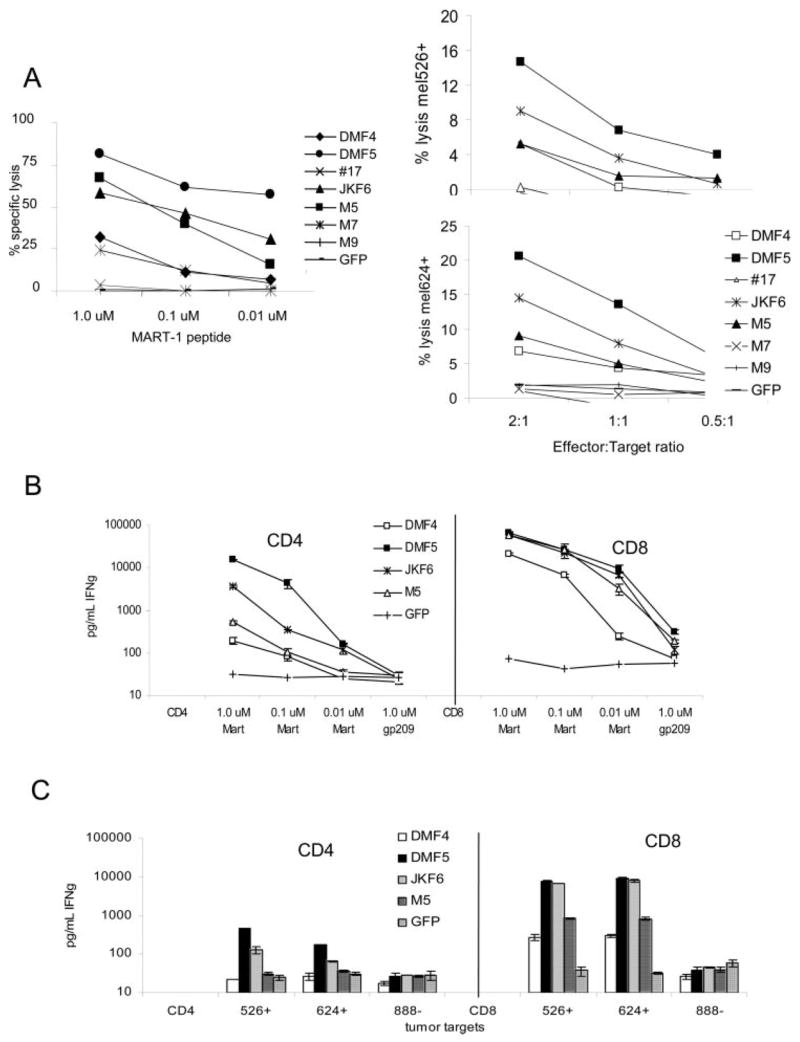
TCR transfer of a high-avidity TCR in vitro can confer antitumor CTL activity and IFN-γ production in a CD8-independent fashion. A, OKT-3 stimulated CD8+ PBMC RNA-electroporated with different avidity MART-1 TCR were evaluated for their ability to lyse 51Cr-labeled target cells in a 4-h assay. CTL were coincubated with (left) MART-1 peptide-pulsed T2 target cells at 2:1 E:T ratio, or (right) at varying E:T ratios against MART-1 expressing tumor cells. B and C, OKT-3-stimulated donor PBMC were separated into CD4+ and CD8+ populations by positive selection using magnetic beads and each of the four highest avidity MART-1 TCR were expressed in each population by RNA electroporation (2 _μ_g/1 × 106 cells). Functional antitumor response was evaluated by coculture with either MART-1-pulsed T2 target cells (B) or MART-1-expressing tumors (C). IFN-γ production was measured in supernatants by ELISA. Values shown are the average of duplicate samples, ±SEM.
PBMC expressing high-avidity TCR are capable of functional recognition of MART-1 target cells, even in the absence of CD8
Since these high-avidity TCRs were chosen in part for their ability to bind MART-1 tetramer independently of CD8, we next determined if this TCR avidity was strong enough to confer CD8-independent recognition of MART-1 Ag on target cells. TCR RNA from the high-avidity receptors DMF5, JKF6, and M5 and medium-avidity DMF4 was electroporated into CD4+-enriched (>95% pure) OKT-3-activated donor PBMC. As assessed by IFN-γ production in response to coculture with titered MART-1 peptide on T2 target cells, each MART-1 TCR in CD4+ cells was able to recognize MART-1 peptide. DMF5 showed the highest response, followed by JKF6 and M5, then DMF4 (Fig. 5_B_), although the overall levels were lower than those observed in CD8+ T cells from the same donor electroporated with the same TCR. In response to MART-1 tumor Ag naturally presented on tumor cells, only those CD4+ cells that expressed the highest-avidity DMF5 or JKF6 receptors were able to recognize and respond to tumors, with DMF5 generating the highest response (Fig. 5_C_).
Nonreactive TIL can be made tumor reactive upon RNA electroporation with a high-avidity MART-1 TCR
There are many differences between the T cells found in TIL and those circulating in PBMC (8). Currently, it is not known whether TIL have unique properties that may make them more effective in treating tumors in vivo than T cells derived from PBMC. To investigate whether it is possible to convert nonreactive TIL to recognize and lyse tumors, we selected previously generated TIL obtained from melanoma patient samples with determined activity only against autologous tumor and not against any known shared tumor Ags such as MART-1. Patient-matched PBMC samples were obtained, and both PBMC and TIL were stimulated similarly, using OKT-3 Ab and irradiated allogeneic feeder PBMC in the presence of high-dose IL-2. CD8+ T cells were isolated and electroporated with TCR RNA from the highest-avidity DMF5 TCR. MART-1 reactivity was assayed by coculture of RNA electroporated T cells with MART-1 peptide-pulsed target cells and tumors, and evaluated for IFN-γ production and CTL target cell lysis (Fig. 6, A and B). It was possible to convert non-MART-1-reactive TIL into reactive TIL using this high-avidity TCR. These RNA electroporated TIL were able to recognize and lyse MART-1-expressing tumors, albeit at lower levels than matched RNA electroporated PBL obtained from the same patient, even though surface MART-1 TCR expression was comparable (based on tetramer staining, data not shown).
FIGURE 6.
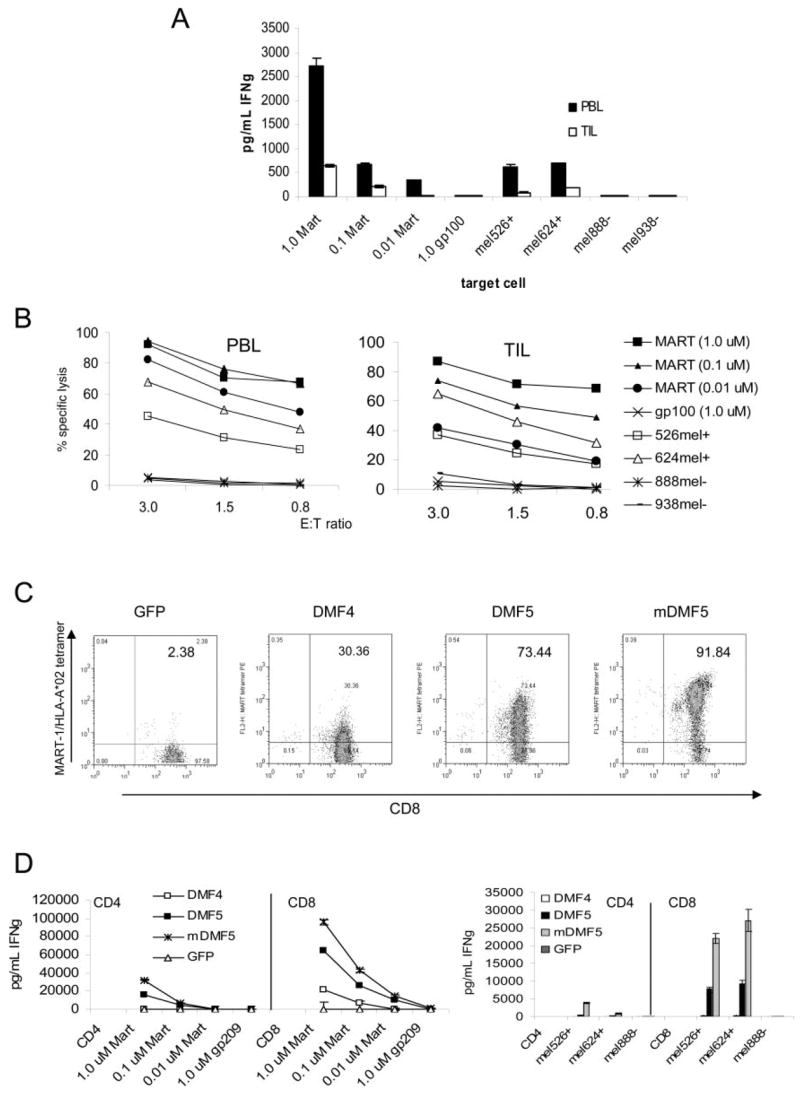
Nonreactive TIL can also be rendered Ag reactive by TCR transfer of the high-avidity TCR DMF5 in vitro, and this TCR can be altered to generate even higher cellular avidity. A and B, Non-tumor-reactive PBL and TIL from the same patient were stimulated using OKT-3 and irradiated feeder cells in the presence of high-dose IL-2 for 3 days before CD8+ T cell enrichment using magnetic beads. These CD8+ T cells were then electroporated with 2 _μ_g/1 × 106 cells TCR RNA from high-avidity DMF5, and cocultured with MART-1 peptide-pulsed T2 target cells or MART-1/HLA-A*0201-expressing melanoma tumors (mel526+, mel624+) or non-HLA-A*0201-expressing tumors (mel888−, mel938−). A, IFN-γ production was measured in supernatants by ELISA. Values shown are the average of duplicate samples, ±SEM. B, TIL and PBL electroporated with DMF5 TCR RNA as in A above, were cocultured against MART-1 peptide pulsed T2 cells or tumors in a 4-h 51Cr release assay. Results are representative of four patient samples tested. C and D, The DMF5 TCR was altered by replacing the constant region with murine TCR constant regions (mDMF5) and compared with the naturally occurring DMF5 and DMF4 TCRs by RNA electroporation in activated CD4+ or CD8+ PBMC. C, CD8 and MART-1/HLA-A*0201 tetramer staining of OKT-3-stimulated, TCR RNA-electroporated CD8+ PBMC. Percentage of live lymphocytes staining double positive is indicated. D, IFN-γ production by 18-h coculture of CD4+ and CD8+ TCR RNA electroporated PBMC with MART-1 peptide pulsed T2 targets (left) or tumor target cells (right). Values shown are the average of duplicate samples, ±SEM.
Avidity of DMF5 TCR-engineered T cells can be further improved by replacing the endogenous TCR constant region with a murine version
One caveat to using TCR gene transfer therapy in T cells is the inherent competition with endogenously expressed α and β TCR genes for appropriate αβ TCR heterodimer pairing. In an attempt to increase the pairing of functional antitumor TCR αβ chains on allogeneic PBMC, modifications were made to the transmembrane constant regions of each TCR chain, replacing them with constant regions derived from the mouse genome (49). These small portions of the TCR vary enough from the human sequence to facilitate preferential pairing, and improve CD3_ζ_ chain recruitment, yet are similar enough that potential immunogenicity should be minimized. We have modified the DMF5 TCR _α_- and _β_-chains with these murine constant regions, and evaluated their ability to confer increased cellular activity against MART-1 peptide-pulsed targets or melanoma target cells. As previously reported for the DMF4 TCR (49), replacing the DMF5 constant regions with the murine sequence (mDMF5) likewise shows an increase in TCR pairing at the cell surface, demonstrated by increased MART-1 tetramer binding by RNA electroporated CD8+ PBMC (Fig. 6_C_). Functional avidity was also increased, as demonstrated by increased IFN-γ production upon coculture with MART-1+ target cells (Fig. 6_D_). This increased activity was particularly apparent when measured against melanoma tumor cells, which normally result in only low TCR-engineered T cell reactivity compared with peptide-pulsed target cells. This modified mDMF5 TCR conferred the highest avidity of any TCR tested, in both CD4+ and CD8+ T cells, against all targets (Fig. 6_D_).
Discussion
The present study was undertaken to determine the relationship between the functional avidity of multiple MART-1-reactive clones, to the properties of the clonal TCR, and to use this information to select TCR that confer high Ag recognition for use in TCR transduction gene therapy protocols. Clones were generated by limiting dilution cloning of day 7–14 in vitro tumor digests in the presence of high-dose IL-2. The properties of the TCR were evaluated by functional assays for IFN-γ secretion in response to MART-1-bearing target cell coculture, combined with the ability of the TCR to bind mutated MART-1:26–35(27L)/HLA-A*0201 tetramers independently of CD8 coreceptor help. α and β TCR genes were isolated from representative clones of high, medium and low avidity using 5′ RACE, and RNA electroporated into activated PBMC T cells from a common donor. IFN-γ production and lytic activity in response to target cell cocultures demonstrated a direct correlation of avidities to those observed in the original CTL clones. Thus, the TCR was the main determinant of cellular avidity during Ag-specific T cell:target cell interaction, and this avidity could be conferred upon allogeneic cells by TCR transfer.
It has recently been shown both in vitro and in an animal model that high-affinity TCR give rise to high avidity T cells and that only high-avidity cells were able to elicit autoimmunity in lymphocytic choriomeningitis virus-infected TCR transgenic mice expressing lymphocytic choriomeningitis virus epitope as “self” in pancreatic islet cells (20). Other animal models have similarly shown that only high-avidity T cells have the ability to mediate tumor regression in spontaneously arising pancreatic tumors in RIP-Tag2-In-sHA transgenic mice (16, 17, 50), or through ACT to treat metastatic B16 melanoma tumors in C57BL/6 mice (15) or HLA-A*0201 transgenic mice engrafted with human melanoma (51). These combined data suggest that to achieve an antitumor immune response in vivo, T cells recognizing tumor Ags with the highest avidity and bearing the highest-affinity TCR provide the most potential to induce an in vivo antitumor response.
Although there is no clear predictive relationship between tetramer binding intensity or tetramer dissociation and functional avidity of T cells (35), it has recently been shown that high avidity T cells can be identified by their ability to bind pMHC tetramers independent of CD8 coreceptor binding (39). In this current study, we used CD8-independent binding of tetramer as a tool to identify higher-affinity TCR for potential clinical TCR gene transfer immunotherapy. The ability to engage pMHC complexes independent of CD8 coreceptor is desirable for TCR destined for transfer into CD4 donor PBMC, or T cells with varying expression levels of CD8. A high-affinity TCR for gene transfer is particularly desirable to compensate for lowered levels of appropriate αβ TCR pairing due to endogenously expressed TCR genes, because overall avidity is determined by a combination of the number of TCR/pMHC interactions as well as the affinity of each interaction. Several groups have used a measure of “dwell-time” between T cells and tetramer complexes in relation to T cell functional avidity, with varying results (52–54). Invoking point mutations in the pMHC-binding region of a vesicular stomatitis virus-specific murine TCR, Kalergis et al. (53) have demonstrated that tetramer dissociation rates are not directly correlated with cellular avidity and that in fact there appears to be an optimal tetramer dissociation rate specific to each TCR/pMHC tetramer combination. TCR mutations that altered this rate above or below a certain level correlated with reduced cellular function, suggesting an optimal level of TCR binding for CTL function. This idea of an “ideal” TCR affinity supports additional recent work demonstrating that HIV-specific CD8+ T cells with a longer TCR:tetramer dwell-time were actually less effective against target cells pulsed with low concentrations of HIV peptide or displaying endogenous HIV Ag, than cells with a shorter TCR:tetramer interaction (52).
Although many high-avidity T cells with high-affinity TCR exist against foreign Ags, thymic selection of CD8+ T cells ensures that no extremely high-affinity T cells against self-Ags are released into the periphery (55). This poses a problem for the development of immunotherapy approaches to treat cancer since many tumor Ags are derived from self proteins (3). The substantial clinical response rate in ACT-based tumor immunotherapy demonstrates human TIL do contain TCR of sufficiently high avidity to eliminate tumors in vivo. In contrast to T cells responding to infection by foreign pathogens, individuals with cancer are unlikely to have naturally occurring antitumor T cells displaying TCR of such high affinity to be functionally detrimental; rather the problem lies in isolating T cells of high enough avidity to induce tumor regression. We have shown here that once isolated, the TCR from these high-avidity T cells can confer not only Ag specificity, but also functional avidity to donor cells.
Of 24 MART-1-specific CTL clones obtained from 5 different patients’ TIL, a diverse variety of cellular avidities was observed, with substantial restriction of the TCR α_-chain to variable region 12-2. This V_α TCR restriction was independent of cellular avidity, with both high- and low-avidity CTL displaying V_α_ 12-2. One particular MART-1-reactive TCR, DMF5, derived from a TIL infusion with demonstrated ability to cause in vivo tumor regression (10), repeatedly exhibited the highest activity against MART-1-expressing tumors in vitro, whether expressed by the native TIL clone, or by gene transfer into donor PBMC. It was possible to improve upon the already high avidity provided by the DMF5 TCR by exchanging the TCR _α_- and _β_-chain constant (C) regions with sequences derived from murine TCR C-regions, resulting in increased TCR surface pairing and functional avidity over the naturally occurring TCR sequence. Although RNA electroporation and gene expression efficiency in PBMC was consistently >95% by GFP controls (data not shown), tetramer staining indicated lower levels of surface TCR expression compared with the original clones. For use in gene therapy, the TCR genes would be incorporated into a retroviral expression system and used to transduce PBMC ex vivo, before reinfusion. The prolonged endogenous expression induced by retroviral transduction would also be likely to increase TCR gene expression levels.
The highest-avidity TCR we identified, CD8-independent DMF5, holds promise as a candidate for allogeneic TCR gene therapy in metastatic melanoma patients. This TCR was sufficient to transform nonreactive donor CD8+ and CD4+ PBMC as well as TIL to recognize MART-1-expressing tumors, produce high levels of multiple immunologically relevant cytokine, and lyse tumor cells in vitro. The only way to determine whether high-avidity TCR have the same effect on human tumors in vivo as they do in mouse models is to test this TCR in a clinical trial, with the goal of inducing tumor regression in patients, and such a clinical trial is being planned.
The patient TIL infusion from which DMF5 was derived resulted in clinical tumor regression, and was comprised primarily of two CD8+ MART-1-reactive T cell clones, DMF4 and DMF5. DMF4 was found to persist in the patient’s peripheral blood at very high levels at more than 1 year after treatment (10). DMF5 was also found in the blood following treatment, but at much reduced levels compared with DMF4. It has recently been shown that the telomere length of TIL is strongly correlated with its ability to persist long-term in vivo following adoptive transfer (9, 56). Further examination of the treatment cells originally administered to the patient revealed that DMF4 cells had long telomeres (8.91 kb) compared to DMF5 with shorter telomeres (4.61 kb), indicative of approaching cell senescence (9). Therefore, although we have shown that the DMF5 TIL had a higher-affinity TCR than DMF4, the actual DMF5 infusion cells may have undergone rapid proliferative exhaustion in response to antigenic tumor stimulation and therefore were unable to persist long-term in vivo. It remains unknown which of these cells (if either) were responsible for the patient’s tumor regression.
Identification of the high avidity TCR DMF5, along with the determination that TCR transfer is sufficient to confer cellular avidity as well as Ag recognition specificity, holds the potential to overcome problems presented by conventional ACT therapy. TCR gene immunotherapy has the potential for use in many types of cancer besides melanoma. Indeed, TCR recognizing the cancer testis Ag NY-ESO1, and p53, expressed in many types of tumors, including melanoma, breast cancer, lung cancer, prostate cancer, and sarcomas, have recently been isolated (11, 12, 49, 57). Since the Ags recognized by antitumor TCR are often comprised of self-peptides, selecting high-avidity TCR may contribute to the induction of autoimmunity; vitiligo and uveitis have been observed in some patients in prior cell transfer trials (4). The use of highly reactive, high-affinity tumor-Ag specific TCR to transduce T cells from cancer patients with poorly reactive or unavailable TIL may have the potential to recognize and eliminate tumors in vivo. The use of autologous cells minimizes any risk of graft vs host disease or cell rejection. Retroviruses encoding antitumor TCR offer an off-the-shelf reagent for use in the immunotherapy of patients with cancer.
Acknowledgments
We thank Y. Li, M. El-Gamil, K. Kerstann, Y. Zhao, Z. Zheng, J. Wunderlich and the TIL lab, S. Farid, and A. Mixon from the National Cancer Institute Surgery Branch for their generous gifts of reagents, their discussions and technical assistance in this study.
Footnotes
1
This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
3
Abbreviations used in this paper: pMHC, peptide bound to MHC class I molecule; ACT, adoptive cell transfer; CU, Cetus unit; STEM, Stemline medium; TIL, tumor-infiltrating lymphocyte; IVT, in vitro transcription.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. 1986;233:1318–1321. doi: 10.1126/science.3489291. [DOI] [PubMed] [Google Scholar]
- 2.Boon T, Coulie PG, Eynde BJ, Bruggen PV. Human T cell responses against melanoma. Annu Rev Immunol. 2006;24:175–208. doi: 10.1146/annurev.immunol.24.021605.090733. [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg SA. Progress in human tumour immunology and immunotherapy. Nature. 2001;411:380–384. doi: 10.1038/35077246. [DOI] [PubMed] [Google Scholar]
- 4.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, Royal RE, Kammula U, White DE, Mavroukakis SA, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 7.Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z, Finkelstein SE, Theoret MR, Rosenberg SA, Restifo NP. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest. 2005;115:1616–1626. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gattinoni L, Powell DJ, Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol. 2006;6:383–393. doi: 10.1038/nri1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou J, Shen X, Huang J, Hodes RJ, Rosenberg SA, Robbins PF. Telomere length of transferred lymphocytes correlates with in vivo persistence and tumor regression in melanoma patients receiving cell transfer therapy. J Immunol. 2005;175:7046–7052. doi: 10.4049/jimmunol.175.10.7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hughes MS, Yu YY, Dudley ME, Zheng Z, Robbins PF, Li Y, Wunderlich J, Hawley RG, Moayeri M, Rosenberg SA, Morgan RA. Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T cell effector functions. Hum Gene Ther. 2005;16:457–472. doi: 10.1089/hum.2005.16.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cohen CJ, Zheng Z, Bray R, Zhao Y, Sherman LA, Rosenberg SA, Morgan RA. Recognition of fresh human tumor by human peripheral blood lymphocytes transduced with a bicistronic retroviral vector encoding a murine anti-p53 TCR. J Immunol. 2005;175:5799–5808. doi: 10.4049/jimmunol.175.9.5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao Y, Zheng Z, Robbins PF, Khong HT, Rosenberg SA, Morgan RA. Primary human lymphocytes transduced with NY-ESO-1 antigen-specific TCR genes recognize and kill diverse human tumor cell lines. J Immunol. 2005;174:4415–4423. doi: 10.4049/jimmunol.174.7.4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morgan RA, Dudley ME, Yu YY, Zheng Z, Robbins PF, Theoret MR, Wunderlich JR, Hughes MS, Restifo NP, Rosenberg SA. High efficiency TCR gene transfer into primary human lymphocytes affords avid recognition of melanoma tumor antigen glycoprotein 100 and does not alter the recognition of autologous melanoma antigens. J Immunol. 2003;171:3287–3295. doi: 10.4049/jimmunol.171.6.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Witte MA, Coccoris M, Wolkers MC, van den Boom MD, Mesman EM, Song JY, van der Valk M, Haanen JB, Schumacher TN. Targeting self antigens through allogeneic TCR gene transfer. Blood. 2006;180:870–877. doi: 10.1182/blood-2005-08-009357. [DOI] [PubMed] [Google Scholar]
- 15.Zeh HJ, III, Perry-Lalley D, Dudley ME, Rosenberg SA, Yang JC. High avidity CTLs for two self-antigens demonstrate superior in vitro and in vivo antitumor efficacy. J Immunol. 1999;162:989–994. [PubMed] [Google Scholar]
- 16.Nugent CT, Morgan DJ, Biggs JA, Ko A, Pilip IM, Pamer EG, Sherman LA. Characterization of CD8+ T lymphocytes that persist after peripheral tolerance to a self antigen expressed in the pancreas. J Immunol. 2000;164:191–200. doi: 10.4049/jimmunol.164.1.191. [DOI] [PubMed] [Google Scholar]
- 17.Lyman MA, Nugent CT, Marquardt KL, Biggs JA, Pamer EG, Sherman LA. The fate of low affinity tumor-specific CD8+ T cells in tumor-bearing mice. J Immunol. 2005;174:2563–2572. doi: 10.4049/jimmunol.174.5.2563. [DOI] [PubMed] [Google Scholar]
- 18.Alexander-Miller MA, Leggatt GR, Berzofsky JA. Selective expansion of high- or low-avidity cytotoxic T lymphocytes and efficacy for adoptive immunotherapy. Proc Natl Acad Sci USA. 1996;93:4102–4107. doi: 10.1073/pnas.93.9.4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Speiser DE, Kyburz D, Stubi U, Hengartner H, Zinkernagel RM. Discrepancy between in vitro measurable and in vivo virus neutralizing cytotoxic T cell reactivities: low T cell receptor specificity and avidity sufficient for in vitro proliferation or cytotoxicity to peptide-coated target cells but not for in vivo protection. J Immunol. 1992;149:972–980. [PubMed] [Google Scholar]
- 20.Gronski MA, Boulter JM, Moskophidis D, Nguyen LT, Holmberg K, Elford AR, Deenick EK, Kim HO, Penninger JM, Odermatt B, et al. TCR affinity and negative regulation limit autoimmunity. Nat Med. 2004;10:1234–1239. doi: 10.1038/nm1114. [DOI] [PubMed] [Google Scholar]
- 21.Corr M, Slanetz AE, Boyd LF, Jelonek MT, Khilko S, al-Ramadi BK, Kim YS, Maher SE, Bothwell AL, Margulies DH. T cell receptor-MHC class I peptide interactions: affinity, kinetics, and specificity. Science. 1994;265:946–949. doi: 10.1126/science.8052850. [DOI] [PubMed] [Google Scholar]
- 22.Krogsgaard M, Davis MM. How T cells ‘see’ antigen. Nat Immunol. 2005;6:239–245. doi: 10.1038/ni1173. [DOI] [PubMed] [Google Scholar]
- 23.Davis MM, Bjorkman PJ. T cell antigen receptor genes and T cell recognition. Nature. 1988;334:395–402. doi: 10.1038/334395a0. [DOI] [PubMed] [Google Scholar]
- 24.Roszkowski JJ, Yu DC, Rubinstein MP, McKee MD, Cole DJ, Nishimura MI. CD8-independent tumor cell recognition is a property of the T cell receptor and not the T cell. J Immunol. 2003;170:2582–2589. doi: 10.4049/jimmunol.170.5.2582. [DOI] [PubMed] [Google Scholar]
- 25.Holler PD, Lim AR, Cho BK, Rund LA, Kranz DM. CD8− T cell transfectants that express a high affinity T cell receptor exhibit enhanced peptide-dependent activation. J Exp Med. 2001;194:1043–1052. doi: 10.1084/jem.194.8.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holler PD, Kranz DM. Quantitative analysis of the contribution of TCR/pepMHC affinity and CD8 to T cell activation. Immunity. 2003;18:255–264. doi: 10.1016/s1074-7613(03)00019-0. [DOI] [PubMed] [Google Scholar]
- 27.Cawthon AG, Alexander-Miller MA. Optimal colocalization of TCR and CD8 as a novel mechanism for the control of functional avidity. J Immunol. 2002;169:3492–3498. doi: 10.4049/jimmunol.169.7.3492. [DOI] [PubMed] [Google Scholar]
- 28.Krogsgaard M, Huppa JB, Purbhoo MA, Davis MM. Linking molecular and cellular events in T-cell activation and synapse formation. Semin Immunol. 2003;15:307–315. doi: 10.1016/j.smim.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 29.Krogsgaard M, Prado N, Adams EJ, He XL, Chow DC, Wilson DB, Garcia KC, Davis MM. Evidence that structural rearrangements and/or flexibility during TCR binding can contribute to T cell activation. Mol Cell. 2003;12:1367–1378. doi: 10.1016/s1097-2765(03)00474-x. [DOI] [PubMed] [Google Scholar]
- 30.Krogsgaard M, Li QJ, Sumen C, Huppa JB, Huse M, Davis MM. Agonist/endogenous peptide-MHC heterodimers drive T cell activation and sensitivity. Nature. 2005;434:238–243. doi: 10.1038/nature03391. [DOI] [PubMed] [Google Scholar]
- 31.Fahmy TM, Bieler JG, Edidin M, Schneck JP. Increased TCR avidity after T cell activation: a mechanism for sensing low-density antigen. Immunity. 2001;14:135–143. [PubMed] [Google Scholar]
- 32.Wooldridge L, van den Berg HA, Glick M, Gostick E, Laugel B, Hutchinson SL, Milicic A, Brenchley JM, Douek DC, Price DA, Sewell AK. Interaction between the CD8 coreceptor and major histocompatibility complex class I stabilizes T cell receptor-antigen complexes at the cell surface. J Biol Chem. 2005;280:27491–27501. doi: 10.1074/jbc.M500555200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wooldridge L, Hutchinson SL, Choi EM, Lissina A, Jones E, Mirza F, Dunbar PR, Price DA, Cerundolo V, Sewell AK. Anti-CD8 antibodies can inhibit or enhance peptide-MHC class I (pMHCI) multimer binding: this is paralleled by their effects on CTL activation and occurs in the absence of an interaction between pMHCI and CD8 on the cell surface. J Immunol. 2003;171:6650–6660. doi: 10.4049/jimmunol.171.12.6650. [DOI] [PubMed] [Google Scholar]
- 34.Valmori D, Dutoit V, Lienard D, Lejeune F, Speiser D, Rimoldi D, Cerundolo V, Dietrich PY, Cerottini JC, Romero P. Tetramer-guided analysis of TCR β-chain usage reveals a large repertoire of melan-A-specific CD8+ T cells in melanoma patients. J Immunol. 2000;165:533–538. doi: 10.4049/jimmunol.165.1.533. [DOI] [PubMed] [Google Scholar]
- 35.Alexander-Miller MA. High-avidity CD8+ T cells: optimal soldiers in the war against viruses and tumors. Immunol Res. 2005;31:13–24. doi: 10.1385/IR:31:1:13. [DOI] [PubMed] [Google Scholar]
- 36.al-Ramadi BK, Jelonek MT, Boyd LF, Margulies DH, Bothwell AL. Lack of strict correlation of functional sensitization with the apparent affinity of MHC/peptide complexes for the TCR. J Immunol. 1995;155:662–673. [PubMed] [Google Scholar]
- 37.Palermo B, Campanelli R, Mantovani S, Lantelme E, Manganoni AM, Carella G, Da Prada G, della Cuna GR, Romagne F, Gauthier L, et al. Diverse expansion potential and heterogeneous avidity in tumor-associated antigen-specific T lymphocytes from primary melanoma patients. Eur J Immunol. 2001;31:412–420. doi: 10.1002/1521-4141(200102)31:2<412::aid-immu412>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 38.Echchakir H, Dorothee G, Vergnon I, Menez J, Chouaib S, Mami-Chouaib F. Cytotoxic T lymphocytes directed against a tumor-specific mutated antigen display similar HLA tetramer binding but distinct functional avidity and tissue distribution. Proc Natl Acad Sci USA. 2002;99:9358–9363. doi: 10.1073/pnas.142308199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choi EM, Chen JL, Wooldridge L, Salio M, Lissina A, Lissin N, Hermans IF, Silk JD, Mirza F, Palmowski MJ, et al. High avidity antigen-specific CTL identified by CD8-independent tetramer staining. J Immunol. 2003;171:5116–5123. doi: 10.4049/jimmunol.171.10.5116. [DOI] [PubMed] [Google Scholar]
- 40.Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Sakaguchi K, Appella E, Yannelli JR, Adema GJ, Miki T, Rosenberg SA. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci USA. 1994;91:6458–6462. doi: 10.1073/pnas.91.14.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kawakami Y, Eliyahu S, Sakaguchi K, Robbins PF, Rivoltini L, Yannelli JR, Appella E, Rosenberg SA. Identification of the immunodominant peptides of the MART-1 human melanoma antigen recognized by the majority of HLA-A2-restricted tumor infiltrating lymphocytes. J Exp Med. 1994;180:347–352. doi: 10.1084/jem.180.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rowe M, Khanna R, Jacob CA, Argaet V, Kelly A, Powis S, Belich M, Croom-Carter D, Lee S, Burrows SR, et al. Restoration of endogenous antigen processing in Burkitt’s lymphoma cells by Epstein-Barr virus latent membrane protein-1: coordinate up-regulation of peptide transporters and HLA-class I antigen expression. Eur J Immunol. 1995;25:1374–1384. doi: 10.1002/eji.1830250536. [DOI] [PubMed] [Google Scholar]
- 43.Hutchinson SL, Wooldridge L, Tafuro S, Laugel B, Glick M, Boulter JM, Jakobsen BK, Price DA, Sewell AK. The CD8 T cell coreceptor exhibits disproportionate biological activity at extremely low binding affinities. J Biol Chem. 2003;278:24285–24293. doi: 10.1074/jbc.M300633200. [DOI] [PubMed] [Google Scholar]
- 44.Trautmann L, Labarriere N, Jotereau F, Karanikas V, Gervois N, Connerotte T, Coulie P, Bonneville M. Dominant TCR V α usage by virus and tumor-reactive T cells with wide affinity ranges for their specific antigens. Eur J Immunol. 2002;32:3181–3190. doi: 10.1002/1521-4141(200211)32:11<3181::AID-IMMU3181>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 45.Mantovani S, Palermo B, Garbelli S, Campanelli R, Robustelli Della Cuna G, Gennari R, Benvenuto F, Lantelme E, Giachino C. Dominant TCR-α requirements for a self antigen recognition in humans. J Immunol. 2002;169:6253–6260. doi: 10.4049/jimmunol.169.11.6253. [DOI] [PubMed] [Google Scholar]
- 46.Sensi M, Traversari C, Radrizzani M, Salvi S, Maccalli C, Mortarini R, Rivoltini L, Farina C, Nicolini G, Wolfel T, et al. Cytotoxic T lymphocyte clones from different patients display limited T cell receptor variable-region gene usage in HLA-A2-restricted recognition of the melanoma antigen Melan-A/MART-1. Proc Natl Acad Sci USA. 1995;92:5674–5678. doi: 10.1073/pnas.92.12.5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ohashi PS, Mak TW, Van den Elsen P, Yanagi Y, Yoshikai Y, Calman AF, Terhorst C, Stobo JD, Weiss A. Reconstitution of an active surface T3/T-cell antigen receptor by DNA transfer. Nature. 1985;316:606–609. doi: 10.1038/316606a0. [DOI] [PubMed] [Google Scholar]
- 48.Schneider U, Schwenk HU, Bornkamm G. Characterization of EBV-genome negative “null” and “T” cell lines derived from children with acute lymphoblastic leukemia and leukemic transformed non-Hodgkin lymphoma. Int J Cancer. 1977;19:621–626. doi: 10.1002/ijc.2910190505. [DOI] [PubMed] [Google Scholar]
- 49.Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan R. Enhanced anti-tumor activity of murine-human hybrid TCR in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 66:8878–8886. doi: 10.1158/0008-5472.CAN-06-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lyman MA, Aung S, Biggs JA, Sherman LA. A spontaneously arising pancreatic tumor does not promote the differentiation of naive CD8+ T lymphocytes into effector CTL. J Immunol. 2004;172:6558–6567. doi: 10.4049/jimmunol.172.11.6558. [DOI] [PubMed] [Google Scholar]
- 51.Bullock TN, Mullins DW, Colella TA, Engelhard VH. Manipulation of avidity to improve effectiveness of adoptively transferred CD8+ T cells for melanoma immunotherapy in human MHC class I-transgenic mice. J Immunol. 2001;167:5824–5831. doi: 10.4049/jimmunol.167.10.5824. [DOI] [PubMed] [Google Scholar]
- 52.Ueno T, Tomiyama H, Fujiwara M, Oka S, Takiguchi M. Functionally impaired HIV-specific CD8 T cells show high affinity TCR-ligand interactions. J Immunol. 2004;173:5451–5457. doi: 10.4049/jimmunol.173.9.5451. [DOI] [PubMed] [Google Scholar]
- 53.Kalergis AM, Boucheron N, Doucey MA, Palmieri E, Goyarts EC, Vegh Z, Luescher IF, Nathenson SG. Efficient T cell activation requires an optimal dwell-time of interaction between the TCR and the pMHC complex. Nat Immunol. 2001;2:229–234. doi: 10.1038/85286. [DOI] [PubMed] [Google Scholar]
- 54.Savage PA, Boniface JJ, Davis MM. A kinetic basis for T cell receptor repertoire selection during an immune response. Immunity. 1999;10:485–492. doi: 10.1016/s1074-7613(00)80048-5. [DOI] [PubMed] [Google Scholar]
- 55.Takahama Y. Journey through the thymus: stromal guides for T-cell development and selection. Nat Rev Immunol. 2006;6:127–135. doi: 10.1038/nri1781. [DOI] [PubMed] [Google Scholar]
- 56.Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, Zhou J, Huang J, Powell DJ, Jr, Rosenberg SA. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol. 2004;173:7125–7130. doi: 10.4049/jimmunol.173.12.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hernandez J, Lee PP, Davis MM, Sherman LA. The use of HLA A2.1/p53 peptide tetramers to visualize the impact of self tolerance on the TCR repertoire. J Immunol. 2000;164:596–602. doi: 10.4049/jimmunol.164.2.596. [DOI] [PubMed] [Google Scholar]