A Draft Sequence of the Neandertal Genome (original) (raw)
. Author manuscript; available in PMC: 2016 Nov 8.
Published in final edited form as: Science. 2010 May 7;328(5979):710–722. doi: 10.1126/science.1188021
Abstract
Neandertals, the closest evolutionary relatives of present-day humans, lived in large parts of Europe and western Asia before disappearing 30,000 years ago. We present a draft sequence of the Neandertal genome composed of more than 4 billion nucleotides from three individuals. Comparisons of the Neandertal genome to the genomes of five present-day humans from different parts of the world identify a number of genomic regions that may have been affected by positive selection in ancestral modern humans, including genes involved in metabolism and in cognitive and skeletal development. We show that Neandertals shared more genetic variants with present-day humans in Eurasia than with present-day humans in sub-Saharan Africa, suggesting that gene flow from Neandertals into the ancestors of non-Africans occurred before the divergence of Eurasian groups from each other.
The morphological features typical of Neandertals first appear in the European fossil record about 400,000 years ago (1–3). Progressively more distinctive Neandertal forms subsequently evolved until Neandertals disappeared from the fossil record about 30,000 years ago (4). During the later part of their history, Neandertals lived in Europe and Western Asia as far east as Southern Siberia (5) and as far south as the Middle East. During that time, Neandertals presumably came into contact with anatomically modern humans in the Middle East from at least 80,000 years ago (6, 7) and subsequently in Europe and Asia.
Neandertals are the sister group of all present-day humans. Thus, comparisons of the human genome to the genomes of Neandertals and apes allow features that set fully anatomically modern humans apart from other hominin forms to be identified. In particular, a Neandertal genome sequence provides a catalog of changes that have become fixed or have risen to high frequency in modern humans during the last few hundred thousand years and should be informative for identifying genes affected by positive selection since humans diverged from Neandertals.
Substantial controversy surrounds the question of whether Neandertals interbred with anatomically modern humans. Morphological features of present-day humans and early anatomically modern human fossils have been interpreted as evidence both for (8, 9) and against (10, 11) genetic exchange between Neandertals and the presumed ancestors of present-day Europeans. Similarly, analysis of DNA sequence data from present-day humans has been interpreted as evidence both for (12, 13) and against (14) a genetic contribution by Neandertals to present-day humans. The only part of the genome that has been examined from multiple Neandertals, the mitochondrial DNA (mtDNA) genome, consistently falls outside the variation found in present-day humans and thus provides no evidence for inter-breeding (15–19). However, this observation does not preclude some amount of interbreeding (14, 19) or the possibility that Neandertals contributed other parts of their genomes to present-day humans (16). In contrast, the nuclear genome is composed of tens of thousands of recombining, and hence independently evolving, DNA segments that provide an opportunity to obtain a clearer picture of the relationship between Neandertals and present-day humans.
A challenge in detecting signals of gene flow between Neandertals and modern human ancestors is that the two groups share common ancestors within the last 500,000 years, which is no deeper than the nuclear DNA sequence variation within present-day humans. Thus, even if no gene flow occurred, in many segments of the genome, Neandertals are expected to be more closely related to some present-day humans than they are to each other (20). However, if Neandertals are, on average across many independent regions of the genome, more closely related to present-day humans in certain parts of the world than in others, this would strongly suggest that Neandertals exchanged parts of their genome with the ancestors of these groups.
Several features of DNA extracted from Late Pleistocene remains make its study challenging. The DNA is invariably degraded to a small average size of less than 200 base pairs (bp) (21, 22), it is chemically modified (21, 23–26), and extracts almost always contain only small amounts of endogenous DNA but large amounts of DNA from microbial organisms that colonized the specimens after death. Over the past 20 years, methods for ancient DNA retrieval have been developed (21, 22), largely based on the polymerase chain reaction (PCR) (27). In the case of the nuclear genome of Neandertals, four short gene sequences have been determined by PCR: fragments of the MC1R gene involved in skin pigmentation (28), a segment of the FOXP2 gene involved in speech and language (29), parts of the ABO blood group locus (30), and a taste receptor gene (31). However, although PCR of ancient DNA can be multiplexed (32), it does not allow the retrieval of a large proportion of the genome of an organism.
The development of high-throughput DNA sequencing technologies (33, 34) allows large-scale, genome-wide sequencing of random pieces of DNA extracted from ancient specimens (35–37) and has recently made it feasible to sequence genomes from late Pleistocene species (38). However, because a large proportion of the DNA present in most fossils is of microbial origin, comparison to genome sequences of closely related organisms is necessary to identify the DNA molecules that derive from the organism under study (39). In the case of Neandertals, the finished human genome sequence and the chimpanzee genome offer the opportunity to identify Neandertal DNA sequences (39, 40).
A special challenge in analyzing DNA sequences from the Neandertal nuclear genome is that most DNA fragments in a Neandertal are expected to be identical to present-day humans (41). Thus, contamination of the experiments with DNA from present-day humans may be mistaken for endogenous DNA. We first applied high-throughput sequencing to Neandertal specimens from Vindija Cave in Croatia (40, 42), a site from which cave bear remains yielded some of the first nuclear DNA sequences from the late Pleistocene in 1999 (43). Close to one million bp of nuclear DNA sequences from one bone were directly determined by high-throughput sequencing on the 454 platform (40), whereas DNA fragments from another extract from the same bone were cloned in a plasmid vector and used to sequence ~65,000 bp (42). These experiments, while demonstrating the feasibility of generating a Neandertal genome sequence, were preliminary in that they involved the transfer of DNA extracts prepared in a clean-room environment to conventional laboratories for processing and sequencing, creating an opportunity for contamination by present-day human DNA. Further analysis of the larger of these data sets (40) showed that it was contaminated with modern human DNA (44) to an extent of 11 to 40% (41). We employed a number of technical improvements, including the attachment of tagged sequence adaptors in the clean-room environment (23), to minimize the risk of contamination and determine about 4 billion bp from the Neandertal genome.
Paleontological samples
We analyzed a total of 21 Neandertal bones from Vindija Cave in Croatia that are of little morphological value. From below the surface of each of these bones, we removed 50 to 100 mg of bone powder using a sterile dentistry drill in our Leipzig clean-room facility. All samples were screened for the presence of Neandertal mtDNA by PCR, and three bones were selected for further analysis (Fig. 1A) [Supporting Online Material (SOM) Text 2]. The first of these bones, Vi33.16 (previously Vi-80) was discovered in stratigraphic layer G3 by Malez and co-workers in 1980 and has been directly dated by carbon-14 accelerator mass spectrometry to 38,310 ± 2,130 years before the present (B.P.) (uncalibrated) (19). It has been previously used for genome sequencing (40, 42) and for the determination of a complete mtDNA sequence (45). The second bone, Vi33.25, comes from layer I, which is deeper and thus older than layer G. A complete mtDNA sequence has been determined from this bone (15). It does not contain enough collagen to allow a direct date. The third bone, Vi33.26, comes from layer G (sublayer unknown) and has not been previously used for large-scale DNA sequencing. It was directly dated to 44,450 ± 550 years B.P. (OxA-V-2291-18, uncalibrated).
Fig. 1.

Samples and sites from which DNA was retrieved. (A) The three bones from Vindija from which Neandertal DNA was sequenced. (B) Map showing the four archaeological sites from which bones were used and their approximate dates (years B.P.).
Sequencing library construction
A total of nine DNA extracts were prepared from the three bones (table S4) using procedures to minimize laboratory contamination that we have developed over the past two decades (22, 41). Samples of each extract were used to construct Roche/454 sequencing libraries that carry the project-specific tag sequence 5′-TGAC-3′ in their 3′-ends. Each library was amplified with the primers used in the 454 sequencing emulsion PCR process. To estimate the percentage of endogenous Neandertal DNA in the extracts, we carried out sequencing runs using the 454 Life Sciences GS FLX platform and mapped the reads against the human, chimpanzee, rhesus, and mouse genomes as well as all nucleotide sequences in GenBank. DNA sequences with a significantly better match to the primate genomes than to any of the other sources of sequences were further analyzed. Mitochondrial DNA contamination from modern humans was estimated by primer extension capture (46) using six biotinylated primers that target informative differences between human and Neandertal mtDNA (45), followed by sequencing on the GS FLX platform. Extracts that contained more than 1.5% hominin DNA relative to other DNA were used to construct further libraries. These were similarly analyzed to assess the percentage of hominin DNA and, if found suitable, were used for production sequencing on the 454 Life Sciences GS FLX/Titanium and Illumina GAII platforms.
Enrichment of Neandertal DNA
Depending on the extract, between 95 and 99% of the DNA sequenced in the libraries was derived from nonprimate organisms, which are presumably derived from microbes that colonized the bone after the death of the Neandertals. To improve the ratio of Neandertal to microbial DNA, we identified restriction enzymes that preferentially cut bacterial DNA sequences in the libraries and treated the libraries with these to increase the relative proportion of Neandertal DNA in the libraries (SOM Text 1). Such enzymes, which have recognition sites rich in the dinucleotide CpG, allowed a 4- to 6-fold increase in the proportion of Neandertal DNA in the libraries sequenced. This is expected to bias the sequencing against GC-rich regions of the genome and is therefore not suitable for arriving at a complete Neandertal genome sequence. However, for producing an overview of the genome at about one-fold coverage, it drastically increases the efficiency of data production without unduly biasing coverage, especially in view of the fact that GC-rich sequences are over-represented in ancient DNA sequencing libraries (23, 45) so that the restriction enzyme treatment may help to counteract this bias.
Sequencing platforms and alignments
In the initial phase of the project, we optimized DNA extraction technology and library construction [e.g., (47)]. In a second phase, we carried out production sequencing on the 454 Life Sciences GS FLX platform from the bones Vi33.16 and Vi33.26 (0.5 Gb and 0.8 Gb of Neandertal sequence, respectively). In the third phase, we carried out production sequencing on the Illumina/Solexa GAII platform from the bones Vi33.16, Vi33.25, and Vi33.26 (1.2 Gb, 1.3 Gb, and 1.5 Gb, respectively) (table S4). Each molecule was sequenced from both ends (SOM Text 2), and bases were called with the machine learning algorithm Ibis (48). All reads were required to carry correct clean-room tags, and previous data where these tags were not used (40, 42) were not included in this study. Except when explicitly stated, the analyses below are based on the largest data sets, generated on the Illumina platform. In total, we generated 5.3 Gb of Neandertal DNA sequence from about 400 mg of bone powder. Thus, methods for extracting and sequencing DNA from ancient bones are now efficient enough to allow genome-wide DNA sequence coverage with relatively minor damage to well-preserved paleontological specimens.
The dominant type of nucleotide misincorporation when ancient DNA is amplified and sequenced is due to deamination of cytosine residues (25). This causes C to T transitions in the DNA sequences, particularly toward the 5′-ends of DNA reads, where at the first position ~40% of cytosine residues can appear as thymine residues. The frequency of C to T misincorporations progressively diminishes further into the molecules. At the 3′-ends, complementary G to A transitions are seen as a result of the enzymatic fill-in procedure in which blunt DNA ends are created before adaptor ligation (23). We implemented an alignment approach that takes these nucleotide misincorporation patterns into account (SOM Text 3) and aligned the Neandertal sequences to either the reference human genome (UCSC hg18), the reference chimpanzee genome ( panTro2), or the inferred human-chimpanzee common ancestral sequence (SOM Text 3).
To estimate the error rate in the Neandertal DNA sequences determined, we compared reads that map to the mitochondrial genomes, which we assembled to 35-, 29- and 72-fold coverage for each of the bones, respectively (15, 45) (SOM Text 4). Although C to T and G to A substitutions, which are caused by deaminated cytosine residues, occur at a rate of 4.5 to 5.9%, other error rates are at most 0.3% (fig. S4). Because we sequence each DNA fragment from both sides, and most fragments more than once (49), the latter error rate is substantially lower than the error rate of the Illumina platform itself (48, 50).
Number of Neandertal individuals
To assess whether the three bones come from different individuals, we first used their mtDNAs. We have previously determined the complete mtDNA sequences from the bones Vi33.16 and Vi33.25 (15, 45), and these differ at 10 positions. Therefore, Vi33.16 and Vi33.25 come from different Neandertal individuals. For the bone Vi33.26, we assembled the mtDNA sequence (SOM Text 4) and found it to be indistinguishable from Vi33.16, suggesting that it could come from the same individual. We analyzed autosomal DNA sequences from the three bones (SOM Text 4) by asking whether the frequency of nucleotide differences between pairs of bones was significantly higher than the frequency of differences within the bones. We find that the within-bone differences are significantly fewer than the between-bone differences for all three comparisons (P ≤ 0.001 in all cases). Thus, all three bones derive from different individuals, although Vi33.16 and Vi33.26 may stem from maternally related individuals.
Estimates of human DNA contamination
We used three approaches that target mtDNA, Y chromosomal DNA, and nuclear DNA, respectively, to gauge the ratio of present-day human relative to Neandertal DNA in the data produced. To analyze the extent of mtDNA contamination, we used the complete mtDNA from each bone to identify positions differing from at least 99% of a worldwide panel of 311 contemporary human mtDNAs, ignoring positions where a substitution in the sequences from the Neandertal library could be due to cytosine deamination (45). For each sequencing library, the DNA fragments that cover these positions were then classified according to whether they appear to be of Neandertal or modern human origin (SOM Text 5 and table S15). For each bone, the level of mtDNA contamination is estimated to be below 0.5% (Table 1).
Table 1.
Estimates of human DNA contamination in the DNA sequences produced. Numbers in bold indicate summary contamination estimates over all Vindija data.
| mtDNAcontamination | Y chromosomalcontamination | Neandertaldiversity (1/2)plus contamination* | Nuclear MLcontamination | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Human | Neandertal | Percent | 95% C.I. | Observed | Expected | Percent | 95% C.I. | Percent | Upper 95% C.I. | Percent(95% C.I.) | |
| Vi33.16 | 56 | 20,456 | 0.27 | 0.21–0.35 | 4 | 255 | 1.57 | 0.43–3.97 | 1.4 | 2.2 | n/a |
| Vi33.25 | 7 | 1,691 | 0.41 | 0.17–0.85 | 0 | 201 | 0.0 | 0.00–1.82 | 1.0 | 1.7 | n/a |
| Vi33.26 | 10 | 4,810 | 0.21 | 0.10–0.38 | 0 | 210 | 0.0 | 0.00–1.74 | 1.1 | 1.9 | n/a |
| All data | 73 | 26,957 | 0.27 | 0.21–0.34 | 4 | 666 | 0.60 | 0.16–1.53 | 1.2 | 1.6 | 0.7 (0.6–0.8) |
Because prior to this study no fixed differences between Neandertal and present-day humans in the nuclear genome were known, we used two alternative strategies to estimate levels of nuclear contamination. In the first strategy, we determined the sex of the bones. For bones derived from female Neandertals, we then estimated modern human male DNA contamination by looking for the presence of Y chromosomal DNA fragments (SOM Text 6). For this purpose, we identified 111,132 nucleotides in the nonrecombining parts of the human reference Y chromosome that are located in contiguous DNA segments of at least 500 nucleotides, carry no repetitive elements, and contain no 30-nucleotide oligomer elsewhere in the genome with fewer than three mismatches. Between 482 and 611 such fragments would be expected for a male Neandertal bone. However, only 0 to 4 fragments are observed (Table 1). We conclude that the three bones are all from female Neandertals and that previous suggestions that Vi33.16 was a male (40, 42) were due to mismapping of autosomal and X chromosomal reads to the Y chromosome. We estimate the extent of DNA contamination from modern human males in the combined data to be about 0.60%, with an upper 95% bound of 1.53%.
In the second strategy, we take advantage of the fact that sites where present-day humans carry a high frequency of a derived allele (i.e., not seen in chimpanzee) while Neandertals carry a high frequency of the ancestral allele (i.e., matching the chimpanzee) provide information about the extent of contamination. To implement this idea, we identified sites where five present-day humans that we sequenced (see below) all differ from the chimpanzee genome by a transversion. We further restricted the analysis to sites covered by two fragments in one Neandertal and one fragment in another Neandertal and where at least one ancestral allele was seen in both individuals. The additional fragment from the first Neandertal then provides an estimate of contamination in combination with heterozygosity at this class of sites (Table 1). Using these data (SOM Text 7), we derive a maximum likelihood estimate of contamination of 0.7% with an upper 95% bound of 0.8%.
In summary, all three measurements of human mtDNA contamination produce estimates of less than 1% contamination. Thus, the vast majority of these data represent bona fide Neandertal DNA sequences.
Average DNA divergence between Neandertals and humans
To estimate the DNA sequence divergence per base pair between the genomes of Neandertals and the reference human genome sequence, we generated three-way alignments between the Neandertal, human, and chimpanzee genomes, filtering out genomic regions that may be duplicated in either humans or chimpanzees (SOM Text 10) and using an inferred genome sequence of the common ancestor of humans and chimpanzees as a reference (51) to avoid potential biases (39). We then counted the number of substitutions specific to the Neandertal, the human, and the chimpanzee genomes (Fig. 2). The overall number of substitutions unique to the Neandertal genome is about 30 times as high as on the human lineage. Because these are largely due to transitions resulting from deamination of cytosine residues in the Neandertal DNA, we restricted the divergence estimates to transversions. We then observed four to six times as many on the Neandertal as on the human lineage, probably due to sequencing errors in the low-coverage Neandertal DNA sequences. The numbers of transversions on the human lineage, as well as those on the lineage from the Neandertal-human ancestor to the chimpanzee, were used to estimate the average divergence between DNA sequences in Neandertals and present-day humans, as a fraction of the lineage from the human reference genome to the common ancestor of Neandertals, humans, and chimpanzees. For autosomes, this was 12.7% for each of the three bones analyzed. For the X chromosome, it was 11.9 to 12.4% (table S26). Assuming an average DNA divergence of 6.5 million years between the human and chimpanzee genomes (52), this results in a point estimate for the average divergence of Neandertal and modern human autosomal DNA sequences of 825,000 years. We caution that this is only a rough estimate because of the uncertainty about the time of divergence of humans and chimpanzees.
Fig. 2.

Nucleotide substitutions inferred to have occurred on the evolutionary lineages leading to the Neandertals, the human, and the chimpanzee genomes. In red are substitutions on the Neandertal lineage, in yellow the human lineage, and in pink the combined lineage from the common ancestor of these to the chimpanzee. For each lineage and each bone from Vindija, the distributions and numbers of substitutions are shown. The excess of C to T and G to A substitutions are due to deamination of cytosine residues in the Neandertal DNA.
Additional Neandertal individuals
To put the divergence of the Neandertal genome sequences from Vindija Cave into perspective with regard to other Neandertals, we generated a much smaller amount of DNA sequence data from three Neandertal bones from three additional sites (SOM Text 8) that cover much of the geographical range of late Neandertals (Fig. 1B): El Sidron in Asturias, Spain, dated to ~49,000 years B.P. (53); Feldhofer Cave in the Neander Valley, Germany, from which we sequenced the type specimen found in 1856 dated to ~42,000 years B.P. (54); and Mezmaiskaya Cave in the Caucasus, Russia, dated to 60,000 to 70,000 years B.P. (55). DNA divergences estimated for each of these specimens to the human reference genome (table S26) show that none of them differ significantly from the Vindija individuals, although these estimates are relatively uncertain due to the limited amount of DNA sequence data. It is noteworthy that the Mezmaiskaya specimen, which is 20,000 to 30,000 years older than the other Neandertals analyzed and comes from the easternmost location, does not differ in divergence from the other individuals. Thus, within the resolution of our current data, Neandertals from across a great part of their range in western Eurasia are equally related to present-day humans.
Five present-day human genomes
To put the divergence of the Neandertal genomes into perspective with regard to present-day humans, we sequenced the genomes of one San from Southern Africa, one Yoruba from West Africa, one Papua New Guinean, one Han Chinese, and one French from Western Europe to 4- to 6-fold coverage on the Illumina GAII platform (SOM Text 9). These sequences were aligned to the chimpanzee and human reference genomes and analyzed using a similar approach to that used for the Neandertal data. Autosomal DNA sequences of these individuals diverged 8.2 to 10.3% back along the lineage leading to the human reference genome, considerably less than the 12.7% seen in Neandertals (SOM Text 10). We note that the divergence estimate for the Yoruba individual to the human genome sequence is ~14% greater than previous estimates for an African American individual (56) and similarly greater than the heterozygosity measured in another Yoruba individual (33). This may be due to differences in the alignment and filtering procedures between this and previous studies (SOM Text 9 and 10). Nevertheless, the divergence of the Neandertal genome to the human reference genome is greater than for any of the present-day human genomes analyzed.
Distributions of DNA divergences to humans
To explore the variation of DNA sequence divergence across the genome, we analyzed the divergence of the Neandertals and the five humans to the reference human genome in 100 kilobase windows for which at least 50 informative transversions were observed. The majority of the Neandertal divergences overlap with those of the humans (Fig. 3), reflecting the fact that Neandertals fall inside the variation of present-day humans. However, the overall divergence is greater for the three Neandertal genomes. For example, their modes are around divergences of ~11%, whereas for the San the mode is ~9% and for the other present-day humans ~8%. For the Neandertals, 13% of windows have a divergence above 20%, whereas this is the case for 2.5% to 3.7% of windows in the current humans.
Fig. 3.
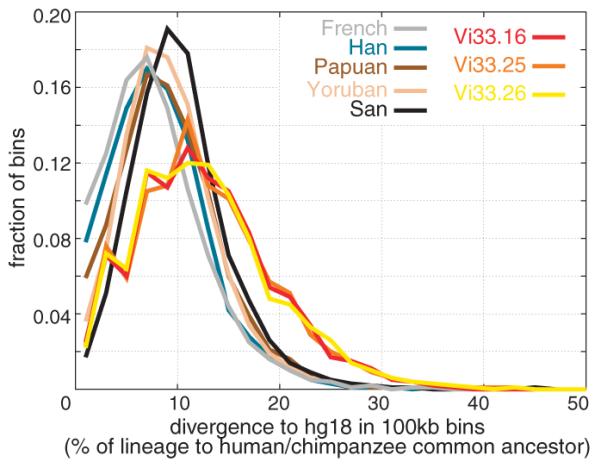
Divergence of Neandertal and human genomes. Distributions of divergence from the human genome reference sequence among segments of 100 kb are shown for three Neandertals and the five present-day humans.
Furthermore, whereas in the French, Han, and Papuan individuals, 9.8%, 7.8%, and 5.9% of windows, respectively, show between 0% and 2% divergence to the human reference genome, in the San and the Yoruba this is the case for 1.7% and 3.7%, respectively. For the three Neandertals, 2.2 to 2.5% of windows show 0% to 2% divergence to the reference genome.
A catalog of features unique to the human genome
The Neandertal genome sequences allow us to identify features unique to present-day humans relative to other, now extinct, hominins. Of special interest are features that may have functional consequences. We thus identified, from whole genome alignments, sites where the human genome reference sequence does not match chimpanzee, orangutan, and rhesus macaque. These are likely to have changed on the human lineage since the common ancestor with chimpanzee. Where Neandertal fragments overlapped, we constructed consensus sequences and joined them into “minicontigs,” which were used to determine the Neandertal state at the positions that changed on the human lineage. To minimize alignment errors and substitutions, we disregarded all substitutions and insertions or deletions (indels) within 5 nucleotides of the ends of minicontigs or within 5 nucleotides of indels.
Among 10,535,445 substitutions and 479,863 indels inferred to have occurred on the human lineage, we have information in the Neandertal genome for 3,202,190 and 69,029, i.e., 30% and 14%, respectively. The final catalog thus represents those sequenced positions where we have high confidence in their Neandertal state (SOM Text 11). As expected, the vast majority of those substitutions and indels (87.9% and 87.3%, respectively) occurred before the Neandertal divergence from modern humans.
Features that occur in all present-day humans (i.e., have been fixed), although they were absent or variable in Neandertals, are of special interest. We found 78 nucleotide substitutions that change the protein-coding capacity of genes where modern humans are fixed for a derived state and where Neandertals carry the ancestral (chimpanzee-like) state (Table 2 and table S28). Thus, relatively few amino acid changes have become fixed in the last few hundred thousand years of human evolution; an observation consistent with a complementary study (57). We found only five genes with more than one fixed substitution changing the primary structure of the encoded proteins. One of these is SPAG17, which encodes a protein important for the axoneme, a structure responsible for the beating of the sperm flagellum (58). The second is PCD16, which encodes fibroblast cadherin-1, a calcium-dependent cell-cell adhesion molecule that may be involved in wound healing (59). The third is TTF1, a transcription termination factor that regulates ribosomal gene transcription (60). The fourth is CAN15, which encodes a protein of unknown function. The fifth is RPTN, which encodes repetin, an extracellular epidermal matrix protein (61) that is expressed in the epidermis and at high levels in eccrine sweat glands, the inner sheaths of hair roots, and the filiform papilli of the tongue.
Table 2.
Amino acid changes that are fixed in present-day humans but ancestral in Neandertals. The table is sorted by Grantham scores (GS). Based on the classification proposed by Li et al. in (87), 5 amino acid substitutions are radical (>150), 7 moderately radical (101 to 150), 33 moderately conservative (51 to 100) and 32 conservative (1 to 50). One substitution creates a stop codon. Genes showing multiple substitutions have bold SwissProt identifiers. (Table S15 shows the human and chimpanzee genome coordinates, additional database identifiers, and the respective bases.) Genes with two fixed amino acids are indicated in bold.
| ID | Pos | AA | GS | Description/function |
|---|---|---|---|---|
| RPTN | 785 | */R | – | Multifunctional epidermal matrix protein |
| GREB1 | 1164 | R/C | 180 | Response gene in estrogen receptor–regulated pathway |
| OR1K1 | 267 | R/C | 180 | Olfactory receptor, family 1, subfamily K, member 1 |
| SPAG17 | 431 | Y/D | 160 | Involved in structural integrity of sperm central apparatus axoneme |
| NLRX1 | 330 | Y/D | 160 | Modulator of innate immune response |
| NSUN3 | 78 | S/F | 155 | Protein with potential SAM-dependent methyl-transferase activity |
| RGS16 | 197 | D/A | 126 | Retinally abundant regulator of G-protein signaling |
| BOD1L | 2684 | G/R | 125 | Biorientation of chromosomes in cell division 1-like |
| CF170 | 505 | S/C | 112 | Uncharacterized protein: C6orf170 |
| STEA1 | 336 | C/S | 112 | Metalloreductase, six transmembrane epithelial antigen of prostate 1 |
| F16A2 | 630 | R/S | 110 | Uncharacterized protein: family with sequence similarity 160, member A2 |
| LTK | 569 | R/S | 110 | Leukocyte receptor tyrosine kinase |
| BEND2 | 261 | V/G | 109 | Uncharacterized protein: BEN domain-containing protein 2 |
| O52W1 | 51 | P/L | 98 | Olfactory receptor, family 52, subfamily W, member 1 |
| CAN15 | 427 | L/P | 98 | Small optic lobes homolog, linked to visual system development |
| SCAP | 140 | I/T | 89 | Escort protein required for cholesterol as well as lipid homeostasis |
| TTF1 | 474 | I/T | 89 | RNA polymerase I termination factor |
| OR5K4 | 175 | H/D | 81 | Olfactory receptor, family 5, subfamily K, member 4 |
| SCML1 | 202 | T/M | 81 | Putative polycomb group (PcG) protein |
| TTL10 | 394 | K/T | 78 | Probable tubulin polyglutamylase, forming polyglutamate side chains on tubulin |
| AFF3 | 516 | S/P | 74 | Putative transcription activator, function in lymphoid development/oncogenesis |
| EYA2 | 131 | S/P | 74 | Tyrosine phosphatase, dephosphorylating “Tyr-142” of histone H2AX |
| NOP14 | 493 | T/R | 71 | Involved in nucleolar processing of pre-18S ribosomal RNA |
| PRDM10 | 1129 | N/T | 65 | PR domain containing 10, may be involved in transcriptional regulation |
| BTLA | 197 | N/T | 65 | B and T lymphocyte attenuator |
| O2AT4 | 224 | V/A | 64 | Olfactory receptor, family 2, subfamily AT, member 4 |
| CAN15 | 356 | V/A | 64 | Small optic lobes homolog, linked to visual system development |
| ACCN4 | 160 | V/A | 64 | Amiloride-sensitive cation channel 4, expressed in pituitary gland |
| PUR8 | 429 | V/A | 64 | Adenylsuccinate lyase (purine synthesis) |
| MCHR2 | 324 | A/V | 64 | Receptor for melanin-concentrating hormone, coupled to G proteins |
| AHR | 381 | V/A | 64 | Aromatic hydrocarbon receptor, a ligand-activated transcriptional activator |
| FAAH1 | 476 | A/G | 60 | Fatty acid amide hydrolase |
| SPAG17 | 1415 | T/A | 58 | Involved in structural integrity of sperm central apparatus axoneme |
| ZF106 | 697 | A/T | 58 | Zinc finger protein 106 homolog / SH3-domain binding protein 3 |
| CAD16 | 342 | T/A | 58 | Calcium-dependent, membrane-associated glycoprotein (cellular recognition) |
| K1C16 | 306 | T/A | 58 | Keratin, type I cytoskeletal 16 (expressed in esophagus, tongue, hair follicles) |
| LIMS2 | 360 | T/A | 58 | Focal adhesion protein, modulates cell spreading and migration |
| ZN502 | 184 | T/A | 58 | Zinc finger protein 502, may be involved in transcriptional regulation |
| MEPE | 391 | A/T | 58 | Matrix extracellular phosphoglycoprotein, putative role in mineralization |
| FSTL4 | 791 | T/A | 58 | Follistatin-related protein 4 precursor |
| SNTG1 | 241 | T/S | 58 | Syntrophin, gamma 1; binding/organizing subcellular localization of proteins |
| RPTN | 735 | K/E | 56 | Multifunctional epidermal matrix protein |
| BCL9L | 543 | S/G | 56 | Nuclear cofactor of beta-catenin signaling, role in tumorigenesis |
| SSH2 | 1033 | S/G | 56 | Protein phosphatase regulating actin filament dynamics |
| PEG3 | 1521 | S/G | 56 | Apoptosis induction in cooperation with SIAH1A |
| DJC28 | 290 | K/Q | 53 | DnaJ (Hsp40) homolog, may have role in protein folding or as a chaperone |
| CLTR2 | 50 | F/V | 50 | Receptor for cysteinyl leukotrienes, role in endocrine and cardiovascular systems |
| KIF15 | 827 | N/S | 46 | Putative kinesin-like motor enzyme involved in mitotic spindle assembly |
| SPOC1 | 355 | Q/R | 43 | Uncharacterized protein: SPOC domain containing 1 |
| TTF1 | 229 | R/Q | 43 | RNA polymerase I termination factor |
| F166A | 134 | T/P | 38 | Uncharacterized protein: family with sequence similarity 166, member A |
| CL066 | 426 | V/L | 32 | Uncharacterized protein: chromosome 12 open reading frame 66 |
| PCD16 | 763 | E/Q | 29 | Calcium-dependent cell-adhesion protein, fibroblasts expression |
| TRPM5 | 1088 | I/V | 29 | Voltage-modulated cation channel (VCAM), central role in taste transduction |
| S36A4 | 330 | H/R | 29 | Solute carrier family 36 (proton/amino acid symporter) |
| GP132 | 328 | E/Q | 29 | High-affinity G-protein couple receptor for lysophosphatidylcholine (LPC) |
| ZFY26 | 237 | H/R | 29 | Zinc finger FYVE domain-containing, associated with spastic paraplegia-15 |
| CALD1 | 671 | I/V | 29 | Actin- and myosin-binding protein, regulation of smooth muscle contraction |
| CDCA2 | 606 | I/V | 29 | Regulator of chromosome structure during mitosis |
| GPAA1 | 275 | E/Q | 29 | Glycosylphosphatidylinositol anchor attachment protein |
| ARSF | 200 | I/V | 29 | Arylsulfatase F precursor, relevant for composition of bone and cartilage matrix |
| OR4D9 | 303 | R/K | 26 | Olfactory receptor, family 4, subfamily D, member 9 |
| EMIL2 | 155 | R/K | 26 | Elastin microfibril interface-located protein (smooth muscle anchoring) |
| PHLP | 216 | K/R | 26 | Putative modulator of heterotrimeric G proteins |
| TKTL1 | 317 | R/K | 26 | Transketolase-related protein |
| MIIP | 280 | H/Q | 24 | Inhibits glioma cells invasion, down-regulates adhesion and motility genes |
| SPTA1 | 265 | N/D | 23 | Constituent of cytoskeletal network of the erythrocyte plasma membrane |
| PCD16 | 777 | D/N | 23 | Calcium-dependent cell-adhesion protein, fibroblasts expression |
| CS028 | 326 | L/F | 22 | Uncharacterized protein: chromosome 19 open reading frame 28 |
| PIGZ | 425 | L/F | 22 | Mannosyltransferase for glycosylphosphatidylinositol-anchor biosynthesis |
| DISP1 | 1079 | V/M | 21 | Segment-polarity gene required for normal Hedgehog (Hh) signaling |
| RNAS7 | 44 | M/V | 21 | Protein with RNase activity for broad-spectrum of pathogenic microorganisms |
| KR241 | 205 | V/M | 21 | Keratin-associated protein, formation of a rigid and resistant hair shaft |
| SPLC3 | 108 | I/M | 10 | Short palate, lung, and nasal epithelium carcinoma-associated protein |
| NCOA6 | 823 | I/M | 10 | Hormone-dependent coactivation of several receptors |
| WWC2 | 479 | M/I | 10 | Uncharacterized protein: WW, C2, and coiled-coil domain containing 2 |
| ASCC1 | 301 | E/D | 0 | Enhancer of NF-kappa-B, SRF, and AP1 transactivation |
| PROM2 | 458 | D/E | 0 | Plasma membrane protrusion in epithelial and nonepithelial cells |
One of the substitutions in RPTN creates a stop codon that causes the human protein to contain 784 rather than 892 amino acids (SOM Text 11). We identified no fixed start codon differences, although the start codon in the gene TRPM1 that is present in Neandertals and chimpanzees has been lost in some present-day humans. TRPM1 encodes melastatin, an ion channel important for maintaining melanocyte pigmentation in the skin. It is intriguing that skin-expressed genes comprise three out of six genes that either carry multiple fixed substitutions changing amino acids or in which a start or stop codon has been lost or gained. This suggests that selection on skin morphology and physiology may have changed on the hominin lineage.
We also identified a number of potential regulatory substitutions that are fixed in present-day humans but not Neandertals. Specifically, we find 42 substitutions and three indels in 5′-untranslated regions, and 190 substitutions and 33 indels in 3′-untranslated regions that have become fixed in humans since they diverged from Neandertals. Of special interest are microRNAs (miRNAs), small RNAs that regulate gene expression by mRNA cleavage or repression of translation. We found one miRNA where humans carry a fixed substitution at a position that was ancestral in Neandertals (hsa-mir-1304) and one case of a fixed single nucleotide insertion where Neandertal is ancestral (AC109351.3). While the latter insertion is in a bulge in the inferred secondary structure of the miRNA that is unlikely to affect folding or putative targets, the substitution in mir-1304 occurs in the seed region, suggesting that it is likely to have altered target specificity in modern humans relative to Neandertals and other apes (fig. S16).
Human accelerated regions (HARs) are defined as regions of the genome that are conserved throughout vertebrate evolution but that changed radically since humans and chimpanzees split from their common ancestor. We examined 2613 HARs (SOM Text 11) and obtained reliable Neandertal sequence for 3259 human-specific changes in HARs. The Neandertals carry the derived state at 91.4% of these, significantly more than for other human-specific substitutions and indels (87.9%). Thus, changes in the HARs tend to predate the split between Neandertals and modern humans. However, we also identified 51 positions in 45 HARs where Neandertals carry the ancestral version whereas all known present-day humans carry the derived version. These represent recent changes that may be particularly interesting to explore functionally.
Neandertal segmental duplications
We analyzed Neandertal segmental duplications by measuring excess read-depth to identify and predict the copy number of duplicated sequences, defined as those with >95% sequence identity (62). A total of 94 Mb of segmental duplications were predicted in the Neandertal genome (table S33), which is in close agreement with what has been found in present-day humans (62) (fig. S18). We identified 111 potentially Neandertal-specific segmental duplications (average size 22,321 bp and total length 1862 kb) that did not overlap with human segmental duplications (fig. S20). Although direct experimental validation is not possible, we note that 81% (90/111) of these regions also showed excess sequence diversity (>3 SD beyond the mean) consistent with their being bona fide duplications (fig. S21). Many of these regions also show some evidence of increased copy number in humans, although they have not been previously classified as duplications (fig. S22). We identified only three putative Neandertal-specific duplications with no evidence of duplication among humans or any other primate (fig. S23), and none contained known genes.
A comparison to any single present-day human genome reveals that 89% of the detected duplications are shared with Neandertals. This is lower than the proportion seen between present-day humans (around 95%) but higher than what is observed when the Neandertals are compared with the chimpanzee (67%) (fig. S19).
Because the Neandertal data set is derived from a pool of three individuals and represents an average sequence coverage of 1.3-fold after filtering, we created two resampled sets from three human genomes (SOM Text 12) at a comparable level of mixture and coverage (table S34 and figs. S24 and S25). The analysis of both resampled sets show a nonsignificant trend toward more duplicated sequences among Neandertals than among present-day humans (88,869 kb, N = 1129 regions for present-day humans versus 94,419 kb, N = 1194 for the Neandertals) (fig. S25).
We also estimated the copy number for Neandertal genes and compared it with those from three previously analyzed human genomes (SOM Text 12). Copy number was correlated between the two groups (_r_2 = 0.91) (fig. S29), with only 43 genes (15 nonredundant genes >10 kb) showing a difference of more than five copies (tables S35 and S36). Of these genes, 67% (29/43) are increased in Neandertals compared with present-day humans, and most of these are genes of unknown function. One of the most extreme examples is the gene PRR20 (NM_198441), for which we predicted 68 copies in Neandertals, 16 in humans, and 58 in the chimpanzee. It encodes a hypothetical proline-rich protein of unknown function. Other genes with predicted higher copy number in humans as opposed to Neandertals included NBPF14 (DUF1220), DUX4 (NM_172239), REXO1L1 (NM_033178), and TBC1D3 (NM_001123391).
A screen for positive selection in early modern humans
Neandertals fall within the variation of present-day humans for many regions of the genome; that is, Neandertals often share derived single-nucleotide polymorphism (SNP) alleles with present-day humans. We devised an approach to detect positive selection in early modern humans that takes advantage of this fact by looking for genomic regions where present-day humans share a common ancestor subsequent to their divergence from Neandertals, and Neandertals therefore lack derived alleles found in present-day humans (except in rare cases of parallel substitutions) (Fig. 4A). Gene flow between Neandertals and modern humans after their initial population separation might obscure some cases of positive selection by causing Neandertals and present-day humans to share derived alleles, but it will not cause false-positive signals.
Fig. 4.

Selective sweep screen. (A) Schematic illustration of the rationale for the selective sweep screen. For many regions of the genome, the variation within current humans is old enough to include Neandertals (left). Thus, for SNPs in present-day humans, Neandertals often carry the derived allele (blue). However, in genomic regions where an advantageous mutation arises (right, red star) and sweeps to high frequency or fixation in present-day humans, Neandertals will be devoid of derived alleles. (B) Candidate regions of selective sweeps. All 4235 regions of at least 25 kb where S (see SOM Text 13) falls below two standard deviations of the mean are plotted by their S and genetic width. Regions on the autosomes are shown in orange and those on the X chromosome in blue. The top 5% by S are shadowed in light blue. (C) The top candidate region from the selective sweep screen contains two genes, ZFP36L2 and THADA. The red line shows the log-ratio of the number of observed Neandertal-derived alleles versus the number of expected Neandertal-derived alleles, within a 100 kilobase window. The blue dots above the panel indicate all SNP positions, and the green dots indicate SNPs where the Neandertal carries the derived allele.
We identified SNPs as positions that vary among the five present-day human genomes of diverse ancestry plus the human reference genome and used the chimpanzee genome to determine the ancestral state (SOM Text 13). We ignored SNPs at CpG sites since these evolve rapidly and may thus be affected by parallel mutations. We identified 5,615,438 such SNPs, at about 10% of which Neandertals carry the derived allele. As expected, SNPs with higher frequencies of the derived allele in present-day humans were more likely to show the derived allele in Neandertals (fig. S31A). We took advantage of this fact to calculate (fig. S31C) the expected number of Neandertal-derived alleles within a given region of the human genome. The observed numbers of derived alleles were then compared with the expected numbers to identify regions where the Neandertal carries fewer derived alleles than expected relative to the human allelic states. A unique feature of this method is that it has more power to detect older selective sweeps where allele frequency spectra in present-day humans have recovered to the point that appreciable derived allele frequencies are observed, whereas it has relatively low power to detect recent selective sweeps where the derived alleles are at low frequencies in present-day humans. It is therefore particularly suited to detect positive selection that occurred early during the history of modern human ancestors in conjunction with, or shortly after, their population divergence from Neandertals (Fig. 4A).
We identified a total of 212 regions containing putative selective sweeps (Fig. 4B and SOM Text 13). The region with the strongest statistical signal contained a stretch of 293 consecutive SNP positions in the first half of the gene AUTS2 where only ancestral alleles are observed in the Neandertals (fig. S34).
We ranked the 212 regions with respect to their genetic width in centimorgans (Fig. 4B, and table S37) because the size of a region affected by a selective sweep will be larger the fewer generations it took for the sweep to reach fixation, as fewer recombination events will then have occurred during the sweep. Thus, the more intense the selection that drove a putative sweep, the larger the affected region is expected to be. Table 3 lists the 20 widest regions and the genes encoded in them. Five of the regions contain no protein-coding genes. These may thus contain structural or regulatory genomic features under positive selection during early human history. The remaining 15 regions contain between one and 12 genes. The widest region is located on chromosome 2 and contains the gene THADA, where a region of 336 kb is depleted of derived alleles in Neandertals. SNPs in the vicinity of THADA have been associated with type II diabetes, and THADA expression differs between individuals with diabetes and healthy controls (63). Changes in THADA may thus have affected aspects of energy metabolism in early modern humans. The largest deficit of derived alleles in Neandertal THADA is in a region where the Neandertals carry ancestral alleles at 186 consecutive human SNP positions (Fig. 4C). In this region, we identified a DNA sequence element of ~700 bp that is conserved from mouse to primates, whereas the human reference genome as well as the four humans for which data are available carry an insertion of 9 bp that is not seen in the Neandertals. We note, however, that this insertion is polymorphic in humans, as it is in dbSNP.
Table 3.
Top 20 candidate selective sweep regions.
| Region (hg18) | S | Width (cM) | Gene(s) |
|---|---|---|---|
| chr2:43265008-43601389 | −6.04 | 0.5726 | ZFP36L2;THADA |
| chr11:95533088-95867597 | −4.78 | 0.5538 | JRKL;CCDC82;MAML2 |
| chr10:62343313-62655667 | −6.1 | 0.5167 | RHOBTB1 |
| chr21:37580123-37789088 | −4.5 | 0.4977 | DYRK1A |
| chr10:83336607-83714543 | −6.13 | 0.4654 | NRG3 |
| chr14:100248177-100417724 | −4.84 | 0.4533 | MIR337;MIR665;DLK1;RTL1;MIR431;MIR493;MEG3;MIR770 |
| chr3:157244328-157597592 | −6 | 0.425 | KCNAB1 |
| chr11:30601000-30992792 | −5.29 | 0.3951 | |
| chr2:176635412-176978762 | −5.86 | 0.3481 | HOXD11;HOXD8;EVX2;MTX2;HOXD1;HOXD10;HOXD13; HOXD4;HOXD12;HOXD9;MIR10B;HOXD3 |
| chr11:71572763-71914957 | −5.28 | 0.3402 | CLPB;FOLR1;PHOX2A;FOLR2;INPPL1 |
| chr7:41537742-41838097 | −6.62 | 0.3129 | INHBA |
| chr10:60015775-60262822 | −4.66 | 0.3129 | BICC1 |
| chr6:45440283-45705503 | −4.74 | 0.3112 | RUNX2;SUPT3H |
| chr1:149553200-149878507 | −5.69 | 0.3047 | SELENBP1;POGZ;MIR554;RFX5;SNX27;CGN;TUFT1;PI4KB; PSMB4 |
| chr7:121763417-122282663 | −6.35 | 0.2855 | RNF148;RNF133;CADPS2 |
| chr7:93597127-93823574 | −5.49 | 0.2769 | |
| chr16:62369107-62675247 | −5.18 | 0.2728 | |
| chr14:48931401-49095338 | −4.53 | 0.2582 | |
| chr6:90762790-90903925 | −4.43 | 0.2502 | BACH2 |
| chr10:9650088-9786954 | −4.56 | 0.2475 |
Mutations in several genes in Table 3 have been associated with diseases affecting cognitive capacities. DYRK1A, which lies in the Down syndrome critical region, is thought to underlie some of the cognitive impairment associated with having three copies of chromsome 21 (64). Mutations in NRG3 have been associated with schizophrenia, a condition that has been suggested to affect human-specific cognitive traits (65, 66). Mutations in CADPS2 have been implicated in autism (67), as have mutations in AUTS2 (68). Autism is a developmental disorder of brain function in which social interactions, communication, activity, and interest patterns are affected, as well as cognitive aspects crucial for human sociality and culture (69). It may thus be that multiple genes involved in cognitive development were positively selected during the early history of modern humans.
One gene of interest may be RUNX2 (CBFA1). It is the only gene in the genome known to cause cleidocranial dysplasia, which is characterized by delayed closure of cranial sutures, hypoplastic or aplastic clavicles, a bell-shaped rib cage, and dental abnormalities (70). Some of these features affect morphological traits for which modern humans differ from Neandertals as well as other earlier hominins. For example, the cranial malformations seen in cleidocranial dysplasia include frontal bossing, i.e., a protruding frontal bone. A more prominent frontal bone is a feature that differs between modern humans and Neandertals as well as other archaic hominins. The clavicle, which is affected in cleidocranial dysplasia, differs in morphology between modern humans and Neandertals (71) and is associated with a different architecture of the shoulder joint. Finally, a bell-shaped rib cage is typical of Neandertals and other archaic hominins. A reasonable hypothesis is thus that an evolutionary change in RUNX2 was of importance in the origin of modern humans and that this change affected aspects of the morphology of the upper body and cranium.
Population divergence of Neandertals and modern humans
A long-standing question is when the ancestral populations of Neandertals and modern humans diverged. Population divergence, defined as the time point when two populations last exchanged genes, is more recent than the DNA sequence divergence because the latter is the sum of the time to population divergence plus the average time to the common ancestors of DNA sequences within the ancestral population. The divergence time of two populations can be inferred from the frequency with which derived alleles of SNPs discovered in one population are seen in the other population. The reason for this is that the older the population divergence, the more likely it is that derived alleles discovered in one population are due to novel mutations in that population. We compared transversion SNPs identified in a Yoruba individual (33) to other humans and used the chimpanzee and orangutan genomes to identify the ancestral alleles. We found that the proportion of derived alleles is 30.6% in the Yoruba, 29.8% in the Han Chinese, 29.7% in the French, 29.3% in the Papuan, 26.3% in the San, and 18.0% in Neandertals. We used four models of Yoruba demographic history to translate derived allele fractions to population divergence (SOM Text 14). All provided similar estimates. Assuming that human-chimpanzee average DNA sequence divergence was 5.6 to 8.3 million years ago, this suggests that Neandertals and present-day human populations separated between 270,000 and 440,000 years ago (SOM Text 14), a date that is compatible with some interpretations of the paleontological and archaeological record (2, 72).
Neandertals are closer to non-Africans than to Africans
To test whether Neandertals are more closely related to some present-day humans than to others, we identified SNPs by comparing one randomly chosen sequence from each of two present-day humans and asking if the Neandertals match the alleles of the two individuals equally often. If gene flow between Neandertals and modern humans ceased before differentiation between present-day human populations began, this is expected to be the case no matter which present-day humans are compared. The prediction of this null hypothesis of no gene flow holds regardless of population expansions, bottlenecks, or substructure that might have occurred in modern human history (SOM Text 15). The reason for this is that when single chromosomes are analyzed in the two present-day populations, differences in demographic histories in the two populations will not affect the results even if they may profoundly influence allele frequencies. Under the alternative model of later gene flow between Neandertals and modern humans, we expect Neandertals to match alleles in individuals from some parts of the world more often than the others.
We restricted this analysis to biallelic SNPs where two present-day humans carry different alleles and where the Neandertals carried the derived allele, i.e., not matching chimpanzee. We measured the difference in the percent matching by a statistic D(H 1, H 2, Neandertal, chimpanzee) (SOM Text 15) that does not differ significantly from zero when the derived alleles in the Neandertal match alleles in the two humans equally often. If D is positive, Neandertal alleles match alleles in the second human (H 2) more often, while if D is negative, Neandertal alleles match alleles in the first human (H 1) more often. We performed this test using eight present-day humans: two European Americans (CEU), two East Asians (ASN), and four West Africans (YRI), for whom sequences have been generated with Sanger technology, with reads of ~750 bp that we mapped along with the Neandertal reads to the chimpanzee genome. We find that the Neandertals are equally close to Europeans and East Asians: D(ASN, CEU, Neandertal, chimpanzee) = –0.53 ± 0.46% (<1.2 SD from 0% or _P_ = 0.25). However, the Neandertals are significantly closer to non-Africans than to Africans: _D(YRI, CEU, Neandertal, chimpanzee)_ = 4.57 ± 0.39% and _D_(_YRI, ASN, Neandertal, chimpanzee_) = 4.81 ± 0.39% (both >11 SD from 0% or P << 10−12) (table S51).
The greater genetic proximity of Neandertals to Europeans and Asians than to Africans is seen no matter how we subdivide the data: (i) by individual pairs of humans (Table 4), (ii) by chromosome, (iii) by substitutions that are transitions or transversions, (iv) by hypermutable CpG versus all other sites, (v) by Neandertal sequences shorter or longer than 50 bp, and (vi) by 454 or Illumina data. It is also seen when we restrict the analysis to A/T and C/G substitutions, showing that our observations are unlikely to be due to biased allele calling or biased gene conversion (SOM Text 15).
Table 4.
Neandertals are more closely related to present-day non-Africans than to Africans. For each pair of modern humans H1 and H2 that we examined, we reported D (_H_1, _H_2, Neandertal, Chimpanzee): the difference in the percentage matching of Neandertal to two humans at sites where Neandertal does not match chimpanzee, with ±1 standard error. Values that deviate significantly from 0% after correcting for 38 hypotheses tested are highlighted in bold (|Z| > 2.8 SD). Neandertal is skewed toward matching non-Africans more than Africans for all pairwise comparisons. Comparisons within Africans or within non-Africans are all consistent with 0%.
| Population comparison | H1 | H2 | % Neandertal matching to H2 –% Neandertal matching to H1(±1 standard error) |
|---|---|---|---|
| ABI3730 sequencing (~750 bp reads) used to discover H1-H2 differences | |||
| African to African | NA18517 (Yoruba) | NA18507 (Yoruba) | −0.1 ± 0.6 |
| NA18517 (Yoruba) | NA19240 (Yoruba) | 1.5 ± 0.7 | |
| NA18517 (Yoruba) | NA19129 (Yoruba) | −0.1 ± 0.7 | |
| NA18507 (Yoruba) | NA19240 (Yoruba) | −0.5 ± 0.6 | |
| NA18507 (Yoruba) | NA19129 (Yoruba) | 0.0 ± 0.5 | |
| NA19240 (Yoruba) | NA19129 (Yoruba) | −0.6 ± 0.7 | |
| African to Non-African | NA18517 (Yoruba) | NA12878 (European) | 4.1 ± 0.8 |
| NA18517 (Yoruba) | NA12156 (European) | 5.1 ± 0.7 | |
| NA18517 (Yoruba) | NA18956 (Japanese) | 2.9 ± 0.8 | |
| NA18517 (Yoruba) | NA18555 (Chinese) | 3.9 ± 0.7 | |
| NA18507 (Yoruba) | NA12878 (European) | 4.2 ± 0.6 | |
| NA18507 (Yoruba) | NA12156 (European) | 5.5 ± 0.6 | |
| NA18507 (Yoruba) | NA18956 (Japanese) | 5.0 ± 0.7 | |
| NA18507 (Yoruba) | NA18555 (Chinese) | 5.8 ± 0.6 | |
| NA19240 (Yoruba) | NA12878 (European) | 3.5 ± 0.7 | |
| NA19240 (Yoruba) | NA12156 (European) | 3.1 ± 0.7 | |
| NA19240 (Yoruba) | NA18956 (Japanese) | 2.7 ± 0.7 | |
| NA19240 (Yoruba) | NA18555 (Chinese) | 5.4 ± 0.9 | |
| NA19129 (Yoruba) | NA12878 (European) | 3.9 ± 0.7 | |
| NA19129 (Yoruba) | NA12156 (European) | 4.9 ± 0.7 | |
| NA19129 (Yoruba) | NA18956 (Japanese) | 5.1 ± 0.8 | |
| NA19129 (Yoruba) | NA18555 (Chinese) | 4.7 ± 0.8 | |
| Non-African to Non-African | NA12878 (European) | NA12156 (European) | −0.5 ± 0.8 |
| NA12878 (European) | NA18956 (Japanese) | 0.4 ± 0.8 | |
| NA12878 (European) | NA18555 (Chinese) | 0.3 ± 0.8 | |
| NA12156 (European) | NA18956 (Japanese) | −0.3 ± 0.8 | |
| NA12156 (European) | NA18555 (Chinese) | 1.3 ± 0.7 | |
| NA18956 (Japanese) | NA18555 (Chinese) | 2.5 ± 0.9 | |
| Illumina GAII sequencing (~76 bp reads) used to discover H1-H2 differences | |||
| African - African | HGDP01029 (San) | HGDP01029 (Yoruba) | −0.1 ± 0.4 |
| African to Non-African | HGDP01029 (San) | HGDP00521 (French) | 4.2 ± 0.4 |
| HGDP01029 (San) | HGDP00542 (Papuan) | 3.9 ± 0.5 | |
| HGDP01029 (San) | HGDP00778 (Han) | 5.0 ± 0.5 | |
| HGDP01029 (Yoruba) | HGDP00521 (French) | 4.5 ± 0.4 | |
| HGDP01029 (Yoruba) | HGDP00542 (Papuan) | 4.4 ± 0.6 | |
| HGDP01029 (Yoruba) | HGDP00778 (Han) | 5.3 ± 0.5 | |
| Non-African to Non-African | HGDP00521 (French) | HGDP00542 (Papuan) | 0.1 ± 0.5 |
| HGDP00521 (French) | HGDP00778 (Han) | 1.0 ± 0.6 | |
| HGDP00542 (Papuan) | HGDP00778 (Han) | 0.7 ± 0.6 |
A potential artifact that might explain these observations is contamination of the Neandertal sequences with non-African DNA. However, the magnitude of contamination necessary to explain the CEU-YRI and ASN-YRI comparisons are both over 10% and thus inconsistent with our estimates of contamination in the Neandertal data, which are all below 1% (Table 1). In addition to the low estimates of contamination, there are two reasons that contamination cannot explain our results. First, when we analyze the three Neandertal bones Vi33.16, Vi33.25, and Vi33.26 separately, we obtain consistent values of the D statistics, which is unlikely to arise under the hypothesis of contamination because each specimen was individually handled and was thus unlikely to have been affected by the same degree of contamination (SOM Text 15). Second, if European contamination explains the skews, the ratio D(H1, H2, Neandertal, chimpanzee)/D(H1, H2, European, chimpanzee) should provide a direct estimate of the contamination proportion α, because the ratio measures how close the Neandertal data are to what would be expected from entirely European contamination. However, when we estimate α for all three population pairs, we obtain statistically inconsistent results: α = 13.9 ± 1.1% for H1-H2 = CEU-YRI, α = 18.9 ± 1.9% for ASN-YRI, and α = −3.9 ± 5.1% for CEU-ASN. This indicates that the skews cannot be explained by a unifying hypothesis of European contamination.
To analyze the relationship of the Neandertals to a more diverse set of modern humans, we repeated the analysis above using the genome sequences of the French, Han, Papuan, Yoruba, and San individuals that we generated (SOM Text 9). Strikingly, no comparison within Eurasia (Papuan-French-Han) or within Africa (Yoruba-San) shows significant skews in D (|Z| < 2 SD). However, all comparisons of non-Africans and Africans show that the Neandertal is closer to the non-African (_D_ from 3.8% to 5.3%, |_Z_| > 7.0 SD) (Table 4). Thus, analyses of present-day humans consistently show that Neandertals share significantly more derived alleles with non-Africans than with Africans, whereas they share equal amounts of derived alleles when compared either to individuals within Eurasia or to individuals within Africa.
Direction of gene flow
A parsimonious explanation for these observations is that Neandertals exchanged genes with the ancestors of non-Africans. To determine the direction of gene flow consistent with the data, we took advantage of the fact that non-Africans are more distantly related to San than to Yoruba (73–75) (Table 4). This is reflected in the fact that D(P, San, Q, chimpanzee) is 1.47 to 1.68 times greater than D(P, Yoruba, Q, chimpanzee), where P and Q are non-Africans (SOM Text 15). Under the hypothesis of modern human to Neandertal gene flow, D(P, San, Neandertal, chimpanzee) should be greater than D(P, Yoruba, Neandertal, chimpanzee) by the same amount, because the deviation of the D statistics is due to Neandertals inheriting a proportion of ancestry from a non-African-like population Q. Empirically, however, the ratio is significantly smaller (1.00 to 1.03, P << 0.0002) (SOM Text 15). Thus, all or almost all of the gene flow detected was from Neandertals into modern humans.
Segments of Neandertal ancestry in non-African genomes
If Neandertal-to-modern human gene flow occurred, we predict that we should find DNA segments with an unusually low divergence to Neandertal in present-day humans. Furthermore, we expect that such segments will tend to have an unusually high divergence to other present-day humans because they come from Neandertals. In the absence of gene flow, segments with low divergence to Neandertals are expected to arise due to other effects, for example, a low mutation rate in a genomic segment since the split from the chimpanzee lineage. However, this will cause present-day humans to tend to have low divergence from each other in such segments, i.e., the opposite effect from gene flow. The qualitative distinction between these predictions allows us to detect a signal of gene flow. To search for segments with relatively few differences between Neandertals and present-day humans, we used haploid human DNA sequences, because in a diploid individual, both alleles would have to be derived from Neandertals to produce a strong signal. To obtain haploid human sequences, we took advantage of the fact that the human genome reference sequence is composed of a tiling path of bacterial artificial chromosomes (BACs), which each represent single human haplotypes over scales of 50 to 150 kb, and we focused on BACs from RPCI11, the individual that contributed about two-thirds of the reference sequence and that has been previously shown to be of about 50% European and 50% African ancestry (SOM Text 16) (76). We then estimated the Neandertal to present-day human divergence and found that in the extreme tail of low-divergence BACs there was a greater proportion of European segments than African segments, consistent with the notion that some genomic segments (SOM Text 16) were exchanged between Neandertals and non-Africans.
To determine whether these segments are unusual in their divergence to other present-day humans, we examined the divergence of each segment to the genome of Craig Venter (77). We find that present-day African segments with the lowest divergence to Neandertals have a divergence to Venter that is 35% of the genome-wide average and that their divergence to Venter increases monotonically with divergence to Neandertals, as would be expected if these segments were similar in Neandertals and present-day humans due to, for example, a low mutation rate in these segments (Fig. 5A). In contrast, the European segments with the lowest divergence to Neandertals have a divergence to Venter that is 140% of the genome-wide average, which drops precipitously with increasing divergence to humans before rising again (Fig. 5A). This nonmonotonic behavior is significant at P < 10−9 and is unexpected in the absence of gene flow from Neandertals into the ancestors of non-Africans. The reason for this is that other causes for a low divergence to Neandertals, such as low mutation rates, contamination by modern non-African DNA, or gene flow into Neandertals, would produce monotonic behaviors. Among the segments with low divergence to Neandertals and high divergence to Venter, 94% of segments are of European ancestry (Fig. 5B), suggesting that segments of likely Neandertal ancestry in present-day humans can be identified with relatively high confidence.
Fig. 5.

Segments of Neandertal ancestry in the human reference genome. We examined 2825 segments in the human reference genome that are of African ancestry and 2797 that are of European ancestry. (A) European segments, with few differences from the Neandertals, tend to have many differences from other present-day humans, whereas African segments do not, as expected if the former are derived from Neandertals. (B) Scatter plot of the segments in (A) with respect to their divergence to the Neandertals and to Venter. In the top left quandrant, 94% of segments are of European ancestry, suggesting that many of them are due to gene flow from Neandertals.
Non-Africans haplotypes match Neandertals unexpectedly often
An alternative approach to detect gene flow from Neandertals into modern humans is to focus on patterns of variation in present-day humans—blinded to information from the Neandertal genome—in order to identify regions that are the strongest candidates for being derived from Neandertals. If these candidate regions match the Neandertals at a higher rate than is expected by chance, this provides additional evidence for gene flow from Neandertals into modern humans.
We thus identified regions in which there is considerably more diversity outside Africa than inside Africa, as might be expected in regions that have experienced gene flow from Neandertals to non-Africans. We used 1,263,750 Perlegen Class A SNPs, identified in individuals of diverse ancestry (78), and found 13 candidate regions of Neandertal ancestry (SOM Text 17). A prediction of Neandertal-to-modern human gene flow is that DNA sequences that entered the human gene pool from Neandertals will tend to match Neandertal more often than their frequency in the present-day human population. To test this prediction, we identified 166 “tag SNPs” that separate 12 of the haplotype clades in non-Africans (OOA) from the cosmopolitan haplotype clades shared between Africans and non-Africans (COS) and for which we had data from the Neandertals. Overall, the Neandertals match the deep clade unique to non-Africans at 133 of the 166 tag SNPs, and 10 of the 12 regions where tag SNPs occur show an excess of OOA over COS sites. Given that the OOA alleles occur at a frequency of much less than 50% in non-Africans (average of 13%, and all less than 30%) (Table 5), the fact that the candidate regions match the Neandertals in 10 of 12 cases (P = 0.019) suggests that they largely derive from Neandertals. The proportion of matches is also larger than can be explained by contamination, even if all Neandertal data were composed of present-day non-African DNA (P = 0.0025) (SOM Text 17).
Table 5.
Non-African haplotypes match Neandertal at an unexpected rate. We identified 13 candidate gene flow regions by using 48 CEU+ASN to represent the OOA population, and 23 African Americans to represent the AFR population. We identified tag SNPs for each region that separate an out-of-Africa specific clade (OOA) from a cosmopolitan clade (COS) and then assessed the rate at which Neandertal matches each of these clades by further subdividing tag SNPs based on their ancestral and derived status in Neandertal and whether they match the OOA-specific clade or not. Thus, the categories are AN (Ancestral Nonmatch), DN (Derived Nonmatch), DM (Derived Match), and AM (Ancestral Match). We do not list the sites where matching is ambiguous.
| Chromo-some | Start of candidateregion in Build 36 | End of candidateregion in Build 36 | Span(bp) | _S_T(estimatedratio ofOOA/AFRgene treedepth) | Averagefrequency oftag in OOAclade | Neandertal(M)atchesOOA-specificcladeAM DM | Neandertal does(N)ot matchOOA-specificcladeAN DN | Qualitativeassessment* | ||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 168,110,000 | 168,220,000 | 110,000 | 2.9 | 6.3% | 5 | 10 | 1 | 0 | OOA |
| 1 | 223,760,000 | 223,910,000 | 150,000 | 2.8 | 6.3% | 1 | 4 | 0 | 0 | OOA |
| 4 | 171,180,000 | 171,280,000 | 100,000 | 1.9 | 5.2% | 1 | 2 | 0 | 0 | OOA |
| 5 | 28,950,000 | 29,070,000 | 120,000 | 3.8 | 3.1% | 16 | 16 | 6 | 0 | OOA |
| 6 | 66,160,000 | 66,260,000 | 100,000 | 5.7 | 28.1% | 6 | 6 | 0 | 0 | OOA |
| 9 | 32,940,000 | 33,040,000 | 100,000 | 2.8 | 4.2% | 7 | 14 | 0 | 0 | OOA |
| 10 | 4,820,000 | 4,920,000 | 100,000 | 2.6 | 9.4% | 9 | 5 | 0 | 0 | OOA |
| 10 | 38,000,000 | 38,160,000 | 160,000 | 3.5 | 8.3% | 5 | 9 | 2 | 0 | OOA |
| 10 | 69,630,000 | 69,740,000 | 110,000 | 4.2 | 19.8% | 2 | 2 | 0 | 1 | OOA |
| 15 | 45,250,000 | 45,350,000 | 100,000 | 2.5 | 1.1% | 5 | 6 | 1 | 0 | OOA |
| 17 | 35,500,000 | 35,600,000 | 100,000 | 2.9 | (no tags) | – | – | – | – | – |
| 20 | 20,030,000 | 20,140,000 | 110,000 | 5.1 | 64.6% | 0 | 0 | 10 | 5 | COS |
| 22 | 30,690,000 | 30,820,000 | 130,000 | 3.5 | 4.2% | 0 | 2 | 5 | 2 | COS |
| Relative tag SNP frequencies in actual data | 34% | 46% | 15% | 5% | ||||||
| Relative tag SNP simulated under a demographic model without introgression | 34% | 5% | 33% | 27% | ||||||
| Relative tag SNP simulated under a demographic model with introgression | 23% | 31% | 37% | 9% |
This analysis shows that some old haplotypes most likely owe their presence in present-day non-Africans to gene flow from Neandertals. However, not all old haplotypes in non-Africans may have such an origin. For example, it has been suggested that the H2 haplotype on chromosome 17 and the D haplotype of the microcephalin gene were contributed by Neandertals to present-day non-Africans (12, 79, 80). This is not supported by the current data because the Neandertals analyzed do not carry these haplotypes.
The extent of Neandertal ancestry
To estimate the proportion of Neandertal ancestry, we compare the similarity of non-Africans to Neandertals with the similarity of two Neandertals, N1 and N2, to each other. Under the assumption that there was no gene flow from Neandertals to the ancestors of modern Africans, the proportion of Neandertal ancestry of non-Africans, f, can be estimated by the ratio S(OOA,AFR,N1,Chimpanzee)/S(N2,AFR,N1,Chimpanzee), where the S statistic is an unnormalized version of the D statistic (SOM Text 18, Eq. S18.4). Using Neandertals from Vindija, as well as Mezmaiskaya, we estimate f to be between 1.3% and 2.7% (SOM Text 18). To obtain an independent estimate of f, we fit a population genetic model to the D statistics in Table 4 and SOM Text 15 as well as to other summary statistics of the data. Assuming that gene flow from Neandertals occurred between 50,000 and 80,000 years ago, this method estimates f to be between 1 and 4%, consistent with the above estimate (SOM Text 19). We note that a previous study found a pattern of genetic variation in present-day humans that was hypothesized to be due to gene flow from Neandertals or other archaic hominins into modern humans (81). The authors of this study estimated the fraction of non-African genomes affected by “archaic” gene flow to be 14%, almost an order of magnitude greater than our estimates, suggesting that their observations may not be entirely explained by gene flow from Neandertals.
Implications for modern human origins
One model for modern human origins suggests that all present-day humans trace all their ancestry back to a small African population that expanded and replaced archaic forms of humans without admixture. Our analysis of the Neandertal genome may not be compatible with this view because Neandertals are on average closer to individuals in Eurasia than to individuals in Africa. Furthermore, individuals in Eurasia today carry regions in their genome that are closely related to those in Neandertals and distant from other present-day humans. The data suggest that between 1 and 4% of the genomes of people in Eurasia are derived from Neandertals. Thus, while the Neandertal genome presents a challenge to the simplest version of an “out-of-Africa” model for modern human origins, it continues to support the view that the vast majority of genetic variants that exist at appreciable frequencies outside Africa came from Africa with the spread of anatomically modern humans.
A striking observation is that Neandertals are as closely related to a Chinese and Papuan individual as to a French individual, even though morphologically recognizable Neandertals exist only in the fossil record of Europe and western Asia. Thus, the gene flow between Neandertals and modern humans that we detect most likely occurred before the divergence of Europeans, East Asians, and Papuans. This may be explained by mixing of early modern humans ancestral to present-day non-Africans with Neandertals in the Middle East before their expansion into Eurasia. Such a scenario is compatible with the archaeological record, which shows that modern humans appeared in the Middle East before 100,000 years ago whereas the Neandertals existed in the same region after this time, probably until 50,000 years ago (82).
It is important to note that although we detect a signal compatible with gene flow from Neandertals into ancestors of present-day humans outside Africa, this does not show that other forms of gene flow did not occur (Fig. 6). For example, we detect gene flow from Neandertals into modern humans but no reciprocal gene flow from modern humans into Neandertals. Although gene flow between different populations need not be bidirectional, it has been shown that when a colonizing population (such as anatomically modern humans) encounters a resident population (such as Neandertals), even a small number of breeding events along the wave front of expansion into new territory can result in substantial introduction of genes into the colonizing population as introduced alleles can “surf” to high frequency as the population expands. As a consequence, detectable gene flow is predicted to almost always be from the resident population into the colonizing population, even if gene flow also occurred in the other direction (83). Another prediction of such a surfing model is that even a very small number of events of interbreeding can result in appreciable allele frequencies of Neandertal alleles in the present-day populations. Thus, the actual amount of interbreeding between Neandertals and modern humans may have been very limited, given that it contributed only 1 to 4% of the genome of present-day non-Africans.
Fig. 6.
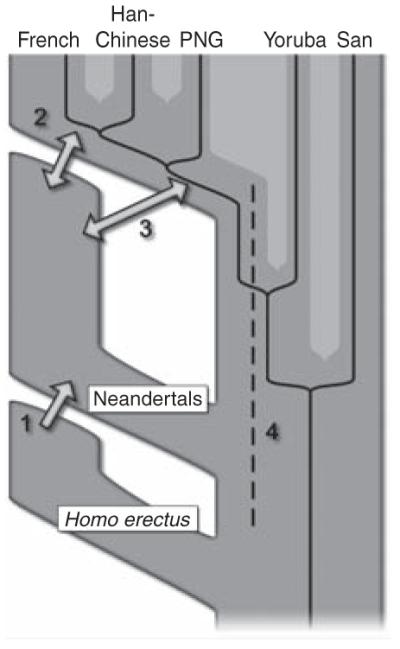
Four possible scenarios of genetic mixture involving Neandertals. Scenario 1 represents gene flow into Neandertal from other archaic hominins, here collectively referred to as Homo erectus. This would manifest itself as segments of the Neandertal genome with unexpectedly high divergence from present-day humans. Scenario 2 represents gene flow between late Neandertals and early modern humans in Europe and/or western Asia. We see no evidence of this because Neandertals are equally distantly related to all non-Africans. However, such gene flow may have taken place without leaving traces in the present-day gene pool. Scenario 3 represents gene flow between Neandertals and the ancestors of all non-Africans. This is the most parsimonious explanation of our observation. Although we detect gene flow only from Neandertals into modern humans, gene flow in the reverse direction may also have occurred. Scenario 4 represents old substructure in Africa that persisted from the origin of Neandertals until the ancestors of non-Africans left Africa. This scenario is also compatible with the current data.
It may seem surprising that we see no evidence for greater gene flow from Neandertals to present-day Europeans than to present-day people in eastern Asia given that the morphology of some hominin fossils in Europe has been interpreted as evidence for gene flow from Neandertals into early modern humans late in Neandertal history [e.g., (84)] (Fig. 6). It is possible that later migrations into Europe, for example in connection with the spread of agriculture, have obscured the traces of such gene flow. This possibility can be addressed by the determination of genome sequences from preagricultural early modern humans in Europe (85). It is also possible that if the expansion of modern humans occurred differently in Europe than in the Middle East, for example by already large populations interacting with Neandertals, then there may be little or no trace of any gene flow in present-day Europeans even if interbreeding occurred. Thus, the contingencies of demographic history may cause some events of past interbreeding to leave traces in present-day populations, whereas other events will leave little or no traces. Obviously, gene flow that left little or no traces in the present-day gene pool is of little or no consequence from a genetic perspective, although it may be of interest from a historical perspective.
Although gene flow from Neandertals into modern humans when they first left sub-Saharan Africa seems to be the most parsimonious model compatible with the current data, other scenarios are also possible. For example, we cannot currently rule out a scenario in which the ancestral population of present-day non-Africans was more closely related to Neandertals than the ancestral population of present-day Africans due to ancient substructure within Africa (Fig. 6). If after the divergence of Neandertals there was incomplete genetic homogenization between what were to become the ancestors of non-Africans and Africans, present-day non-Africans would be more closely related to Neandertals than are Africans. In fact, old population substructure in Africa has been suggested based on genetic (81) as well as paleontological data (86).
In conclusion, we show that genome sequences from an extinct late Pleistocene hominin can be reliably recovered. The analysis of the Neandertal genome shows that they are likely to have had a role in the genetic ancestry of present-day humans outside of Africa, although this role was relatively minor given that only a few percent of the genomes of present-day people outside Africa are derived from Neandertals. Our results also point to a number of genomic regions and genes as candidates for positive selection early in modern human history, for example, those involved in cognitive abilities and cranial morphology. We expect that further analyses of the Neandertal genome as well as the genomes of other archaic hominins will generate additional hypotheses and provide further insights into the origins and early history of present-day humans.
Supplementary Material
Supplement
Acknowledgments
We thank E. Buglione, A. Burke, Y.-J. Chen, J. Salem, P. Schaffer, E. Szekeres, and C. Turcotte at 454 Life Sciences Corp. for production sequencing on the 454 platform; S. Fisher, J. Wilkinson, J. Blye, R. Hegarty, A. Allen, S. K. Young, and J. L. Chang for nine Illumina sequencing runs performed at the Broad Institute; J. Rothberg and E. Rubin for input leading up to this project; O. Bar-Yosef, L. Excoffier, M. Gralle, J.-J. Hublin, D. Lieberman, M. Stoneking, and L. Vigilant for constructive criticism; I. Janković for assistance with the Vindija collection; S. Ptak, M. Siebauer, and J. Visagie for help with data analysis, M. Richards and S. Talamo for carbon dating; J. Dabney for editorial assistance; the Genome Center at Washington University for prepublication use of the orangutan genome assembly; and K. Finstermeier for expert graphical design. Neandertal bone extract sequence data have been deposited at European Bioinformatics Institute under STUDY accession ERP000119, alias Neandertal Genome project. HGDP sequence data have been deposited at EBI under STUDY accession ERP000121, alias Human Genome Diversity Project. We are grateful to the Max Planck Society, and particularly the Presidential Innovation Fund, for making this project possible. C.L.-F. was supported by a grant from the Ministerio de Ciencia e Innovación; E.Y.D. and M.S. were supported in part by grant GM40282; A.-S.M. was supported by a Janggen-Pöhn fellowship; N.F.H. and J.C.M. were supported in part by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health; and D.R. by a Burroughs Wellcome Career Development Award in the Biomedical Sciences.
Footnotes
Author contributions: S.P. conceived and coordinated the project; D.R. coordinated population genetic analyses; R.E.G. and J.Ke. coordinated bioinformatic aspects; R.E.G., J.Kr., A.W.B., M.E., and S.P. developed the initial project strategies; J.Kr. and T.M. collected and analyzed fossil samples; J.Kr., T.M., A.W.B., and M.M. developed the DNA extraction and library preparation protocols and performed laboratory work prior to sequencing; K.P. designed the restriction enzyme enrichment method; A.A.-P., A.B., B.Hb., B.Hff., M.Sg., R.S., A.W., J.A., M.E., and M.K. performed and coordinated DNA sequencing on the 454 and Illumina platforms; J.A. and M.E. organized and coordinated sequence production on the 454 platform; C.N., E.S.L., C.R., and N.N. organized and performed nine sequencing runs on the Illumina platform at the Broad Institute; M.K. and J.Ke. compiled the catalog of human-specific genomic features; U.S., M.K., N.H., J.M., J.Ke., K.P., and R.E.G. developed and implemented the primary sequence alignment and analysis methodologies; R.E.G., U.S., J.Kr., A.W.B., H.B., P.L.F.J. and M.L. developed and implemented the wet lab and bioinformatic assays for human DNA contamination; C.A., T.M.-B., and E.E.E. performed structural variation analyses; H.L., J.M., and D.R. designed and implemented analyses of population divergences; R.E.G., N.P., W.Z., J.M., H.L., M.H.-Y.F., E.Y.D., A.S.-M., P.L.F.J., J.J., J.G., M.L., D.F., M.S., E.B., R.N., S.P., and D.R. developed and implemented population genetics comparisons; R.E.G., M.L., J.G., D.F., J.D.J., D.R., and S.P. designed and implemented the screen for selective sweeps; P.R., D.B., Z.K., I.G., C.V., V.B.D., L.V.G., C.L.-F., M.R., J.F., A.R., and R.S. provided samples, analyses, and paleontological expertise; D.R. and S.P. edited the manuscript.
References and Notes
- 1.Bischoff JL, et al. High-Resolution U-Series Dates from the Sima de los Huesos Hominids Yields 600+/−66 kyrs: Implications for the Evolution of the Early Neanderthal Lineage. Vol. 34. Elsevier; Amsterdam, PAYS-BAS: 2007. [Google Scholar]
- 2.Hublin JJ. Proc. Natl. Acad. Sci. U.S.A. 2009;106:16022. doi: 10.1073/pnas.0904119106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stringer CB, Hublin J. J. Hum. Evol. 1999;37:873. doi: 10.1006/jhev.1999.0367. [DOI] [PubMed] [Google Scholar]
- 4.Finlayson C, et al. Nature. 2006;443:850. doi: 10.1038/nature05195. [DOI] [PubMed] [Google Scholar]
- 5.Krause J, et al. Nature. 2007;449:902. doi: 10.1038/nature06193. [DOI] [PubMed] [Google Scholar]
- 6.Grün R, et al. J. Hum. Evol. 2005;49:316. doi: 10.1016/j.jhevol.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 7.Mercier N, Valladas H. In: Late Quaternary Chronology and Palaeoclimate of the Eastern Mediterranean, Radiocarbon. Bar-Yosef O, Kra R, editors. 1994. pp. 13–20. [Google Scholar]
- 8.Trinkaus E, et al. Proc. Natl. Acad. Sci. U.S.A. 2003;100:11231. doi: 10.1073/pnas.2035108100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zilhão J, Trinkaus E. Trabalhos de Arqueologia. Vol. 22. Instituto Português de Arqueologia; Lisbon: 2002. [Google Scholar]
- 10.Bailey SE, Weaver TD, Hublin JJ. J. Hum. Evol. 2009;57:11. doi: 10.1016/j.jhevol.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 11.Bräuer G, Broeg H, Stringer C. Neanderthals Revisited: New Approaches and Perspectives. 2006:269–279. [Google Scholar]
- 12.Evans PD, Mekel-Bobrov N, Vallender EJ, Hudson RR, Lahn BT. Proc. Natl. Acad. Sci. U.S.A. 2006;103:18178. doi: 10.1073/pnas.0606966103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wall JD, Hammer MF. Curr. Opin. Genet. Dev. 2006;16:606. doi: 10.1016/j.gde.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 14.Currat M, Excoffier L. PLoS Biol. 2004;2:e421. doi: 10.1371/journal.pbio.0020421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Briggs AW, et al. Science. 2009;325:318. doi: 10.1126/science.1174462. [DOI] [PubMed] [Google Scholar]
- 16.Krings M, et al. Cell. 1997;90:19. doi: 10.1016/s0092-8674(00)80310-4. [DOI] [PubMed] [Google Scholar]
- 17.Orlando L, et al. Curr. Biol. 2006;16:R400. doi: 10.1016/j.cub.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 18.Ovchinnikov IV, et al. Nature. 2000;404:490. doi: 10.1038/35006625. [DOI] [PubMed] [Google Scholar]
- 19.Serre D, et al. PLoS Biol. 2004;2:E57. doi: 10.1371/journal.pbio.0020057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pääbo S. Trends Cell Biol. 1999;9:M13. [PubMed] [Google Scholar]
- 21.Pääbo S. Proc. Natl. Acad. Sci. U.S.A. 1989;86:1939. doi: 10.1073/pnas.86.6.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pääbo S, et al. Annu. Rev. Genet. 2004;38:645. doi: 10.1146/annurev.genet.37.110801.143214. [DOI] [PubMed] [Google Scholar]
- 23.Briggs AW, et al. Proc. Natl. Acad. Sci. U.S.A. 2007;104:14616. [Google Scholar]
- 24.Brotherton P, et al. Nucleic Acids Res. 2007;35:5717. doi: 10.1093/nar/gkm588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hofreiter M, Jaenicke V, Serre D, von Haeseler A, Pääbo S. Nucleic Acids Res. 2001;29:4793. doi: 10.1093/nar/29.23.4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Höss M, Jaruga P, Zastawny TH, Dizdaroglu M, Pääbo S. Nucleic Acids Res. 1996;24:1304. doi: 10.1093/nar/24.7.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saiki RK, et al. Science. 1985;230:1350. doi: 10.1126/science.2999980. [DOI] [PubMed] [Google Scholar]
- 28.Lalueza-Fox C, et al. Science. 2007;318:1453. [Google Scholar]
- 29.Krause J, et al. Curr. Biol. 2007;17:1908. doi: 10.1016/j.cub.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 30.Lalueza-Fox C, et al. BMC Evol. Biol. 2008;8:342. doi: 10.1186/1471-2148-8-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lalueza-Fox C, Gigli E, de la Rasilla M, Fortea J, Rosas A. Biol. Lett. 2009;5:809. doi: 10.1098/rsbl.2009.0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krause J, et al. Nature. 2006;439:724. doi: 10.1038/nature04432. [DOI] [PubMed] [Google Scholar]
- 33.Bentley DR, et al. Nature. 2008;456:53. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Margulies M, et al. Nature. 2005;437:376. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poinar HN, et al. Science. 2006;311:392. doi: 10.1126/science.1123360. [DOI] [PubMed] [Google Scholar]
- 36.Rasmussen M, et al. Nature. 2010;463:757. doi: 10.1038/nature08835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stiller M, et al. Proc. Natl. Acad. Sci. U.S.A. 2006;103:13578. doi: 10.1073/pnas.0605327103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller W, et al. Nature. 2008;456:387. doi: 10.1038/nature07446. [DOI] [PubMed] [Google Scholar]
- 39.Prüfer K, et al. Genome Biol. 2010;11:R47. doi: 10.1186/gb-2010-11-5-r47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Green RE, et al. Nature. 2006;444:330. doi: 10.1038/nature05336. [DOI] [PubMed] [Google Scholar]
- 41.Green RE, et al. EMBO J. 2009;28:2494. doi: 10.1038/emboj.2009.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Noonan JP, et al. Science. 2006;314:1113. doi: 10.1126/science.1131412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Greenwood AD, Capelli C, Possnert G, Pääbo S. Mol. Biol. Evol. 1999;16:1466. doi: 10.1093/oxfordjournals.molbev.a026058. [DOI] [PubMed] [Google Scholar]
- 44.Wall JD, Kim SK. PLoS Genet. 2007;3:e175. doi: 10.1371/journal.pgen.0030175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Green RE, et al. Cell. 2008;134:416. doi: 10.1016/j.cell.2008.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Briggs AW, et al. J. Vis. Exp. 2009;2009:1573. doi: 10.3791/1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maricic T, Pääbo S. Biotechniques. 2009;46:51, 54. doi: 10.2144/000113042. [DOI] [PubMed] [Google Scholar]
- 48.Kircher M, Stenzel U, Kelso J. Genome Biol. 2009;10:R83. doi: 10.1186/gb-2009-10-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Briggs AW, et al. Nucleic Acids Res. 2010;38:e87. doi: 10.1093/nar/gkp1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dohm JC, Lottaz C, Borodina T, Himmelbauer H. Nucleic Acids Res. 2008;36:e105. doi: 10.1093/nar/gkn425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paten B, et al. Genome Res. 2008;18:1829. doi: 10.1101/gr.076521.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goodman M. Am. J. Hum. Genet. 1999;64:31. doi: 10.1086/302218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.de Torres T, et al. Archaeometry. doi: 10.1111/j.1475-4754.2009.00491.x. published online 29 October 2009. [DOI] [Google Scholar]
- 54.Schmitz RW, et al. Proc. Natl. Acad. Sci. U.S.A. 2002;99:13342. doi: 10.1073/pnas.192464099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Skinner AR, et al. Appl. Radiat. Isot. 2005;62:219. doi: 10.1016/j.apradiso.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 56.Patterson N, Richter DJ, Gnerre S, Lander ES, Reich D. Nature. 2006;441:1103. doi: 10.1038/nature04789. [DOI] [PubMed] [Google Scholar]
- 57.Burbano HA, et al. Science. 2010;328:723. doi: 10.1126/science.1188046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Z, et al. Mol. Cell. Proteomics. 2005;4:914. doi: 10.1074/mcp.M400177-MCP200. [DOI] [PubMed] [Google Scholar]
- 59.Matsuyoshi N, Imamura S. Biochem. Biophys. Res. Commun. 1997;235:355. doi: 10.1006/bbrc.1997.6707. [DOI] [PubMed] [Google Scholar]
- 60.Richard P, Manley JL. Genes Dev. 2009;23:1247. doi: 10.1101/gad.1792809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huber M, et al. J. Invest. Dermatol. 2005;124:998. doi: 10.1111/j.0022-202X.2005.23675.x. [DOI] [PubMed] [Google Scholar]
- 62.Alkan C, et al. Nat. Genet. 2009;41:1061. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Parikh H, Lyssenko V, Groop LC. BMC Med. Genomics. 2009;2:72. doi: 10.1186/1755-8794-2-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hämmerle B, Elizalde C, Galceran J, Becker W, Tejedor FJ. J. Neural Transm. Suppl. 2003;2003:129. doi: 10.1007/978-3-7091-6721-2_11. [DOI] [PubMed] [Google Scholar]
- 65.Crow TJ. Eur. Neuropsychopharmacol. 1995;5(suppl):59. doi: 10.1016/0924-977x(95)00032-k. [DOI] [PubMed] [Google Scholar]
- 66.Khaitovich P, et al. Genome Biol. 2008;9:R124. doi: 10.1186/gb-2008-9-8-r124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sadakata T, et al. J. Clin. Invest. 2007;117:931. doi: 10.1172/JCI29031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sultana R, et al. Genomics. 2002;80:129. doi: 10.1006/geno.2002.6810. [DOI] [PubMed] [Google Scholar]
- 69.Tomasello M, Carpenter M, Call J, Behne T, Moll H. Behav. Brain Sci. 2005;28:675. doi: 10.1017/S0140525X05000129. discussion 691. [DOI] [PubMed] [Google Scholar]
- 70.Mundlos S, et al. Cell. 1997;89:773. doi: 10.1016/s0092-8674(00)80260-3. [DOI] [PubMed] [Google Scholar]
- 71.Voisin JL. J. Hum. Evol. 2008;55:438. doi: 10.1016/j.jhevol.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 72.Weaver TD, Roseman CC, Stringer CB. Proc. Natl. Acad. Sci. U.S.A. 2008;105:4645. doi: 10.1073/pnas.0709079105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Behar DM, et al. Am. J. Hum. Genet. 2008;82:1130. doi: 10.1016/j.ajhg.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sun JX, Mullikin JC, Patterson N, Reich DE. Mol. Biol. Evol. 2009;26:1017. doi: 10.1093/molbev/msp025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wood ET, et al. Eur. J. Hum. Genet. 2005;13:867. doi: 10.1038/sj.ejhg.5201408. [DOI] [PubMed] [Google Scholar]
- 76.Reich D, et al. PLoS Genet. 2009;5:e1000360. doi: 10.1371/journal.pgen.1000360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Levy S, et al. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hinds DA, et al. Science. 2005;307:1072. doi: 10.1126/science.1105436. [DOI] [PubMed] [Google Scholar]
- 79.Hardy J, et al. Biochem. Soc. Trans. 2005;33:582. doi: 10.1042/BST0330582. [DOI] [PubMed] [Google Scholar]
- 80.Stefansson H, et al. Nat. Genet. 2005;37:129. doi: 10.1038/ng1508. [DOI] [PubMed] [Google Scholar]
- 81.Wall JD, Lohmueller KE, Plagnol V. Mol. Biol. Evol. 2009;26:1823. doi: 10.1093/molbev/msp096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bar-Yosef O. In: Neandertals and Modern Humans in Western Asia. Akazawa T, Aoki K, Bar-Yosef O, editors. Plenum; New York: 1999. pp. 39–56. [Google Scholar]
- 83.Currat M, Ruedi M, Petit RJ, Excoffier L. Evolution. 2008;62:1908. doi: 10.1111/j.1558-5646.2008.00413.x. [DOI] [PubMed] [Google Scholar]
- 84.Zilhão J, et al. PLoS ONE. 2010;5:e8880. doi: 10.1371/journal.pone.0008880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Krause J, et al. Curr. Biol. 2010;20:231. doi: 10.1016/j.cub.2009.11.068. [DOI] [PubMed] [Google Scholar]
- 86.Gunz P, et al. Proc. Natl. Acad. Sci. U.S.A. 2009;106:6094. doi: 10.1073/pnas.0808160106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li WH, Wu CI, Luo CC. Mol. Biol. Evol. 1985;2:150. doi: 10.1093/oxfordjournals.molbev.a040343. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement