Role of the normal gut microbiota (original) (raw)
Abstract
Relation between the gut microbiota and human health is being increasingly recognised. It is now well established that a healthy gut flora is largely responsible for overall health of the host. The normal human gut microbiota comprises of two major phyla, namely Bacteroidetes and Firmicutes. Though the gut microbiota in an infant appears haphazard, it starts resembling the adult flora by the age of 3 years. Nevertheless, there exist temporal and spatial variations in the microbial distribution from esophagus to the rectum all along the individual’s life span. Developments in genome sequencing technologies and bioinformatics have now enabled scientists to study these microorganisms and their function and microbe-host interactions in an elaborate manner both in health and disease. The normal gut microbiota imparts specific function in host nutrient metabolism, xenobiotic and drug metabolism, maintenance of structural integrity of the gut mucosal barrier, immunomodulation, and protection against pathogens. Several factors play a role in shaping the normal gut microbiota. They include (1) the mode of delivery (vaginal or caesarean); (2) diet during infancy (breast milk or formula feeds) and adulthood (vegan based or meat based); and (3) use of antibiotics or antibiotic like molecules that are derived from the environment or the gut commensal community. A major concern of antibiotic use is the long-term alteration of the normal healthy gut microbiota and horizontal transfer of resistance genes that could result in reservoir of organisms with a multidrug resistant gene pool.
Keywords: Normal gut microbiota, Bioinformatics, Health, Immunomodulation, Metabolic function
Core tip: In this review we present an up-to-date overview of the normal gut microbiota, their functional implications in health, and the mechanistic insights that orchestrate these functions. We also discuss the characteristics that define a healthy gut microbiota and factors that shape and perturb the gut microbial diversity and functions. The evidence that we present here is a composite of observational and experimental studies on humans, germ free and humanized mice.
INTRODUCTION
Microbiota refers to the entire population of microorganisms that colonizes a particular location; and includes not just bacteria, but also other microbes such as fungi, archaea, viruses, and protozoans[1]. Significant interest have evolved on the gut microbiota in the recent years within the scientific community; and the gut microbiota have been associated with a large array of human diseases ranging from luminal diseases such as inflammatory bowel diseases (IBD)[2] and irritable bowel syndrome (IBS)[3], metabolic diseases such obesity and diabetes[4], allergic disease[5] to neurodevelopmental illnesses, though the strength of evidence is not robust with many of them. It has been speculated since long that the gut microbiota bear significant functional role in maintaining the gut in the normal individual and human health as a whole. There is now mounting evidence resulting from studies on humans and germ free mice that supports these speculations. Several high quality data from the US Human Microbiome Project (HMP)[6], European Metagenomics of the Human Intestinal Tract (MetaHIT)[7] and several other studies have now demonstrated the beneficial functions of the normal gut flora on health down to the genetic level. For example, studies have now identified several gut microbial genes, such as the HMO-related gene cluster 1 that is responsible for human milk oligosaccharide digestion.
From an immunological perspective, microorganisms are viewed as pathogens by the host immune system that recognizes and eliminates them. However, majority of the gut bacteria are non-pathogenic and, co-habit with the enterocytes in a symbiotic relationship. The gut commensals predominantly aid in nutrient metabolism, drug metabolism, prevention of colonization of pathogenic microorganisms and in intestinal barrier function. At the same time, the immune system has co-evolved to live in a collaborative relationship with the healthy microbiota, while serving its function to fight off invasive pathogenic microorganisms.
The purpose of this manuscript is to review the recent evidence on the functions of the normal gut microbiota and the mechanistic insights into the execution of these pro-health functions. Data presented in this review is a composite of both observational and experimental studies on humans, germ free and humanized mice. The implications of the gut microbiota in disease states are out of the scope of this review.
CURRENT METHODS TO STUDY GUT MICROBIOTA
To study the gut microbiota, stool samples have to be collected from individuals and DNA from stool is isolated. Isolation, identification and enumeration of the vast majority of gastrointestinal microorganisms using conventional culture based techniques is an arduous task. Earlier, using culture based techniques, scientists were able to isolate only 10%-25% of the microbiota, and this was because most of the microorganisms in the gut are anaerobic. Later, with the improvements in the anaerobic culturing techniques, dominant genera were identified such as, Bacteroides, Clostridium, Bifidobacterium etc. The major drawback in using these techniques is the difficulty in studying the culture characteristics of various colonies on a petri plate. Secondly, it is time consuming[8-10].
With the availability of high throughput gene sequencing technology, study of the gut microbiota currently consists of two major stages: (1) 16S rRNA based sequencing of bacterial gene; and (2) bioinformatics analysis. Metabolomics is another rapidly expanding field of gut microbiota research that evaluates small molecules associated with the interrelationship of host-bacterial metabolism that has implications in health and disease. Composite data from the gut microbiota and the metabolome currently provides the most powerful evidence that can demonstrate the closest association with health and diseased states.
Bacterial gene sequencing
Sequencing of bacterial genes involves metagenomic analysis of DNA that codes for the 16S rRNA. The 16S region of bacterial gene is small (1.5 Kb size) and highly conserved, with 9 hyper variable sites that are sufficient to differentiate various bacterial species[11]. Common regions for bacterial identification in 16S rRNA are the V3, V4, V6 and V8[12]. With the development of biomedical technology, bacterial gene sequencing has rapidly evolved from Sanger’s sequencing to several variations of next-generation sequencing (NGS). Even though NGS could provide voluminous data with fair to good accuracy, they are not free from problems. A recent study have shown that sequencing could be prone to errors that most likely results from the library preparation methods and choice of primers[13]. The other issue of concern in 16S rRNA based sequencing is the variability of results across different sequencing centers, both for predominant and minor taxa. This variation again could be result of differences in primers used to generate the amplicon libraries[14]. Table 1 presents the accuracy, advantages and disadvantages of the currently available sequencing techniques[15,16].
Table 1.
Advantages and disadvantages of few of the currently available next generation sequencing techniques[15,16]
| Techniques used in next generation sequencing | Accuracy | Advantages | Disadvantages |
|---|---|---|---|
| 454 Pyrosequencing | 99.9% | Less amount of sample, long read lengths, large number of samples can be easily read | Homopolymer errors Expensive |
| Shot gun Sequencing | 98% | Short reads in short time | Assembly process is computationally expensive |
| Illumina Sequencing (Sequencing by synthesis) | 98% | Accurate, quicker, reliable and cheap | Expensive |
| Pacific Bio Sequencing (single molecule real-time sequencing) | 99.9% | Fast and provides long read length | Expensive equipment |
| Ion Torrent Sequencing (Ion semiconductor) | 98% | Fast and less expensive equipment | Multiple monomer errors |
| SOLiD (Sequencing by Ligation) Sequencing | 99.9% | Less expensive when compared to other methods | Slow and difficult to sequence palindromes |
BIOINFORMATICS ANALYSIS
The data obtained from sequencing is often voluminous, fragmented, noisy, overlapping, and contaminated. Bioinformatics analysis enables cleaning up the data and the identification of the bacterial taxa. This can also be extended to obtaining information also on metabolic functions using a wide array of bioinformatics platforms. Furthermore, statistical analysis of the sequence data also help in identifying alpha diversity (diversity of species within the same individual), beta diversity (inter-individual species diversity), relative abundance, and several other parameters related to the organisms. Figure 1 shows the workflow of study of the gut microbiota.
Figure 1.
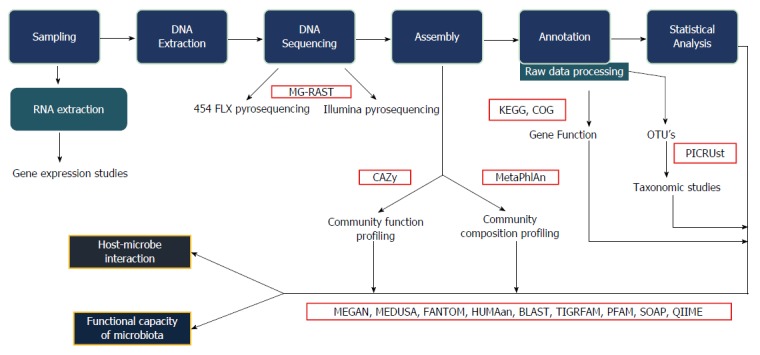
Bioinformatics work flow. This figure explains the various steps involved in the bioinformatics analysis, starting from collection of samples, extraction, sequencing and statistical analysis. The interaction between host and microbes along with the functional capacity of the microbiota can be studied. MG-RAST: Metagenomics rapid annotation using subsystem technology; CAZy: Carbohydrate active-enzymes; MetaPhlAn: Metagenomic phylogenetic analysis; KEGG: Kyoto encyclopaedia for genes and genomics; COG: Clusters of orthologous group; PICRUst: Phylogenetic investigation of communities by reconstruction of unobserved states; MEGAN: Meta genome analyzer; MEDUSA: Metagenomic data utilization and analysis; FANTOM: Functional annotation and taxonomic analysis of metagenomes; HUMAan: Human microbiome project unified metabolic analysis network; BLAST: Basic local alignment search tool; TIGRFAM: Protein sequence classification; PFAM: Protein families; SOAP: Short oligonucleotide analysis package; QIIME: Quantitative insights into microbial ecology.
COMPOSITION OF THE NORMAL GUT MICROBIOTA
Even though it was earlier thought that the gut microbiota comprised of 500-1000 species of microbes[17] a recent large scale study has estimated that the collective human gut microflora is composed of over 35000 bacterial species[18]. Furthermore, if defined from a perspective of total bacterial genes, the Human Microbiome Project and the Metagenome of the Human Intestinal tract (MetaHIT) studies suggest that there could be over 10 million non-redundant genes in the human microbiome. A Danish study of the gut microbiome and their function involving 123 non-obese and 169 obese individuals resulted in the concept of high gene count (HGC) and low gene count (LGC), both of which have implications in health and disease[19]. The HGC microbiome includes Anaerotruncus colihominis, Butyrivibrio crossotus, Akkermansia sp., and Fecalibacterium sp.; with a high Akkermansia (Verrucomicrobia): Ruminococcus torque/gnavus ratio. The defining features of HGC microbiome in favour of a digestive health includes increased proportion of butyrate producing organisms, increased propensity for hydrogen production, development of a methanogenic/acetogenic ecosystem and reduced production of hydrogen sulfide[19]. The HGC individuals have a functionally much robust gut microbiome and lower prevalence of metabolic disorders and obesity. On the other hand, LGC individuals harbor a higher proportion of pro-inflammatory bacteria such as Bacteroides and Ruminococcus gnavus, both of which are known to be associated inflammatory bowel disease[20,21]. Other members of LGC bacteria include Parabacteroides, Campylobacter, Dialister, Porphyromonas, Staphylococcus and Anaerostipes. In addition, few of the key bacterial metabolites in LGC individuals include modules for β-glucuronide degradation, degradation of aromatic amino acids, and dissimilatory nitrite reduction, all of which are known to have deleterious effects.
Overall, the healthy gut microbiota is predominantly constituted by the phyla Firmicutes and Bacteroidetes. This is followed by the phyla Actinobacteria and Verrucomicrobia. Even though this general profile remains constant, gut microbiota exhibits both temporal and spatial differences in distribution at the genus level and beyond. As one travels from the esophagus distally to the rectum, there will be a marked difference in diversity and number of bacteria ranging from 101 per gram of contents in the esophagus and stomach to 1012 per gram of contents in the colon and distal gut[22]. Figure 2 depicts the temporal diversity of the gut microbiota as one travels from the esophagus distally to the colon. Streptococcus appears to be the dominant genus in the distal esophagus, duodenum and jejunum[23,24]. Helicobacter is the dominant genera present in the stomach and determines the entire microbial landscape of the gastric flora, i.e., when Helicobacter pylori (H. pylori) inhabits the stomach as a commensal, there is a rich diversity constituted by other dominant genus such as Streptococcus (most dominant), Prevotella, Veillonella and _Rothia_[25,26]. This diversity shrinks once H. pylori acquire a pathogenic phenotype. The large intestine constitutes of over 70% of the all microbes found in the body, and gut flora that is generally discussed in the context of disease state by and large implies the colonic flora (especially those derived from stool metagenomic data). The predominant phyla that inhabit the large intestine include Firmicutes and Bacteroidetes. Traditionally, the Firmicutes: Bacteroidetes ratio has been implicated in predisposition to disease states[27]. However, the significant variability even in healthy individuals that has been observed across recent studies makes the relevance of this ratio debatable. Besides genera from phyla Firmicutes and Bacteroidetes, human colon also harbors primary pathogens, e.g., species such as Campylobacter jejuni, Salmonella enterica, Vibrio cholera and Escherichia coli (E. coli), and Bacteroides fragilis, but with a low abundance (0.1% or less of entire gut microbiome)[6,28]. The abundance of the phylum Proteobacteria is markedly low; and its absence along with high abundance of signature genera such as Bacteroides, Prevotella and Ruminococcus suggests a healthy gut microbiota[29]. Besides this longitudinal difference, there also exists an axial difference from the lumen to the mucosal surface of the intestine. While Bacteroides, Bifidobacterium, Streptococcus, Enterobacteriacae, Enterococcus, Clostridium, Lactobacillus and Ruminococcus are the predominant luminal microbial genera (can be identified in stool), only Clostridium, Lactobacillus, Enterococcus and Akkermansia are the predominant mucosa and mucus associated genera (detected in the mucus layer and epithelial crypts of the small intestine)[30].
Figure 2.
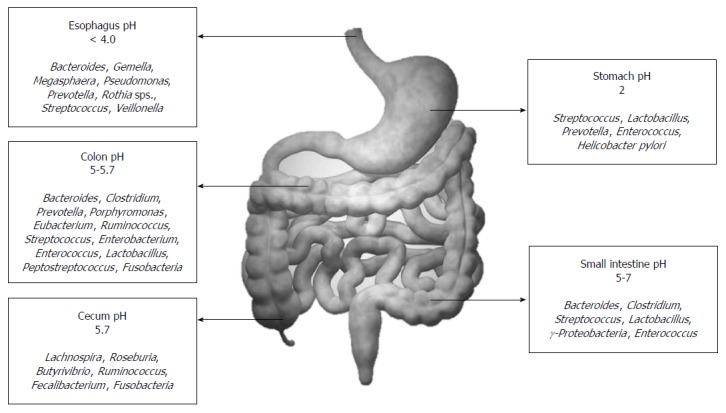
Distribution of the normal human gut flora.
The other way of classifying the gut flora, as proposed by the MetaHIT Consortium[31], is based on species composition which cluster into well-balanced host-microbial symbiotic states that is stable over geography and gender, but can respond differently to diet and drugs. These clusters have been named enterotypes. Interestingly, the abundance of molecular functions however may not correlate with abundance of species within the enterotypes. Furthermore, as shown in a recent study on the association of gut microbiome with atherosclerosis, there may not be significant changes in the enterotype observed in disease conditions[32]. There are broadly three enterotypes[29], namely: Enterotype 1, which has a high abundance of Bacteroides; Enterotype 2, which has high abundance of Prevotella; and Enterotype 3 which has high abundance of Ruminococcus. The bacteria belonging to Enterotype 1 have a wide saccharolytic potential, as evidenced by the presence of genes that code for enzymes such as proteases, hexoaminidases and galactosidases. In view of these set of enzymatic potential, it appears likely that these organisms derive energy from dietary carbohydrates and proteins. Enterotype 2 behave predominantly as a degrader of the mucin glycoproteins that line the gut mucosal layer. Enterotype 3 also is associated with mucin degradation, in addition to membrane transport of sugars. The enterotypes also possess other specific metabolic functions. For instance, biotin, riboflavin, pantothenate and ascorbate synthesis are more abundantly seen in enterotype 1 while thiamine and folate synthesis are more predominant in enterotype 2. However, the concept of enterotyping does not explain the relative distribution of different classes of organisms in different individuals. Since Bacteroides and Prevotella do not exist in equal proportion in the gut, the concept of enterogradient based upon the dominance of either of these two organisms could be another defining concept. This could explain the inter-individual distribution at the class level in a better way[33].
FUNCTIONAL ASPECTS OF THE NORMAL GUT MICROBIOTA
The gut microbiota maintains a symbiotic relationship with the gut mucosa and imparts substantial metabolic, immunological and gut protective functions in the healthy individual. The gut microbiota, which derives its nutrient from host dietary components and shed epithelial cells, is an organ by itself with an extensive metabolic capability and substantial functional plasticity[34]. These characteristics of the gut microbiome have been rapidly shifting the research focus from the abundance and diversity of the microbial members to the functional aspects. This section provides a brief overview of the major functions of the normal gut microbiota.
Nutrient metabolism
The gut microbiota largely derives their nutrients from dietary carbohydrates. Fermentation of the carbohydrates that escaped proximal digestion and indigestible oligosaccharides by colonic organisms such as Bacteroides, Roseburia, Bifidobacterium, Fecalibacterium, and Enterobacteria result in the synthesis of short chain fatty acids (SCFA) such as butyrate, propionate and acetate, which are rich sources of energy for the host[35,36]. This host energy balance is believed to be mediated via a ligand-receptor interaction of the SCFAs with a G protein-coupled receptor Gpr41. Another enteroendocrine hormone PYY (Peptide Tyrosine Tyrosine/Pancreatic Peptide YY3-36) has also been implicated in this action[37]. Furthermore, butyrate can prevent the accumulation of toxic metabolic by-products such as D-lactate[38]. Members of the genus Bacteroides, which are the predominant organisms that participate in carbohydrate metabolism, perform this by expressing enzymes such as glycosyl transferases, glycoside hydrolases and polysaccharide lyases. The best example among these organisms is Bacteroides thetaiotaomicron that is endowed with a genome that codes for over 260 hydrolases, which is far more than the number encoded by the human genome[39]. The oxalate that is synthesized in the intestine as a result of carbohydrate fermentation and bacterial metabolism is countered by organisms such as Oxalobacter formigenes, Lactobacillus species, and Bifidobacterium species thereby reducing the risk of formation of oxalate stone in the kidney[40,41].
The gut microbiota has also been shown to impart a positive impact on lipid metabolism by suppressing the inhibition of lipoprotein lipase activity in adipocytes. Furthermore, Bacteroides thetaiotaomicron is demonstrated to augment the efficiency of lipid hydrolysis by up regulating expression of a colipase that is required by pancreatic lipase for lipid digestion[42].
The gut microbiota is also enriched with an efficient protein metabolizing machinery that function via the microbial proteinases and peptidases in tandem with human proteinases. Several amino acid transporters on the bacterial cell wall facilitate amino acid entry from the intestinal lumen into the bacteria, wherein several gene products convert the amino acids into small signaling molecules and antimicrobial peptides (bacteriocins). Important examples include conversion of L-histidine to histamine by the bacterial enzyme histamine decarboxylase, which is coded by the bacterial hdcA genes[43]; and glutamate to γ-amino butyric acid (GABA) by glutamate decarboxylases, which are coded by the bacterial gadB genes[44].
Synthesis of vitamin K and several components of vitamin B is another major metabolic function of the gut microbiota. Members of genus Bacteroides have been shown to synthesize conjugated linoleic acid (CLA) that is known to be antidiabetic, antiatherogenic, antiobesogenic, hypolipidemic and have immunomodulatory properties[45-47]. The gut microbiota, especially Bacteroides intestinalis, and to a certain extent Bacteroides fragilis and E. coli, also has the capacity to deconjugate and dehydrate the primary bile acids and convert them into the secondary bile acids deoxycholic and lithocolic acids in the human colon[48]. The normal gut microbiota has also been shown to impart a healthy metabolome in the serum by increasing the concentrations of pyruvic acid, citric acid, fumaric acid and malic acid, all of which are indicators of higher energy metabolism[49].
Recent studies have shown that human gut microbiota is also involved in breakdown of various polyphenols (phenolic compounds) that are consumed in the diet. Polyphenolic secondary metabolites are found in a variety of plants, fruits and plant derived products (tea, cocoa, wine), for example, flavanols, flavanones, flavan-3-ols, anthocyanidins, isoflavones, flavones, tannins, lignans and chlorogenic acids. Of these, flavanoids and flavanoid sub-families are most commonly absorbed by the intestine. Polyphenols exist as glycosylated derivatives bounded with sugars such as glucose, galactose, rhamnose, ribulose, arabinopyrinose and arabinofuranose. Polyphenols, which usually remain inactive in diet are biotransformed to active compounds after removal of the sugar moiety by the gut microbiota, among other factors. Structural specificity of polyphenol and individual richness of microbiota determines the level of biotransformation that occur in the intestine. The final active products are absorbed by the portal vein and travel to other tissues and organs, thereby providing antimicrobial and other metabolic action. This can be exemplified by the conversion of inactive isoflavones to the aglycon equol, which has anti-androgenic and hypolipidemic effects[50]. Table 2 shows an elaborate list of the dietary polyphenols and the gut microbiota involved in its transformation[51-69].
Table 2.
Types of dietary polyphenols present in various foods and the types of microorganisms those are responsible for the degradation
| Polyphenolic compounds | Classes involved | Foods containing polyphenols | Gut bacteria |
|---|---|---|---|
| Flavanols | Kaempferol[51], Quercetin[53], Myricetin[52] | Onions, capers, apples, broccoli, grapes and plums | Bacteroides distasonis, Bacteroides uniformis, Enterococcus casseliflavus and Eubacterium ramulus |
| Flavanones | Hesperetin, Naringenin[54] | Citrus fruits and tomatoes | Clostridium sps, E. ramulus |
| Flavan-3-ols | Catechin[55], Epicatechin[56], Gallocatechin[57,58] | Green tea, cocoa, kola, banana, pomegranate | Bifidobacterium infantis and Clostridium coccides |
| Anthocyanidins | Cyanidin[59], Pelagonidin, Malvidin[60] | Bilberries and all red, blue and purple fruits (especially berries) | Lactobacillus plantarum, L. casei, L. acidophilus and Bifidobacterium longum |
| Isoflavones | Daidzein[61,62], Geinstein[63], Formononentin[64] | Soy, beans, lentils, chickpea (Fabaceae family) | Lactobacillus and Bifidobacterium |
| Flavones | Luteolin[65], Apigenin[66] | Cereals, parsley, thyme, celery and citrus fruits | C.orbiscinden, Enterococcus avium |
| Tannins | Gallo tannins, Ellagitannins[67] | Raseberries, cranberries, strawberries, walnuts, grapes and pomegranate | Butyrivibrio sps |
| Lignins | Secoisolariciesinol, metaresinol, pinoresinol, larciresinol, isolarciresinol, syringiresinol[68] | Flax seeds, cereals, strawberries, and apricots | Species of Bacteroides, Clostridium, Peptostreptococcus and Eubacterium |
| Chlorogenic acids | Caffeic acid, feruic acid[69] | Peach, plums and coffee | E. coli, Bifidobacterium sps and L. gasseri |
Xenobiotic and drug metabolism
The capability of the gut microbiome to metabolize xenobiotics and drugs was first recognized over 40 years back. An increasing body of evidence has now provided sufficient insights on the role of the gut microbiota on xenobiotic metabolism, which could have profound impact on therapy for various diseases in future. Recent studies by Clayton et al[70] have shown that a gut microbial metabolite p-cresol can reduce the capacity of the liver to metabolize acetaminophen due to competitive inhibition of hepatic sulfotransferases. Furthermore, cardiac glycosides like digoxin have been recently shown to up-regulate a cytochrome containing operon in the common organism Eggerthella lenta from the Actinobacteria phyla, which results in inactivation of digoxin[71]. Another interesting example of microbiome induced drug metabolism is the microbial β-glucoronidase induced deconjugation of the anticancer drug irinotecan that can contribute to its toxicities such as diarrhea, inflammation and anorexia[72].
Antimicrobial protection
The requirement of a healthy gut microbiota for normal homeostasis puts the gut mucosal immune system in a challenging situation in that it needs to be tolerant to the beneficial commensals and yet prevent overgrowth of the resident pathogens. One of the simplest mechanisms of antimicrobial protection is the presence of the two-tiered mucus layer, which keeps luminal microbes away from epithelial contact, predominantly in the large intestine. Mucus is constituted of a variety of mucin glycoproteins that are secreted by the intestinal goblet cells and extend up to 150 μm away from the colonic epithelium[73,74]. The inner layer is denser and does not contain any organism, while the outer layer is more dynamic and provides glycans as a source of nutrition for the organisms[75]. Other than the mucin glycoproteins, the goblet cells also produce factors like trefoil-factor and the resistin-like molecule-β that can stabilize mucin polymers and thereby maintain barrier integrity[76,77].
Contrary to the large intestine where the mucus plays an important role, antimicrobial proteins play a larger role in the small intestine since the mucus layer here is discontinuous and inadequate. The gut microbiota, via its structural components and metabolites, has been shown to induce synthesis of antimicrobial proteins (AMP) such as cathelicidins, C-type lectins, and (pro)defensins by the host Paneth cells via a pattern recognition receptor (PRR) mediated mechanism[78,79]. The PRR family includes the membrane associated TLRs, C-type lectin receptors (CLRs) such as Dectin-1, and the cytosolic nucleotide-binding and oligomerisation domains (NOD) like receptors (NLRs)[80]. The PRRs in turn are activated by organism specific microbe-associate molecular patterns (MAMPs), which includes various microbial components such as peptidoglycan, LPS, lipid A, flagella and bacterial RNA/DNA, fungal cell wall β-glucans[80,81]. PRR-MAMP (pattern recognition receptor- Microbe Associated Molecular Patterns) cross-talk results in activation of several signaling pathways that are essential for promoting mucosal barrier function, and production of AMPs, mucin glycoproteins and IgA. Since the Paneth cells reside in the base of the small intestinal crypts, concentration of the AMPs are maximal at this location. Even though the composite healthy microbiota appears to be a prerequisite for AMP production, Bacteroides thetaiotaomicron and Lactobacillus innocua appear to be among the key individual species that drive this production[82,83]. The organism Bacteroides thetaiotaomicron has also been shown to induce expression of the matrix metalloproteinase matrilysin from the Paneth cells, which subsequently cleaves prodefensin to form active defensin[84]. Another example of microbiota-host interaction in providing antimicrobial protection is the capability of Lactobacillus sp. to produce lactic acid, which can augment the antimicrobial activity of host lysozyme by disrupting the outer membrane of the bacterial cell wall[85]. Besides this two-way interactive mechanism of AMP expression, bacterial metabolic products such as SCFAs and lithocholic acid have also been shown to induce the expression of cathelicidin by mechanisms involving histone deacetylation and MEK/ERK (Mitogen activated protein kinase/Extracellular signal regulated kinases) pathway[86-88]. The AMPs primarily act by disrupting the surface structures of both commensals and pathogens.
The other mechanism that the gut microbiota has evolved is to keep a check on the overgrowth of pathogenic strains by inducing local immunoglobulins. The gut microbiota, especially Gram-negative organisms like Bacteroides are shown to activate intestinal dendritic cells (DCs), which induces plasma cells in the intestinal mucosa to express secretory IgA (sIgA)[89]. The sIgA can in turn coat the gut microbiota. The sIgA that coats the microbiota are predominantly of sIgA2 subclass, which is more resistant to degradation by bacterial proteases. Furthermore, the intestinal epithelial cells (IECs) can produce a proliferation-inducing ligand (APRIL) in a TLR-mediated bacterial sensing mechanism that can induce class switching from a systemic sIgA1 phenotype to the intestinal mucosal sIgA2[90]. These mechanisms restrict the translocation of the microbiota from the intestinal lumen to the circulation, thereby preventing a systemic immune response.
Immunomodulation
The gut microbiota contribute to gut immunomodulation in tandem with both the innate and adaptive immune systems. The components and the cell types from the immune system that participate in the immunomodulatory process includes the gut associated lymphoid tissues (GALT), effector and regulatory T cells, IgA producing B (plasma) cells, Group 3 innate lymphoid cells, and, resident macrophages and dendritic cells in the lamina propria (Figure 3).
Figure 3.
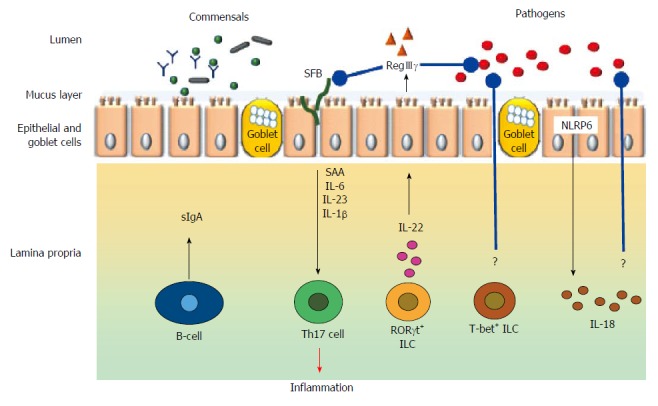
Broad schematic representation of cell types and mediators involved in immunomodulation in the gut. Black arrow indicate either physiological secretion or activation; Red arrow indicates pathological event; Blue arrows with rounded ends indicates pathogen inhibition; ? indicates unknown mechanisms; SFB indicates short filamentous bacteria.
The role of gut microbiota in shaping a normal GALT is implied by the impaired development of the Peyer’s patches and isolated lymphoid follicles that are marked by the abundance of IgE+ B cells instead of the normally seen IgA+ B cells[91]. The effector T cell responses in the intestine have also been shown to be primarily controlled by Th2 responses as opposed to the Th1 responses[92]. The latter is primarily mediated by Th1 and Th17 cells under a physiological milieu; and gut commensals are believed to result in TLR-MyD88 signaling mediated activation of IL1β which in turn promote development of IL17[93].
Intestinal microbiota is also essential for the normal development and function of Foxp3+ T regulatory (Treg) cells. However, the mechanism by which this is mediated is still not clear. For example, in the case of certain Clostridium clusters it could be either independent of PRRs or dependent on My-D88 dependent mechanisms[94]. In the case of Bacillus fragilis, induction of Tregs appear to be mediated by TLR2 signaling by polysaccharide A[95]. SCFAs, especially butyrate have also been implicated in the development and function of Tregs. SCFAs are shown to activate G-protein coupled receptors expressed by the IECs and regulate Treg by epigenetic regulation (increased acetylation) of the Foxp3 locus[96-98].
As mentioned in the previous section, mucosal plasma cells produce secretory IgA upon induction by DCs. Though the mechanisms are not clear, it is speculated that this function is mediated by My-D88 signaling in lamina propria and follicular DCs. My-D88 signaling can be activated by the gut microbiota. Furthermore, in addition to class switching of sIgA by APRIL mediated stimulation, the gut microbiota also stimulate DCs in the Peyer’s patches to secrete TGF-β, CXCL13, and B-cell activating protein (BAFF), which leads to IgA production and class switching[99].
Another set of innate immune cells, namely the innate lymphoid cells (ILCs) are capable of responding rapidly to epithelium-derived cytokine signals[100]. ILCs arise from common lymphoid precursors and have a cytokine expression pattern that is similar to that of T helper subsets (particularly Th17 cells); but the differentiation is more dependent on microbial composition rather than somatic recombination[101]. Based on the functional properties, ILCs can be divided into three groups, namely, group 1 [T box expressed in T cells (T-bet)+], Group 2 [Gata binding protein 3 (GATA-3)+], and group 3 [retinoid-related orphan receptor gamma t (RORγt)+]. Of these, RORγt+ ILCs appear to be most closely associated with regulation of gut immunity[102]. Even though the precise mechanisms are unclear, it is speculated that gut microbes could regulate ILCs both directly and indirectly. Evidence in favor of the former is provided by the observation that the bacterial metabolite indole-3-aldehyde stimulates ILC via the aryl hydrocarbon receptor to induce synthesis of IL22[103]. Indirect mechanism of ILC regulation, on the other hand, is via the recruitment of other immune cells such as the CX3CR1+ intestinal macrophages[104].
The immunomodulatory action of resident macrophages in the lamina propria is to express pro-IL1β in the steady state, which aids in the rapid production of mature IL1β in response to pathogen invasion. MyD-88 dependent mechanisms induced by commensal flora is essential for this action; while the microbiota regulated IL-10 production by the macrophages entail MyD-88 independent mechanisms[105,106].
Apart from the gut microbiota, other factors also play a role in modulation of the gut immune system. For example, the IECs secrete an isoform of alkaline phosphatase (intestinal alkaline phosphatase) that dephosphorylates the LPS endotoxin[107]. Another example is the reduced neutrophil recruitment into the intestinal lumen in response to tumour necrosis factor-α (TNF-α). This action is mediated by the intestinal alkaline phosphatase[107]. Furthermore, an immunoprotective mechanism that is acquired at birth and is seen predominantly with vaginal delivery is the down regulation of IL-1 receptor-associated kinase (IRAK-1), which acts through TLR4[108].
Integrity of the gut barrier and structure of the gastrointestinal tract
Currently there is a convincing body of evidence that supports the role of the gut microbiota in maintaining the structure and function of the gastrointestinal tract. Bacteroides thetaiotaomicron is reported to induce expression of the small proline-rich protein 2A (sprr2A), which is required for maintenance of desmosomes at the epithelial villus[109]. Another mechanism that maintains the tight junctions is by TLR2 mediated signaling that is stimulated by the microbial cell wall peptidoglycan[110]. Furthermore, the Lactobacillus rhamnosus GG strain produces two soluble proteins namely p40 and p75 that can prevent cytokine induced apoptosis of the intestinal epithelial cells in an epithelial growth factor receptor (EGFR) and protein kinase C (PKC) pathway dependent manner[111]. The endocannabinoid system is yet another entity that regulates gut microbiota mediated maintenance of the gut barrier function. E.g., the Gram negative bacteria Akkermansia muciniphilia can increase the levels of endocannabinoids that control gut barrier functions by decreasing metabolic endotoxemia[112].
The gut microbiota contributes to structural development of the gut mucosa by inducing the transcription factor angiogenin-3, which has been implicated in the development of intestinal microvasculature[113]. This is also supported by a significant reduction of villus capillary network in germ-free (GF) mice, which in turn can impair nutrient digestion and absorption. Other evidence that support role of gut microbiota in maintaining structure and function is obtained from GF mice that have a lower intestinal surface area[114], thin villi (secondary to lower regeneration)[115], increase cell cycle time[116] and impaired peristalsis[117]. The gut microbiota can also modulate mucosal glycosylation patterns that are microbial attachment sites both at the cell surface and subcellular levels. For example, a signaling molecule secreted by the organism Bacteroides thetaiotaomicron can stimulate expression of the carbohydrate moiety fucose on the cell surface glycoconjugates[118].
FACTORS AFFECTING VARIATIONS IN THE NORMAL GUT MICROBIOTA
Several factors contribute to the shaping of the healthy gut microbiota; and this continues dynamically all throughout the life of an individual.
Age
Even though it is widely believed that the gut gets colonized by microbes immediately after birth, there is emerging evidence that the infant gut could be colonized by organisms even _in utero_[119]. 16S rRNA based sequencing studies have revealed that the first meconium is rich in genera such as Escherichia-Shigella, Enterococcus, Leuconostoc, Lactococcus, and _Streptococcus_[120]. Nevertheless, it is now clear that the first microbiota profile is largely shaped by the mode of delivery. The intestines of infants born vaginally are initially colonized by organisms from the maternal vagina, which is best exemplified by the organisms from the genera Lactobacillus and _Prevotella_[121]. On the contrary, in cesarean delivery mostly the maternal skin flora colonizes the infant’s intestine, as exemplified by the dominance of Streptococcus, Corynebacterium, and _Propionibacterium_[119,121]. The initial milieu of the infant’s gut microbiota after primary inoculation appears unstable and devoid of diversity; but with time it stabilizes, diversifies, and acquires 40%-60% similarity with the adult microbiota by the age of 3 years[122]. On the contrary, studies have also shown that young children and adolescents could demonstrate significant differences in proportions of Bacteroides and Bifidobacterium compared to adults[123,124]. The gut microbiota by and large rest in a stable state from the 3rd to the 7th decade of life, even though proportions of Bifidobacteria, Firmicutes, and Fecalibacterium prausnitzii tend to decrease with an increase in E. coli, Proteobacteria and _Staphylococcus_[125-127]. Few of the functional impacts of the temporal alteration in the normal gut flora include a reduced capability to synthesize vitamin B12, reduced activities of microbial reductases, increased tendency for DNA alterations, elevated stress response, and immune dysfunction[128]. Although the initially developing microbiota is largely influenced by the type of feed (breast milk or formula feeds) after primary inoculation, the temporal alteration is affected by dietary patterns, lifestyle, life events, and environmental factors including antibiotic use[1].
In pre-term infants, bacteria that colonize the gut include Bifidobacterium and Lactobacillus and basically, these differ depending on the type of feeding habits. In formula-fed infants, Enterococcus, Enterobacteria, Bacteroides, Clostridia, and other anaerobic Streptococcus dominates the gut niche; whereas, in breast-fed infants Bifidobacterium and Lactobacillus dominates. Breast milk contains indigestible glycans termed as human milk oligosaccharides (HMO) which are easily broken down by these bacteria. Pre-term microbiota are said to maintain the gut associated lymphoid tissue (GALT), and is involved in generating the innate immunity during development. Therefore, abnormal colonization of the gut microbiota may result in pediatric diseases because of poor immunity[129,130].
Diet
The earliest effect on the gut microbiota, after the mode of delivery, is the early infant diet, i.e., breast milk and formula feeds. Several studies have shown substantial differences in the gut microbial composition between breast-fed and formula-fed infants. It is important to understand the effect of breast milk and formula feeds on the gut microbiota since there has been an increasing trend of moving away from breast-feeding by modern day mothers. Besides meeting the nutritional and physiological demands of the infant, breast milk also contains several bioactive compounds that are not available in formula-feeds. These compounds have a significant role in nutrient digestion and absorption, immune protection and anti-microbial defense[131,132]. HMOs provide nutrition to the colonic bacteria of the infant, thereby providing a selective growth advantage for Bifidobacterium sp.[133]. This has been observed at a significantly higher abundance in breast-fed infants compared to that in formula-fed infants. These organisms ferment dietary oligosaccharides resulting in health promoting SCFAs such as butyrate, and modulate the host immune system to express IgG[134]. Studies have shown that several strains of Bifidobacterium, especially the Bifidobacterium longus subs infantis contain unique gene cluster (HMO-related gene cluster 1) that codes for difference glycosidases (sialidase, fucosidase, hexosaminidase and galactosidase) and carbohydrate transporters that are capable of importing and metabolizing HMOs[134]. On the contrary, the abundance of anaerobic organisms like Bacteroides sp. and Clostridium sp. is lower in breast-fed infants as compared to formula-fed ones[135-137]. Even though Bacteroides sp. can also digest HMO, the abundance of Bifidobacterium is higher in breast-fed infants, thus pointing towards competitive relationship between these two organisms in favor of Bifidobacterium in breast-fed infants.
Diet continues to be the most important determinant in shaping the composition, diversity and richness even throughout adulthood. In general, intake of diet rich in fruits, vegetables and fibers is associated with a higher richness and diversity of the gut microbiota. Individuals consuming this kind of a diet have a higher abundance of the insoluble carbohydrate metabolizing organisms of the Firmicutes phylum such as Ruminococcus bromii, Roseburia and _Eubacterium rectale_[138]. It was recently shown that a 4-d administration of animal-based diet resulted in a decrease in the abundance of Firmicutes; and an increase in that of bile-tolerant organisms such as Alistipes sp. and Bacteroides sp. from the phylum Bacteroidetes and Bilophila sp. from the phylum Proteobacteria. This indicates that even very short dietary manipulations can have substantial impact on the gut microbiota[139].
Several studies have shown that there are significant geographic and seasonal variations in the gut microbiome. However, these differences were also associated with a difference in dietary patterns. For example, it was demonstrated that rural African children had a higher abundance of Prevotella, while children from Europe had higher proportions of _Bacteroides_[140]. Even though Prevotella and Bacteroides are taxonomically and functionally similar, higher abundance of Prevotella indicates an agrarian diet that was consumed by the African children. On the contrary, the children from Europe consumed a western diet rich in animal protein, sugar, starch and poor in fibers, which is marked by the higher abundance of Bacteroides. Furthermore, it was also shown that the relative abundance of the phylum Actinobacteria was significantly higher in Hutterites during winter season compared to that during summers. This could be ascribed to the higher intake of meat-based diet in winter when compared to the fresh, carbohydrate and fiber rich diet that was consumed during summer[141].
Dietary polyphenols, besides their systemic antimicrobial and metabolic functions, also play a role in the inhibition of gut bacteria. While the polyphenolic compound querectin is degraded by Bacteroides distasonis, Bacteroides uniformis, Bacteroides ovatus, Enterococcus casseliflavus, and _Eubacterium ramulus_are the compound that degrade this flavanol, hesperetine (a rutinoside containing aglycon), is poorly degraded by the colonic microbiota. This aglycon has an inhibitory activity against vancomycin- intermediate Staphylococcus aureus and _H. pylori_[142].
Seaweeds are active resources with bioactive compounds with various biological activities such as antibacterial, anti-oxidant, anti-inflammatory, anti-coagulant, anti-viral and apoptotic activity. They are rich source of fiber with nearly 50%-60% of water soluble fibers, and are also rich in sulfated polysaccharides such as porphyrans, and agarases. Few species of red sea weeds like Palmaria decipiens and Pterocladiella capillacea contains sulfated polysaccharides and uronic acids (i.e., xylans and xylogalactans) respectively[143]. Several human and rat studies have demonstrated a significant shift in the gut microbiota upon the use of seaweeds as a food supplement. In humans, supplementation of Gelidium seaweed has significantly increased the expression of Bifidobacterium genera, without any change in the others. There was also an increase in the production of the SCFA’s[144]. Another study conducted on Japanese populations explained the transfer of porphyranases and agarases to the gut bacteria Bacteroides plebius through carbohydrate active enzymes (CAZymes)[145]. These studies points towards the feasibility the use of sea weeds as a potential prebiotic.
Antibiotics
Even though study on antibiotics in general have centered around their bactericidal and bacteriostatic activities against pathogens, recent years have seen several studies on their effect on gut bacterial ecology in a holistic manner. A strong body of evidence has now clearly demonstrated that use of antibiotics does have several short and long-term implications in the ecology of the normal gut microbiota. It has been shown that multi-drug resistant bacterial genes have been prevalent for thousands of years before the advent of antibiotics, indicating an influence of exposure to small molecules from the environment with growth inhibitory properties[146]. This could also be secondary to a dysbiotic commensal microbiota that could further augment the development of resistance genes[147]. This culminates in uncoupling of the mutualistic relationship between the healthy gut microbiota and the host intestinal milieu.
One of the major properties of the healthy gut microbiota against pathogen is the capability to cause competitive exclusion[148]. It was demonstrated around four decades ago that antibiotics could result in disruption of the competitive exclusion machinery that resulted in Salmonella infection immediately after antibiotic therapy. One of the possible mechanisms of this kind of event could be a loss of the wide network of interspecies interactions within the microbiota that increase the abundance of host-derived sialic acid which is growth promoting for pathogens such as Salmonella typhimurium and _Clostridium difficile_[149]. Major changes in the gut microbiota in response to antibiotics include diminished taxonomic diversity and persistence of the changes in a substantial proportion of individuals. It has been shown that the effect of even short-term use (7 d) of broad-spectrum antibiotics with predominant anaerobic coverage (e.g., Clindamycin) could last up to 2 year, with a persistent non-recovery of the diversity of _Bacteroides_[150]. Similarly, a short course H. pylori eradication with clarithromycin containing triple therapy resulted in a dramatic reduction in the diversity of Actinobacteria with a thousand-fold increase in the ermB resistance gene[151]. This persisted for over 4-years in a proportion of these patients, while it recovered in the others. The effect of ciprofloxacin, which has predominantly Gram-positive coverage, is relatively short-lived with abrupt reduction of Ruminococcus sps.[152]. Another recent study that evaluated the role of short course (7 d) of ciprofloxacin and beta-lactams indicated the reduction of microbial diversity by 25% and the core taxa from 29 to 12 with an increase in the Bacteroidetes: Firmicutes ratio[153]. The major concern that stems out of use of broad-spectrum antibiotics, besides alteration of the normal gut microbial diversity, is the phenomenon of propagating the resistance strain via horizontal gene transfer[154,155]. Bacterial species are capable of transferring mutant genetic information across different species through mechanisms such as conjugation, phage transduction and natural transformation. The gene transfer could also be via transposons and integrin. Interestingly, it has been shown that among different environments, the human gut associated microbiota has 25 times more likelihood of having horizontal gene transfer[156]. This would result in development of a reservoir state of resistance genes, and therefore mandates extreme care in the use of broad spectrum antibiotics.
PROBIOTICS, PREBIOTICS AND SYNBIOTICS
The World Health Organization defines probiotics as live microorganisms that can provide benefits to human health when administered in adequate amounts. Several species such as Lactobacillus casei, Lactobacillus planatarum, Lactobacillus bulgaricus, Lactobacillus acidophilus, Bifidobacterium longum, Bifidobacterium infantis, Streptococcus thermophilus, E. coli strain Nissle 1917, to name a few have been shown to impart immunomodulatory and gut barrier functions. These and several others have been used commercially in the management of human illnesses e.g., IBD and antibiotic associated diarrhea. The fundamental concept of using these organisms in the treatment armamentarium is mimicking the physiological health promoting functions of the “good” bacteria. Addition of a prebiotic could possibly augment the effect of the probiotics. Prebiotics are defined as food ingredients that contain non-digestible oligosaccharides (e.g., galactooligosaccharides and inulin); and a probiotic and prebiotic are together called a synbiotic. The gut bacteria selectively ferment these fibers resulting in the synthesis of SCFAs, which in turn imparts the pro-health effects (vide supra). A detailed discussion on pro- and prebiotics is out of the scope of this review since it deals predominantly on a normal gut microbiota. Nevertheless, we believe that even though dietary fibers and healthy gut microbiota are known to promote health, use of synbiotics for maintenance of health needs to be studied with much robustness before using them commercially as health promoters[157,158].
Footnotes
Conflict-of-interest statement: The authors declare no conflicts of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: November 17, 2014
First decision: March 26, 2015
Article in press: July 3, 2015
P- Reviewer: Berrington JE, Duda-Chodak A, Duryee MJ, Lagier JC S- Editor: Ma YJ L- Editor: A E- Editor: Liu XM
References
- 1.Sekirov I, Russell SL, Antunes LC, Finlay BB. Gut microbiota in health and disease. Physiol Rev. 2010;90:859–904. doi: 10.1152/physrev.00045.2009. [DOI] [PubMed] [Google Scholar]
- 2.Ferreira CM, Vieira AT, Vinolo MA, Oliveira FA, Curi R, Martins Fdos S. The central role of the gut microbiota in chronic inflammatory diseases. J Immunol Res. 2014;2014:689492. doi: 10.1155/2014/689492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kennedy PJ, Cryan JF, Dinan TG, Clarke G. Irritable bowel syndrome: a microbiome-gut-brain axis disorder? World J Gastroenterol. 2014;20:14105–14125. doi: 10.3748/wjg.v20.i39.14105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karlsson F, Tremaroli V, Nielsen J, Bäckhed F. Assessing the human gut microbiota in metabolic diseases. Diabetes. 2013;62:3341–3349. doi: 10.2337/db13-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bisgaard H, Li N, Bonnelykke K, Chawes BL, Skov T, Paludan-Müller G, Stokholm J, Smith B, Krogfelt KA. Reduced diversity of the intestinal microbiota during infancy is associated with increased risk of allergic disease at school age. J Allergy Clin Immunol. 2011;128:646–652.e1-5. doi: 10.1016/j.jaci.2011.04.060. [DOI] [PubMed] [Google Scholar]
- 6.Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lagier JC, Hugon P, Khelaifia S, Fournier PE, La Scola B, Raoult D. The rebirth of culture in microbiology through the example of culturomics to study human gut microbiota. Clin Microbiol Rev. 2015;28:237–264. doi: 10.1128/CMR.00014-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lagier JC, Armougom F, Million M, Hugon P, Pagnier I, Robert C, Bittar F, Fournous G, Gimenez G, Maraninchi M, et al. Microbial culturomics: paradigm shift in the human gut microbiome study. Clin Microbiol Infect. 2012;18:1185–1193. doi: 10.1111/1469-0691.12023. [DOI] [PubMed] [Google Scholar]
- 10.Rajilić-Stojanović M, de Vos WM. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol Rev. 2014;38:996–1047. doi: 10.1111/1574-6976.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peterson DA, Frank DN, Pace NR, Gordon JI. Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host Microbe. 2008;3:417–427. doi: 10.1016/j.chom.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R. Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat Methods. 2008;5:235–237. doi: 10.1038/nmeth.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schirmer M, Ijaz UZ, D’Amore R, Hall N, Sloan WT, Quince C. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res. 2015;43:e37. doi: 10.1093/nar/gku1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim M, Yu Z. Variations in 16S rRNA-based microbiome profiling between pyrosequencing runs and between pyrosequencing facilities. J Microbiol. 2014;52:355–365. doi: 10.1007/s12275-014-3443-3. [DOI] [PubMed] [Google Scholar]
- 15.Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, Bertoni A, Swerdlow HP, Gu Y. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics. 2012;13:341. doi: 10.1186/1471-2164-13-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu L, Li Y, Li S, Hu N, He Y, Pong R, Lin D, Lu L, Law M. Comparison of next-generation sequencing systems. J Biomed Biotechnol. 2012;2012:251364. doi: 10.1155/2012/251364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramakrishna B, Krishnan S. The normal bacterial flora of the human intestine and its regulation. J Clin Gastroenterol. 2007;41:S2–S6. [Google Scholar]
- 18.Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto JM, Kennedy S, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500:541–546. doi: 10.1038/nature12506. [DOI] [PubMed] [Google Scholar]
- 20.Swidsinski A, Weber J, Loening-Baucke V, Hale LP, Lochs H. Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J Clin Microbiol. 2005;43:3380–3389. doi: 10.1128/JCM.43.7.3380-3389.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joossens M, Huys G, Cnockaert M, De Preter V, Verbeke K, Rutgeerts P, Vandamme P, Vermeire S. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut. 2011;60:631–637. doi: 10.1136/gut.2010.223263. [DOI] [PubMed] [Google Scholar]
- 22.O’Hara AM, Shanahan F. The gut flora as a forgotten organ. EMBO Rep. 2006;7:688–693. doi: 10.1038/sj.embor.7400731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pei Z, Bini EJ, Yang L, Zhou M, Francois F, Blaser MJ. Bacterial biota in the human distal esophagus. Proc Natl Acad Sci USA. 2004;101:4250–4255. doi: 10.1073/pnas.0306398101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Justesen T, Nielsen OH, Jacobsen IE, Lave J, Rasmussen SN. The normal cultivable microflora in upper jejunal fluid in healthy adults. Scand J Gastroenterol. 1984;19:279–282. [PubMed] [Google Scholar]
- 25.Blaser MJ. Hypothesis: the changing relationships of Helicobacter pylori and humans: implications for health and disease. J Infect Dis. 1999;179:1523–1530. doi: 10.1086/314785. [DOI] [PubMed] [Google Scholar]
- 26.Andersson AF, Lindberg M, Jakobsson H, Bäckhed F, Nyrén P, Engstrand L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS One. 2008;3:e2836. doi: 10.1371/journal.pone.0002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 28.Gillespie JJ, Wattam AR, Cammer SA, Gabbard JL, Shukla MP, Dalay O, Driscoll T, Hix D, Mane SP, Mao C, et al. PATRIC: the comprehensive bacterial bioinformatics resource with a focus on human pathogenic species. Infect Immun. 2011;79:4286–4298. doi: 10.1128/IAI.00207-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hollister EB, Gao C, Versalovic J. Compositional and functional features of the gastrointestinal microbiome and their effects on human health. Gastroenterology. 2014;146:1449–1458. doi: 10.1053/j.gastro.2014.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swidsinski A, Loening-Baucke V, Lochs H, Hale LP. Spatial organization of bacterial flora in normal and inflamed intestine: a fluorescence in situ hybridization study in mice. World J Gastroenterol. 2005;11:1131–1140. doi: 10.3748/wjg.v11.i8.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, et al. Enterotypes of the human gut microbiome. Nature. 2011;473:174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karlsson FH, Fåk F, Nookaew I, Tremaroli V, Fagerberg B, Petranovic D, Bäckhed F, Nielsen J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat Commun. 2012;3:1245. doi: 10.1038/ncomms2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bäckhed F, Fraser CM, Ringel Y, Sanders ME, Sartor RB, Sherman PM, Versalovic J, Young V, Finlay BB. Defining a healthy human gut microbiome: current concepts, future directions, and clinical applications. Cell Host Microbe. 2012;12:611–622. doi: 10.1016/j.chom.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 34.Sonnenburg JL, Xu J, Leip DD, Chen CH, Westover BP, Weatherford J, Buhler JD, Gordon JI. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science. 2005;307:1955–1959. doi: 10.1126/science.1109051. [DOI] [PubMed] [Google Scholar]
- 35.Macfarlane S, Macfarlane GT. Regulation of short-chain fatty acid production. Proc Nutr Soc. 2003;62:67–72. doi: 10.1079/PNS2002207. [DOI] [PubMed] [Google Scholar]
- 36.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–594. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 37.Samuel BS, Shaito A, Motoike T, Rey FE, Backhed F, Manchester JK, Hammer RE, Williams SC, Crowley J, Yanagisawa M, et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc Natl Acad Sci USA. 2008;105:16767–16772. doi: 10.1073/pnas.0808567105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bourriaud C, Akoka S, Goupry S, Robins R, Cherbut C, Michel C. Butyrate production from lactate by human colonic microflora. Reprod Nutr Dev. 2002;42:S55. [Google Scholar]
- 39.Cantarel BL, Lombard V, Henrissat B. Complex carbohydrate utilization by the healthy human microbiome. PLoS One. 2012;7:e28742. doi: 10.1371/journal.pone.0028742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sidhu H, Hoppe B, Hesse A, Tenbrock K, Brömme S, Rietschel E, Peck AB. Absence of Oxalobacter formigenes in cystic fibrosis patients: a risk factor for hyperoxaluria. Lancet. 1998;352:1026–1029. doi: 10.1016/S0140-6736(98)03038-4. [DOI] [PubMed] [Google Scholar]
- 41.Magwira CA, Kullin B, Lewandowski S, Rodgers A, Reid SJ, Abratt VR. Diversity of faecal oxalate-degrading bacteria in black and white South African study groups: insights into understanding the rarity of urolithiasis in the black group. J Appl Microbiol. 2012;113:418–428. doi: 10.1111/j.1365-2672.2012.05346.x. [DOI] [PubMed] [Google Scholar]
- 42.Hooper LV, Wong MH, Thelin A, Hansson L, Falk PG, Gordon JI. Molecular analysis of commensal host-microbial relationships in the intestine. Science. 2001;291:881–884. doi: 10.1126/science.291.5505.881. [DOI] [PubMed] [Google Scholar]
- 43.Thomas CM, Hong T, van Pijkeren JP, Hemarajata P, Trinh DV, Hu W, Britton RA, Kalkum M, Versalovic J. Histamine derived from probiotic Lactobacillus reuteri suppresses TNF via modulation of PKA and ERK signaling. PLoS One. 2012;7:e31951. doi: 10.1371/journal.pone.0031951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Biase D, Pennacchietti E. Glutamate decarboxylase-dependent acid resistance in orally acquired bacteria: function, distribution and biomedical implications of the gadBC operon. Mol Microbiol. 2012;86:770–786. doi: 10.1111/mmi.12020. [DOI] [PubMed] [Google Scholar]
- 45.Baddini Feitoza A, Fernandes Pereira A, Ferreira da Costa N, Gonçalves Ribeiro B. Conjugated linoleic acid (CLA): effect modulation of body composition and lipid profile. Nutr Hosp. 2009;24:422–428. [PubMed] [Google Scholar]
- 46.Devillard E, McIntosh FM, Duncan SH, Wallace RJ. Metabolism of linoleic acid by human gut bacteria: different routes for biosynthesis of conjugated linoleic acid. J Bacteriol. 2007;189:2566–2570. doi: 10.1128/JB.01359-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Devillard E, McIntosh FM, Paillard D, Thomas NA, Shingfield KJ, Wallace RJ. Differences between human subjects in the composition of the faecal bacterial community and faecal metabolism of linoleic acid. Microbiology. 2009;155:513–520. doi: 10.1099/mic.0.023416-0. [DOI] [PubMed] [Google Scholar]
- 48.Fukiya S, Arata M, Kawashima H, Yoshida D, Kaneko M, Minamida K, Watanabe J, Ogura Y, Uchida K, Itoh K, et al. Conversion of cholic acid and chenodeoxycholic acid into their 7-oxo derivatives by Bacteroides intestinalis AM-1 isolated from human feces. FEMS Microbiol Lett. 2009;293:263–270. doi: 10.1111/j.1574-6968.2009.01531.x. [DOI] [PubMed] [Google Scholar]
- 49.Velagapudi VR, Hezaveh R, Reigstad CS, Gopalacharyulu P, Yetukuri L, Islam S, Felin J, Perkins R, Borén J, Oresic M, et al. The gut microbiota modulates host energy and lipid metabolism in mice. J Lipid Res. 2010;51:1101–1112. doi: 10.1194/jlr.M002774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marín L, Miguélez EM, Villar CJ, Lombó F. Bioavailability of dietary polyphenols and gut microbiota metabolism: antimicrobial properties. Biomed Res Int. 2015;2015:905215. doi: 10.1155/2015/905215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Winter J, Moore LH, Dowell VR, Bokkenheuser VD. C-ring cleavage of flavonoids by human intestinal bacteria. Appl Environ Microbiol. 1989;55:1203–1208. doi: 10.1128/aem.55.5.1203-1208.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rechner AR, Smith MA, Kuhnle G, Gibson GR, Debnam ES, Srai SK, Moore KP, Rice-Evans CA. Colonic metabolism of dietary polyphenols: influence of structure on microbial fermentation products. Free Radic Biol Med. 2004;36:212–225. doi: 10.1016/j.freeradbiomed.2003.09.022. [DOI] [PubMed] [Google Scholar]
- 53.Schneider H, Simmering R, Hartmann L, Pforte H, Blaut M. Degradation of quercetin-3-glucoside in gnotobiotic rats associated with human intestinal bacteria. J Appl Microbiol. 2000;89:1027–1037. doi: 10.1046/j.1365-2672.2000.01209.x. [DOI] [PubMed] [Google Scholar]
- 54.Manach C, Williamson G, Morand C, Scalbert A, Rémésy C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am J Clin Nutr. 2005;81:230S–242S. doi: 10.1093/ajcn/81.1.230S. [DOI] [PubMed] [Google Scholar]
- 55.Monagas M, Urpi-Sarda M, Sánchez-Patán F, Llorach R, Garrido I, Gómez-Cordovés C, Andres-Lacueva C, Bartolomé B. Insights into the metabolism and microbial biotransformation of dietary flavan-3-ols and the bioactivity of their metabolites. Food Funct. 2010;1:233–253. doi: 10.1039/c0fo00132e. [DOI] [PubMed] [Google Scholar]
- 56.Tzounis X, Vulevic J, Kuhnle GG, George T, Leonczak J, Gibson GR, Kwik-Uribe C, Spencer JP. Flavanol monomer-induced changes to the human faecal microflora. Br J Nutr. 2008;99:782–792. doi: 10.1017/S0007114507853384. [DOI] [PubMed] [Google Scholar]
- 57.Schneider H, Blaut M. Anaerobic degradation of flavonoids by Eubacterium ramulus. Arch Microbiol. 2000;173:71–75. doi: 10.1007/s002030050010. [DOI] [PubMed] [Google Scholar]
- 58.Yamakoshi J, Tokutake S, Kikuchi M, Kubota Y, Konishi H, Mitsuoka T. Effect of proanthocyanidin-rich extract from grape seeds on human fecal flora and fecal odor. Microb Ecol Health Dis. 2001;13:25–31. [Google Scholar]
- 59.Miladinović B, Kostić M, Šavikin K, Đorđević B, Mihajilov-Krstev T, Živanović S, Kitić D. Chemical profile and antioxidative and antimicrobial activity of juices and extracts of 4 black currants varieties (Ribes nigrum L.) J Food Sci. 2014;79:C301–C309. doi: 10.1111/1750-3841.12364. [DOI] [PubMed] [Google Scholar]
- 60.Burdulis D, Sarkinas A, Jasutiené I, Stackevicené E, Nikolajevas L, Janulis V. Comparative study of anthocyanin composition, antimicrobial and antioxidant activity in bilberry (Vaccinium myrtillus L.) and blueberry (Vaccinium corymbosum L.) fruits. Acta Pol Pharm. 2009;66:399–408. [PubMed] [Google Scholar]
- 61.Minamida K, Tanaka M, Abe A, Sone T, Tomita F, Hara H, Asano K. Production of equol from daidzein by gram-positive rod-shaped bacterium isolated from rat intestine. J Biosci Bioeng. 2006;102:247–250. doi: 10.1263/jbb.102.247. [DOI] [PubMed] [Google Scholar]
- 62.Wiseman H, Casey K, Bowey EA, Duffy R, Davies M, Rowland IR, Lloyd AS, Murray A, Thompson R, Clarke DB. Influence of 10 wk of soy consumption on plasma concentrations and excretion of isoflavonoids and on gut microflora metabolism in healthy adults. Am J Clin Nutr. 2004;80:692–699. doi: 10.1093/ajcn/80.3.692. [DOI] [PubMed] [Google Scholar]
- 63.Coldham NG, Darby C, Hows M, King LJ, Zhang AQ, Sauer MJ. Comparative metabolism of genistin by human and rat gut microflora: detection and identification of the end-products of metabolism. Xenobiotica. 2002;32:45–62. doi: 10.1080/00498250110085809. [DOI] [PubMed] [Google Scholar]
- 64.Heinonen SM, Wähälä K, Adlercreutz H. Identification of urinary metabolites of the red clover isoflavones formononetin and biochanin A in human subjects. J Agric Food Chem. 2004;52:6802–6809. doi: 10.1021/jf0492767. [DOI] [PubMed] [Google Scholar]
- 65.Hanske L, Loh G, Sczesny S, Blaut M, Braune A. The bioavailability of apigenin-7-glucoside is influenced by human intestinal microbiota in rats. J Nutr. 2009;139:1095–1102. doi: 10.3945/jn.108.102814. [DOI] [PubMed] [Google Scholar]
- 66.Odenyo AA, Bishop R, Asefa G, Jamnadass R, Odongo D, Osuji P. Characterization of tannin-tolerant bacterial isolates from East African ruminants. Anaerobe. 2001;7:1, 5–15. [Google Scholar]
- 67.Heinonen S, Nurmi T, Liukkonen K, Poutanen K, Wähälä K, Deyama T, Nishibe S, Adlercreutz H. In vitro metabolism of plant lignans: new precursors of mammalian lignans enterolactone and enterodiol. J Agric Food Chem. 2001;49:3178–3186. doi: 10.1021/jf010038a. [DOI] [PubMed] [Google Scholar]
- 68.Gonthier MP, Remesy C, Scalbert A, Cheynier V, Souquet JM, Poutanen K, Aura AM. Microbial metabolism of caffeic acid and its esters chlorogenic and caftaric acids by human faecal microbiota in vitro. Biomed Pharmacother. 2006;60:536–540. doi: 10.1016/j.biopha.2006.07.084. [DOI] [PubMed] [Google Scholar]
- 69.Couteau D, McCartney AL, Gibson GR, Williamson G, Faulds CB. Isolation and characterization of human colonic bacteria able to hydrolyse chlorogenic acid. J Appl Microbiol. 2001;90:873–881. doi: 10.1046/j.1365-2672.2001.01316.x. [DOI] [PubMed] [Google Scholar]
- 70.Clayton TA, Baker D, Lindon JC, Everett JR, Nicholson JK. Pharmacometabonomic identification of a significant host-microbiome metabolic interaction affecting human drug metabolism. Proc Natl Acad Sci USA. 2009;106:14728–14733. doi: 10.1073/pnas.0904489106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saha JR, Butler VP, Neu HC, Lindenbaum J. Digoxin-inactivating bacteria: identification in human gut flora. Science. 1983;220:325–327. doi: 10.1126/science.6836275. [DOI] [PubMed] [Google Scholar]
- 72.Wallace BD, Wang H, Lane KT, Scott JE, Orans J, Koo JS, Venkatesh M, Jobin C, Yeh LA, Mani S, et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science. 2010;330:831–835. doi: 10.1126/science.1191175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci USA. 2008;105:15064–15069. doi: 10.1073/pnas.0803124105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim YS, Ho SB. Intestinal goblet cells and mucins in health and disease: recent insights and progress. Curr Gastroenterol Rep. 2010;12:319–330. doi: 10.1007/s11894-010-0131-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Johansson ME, Larsson JM, Hansson GC. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc Natl Acad Sci USA. 2011;108 Suppl 1:4659–4665. doi: 10.1073/pnas.1006451107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Podolsky DK, Lynch-Devaney K, Stow JL, Oates P, Murgue B, DeBeaumont M, Sands BE, Mahida YR. Identification of human intestinal trefoil factor. Goblet cell-specific expression of a peptide targeted for apical secretion. J Biol Chem. 1993;268:6694–6702. [PubMed] [Google Scholar]
- 77.Artis D, Wang ML, Keilbaugh SA, He W, Brenes M, Swain GP, Knight PA, Donaldson DD, Lazar MA, Miller HR, et al. RELMbeta/FIZZ2 is a goblet cell-specific immune-effector molecule in the gastrointestinal tract. Proc Natl Acad Sci USA. 2004;101:13596–13600. doi: 10.1073/pnas.0404034101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hooper LV. Do symbiotic bacteria subvert host immunity? Nat Rev Microbiol. 2009;7:367–374. doi: 10.1038/nrmicro2114. [DOI] [PubMed] [Google Scholar]
- 79.Salzman NH, Underwood MA, Bevins CL. Paneth cells, defensins, and the commensal microbiota: a hypothesis on intimate interplay at the intestinal mucosa. Semin Immunol. 2007;19:70–83. doi: 10.1016/j.smim.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 80.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 81.Carvalho FA, Aitken JD, Vijay-Kumar M, Gewirtz AT. Toll-like receptor-gut microbiota interactions: perturb at your own risk! Annu Rev Physiol. 2012;74:177–198. doi: 10.1146/annurev-physiol-020911-153330. [DOI] [PubMed] [Google Scholar]
- 82.Cash HL, Whitham CV, Behrendt CL, Hooper LV. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science. 2006;313:1126–1130. doi: 10.1126/science.1127119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hooper LV, Stappenbeck TS, Hong CV, Gordon JI. Angiogenins: a new class of microbicidal proteins involved in innate immunity. Nat Immunol. 2003;4:269–273. doi: 10.1038/ni888. [DOI] [PubMed] [Google Scholar]
- 84.López-Boado YS, Wilson CL, Hooper LV, Gordon JI, Hultgren SJ, Parks WC. Bacterial exposure induces and activates matrilysin in mucosal epithelial cells. J Cell Biol. 2000;148:1305–1315. doi: 10.1083/jcb.148.6.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Alakomi HL, Skyttä E, Saarela M, Mattila-Sandholm T, Latva-Kala K, Helander IM. Lactic acid permeabilizes gram-negative bacteria by disrupting the outer membrane. Appl Environ Microbiol. 2000;66:2001–2005. doi: 10.1128/aem.66.5.2001-2005.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kida Y, Shimizu T, Kuwano K. Sodium butyrate up-regulates cathelicidin gene expression via activator protein-1 and histone acetylation at the promoter region in a human lung epithelial cell line, EBC-1. Mol Immunol. 2006;43:1972–1981. doi: 10.1016/j.molimm.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 87.Schauber J, Svanholm C, Termén S, Iffland K, Menzel T, Scheppach W, Melcher R, Agerberth B, Lührs H, Gudmundsson GH. Expression of the cathelicidin LL-37 is modulated by short chain fatty acids in colonocytes: relevance of signalling pathways. Gut. 2003;52:735–741. doi: 10.1136/gut.52.5.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Termén S, Tollin M, Rodriguez E, Sveinsdóttir SH, Jóhannesson B, Cederlund A, Sjövall J, Agerberth B, Gudmundsson GH. PU.1 and bacterial metabolites regulate the human gene CAMP encoding antimicrobial peptide LL-37 in colon epithelial cells. Mol Immunol. 2008;45:3947–3955. doi: 10.1016/j.molimm.2008.06.020. [DOI] [PubMed] [Google Scholar]
- 89.He B, Xu W, Santini PA, Polydorides AD, Chiu A, Estrella J, Shan M, Chadburn A, Villanacci V, Plebani A, et al. Intestinal bacteria trigger T cell-independent immunoglobulin A(2) class switching by inducing epithelial-cell secretion of the cytokine APRIL. Immunity. 2007;26:812–826. doi: 10.1016/j.immuni.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 90.Macpherson AJ, Uhr T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science. 2004;303:1662–1665. doi: 10.1126/science.1091334. [DOI] [PubMed] [Google Scholar]
- 91.Durkin HG, Bazin H, Waksman BH. Origin and fate of IgE-bearing lymphocytes. I. Peyer’s patches as differentiation site of cells. Simultaneously bearing IgA and IgE. J Exp Med. 1981;154:640–648. doi: 10.1084/jem.154.3.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chung H, Pamp SJ, Hill JA, Surana NK, Edelman SM, Troy EB, Reading NC, Villablanca EJ, Wang S, Mora JR, et al. Gut immune maturation depends on colonization with a host-specific microbiota. Cell. 2012;149:1578–1593. doi: 10.1016/j.cell.2012.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hasegawa M, Osaka T, Tawaratsumida K, Yamazaki T, Tada H, Chen GY, Tsuneda S, Núñez G, Inohara N. Transitions in oral and intestinal microflora composition and innate immune receptor-dependent stimulation during mouse development. Infect Immun. 2010;78:639–650. doi: 10.1128/IAI.01043-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Geuking MB, Cahenzli J, Lawson MA, Ng DC, Slack E, Hapfelmeier S, McCoy KD, Macpherson AJ. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity. 2011;34:794–806. doi: 10.1016/j.immuni.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 95.Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, Mazmanian SK. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science. 2011;332:974–977. doi: 10.1126/science.1206095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, Glickman JN, Garrett WS. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–450. doi: 10.1038/nature12721. [DOI] [PubMed] [Google Scholar]
- 98.Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504:451–455. doi: 10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Suzuki K, Maruya M, Kawamoto S, Sitnik K, Kitamura H, Agace WW, Fagarasan S. The sensing of environmental stimuli by follicular dendritic cells promotes immunoglobulin A generation in the gut. Immunity. 2010;33:71–83. doi: 10.1016/j.immuni.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 100.Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol. 2011;12:21–27. doi: 10.1038/ni.1962. [DOI] [PubMed] [Google Scholar]
- 101.Spits H, Cupedo T. Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annu Rev Immunol. 2012;30:647–675. doi: 10.1146/annurev-immunol-020711-075053. [DOI] [PubMed] [Google Scholar]
- 102.Sonnenberg GF, Artis D. Innate lymphoid cell interactions with microbiota: implications for intestinal health and disease. Immunity. 2012;37:601–610. doi: 10.1016/j.immuni.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zelante T, Iannitti RG, Cunha C, De Luca A, Giovannini G, Pieraccini G, Zecchi R, D’Angelo C, Massi-Benedetti C, Fallarino F, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity. 2013;39:372–385. doi: 10.1016/j.immuni.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 104.Manta C, Heupel E, Radulovic K, Rossini V, Garbi N, Riedel CU, Niess JH. CX(3)CR1(+) macrophages support IL-22 production by innate lymphoid cells during infection with Citrobacter rodentium. Mucosal Immunol. 2013;6:177–188. doi: 10.1038/mi.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Franchi L, Kamada N, Nakamura Y, Burberry A, Kuffa P, Suzuki S, Shaw MH, Kim YG, Núñez G. NLRC4-driven production of IL-1β discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat Immunol. 2012;13:449–456. doi: 10.1038/ni.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rivollier A, He J, Kole A, Valatas V, Kelsall BL. Inflammation switches the differentiation program of Ly6Chi monocytes from antiinflammatory macrophages to inflammatory dendritic cells in the colon. J Exp Med. 2012;209:139–155. doi: 10.1084/jem.20101387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bates JM, Akerlund J, Mittge E, Guillemin K. Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe. 2007;2:371–382. doi: 10.1016/j.chom.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lotz M, Gütle D, Walther S, Ménard S, Bogdan C, Hornef MW. Postnatal acquisition of endotoxin tolerance in intestinal epithelial cells. J Exp Med. 2006;203:973–984. doi: 10.1084/jem.20050625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lutgendorff F, Akkermans LM, Söderholm JD. The role of microbiota and probiotics in stress-induced gastro-intestinal damage. Curr Mol Med. 2008;8:282–298. doi: 10.2174/156652408784533779. [DOI] [PubMed] [Google Scholar]
- 110.Cario E, Gerken G, Podolsky DK. Toll-like receptor 2 controls mucosal inflammation by regulating epithelial barrier function. Gastroenterology. 2007;132:1359–1374. doi: 10.1053/j.gastro.2007.02.056. [DOI] [PubMed] [Google Scholar]
- 111.Yan F, Cao H, Cover TL, Washington MK, Shi Y, Liu L, Chaturvedi R, Peek RM, Wilson KT, Polk DB. Colon-specific delivery of a probiotic-derived soluble protein ameliorates intestinal inflammation in mice through an EGFR-dependent mechanism. J Clin Invest. 2011;121:2242–2253. doi: 10.1172/JCI44031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cani PD, Possemiers S, Van de Wiele T, Guiot Y, Everard A, Rottier O, Geurts L, Naslain D, Neyrinck A, Lambert DM, et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut. 2009;58:1091–1103. doi: 10.1136/gut.2008.165886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Stappenbeck TS, Hooper LV, Gordon JI. Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc Natl Acad Sci USA. 2002;99:15451–15455. doi: 10.1073/pnas.202604299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gordon HA, Bruckner-kardoss E. Effect of normal microbial flora on intestinal surface area. Am J Physiol. 1961;201:175–178. doi: 10.1152/ajplegacy.1961.201.1.175. [DOI] [PubMed] [Google Scholar]
- 115.Banasaz M, Norin E, Holma R, Midtvedt T. Increased enterocyte production in gnotobiotic rats mono-associated with Lactobacillus rhamnosus GG. Appl Environ Microbiol. 2002;68:3031–3034. doi: 10.1128/AEM.68.6.3031-3034.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Alam M, Midtvedt T, Uribe A. Differential cell kinetics in the ileum and colon of germfree rats. Scand J Gastroenterol. 1994;29:445–451. doi: 10.3109/00365529409096836. [DOI] [PubMed] [Google Scholar]
- 117.Husebye E, Hellström PM, Midtvedt T. Intestinal microflora stimulates myoelectric activity of rat small intestine by promoting cyclic initiation and aboral propagation of migrating myoelectric complex. Dig Dis Sci. 1994;39:946–956. doi: 10.1007/BF02087542. [DOI] [PubMed] [Google Scholar]
- 118.Hooper LV, Gordon JI. Commensal host-bacterial relationships in the gut. Science. 2001;292:1115–1118. doi: 10.1126/science.1058709. [DOI] [PubMed] [Google Scholar]
- 119.Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, Knight R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci USA. 2010;107:11971–11975. doi: 10.1073/pnas.1002601107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gosalbes MJ, Llop S, Vallès Y, Moya A, Ballester F, Francino MP. Meconium microbiota types dominated by lactic acid or enteric bacteria are differentially associated with maternal eczema and respiratory problems in infants. Clin Exp Allergy. 2013;43:198–211. doi: 10.1111/cea.12063. [DOI] [PubMed] [Google Scholar]
- 121.Mackie RI, Sghir A, Gaskins HR. Developmental microbial ecology of the neonatal gastrointestinal tract. Am J Clin Nutr. 1999;69:1035S–1045S. doi: 10.1093/ajcn/69.5.1035s. [DOI] [PubMed] [Google Scholar]
- 122.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ringel-Kulka T, Cheng J, Ringel Y, Salojärvi J, Carroll I, Palva A, de Vos WM, Satokari R. Intestinal microbiota in healthy U.S. young children and adults--a high throughput microarray analysis. PLoS One. 2013;8:e64315. doi: 10.1371/journal.pone.0064315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Agans R, Rigsbee L, Kenche H, Michail S, Khamis HJ, Paliy O. Distal gut microbiota of adolescent children is different from that of adults. FEMS Microbiol Ecol. 2011;77:404–412. doi: 10.1111/j.1574-6941.2011.01120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Biagi E, Nylund L, Candela M, Ostan R, Bucci L, Pini E, Nikkïla J, Monti D, Satokari R, Franceschi C, et al. Through ageing, and beyond: gut microbiota and inflammatory status in seniors and centenarians. PLoS One. 2010;5:e10667. doi: 10.1371/journal.pone.0010667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Claesson MJ, Cusack S, O’Sullivan O, Greene-Diniz R, de Weerd H, Flannery E, Marchesi JR, Falush D, Dinan T, Fitzgerald G, et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci USA. 2011;108 Suppl 1:4586–4591. doi: 10.1073/pnas.1000097107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mariat D, Firmesse O, Levenez F, Guimarăes V, Sokol H, Doré J, Corthier G, Furet JP. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 2009;9:123. doi: 10.1186/1471-2180-9-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lan Y, Kriete A, Rosen GL. Selecting age-related functional characteristics in the human gut microbiome. Microbiome. 2013;1:2. doi: 10.1186/2049-2618-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Groer MW, Luciano AA, Dishaw LJ, Ashmeade TL, Miller E, Gilbert JA. Development of the preterm infant gut microbiome: a research priority. Microbiome. 2014;2:38. doi: 10.1186/2049-2618-2-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sherman MP, Zaghouani H, Niklas V. Gut microbiota, the immune system, and diet influence the neonatal gut-brain axis. Pediatr Res. 2015;77:127–135. doi: 10.1038/pr.2014.161. [DOI] [PubMed] [Google Scholar]
- 131.Albenberg LG, Wu GD. Diet and the intestinal microbiome: associations, functions, and implications for health and disease. Gastroenterology. 2014;146:1564–1572. doi: 10.1053/j.gastro.2014.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Brown EM, Sadarangani M, Finlay BB. The role of the immune system in governing host-microbe interactions in the intestine. Nat Immunol. 2013;14:660–667. doi: 10.1038/ni.2611. [DOI] [PubMed] [Google Scholar]
- 133.Zivkovic AM, German JB, Lebrilla CB, Mills DA. Human milk glycobiome and its impact on the infant gastrointestinal microbiota. Proc Natl Acad Sci USA. 2011;108 Suppl 1:4653–4658. doi: 10.1073/pnas.1000083107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ouwehand A, Isolauri E, Salminen S. The role of the intestinal microflora for the development of the immune system in early childhood. Eur J Nutr. 2002;41 Suppl 1:I32–I37. doi: 10.1007/s00394-002-1105-4. [DOI] [PubMed] [Google Scholar]
- 135.Yoshioka H, Iseki K, Fujita K. Development and differences of intestinal flora in the neonatal period in breast-fed and bottle-fed infants. Pediatrics. 1983;72:317–321. [PubMed] [Google Scholar]
- 136.Harmsen HJ, Wildeboer-Veloo AC, Raangs GC, Wagendorp AA, Klijn N, Bindels JG, Welling GW. Analysis of intestinal flora development in breast-fed and formula-fed infants by using molecular identification and detection methods. J Pediatr Gastroenterol Nutr. 2000;30:61–67. doi: 10.1097/00005176-200001000-00019. [DOI] [PubMed] [Google Scholar]
- 137.Stark PL, Lee A, Parsonage BD. Colonization of the large bowel by Clostridium difficile in healthy infants: quantitative study. Infect Immun. 1982;35:895–899. doi: 10.1128/iai.35.3.895-899.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Walker AW, Ince J, Duncan SH, Webster LM, Holtrop G, Ze X, Brown D, Stares MD, Scott P, Bergerat A, et al. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2011;5:220–230. doi: 10.1038/ismej.2010.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, Lionetti P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci USA. 2010;107:14691–14696. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Davenport ER, Mizrahi-Man O, Michelini K, Barreiro LB, Ober C, Gilad Y. Seasonal variation in human gut microbiome composition. PLoS One. 2014;9:e90731. doi: 10.1371/journal.pone.0090731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Dueñas M, Muñoz-González I, Cueva C, Jiménez-Girón A, Sánchez-Patán F, Santos-Buelga C, Moreno-Arribas MV, Bartolomé B. A survey of modulation of gut microbiota by dietary polyphenols. Biomed Res Int. 2015;2015:850902. doi: 10.1155/2015/850902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.O’Sullivan L, Murphy B, McLoughlin P, Duggan P, Lawlor PG, Hughes H, Gardiner GE. Prebiotics from marine macroalgae for human and animal health applications. Mar Drugs. 2010;8:2038–2064. doi: 10.3390/md8072038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ramnani P, Chitarrari R, Tuohy K, Grant J, Hotchkiss S, Philp K, Campbell R, Gill C, Rowland I. In vitro fermentation and prebiotic potential of novel low molecular weight polysaccharides derived from agar and alginate seaweeds. Anaerobe. 2012;18:1–6. doi: 10.1016/j.anaerobe.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 145.Hehemann JH, Correc G, Barbeyron T, Helbert W, Czjzek M, Michel G. Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature. 2010;464:908–912. doi: 10.1038/nature08937. [DOI] [PubMed] [Google Scholar]
- 146.Dethlefsen L, McFall-Ngai M, Relman DA. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature. 2007;449:811–818. doi: 10.1038/nature06245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kersten RD, Yang YL, Xu Y, Cimermancic P, Nam SJ, Fenical W, Fischbach MA, Moore BS, Dorrestein PC. A mass spectrometry-guided genome mining approach for natural product peptidogenomics. Nat Chem Biol. 2011;7:794–802. doi: 10.1038/nchembio.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bohnhoff M, Miller CP. Enhanced susceptibility to Salmonella infection in streptomycin-treated mice. J Infect Dis. 1962;111:117–127. doi: 10.1093/infdis/111.2.117. [DOI] [PubMed] [Google Scholar]
- 149.Ng KM, Ferreyra JA, Higginbottom SK, Lynch JB, Kashyap PC, Gopinath S, Naidu N, Choudhury B, Weimer BC, Monack DM, et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature. 2013;502:96–99. doi: 10.1038/nature12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Jernberg C, Löfmark S, Edlund C, Jansson JK. Long-term ecological impacts of antibiotic administration on the human intestinal microbiota. ISME J. 2007;1:56–66. doi: 10.1038/ismej.2007.3. [DOI] [PubMed] [Google Scholar]
- 151.Jakobsson HE, Jernberg C, Andersson AF, Sjölund-Karlsson M, Jansson JK, Engstrand L. Short-term antibiotic treatment has differing long-term impacts on the human throat and gut microbiome. PLoS One. 2010;5:e9836. doi: 10.1371/journal.pone.0009836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008;6:e280. doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Panda S, El khader I, Casellas F, López Vivancos J, García Cors M, Santiago A, Cuenca S, Guarner F, Manichanh C. Short-term effect of antibiotics on human gut microbiota. PLoS One. 2014;9:e95476. doi: 10.1371/journal.pone.0095476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Frost LS, Leplae R, Summers AO, Toussaint A. Mobile genetic elements: the agents of open source evolution. Nat Rev Microbiol. 2005;3:722–732. doi: 10.1038/nrmicro1235. [DOI] [PubMed] [Google Scholar]
- 155.Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405:299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- 156.Smillie CS, Smith MB, Friedman J, Cordero OX, David LA, Alm EJ. Ecology drives a global network of gene exchange connecting the human microbiome. Nature. 2011;480:241–244. doi: 10.1038/nature10571. [DOI] [PubMed] [Google Scholar]
- 157.Hemarajata P, Versalovic J. Effects of probiotics on gut microbiota: mechanisms of intestinal immunomodulation and neuromodulation. Therap Adv Gastroenterol. 2013;6:39–51. doi: 10.1177/1756283X12459294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Binns N. Probiotics, Prebiotics and the gut microbiota. In: Gibson GR, Sanders ME, editors. Health effects of Prebiotics and Probiotics, Intestinal Life Science Institute (ILSI). D/2013/10.996/36. Belgium: ILSI; 2013. pp. 16–20. [Google Scholar]