Selection, Transmission, and Reversion of an Antigen-Processing Cytotoxic T-Lymphocyte Escape Mutation in Human Immunodeficiency Virus Type 1 Infection (original) (raw)
Abstract
Numerous studies now support that human immunodeficiency virus type 1 (HIV-1) evolution is influenced by immune selection pressure, with population studies showing an association between specific HLA alleles and mutations within defined cytotoxic T-lymphocyte epitopes. Here we combine sequence data and functional studies of CD8 T-cell responses to demonstrate that allele-specific immune pressures also select for mutations flanking CD8 epitopes that impair antigen processing. In persons expressing HLA-A3, we demonstrate consistent selection for a mutation in a C-terminal flanking residue of the normally immunodominant Gag KK9 epitope that prevents its processing and presentation, resulting in a rapid decline in the CD8 T-cell response. This single amino acid substitution also lies within a second HLA-A3-restricted epitope, with the mutation directly impairing recognition by CD8 T cells. Transmission of the mutation to subjects expressing HLA-A3 was shown to prevent the induction of normally immunodominant acute-phase responses to both epitopes. However, subsequent in vivo reversion of the mutation was coincident with delayed induction of new CD8 T-cell responses to both epitopes. These data demonstrate that mutations within the flanking region of an HIV-1 epitope can impair recognition by an established CD8 T-cell response and that transmission of these mutations alters the acute-phase CD8+ T-cell response. Moreover, reversion of these mutations in the absence of the original immune pressure reveals the potential plasticity of immunologically selected evolutionary changes.
It is well established that human immunodeficiency virus type 1 (HIV-1)-specific CD8+ T-cell responses are important to the control of HIV-1 replication, in particular during the acute phase of infection (3, 10, 15, 34, 40, 56). A number of mechanisms that HIV-1 uses to evade these host immune responses have now been described, including impairment of HIV-1-specific CD4+ and CD8+ T-cell function (8, 21), down-regulation of HLA class I molecules (25, 57, 65), and mutations within defined cytotoxic T-lymphocyte (CTL) epitopes (3, 10, 31). The role of sequence variation within epitopes as a means of immune escape has been well documented in infected persons as well as in vaccine challenge studies, and recent studies now support a role for CD8+ T-cell selective pressures in also shaping the global sequence diversity of HIV-1 (44, 68). Transmission of such evolving CD8 escape mutations can, therefore, have a direct impact on the ability of newly infected individuals to mount effective CD8+ T-cell responses against particular HIV-1 epitopes during acute infection, as illustrated in the case of mother-to-child transmission (32). This finding is further supported by a recent study illustrating that individuals expressing rare HLA alleles within a population, against which less immune selection pressure is likely to have been applied at the population level, may have a selective advantage in containing HIV-1 replication (61).
Viral escape from CTL responses due to mutation of residues within the targeted epitope has been described for both HIV-1 and simian immunodeficiency virus (3, 10, 16, 26, 30, 33, 35, 38, 45, 48, 49, 63, 64). Such mutations either directly impact the ability of the epitope to bind and be presented on the surface of a cell by the major histocompatibility complex (MHC) class I molecule or alter the ability of the T-cell receptor (TCR) to properly engage and recognize the MHC-peptide complex. Mutations within flanking regions of epitopes can also potentially inhibit CD8+ T-cell responses by interfering with the proper processing of these antigens, but this is yet to be shown during the natural course of human viral infections. There are multiple steps required for the successful processing of a CD8 epitope which could be sensitive to sequence variations both within and flanking an epitope, including proteasomal cleavage of endogenously synthesized proteins (14, 47, 60, 68), transporter associated with antigen-processing (TAP) activity (1), and aminopeptidase N-terminal trimming (13, 29, 39, 42, 60). Although the impact of mutations flanking epitopes has been shown for experimentally induced mutations (11, 19, 22, 24, 46), whether specific CD8+ T-cell responses play a role in the selection of these mutations during natural infection remains unknown (19).
Here, we study the rapidly mutating retrovirus HIV-1 and show evolution of a single amino acid mutation distal to the C terminus of an epitope which inhibits normal processing and presentation of this typically immunodominant epitope. The same mutation also lies within a second CTL epitope and leads to impaired MHC-peptide-TCR interaction. Additional HLA-A3-positive subjects infected with circulating strains of HIV-1 harboring mutations in this position were unable to mount acute-phase responses against these normally immunodominant epitopes (67), demonstrating not only that transmission of these mutations is occurring, but also that they can impair the critical early cellular immune responses.
MATERIALS AND METHODS
Subjects.
Twenty-two HIV-1-infected subjects were enrolled from the Boston Acute Infection Cohort. Thirteen individuals expressed the HLA class I allele A3, and an additional nine individuals were HLA-A3 negative. All study subjects were identified during primary HIV-1 infection and were successfully treated with highly active antiretroviral therapy, with the exception of subject AC-38, who remained untreated. Longitudinal studies were performed on HLA-A3-positive subjects AC-38, AC-33, and AC-14. Subject AC-38 was diagnosed with early HIV-1 infection following high-risk exposure. An HIV-1 p24 Gag enzyme-linked immunosorbent assay (ELISA) 49 days prior to presentation had been negative but was positive by day 14 postpresentation. At the earliest measurement on day 59, viral loads were 1,500 copies of HIV-1 RNA per ml of plasma with a CD4+ T-cell count of 697 cells per μl. HIV-1 viremia remained controlled to levels below 2,000 copies/ml for the first year of infection, with a rise in viral load to 9,020 copies/ml at day 435. The subject subsequently moved and was temporarily lost to follow-up until 3 years postinfection (day 1073), when viral loads had risen to 60,000 copies/ml. By day 1213 and day 1510, viral loads were slightly lower at 11,200 copies/ml and 19,700 copies/ml, respectively. Subjects AC-33 and AC-14 were both treated during acute HIV-1 infection with viral loads greater than 500,000 RNA copies/ml and 95,000 RNA copies/ml, respectively, and underwent one or more structured treatment interruptions with peak viral rebounds between 15,000 and 110,000 RNA copies/ml.
IFN-γ ELISPOT assay.
HIV-1-specific CD8+ T-cell responses were quantified by gamma interferon (IFN-γ) ELISPOT assay as previously described (67), using overlapping peptides (15- to 18-mer peptides overlapping by 10 amino acids) spanning the entire expressed HIV-1 clade B 2001 consensus sequence (http://hiv-web.lanl.gov), as well as peptides corresponding to optimal described clade B CTL epitopes (18) and autologous virus sequences when available. The number of specific IFN-γ-secreting T cells were counted by automated reader, calculated by subtracting the negative control value and expressed as spot-forming cells (SFC) per 106 input cells. A response was considered positive if there were ≥50 SFC per 106 cells and at least three times greater than mean background activity. CD8+ T-cell dependence of all responses was confirmed by CD4+/CD8+ T-cell depletion-enrichment studies using magnetic beads (MACS; Miltenyi Biotech, Bergisch Gladbach, Germany) (67). Comparison of autologous and variant epitopes was performed using log dilutions of peptides as described elsewhere (6).
Flow cytometric detection of antigen-induced intracellular IFN-γ.
Intracellular cytokine staining assays were performed as described previously (4). Cells were analyzed on a FACSort flow cytometer (Becton Dickinson Immunocytometry Systems, San Jose, Calif.). Vaccinia virus experiments, utilizing constructs vT135 and vT142 (NIH AIDS Reagent Program), were performed by infecting BLCL target cells overnight with a multiplicity of infection of 3:1, as described previously (4). Target cells were then washed three times and used in the assay at an effector-to-target ratio of 10:1.
Sequencing of autologous virus.
Nested PCR for p17 and p24 on proviral DNA or plasma viral RNA was performed as previously described (6). PCR fragments were population sequenced to identify regions of sequence variation and where necessary were additionally cloned (TOPO TA; Invitrogen, Carlsbad, Calif.) and DNA mini-prepped (QiaPrep Turbo mini-prep). All fragments were sequenced bidirectionally on an ABI 3100 PRISM automated sequencer. Sequencher (Gene Codes Corp., Ann Arbor, Mich.) and MacVector 4.1 (Oxford Molecular) were used to edit and align sequences.
HLA-A*0301 peptide binding assays.
Quantitative assays for the binding of peptides to soluble HLA-A*0301 molecules based on the inhibition of binding of a radiolabeled standard probe peptide to HLA-A*0301 molecules were performed as previously described (59). Binding values were then calculated relative to the 50% inhibitory concentration (IC50) of the standard probe peptide.
Statistical analysis.
Statistical analysis and graphical presentation were done using SigmaPlot 5.0 (SPSS Inc., Chicago, Ill.). Statistical analysis of significance (P values) was based on the chi-square test.
Nucleotide sequence accession numbers.
Sequence data are available from GenBank under accession numbers AY611238 through AY611385.
RESULTS
Loss of two immunodominant HLA-A3-restricted CD8+ T-cell responses is associated with sequence variation at p17 residue K28.
Previous studies have shown that HLA-A3-restricted CD8+ T-cell responses directed against two overlapping immunodominant epitopes in p17 Gag (KIRLRPGGK18-26 [KK9] and RLRPGGKKK20-28 [RK9]) are frequently involved in acute-phase responses to primary HIV-1 infection (7, 27, 67). To determine the effects of CD8+ T-cell-mediated immune selection pressure on subsequent viral evolution within these epitopes, HIV-1 Gag-specific CD8+ T-cell responses in eight HLA-A3 subjects mounting both RK9- and KK9-specific responses during acute HIV-1 infection (AC-38, -14, -06, -01, -03, -15, -21, and -23) were studied with respect to magnitude of CD8 T-cell response and sequence of autologous virus within the targeted region. These subjects were followed longitudinally by IFN-γ ELISPOT assay using described HIV-1 Gag epitope peptides (18).
The first of these subjects (AC-38), who never initiated antiretroviral therapy, exhibited a CD8+ T-cell response directed against six HIV-1 Gag CD8 epitopes at the first available time point 64 days following presentation with primary HIV-1 infection (negative HIV-1 p24 Gag ELISA 49 days prior to presentation and indeterminant ELISA at presentation, becoming positive at day 14). The initially immunodominant CD8+ T-cell response was directed against the HLA-B57-restricted epitope TW10 (TSTLQEQIGW) (Fig. 1), but this response declined following viral escape (TS_N_LQEQIGW) by as early as day 64 (data not shown). Other CD8+ T-cell responses, reaching T-cell frequencies of >1,000 SFC/106 peripheral blood mononuclear cells (PBMC), were generated against the two overlapping HLA-A3-restricted epitopes in Gag, KK9 and RK9, as well as two other HLA-B57-restricted epitopes, IW9 (ISPRTLNAW147-155) and KF11 (KAFSPEVIPMF162-172). Finally, a subdominant response was directed against the HLA-A1-restricted GY9 epitope (GSEELRSLY71-79). Longitudinal assessment of the four immunodominant responses from early throughout chronic infection in AC-38 revealed that while the two immunodominant HLA-B57-restricted responses were maintained, the two HLA-A3-restricted responses declined (Fig. 1). In order to determine whether viral sequence evolution and immune escape could be the cause of this decline, bulk and clonal sequencing of these A3-restricted epitopes were undertaken in this subject. These data demonstrated that at p17 residue K28 a lysine-to-glutamine (K28Q) substitution appeared between day 435 and day 1073 after infection (Table 1), coincident with the decline in the KK9 and RK9 responses. Transient sequence variations over time at two other residues in this region (R20W and K26I) were also present prior to the K28Q mutation (Table 2) becoming fixed, but these did not persist.
FIG. 1.
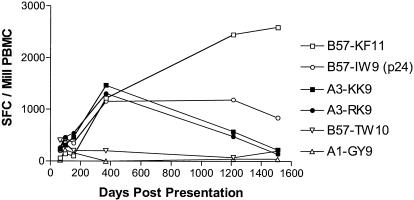
Decline of HLA-A3-restricted RK9 and KK9 CD8 T-cell responses in subject AC-38. HIV-1 Gag-specific CD8+ T-cell responses were followed longitudinally in HLA-A3-positive subject AC-38 during untreated HIV-1 infection using the IFN-γ ELISPOT assay and described optimal epitope peptides. The magnitude of these responses is indicated in SFC per 106 PBMC.
TABLE 1.
Viral evolution within the HLA-A3-restricted p17 KK9 and RK9 epitopes in subject AC-38: clonal sequencing
| Days post- presen- tation | Clonal sequencea | Clonal fre- quencyb |
|---|---|---|
| KIRLRPGGK | ||
| RLRPGGKKK | ||
| 10203040 | ||
| MGARASVLSGGELDRWEKIRLRPGGKKKYKLKHIVWASRE | ||
| 64 | .............................R.......... | 8/17 |
| ...................W.........R.......... | 9/17 | |
| 435 | .............................R.......... | 3/5 |
| .........................I...R.......... | 2/5 | |
| 1073 | ...........................Q.R.......... | 6/11 |
| ...........................Q.R....I..... | 5/11 | |
| 1213 | ...........................Q.R.......... | 11/14 |
| .............G.............Q.R.......... | 3/14 |
TABLE 2.
Vaccinia virus constructs
| Vaccinia virus constructb | Sequencea |
|---|---|
| KIRLRPGGK | |
| RLRPGGKKK | |
| 10203040 | |
| MGARASVLSGGELDRWEKIRLRPGGKKKYKLKHIVWASRE | |
| vT135 (WT) | ...........K..A..............R...L....... |
| vT142 (K28Q) | ......I....K..A............Q.RM..L....... |
Additional longitudinal characterization of p17 RK9- and KK9-specific CD8+ T-cell responses was performed in the remaining seven HLA-A3 subjects in whom acute-phase responses to KK9 and RK9 were detected. These subjects were identified and treated during acute HIV-1 infection but underwent subsequent treatment interruptions associated with substantial viral replication (between 18,000 and 78,000 RNA copies/ml in seven of eight subjects). Of these subjects, AC-14 also exhibited a significant decline in both the RK9- and KK9-specific responses associated with the development of both K26R and K28Q mutations (4.5 years postpresentation) (Table 3). Similarly, subject AC-06, who was superinfected with a second strain of virus (6), also experienced a decline in magnitude of CD8 T-cell responses to both epitopes, even though the only sequence change was in position K28 (K28T) within epitope RK9, with epitope KK9 unchanged. In the remaining five HLA-A3 subjects who demonstrated acute-phase responses to these epitopes, no significant decline in the RK9 and KK9 responses over time was observed (range, 50 to 69 months follow-up) in the presence of ongoing viral replication following treatment interruption (Table 3 and data not shown). Thus, of a total of eight HLA-A3-expressing persons examined, three experienced a loss of CD8+ T-cell responses against both immunodominant HLA-A3-restricted p17 Gag epitopes associated with amino acid substitutions at residue K28.
TABLE 3.
Evolving or transmitted HIV-1 p17 K28 mutations impair mounting of HLA-A3-restricted RK9 and KK9 responses
| Subject | HLA | Time point (days postpresentation) | SequenceaKIRLRPGGK RLRPGGKKK | ELISPOTb | Clonal frequencyc (%) | |
|---|---|---|---|---|---|---|
| KK9 | RK9 | |||||
| K28 mutations are associated with loss of KK9/RK9 responses: | ||||||
| AC-38 | A3 | Acute (64) | ........... | 1,470 | 1,310 | |
| Chronic (1213) | ..........Q | 220 | 140 | |||
| AC-14 | A3 | Acute (26) | R.......... | 1,200 | 630 | |
| Chronic (1660) | R.......R.Q | 100 | 210 | |||
| AC-06d | A3 | Virus A (18) | ........... | 1,100 | 1,000 | |
| Virus B (1170) | ..........T | 150 | 120 | |||
| AC-01 | A3 | Acute (16) | ........... | 130 | 480 | |
| AC-03 | A3 | Acute (30) | ........... | 60 | 250 | |
| AC-15 | A3 | Acute (10) | ........... | 440 | 660 | |
| AC-21 | A3 | Acute (4) | ........... | 120 | 800 | |
| AC-23 | A3 | Acute (20) | ........... | 0 | 170 | |
| Acute acquired K28 mutations impair mounting of KK9/RK9 responses in HLA-A3 subjects | ||||||
| AC-05 | A3 | Acute (24) | ........R.Q | 0 | 0 | |
| AC-20 | A3 | Acute (8) | .......S..R | 0 | 0 | |
| AC-24 | A3 | Acute (1) | .......R..Q | 0 | 0 | |
| AC-33 | A3 | Acute (1) | .......S..R | 0 | 0 | |
| AC-58 | A3 | Acute (6) | ..........Q | 0 | 0 | |
| Frequency of transmission of K28 variants in non-HLA-A3 subjects | ||||||
| AC-02 | Non-A3 | Acute (27) | ........R.Q | ND | ND | |
| AC-09 | Non-A3 | Acute (1) | ........... | ND | ND | |
| AC-10 | Non-A3 | Acute (5) | ........... | ND | ND | |
| AC-16 | Non-A3 | Acute (1) | ........... | ND | ND | |
| AC-25 | Non-A3 | Acute (1) | R.........Q | ND | ND | |
| AC-26 | Non-A3 | Acute (1) | ........... | ND | ND | |
| AC-27 | Non-A3 | Acute (0) | ..........Q | ND | ND | |
| AC-45 | Non-A3 | Acute (9) | ..........Q | ND | ND | |
| AC-59 | Non-A3 | Acute (1) | ........... | ND | ND | |
| Reversion of a transmitted K28R variant enables mounting of KK9/RK9 responses | ||||||
| AC-33 | A3 | 0 | .......S..R | 0 | 0 | 14/14 (100) |
| .......S... | 0/14 (0) | |||||
| 482 | .......S..R | 0 | 0 | 21/24 (87) | ||
| .......S... | 3/24 (13) | |||||
| 811 | .......S..R | 810 | 1,370 | 9/12 (75) | ||
| .......S... | 3/12 (25) | |||||
| 1006 | .......S..R | ND | ND | 9/15 (60) | ||
| .......S... | 6/15 (40) | |||||
| 1413 | .......S..R | 1,120 | 1,020 | 0/21 (0) | ||
| .......S... | 21/21 (100) | |||||
| 1534 | .......S..R | 310 | 590 | 5/9 (56) | ||
| .......S... | 4/9 (44) |
Impact of the p17 Gag K28 mutation on epitope processing and recognition by KK9- and RK9-specific CD8+ T-cell lines.
We next examined the effects of the K28 mutation on processing, presentation, and recognition of the two HLA-A3-restricted epitopes. As the p17 K28 mutation is located at the C-terminal position of the HLA-A3-restricted RK9 epitope, we initially assessed the impact of this mutation on CD8+ T-cell responses directed against this epitope by comparing the wild-type peptide and the K28Q variant peptide for their abilities to bind to HLA-A3 and be effectively recognized by RK9-specific T-cell lines. The K28Q mutation reduced the binding of the RK9 epitope to the HLA-A3 molecule from an IC50 of 21 nM to 162 nM and inhibited recognition by RK9-specific CD8+ T-cell lines when target cells were directly labeled with the variant epitope peptide (Fig. 2A). Each of the sequence variations within the RK9 epitope that arose in subject AC-38, shown in Table 1, was also tested using nonexpanded autologous PBMC from this individual. These variant peptides all dramatically reduced recognition (Fig. 2B). These data suggest that the C-terminal K28Q mutation represents an escape mutation within the HLA-A3-restricted RK9 epitope impacting peptide-MHC-TCR interactions.
FIG. 2.
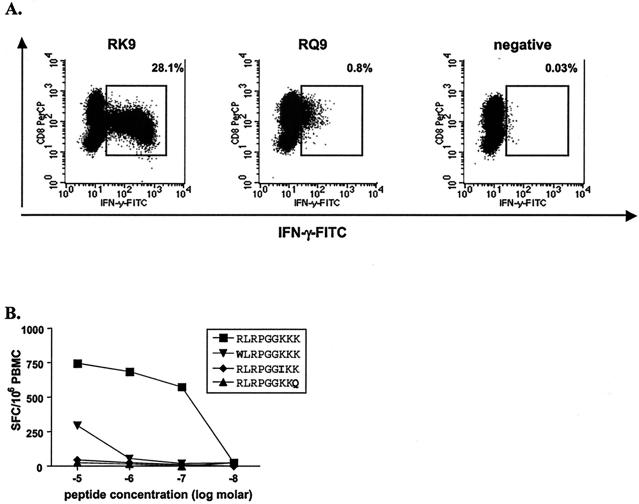
Impaired recognition of variant RK9 epitope in subject AC-38. (A) Peptide-specific responses exhibited by RK9-specific CD8+ T-cell lines were quantified by flow cytometry using intracellular IFN-γ staining. The wild-type peptide (RK9) was recognized by 28.1% of CD8+ T cells, while the RQ9 variant peptide was poorly recognized (0.8%). (B) Peptide-specific responses exhibited by autologous unstimulated PBMC from subject AC-38 were quantified by IFN-γ ELISPOT. Each of the variant RK9 peptides showed reduced reactivity compared to the autologous peptide (RLRPGGKKK).
The loss of responses to the KK9 epitope, in which there were no mutations to affect peptide-TCR-MHC interactions, suggested that processing of the KK9 epitope may have been impaired by the K28Q mutation in the C-terminal flanking region. To address this, B cells were infected with HIV-1 p55-expressing vaccinia virus constructs encoding either the wild-type epitope (Vac T135; K28) or the variant epitope (Vac T142; K28Q) (Table 2). Intracellular cytokine staining assays revealed that while both the KK9- and RK9-specific CD8+ T-cell lines were capable of recognizing target cells infected with the autologous T135 vaccinia virus, neither line was capable of recognizing target cells infected with the T142 variant vaccinia virus expressing the K28Q mutation (Fig. 3). A control CD8+ T-cell line against the HLA-B14-restricted DA9 epitope (DRFYKTLRA298-306) in p24 that was conserved within both vaccinia virus constructs was able to efficiently recognize target cells infected with either vaccinia virus. Western blot quantitation of lysates from both wild-type-infected and variant vaccinia virus-infected cells supported proper p17 expression by all vaccinia viruses (data not shown), ensuring that these results were not due to defective protein expression by the T142 variant vaccinia virus, but rather to impaired presentation of the KK9 epitope to the respective T-cell lines. Taken together, these data indicate that the K28Q mutation resulted in impaired processing and recognition of the KK9 epitope by epitope-specific CD8+ T cells.
FIG. 3.
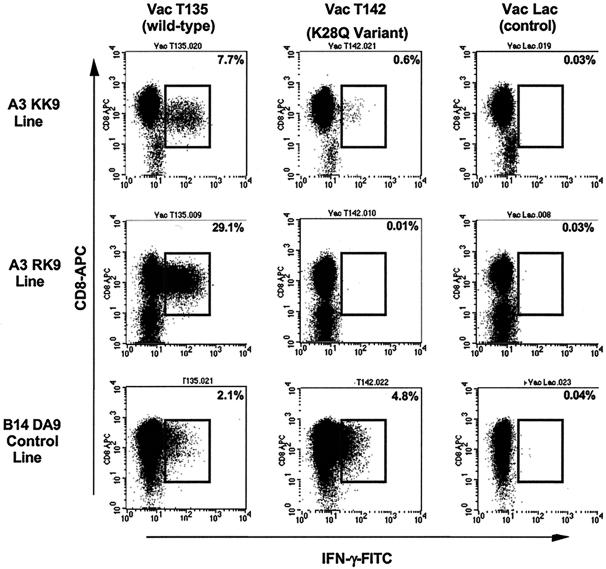
KK9- and RK9-specific CD8 T-cell lines fail to recognize target cells infected with vaccinia virus expressing the K28Q variant. Cell lines were tested for recognition of target cells infected with vaccinia virus expressing either wild-type sequences of RK9 and KK9 (Vac T135) or the K28Q variant sequence (Vac T142). KK9- and RK9-specific cell lines failed to recognize target cells infected with vaccinia virus expressing the K28Q variant, while target cells infected with the wild-type vaccinia virus induced strong IFN-γ production. A control CD8+ T-cell line specific for the HLA-B14-restricted p24 Gag epitope DRFYKTLRA (DA9) that was conserved in both vaccinia viruses (T135 and T142) recognized infected target cells equally well. Target cells infected with the control vaccinia virus Lac were used as negative controls.
As proteasomes have been shown to be important in the generation of many CD8 epitopes (11, 19, 22, 24, 46), we used a proteasomal cleavage prediction program which models in vivo cleavage to determine the potential of this mutation to alter antigen processing. The proteasomal cleavage prediction program NetChop C-term 2.0 (36) was utilized to determine whether any of the K28 variants of p17 would be predicted to be less effectively processed than the wild-type sequence. This program is trained against a database of MHC class I ligands and designed to predict cleavage sites of the human proteasome (36, 68). This analysis revealed that the probability for cleavage at residue 28 dropped dramatically from 59.8% for the wild-type sequence to 1.3, 1.2, and 11.3% for the K28Q, K28T, and K28R variants, respectively, when tested using an HIV-1 clade B consensus p17 sequence. This program also suggested that the only other two sequence variations between the vaccinia virus constructs in the vicinity of the A3 epitopes, namely, positions 7 (V7I) and 31 (L31M), would not be predicted to influence processing of the KK9 epitope (data not shown). These data support a role for the K28Q mutation in impairing the proper antigen processing of the KK9 epitope by altering a proteasomal cleavage site.
Finally, analysis of a more extensive Western Australia Sequence database (180 sequences from HLA-defined chronic HIV-infected subjects [44]) supported the impact of immune pressure upon this region of Gag. A strong HLA-A*0301-associated selection at residue K28 for glutamine (Q; 28%) or arginine (R; 29%) substitutions was detected, with the overall odds ratio for selective pressures upon this residue in HLA-A*0301 subjects versus non-A*0301 subjects being 6.3 (P < 0.0001). This association remained significant (odds ratio of 6.0; P < 0.0001) after adjusting for other HLA-A, -B, -C, and -DR alleles. Taken together, these data demonstrate that mutations in Gag position K28 can impair both the recognition of the HLA-A3-restricted RK9 epitope and the processing of the HLA-A3-restricted KK9 epitope, resulting in an HLA-A3 “footprint” on the sequence of HIV-1 in this position.
Transmission of HIV-1 p17 K28 variants impairs induction of HLA-A3-restricted CD8 T-cell responses.
It was previously observed that not all individuals expressing HLA-A3 mounted KK9- and RK9-specific CD8+ T-cell responses during acute HIV-1 infection (67). Therefore, we were interested in assessing whether infection with viruses harboring p17 K28 mutations might explain the inability of some HLA-A3-expressing subjects to mount these responses. Five additional HLA-A3 subjects (AC-05, -20, -24, -33, and -58) were chosen based on their inability to mount acute-phase KK9 or RK9 responses (Table 3). Sequencing of acute-phase autologous virus indicated that viruses from all five subjects exhibited an amino acid substitution in position K28 (P = 0.001 compared to eight individuals mounting responses), as well as in some cases additional mutations (Table 3). These data suggest that infection with the K28 variant impairs the ability of HLA-A3-expressing individuals to mount primary CD8+ T-cell responses against these otherwise immunodominant epitopes during acute infection.
Given the relatively high allele frequency of HLA-A3 (13.2%) in U.S. Caucasians (20) and the development of this escape mutation in persons expressing this allele, we next examined viruses obtained from HIV-1-infected persons identified and treated during acute infection who did not express HLA-A3 for the presence of this mutation. Sequence analysis of KK9 and RK9 epitopes in these nine additional HLA-A3-negative HIV-1-infected subjects during acute infection revealed K28 mutations in four of these subjects (Table 3). Given that these viruses were sequenced during acute infection and before significant immune selective pressures could develop, these mutations are likely to have been transmitted. Together with the population data presented above, showing a strong association between this K28 mutation and expression of HLA-A3, the data suggest that these persons acquired viruses exposed to prior HLA-A3-specific positive selection pressures.
Reversion of the p17 K28 mutation is associated with delayed mounting of KK9- and RK9-specific CD8+ T-cell responses.
The above data demonstrate that immune pressure can result in escape mutations at residue K28 in p17 Gag and that the transmission of these variant viruses to other individuals expressing HLA-A3 inhibits the development of these normally immunodominant responses in acute infection. We next studied the genetic stability of this escape variant following transmission. Of the five HLA-A3 individuals infected with a K28 mutation who lacked KK9- and RK9-specific T-cell responses, one subject (AC-33) was able to mount delayed responses to both epitopes. This individual was infected with a strain of HIV-1 encoding two variants within this region of p17 Gag, including a K28R mutation (Table 3). The earliest detectable CD8+ T-cell response in this subject was directed against the HLA-B44-restricted p24 CD8+ T-cell epitope AW11 (AEQASQDVKNW174-184; 220 SFC/106 PBMC), which arose during acute HIV-1 infection prior to initiation of therapy (data not shown). No HLA-A3-restricted responses against the KK9 or RK9 epitopes were detectable at this time. However, 811 days postinfection (387 days after stopping therapy) both KK9- and RK9-specific responses were detected, and these responses became the dominant HIV-1-specific CD8+ T-cell responses in this individual (810 and 1,370 SFC/106 PBMC, respectively). While the analysis of immune responses directed against the variant RK9 epitope (RLRPGSKKR) using autologous unstimulated PBMC from this time point confirmed impaired recognition of this transmitted variant, the reverted “wild-type” epitope, RLRPGSKKK, was very well recognized (Fig. 4). Sequence analysis of autologous HIV-1 demonstrated full reversion of this K28R variant back to wild type by day 1413 (Table 3), consistent with the late appearance of these two responses and suggesting that the switch to wild-type sequence was due to epitope reversion in the absence of the immune pressure that selected for this mutation. Interestingly, analysis of virus sequences and CD8+ T-cell responses in subject AC-33 at the latest time point available (day 1534) indicated a partial return of the K28R variant in plasma virus (56% of clones assessed) and a partial decline of RK9- and KK9-specific T-cell responses. These data support immune pressure mediated by these HLA-A3-restricted responses on this epitope (Table 3) and reflect a remarkable dynamic interplay between immune responses and viral sequence variation.
FIG. 4.
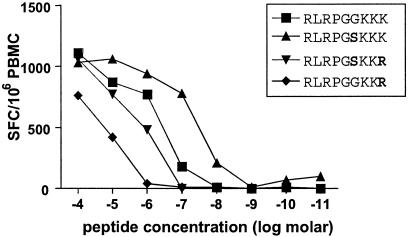
Impaired recognition of variant RK9 epitope in subject AC-33. Peptide-specific responses exhibited by autologous unstimulated PBMC from subject AC-33 were quantified by IFN-γ ELISPOT. Each of the variant RK9 peptides showed reduced reactivity compared to the autologous peptide (RLRPGSKKK).
DISCUSSION
CD8+ T-cell responses play a critical role in the containment of HIV-1. Escape from these responses during the course of HIV-1 infection through mutations within targeted epitopes has been observed and may represent a significant challenge to long-term immune control of HIV-1 replication. Herein we describe an additional mechanism of immune escape that would not have been apparent from simply sequencing the targeted epitopes. In this case, a Gag epitope that is frequently targeted in persons expressing HLA-A3 was impacted by a single amino acid mutation in its C-terminal flanking region which inhibited the processing and presentation of the epitope. The same mutation also lies within a second partially overlapping HLA-A3 epitope, inhibiting immune recognition of this epitope by the well-characterized escape mechanism involving impairment of peptide-TCR-HLA interactions. The observation that other HLA-A3 subjects infected with HIV-1 strains encoding similar mutations were unable to mount responses against either of these epitopes during acute infection illustrates not only that horizontal transmission of antigen-processing mutations occurs, but also that these mutations can undermine the development of otherwise immunodominant CD8+ T-cell responses against HIV-1.
Many factors impact the selection and immunodominance of CD8+ T-cell responses. These include the kinetics of pathogen protein production (37, 50, 54), proteasome specificity (14, 47, 60, 68), TAP transport (1), N-terminal trimming (13, 29, 39, 42, 60), MHC binding (52, 58), and TCR availability and affinity (12, 23, 28). A recent study by Yusim et al. supports an important role for proteasome processing sites in the selection of CD8 epitopes in HIV-1 (68). Here, we have illustrated the development of an antigen-processing escape mutation in HIV-1 as a consequence of an HLA-A3-restricted virus-specific CD8+ T-cell response. Three out of the eight HLA-A3-expressing subjects studied developed an amino acid substitution at residue K28 of p17 that was associated with the loss of CD8+ T-cell responses against both immunodominant HLA-A3-restricted p17 Gag epitopes. It is interesting that in subject AC-38 the K28Q mutation ultimately evolved, despite the fact that the transient R20W and K26I mutations effectively impacted recognition by RK9-specific responses. These transient R20W and K26I mutations were also found to strongly impact recognition of KK9-specific responses (data not shown). These data may suggest preferential selection for the K28 processing mutation in the evasion from these HLA-A3-restricted CD8+ T-cell responses. Alignments of the KK9 and RK9 epitopes from the Los Alamos HIV Sequence Database (http://hiv-web.lanl.gov) also indicate a higher degree of sequence variation at residue K28 with the K28Q (8%), K28R (6%), and K28T (6%) mutations occurring most frequently, and data from the Western Australia Sequence database (44) analyzed here support the impact of immune pressure upon this region of Gag in HLA-A3-infected individuals.
Identification of a CD8+ T-cell response as the underlying driving force for selection of an antigen-processing mutation has not been previously described. However, single amino acid residue variations between different murine leukemia virus and HIV-1 isolates in regions flanking CD8 epitopes have previously been shown to alter proper epitope processing (11, 22, 24). Similarly, mutations within defined CD8 epitopes between different murine leukemia virus and HIV-1 strains have also been shown to impact antigen processing (46, 66). Together, these studies suggest that understanding the impact of HIV-1 sequence variation on recognition by CD8+ T-cell responses will not be limited to mutations within defined CD8 epitopes and that evasion of host CD8+ T-cell responses by HIV-1 through processing mutations may be more common than previously appreciated.
Selection for CD8 epitope antigen-processing mutations may provide a unique advantage to HIV-1 in the setting of transmission. While transmitted mutations within an epitope would presumably only benefit the virus if the subsequent host expressed the same restricting HLA molecule, antigen-processing mutations that prevent proper presentation of a larger region of HIV-1 could prove beneficial regardless of the recipient's HLA molecules. Considering that some conserved regions of gag and hydrophobic stretches of nef encode substantial numbers of clustered CD8 epitopes (5, 43, 68), impaired antigen processing within these regions could have a significant impact on the ability of the newly infected host to mount effective CD8 responses. The impact of such mutations may be analogous to the impact that alterations in glycosylation sites have upon evasion of numerous neutralizing antibody responses (2, 17, 55, 62). The observation that some HLA-A3 subjects fail to mount responses against these normally immunodominant HIV-1-specific CD8+ T-cell epitopes due to infection with viruses containing K28 variants indicates that at the population level transmitted mutations can impair a host's ability to mount immunodominant CD8+ T-cell responses. As HLA-A3 represents a frequent HLA allele expressed within Caucasian populations, our data illustrating transmission of an HLA-A3-restricted CD8 escape processing mutation may provide some experimental explanation for the recent report that individuals expressing HLA class I alleles that are frequent in a population experience a more rapid HIV-1 disease progression (61).
The degree to which reversion of CTL escape mutations is occurring after transmission into a new host remains unknown but will be crucial for determining the rate by which these mutations are accumulating within circulating HIV-1 strains. Reversion is likely dependent upon the impact of the mutation on viral fitness or replicative capacity (9, 41, 51, 53). Transmitted mutations no longer required to evade host CD8+ T-cell responses, or which compromise the structure or function of an HIV-1 protein, may be more likely to revert. The observed reversion of the K28R mutation to wild-type sequence in subject AC-33 may suggest that a fitness cost to the virus may be associated with maintenance of such mutations. Therefore, a balance between escape and reversion within HIV-1-specific CD8 T-cell epitopes, influenced by the frequency of HLA alleles within a population (44), may determine the overall integrity and conservation of CD8 T-cell epitopes in the viral population.
The observed escape from an immunodominant CD8+ T-cell response through an antigen-processing mutation further highlights the approaches available to HIV-1 to evade host immune responses. As these processing mutations may represent an effective way for HIV-1 to escape from multiple host CD8+ T-cell responses, a better understanding of the extent to which these mutations develop, are being transmitted, and may be reverting will be important not only to our understanding of HIV-1 pathogenesis but also to the selection and design of antigens capable of effectively inducing immune responses.
Acknowledgments
We thank the subjects participating in this study and Scott Southwood and Stephani Stewart for performing the HLA class I binding assay.
This study was supported by the Doris Duke Charitable Foundation (M.A., E.S.R., and B.D.W.), the National Institutes of Health (T.M.A., M.A., E.S.R., and B.D.W.), the Foundation for AIDS and Immune Research (X.G.Y. and M.A.), the Partners/Fenway/Shattuck Center for AIDS Research (T.M.A., M.A., and X.G.Y.), the Deutsche Forschungsgemeinschaft (M.L.), and the National Institute of Allergy and Infectious Diseases (A.S.). B.D.W. is the recipient of a Doris Duke Distinguished Clinical Scientist Award.
REFERENCES
- 1.Abele, R., and R. Tampe. 1999. Function of the transport complex TAP in cellular immune recognition. Biochim. Biophys. Acta 1461**:**405-419. [DOI] [PubMed] [Google Scholar]
- 2.Albert, J., B. Abrahamsson, K. Nagy, E. Aurelius, H. Gaines, G. Nystrom, and E. M. Fenyo. 1990. Rapid development of isolate-specific neutralizing antibodies after primary HIV-1 infection and consequent emergence of virus variants which resist neutralization by autologous sera. AIDS 4**:**107-112. [DOI] [PubMed] [Google Scholar]
- 3.Allen, T. M., D. H. O'Connor, P. Jing, J. L. Dzuris, B. R. Mothe, T. U. Vogel, E. Dunphy, M. E. Liebl, C. Emerson, N. Wilson, K. J. Kunstman, X. Wang, D. B. Allison, A. L. Hughes, R. C. Desrosiers, J. D. Altman, S. M. Wolinsky, A. Sette, and D. I. Watkins. 2000. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature 407**:**386-390. [DOI] [PubMed] [Google Scholar]
- 4.Altfeld, M., M. M. Addo, R. L. Eldridge, X. G. Yu, S. Thomas, A. Khatri, D. Strick, M. N. Phillips, G. B. Cohen, S. A. Islam, S. A. Kalams, C. Brander, P. J. Goulder, E. S. Rosenberg, and B. D. Walker. 2001. Vpr is preferentially targeted by CTL during HIV-1 infection. J. Immunol. 167**:**2743-2752. [DOI] [PubMed] [Google Scholar]
- 5.Altfeld, M., M. M. Addo, R. Shankarappa, P. K. Lee, T. M. Allen, X. G. Yu, A. Rathod, J. Harlow, K. O'Sullivan, M. N. Johnston, P. J. Goulder, J. I. Mullins, E. S. Rosenberg, C. Brander, B. Korber, and B. D. Walker. 2003. Enhanced detection of human immunodeficiency virus type 1-specific T-cell responses to highly variable regions by using peptides based on autologous virus sequences. J. Virol. 77**:**7330-7340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Altfeld, M., T. M. Allen, X. G. Yu, M. N. Johnston, D. Agrawal, B. T. Korber, D. C. Montefiori, D. H. O'Connor, B. T. Davis, P. K. Lee, E. L. Maier, J. Harlow, P. J. Goulder, C. Brander, E. S. Rosenberg, and B. D. Walker. 2002. HIV-1 superinfection despite broad CD8+ T-cell responses containing replication of the primary virus. Nature 420**:**434-439. [DOI] [PubMed] [Google Scholar]
- 7.Altfeld, M., E. S. Rosenberg, R. Shankarappa, J. S. Mukherjee, F. M. Hecht, R. L. Eldridge, M. M. Addo, S. H. Poon, M. N. Phillips, G. K. Robbins, P. E. Sax, S. Boswell, J. O. Kahn, D. Brander, P. J. R. Goulder, J. A. Levy, J. I. Mullins, and B. D. Walker. 2001. Cellular immune responses and viral diversity in individuals treated during acute and early HIV-1 infection. J. Exp. Med. 193**:**169-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Appay, V., P. R. Dunbar, M. Callan, P. Klenerman, G. M. Gillespie, L. Papagno, G. S. Ogg, A. King, F. Lechner, C. A. Spina, S. Little, D. V. Havlir, D. D. Richman, N. Gruener, G. Pape, A. Waters, P. Easterbrook, M. Salio, V. Cerundolo, A. J. McMichael, and S. L. Rowland-Jones. 2002. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat. Med. 8**:**379-385. [DOI] [PubMed] [Google Scholar]
- 9.Arts, E. J., and M. E. Quinones-Mateu. 2003. Sorting out the complexities of HIV-1 fitness. AIDS 17**:**780-781. [DOI] [PubMed] [Google Scholar]
- 10.Barouch, D. H., J. Kunstman, M. J. Kuroda, J. E. Schmitz, S. Santra, F. W. Peyerl, G. R. Krivulka, K. Beaudry, M. A. Lifton, D. A. Gorgone, D. C. Montefiori, M. G. Lewis, S. M. Wolinsky, and N. L. Letvin. 2002. Eventual AIDS vaccine failure in a rhesus monkey by viral escape from cytotoxic T lymphocytes. Nature 415**:**335-339. [DOI] [PubMed] [Google Scholar]
- 11.Beekman, N. J., P. A. van Veelen, T. van Hall, A. Neisig, A. Sijts, M. Camps, P. M. Kloetzel, J. J. Neefjes, C. J. Melief, and F. Ossendorp. 2000. Abrogation of CTL epitope processing by single amino acid substitution flanking the C-terminal proteasome cleavage site. J. Immunol. 164**:**1898-1905. [DOI] [PubMed] [Google Scholar]
- 12.Belz, G. T., P. G. Stevenson, and P. C. Doherty. 2000. Contemporary analysis of MHC-related immunodominance hierarchies in the CD8+ T cell response to influenza A viruses. J. Immunol. 165**:**2404-2409. [DOI] [PubMed] [Google Scholar]
- 13.Beninga, J., K. L. Rock, and A. L. Goldberg. 1998. Interferon-gamma can stimulate post-proteasomal trimming of the N terminus of an antigenic peptide by inducing leucine aminopeptidase. J. Biol. Chem. 273**:**18734-18742. [DOI] [PubMed] [Google Scholar]
- 14.Bochtler, M., L. Ditzel, M. Groll, C. Hartmann, and R. Huber. 1999. The proteasome. Annu. Rev. Biophys. Biomol. Struct. 28**:**295-317. [DOI] [PubMed] [Google Scholar]
- 15.Borrow, P., H. Lewicki, B. H. Hahn, G. M. Shaw, and M. B. Oldstone. 1994. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J. Virol. 68**:**6103-6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borrow, P., H. Lewicki, X. P. Wei, M. S. Horwitz, N. Peffer, H. Meyers, J. A. Nelson, J. E. Gairin, B. H. Hahn, M. B. A. Oldstone, and G. M. Shaw. 1997. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat. Med. 3**:**205-211. [DOI] [PubMed] [Google Scholar]
- 17.Bradney, A. P., S. Scheer, J. M. Crawford, S. P. Buchbinder, and D. C. Montefiori. 1999. Neutralization escape in human immunodeficiency virus type 1-infected long-term nonprogressors. J. Infect. Dis. 179**:**1264-1267. [DOI] [PubMed] [Google Scholar]
- 18.Brander, C., and P. J. R. Goulder. 2000. The evolving field of HIV CTL epitope mapping: new approaches to the identification of novel epitopes. In B. Korber (ed.), HIV molecular immunology database 2000. Los Alamos National Laboratory, Los Alamos, N.Mex.
- 19.Brander, C., O. O. Yang, N. G. Jones, Y. Lee, P. Goulder, R. P. Johnson, A. Trocha, D. Colbert, C. Hay, S. Buchbinder, C. C. Bergmann, H. J. Zweerink, S. Wolinsky, W. A. Blattner, S. A. Kalams, and B. D. Walker. 1999. Efficient processing of the immunodominant, HLA-A*0201-restricted human immunodeficiency virus type 1 cytotoxic T-lymphocyte epitope despite multiple variations in the epitope flanking sequences. J. Virol. 73**:**10191-10198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao, K., J. Hollenbach, X. Shi, W. Shi, M. Chopek, and M. A. Fernandez-Vina. 2001. Analysis of the frequencies of HLA-A, B, and C alleles and haplotypes in the five major ethnic groups of the United States reveals high levels of diversity in these loci and contrasting distribution patterns in these populations. Hum. Immunol. 62**:**1009-1030. [DOI] [PubMed] [Google Scholar]
- 21.Champagne, P., G. S. Ogg, A. S. King, C. Knabenhans, K. Ellefsen, M. Nobile, V. Appay, G. P. Rizzardi, S. Fleury, M. Lipp, R. Forster, S. Rowland-Jones, R. P. Sekaly, A. J. McMichael, and G. Pantaleo. 2001. Skewed maturation of memory HIV-specific CD8 T lymphocytes. Nature 410**:**106-111. [DOI] [PubMed] [Google Scholar]
- 22.Chassin, D., M. Andrieu, W. Cohen, B. Culmann-Penciolelli, M. Ostankovitch, D. Hanau, and J. G. Guillet. 1999. Dendritic cells transfected with the nef genes of HIV-1 primary isolates specifically activate cytotoxic T lymphocytes from seropositive subjects. Eur. J. Immunol. 29**:**196-202. [DOI] [PubMed] [Google Scholar]
- 23.Chen, W., L. C. Anton, J. R. Bennink, and J. W. Yewdell. 2000. Dissecting the multifactorial causes of immunodominance in class I-restricted T cell responses to viruses. Immunity 12**:**83-93. [DOI] [PubMed] [Google Scholar]
- 24.Cohen, W. M., A. Bianco, F. Connan, L. Camoin, M. Dalod, G. Lauvau, E. Ferries, B. Culmann-Penciolelli, P. M. van Endert, J. P. Briand, J. Choppin, and J. G. Guillet. 2002. Study of antigen-processing steps reveals preferences explaining differential biological outcomes of two HLA-A2-restricted immunodominant epitopes from human immunodeficiency virus type 1. J. Virol. 76**:**10219-10225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Collins, K. L., B. K. Chen, S. A. Kalams, B. D. Walker, and D. Baltimore. 1998. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 391**:**397-401. [DOI] [PubMed] [Google Scholar]
- 26.Couillin, I., B. Culmann-Penciolelli, E. Gomard, J. Choppin, J. P. Levy, J. G. Guillet, and S. Saragosti. 1994. Impaired cytotoxic T lymphocyte recognition due to genetic variations in the main immunogenic region of the human immunodeficiency virus 1 NEF protein. J. Exp. Med. 180**:**1129-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dalod, M., M. Dupuis, J. C. Deschemin, C. Goujard, C. Deveau, L. Meyer, N. Ngo, C. Rouzioux, J. G. Guillet, J. F. Delfraissy, M. Sinet, and A. Venet. 1999. Weak anti-HIV CD8+ T-cell effector activity in HIV primary infection. J. Clin. Investig. 104**:**1431-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Daly, K., P. Nguyen, D. L. Woodland, and M. A. Blackman. 1995. Immunodominance of major histocompatibility complex class I-restricted influenza virus epitopes can be influenced by the T-cell receptor repertoire. J. Virol. 69**:**7416-7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elliott, T., A. Willis, V. Cerundolo, and A. Townsend. 1995. Processing of major histocompatibility class I-restricted antigens in the endoplasmic reticulum. J. Exp. Med. 181**:**1481-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Evans, D. T., D. H. O'Connor, P. Jing, D. J. L., J. Sydney, J. da Silva, T. M. Allen, H. Horton, J. E. Venham, R. A. Rudersdorf, C. D. Pauza, R. E. Bontrop, R. DeMars, A. Sette, A. L. Hughes, and D. I. Watkins. 1999. Virus-specific CTL responses select for amino acid variation in SIV Env and Nef. Nat. Med. 5**:**1270-1276. [DOI] [PubMed] [Google Scholar]
- 31.Goulder, P. J., C. Brander, Y. Tang, C. Tremblay, R. A. Colbert, M. M. Addo, E. S. Rosenberg, T. Nguyen, R. Allen, A. Trocha, M. Altfeld, S. He, M. Bunce, R. Funkhouser, S. I. Pelton, S. K. Burchett, K. McIntosh, B. T. Korber, and B. D. Walker. 2001. Evolution and transmission of stable CTL escape mutations in HIV infection. Nature 412**:**334-338. [DOI] [PubMed] [Google Scholar]
- 32.Goulder, P. J., C. Pasquier, E. C. Holmes, B. Liang, Y. Tang, J. Izopet, K. Saune, E. S. Rosenberg, S. K. Burchett, K. McIntosh, M. Barnardo, M. Bunce, B. D. Walker, C. Brander, and R. E. Phillips. 2001. Mother-to-child transmission of HIV infection and CTL escape through HLA-A2-SLYNTVATL epitope sequence variation. Immunol. Lett. 79**:**109-116. [DOI] [PubMed] [Google Scholar]
- 33.Goulder, P. J. R., R. E. Phillips, R. A. Colbert, S. McAdam, G. Ogg, M. A. Nowak, P. Giangrande, G. Luzzi, B. Morgan, A. Edwards, A. J. McMichael, and S. Rowland Jones. 1997. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat. Med. 3**:**212-217. [DOI] [PubMed] [Google Scholar]
- 34.Jin, X., D. E. Bauer, S. E. Tuttleton, S. Lewin, A. Gettie, J. Blanchard, C. E. Irwin, J. T. Safrit, J. Mittler, L. Weinberger, L. G. Kostrikis, L. Q. Zhang, A. S. Perelson, and D. D. Ho. 1999. Dramatic rise in plasma viremia after CD8+ T cell depletion in simian immunodeficiency virus-infected macaques. J. Exp. Med. 189**:**991-998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson, R. P., A. Trocha, T. M. Buchanan, and B. D. Walker. 1992. Identification of overlapping HLA class I-restricted cytotoxic T cell epitopes in a conserved region of the human immunodeficiency virus type 1 envelope glycoprotein: definition of minimum epitopes and analysis of the effects of sequence variation. J. Exp. Med. 175**:**961-971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kesmir, C., A. K. Nussbaum, H. Schild, V. Detours, and S. Brunak. 2002. Prediction of proteasome cleavage motifs by neural networks. Protein Eng. 15**:**287-296. [DOI] [PubMed] [Google Scholar]
- 37.Kim, S. Y., R. Byrn, J. Groopman, and D. Baltimore. 1989. Temporal aspects of DNA and RNA synthesis during human immunodeficiency virus infection: evidence for differential gene expression. J. Virol. 63**:**3708-3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koenig, S., A. J. Conley, Y. A. Brewah, G. M. Jones, S. Leath, L. J. Boots, V. Davey, G. Pantaleo, J. F. Demarest, C. Carter, C. Wannebo, J. R. Yannelli, S. A. Rosenberg, and H. C. Lane. 1995. Transfer of HIV-1-specific cytotoxic T lymphocytes to an AIDS patient leads to selection for mutant HIV variants and subsequent disease progression. Nat. Med. 1**:**330-336. [DOI] [PubMed] [Google Scholar]
- 39.Komlosh, A., F. Momburg, T. Weinschenk, N. Emmerich, H. Schild, E. Nadav, I. Shaked, and Y. Reiss. 2001. A role for a novel luminal endoplasmic reticulum aminopeptidase in final trimming of 26S proteasome-generated major histocompatability complex class I antigenic peptides. J. Biol. Chem. 276**:**30050-30056. [DOI] [PubMed] [Google Scholar]
- 40.Koup, R. A., J. T. Safrit, Y. Cao, C. A. Andrews, G. McLeod, W. Borkowsky, C. Farthing, and D. D. Ho. 1994. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 68**:**4650-4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leigh Brown, A. J., S. D. Frost, W. C. Mathews, K. Dawson, N. S. Hellmann, E. S. Daar, D. D. Richman, and S. J. Little. 2003. Transmission fitness of drug-resistant human immunodeficiency virus and the prevalence of resistance in the antiretroviral-treated population. J. Infect. Dis. 187**:**683-686. [DOI] [PubMed] [Google Scholar]
- 42.Levy, F., L. Burri, S. Morel, A. L. Peitrequin, N. Levy, A. Bachi, U. Hellman, B. J. Van den Eynde, and C. Servis. 2002. The final N-terminal trimming of a subaminoterminal proline-containing HLA class I-restricted antigenic peptide in the cytosol is mediated by two peptidases. J. Immunol. 169**:**4161-4171. [DOI] [PubMed] [Google Scholar]
- 43.Lucchiari-Hartz, M., V. Lindo, N. Hitziger, S. Gaedicke, L. Saveanu, P. M. van Endert, F. Greer, K. Eichmann, and G. Niedermann. 2003. Differential proteasomal processing of hydrophobic and hydrophilic protein regions: contribution to cytotoxic T lymphocyte epitope clustering in HIV-1-Nef. Proc. Natl. Acad. Sci. USA 100**:**7755-7760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moore, C. B., M. John, I. R. James, F. T. Christiansen, C. S. Witt, and S. A. Mallal. 2002. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science 296**:**1439-1443. [DOI] [PubMed] [Google Scholar]
- 45.O'Connor, D. H., T. M. Allen, T. U. Vogel, P. Jing, I. P. DeSouza, E. Doods, E. Dunphy, C. Melsaether, B. Mothe, H. Horton, A. L. Hughes, and D. I. Watkins. 2002. Acute phase CTL escape is a hallmark of SIV infection. Nat. Med. 8**:**493-499. [DOI] [PubMed] [Google Scholar]
- 46.Ossendorp, F., M. Eggers, A. Neisig, T. Ruppert, M. Groettrup, A. Sijts, E. Mengede, P. M. Kloetzel, J. Neefjes, U. Koszinowski, and C. Melief. 1996. A single residue exchange within a viral CTL epitope alters proteasome-mediated degradation resulting in lack of antigen presentation. Immunity 5**:**115-124. [DOI] [PubMed] [Google Scholar]
- 47.Pamer, E., and P. Cresswell. 1998. Mechanisms of MHC class I-restricted antigen processing. Annu. Rev. Immunol. 16**:**323-358. [DOI] [PubMed] [Google Scholar]
- 48.Phillips, R. E., S. Rowland-Jones, D. F. Nixon, F. M. Gotch, J. P. Edwards, A. O. Ogunlesi, J. G. Elvin, J. A. Rothbard, C. R. Bangham, C. R. Rizza, et al. 1991. Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature 354**:**453-459. [DOI] [PubMed] [Google Scholar]
- 49.Price, D. A., P. J. Goulder, P. Klenerman, A. K. Sewell, P. J. Easterbrook, M. Troop, C. R. Bangham, and R. E. Phillips. 1997. Positive selection of HIV-1 cytotoxic T lymphocyte escape variants during primary infection. Proc. Natl. Acad. Sci. USA 94**:**1890-1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Probst, H. C., K. Tschannen, A. Gallimore, M. Martinic, M. Basler, T. Dumrese, E. Jones, and M. F. van den Broek. 2003. Immunodominance of an antiviral cytotoxic T cell response is shaped by the kinetics of viral protein expression. J. Immunol. 171**:**5415-5422. [DOI] [PubMed] [Google Scholar]
- 51.Quinones-Mateu, M. E., S. C. Ball, A. J. Marozsan, V. S. Torre, J. L. Albright, G. Vanham, G. van Der Groen, R. L. Colebunders, and E. J. Arts. 2000. A dual infection/competition assay shows a correlation between ex vivo human immunodeficiency virus type 1 fitness and disease progression. J. Virol. 74**:**9222-9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rammensee, H. G., T. Friede, and S. Stevanoviic. 1995. MHC ligands and peptide motifs: first listing. Immunogenetics 41**:**178-228. [DOI] [PubMed] [Google Scholar]
- 53.Rangel, H. R., J. Weber, B. Chakraborty, A. Gutierrez, M. L. Marotta, M. Mirza, P. Kiser, M. A. Martinez, J. A. Este, and M. E. Quinones-Mateu. 2003. Role of the human immunodeficiency virus type 1 envelope gene in viral fitness. J. Virol. 77**:**9069-9073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ranki, A., A. Lagerstedt, V. Ovod, E. Aavik, and K. J. Krohn. 1994. Expression kinetics and subcellular localization of HIV-1 regulatory proteins Nef, Tat and Rev in acutely and chronically infected lymphoid cell lines. Arch. Virol. 139**:**365-378. [DOI] [PubMed] [Google Scholar]
- 55.Richman, D. D., T. Wrin, S. J. Little, and C. J. Petropoulos. 2003. Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proc. Natl. Acad. Sci. USA 100**:**4144-4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schmitz, J. E., M. J. Kuroda, S. Santra, V. G. Sasseville, M. A. Simon, M. A. Lifton, P. Racz, K. Tenner-Racz, M. Dalesandro, B. J. Scallon, J. Ghrayeb, M. A. Forman, D. C. Montefiori, E. P. Rieber, N. L. Letvin, and K. A. Reimann. 1999. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science 283**:**857-860. [DOI] [PubMed] [Google Scholar]
- 57.Schwartz, O., V. Marechal, S. Le Gall, F. Lemonnier, and J. M. Heard. 1996. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med. 2**:**338-342. [DOI] [PubMed] [Google Scholar]
- 58.Sette, A., and J. Sidney. 1999. Nine major HLA class I supertypes account for the vast preponderance of HLA-A and -B polymorphism. Immunogenetics 50**:**201-212. [DOI] [PubMed] [Google Scholar]
- 59.Sidney, J., S. Southwood, C. Oseroff, M.-F. del Guercio, A. Sette, and H. M. Grey. 1998. Measurement of MHC/peptide interactions by gel filtration, p. 18.3.1-18.3.19. In J. E. Coligan, A. M. Kruisbeek, D. M. Margulies, E. M. Shevach, and W. Strober (ed.), Current protocols in immunology. Wiley Interscience, Hoboken, N.J.
- 60.Stoltze, L., M. Schirle, G. Schwarz, C. Schroter, M. W. Thompson, L. B. Hersh, H. Kalbacher, S. Stevanovic, H. G. Rammensee, and H. Schild. 2000. Two new proteases in the MHC class I processing pathway. Nat. Immunol. 1**:**413-418. [DOI] [PubMed] [Google Scholar]
- 61.Trachtenberg, E., B. Korber, C. Sollars, T. B. Kepler, P. T. Hraber, E. Hayes, R. Funkhouser, M. Fugate, J. Theiler, Y. S. Hsu, K. Kunstman, S. Wu, J. Phair, H. Erlich, and S. Wolinsky. 2003. Advantage of rare HLA supertype in HIV disease progression. Nat. Med. 9**:**928-935. [DOI] [PubMed] [Google Scholar]
- 62.Wei, X., J. M. Decker, S. Wang, H. Hui, J. C. Kappes, X. Wu, J. F. Salazar-Gonzalez, M. G. Salazar, J. M. Kilby, M. S. Saag, N. L. Komarova, M. A. Nowak, B. H. Hahn, P. D. Kwong, and G. M. Shaw. 2003. Antibody neutralization and escape by HIV-1. Nature 422**:**307-312. [DOI] [PubMed] [Google Scholar]
- 63.Wilson, C. C., R. C. Brown, B. T. Korber, B. M. Wilkes, D. J. Ruhl, D. Sakamoto, K. Kunstman, K. Luzuriaga, I. C. Hanson, S. M. Widmayer, A. Wiznia, S. Clapp, A. J. Ammann, R. A. Koup, S. M. Wolinsky, and B. D. Walker. 1999. Frequent detection of escape from cytotoxic T-lymphocyte recognition in perinatal human immunodeficiency virus (HIV) type 1 transmission: the Ariel project for the prevention of transmission of HIV from mother to infant. J. Virol. 73**:**3975-3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wolinsky, S. M., B. T. Korber, A. U. Neumann, M. Daniels, K. J. Kunstman, A. J. Whetsell, M. R. Furtado, Y. Cao, D. D. Ho, and J. T. Safrit. 1996. Adaptive evolution of human immunodeficiency virus-type 1 during the natural course of infection. Science 272**:**537-542. [DOI] [PubMed] [Google Scholar]
- 65.Yang, O. O., P. T. Nguyen, S. A. Kalams, T. Dorfman, H. G. Gottlinger, S. Stewart, I. S. Chen, S. Threlkeld, and B. D. Walker. 2002. Nef-mediated resistance of human immunodeficiency virus type 1 to antiviral cytotoxic T lymphocytes. J. Virol. 76**:**1626-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yokomaku, Y., H. Miura, H. Tomiyama, A. Kawana-Tachikawa, M. Takiguchi, A. Kojima, Y. Nagai, A. Iwamoto, Z. Matsuda, and K. Ariyoshi. 2004. Impaired processing and presentation of cytotoxic-T-lymphocyte (CTL) epitopes are major escape mechanisms from CTL immune pressure in human immunodeficiency virus type 1 infection. J. Virol. 78**:**1324-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu, X. G., M. M. Addo, E. S. Rosenberg, W. R. Rodriguez, P. K. Lee, C. A. Fitzpatrick, M. N. Johnston, D. Strick, P. J. Goulder, B. D. Walker, and M . Altfeld. 2002. Consistent patterns in the development and immunodominance of human immunodeficiency virus type 1 (HIV-1)-specific CD8+ T-cell responses following acute HIV-1 infection. J. Virol. 76**:**8690-8701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yusim, K., C. Kesmir, B. Gaschen, M. M. Addo, M. Altfeld, S. Brunak, A. Chigaev, V. Detours, and B. T. Korber. 2002. Clustering patterns of cytotoxic T-lymphocyte epitopes in human immunodeficiency virus type 1 (HIV-1) proteins reveal imprints of immune evasion on HIV-1 global variation. J. Virol. 76**:**8757-8768. [DOI] [PMC free article] [PubMed] [Google Scholar]