Spatial organization and signal transduction at intercellular junctions (original) (raw)
. Author manuscript; available in PMC: 2013 Jun 26.
Published in final edited form as: Nat Rev Mol Cell Biol. 2010 Mar 31;11(5):342–352. doi: 10.1038/nrm2883
Abstract
The coordinated organization of cell membrane receptors into diverse micrometre-scale spatial patterns is emerging as an important theme of intercellular signalling, as exemplified by immunological synapses. Key characteristics of these patterns are that they transcend direct protein–protein interactions, emerge transiently and modulate signal transduction. Such cooperativity over multiple length scales presents new and intriguing challenges for the study and ultimate understanding of cellular signalling. As a result, new experimental strategies have emerged to manipulate the spatial organization of molecules inside living cells. The resulting spatial mutations yield insights into the interweaving of the spatial, mechanical and chemical aspects of intercellular signalling.
Cell-to-cell communication is mediated by various methods. Endocrine signals are secreted and reach distant target cells, paracrine signals are secreted and reach targets in the vicinity, and autocrine signals are secreted and received by the same cell. By contrast, in juxtacrine signalling, surfaces of interacting cells come into direct contact and receptor–ligand recognition at this interface triggers intracellular signalling. Cell–cell interactions involve multiple adhesion and signalling molecules, the collective behaviour of which regulates signal transduction1. Also intrinsic to juxtacrine signalling configurations are large physical constraints on molecular movement and assembly. Genetic and biochemical approaches have been invaluable in identifying the molecular components of signal transduction pathways in juxtacrine signalling and in characterizing the biochemical interactions among them. Despite this wealth of information, in many cases it remains impossible to describe the behaviour of a signalling system in terms of the individual molecular properties of its components. Protein–protein inter actions and the formation of molecular clusters are widely implicated in signal transduction and contribute to a first level of cooperativity2–4. Recently, the coordinated organization of cell membrane receptors into micrometre-scale patterns has emerged as a broadly important theme of intercellular signalling1,5–9.
A paradigm for the interplay of spatial patterns and signal transduction is the junction between T cells and their target cells, termed the immunological synapse8–13. Spatial patterns of proteins at the cell–cell interface develop as hundreds of receptors recognize their cognate ligands on the apposed cell membrane. Multiple signalling and adhesion molecules also become organized into distinctive spatial patterns at the interface between the two cells8,9,13,14 (FIG. 1). The patterns create long-range interactions and seem to have specific purposes in signal transduction8,9. They host the local enrichment or depletion of key signalling components, which can bias biochemical cascades towards different functional outcomes. For example, this can result in location-specific signalling of identical receptors. Recent evidence suggests that the spatial organization of the immunological synapse has an active role in regulating the signalling state of individual molecular components, and thus can alter long-term cell activation15–17. Various different spatial arrangements can occur between different types of immune cells and their respective targets, correlating with different functions8,13. The protein patterns are not static on the cell surface18,19. Instead, they evolve on the timescale of signalling, usually over the course of minutes, and can change depending on the cell signalling state15,16. Here, we highlight recent evidence suggesting that the spatial organization of proteins at cell–cell interfaces may be a widespread regulatory mechanism of intercellular signal transduction.
Figure 1. Micrometre-scale protein patterns in the immunological synapse.
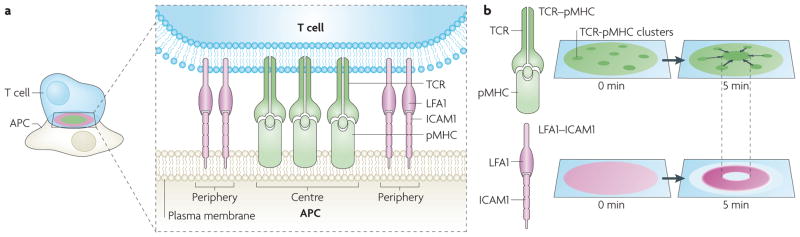
a. The intercellular junction between a T cell and an antigen presenting cell (APC) is known as the immunological synapse. Micrometre-scale protein patterns emerge at the interface between the two cells. A top down and en face view of the immunological synapse reveals highly organized, concentric protein regions. T cell receptors (TCRs) bound to major histocompatibility complexes displaying an antigenic peptide (pMHCs) localize at the central (green) region, and the T cell leukocyte function-associated antigen (LFA1; also known as αLβ2 integrin) bound to intercellular adhesion molecule 1 (ICAM1) localizes to the peripheral (purple) region. b. Formation of micrometre-scale patterns from the time point of contact with an activating APC. TCRs recognize pMHCs and form small clusters (dark green) that are driven by the actin cytoskeleton to the centre of the immunological synapse (top). After 5 minutes, most of the TCRs are in the central zone of the immunological synapse. The T cell integrin LFA1 recognizes ICAM1 and the conjugates form an enriched ring, peripheral to the TCR central zone (bottom).
This Review focuses on the relationship between protein spatial organization and signalling in intercellular junctions, highlighting examples primarily from the immunological synapse. We discuss two structures that are largely responsible for physically controlling this spatial organization: the cell membrane and the actin cytoskeleton. Finally, we review emerging experimental strategies to study and manipulate spatial organization and signalling in living cells.
Micrometre-scale signalling patterns
At the front-line of adaptive immunity, T cells recognize pathogen-derived peptides on the surface of antigen presenting cells (APCs) at the immunological synapse20. Activation of T cells is triggered when T cell receptors (TCRs) recognize their ligand — major histocompatibility complexes (MHCs) displaying the appropriate antigenic peptide (pMHCs). Within 5 minutes of contact, pMHC–TCR complexes form molecular clusters containing tens to hundreds of molecules that are driven by the actin cytoskeleton from the periphery to the centre of the immunological synapse21. Concurrently, the adhesion molecule leukocyte function-associated antigen 1 (LFA1; also known as αLβ2 integrin), on the surface of T cells, ligates intercellular adhesion molecule 1 (ICAM1) on the APC. ICAM1 becomes enriched in a peripheral ring surrounding the central accumulation of TCR within 5 minutes of contact. The TCR and ICAM1 patterns can span the interface of the cell–cell contact zone, which is about 5–10 micrometres in diameter (FIG. 1). This organization was first seen more than a decade ago, triggering intense interest in its possible roles in T cell signalling5,22. Since then, the spatial and temporal complexity of immunological synapses has been explored by a combination of biochemical, genetic, imaging and patterning approaches. Many other signalling and adhesion molecules reorganize as well, and the patterns have functional consequences8–10,12–14,23. In light of this preponderance of observations, we argue that the consideration of spatial organization is indispensable to understanding signal transduction at this inter cellular junction.
Here, in an effort to highlight the best understood examples of how spatial organization can affect signalling, we focus on the classical protein pattern of the T cell immunological synapse: TCRs in the centre and adhesion molecules in a peripheral ring. Our discussion is not intended to be a comprehensive review of immunological synapse signalling, which is well described elsewhere24–27. It is important to note that a diversity of protein patterns besides the canonical one form for different subsets of T cells at different developmental stages and for other immune cell–cell interactions8. The developmental precursors of T cells, thymocytes, form multifocal TCR synapses28. Another subset of immune cells, natural killer (NK) cells, also form organized signalling junctions with their target cells29–31. Their receptors recognize MHCs on other cells, but trigger an inhibitory signal that protects the target cells from the cytotoxic activity of the NK cell. These receptors also reorganize into different patterns at the junction with the target cells: multifocal, homogeneous or homogeneous with regions in which other molecules are excluded. An intriguing configuration is the ring-like enrichment of the NK cell receptors around a central zone of ICAM1, which resembles an inverted T cell synapse29,30. The mechanisms of pattern establishment and control in NK cells seem to be different from those in T cells. NK cells do not require cytoskeleton- or ATP-dependent processes, and the pattern morphology depends on MHC surface density and the balance of activating and inhibitory receptors that are engaged30–32. The exact relationships between spatial organization and signalling in thymocyte and NK cell synapses are less understood than in T cells, but their variety underscores the diversity of protein patterns at intercellular junctions.
The signalling state is location-dependent
The immunological synapse provides several striking examples of how spatial patterns can influence signalling activity. Within five minutes of cell–cell contact, TCR clusters are transported by the actin cytoskeleton to a micrometre- scale zone at the centre of the contact interface5. It is well-established that TCRs in this central zone are dephosphorylated and internalized5,23 (FIG. 1; FIG. 2a). TCR signalling is determined not just by engagement to its ligand pMHC but also by its spatial position17,23,33. Recently, the location-specific signalling state of TCRs at a late signalling stage (about an hour after contact) was addressed16 (FIG. 2b). TCRs in an initial phosphorylation state are localized to the periphery of the immunological synapse, whereas TCRs in a terminal phosphorylation state are at the centre15,16. At both time points, the signalling state of TCRs is altered as a function of their location at the centre or the periphery of the cell–cell interface.
Figure 2. Signalling states are location-dependent in the immunological synapse.

T cell receptors (TCRs) are distributed throughout the immunological synapse; however, their signalling state depends on their location and the time point from their contact with an antigen presenting cell (APC). a. At early signalling (< 20 minutes from APC contact), TCR clusters form and signal in the periphery21. These clusters are transported by the actin cytoskeleton to the centre, where they are downregulated5,8,22,23. **b.** At late signalling (> 40 minutes from APC contact), non-signalling (or low-signalling) TCR clusters are detected in the periphery. Signalling TCR clusters that are fully phosphorylated on all sites are seen in the centre16.
Patterns in immunological synapses are dynamic structures that are actively controlled by and able to adapt to signalling activity. For example, the strength of TCR activation can determine TCR organization and signalling. High stimulation of T cells (by many or strong TCR ligands) results in the aforementioned accumulation of TCRs at the centre of the cell–cell interface, where TCRs become dephosphorylated and internalized23. However, at low stimulation (by fewer or weaker TCR ligands), or at later time points of strong stimulation, there is evidence that the central zone assumes an opposite role and becomes the site of sustained phosphorylation15,16 (FIG. 2). These opposing behaviours suggest that the central and peripheral zones do not have fixed roles as stimulatory or inhibitory regions. On the contrary, their specific effect on signalling depends on the overall signalling context. The immunological synapse is a “molecular machine controlling T cell activation” (REF. 5) that uses spatial organization to balance its signalling outcome23.
Altered spatial organization and collective signalling
At high stimulation, as described above, TCRs are transported by the actin cytoskeleton to the centre of the immunological synapse, where they are down regulated. Because TCR deactivation coincides with TCR transport to the centre of the immunological synapse, it was initially unclear whether TCR activity is spatially or temporally regulated. This ambiguity was resolved by an experiment in which some TCR clusters were physically constrained in the periphery at all time points, including when they would otherwise be in the centre17 (FIG. 3). The experimental strategy to achieve such selective control over TCR spatial organization is discussed later. The net result of these experiments was to reveal that at the same time point the peripheral TCR clusters remain phosphorylated while the central TCR clusters are dephosphorylated and inactivated. The simultaneous presence of signalling and non-signalling TCRs provides direct evidence that the TCR signalling state is influenced by its radial location irrespective of the time point of signalling. Furthermore, the altered TCR localization and signalling increased the T cell calcium flux, a downstream response that triggers transcription factor activation, and thus altered the signalling outcome of the whole T cell.
Figure 3. Spatial organization influences cell signalling in the immunological synapse.

The spatial organization of signalling and non-signalling T cell receptors (TCRs) changes with different levels of cell stimulation and perturbing the spatial organization of TCRs by blocking their transport modifies the overall response of the cell. a. At high T cell activation (by strong or many agonists), signalling TCRs are located in the periphery and are transported to the centre, where they are downregulated. The T cell response is strong5,8,22,23. b. Physical barriers block the transport of TCRs to the centre at high T cell activation, and TCR clusters are constrained to the periphery, where they continue to signal. As a result, the T cell response is prolonged and higher than in part a17. c. At low T cell activation (by weak or few agonists), signalling TCRs are undetectable and non-signalling TCRs are in the periphery. The T cell response is weak. d. When TCR transport to the centre is artificially induced at low levels, TCR signalling can be detected in the centre and the response of the T cell is higher than in part c16.
In the above example17, the translocation of TCRs from the centre to the periphery was constrained by physical barriers (FIG. 3b; see also later section on experimental organization control). This perturbation allowed the comparison of biochemically identical cells, in which only the localization of a specific receptor is altered. We refer to these perturbations as ‘spatial mutations’. The term ‘mutation’ refers originally to genetic alterations and here its definition is extended to include the alteration of spatial parameters.
The previous example illustrates how, at high activation, constrained TCR transport to the centre decreases TCR signalling downregulation. Conversely, at low stimulation, there is evidence that enhanced TCR transport to the centre boosts TCR signalling15. Immunofluorescence shows that the centre of the immunological synapse is the site of sustained signalling under low stimulatory conditions15,16. This observation raised the question whether enhancing TCR transport at such conditions would also alter T cell signalling. Indeed, artificially induced central accumulation of TCRs, in cases where it would not occur naturally, resulted in enhanced long-term cell signalling16 (FIG. 3c,d).
The regulatory function of spatial organization is further exemplified by experiments that test the cellular response to spatially inhomogeneous stimuli. Besides physical barriers that alter the spatial reorganization of proteins driven by a cell17, pre-patterned arrangements of different surface receptor ligands or antibodies can impose spatial organization on a cell. These are another version of our definition of spatial mutation, and experimental methodologies to achieve this are discussed later. In one striking example of this strategy, researchers recreated the pattern of the canonical immunological synapse, with TCRs in the centre surrounded by adhesion molecules, and successfully activated the T cells (see also later section on experimental organization control)33. More importantly, when the pre-programmed geometry differed from the natural pattern, T cell signalling was noticeably altered. Different geometrical orientations were tested: one of inversion, whereby TCRs are in a peripheral ring and adhesion molecules are in the centre, and another of breaking up the localization of TCRs into multiple foci. After several hours of activation by the different patterns, the secretion levels of a proliferative cytokine were similar. However, secretion of the targeted cytokine was decreased in cells responding to the inverted pattern.
The spatial organization of co-stimulatory molecules also affects signalling. Recent work using immobilized patterned antibodies to stimulate cells shows that T cells are maximally co-stimulated with CD28, but only if CD28 is in the periphery with respect to TCRs34. If CD28 is forced to the centre or CD28 and TCR colocalize, cell signalling decreases (see also later section on experimental manipulation of organization). In this case, TCR and CD28 signalling need to be spatially segregated, and the CD28 has to be peripheral to TCRs for optimal co-stimulation. This work uses fixed patterns that do not fully reflect the dynamic association of TCRs and CD28 in microclusters in the periphery and their accumulation in separate compartments in the centre of the immunological synapse27,35. However, it tests and emphasizes the difference in signalling in the two synapse zones and its effect on long term T cell activity.
Spatial organization in biochemical signalling cascades
Intracellular biochemical signalling cascades operate far from reaction equilibrium and homogeneity. As such, many simple chemical rules of thumb no longer apply. For example, classical chemical kinetic rate equations are predicated on the assumption that all species are fully mixed and, therefore, randomly distributed in solution. This is not true for any of the signalling systems we consider here, thus the measured kinetic rates for specific protein interactions in solution do not apply36. Although it is clear that the spatial organization of molecular components can bias reaction outcomes, specific and quantitative analysis of these effects is often quite difficult by human intuition alone23,37–40. Nevertheless, this seems to still be the predominant method used to deduce biological signalling mechanisms. However, computational modelling is gaining ground as an important aspect of the study and understanding of spatially regulated signalling cascades. In one example of this, a computational model was developed that specifically included the spatial position of reactants23. Simulations revealed how the synapse centre can be a site for both enhanced signalling and downregulation. Although the model is simplified and can not capture the full intricacy of T cell biology, it provided a substantial advance in the mechanistic understanding of TCR signalling regulation. Better and more inclusive models continue to emerge37–41, as do more quantitative experiments to measure spatial organization and function in cellular signalling systems.
Another revealing example of the spatial sensitivity of a simple reaction network is the initiation of blood clotting, which involves a cascade of tens of biochemical reactions. Competition between catalytic product formation and diffusive mixing controls the progress of the whole signalling cascade and exhibits distinctive sensitivity to spatial organization. If the reactants are patterned on a surface, the initiation of the clotting reaction depends not on the overall amount of reactant but on the dimensions and spacing of regions of reactant42,43. This dependence can be reproduced in a synthetic signalling cascade that has similar competition between product formation and dispersion by diffusion. The in vitro sensitivity of the blood clotting reaction cascade to spatial organization is physiologically relevant in vivo. Bacteria in the blood stream release factors that can initiate blood clotting. In mice, clotting caused by bacterial infection occurs only in regions where bacteria are clustered and yield a high local concentration of reactant44.
Cellular organization mechanisms
The physical mechanisms that establish and regulate the spatial organization of signalling molecules are equally as important as the chemical reactions themselves. Here, we highlight key roles of the cell membrane and the actin cytoskeleton.
Cell membrane
The cell membrane is a spatially heterogeneous yet liquid mixture of lipids, proteins and other molecules that provides the environment in which nearly all signal transduction processes occur. Protein–protein interactions in the membrane clearly have a central role in defining the assembly and composition of receptor signalling clusters. The lipid and cholesterol components of the cell membrane also exhibit clear miscibility phase separation in vitro, and this is thought to contribute to membrane organization in vivo44. Membrane domains formed by phase separation are widely referred to as lipid rafts. Although a significant role for lipid composition on membrane organization in vivo is almost a certainty, specifics of the raft hypothesis remain hotly debated. This subject is comprehensively reviewed elsewhere and is not discussed in detail here45.
What is clear is that many signalling systems, such as through TCRs, signal from clusters that form dynamically in the cell membrane (probably through a combination of protein and lipid interactions). These clusters are sometimes referred to as rafts. What is not clear is to what degree the composition and structure of these clusters varies in time, from one to another and on the same cell under different signalling conditions. Indeed, much of the controversy surrounding the term ‘membrane raft’ stems from the difficulty in defining structures that are so dynamic and variable. There may well be certain characteristics common to many types of molecular clusters occurring in the cell membrane. From a signalling perspective, however, we suggest that any such commonality is not the most relevant feature. It is the specific functionality resulting from individual details of the composition and distribution of signalling clusters that we must elucidate in order to understand signalling mechanisms at the cell membrane.
Recent work has revealed a specific example of the functional organization of the membrane signalling proteins in T cells46. TCRs and LAT are organized in protein clusters (sometimes referred to as islands) of 7–30 molecules in quiescent cells. On activation, these clusters coalesce and form larger structures of a few hundred molecules that have been referred to as microclusters (all of these structures could perhaps be considered as different types of raft)21. This study combines three different experimental approaches — hsPALM, transmission electron microscopy (TEM) and dcFFCS — to observe different aspects of the protein cluster arrangements, yielding an unprecedented high-resolution and dynamic view of membrane organization. This observation of independent dynamic domains of TCRs and LAT and their concatenation on activation reveals an elegant choreography of membrane organization that evolves dynamically during signalling. We suggest that substantial breakthroughs in understanding signalling processes may be achieved by revealing the rich repertoire of membrane organizational states.
Juxtacrine signalling at intercellular junctions, such as the immunological synapse, leads to some interesting membrane mechanical effects that can also contribute to protein clustering1,47. The dissociation constant for an intermembrane protein complex is different from that in solution because the probability of interactions depends on the intermembrane separation and the protein surface density46,48 (FIG. 4a). Although membranes are flexible, high bending is energetically unfavourable. In intercellular junctions, a protein-binding pair from opposite membranes can effectively prevent the interaction of a neighbouring protein-binding pair if the intermembrane spacings of the pairs are very different49 (FIG. 4b) or can induce clustering among similar proteins50. An important consequence of this is that intermembrane protein binding can exhibit cooperative effects between remote pairs of proteins that are not in direct contact. The proximity of short and long intermembrane pairs requires that the membranes bend to accommodate the size difference. In the fluid environment of the cell membrane, this intermembrane size selection can lead to spatial segregation into patterns that guide signalling37,49,51–53 (FIG. 4c). For example, experiments54 have shown that protein intermembrane spacing is crucial for TCR signalling and it has been proposed that the exclusion of long phosphatases from TCR regions of short intermembrane spacing is the reason for TCR triggering51,55. Re-engineered pMHC–TCR complexes with elongated intermembrane spacing disrupt signalling in the T cell without altering the intrinsic complex interactions.
Figure 4. Cellular mechanisms controlling spatial organization in intercellular junctions.

The interplay of the cell membrane and cytoskeleton at intercellular junctions yields short- and long-range spatial organizations of proteins, which transcend direct protein–protein interactions. a. The probability of a binding interaction between two membrane proteins is much higher when their orientation is pre-aligned by the membrane, compared to proteins in solution. Therefore, weak interactions are effectively strengthened. b. Binding across the intercellular space is governed by intermembrane spacing, which is determined by established protein-binding pairs. c. The binding of pairs that create different sizes of intermembrane spacing are segregated (blue versus green binding pairs) to minimize membrane bending. Additionally, large proteins (red) can enter wide intermembrane spacing regions, but are excluded from entering regions of tight intermembrane spacing. d. The moving actin cytoskeleton, through multiple weak associations with adaptor molecules, can selectively transport membrane molecules (green) and establish long-range protein organization. The force applied by actin can depend on protein cluster size63.
Actin cytoskeleton
The actin cytoskeleton has long been implicated in controlling the dynamic spatial organization of the immunological synapse. It mediates long-range interactions by physically transporting surface molecules, such as TCRs and the integrin LFA1, and directly interacts with the components of signalling cascades21,25,55–57. The actin cytoskeleton itself is a dynamic structure with a highly heterogeneous composition and mechanical properties. There are regions of lower or higher branching and different polymerization and depolymerization rates53,56,58–60. For example, in the immunological synapse, the outer periphery of the cell-to-cell interface displays a dense network of filamentous actin (F-actin), whereas the centre of the interface is either depleted of F-actin or F-actin is present at a lower density24,56. TCR signalling leads to the activation of multiple regulators of actin polymerization, such as Cdc42, WAVE2 (also known as WASF2), and Wiskott–Aldrich syndrome protein (WASP), which themselves can remodel the local cytoskeleton55.
The regulation of protein transport is key to controlling the signalling activity of individual proteins and the whole cell. Adaptor molecules25,58,61, such as talin for LFA1 (REF. 62), mediate selective interactions between signalling molecules and actin. As spatial organization varies with the overall signalling context of the cell, it is plausible that these adaptor molecules are not just constant links but are actively regulated. The adaptor proteins for TCR transport remain elusive. The ezrin, radixin and moesin (ERM) proteins, which can bind both TCRs (or other signalling molecules) and F-actin, are implicated but their role is still controversial and they do not seem to be sufficient for TCR transport25,55.
The mechanisms by which the cytoskeleton physically and selectively transports molecules are under investigation. Although it is clear that receptor transport in the immunological synapse is driven by actin, the mechanism of differential protein sorting remains unresolved. TCRs and LFA1 are driven towards the centre of the immunological synapse, but their final destinations are micrometres apart56,63 (FIG. 1). Recent experiments in which the clustering state of LFA1 was externally manipulated have begun to shed more light on such processing of actin-mediated sorting. LFA1 does not form the large clusters that TCRs do; however, its cluster size can be experimentally controlled through the bivalent and tetravalent antibody cross-linking of LFA1 or its ICAM1 ligand63. The higher the degree of cross-linking, the closer LFA1 is brought to the centre of the immunological synapse, where TCRs would be. With the tetravalent cross-linking, LFA1 reaches the centre of the immunological synapse, sharing it with the TCR clusters. This observation suggests that sorting of proteins in the immunological synapse can be accomplished by the regulation of cluster size and cluster interactions with the driving actin cytoskeleton (FIG. 4d).
The physical interaction of TCR clusters with actin is further revealed in experiments that alter the native path of TCR clusters to the centre of the immunological synapse57. The experimental strategy to introduce selective barriers to TCR transport is discussed later. When TCR clusters encounter a physical barrier en route to the centre, they do not always stop. If they approach a barrier at an angle with respect to the driving actin flow, they continue their motion tangential to the barrier. They remain driven by the actin cytoskeleton, although their apparent directions differ. This behaviour strongly suggests that multiple, dynamic and weak interactions couple TCR clusters to the moving actin flow, similar to a drag force. This mechanism allows TCR clusters to divert around obstacles and still be driven by actin without building up substantial elastic energy. The mechanism is also easily extended to sort many molecules on the cell surface.
Multiple length scales of spatial organization
So far we have primarily discussed micrometre-scale organization of proteins in the immunological synapse. At least for TCRs, this pattern is a result of the accumulation of multiple smaller clusters of tens to hundreds of molecules. Such clusters are observed for many other molecules in the immunological synapse, including LAT64, ζ-chain-associated protein kinase 70 (ZAP70)64, SH2 domain containing leukocyte protein of 76kDa (SLP76; also known as LCP2)65, CD28 (REF. 35) and CD2 (REF. 53). Besides those seen in immune cells, smaller clusters have long been observed for many other membrane proteins, such as ErbB family receptors66, and even for intracellular signalling molecules such as Ras67. A recent study of the ephrin type A receptor 2 (EPHA2) receptor Tyr kinase suggests that clustering and micrometre-scale spatial translocation of the receptor clusters can lead to unanticipated emergent properties such as mechanical-force sensing68. In chemotaxis, bacterial receptors also exist in an organized manner at the cell poles, with exponentially distributed cluster sizes from tens to thousands of receptors69. In these clusters, different receptors functionally cooperate70,71. Although in this review we focus on micrometre-scale organization, there is a continuum of protein spatial organizations, from molecular complexes to micrometre-scale domains that span the cell-to-cell interface72. We should anticipate that organization at all these different levels feeds back to regulate specific protein and collective cell signalling.
Experimental organization control
Studies of the role of spatial organization in signal transduction require experimental approaches that directly manipulate the spatial component of cellular signalling systems. In response to this need, several strategies have recently been introduced. These are based largely on surface chemistry and material fabrication techniques that are not typically included in a classical cell biology repertoire. A more detailed discussion of these methodologies as they apply to cell biology follows.
Hybrid live cell–supported bilayer junctions
Hybrid live cell–supported lipid membrane junctions can reconstitute much of the micrometre- and molecular-scale re organization that occurs in natural intercellular junctions. Aspects of the immunological and neuronal synapse can be reconstituted between live cells and synthetic surfaces5,73 (FIG. 5a). Recently, a complex signalling interaction between the EPHA2 receptor Tyr kinase on live epithelial cells and its membrane surface ephrin A1 ligand in supported membranes has also been achieved68. The supported membrane is a continuous fluid lipid bilayer that forms by the spontaneous self-assembly of liposomes or proteoliposomes on clean glass surfaces74. Proteins with membrane anchors can be tethered to the bilayer and experience the free lateral mobility that is inherent to the native cell membrane75,76. Other advantages of this system are control over identity and quantity of the components77. Additionally, the well-defined planar interface facilitates high resolution imaging by fluorescence microscopy.
Figure 5. Experimental manipulation of spatial organization in intercellular junctions.
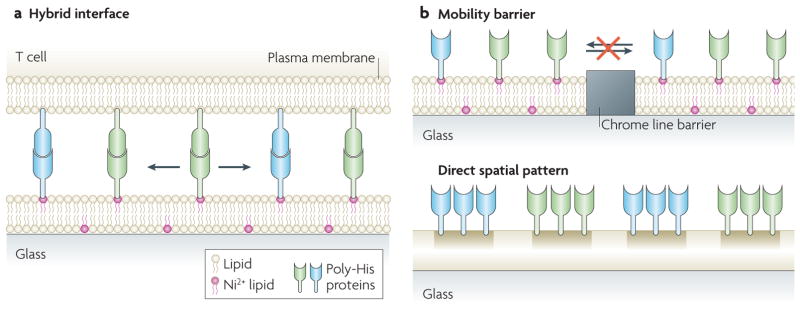
a. Cell-to-cell interactions are reconstituted in hybrid interfaces between living cells and functionalized surfaces. The glass coverslip is functionalized with a supported lipid bilayer that is stably adhered to the surface, while exhibiting free diffusion (arrows) of its lipid components. Proteins are tethered to the fluid bilayer, for example by poly-histidine tags that bind Ni2+-chelating lipids. Cells recognize their membrane-anchored ligands and can rearrange their organization. b. The surface can be patterned with subcellular features that alter the native spatial organization of membrane proteins. Chrome lines are barriers to lipid mobility and the transport of membrane-tethered proteins and any cell proteins engaged with them (top). They can restrict the reorganization of cell surface proteins initiated by the cell. Different proteins (green and blue) can be immobilized to the surface in any pre-set configuration, forcing cell ligands to also reorganize according to the presented arrangement (bottom).
Solid-state structures patterned on the glass surface can act as barriers to the lateral diffusion and transport of lipids and proteins in the supported lipid bilayer75,76,78 (FIG. 5b). As receptors engage their ligands in the bilayer, they are also selectively restricted by the pattern on the substrate. Crucially, only the proteins interacting directly with the membrane components are subjected to the constraints; the rest of the cell is free to rearrange. Different configurations of metal lines pre-patterned on the surface (for example, by electron beam lithography (BOX 1)) can alter the geometric pattern of cell surface receptors and associated signalling molecules17,57,63 (FIG. 6a). A key feature of a supported membrane with patterns of mobility barriers is that molecular-level clustering is allowed to proceed naturally but large-scale organization is selectively under direct control. This allows analysis of large-scale spatial pattern and clustering effects without the side effects caused by the altered molecular-scale assembly of signalling complexes (which would occur on purely solid surfaces).
Box 1. Patterning strategies.
Functionalized surface–live cell interactions are a powerful model system for the study of intercellular signalling. The surface can be patterned with regions of metals, proteins or lipid bilayer in different configurations by several methods. The different strategies vary in the size and precision of the features, the area that can be patterned and the ease of use80,82.
Microfluidics
Several channels with different proteins flow unmixed over the surface and deposit juxtaposed patterns83,84,107. Pattern geometry is limited to permissible flow patterns but allows for multiple proteins to be co-patterned. The resolution is < 10 μm.
Photolithography
The surface is initially coated with a light-sensitive polymer. Illumination with visible or ultraviolet light through a patterned mask destroys the polymer and allows for metals or proteins to be deposited. After removal of the rest of the polymer, the rest of the surface can be filled with another protein or lipid bilayer81,85. The resolution is ~ 1μm.
Electron-beam lithography
Similar to photolithography, but an electron beam is used to draw patterns that destroy the polymer and create regions for differential deposition17,78. The resolution is ~ 20nm.
Microcontact printing and nanoimprinting
The surface is patterned, by either method, and physically etched according to the pattern to create a template or master three-dimensional (3D) surface82,86. The pattern can be further replicated by deposition of polydimethylsiloxane (PDMS), which solidifies according to the 3D surface. Either the template surface or the separated PDMS block are absorbed with protein and stamped on the target surface, thus maintaining the patterned configuration. Typically, nanoimprinting resembles embossing more than stamping, recreating a 3D effect in the target surface. The resolution for microcontact printing is ~ 1 μm and for nanoimprinting is < 20 nm.
Dip-pen nanolithography
An atomic force microscopy (AFM) tip loaded with a small molecule or a protein solution as ink is used to write on the surface87. Any pattern is possible, but deposition is serial. The resolution is < 50 nm.
Block co-polymer micelle nanolithography
Polymers self-assemble over large areas of the surface into regular patterns that are used as templates for further functionalization88. The resolution is < 20 nm.
Figure 6. Immunological synapse spatial mutations.

a. Physical barriers to protein transport on a fluid supported lipid bilayer. Thin chrome lines create barriers to the diffusion of bilayer-tethered proteins (such as major histocompatibility complexes displaying an antigenic peptide (pMHC)) and cellular proteins (such as T cell receptors (TCRs)) interacting with them (left). The spatial organization of the immunological synapse (TCRs (green) and intercellular adhesion molecule 1 (ICAM1; red)) without (1) and with (2–4) barriers of different geometries (right). b. Subcellular size protein patterns functionalized on a surface. A TCR-activating antibody (anti-CD3; green) and an adhesion molecule (ICAM1; purple) are patterned according to the immunological synapse pattern: anti-CD3 is central to the surrounding adhesion molecules (left). Anti-CD3, shown in schematics and cell overlays (in which anti-CD3 is blue) can be seen in a wild-type central zone pattern or in two variant patterns: multifocal and a peripheral ring (right). c. The subcellular pattern of a TCR-activating antibody (anti-CD3ε; green) and a co-stimulatory antibody (anti-CD28; blue) on an adhesion molecule (ICAM1; purple)-rich surface (left). Different patterns are tested for their effect on T cell activation: TCR and CD28 follow the pattern of anti-CD3 and anti-CD28 antibodies, respectively, which can be either co-localized or segregated. Images in part a are reproduced, with permission, from REF. 17 © (2005) American Association for the Advancement of Science. Images in part b are reproduced, with permission, from REF. 33 © (2006) National Academy of Sciences. Images in part c are reproduced, with permission, from REF. 34 © (2008) National Academy of Sciences.
Direct spatial pattern on solid surfaces
The geometry of the hybrid signalling junction can also be altered by direct patterning of proteins on solid surfaces (FIG. 5b; FIG. 6b,c). Strategies inspired by semiconductor fabrication and polymer chemistry have emerged to manipulate the spatial organization of molecules on the surface and therefore inside living cells79–82 (BOX 1). The surface is patterned with subcellular features that localize cellular ligands in pre-set configurations (FIG. 5b). The pattern feature size, geometry, area patterned and diversity of proteins that can be patterned at the same time vary greatly between different techniques. The patterning strategies include microfluidics83,84, photolithography81,85, electron beam lithography, microcontact printing86, nanoimprinting82, dip-pen nanolithography87 and block–co-polymer micelle nanolithography88. Some of these patterning strategies can be combined with supported fluid lipid bilayers on the same surface, providing both mobile and immobile stimuli17,57,75,81,89–91. The cell biological application of these techniques was originally pioneered for the study of cell adhesion and focal adhesion sites80,92–94. More recent developments have emphasized control of signalling specificity along with spatial organization17,33,34,75,91,95,96. Their application in cell studies meets some biology-inherent challenges. At the very least, patterns need to be biocompatible with subcellular and even molecular dimensions17,33,34,93.
In the studies of TCR localization to the centre or the periphery, antibodies to TCRs and adhesion molecules were patterned with features as small as a few micrometres33. The presentation of anti-TCR antibodies in a peripheral ring constrained TCR triggering and signalling to this specific peripheral configuration and prevented TCRs from forming a central cluster (FIG. 6b). In studies of CD28 co-stimulation, anti-TCR and anti-CD28 antibodies were added to the adhesion molecule-rich surface, thus offering a pre-set orientation for activated TCRs and CD28 (REF. 34) (FIG. 6c). Such inhomogeneous ligand presentations have revealed the importance of spatial organization to specific molecules or signalling pathways.
Control of spatial organization can also be achieved by the optical control of protein or activity. Light-sensitive97–99 photo-switchable molecules98,100,101 or local uncaging102–104 combined with diffraction-limited lasers enable high spatial and temporal control over protein activity. Optically controlled uncaging of T cell-activating peptide can stimulate the cells with high temporal and possibly spatial resolution102,103. Both of these types of approach can cause spatially inhomogeneous activation of cells and, when combined with readouts of signalling105,106, can elucidate mechanisms of the spatial control of signalling.
Conclusion
The immunological synapse provides vivid examples of how signalling reactions in biology use spatial organization on multiple length scales. The proteins in this intercellular junction are highly organized into micrometre-scale patterns that control signal transduction. The spatial organization of the interface determines the signalling state of individual molecules and the signalling state of a cell determines its specific spatial organization. The immunological synapse provides examples of how altered spatial organization changes the signalling outcome of a single molecule or the cell as a whole. Computational modelling and patterning of simple reaction networks illustrate how complex biochemical cascades depend strictly on the spatial organization of their components. We argue that spatial organization is an integral part of signal transduction regulation that is probably important for other juxtacrine signalling systems as well.
Spatial organization is physically controlled by the interplay between the cell membrane and the actin cytoskeleton. New experimental strategies have emerged that draw inspiration from fields such as semiconductor fabrication and polymer chemistry and apply these concepts to manipulate the spatial organization of molecules inside living cells. These approaches are becoming indispensable to understanding the control of spatial organization and exploring its role in signalling.
Acknowledgments
The hybrid live cell–supported membrane component of this work was supported by the Director, Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences and Biosciences Division of the U.S. Department of Energy under contract number DE-AC02-05CH11231. General support for this project was also provided by a National Science Foundation CAREER award MCB-0448614 to J.T.G.
Glossary
Cytokine
A member of a large family of immunomodulating secreted proteins that interact with cellular receptors. Cytokine production results in the activation of an intracellular signalling cascade that commonly regulates processes such as inflammation
Miscibility phase separation
The partitioning of lipid components (in the context of membranes) into domains that have different chemical compositions and physical properties
LAT
(Linker for activation of T cells). A transmembrane protein that on TCR activation becomes rapidly phosphorylated and binds multiple adaptor molecules and indirectly recruits others
hsPALM
(High speed photoactivated localization microscopy). A fluorescence imaging technique in which sequential activation, localization and bleaching of fluorescent reporter proteins yields an image with a resolution of a few tens of nanometers, well below the diffraction limit
dcFFCS
(Dual colour fluorescence cross-correlation spectroscopy). A technique that analyses the dynamics and association of two different diffusing fluorescent proteins
Chemotaxis
Directed cell movement according to chemical stimuli
Liposome or proteoliposome
A vesicle made of lipid bilayer in an aqueous environment. Membrane proteins can be incorporated in the bilayer
Photo-switchable molecule
A molecule with a functionality (ligand binding, conformational change or absorption spectrum) that is controlled by light and in some cases can be toggled on and off
Uncaging
The light-controlled release of a functional group that hides (cages) another functional group
Footnotes
Competing interests statement
The authors declare no competing financial interests.
Contributor Information
Boryana N. Manz, Email: boryana@berkeley.edu.
Jay T. Groves, Email: jtgroves@lbl.gov.
References
- 1.Groves JT. Molecular organization and signal transduction at intermembrane junctions. Angew Chem Int Ed Engl. 2005;44:3524–3538. doi: 10.1002/anie.200461014. [DOI] [PubMed] [Google Scholar]
- 2.Reich Z, et al. Ligand-specific oligomerization of T-cell receptor molecules. Nature. 1997;387:617–620. doi: 10.1038/42500. [DOI] [PubMed] [Google Scholar]
- 3.Bray D, Levin MD, Morton-Firth CJ. Receptor clustering as a cellular mechanism to control sensitivity. Nature. 1998;393:85–88. doi: 10.1038/30018. [DOI] [PubMed] [Google Scholar]
- 4.Maheshwari G, Brown G, Lauffenburger DA, Wells A, Griffith LG. Cell adhesion and motility depend on nanoscale RGD clustering. J Cell Sci. 2000;113:1677–1686. doi: 10.1242/jcs.113.10.1677. [DOI] [PubMed] [Google Scholar]
- 5.Grakoui A, et al. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285:221–227. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- 6.Delon J, Germain RN. Information transfer at the immunological synapse. Curr Biol. 2000;10:R923–R933. doi: 10.1016/s0960-9822(00)00870-8. [DOI] [PubMed] [Google Scholar]
- 7.Davis MM, et al. Dynamics of cell surface molecules during T cell recognition. Annu Rev Biochem. 2003;72:717–742. doi: 10.1146/annurev.biochem.72.121801.161625. [DOI] [PubMed] [Google Scholar]
- 8.Davis DM, Dustin ML. What is the importance of the immunological synapse? Trends Immunol. 2004;25:323–327. doi: 10.1016/j.it.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 9.Singleton KL, et al. Spatiotemporal patterning during T cell activation is highly diverse. Sci Signal. 2009;2:ra15. doi: 10.1126/scisignal.2000199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Der Merwe PA, Davis SJ. Immunology. The immunological synapse — a multitasking system. Science. 2002;295:1479–1480. doi: 10.1126/science.1069896. [DOI] [PubMed] [Google Scholar]
- 11.Huppa JB, Davis MM. T-cell-antigen recognition and the immunological synapse. Nature Rev Immunol. 2003;3:973–983. doi: 10.1038/nri1245. [DOI] [PubMed] [Google Scholar]
- 12.Dustin ML, Colman DR. Neural and immunological synaptic relations. Science. 2002;298:785–789. doi: 10.1126/science.1076386. [DOI] [PubMed] [Google Scholar]
- 13.Friedl P, den Boer AT, Gunzer M. Tuning immune responses: diversity and adaptation of the immunological synapse. Nature Rev Immunol. 2005;5:532–545. doi: 10.1038/nri1647. [DOI] [PubMed] [Google Scholar]
- 14.Kao H, Lin J, Littman DR, Shaw AS, Allen PM. Regulated movement of CD4 in and out of the immunological synapse. J Immunol. 2008;181:8248–8257. doi: 10.4049/jimmunol.181.12.8248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cemerski S, et al. The stimulatory potency of T cell antigens is influenced by the formation of the immunological synapse. Immunity. 2007;26:345–355. doi: 10.1016/j.immuni.2007.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cemerski S, et al. The balance between T cell receptor signaling and degradation at the center of the immunological synapse is determined by antigen quality. Immunity. 2008;29:414–422. doi: 10.1016/j.immuni.2008.06.014. Reports that the centre of the immunological synapse can be a site of sustained signalling for low doses of, or weak, antigens. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mossman KD, Campi G, Groves JT, Dustin ML. Altered TCR signaling from geometrically repatterned immunological synapses. Science. 2005;310:1191–1193. doi: 10.1126/science.1119238. TCRs were physically restricted in the periphery of the synapse and their radial localization was shown to regulate their signalling. [DOI] [PubMed] [Google Scholar]
- 18.Sims TN, et al. Opposing effects of PKCτ and WASp on symmetry breaking and relocation of the immunological synapse. Cell. 2007;129:773–785. doi: 10.1016/j.cell.2007.03.037. [DOI] [PubMed] [Google Scholar]
- 19.Dustin ML. Cell adhesion molecules and actin cytoskeleton at immune synapses and kinapses. Curr Opin Cell Biol. 2007;19:529–533. doi: 10.1016/j.ceb.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paul WE, Seder RA. Lymphocyte-responses and cytokines. Cell. 1994;76:241–251. doi: 10.1016/0092-8674(94)90332-8. [DOI] [PubMed] [Google Scholar]
- 21.Varma R, Campi G, Yokosuka T, Saito T, Dustin ML. T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity. 2006;25:117–127. doi: 10.1016/j.immuni.2006.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monks CR, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 1998;395:82–86. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- 23.Lee KH, et al. The immunological synapse balances T cell receptor signaling and degradation. Science. 2003;302:1218–1222. doi: 10.1126/science.1086507. Combines experiments and computer modelling to reveal the dual capacity of the immunological synapse to act as an enhancer and a downregulator of TCR signalling. [DOI] [PubMed] [Google Scholar]
- 24.Dustin ML. T-cell activation through immunological synapses and kinapses. Immunol Rev. 2008;221:77–89. doi: 10.1111/j.1600-065X.2008.00589.x. [DOI] [PubMed] [Google Scholar]
- 25.Gomez TS, Billadeau DD. T cell activation and the cytoskeleton: you can’t have one without the other. Adv Immunol. 2008;97:1–64. doi: 10.1016/S0065-2776(08)00001-1. [DOI] [PubMed] [Google Scholar]
- 26.Orange JS. Formation and function of the lytic NK-cell immunological synapse. Nature Rev Immunol. 2008;8:713–725. doi: 10.1038/nri2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yokosuka T, Saito T. Dynamic regulation of T-cell costimulation through TCR-CD28 microclusters. Immunol Rev. 2009;229:27–40. doi: 10.1111/j.1600-065X.2009.00779.x. [DOI] [PubMed] [Google Scholar]
- 28.Hallman E, Burack WR, Shaw AS, Dustin ML, Allen PM. Immature CD4+CD8+ thymocytes form a multifocal immunological synapse with sustained tyrosine phosphorylation. Immunity. 2002;16:839–848. doi: 10.1016/s1074-7613(02)00326-6. [DOI] [PubMed] [Google Scholar]
- 29.Davis DM, et al. The human natural killer cell immune synapse. Proc Natl Acad Sci USA. 1999;96:15062–15067. doi: 10.1073/pnas.96.26.15062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Almeida CR, Davis DM. Segregation of HLA-C from ICAM-1 at NK cell immune synapses is controlled by its cell surface density. J Immunol. 2006;177:6904–6910. doi: 10.4049/jimmunol.177.10.6904. [DOI] [PubMed] [Google Scholar]
- 31.Culley FJ, et al. Natural killer cell signal integration balances synapse symmetry and migration. PLoS Biol. 2009;7:e1000159. doi: 10.1371/journal.pbio.1000159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Masilamani M, Nguyen C, Kabat J, Borrego F, Coligan JE. CD94/NKG2A inhibits NK cell activation by disrupting the actin network at the immunological synapse. J Immunol. 2006;177:3590–3596. doi: 10.4049/jimmunol.177.6.3590. [DOI] [PubMed] [Google Scholar]
- 33.Doh J, Irvine DJ. Immunological synapse arrays: patterned protein surfaces that modulate immunological synapse structure formation in T cells. Proc Natl Acad Sci USA. 2006;103:5700–5705. doi: 10.1073/pnas.0509404103. Developed a surface recreating canonical, multifocal and inverted synapses, and showed that some downstream T cell signalling pathways are altered by the new geometries. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen K, Thomas VK, Dustin ML, Kam LC. Micropatterning of costimulatory ligands enhances CD4+ T cell function. Proc Natl Acad Sci USA. 2008;105:7791–7796. doi: 10.1073/pnas.0710295105. Reveals that segregation, colocalization and relative radial juxtaposition alter how CD28 co-stimulates TCR signalling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yokosuka T, et al. Spatiotemporal regulation of T cell costimulation by TCR-CD28 microclusters and protein kinase Cτ translocation. Immunity. 2008;29:589–601. doi: 10.1016/j.immuni.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huppa JB, et al. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature. 2010;463:963–967. doi: 10.1038/nature08746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weikl TR, Lipowsky R. Pattern formation during T-cell adhesion. Biophys J. 2004;87:3665–3678. doi: 10.1529/biophysj.104.045609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Altan-Bonnet G, Germain RN. Modeling T cell antigen discrimination based on feedback control of digital ERK responses. Plos Biol. 2005;3:1925–1938. doi: 10.1371/journal.pbio.0030356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wylie DC, Das J, Chakraborty AK. Sensitivity of T cells to antigen and antagonism emerges from differential regulation of the same molecular signaling module. Proc Natl Acad Sci USA. 2007;104:5533–5538. doi: 10.1073/pnas.0611482104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feinerman O, Germain RN, Altan-Bonnet G. Quantitative challenges in understanding ligand discrimination by αβ T cells. Mol Immunol. 2008;45:619–631. doi: 10.1016/j.molimm.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chakraborty AK, Das J. Pairing computation with experimentation: a powerful coupling for understanding T cell signalling. Nature Rev Immunol. 2010;10:59–71. doi: 10.1038/nri2688. [DOI] [PubMed] [Google Scholar]
- 42.Kastrup CJ, Runyon MK, Shen F, Ismagilov RF. Modular chemical mechanism predicts spatiotemporal dynamics of initiation in the complex network of hemostasis. Proc Natl Acad Sci USA. 2006;103:15747–15752. doi: 10.1073/pnas.0605560103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kastrup CJ, Shen F, Runyon MK, Ismagilov RF. Characterization of the threshold response of initiation of blood clotting to stimulus patch size. Biophys J. 2007;93:2969–2977. doi: 10.1529/biophysj.107.109009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Veatch SL, et al. Critical fluctuations in plasma membrane vesicles. ACS Chem Biol. 2008;3:287–293. doi: 10.1021/cb800012x. [DOI] [PubMed] [Google Scholar]
- 45.Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- 46.Dustin ML, Ferguson LM, Chan PY, Springer TA, Golan DE. Visualization of CD2 interaction with LFA-3 and determination of the two-dimensional dissociation constant for adhesion receptors in a contact area. J Cell Biol. 1996;132:465–474. doi: 10.1083/jcb.132.3.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Groves JT. Bending mechanics and molecular organization in biological membranes. Annu Rev Phys Chem. 2007;58:697–717. doi: 10.1146/annurev.physchem.56.092503.141216. [DOI] [PubMed] [Google Scholar]
- 48.Dustin ML. Adhesive bond dynamics in contacts between T lymphocytes and glass-supported planar bilayers reconstituted with the immunoglobulin-related adhesion molecule CD58. J Biol Chem. 1997;272:15782–15788. doi: 10.1074/jbc.272.25.15782. [DOI] [PubMed] [Google Scholar]
- 49.Qi SY, Groves JT, Chakraborty AK. Synaptic pattern formation during cellular recognition. Proc Natl Acad Sci USA. 2001;98:6548–6553. doi: 10.1073/pnas.111536798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weikl TR, Asfaw M, Krobath H, Rozycki B, Lipowsky R. Adhesion of membranes via receptor-ligand complexes: domain formation, binding cooperativity, and active processes. Soft Matter. 2009;5:3213–3224. [Google Scholar]
- 51.Burroughs NJ, Lazic Z, van der Merwe PA. Ligand detection and discrimination by spatial relocalization: a kinase-phosphatase segregation model of TCR activation. Biophys J. 2006;91:1619–1629. doi: 10.1529/biophysj.105.080044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Milstein O, et al. Nanoscale increases in CD2-CD48-mediated intermembrane spacing decrease adhesion and reorganize the immunological synapse. J Biol Chem. 2008;283:34414–34422. doi: 10.1074/jbc.M804756200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaizuka Y, Douglass AD, Vardhana S, Dustin ML, Vale RD. The coreceptor CD2 uses plasma membrane microdomains to transduce signals in T cells. J Cell Biol. 2009;185:521–534. doi: 10.1083/jcb.200809136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Choudhuri K, Wiseman D, Brown MH, Gould K, van der Merwe PA. T-cell receptor triggering is critically dependent on the dimensions of its peptide-MHC ligand. Nature. 2005;436:578–582. doi: 10.1038/nature03843. [DOI] [PubMed] [Google Scholar]
- 55.Billadeau DD, Nolz JC, Gomez TS. Regulation of T-cell activation by the cytoskeleton. Nature Rev Immunol. 2007;7:131–143. doi: 10.1038/nri2021. [DOI] [PubMed] [Google Scholar]
- 56.Kaizuka Y, Douglass AD, Varma R, Dustin ML, Vale RD. Mechanisms for segregating T cell receptor and adhesion molecules during immunological synapse formation in Jurkat T cells. Proc Natl Acad Sci USA. 2007;104:20296–20301. doi: 10.1073/pnas.0710258105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.DeMond AL, Mossman KD, Starr T, Dustin ML, Groves JT. T cell receptor microcluster transport through molecular mazes reveals mechanism of translocation. Biophys J. 2008;94:3286–3292. doi: 10.1529/biophysj.107.119099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burkhardt JK, Carrizosa E, Shaffer MH. The actin cytoskeleton in T cell activation. Annu Rev Immunol. 2008;26:233–259. doi: 10.1146/annurev.immunol.26.021607.090347. [DOI] [PubMed] [Google Scholar]
- 59.Ponti A, Machacek M, Gupton SL, Waterman-Storer CM, Danuser G. Two distinct actin networks drive the protrusion of migrating cells. Science. 2004;305:1782–1786. doi: 10.1126/science.1100533. [DOI] [PubMed] [Google Scholar]
- 60.Chhabra ES, Higgs HN. The many faces of actin: matching assembly factors with cellular structures. Nature Cell Biol. 2007;9:1110–1121. doi: 10.1038/ncb1007-1110. [DOI] [PubMed] [Google Scholar]
- 61.Koretzky GA, Myung PS. Positive and negative regulation of T-cell activation by adaptor proteins. Nature Rev Immunol. 2001;1:95–107. doi: 10.1038/35100523. [DOI] [PubMed] [Google Scholar]
- 62.Smith A, et al. A talin-dependent LFA-1 focal zone is formed by rapidly migrating T lymphocytes. J Cell Biol. 2005;170:141–151. doi: 10.1083/jcb.200412032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hartman NC, Nye JA, Groves JT. Cluster size regulates protein sorting in the immunological synapse. Proc Natl Acad Sci USA. 2009;106:12729–12734. doi: 10.1073/pnas.0902621106. Proposes that frictional coupling to the actin cytoskeleton is a mechanism that controls the spatial patterning of membrane proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bunnell SC, et al. T cell receptor ligation induces the formation of dynamically regulated signaling assemblies. J Cell Biol. 2002;158:1263–1275. doi: 10.1083/jcb.200203043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nguyen K, Sylvain NR, Bunnell SC. T cell costimulation via the integrin VLA-4 inhibits the actin-dependent centralization of signaling microclusters containing the adaptor SLP-76. Immunity. 2008;28:810–821. doi: 10.1016/j.immuni.2008.04.019. [DOI] [PubMed] [Google Scholar]
- 66.Warren CM, Landgraf R. Signaling through ERBB receptors: multiple layers of diversity and control. Cell Signal. 2006;18:923–933. doi: 10.1016/j.cellsig.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 67.Tian T, et al. Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nature Cell Biol. 2007;9:905–914. doi: 10.1038/ncb1615. [DOI] [PubMed] [Google Scholar]
- 68.Salaita K, et al. Restriction of receptor movement alters cellular response: physical force sensing by EphA2. Science. 2010;327:1380–1385. doi: 10.1126/science.1181729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Greenfield D, et al. Self-organization of the Escherichia coli chemotaxis network imaged with super-resolution light microscopy. Plos Biol. 2009;7:e1000137. doi: 10.1371/journal.pbio.1000137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gestwicki JE, Kiessling LL. Inter-receptor communication through arrays of bacterial chemoreceptors. Nature. 2002;415:81–84. doi: 10.1038/415081a. [DOI] [PubMed] [Google Scholar]
- 71.Sourjik V, Berg HC. Functional interactions between receptors in bacterial chemotaxis. Nature. 2004;428:437–441. doi: 10.1038/nature02406. [DOI] [PubMed] [Google Scholar]
- 72.Lillemeier BF, et al. TCR and LAT are expressed on separate protein islands on T cell membranes and concatenate during activation. Nature Immunol. 2010;11:90–96. doi: 10.1038/ni.1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pautot S, Lee H, Isacoff EY, Groves JT. Neuronal synapse interaction reconstituted between live cells and supported lipid bilayers. Nature Chem Biol. 2005;1:283–289. doi: 10.1038/nchembio737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sackmann E. Supported membranes: scientific and practical applications. Science. 1996;271:43–48. doi: 10.1126/science.271.5245.43. [DOI] [PubMed] [Google Scholar]
- 75.Groves JT, Dustin ML. Supported planar bilayers in studies on immune cell adhesion and communication. J Immunol Methods. 2003;278:19–32. doi: 10.1016/s0022-1759(03)00193-5. [DOI] [PubMed] [Google Scholar]
- 76.Groves JT. Spatial mutation of the T cell immunological synapse. Curr Opin Chem Biol. 2006;10:544–550. doi: 10.1016/j.cbpa.2006.10.021. [DOI] [PubMed] [Google Scholar]
- 77.Nye JA, Groves JT. Kinetic control of histidine-tagged protein surface density on supported lipid bilayers. Langmuir. 2008;24:4145–4149. doi: 10.1021/la703788h. [DOI] [PubMed] [Google Scholar]
- 78.Groves JT, Ulman N, Boxer SG. Micropatterning fluid lipid bilayers on solid supports. Science. 1997;275:651–653. doi: 10.1126/science.275.5300.651. [DOI] [PubMed] [Google Scholar]
- 79.Falconnet D, Csucs G, Grandin HM, Textor M. Surface engineering approaches to micropattern surfaces for cell-based assays. Biomaterials. 2006;27:3044–3063. doi: 10.1016/j.biomaterials.2005.12.024. [DOI] [PubMed] [Google Scholar]
- 80.Whitesides GM, Ostuni E, Takayama S, Jiang X, Ingber DE. Soft lithography in biology and biochemistry. Annu Rev Biomed Eng. 2001;3:335–373. doi: 10.1146/annurev.bioeng.3.1.335. [DOI] [PubMed] [Google Scholar]
- 81.Mossman K, Groves J. Micropatterned supported membranes as tools for quantitative studies of the immunological synapse. Chem Soc Rev. 2007;36:46–54. doi: 10.1039/b605319j. [DOI] [PubMed] [Google Scholar]
- 82.Nie Z, Kumacheva E. Patterning surfaces with functional polymers. Nature Mater. 2008;7:277–290. doi: 10.1038/nmat2109. [DOI] [PubMed] [Google Scholar]
- 83.Sia SK, Whitesides GM. Microfluidic devices fabricated in poly(dimethylsiloxane) for biological studies. Electrophoresis. 2003;24:3563–3576. doi: 10.1002/elps.200305584. [DOI] [PubMed] [Google Scholar]
- 84.Delamarche E, Juncker D, Schmid H. Microfluidics for processing surfaces and miniaturizing biological assays. Adv Mater. 2005;17:2911–2933. [Google Scholar]
- 85.Irvine DJ, Doh J, Huang B. Patterned surfaces as tools to study ligand recognition and synapse formation by T cells. Curr Op Immunol. 2007;19:463–469. doi: 10.1016/j.coi.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 86.Ruiz SA, Chen CS. Microcontact printing: a tool to pattern. Soft Matter. 2007;3:168–177. doi: 10.1039/b613349e. [DOI] [PubMed] [Google Scholar]
- 87.Salaita K, Wang Y, Mirkin CA. Applications of dip-pen nanolithography. Nature Nanotechnol. 2007;2:145–155. doi: 10.1038/nnano.2007.39. [DOI] [PubMed] [Google Scholar]
- 88.Girard PP, Cavalcanti-Adam EA, Kemkemer R, Spatz JP. Cellular chemomechanics at interfaces: sensing, integration and response. Soft Matter. 2007;3:307–326. doi: 10.1039/b614008d. [DOI] [PubMed] [Google Scholar]
- 89.Sackmann E, Tanaka M. Supported membranes on soft polymer cushions: fabrication, characterization and applications. Trends Biotechnol. 2000;18:58–64. doi: 10.1016/s0167-7799(99)01412-2. [DOI] [PubMed] [Google Scholar]
- 90.Tanaka M, Sackmann E. Polymer-supported membranes as models of the cell surface. Nature. 2005;437:656–663. doi: 10.1038/nature04164. [DOI] [PubMed] [Google Scholar]
- 91.Wu M, Holowka D, Craighead HG, Baird B. Visualization of plasma membrane compartmentalization with patterned lipid bilayers. Proc Natl Acad Sci USA. 2004;101:13798–13803. doi: 10.1073/pnas.0403835101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Geometric control of cell life and death. Science. 1997;276:1425–1428. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- 93.Arnold M, et al. Activation of integrin function by nanopatterned adhesive interfaces. Chemphyschem. 2004;5:383–388. doi: 10.1002/cphc.200301014. [DOI] [PubMed] [Google Scholar]
- 94.Geiger B, Spatz JP, Bershadsky AD. Environmental sensing through focal adhesions. Nature Rev Mol Cell Biol. 2009;10:21–33. doi: 10.1038/nrm2593. [DOI] [PubMed] [Google Scholar]
- 95.Torres AJ, Wu M, Holowka D, Baird B. Nanobiotechnology and cell biology: micro- and nanofabricated surfaces to investigate receptor-mediated signaling. Annu Rev Biophys. 2008;37:265–288. doi: 10.1146/annurev.biophys.36.040306.132651. [DOI] [PubMed] [Google Scholar]
- 96.Schwarzenbacher M, et al. Micropatterning for quantitative analysis of protein-protein interactions in living cells. Nature Methods. 2008;5:1053–1060. doi: 10.1038/nmeth.1268. [DOI] [PubMed] [Google Scholar]
- 97.Airan RD, Thompson KR, Fenno LE, Bernstein H, Deisseroth K. Temporally precise in vivo control of intracellular signalling. Nature. 2009;458:1025–1029. doi: 10.1038/nature07926. [DOI] [PubMed] [Google Scholar]
- 98.Wu YI, et al. A genetically encoded photoactivatable Rac controls the motility of living cells. Nature. 2009;461:104–108. doi: 10.1038/nature08241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Levskaya A, Weiner OD, Lim WA, Voigt CA. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature. 2009;461:997–1001. doi: 10.1038/nature08446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gorostiza P, Isacoff EY. Optical switches for remote and noninvasive control of cell signaling. Science. 2008;322:395–399. doi: 10.1126/science.1166022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Szobota S, et al. Remote control of neuronal activity with a light-gated glutamate receptor. Neuron. 2007;54:535–545. doi: 10.1016/j.neuron.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 102.DeMond AL, Starr T, Dustin ML, Groves JT. Control of antigen presentation with a photoreleasable agonist peptide. J Am Chem Soc. 2006;128:15354–15355. doi: 10.1021/ja065304l. [DOI] [PubMed] [Google Scholar]
- 103.Huse M, et al. Spatial and temporal dynamics of T cell receptor signaling with a photoactivatable agonist. Immunity. 2007;27:76–88. doi: 10.1016/j.immuni.2007.05.017. [DOI] [PubMed] [Google Scholar]
- 104.Guo YM, et al. Imaging dynamic cell-cell junctional coupling in vivo using Trojan-LAMP. Nature Methods. 2008;5:835–841. doi: 10.1038/nmeth.1238. [DOI] [PubMed] [Google Scholar]
- 105.Meyer T, Teruel MN. Fluorescence imaging of signaling networks. Trends Cell Biol. 2003;13:101–106. doi: 10.1016/s0962-8924(02)00040-5. [DOI] [PubMed] [Google Scholar]
- 106.Sabouri-Ghomi M, Wu Y, Hahn K, Danuser G. Visualizing and quantifying adhesive signals. Curr Opin Cell Biol. 2008;20:541–550. doi: 10.1016/j.ceb.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Takayama S, et al. Patterning cells and their environments using multiple laminar fluid flows in capillary networks. Proc Natl Acad Sci USA. 1999;96:5545–5548. doi: 10.1073/pnas.96.10.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]