Pain and immunity: implications for host defence (original) (raw)
. Author manuscript; available in PMC: 2020 Jan 1.
Published in final edited form as: Nat Rev Immunol. 2019 Jul;19(7):433–447. doi: 10.1038/s41577-019-0147-2
Abstract
Pain is a hallmark of tissue injury, inflammatory diseases, pathogen invasion and neuropathy. It is mediated by nociceptor sensory neurons that innervate the skin, joints, bones, muscles and mucosal tissues and protects organisms from noxious stimuli. Nociceptors are sensitized by inflammatory mediators produced by the immune system, including cytokines, lipid mediators and growth factors, and can also directly detect pathogens and their secreted products to produce pain during infection. Upon activation, nociceptors release neuropeptides from their terminals that potently shape the function of innate and adaptive immune cells. For some pathogens, neuron–immune interactions enhance host protection from infection, but for other pathogens neuron–immune signalling pathways can be exploited to facilitate pathogen survival. Here, we discuss the role of nociceptor interactions with the immune system in pain and infection and how understanding these pathways could produce new approaches to treat infectious diseases and chronic pain.
Introduction
Pain is an unpleasant sensation that often accompanies tissue injury, infection, cancer and inflammatory diseases. Chronic disease conditions including diabetic neuropathy, rheumatoid arthritis, osteoarthritis, irritable bowel syndrome, and ulcerative colitis are also characterized by pain. Celsus defined pain (dolor) as one of the four cardinal signs of inflammation (60 A.D). Pain as experienced by mammals is a critical defensive mechanism leading to behavioral withdrawal from environmental dangers and noxious stimuli. While acute pain represents a mechanism to avoid further injury, chronic pain can be maladaptive and pathologic, constituting a major burden on society1.
Pain is initiated by the activation of somatosensory neurons called nociceptors [G] that innervate the skin, cornea, genitourinary tract, gastrointestinal tract, joints, bones, muscles and deep visceral tissues1,2. Nociceptors express molecular sensors at their peripheral nerve terminals, such as transient receptor potential (TRP) channels and G-protein coupled receptors (GPCRs)1,3,4. These sensors detect noxious inflammatory stimuli including reactive chemicals, damaging temperatures (either heat or cold), mechanical injury and ATP and immune mediators, including bradykinin, histamine and cytokines3,4. Recent studies have shown that nociceptors also detect bacterial pathogens and microbial products, such as pore-forming toxins, during infection5. Upon sensing these noxious stimuli, action potentials are generated at nociceptor terminals that are transduced to the dorsal horn of the spinal cord and relayed to the brain to be processed and perceived as pain3. Given the shared role of nociceptor neurons and the immune system in protecting organisms from danger, coordinating pain signalling with the immune response may be physiologically advantageous.
Recent work has shown that neuroimmune interactions in pain are bidirectional. Immune cells release cytokines, lipids and growth factors that act on peripheral nociceptors and central nervous system (CNS) neurons to sensitize pain (Box 1). In turn, nociceptors actively release neuropeptides from their peripheral nerve terminals that modulate the activity of innate and adaptive immune cells4,6,7. Nociceptors regulate psoriasis-like inflammation8, host responses to candidal infections9, inflammatory bowel disease10, fungal oesteoinflammation11 and Streptococcus pyogenes skin infections12. Enhancing our knowledge of nociceptor-immune communication could therefore increase our understanding of the pathophysiology of multiple diseases and lead to new therapies for chronic pain and, more broadly, pathological inflammation.
Box1. Peripheral and central pain sensitization.
Neuroimmune interactions modulate both peripheral and central mechanisms of pain. Pain sensitization results in allodynia — which is defined as an increased sensitivity to normally innocuous stimuli (for example, touch) — and hyperalgesia, which is defined as an increased response to painful stimuli (such as noxious mechanical force and heat). Peripheral sensitization of pain occurs when inflammatory cells and mediators induce signalling pathways in afferent nociceptorsthat increase sensitivity to mechanical or thermal stimuli. The peripheral nerve terminals of nociceptors have been found to express a wide range of receptors that detect immune cell-derived mediators6,16. Immune receptor signalling in neurons leads to modification of membrane conductance properties, and gating of transient receptor potential (TRP) and voltage-gated ion channels. Central sensitization of pain occurs when molecular and biochemical changes occur in the central nervous system (CNS) that augment the nociceptive afferent input leading to pain hypersensitivity15,148. In neuropathic pain disorders, central sensitization is a major driving force of the persistence of pain that arises spontaneously even without the presence of noxious triggers13,148. In the spinal cord dorsal horn, resident microglia, astrocytes and infiltrating T cells act on presynaptic terminals from first order dorsal root ganglion (DRG) neurons and post-synaptic sites on second order neurons to modulate membrane excitability and synaptic transmission.
In this Review, we discuss the crosstalk that occurs between nociceptors and the immune system during pain processes and consider the therapeutic potential of targeting these pathways. We review molecular mechanisms by which immune cells and microbial pathogens signal to nociceptors to modulate pain. We do not go in depth into central sensitization mechanisms or microglial cells, although we do discuss their significance in pain. Finally, we discuss the important role of nociceptors in regulating the function of innate and adaptive immune cells, with a focus on host defence.
Immune system contributions to pain
Pain is often categorized into neuropathic or inflammatory pain. Neuropathic pain is caused by direct nerve damage or lesions in the neural circuitry that mediates pain13–15. By contrast, inflammatory pain is driven by immune-associated stimuli and can be modelled in animals using wound incision or injection of inflammatory stimuli, such as complete Freund’s adjuvant (CFA), lipopolysaccharides (LPS) or carrageenan6,16. Despite the nomenclature, immune cells contribute significantly to both neuropathic and inflammatory pain. Distinct immune cell-types release mediators that act on the terminals of nociceptors to drive peripheral sensitization (Figure 1a). In the CNS, microglia and T cells modulate neurotransmission and spinal cord pain circuitry to drive central sensitization (Figure 1b). In acute inflammation, pain often parallels the immune response and diminishes with resolution of inflammation. In chronic diseases such as rheumatoid arthritis and colitis, persistent immune triggers such as cytokines mediate long-lasting pain.
Figure 1. Neuro-immune interactions at peripheral nerve terminals and spinal cord in pain.
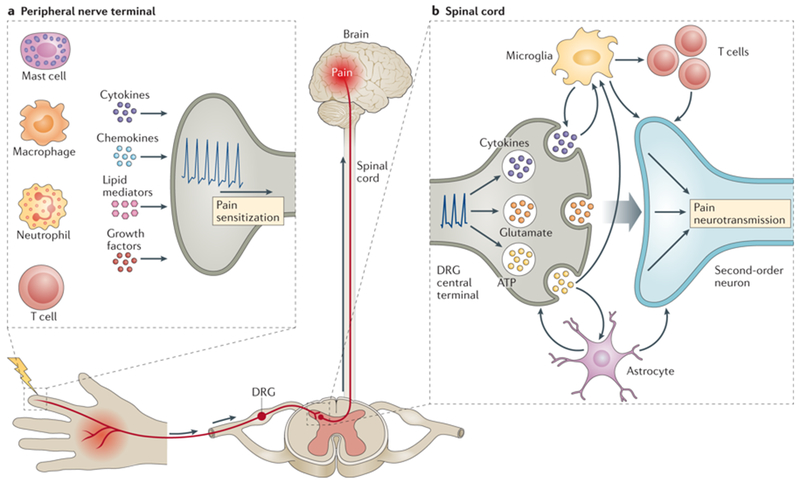
a) During inflammation, immune cells (mast cells, macrophages/monocytes, neutrophils and T cells) release mediators (cytokines, chemokines, lipid mediators and growth factors) that act on peripheral nerve terminals of nociceptor neurons. Action potentials are transduced via the dorsal root ganglia (DRG) to the spinal cord and relayed to the brain to be processed as pain. b) In the spinal cord dorsal horn, neuroimmune interactions contribute to central mechanisms of pain. Primary DRG nociceptive afferents (presynaptic) release glutamate, ATP and chemokines from their central terminals, mediating neurotransmission to second order postsynaptic neurons [G] that relay signals to the brain. T cells, microglia and astrocytes also produce pro-inflammatory cytokines and growth factors that act on both presynaptic and postsynaptic nerve terminals to increase neurotransmission and mediating central pain sensitization.
Cytokines and pain
Cytokines released by immune cells and non-neuronal cells (for example, epithelial cells) can directly act on nociceptor neurons to sensitize pain pathways (Figure 2). Multiple studies have shown that cytokine and chemokine signalling pathways impact pain-like behaviors in animal models of inflammatory and neuropathic pain16,17. Pro-inflammatory cytokines, such as tumour necrosis factor (TNF) and interleukin-1β (IL-1β) are pro-algesic by directly driving nociceptor activation6,16. There is also emerging interest in the role of chemokines in pain, which we discuss in Box 2. Our discussion of specific cytokines below is not exhaustive, and further molecular profiling and investigations of neurons are needed to accurately determine exact immune molecules that act on nociceptors16.
Figure 2. Molecular mechanisms of immune-driven pain.
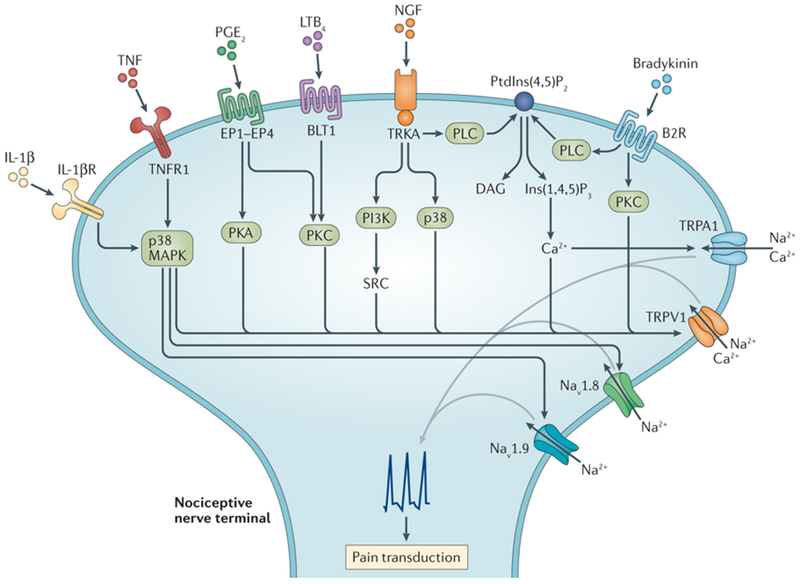
Immune cells release mediators that are directly sensed by nociceptor terminals to modulate neuronal excitation and pain transduction. Interleukin 1β (IL-1 β), tumor necrosis factor (TNF), nerve growth factor (NGF), prostaglandin E2 (PGE2), leukotriene B4 (LTB4) and bradykinin bind their cognate receptors expressed by nociceptor terminals to mediate neuronal firing. Receptor mediated signaling cascades through PI3K, p38, PKA and SRC lead to phosphorylation of TRP ion channels TRPV1 and TRPA1, as well as the voltage-gated sodium channels (Nav1.8 and Nav1.9), leading to changes in gating properties of these ion channels. Phospholipase C (PLC) is activated downstream of TrkA or B2R, mediating hydrolysis of the inhibitory molecule phosphatidylinositol-4,5-bisphosphate (PIP2) into inositol trisphosphate (IP3) and diacylglycerol (DAG). IP3 in turn mediates intracellular calcium release that sensitizes TRPV1 and TRPA1 activity. Immune mediators can also induce the upregulation of trafficking of ion channels to the membrane or increased transcriptional expression of these ion channels. The overall result of these immune-mediated pathways in nociceptors is the lowering of the threshold for responses to mechanical or thermal stimuli leading to increased pain sensitivity.
Box 2: Chemokines and pain.
Chemokines are mediators of neuroimmune communication in pain involved in both peripheral and central pain sensitization. CXC-chemokine ligand 1 (CXCL1) and CC-chemokine ligand 2 (CCL2, also known as MCP1) are two chemokines associated with pain. Mouse dissociated DRG neurons express CXCR2, the cognate receptor for CXCL129, and release the neuropeptide CGRP upon stimulation with CXCL1149. In CFA-induced inflammatory pain, CXCL1 and CXCR2 are induced in DRG neurons, and silencing CXCR2 expression in neurons reduced mechanical allodynia and heat hyperalgesia150. In a knee osteoarthritis pain model, mechanical allodynia was reversed by CCR2 receptor blockade151. CCL17 also critically drives pain in models of arthritis and joint inflammation downstream of GM-CSF-induced macrophage activation42. CCL2 and CXCL1 were also shown to be involved in neuronal signalling in the spinal cord via engagement with their receptors in astrocytes to drive neuropathic pain152,153. CXCL13, CCL21 and CX3CL1 (also known as fractalkine) also modulate central pain sensitization by acting through microglia and astrocytes in the spinal cord. After spinal nerve ligation (SNL), CXCL13 levels were increased in spinal dorsal horn neurons; intrathecal injection of CXCL13 also induced pain and astrocyte activation 154. CXCL13-induced pain depended on expression of its cognate receptor CXCR5 by astrocytes and activation of downstream ERK signalling pathway154. After nerve injury, neuronal CCL21 is upregulated in nociceptor afferents and transported to presynaptic [G] terminals in the dorsal horn where it causes microglial P2X4 receptor upregulation and mediates tactile allodynia155. Spinal administration of CX3CL1 dose-dependently evoked mechanical allodynia and thermal hyperalgesia in neuropathic pain models and blocking spinal CX3CR1 signalling reduced both nociceptive responses156. CX3CL1 is expressed by neurons, and its receptor CX3CR1 is expressed by microglia and upregulated after neuropathy157.
TNF and pain.
TNF mediates pain sensitization mainly through its receptor TNF receptor 1 (TNFR1), the role of another receptor TNFR2 in pain is less clear16,18,19. TNF application to dorsal root ganglia (DRG) [G] cell bodies induces increased neuronal excitability that is dependent on p38 signaling downstream of TNFR1 and tetrodotoxin-resistant sodium channels20. In a mouse model of carrageenan-induced inflammatory pain, mechanical hyperalgesia [G] was abolished in TNFR1-deficient mice21. In antigen-induced arthritis (AIA) and collagen-induced arthritis (CIA) models, systemic neutralization of TNF attenuated mechanical and thermal hypersensitivity in the affected joints18,22. Direct injection of TNF into the joint induces mechanical hypersensitivity, and this hyperalgesic effect was prevented by blockade of p38 signaling19. In the CNS, microglia and astrocytes also release TNF which acts on CNS neural circuitry to modulate pain signaling15. TNF increases the excitatory postsynaptic current frequencies and synaptic transmission in the spinal dorsal horn after intrathecal injection23. TNF blockade in human patients with rheumatoid arthritis also significantly reduced pain, providing not only symptomatic relief, but also nociceptive signal changes in the thalamus and somatosensory cortex involved in pain perception24.
IL-1β band pain.
IL-1β is a cytokine produced downstream of the inflammasome and is involved driving pain sensitivity in a number of diseases including gout and rheumatoid arthritis. IL-1β was one of the first cytokines demonstrated to cause hyperalgesia upon both local challenge and systemic administration in mice25. Studies in mice and rats have shown its hyperalgesic action in inflammatory pain models21,25,26. IL-1β-mediated mechanical hyperalgesia in carrageenan-induced pain depends on downstream production of prostaglandins by cyclooxygenases21,25. IL-1β-induced thermal hyperalgesia in arthritic joint pain also depends on the upregulation of transient receptor potential cation channel subfamily V member 1 (TRPV1)26. IL-1β neutralization reduces carrageenan- and IL-1β-induced mechanical hyperalgesia21,25,26. IL-1β was found to increase action potential generation in DRG nociceptors in response to both mechanical and thermal stimuli in rats27. IL-1β signalling in nociceptors leads to sensitization of Nav1.8 sodium channels and increased excitability downstream of p38 MAPK signalling28. In the spinal cord, microglia produce IL-1β which acts on receptors expressed on dorsal horn neurons to mediate central sensitization15.
IL-6 and pain.
IL-6 is considered an important mediator of the pain response. Transcriptional analyses show high expression of gp130, a component of the IL-6 receptor, in DRG neurons29. Compared with wild-type mice, IL-6−/− mice showed attenuated hyperalgesia to mechanical and thermal stimuli after footpad injection of carrageenan16,30. Specific loss of gp130 from nociceptors in mice attenuated mechanical hyperalgesia during inflammation due to diminished JAK-STAT-dependent expression of the TRPA1 ion channel31. It has been shown that spinal IL-6 contributes to central sensitization and hyperalgesia in joint inflammation, and that application of soluble gp130 to the spinal cord to bind IL-6 dampens the central sensitization32.
IL-17 and pain.
IL-17A drives inflammation and pathology in several autoimmune and inflammatory diseases. Intraplantar injection of recombinant IL-17A was shown to induce hyperalgesia in mice, and this effect was dependent on neutrophil recruitment and TNF-TNFR signalling33. In an antigen-induced arthritis model of pain, IL-17A levels were increased after induction of arthritis and correlated with mechanical hyperalgesia34. Neutralizing endogenous IL-17A prevented pain-like behaviours in mice, with this effect mediated via the expression of TNF, IL-1β, and CXCL1 and neutrophil recruitment34. IL-17A was also observed to directly induce rapid phosphorylation of protein kinase B and ERK in DRG cell cultures which contain nociceptors35.
IL-10 and pain.
IL-10 is a key anti-inflammatory cytokine that also possesses anti-nociceptive properties. Several immune cell-types are able to produce IL-10, including regulatory T cells and macrophages. Previous studies using IL-10−/− mice or neutralization of spinal IL-10 activity prolonged the duration of paclitaxel-induced mechanical allodynia36. Moreover, exogenous IL-10 injection into the spinal cord decreased paclitaxel-induced pain36. In the chronic constriction injury model of chronic pain, intrathecal injection of IL-10 and adenovirus-mediated spinal IL-10 delivery both prevented mechanical allodynia and thermal hyperalgesia37. Addition of IL-10 to DRG cultures from paclitaxel-treated mice suppressed spontaneous neuronal discharges36. Electrophysiological studies in rat DRGs showed that IL-10 directly mediated the down-regulation of nociceptor expression of voltage gated sodium channels38. Thus, IL-10-mediated suppression of pain may involve both its inhibitory effect on the expression of pro-algesic cytokines such as TNF, IL-6 and IL-1β, and also a direct effect on neurons.
GM-CSF and G-CSF in pain.
Granulocyte macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF) and macrophage colony-stimulating factor (M-CSF, also known as CSF1) play key roles in myeloid immune cell function and recently have been found to mediate pain16. GM-CSF and G-CSF signalling mediates cancer-evoked pain in a JAK-MAPK-dependent manner39. GM-CSF mediates CFA-induced inflammatory pain as well as pain in experimental arthritis models40. M-CSF has also been shown to act downstream of TNF- and GM-CSF-driven inflammatory pain41. Mechanistically, GM-CSF mediates pain through the induction of interferon regulated factor 4 (IRF4) and subsequent CCL17 production by macrophages; CCL17-deficient mice and IRF4-deficient mice showed attenuated pain responses to GM-CSF and in other inflammatory pain models42.
Lipid mediators.
Lipid mediators released during inflammation play a major role in pain signalling. Prostaglandins and leukotrienes drive pain sensitivity, whereas pro-resolving lipids such as resolvins and maresins have analgesic actions6,43. The analgesic effects of non-steroidal inflammatory drugs (NSAIDs) work by blockade of cyclooxygenase 1 (COX1) and COX2, which convert arachidonic acid to prostanoid precursors that are then converted to prostaglandins and thromboxanes44. Five bioactive prostanoids are generated by this pathway, namely PGE2, PGI2, PGD2, PGF2α and thromboxane44. Prostaglandin-driven sensitization of peripheral nerve terminals is a major cause of inflammatory pain6. A seminal study in humans showed that PGE2 injection in human volunteers caused hyperalgesia45. Peripheral nociceptive terminals express GPCRs for several prostanoids: EP1-EP4 which bind PGE2, DP1 and DP2 which bind PGD2, and the prostacyclin receptor (IP) which binds PGI244. Activation of the DP1, EP2, EP4 and IP receptors mediates an increase in neuronal cAMP levels, while EP1 receptor activation induces calcium mobilization44. Peripheral inflammation also induces COX2 expression in CNS neurons, leading to accumulation of PGE2 levels in cerebrospinal fluid and modulation of pain via central mechanisms46. 47Leukotrienes are also generated downstream of arachidonic acid metabolism and drive pain sensitization. Intradermal injection of leukotriene B4 (LTB4), but not LTD4, induced hyperalgesia within 20 minutes in the rat footpad48. LTB4 is a potent chemoattractant of neutrophils, and it was shown that LTB4-mediated hyperalgesia depended on neutrophil influx48. LTB4 acts through BLT1 (Figure 2), its high affinity receptor, and BLT2, a lower affinity receptor. At low LTB4 doses, BLT1 drives PKC-dependent TRPV1 sensitization and thus thermal hyperalgesia, however at higher LTB4 doses, BLT2 is activated and produces analgesia49.
Growth factors.
Neurotrophic growth factors including nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) can be released by immune cells during inflammation, which can induce sprouting of peripheral nerves and nociceptive signal transduction that modulates pain. NGF causes peripheral pain sensitization by binding tropomyosin receptor kinase A (TrkA) (Figure 2), which is highly expressed by nociceptor sensory afferents6,43. During inflammation, NGF is produced by mast cells and macrophages6. In animal models and human studies, cutaneous injection of NGF produces pain and hyperalgesia within 20 minutes of injection50,51. Increased NGF levels have been shown in the synovium of patients with inflammatory joint diseases52. Treatment with a monoclonal antibody against NGF in patients with advanced knee osteoarthritis resulted in significant reduction in pain53; however adverse side effects such as joint destruction and neuropathies were also often observed in the treated patients54. NGF sensitization leads to PI3K-SRC and p38-MAPK signalling pathways in nociceptors that increases the trafficking of the TRPV1 ion channel to the outer membrane of nociceptors, leading to increased thermal sensitivity50,55,56. NGF also drives Nav1.8 sensitization, and NGF fails to produce thermal hyperalgesia in Nav1.8-deficient mice57. In neuropathic pain conditions, microglia in the CNS release BDNF which acts on its receptor TrkB expressed on synapse neurons to cause a shift in the anion gradient, leading to pain disinhibition58. Intrathecal administration of BDNF or ATP-stimulated microglia evokes depolarization of spinal L1 neurons and allodynia [G] in a TrkB-dependent manner58. Recently, work has shown that microglia and adaptive immune cells differentially modulate neuropathic pain in a gender-dependent fashion59. In male mice, microglia drive neuropathic pain after spared nerve injury (SNI) through BDNF-dependent signalling; however, in female mice, SNI-pain is driven by adaptive immune cells independently of BDNF59.
Immune sensitization of nociceptor ion channels
A key mechanism though which immune-derived mediators signal in nociceptors to drive pain is by modifying ion channel activity. Below, we describe how specific cytokines and inflammatory mediators lead to sensitization of TRP channels (TRPV1, TRPA1, TRPV4) and voltage-gated sodium channels (Nav1.7, Nav1.8, Nav1.9).
TRPV1.
TRPV1 is a large-pore cation channel that mediates sensation of noxious heat (≥43°C), protons, and capsaicin [G], and mediates thermal hyperalgesia during inflammation60. During tissue inflammation or damage, cytokines, prostaglandins, NGF and bradykinin signal in neurons to increase TRPV1 expression and/or TRPV1 activity6,60. Immune mediators activate mitogen activated protein kinase (MAPK), protein kinase C (PKC) and protein kinase A (PKA), which phosphorylate the membrane or cytoplasmic residues of TRPV1 to change its gating properties and facilitate its opening61. NGF and bradykinin mediated potentiation of TRPV1 activity involves the PLC-dependent hydrolysis of inhibitory molecule phosphatidylinositol-4,5-bisphosphate into inositol trisphosphate (IP3) and diacylglycerol62,63. IP3 in turn mediates the intracellular calcium release and thus sensitizes TRPV1 as well as TRPA1 channel62–64. 65PKA and PKC-dependent sensitization of TRPV1 depends upon binding of scaffolding protein AKAP79 with the C-terminal region of TRPV166. TNF and IL-6 elevate gene expression or membrane expression of TRPV167,68.
TRPA1.
TRPA1 is a nociceptive ion channel that responds to electrophilic reactive chemicals such as allyl isothiocyanates and to inflammatory products, including lipid peroxidation products, such as 4-hydroxy-2-nonenal, and oxidized lipids, such as 15-deoxy-prostaglandin J260,69. TRPA1 is predominantly expressed by nociceptor sensory neurons of vagal ganglia, trigeminal and dorsal root ganglia in both peptidergic and non-peptidergic neurons60. Bradykinin produced during inflammation activates TRPA1 channels downstream of the bradykinin receptor B2R signalling64. B2R activation of PLC releases the intermediates diacylglycerol and inositol triphosphate (IP3) from phosphatidylinositol 4,5, biphosphate, which in turn gates TRPA1 opening64. CFA-induced peripheral inflammation and nerve injury via spinal nerve ligation (SNL) can both increase TRPA1 mRNA expression in nociceptors, downstream of TrkA/p38 MAPK signalling, which drives cold pain sensitization70. Inflammatory stimuli can also increase membrane expression of TRPA1 downstream of PKA and PLC signalling to sensitize nociceptors71.
TRPV4.
TRPV4 is a Ca2+-permeable ion channel that is activated by warm temperatures (>25°C), hypotonicity and acidic pH. Besides its key role in osmosensation and hypotonicity-induced nociception72, TRPV4 also mediates pain-related behaviours in response to inflammatory mediators73 and in chemotherapy-induced neuropathy74. Increased temperature and reduced pH, which are both characteristic of tissue inflammation, activate TRPV4. TRPV4 contributes to mechanical hyperalgesia induced by carrageenan or an inflammatory soup containing bradykinin, substance P [G], PGE2, serotonin and histamine involving cAMP–PKA and PKC signalling75. In taxol-induced neuropathic pain, TRPV4 mediated nociceptive behavioral responses to mechanical and hypotonic stimulation via integrin–SRC tyrosine kinase signalling74.
Nav1.7, Nav1.8 and Nav1.9.
The voltage-gated sodium channels Nav1.7, Nav1.8 and Nav1.9 are highly enriched in nociceptors that mediate action potential generation76. Similarly to TRP channels, several pro-inflammatory mediators including IL-1β and TNF signal through kinases to phosphorylate these sodium channels to facilitate their opening20,28,43. IL-1β induces a p38-mediated change in Nav1.8 activity that involves distinct phosphorylation residues than PKA/PKC kinases20,28. Inflammation also modulates the expression of sodium channels. Nav1.7 and Nav1.8 are upregulated in the dorsal root ganglia following carrageenan or CFA injection into the rat hind paw77 and in rat knee joints after induction of inflammation78
Microbial infection and pain
Pain is the herald and hallmark of many infections. Increasingly infections are appreciated as triggers for chronic pain and downstream remodeling of the nervous system. Pain during infection was thought to be the by-product of immune responses to pathogens. However, recent studies have demonstrated that nociceptors can directly sense microbes and their products (Figure 3).
Figure 3. Molecular mechanisms of microbial-driven pain.
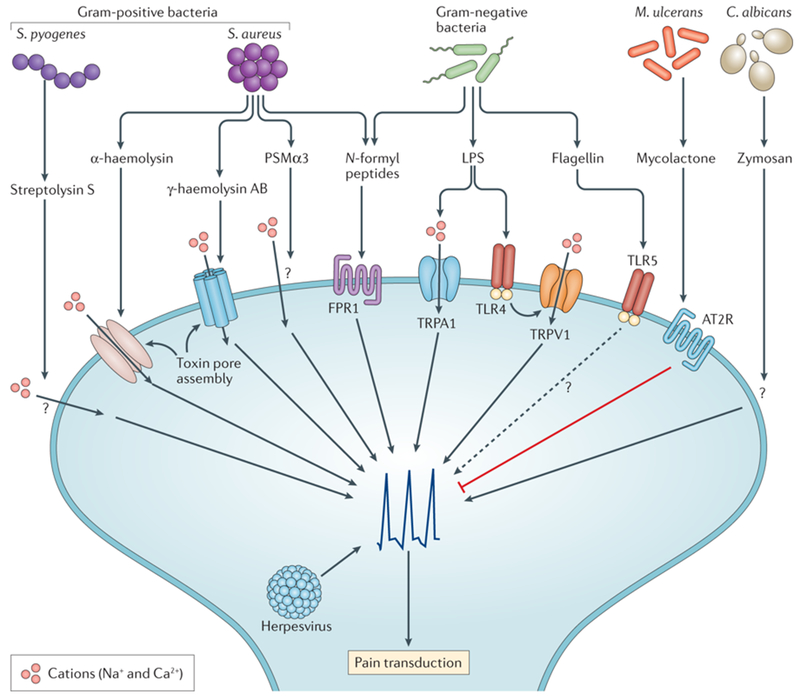
Nociceptors can directly detect microbial pathogens and their products. S. aureus directly activates nociceptors through N-formyl peptides and the pore forming toxins (PFTs) α-haemolysin, PSMα3 and γ-haemolysin AB. Streptococcus pyogenes activates neurons through streptolysin S, which is a peptide toxin that also mediates cell lysis. In neurons, PFTs induce rapid cation influx and subsequent depolarization leading to pain production. Lipopolysaccharides (LPS), a major cell wall component of Gram-negative bacteria, also activates nociceptors through either neuronal TLR4 or directly gating of the TRPA1 ion channel. A subset of sensory neurons also expresses TLR5, whose cognate ligand is bacterial flagellin expressed by gram-negative bacteria. Candida albicans, a fungal pathogen, activates nociceptors through zymosan. Zymosan is a major cell wall component of fungi. Mycobacterium ulcerans, a skin pathogen that causes Buruli ulcer, silences pain by secreting a mycolactone that cause neuronal hyperpolarization by inducing angiotensin II receptor signaling. Herpes viruses invade sensory neurons and can cause significant pain following reactivation in syndromes such as post-herpetic neuralgia. Many molecular mechanisms remain to be determined for pathogen-induced pain though it is now clear that nociceptors, like immune cells, are able to directly respond to pathogen-derived molecules.
Bacterial infections and pain
Direct sensing of pathogens by nociceptors was first appreciated in the context of bacterial infection. The major human pathogen Staphylococcus aureus is a Gram-positive coccus which causes a variety of pyogenic and typically painful infections. In a mouse model of cutaneous S. aureus infection, acute pain correlated with bacterial population expansion rather than with tissue swelling or myeloid cell recruitment79. The S. aureus pore-forming toxin (PFT) α-hemolysin (αHL) directly caused calcium influx and action potential generation in nociceptors and was sufficient to induce pain-like behaviours when injected in mice79. Two other S. aureus PFTs — the bicomponent leukocidin γ-hemolysin AB and the phenol soluble modulin PSMα3 — are also able to directly induce neuronal firing and produce pain79,80. The mechanism of action of PFTs is through their assembly into membrane-perforating pores that produce neuronal depolarization, while N-formyl peptides likely act via the GPCR formyl peptide receptor 1 (FPR1)79.
Another Gram-positive human pathogen, Streptococcus pyogenes, is the most frequent cause of necrotizing fasciitis (also referred to as ‘flesh-eating disease’), which is a rapidly progressive infection of deep soft tissues. Necrotizing fasciitis is characterized by early pain out of proportion with other signs of inflammation, such as noticeable redness or swelling. In mouse models of necrotizing fasciitis, S. pyogenes produces pain by secreting the PFT streptolysin S (SLS), which directly acts on nociceptors to cause neuronal activation and release of the neuropeptide calcitonin gene-related peptide (CGRP) [G]12. Two clinical isolates of S. pyogenes caused spontaneous acute nociceptive responses and mechanical hyperalgesia within an hour of inoculation12. Bacteria lacking SLS did not produce pain, and an SLS neutralizing antibody also significantly reduced pain during infection12.
Gram-negative bacterial infections are also frequently painful. Lipopolysaccharide (LPS), which is a major component of Gram-negative bacterial cell walls, induces pain when injected into mice. Recent work suggests that LPS can directly act on nociceptors81,82, as nociceptors express Toll-like receptor 4 (TLR4) . LPS derived from the odontogenic pathogen Porphyromonas gingivalis elicits dose-dependent calcium influx and inward currents, sensitizes TRPV1 to capsaicin, and stimulates release of CGRP in cultured trigeminal neurons, with inhibition of these effects by TLR4 antagonism81. In a mouse model of urinary tract infection (UTI) with uropathogenic E. coli, UTI-associated pelvic pain was driven by LPS in a TLR4-dependent fashion independent of bacterial colonization or inflammation83. In contrast, another study found that LPS from multiple Gram-negative pathogens, including Salmonella, Klebsiella, and Pseudomonas species, was able to directly induce calcium influx and CGRP release from cultured nociceptors independent of TLR4 but through direct action on the ion channel TRPA182. Acute pain responses and mechanical hyperalgesia in response to LPS were attenuated in TRPA1−/− mice but preserved in TLR4−/− mice82. CGRP release and local vasodilation was also diminished in TRPA1−/− mice, but systemic inflammatory responses to LPS were preserved82. Recent work also showed that LPS can activate TRPV4 and increase intracellular calcium in respiratory epithelial cells independent of TLR4 signaling84, suggesting another potential mechanism of LPS activation of nociceptors.
Other factors produced by Gram-negative bacteria may also contribute to pain induction. A subset of Aβ DRG neurons were found to express mRNA encoding TLR5 (which detects flagellin from bacterial flagella) and co-application of flagellin with the analgesic compound QX-314 to induce TLR5-dependent blockade of Aβ fibers was exploited to inhibit neuropathic pain85. Nociceptors may be able to distinguish among closely related bacteria; approximately 70% of cultured DRG neurons showed calcium influxes to the symbiont Clostridium ramosum but DRG neurons showed a minimal response to the pathogenic Clostridial bacteria Peptostreptococcus magnus86. One possible explanation is that LPS from different Gram-negative bacteria may induce differential activation of TRPA1, with the degree of activation being shaped by the lipid A moiety of LPSs84.
Fungal infection and pain
Like bacterial infections, fungal infections are frequently characterized by pain. A prototypical example is Candida albicans, an opportunistic yeast which causes painful mucocutaneous disease along with invasive infections. Zymosan and β-glucan are yeast cell wall components that are recognized by host cells through TLR2 and dectin 1. Recent work suggests nociceptors directly sense fungal pathogens using similar pathways9,11,87. In cultured DRG neurons, zymosan or heat-killed C. albicans can induce rapid calcium influx and CGRP release9. Similarly, β-glucan directly activates nociceptors and triggers CGRP release in DRG cultures in a dectin 1-dependent fashion11. Nociceptor activation also involved TRPV1 and TRPA1 channels, as deficiency in either ion channel led to partial impairment of their activation in response to β-glucan or heat-killed C. albicans, while deficiency in both ion channels severely impaired their activation11. Further experiments in mice demonstrated that subcutaneous infection with live C. albicans or the administration of β-glucan elicited dectin 1-mediated mechanical allodynia in a manner that was independent of the innate and adaptive immune response but dependent upon ATP signalling via the P2X3 receptor87. In response to heat-killed C. albicans, acute pain behaviour was inhibited in TRPV1- or TRPA1-deficient mice, and TRPV1/TRPA1 double deficient mice also had inhibition of mechanical allodynia87. As zymosan and β-glucan are cell wall components of many other clinically significant pathogens, including Aspergillus and Pneumocystis, similar mechanisms may be at play in other fungal diseases.
Herpes virus infection and pain
Viral modulation of nociceptors has long been appreciated due to the well-characterized neuroinvasive herpes viruses. These three pathogens, namely herpes simplex virus 1 (HSV1), HSV2 and varicella zoster virus (VZV), gain access to sensory nerve endings after initial mucocutaneous disease, establish persistent infection in neuronal cell bodies in dorsal root or trigeminal ganglia and, under certain conditions, cause recrudescent disease generally limited to the area innervated by infected sensory ganglia88,89. 89During infection of nociceptors, lesions are classically exquisitely painful. Post-herpetic neuralgia (PHN) which occurs following reactivation of VZV is characterized by persistent neuropathic pain88. The molecular mechanism underlying pain production during infection and after viral clearance remain to be elucidated, but PHN is thought to result from abnormal hyperactivity of damaged primary afferents versus deafferentation with generation of abnormal connections88. Many other viral infections are also characteristically painful, and deciphering nociceptor responses during viral infection is an area that warrants further investigation.
Analgesic infections
In contrast to typically painful infections, some pathogens may exploit analgesia to facilitate their spread. Mycobacterium ulcerans is the etiologic agent of Buruli ulcer, a disease characterized by destructive yet painless ulceration. These disparate manifestations are caused by the secreted polyketide toxin mycolactone90. Mycolactone hyperpolarizes sensory neurons by activation of the angiotensin II receptor pathway and subsequent activation of the TRAAK potassium channel90. Another species of mycobacteria M. leprae, the causative agent of leprosy, causes similar hypoesthesia presumably secondary to the extensive nerve destruction seen in leprosy. Other pathogens also cause painless lesions despite significant tissue inflammation and destruction (for example, Treponema pallidum and the chancre of primary syphilis), and direct action on sensory neurons offers a possible mechanism that could explain disassociation of pain from inflammation.
Modulation of the immune response by nociceptors
As pain was being recognized as a key feature of inflammation and infection, it was appreciated that it plays a role incontributes to the response to noxious stimuli. However, the contribution of pain to host defence was traditionally thought to be limited to higher order cognitive responses that would motivate removal from a harmful environment, protect damaged tissues from further injury, and drive sickness behaviour allowing for rest and recovery. Seminal experiments in the 19th and early 20th century demonstrated that neuronal signalling via secreted neurotransmitters CGRP and substance P could generate immediate vasodilation, increasing blood flow and edema independently of the immune response in a process called ‘neurogenic inflammation’ (Box 3). Innate and adaptive immune cells also express receptors for CGRP, substance P and other neurotransmitters. Increasing evidence points to a role for complex neural-immune communication through these neurotransmitters to potently regulate innate and adaptive immunity (Figure 4). Here, we will review the modulation of immune cells by nociceptor activation, particularly in the context of pathogen response. Defining the molecular mechanisms of these neuroimmune interactions could reshape our understanding of host defense.
Box 3: Neurogenic inflammation.
Seminal experiments in the 19th and early 20th century by Goltz (1874), Strickler (1876), Bayliss (1901) and others demonstrated that stimulation of sensory nerves generates immediate vasodilation, increased blood flow and edema independent of the immune response, a process termed neurogenic inflammation4. Extensive later work demonstrated that the underlying mechanism was release of neuronal mediators from nociceptors into the peripheral field via a local axon reflex. In addition to the orthodromic [G] propagation of action potentials to the spinal cord to transmit pain centrally, generated action potentials also back-propagate from axonal branch points in an antidromic [G] fashion and cause calcium influx and vesicle release from neighboring nerve terminals, leading to tightly localized and rapid release of neuronal effectors into affected tissue4. A variety of neurogenic inflammatory mediators have been identified, among which the most potent are neuropeptides calcitonin gene related protein (CGRP) and Substance P. Substance P acts through its receptor NK1R on endothelial cells to drive vascular permeability4. CGRP acts via its cognate receptor on vascular and lymphatic smooth muscle cells to drive vasodilation and lymph flow. CGRP can also promote vasodilation via release of nitric oxide (NO) from endothelial cells4. Nociceptors release many other neuropeptides, such as pituitary adenylate cyclase-activating polypeptide (PACAP), and other mediators, such as prostaglandins and NO, which have also been implicated in neurogenic inflammation4. Taken together, this leads to the warmth, erythema, and edema of inflammation. This process first suggested a direct role of nociceptors in not only sensing, but also regulating and targeting response to noxious stimuli, including to pathogens.
Figure 4. Nociceptors regulate inflammation through neural-vascular and neuro-immune interactions.
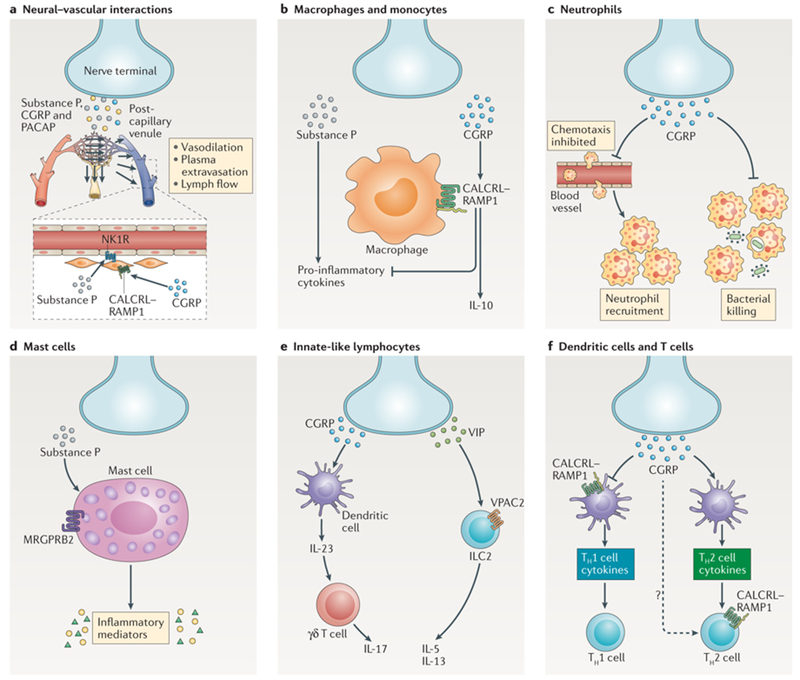
a) Nociceptors release neuropeptides (such as substance P, CGRP and PACAP) from peripheral nerve terminals that act on the vasculature to drive ‘neurogenic inflammation’. Substance P activates NK1R on endothelial cells to promote vascular permeability and edema. CGRP acts via the CALCRL-RAMP1 receptor complex on vascular and lymphatic smooth muscle cells to promote relaxation and acts on endothelial cells to release nitric oxide, leading to vasodilation. b) Nociceptors release substance P, which acts on monocytes and macrophages via NK1R and downstream ERK-p38 MAPK-mediated NF-κB activation to drive expression of pro-inflammatory cytokines (left). CGRP acts on CALCRL-RAMP1 to drive cAMP-PKA-dependent modulation of CREB to upregulate IL-10 and inducible cAMP early repressor (ICER)-dependent transcriptional repression of pro-inflammatory cytokines (right). c) Nociceptors release CGRP, which decreases neutrophil recruitment to the lungs and skin and inhibits the ability of neutrophils to opsonophagocytose and kill bacteria. d) Substance P activates the Mgprb2 receptor on mast cells leading to mast cell degranulation and proinflammatory mediator release. e) CGRP from nociceptive neurons drives IL-23 production by dermal dendritic cells which in turn leads to activation of γδ T cells and IL-17 production (left). The neuropeptide VIP activates its receptor VPAC2 on Type II innate lymphoid cells (ILC2) to drive production of IL-5 and IL-13 (right). f) CGRP binds RAMP1 on dendritic cells to inhibit dendritic cell migration and activation, decreasing Th1 cell differentiation. In contrast, CGRP potentiates dendritic cell polarization of Th2 cell differentiation. CGRP can also acts via RAMP1 to increase IL-4 production by T cells directly to amplify the Th2 response.
Monocytes and macrophages
Substance P has a robust effect on monocytes and macrophages (Figure 4b) and induces their release of pro-inflammatory cytokines, including IL-1, TNF and IL-691, via ERK/p38 MAPK-mediated NF-κB activation92. Recent work supports additional roles for substance P, with another study suggesting that substance P is able to polarize already activated macrophages to a tissue-repairing M2 macrophage [G] phenotype93. Increased M2 macrophage numbers, in association with decreased inflammation, were also seen upon substance P administration in a murine model of inflammatory arthritis94.
CGRP has a predominantly anti-inflammatory effect on myeloid cells, inducing the down-regulation of cytokine production, oxidative burst synthesis and antigen presentation, and the up-regulation of the anti-inflammatory cytokine IL-10 in macrophages and dendritic cells (DCs)95–97. CGRP signals through its receptor complex, which is a heterodimer of the GPCRs calcitonin gene-related peptide type 1 receptor (CALCRL) and receptor activity-modifying protein 1 (RAMP1). In myeloid cells, RAMP1-CALCRL signalling leads to cAMP/PKA-dependent modulation of CREB to upregulate IL-10, and inducible cAMP early repressor (ICER)-dependent transcriptional repression of pro-inflammatory cytokines95,98,99. CGRP administration protected mice from development of fatal shock response to endotoxin administration via inhibition of TNF release from peritoneal macrophages100, suggesting that CGRP signalling to macrophages could limit inflammatory responses and prevent systemic propagation from localized inflammation. In septic peritonitis, RAMP1-deficient mice had improved early anti-bacterial defense with reduced levels of IL-10101, suggesting similar activity of CGRP in limiting inflammatory response in this model of polymicrobial sepsis.
Other neuropeptides can also act on macrophages, with vasoactive intestinal peptide (VIP) [G] and pituitary adenylate cyclase-activating polypeptide (PACAP) [G] able to inhibit macrophage production of cytokines such as TNF and IL-6 through a cAMP-dependent pathway102,103. The opposing activities of various neuropeptides may be context dependent, suggesting an ability of nociceptors to have varied effects based on the type of infection or immune response. During bacterial infection, neural inhibition of macrophage activation may predominate based on limited studies of nociceptor silencing. With Nav1.8+ neuron ablation, S. aureus infection was accompanied by increased monocyte recruitment and cytokine production, with CGRP injection in nociceptor deficient mice reversing observed increased lymphadenopathy79. However, when subjected to overwhelming sepsis secondary to cecal ligation and puncture, TRPV1−/− mice developed worsehad worsened markers of sepsis and accelerated organ damage; this was associated with impaired macrophage function, with ex vivo peritoneal macrophages showing increased cytokine release but reduced phagocytosis. This suggests that absence of TRPV1 activity leads to an increased but less effective inflammatory response in this model104. Taken together, these data suggest that nociceptors potently regulate macrophages during infection, with in vivo studies supporting a predominantly anti-inflammatory effect of nocieceptors on macrophage function in the models of bacterial infection studied.
One intriguing area that remains to be characterized is the impact that the mechanisms described above have on pain, as bidirectional communication between nociceptors and immune cells would be expected to feedback on nociceptor activation and perceived pain. For example, CGRP mediated down-regulation of macrophage activation and release of TNF and other proalgesic cytokines would be expected to reduce further nociceptor activation and sensitization, thus serving as a negative control mechanism to diminish the inciting painful stimuli and ultimately resolve the inflammatory response. In contrast, a dominant substance P-mediated nociceptive response with downstream release of proalgesic cytokines could act to amplify and sustain both pain and inflammation.
Neutrophils
Nociceptor interactions with neutrophils have been less well characterized, but recent studies show a key role for nociceptors in regulating neutrophil recruitment and activation (Figure 4c). Elegant earlier work in arthritis models showed that nociceptor signalling induced shedding of L-selectin from neutrophils, leading to decreased neutrophil recruitment into the inflamed joint105. In Streptococcus pyogenes-induced necrotizing fasciitis, ablation of nociceptors led to increased neutrophil recruitment in association with more well-circumscribed abscesses, smaller necrotic lesions and improved control of infection12. Similarly, in cutaneous S. aureus infection, nociceptor silencing led to increased infiltration of myeloid cells including neutrophils at infection sites and in draining lymph nodes79. This effect is not limited to cutaneous nociceptors, as TRPV1+ neurons that innervate the respiratory tract were demonstrated to suppress pulmonary neutrophil recruitment in S. aureus pneumonia106. TRPV1−/− mice also show increased neutrophil recruitment in mouse models of post-myocardial infarction inflammation107. Furthermore, analysis of dynamic neutrophil behaviour via live imaging in the S. aureus pneumonia model showed enhanced neutrophil function in the absence of nociceptors106. In summation, these studies suggest marked inhibition of neutrophil recruitment and activity by nociceptors in multiple models of infection and inflammation. Given the ability of substance P and CGRP to induce vasodilation and microvascular plasma extravasation, it may be expected that nociceptive signalling would potentiate immune cell accumulation in inflamed tissues. It is possible that the vascular edema and classic neurogenic inflammation occurs at earlier time points following nociceptor stimulation (that is, within minutes) whereas the same signals suppress neutrophil extravasation at later time points (that is, within hours).
Nociceptor effects on neutrophils are likely at least partially mediated by neuronal production of CGRP, as nociceptor ablation led to significant decreases in CGRP levels in the bronchoalveolar lavage fluid during S. aureus lung infections106, and CGRP inhibited murine neutrophil killing of both S. aureus106 and S. pyogenes12, potentially by shutting down myeloperoxidase (MPO) production12. Nociceptors also suppressed the expression of both TNF and CXCL1, two key cytokines that mediate neutrophil activation and chemoattraction106. CGRP has also been shown to decrease neutrophil recruitment by inhibiting chemokine production by endothelial cells108. Other work has also suggested CGRP acts to promote neutrophil adherence to endothelium109 and modulate neutrophil degranulation110, suggesting complex actions of CGRP on various aspects of neutrophil function. Of note, CGRP is also expressed by non-nociceptor sources including neuroepithelial bodies in the respiratory tract and enteric neurons in the gastrointestinal tract 111,112.
Mast cells
A tight interplay between nociceptors and mast cells has long been postulated due to their anatomic colocalization (Figure 4d). Mast cells are found in close apposition to nerve fibres in barrier tissues, particularly CGRP- and substance P-positive neurons113–115. Whether neurons form ‘synapses’ with mast cells is unknown in vivo, but cultured CADM1-expressing mast cells have been demonstrated to form direct attachments to DRG sensory neurites116. Increases in the number of nerve-mast cell contacts has been reported in models of inflammatory and allergic diseases117, as well as in parasitic infection of the colon114. Substance P alone is sufficient to cause mast cell activation, degranulation, and release of inflammatory mediators including histamine, leukotrienes, tryptases, prostaglandin D2 and TNF118. Substance P-mediated activation of mast cells is mediated by the newly identified Mas-related G-protein coupled receptor member B2 (MRGPRB2), which is highly enriched in mast cells119,120. This contrasts to in other cell types, where substance P is detected by NK1R (also known as TACR1). Rodent mast cells also express receptors for CGRP, and CGRP can potentiate mast cell activation and degranulation121,122. CGRP-mediated vasodilation and mast cell de-granulation is a pathophysiological mechanism in mouse models of migraine123, although the physiological contribution of mast cell activation in human migraine remains less well understood124. Although mast cell-neuronal communication has been best studied in allergic diseases, similar mast cell-based responses are implicated in innate immune responses to various pathogens, particularly parasites, and the contribution of nociceptors and mast cell interactions in host defense is a promising area of investigation.
Dendritic Cells
Like mast cells, DCs have been found to be in close contact with nociceptors in peripheral tissues (Figure 4f)8. Nociceptor regulation of DCs has an important role in modulating cutaneous immunity. In an imiquimod-induced psoriasis model, ablation of nociceptors suppressed production of IL-23 by DCs, production of IL-17 by γδ T cells and recruitment of other inflammatory cells8. An analogous role in infectious response was demonstrated in a mouse model of cutaneous Candida albicans infection where nociceptive neurons were required for IL-23 production by CD301b+ dermal DCs; absence of TRPV1+ nociceptors resulted in decreased downstream activation of γδ T cells and IL-17A production, leading to worse fungal infection9.
Nociceptor modulation of DCs is likely to be varied based on context. In vitro and in vivo studies in contact hypersensitivity models shows that CGRP acts via a cAMP/PKA pathway to inhibit DC production of IL-12, expression of CCR2 and to decrease DC activation and migration, leading to suppressed Th1 cell responses125. In contrast, CGRP exposure enhances Langerhans cell polarization towards a Th2 cell phenotype by stimulating production of IL-4 and the Th2-type chemokines CCL17 and CCL22126. Like CGRP, the neuropeptides PACAP and VIP can also enhance Langerhans cells antigen presentation for Th2 responses as well as Th17 cell responses127,128,129. Work with experimental colitis and airway inflammation models show similar interactions occur in the gut and airways130,131.
Innate-like Lymphocytes
Innate-like lymphocytes, including innate lymphoid cells (ILCs) and γδ T cells, play key roles in bridging innate and adaptive immune responses (Figure 4e). These cells have a tropism for densely innervated mucosal and epithelial barrier tissues, and neuronal interactions with innate-like lymphocytes are an emerging aspect of their biology. In cutaneous Candida albicans infection, absence of TRPV1+ neurons led to decreased numbers of skin-resident γδ T cells and a corresponding increase in fungal burden9. In contrast, nociceptor-deficient mice demonstrated higher absolute numbers of lung-resident γδ T cells at homeostasis106. In S. aureus pneumonia, loss of nociceptors led to improved outcomes of infection, but this protection was abrogated in γδ T cell-deficient mice, suggesting a mechanism of neuronal suppression of host defense via γδ T cells. Given the diversity of γδ T cell subsets in different barrier sites, it would be interesting to tease apart the distinct neural– γδ T cell interactions in different tissues.
ILCs, particularly Type 2 ILCs (ILC2s), have also been shown to interact with sensory neurons and their neuropeptides to coordinate immune response. ILC2s specifically express the receptor for the neuropeptide neuromedin U (NMU)132, which is secreted by mucosal neurons in the enteric nervous system and respiratory tract. NMU was shown to drive protective ILC2 responses in mice infected with the helminth Nippostrongylus brasiliensis, with loss of the NMU receptor impairing control of parasitic infection133. NMU also amplifies IL-5 production by pulmonary ILC2s in house dust mite (HDM)-induced allergic airway hypersensitivity132. CGRP has been shown to stimulate ILC2 production of IL-5 in vitro, with _Calcrl_−/− mice showing dampened ILC2-induced immune cell recruitment in a murine model of asthma112. VIP is another important neuropeptide that modulates ILCs. Lung and intestinal ILC2s also express the VIP receptor VPAC2; VIP induces expression of IL-5 by ILC2s in a VPAC2-dependent manner134.
B cells and T cells
Given that nociceptors can modulate the functions of antigen-presenting cells and innate-like lymphocytes, it follows that neurons may also significantly affect the adaptive immune response (Figure 4f). Distinct neuropeptides are able to drive differential polarization of T cell responses, whether through regulation of antigen-presenting cells as described above or via direct modulation of lymphocytes. In models of contact dermatitis, CGRP inhibited Th1-type contact hypersensitivity but promoted Th2-type contact hypersensitivity135. Inin vitro studies, CGRP treatment of dermal DCs led to enhanced generation of Th17 cells and increased IL-17A and IL-22 production136. This effect is not unique to cutaneous nociceptors, as in a model of ovalbumin and HDM-induced airway inflammation, nociceptors stimulated Th2-type effector cytokine production from resident ILC2s and this drove downstream CD4+ T cell recruitment and activation137. This effect was thought to be at least partially mediated by VIP. This study and others demonstrate that VIP promotes Th2-type responses and inhibits Th1-type responses with beneficial effects on Th1-type immunopathology138. Work in experimental colitis suggests a pro-inflammatory effect of substance P from nociceptors mediated by signalling to mucosal Th1 cells10,139. Substance P also drives polarization of Th17 cells in in vitro assays in a monocyte-dependent fashion, and in vivo in a murine model of inflammatory arthritis94,140.
Nociceptive signalling can also modulate B cell responses and humoral immunity. In a model of Salmonella infection and vaccination, mice deficient in the substance P receptor NK1R had increased IgA responses due to increased cytokine production from Th2 cells with corresponding protection from infection141. In a mouse fracture model of regional pain, inhibition of substance P or CGRP signalling blocked development of IgM autoantibody responses142. The extent to which these effects are mediated directly through modulation of lymphocytes versus indirectly via antigen-presenting cells remains to be determined. T cells and B cells do express neuropeptide receptors and in vitro experiments suggest that neuropeptides may directly modulate their functions143–146. Nonetheless, in vivo experiments using conditional ablation of neurotransmitters and neuropeptide receptors in lymphocytes will be necessary to properly understand these pathways.
Clinical implications and future directions
Pain is a major hallmark of many disparate diseases. Increasing evidence points to a role for neuroimmune interactions in the initiation, propagation and chronicity of pain associated with inflammatory and infectious diseases in diverse tissues. Modulation of these interactions may offer new ways to target both pain and the underlying disease. For example, inhibition of pro-inflammatory cytokines that drive chronic pain sensitization or stimulation of antinociceptive cytokine signalling could be exploited to block pain. Despite the association of a number of immune mediators with nociceptor sensitization and pain responses in pre-clinical studies, little of this knowledge has been translated into pain-relieving therapies. This might be due to the failure of several promising clinical trials in humans due to adverse side effects. For example, NGF antagonism showed promise in alleviating pain in animal models, but was associated with developmental defects, joint destruction and neuropathies in human subjects54. Clinical trials using TRPV1 antagonists were also limited by off-target effects, including hyperthermia147. Given the role of some major cytokine candidates (such as TNF and GM-CSF) in protective immune responses against pathogens, it is also important that pain-targeting therapies consider the adverse effects of immunosuppression, especially in chronic settings. In the future, more specific targeting of immune mediators and their signalling pathways in nociceptors may offer alternative pain treatments.
An alternative approach is suggested by the emerging studies which highlight direct activation of nociceptors by microbial ligands and microorganisms. This could include both contributions from pathogens as well as commensal microbes in the skin and gut microbiota. One challenge is to identify the key factors released by microbes that contribute to different painful syndromes, as targeting these factors could be used to treat pain. An example of the power of this approach can be seen from our work on the PFTs from S. aureus, which mediate nociceptor depolarization and acute pain during skin infection; we showed that QX-314, a membrane-impermeable sodium channel blocker, alleviates pain more potently than ibuprofen after S. aureus infection80. The putative mechanism is that pores formed by PFTs allow for specific delivery of QX-314 into nociceptors, where it acts to inhibit sodium channel activity and neuronal firing. Thus, QX-314 can potentially be used for targeting pain not only in S. aureus infection but pain caused by other pathogens that produce PFTs. A greater understanding of the molecular basis of pain associated with the microbiota and pathogens could enable new approaches to treat pain in infectious illnesses, as well as to treat painful conditions where the gut microbiota modulates pain signalling.
Our emerging understanding of how nociceptors signal to the immune system could also lead to new therapeutic strategies for acute infectious diseases and chronic inflammatory conditions. We now appreciate that neuroimmune signalling can be a double-edged sword in the context of host defence. Pathogens may silence pain to facilitate their spread from host to host or activate pain to suppress the immune system. In S. pyogenes-induced necrotizing fasciitis in mice, blocking nociceptor–immune signalling by administration of botulinum toxin to block local neuropeptide release resulted in improved control of infection12. In contrast, induction of nociceptor-mediated anti-inflammatory responses could be used in infections where morbidity results more from pathological an outsized inflammation or to treat autoimmune disorders, possibly with more localized activity and less systemic immunosuppression than conventional approaches. Nociceptor signalling is beneficial for host defence in certain infectious diseases and is silenced by some pathogens, presumably to facilitate disease. Therefore, a better understanding of the underlying processes could help to develop therapeutic approaches for a diverse spectrum of illnesses.
Glossary terms:
nociceptor
Nociceptors are sensory nerves that are activated by noxious or potentially damaging stimuli. Two classes of nerve fibers, Aδ and C fibers, make up nociceptors in humans, and nociceptors are classified according to their ability to respond to mechanical, thermal, and chemical stimuli. Free nerve endings in the periphery serving as receptive sites extend from neuronal cell bodies in the dorsal root or cranial nerve ganglia.
dorsal root ganglia (DRG)
Dorsal root ganglia (DRG) contain clusters of sensory neurons that reside adjacent to the spinal cord, which include nociceptors. These neurons are pseudo-unipolar in nature, meaning that they have one axon with two processes: one peripheral axonal branch that innervates the tissues of the body to receive sensory information, and one axonal branch that sends nerve impulses to the spinal cord. DRG also house satellite glia and macrophages that can modulate the function of sensory neurons.
allodynia
Allodynia is a painful sensation caused by a normally innocuous stimuli. For example, mechanical allodynia can be caused by light touch or stroking.
hyperalgesia
Hyperalgesia is an enhanced sensitivity to a normally painful mechanical or thermal stimuli.
Capsaicin
Capsaicin is the pungent ingredient from chili peppers that elicits the burning sensation of pain. Capsaicin is a ligand that binds to the transient receptor potential subfamily V member 1 (TRPV1) ion channel on nociceptors to drive pain sensation.
Substance P
Substance P is an 11 amino acid neuropeptide belonging to the tachykinin family. It is formed by differential splicing of the preprotachykinin A gene (TAC1). Substance P is widely distributed throughout the nervous system but has been best appreciated as an important neurotransmitter in nociceptive pathways.
CGRP
Calcitonin gene-related peptide (CGRP) is a neuropeptide formed by the alternative splicing of the calcitonin gene. It has two isoforms, α-CGRP and β-CGRP, which differ in three amino acids and are transcribed from distinct genes (Calca and Caleb). Classically, α-CGRP has been thought to be the primary form expressed in the peripheral and central nervous systems, while β-CGRP is mainly found in the enteric nervous system. Of note, while the primary association of CGRP is with the nervous system and particularly with nociceptive signaling, expression has been reported in non-neuronal cells as well.
M2 macrophage
M2 macrophages are known as ‘alternatively activated’ or anti-inflammatory macrophages. Multiple anti-inflammatory cytokines such as IL-4 , IL-13, or IL-10 drive M2 macrophage polarization. While M1 macrophages drive host inflammation via release of pro-inflammatory cytokines, M2 macrophages mediate the resolution of inflammation and the wound healing.
Vasoactive intestinal polypeptide (VIP)
VIP is a 28 amino-acid peptide belonging to the glucagon/secretin family and encoded by the VIP gene in humans. VIP acts via the G-protein coupled receptors VPAC1 and VPAC2. It is broadly expressed throughout the nervous system and peripheral tissues and has been best appreciated as a neurotransmitter implicated in gastrointestinal motility.
Pituitary adenylate cyclase-activating polypeptide (PACAP)
PACAP is a peptide with close homology to VIP encoded by the Adcyapl gene in humans and has two biologically active forms, PACAP-27 or PACAP-38. PACAP has high affinity for three classes of G-protein coupled receptors: VPAC1, VPAC2, and PAC1. Like VIP, PACAP is broadly expressed in the nervous system and peripheral tissues.
orthodromic
Orthodromic refers to the traditional direction of action potentials in nerves, running along the axon away from the neuronal soma. For a peripheral sensory neuron, orthodromic means propagation of action potentials towards central nerve terminals and the spinal cord.
antidromic
Antidromic refers to the opposite direction of traditional nerve impulses. For a peripheral sensory neuron, antidromic refers to back-propagation of action potentials towards peripheral nerve endings.
Presynaptic neuron
Anatomically, the neuron before the synapse which delivers the chemical neurotransmitter to the postsynaptic neuron. In Figure 1, presynaptic refers to peripheral sensory neurons projecting from the peripheral tissues to spinal cord whose cell bodies are located in the dorsal root ganglia. These presynaptic neurons transmit pain signals from the periphery to the spinal cord.
Postsynaptic neuron
The neuron after the synapse which receives the chemical transmitter from the presynaptic neuron. In Figure 1, postsynaptic refers to second order neurons in the dorsal horn of the spinal cord that project to the brain. These neurons transmit the nociceptive signals received from presynaptic neurons.
References
- 1.Scholz J & Woolf CJ Can we conquer pain? Nat Neurosci 5 Suppl, 1062–1067, doi: 10.1038/nn942 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Julius D & Basbaum AI Molecular mechanisms of nociception. Nature 413, 203–210, doi: 10.1038/35093019 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Basbaum AI, Bautista DM, Scherrer G & Julius D Cellular and molecular mechanisms of pain. Cell 139, 267–284 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiu IM, von Hehn CA & Woolf CJ Neurogenic inflammation and the peripheral nervous system in host defense and immunopathology. Nat Neurosci 15, 1063–1067, doi: 10.1038/nn.3144 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiu IM, Pinho-Ribeiro FA & Woolf CJ Pain and infection: pathogen detection by nociceptors. Pain 157, 1192–1193, doi: 10.1097/j.pain.0000000000000559 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinho-Ribeiro FA, Verri WA Jr. & Chiu IM Nociceptor Sensory Neuron-Immune Interactions in Pain and Inflammation. Trends Immunol 38, 5–19, doi: 10.1016/j.it.2016.10.001 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baral P, Mills K, Pinho-Ribeiro FA & Chiu IM Pain and Itch: Beneficial or Harmful to Antimicrobial Defense? Cell Host Microbe 19, 755–759, doi: 10.1016/j.chom.2016.05.010 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Riol-Blanco L et al. Nociceptive sensory neurons drive interleukin-23-mediated psoriasiform skin inflammation. Nature 510, 157–161, doi: 10.1038/nature13199 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kashem SW et al. Nociceptive Sensory Fibers Drive Interleukin-23 Production from CD301b+ Dermal Dendritic Cells and Drive Protective Cutaneous Immunity. Immunity 43, 515–526, doi: 10.1016/j.immuni.2015.08.016 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engel MA et al. TRPA1 and substance P mediate colitis in mice. Gastroenterology 141, 1346–1358, doi: 10.1053/j.gastro.2011.07.002 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Maruyama K et al. Nociceptors Boost the Resolution of Fungal Osteoinflammation via the TRP Channel-CGRP-Jdp2 Axis. Cell Rep 19, 2730–2742, doi: 10.1016/j.celrep.2017.06.002 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Pinho-Ribeiro FA et al. Blocking Neuronal Signaling to Immune Cells Treats Streptococcal Invasive Infection. Cell 173, 1083–1097 e1022, doi: 10.1016/j.cell.2018.04.006 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Costigan M, Scholz J & Woolf CJ in Annu Rev Neurosci Vol. 32 1–32 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Latremoliere A & Woolf CJ Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain 10, 895–926, doi: 10.1016/j.jpain.2009.06.012 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ji RR, Xu ZZ & Gao YJ Emerging targets in neuroinflammation-driven chronic pain. Nat Rev Drug Discov 13, 533–548, doi: 10.1038/nrd4334 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cook AD, Christensen AD, Tewari D, McMahon SB & Hamilton JA Immune Cytokines and Their Receptors in Inflammatory Pain. Trends Immunol 39, 240–255, doi: 10.1016/j.it.2017.12.003 (2018). [DOI] [PubMed] [Google Scholar]
- 17.White FA, Bhangoo SK & Miller RJ Chemokines: integrators of pain and inflammation. Nat Rev Drug Discov 4, 834–844, doi: 10.1038/nrd1852 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boettger MK et al. Antinociceptive effects of tumor necrosis factor alpha neutralization in a rat model of antigen-induced arthritis: evidence of a neuronal target. Arthritis Rheum 58, 2368–2378, doi: 10.1002/art.23608 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Richter F et al. Tumor necrosis factor causes persistent sensitization of joint nociceptors to mechanical stimuli in rats. Arthritis Rheum 62, 3806–3814, doi: 10.1002/art.27715 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Jin X & Gereau RW t. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. J Neurosci 26, 246–255, doi: 10.1523/JNEUROSCI.3858-05.2006 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cunha TM et al. A cascade of cytokines mediates mechanical inflammatory hypernociception in mice. Proc Natl Acad Sci U S A 102, 1755–1760, doi: 10.1073/pnas.0409225102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inglis JJ et al. Collagen-induced arthritis as a model of hyperalgesia: functional and cellular analysis of the analgesic actions of tumor necrosis factor blockade. Arthritis Rheum 56, 4015–4023, doi: 10.1002/art.23063 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Zhang L et al. TNF-alpha contributes to spinal cord synaptic plasticity and inflammatory pain: distinct role of TNF receptor subtypes 1 and 2. Pain 152, 419–427, doi: 10.1016/j.pain.2010.11.014 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hess A et al. Blockade of TNF-alpha rapidly inhibits pain responses in the central nervous system. Proc Natl Acad Sci U S A 108, 3731–3736, doi: 10.1073/pnas.1011774108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferreira SH, Lorenzetti BB, Bristow AF & Poole S Interleukin-1 beta as a potent hyperalgesic agent antagonized by a tripeptide analogue. Nature 334, 698–700, doi: 10.1038/334698a0 (1988). [DOI] [PubMed] [Google Scholar]
- 26.Ebbinghaus M et al. The role of interleukin-1beta in arthritic pain: main involvement in thermal, but not mechanical, hyperalgesia in rat antigen-induced arthritis. Arthritis Rheum 64, 3897–3907, doi: 10.1002/art.34675 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Fukuoka H, Kawatani M, Hisamitsu T & Takeshige C Cutaneous hyperalgesia induced by peripheral injection of interleukin-1 beta in the rat. Brain Res 657, 133–140 (1994). [DOI] [PubMed] [Google Scholar]
- 28.Binshtok AM et al. Nociceptors are interleukin-1beta sensors. J Neurosci 28, 14062–14073, doi: 10.1523/JNEUROSCI.3795-08.2008 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thakur M et al. Defining the nociceptor transcriptome. Front Mol Neurosci 7, 87, doi: 10.3389/fnmol.2014.00087 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu XJ et al. Nociceptive responses in interleukin-6-deficient mice to peripheral inflammation and peripheral nerve section. Cytokine 9, 1028–1033, doi: 10.1006/cyto.1997.0243 (1997). [DOI] [PubMed] [Google Scholar]
- 31.Malsch P et al. Deletion of interleukin-6 signal transducer gp130 in small sensory neurons attenuates mechanonociception and down-regulates TRPA1 expression. J Neurosci 34, 9845–9856, doi: 10.1523/JNEUROSCI.5161-13.2014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vazquez E et al. Spinal interleukin-6 is an amplifier of arthritic pain in the rat. Arthritis Rheum 64, 2233–2242, doi: 10.1002/art.34384 (2012). [DOI] [PubMed] [Google Scholar]
- 33.McNamee KE et al. IL-17 induces hyperalgesia via TNF-dependent neutrophil infiltration. Pain 152, 1838–1845, doi: 10.1016/j.pain.2011.03.035 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Pinto LG et al. IL-17 mediates articular hypernociception in antigen-induced arthritis in mice. Pain 148, 247–256, doi: 10.1016/j.pain.2009.11.006 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Richter F et al. Interleukin-17 sensitizes joint nociceptors to mechanical stimuli and contributes to arthritic pain through neuronal interleukin-17 receptors in rodents. Arthritis Rheum 64, 4125–4134, doi: 10.1002/art.37695 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Krukowski K et al. CD8+ T Cells and Endogenous IL-10 Are Required for Resolution of Chemotherapy-Induced Neuropathic Pain. J Neurosci 36, 11074–11083, doi: 10.1523/JNEUROSCI.3708-15.2016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Milligan ED et al. Controlling pathological pain by adenovirally driven spinal production of the anti-inflammatory cytokine, interleukin-10. Eur J Neurosci 21, 2136–2148, doi: 10.1111/j.1460-9568.2005.04057.x (2005). [DOI] [PubMed] [Google Scholar]
- 38.Shen KF et al. Interleukin-10 down-regulates voltage gated sodium channels in rat dorsal root ganglion neurons. Exp Neurol 247, 466–475, doi: 10.1016/j.expneurol.2013.01.018 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Schweizerhof M et al. Hematopoietic colony-stimulating factors mediate tumor-nerve interactions and bone cancer pain. Nat Med 15, 802–807, doi: 10.1038/nm.1976 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Cook AD et al. Granulocyte-macrophage colony-stimulating factor is a key mediator in inflammatory and arthritic pain. Ann Rheum Dis 72, 265–270, doi: 10.1136/annrheumdis-2012-201703 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Saleh R et al. CSF-1 in Inflammatory and Arthritic Pain Development. J Immunol 201, 2042–2053, doi: 10.4049/jimmunol.1800665 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Achuthan A et al. Granulocyte macrophage colony-stimulating factor induces CCL17 production via IRF4 to mediate inflammation. J Clin Invest 126, 3453–3466, doi: 10.1172/JCI87828 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Talbot S, Foster SL & Woolf CJ Neuroimmunity: Physiology and Pathology. Annu Rev Immunol 34, 421–447, doi: 10.1146/annurev-immunol-041015-055340 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Chen L, Yang G & Grosser T Prostanoids and inflammatory pain. Prostaglandins Other Lipid Mediat 104-105, 58–66, doi: 10.1016/j.prostaglandins.2012.08.006 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Ferreira SH Prostaglandins, aspirin-like drugs and analgesia. Nat New Biol 240, 200–203 (1972). [DOI] [PubMed] [Google Scholar]
- 46.Samad TA et al. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature 410, 471–475, doi: 10.1038/35068566 (2001). [DOI] [PubMed] [Google Scholar]
- 47.Baba H, Kohno T, Moore KA & Woolf CJ Direct activation of rat spinal dorsal horn neurons by prostaglandin E2. J Neurosci 21, 1750–1756 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levine JD, Lau W, Kwiat G & Goetzl EJ Leukotriene B4 produces hyperalgesia that is dependent on polymorphonuclear leukocytes. Science 225, 743–745 (1984). [DOI] [PubMed] [Google Scholar]
- 49.Zinn S et al. The leukotriene B4 receptors BLT1 and BLT2 form an antagonistic sensitizing system in peripheral sensory neurons. J Biol Chem 292, 6123–6134, doi: 10.1074/jbc.M116.769125 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Denk F, Bennett DL & McMahon SB Nerve Growth Factor and Pain Mechanisms. Annu Rev Neurosci 40, 307–325, doi: 10.1146/annurev-neuro-072116-031121 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Mizumura K & Murase S Role of nerve growth factor in pain. Handb Exp Pharmacol 227, 57–77, doi: 10.1007/978-3-662-46450-2_4 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Halliday DA, Zettler C, Rush RA, Scicchitano R & McNeil JD Elevated nerve growth factor levels in the synovial fluid of patients with inflammatory joint disease. Neurochem Res 23, 919–922 (1998). [DOI] [PubMed] [Google Scholar]
- 53.Lane NE et al. Tanezumab for the treatment of pain from osteoarthritis of the knee. N Engl J Med 363, 1521–1531, doi: 10.1056/NEJMoa0901510 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bannwarth B & Kostine M Nerve Growth Factor Antagonists: Is the Future of Monoclonal Antibodies Becoming Clearer? Drugs 77, 1377–1387, doi: 10.1007/s40265-017-0781-6 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Ji RR, Samad TA, Jin SX, Schmoll R & Woolf CJ p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron 36, 57–68 (2002). [DOI] [PubMed] [Google Scholar]
- 56.Zhang X, Huang J & McNaughton PA NGF rapidly increases membrane expression of TRPV1 heat-gated ion channels. EMBO J 24, 4211–4223, doi: 10.1038/sj.emboj.7600893 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kerr BJ, Souslova V, McMahon SB & Wood JN A role for the TTX-resistant sodium channel Nav 1.8 in NGF-induced hyperalgesia, but not neuropathic pain. Neuroreport 12, 3077–3080 (2001). [DOI] [PubMed] [Google Scholar]
- 58.Coull JA et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438, 1017–1021, doi: 10.1038/nature04223 (2005). [DOI] [PubMed] [Google Scholar]
- 59.Sorge RE et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci 18, 1081–1083, doi: 10.1038/nn.4053 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Julius D TRP channels and pain. Annu Rev Cell Dev Biol 29, 355–384, doi: 10.1146/annurev-cellbio-101011-155833 (2013). [DOI] [PubMed] [Google Scholar]
- 61.Cheng JK & Ji RR Intracellular signaling in primary sensory neurons and persistent pain. Neurochem Res 33, 1970–1978, doi: 10.1007/s11064-008-9711-z (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chuang HH et al. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature 411, 957–962, doi: 10.1038/35082088 (2001). [DOI] [PubMed] [Google Scholar]
- 63.Prescott ED & Julius D A modular PIP2 binding site as a determinant of capsaicin receptor sensitivity. Science 300, 1284–1288, doi: 10.1126/science.1083646 (2003). [DOI] [PubMed] [Google Scholar]
- 64.Bandell M et al. Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron 41, 849–857 (2004). [DOI] [PubMed] [Google Scholar]
- 65.Premkumar LS & Ahern GP Induction of vanilloid receptor channel activity by protein kinase C. Nature 408, 985–990, doi: 10.1038/35050121 (2000). [DOI] [PubMed] [Google Scholar]
- 66.Zhang X, Li L & McNaughton PA Proinflammatory mediators modulate the heat-activated ion channel TRPV1 via the scaffolding protein AKAP79/150. Neuron 59, 450–461, doi: 10.1016/j.neuron.2008.05.015 (2008). [DOI] [PubMed] [Google Scholar]
- 67.Khan AA et al. Tumor necrosis factor alpha enhances the sensitivity of rat trigeminal neurons to capsaicin. Neuroscience 155, 503–509, doi: 10.1016/j.neuroscience.2008.05.036 (2008). [DOI] [PubMed] [Google Scholar]
- 68.Fang D et al. Interleukin-6-mediated functional upregulation of TRPV1 receptors in dorsal root ganglion neurons through the activation of JAK/PI3K signaling pathway: roles in the development of bone cancer pain in a rat model. Pain 156, 1124–1144, doi: 10.1097/j.pain.0000000000000158 (2015). [DOI] [PubMed] [Google Scholar]
- 69.Viana F TRPA1 channels: molecular sentinels of cellular stress and tissue damage. J Physiol 594, 4151–4169, doi: 10.1113/JP270935 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Obata K et al. TRPA1 induced in sensory neurons contributes to cold hyperalgesia after inflammation and nerve injury. J Clin Invest 115, 2393–2401, doi: 10.1172/JCI25437 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schmidt M, Dubin AE, Petrus MJ, Earley TJ & Patapoutian A Nociceptive signals induce trafficking of TRPA1 to the plasma membrane. Neuron 64, 498–509, doi: 10.1016/j.neuron.2009.09.030 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Alessandri-Haber N et al. Hypotonicity induces TRPV4-mediated nociception in rat. Neuron 39, 497–511 (2003). [DOI] [PubMed] [Google Scholar]
- 73.Alessandri-Haber N, Dina OA, Joseph EK, Reichling D & Levine JD A transient receptor potential vanilloid 4-dependent mechanism of hyperalgesia is engaged by concerted action of inflammatory mediators. J Neurosci 26, 3864–3874, doi: 10.1523/JNEUROSCI.5385-05.2006 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Alessandri-Haber N et al. Transient receptor potential vanilloid 4 is essential in chemotherapy-induced neuropathic pain in the rat. J Neurosci 24, 4444–4452, doi: 10.1523/JNEUROSCI.0242-04.2004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Todaka H, Taniguchi J, Satoh J, Mizuno A & Suzuki M Warm temperature-sensitive transient receptor potential vanilloid 4 (TRPV4) plays an essential role in thermal hyperalgesia. J Biol Chem 279, 35133–35138, doi: 10.1074/jbc.M406260200 (2004). [DOI] [PubMed] [Google Scholar]
- 76.Dib-Hajj SD, Cummins TR, Black JA & Waxman SG Sodium channels in normal and pathological pain. Annu Rev Neurosci 33, 325–347, doi: 10.1146/annurev-neuro-060909-153234 (2010). [DOI] [PubMed] [Google Scholar]
- 77.Black JA, Liu S, Tanaka M, Cummins TR & Waxman SG Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain 108, 237–247, doi: 10.1016/j.pain.2003.12.035 (2004). [DOI] [PubMed] [Google Scholar]
- 78.Strickland IT et al. Changes in the expression of Nav1.7, Nav1.8 and Nav1.9 in a distinct population of dorsal root ganglia innervating the rat knee joint in a model of chronic inflammatory joint pain. Eur J Pain 12, 564–572, doi: 10.1016/j.ejpain.2007.09.001 (2008). [DOI] [PubMed] [Google Scholar]
- 79.Chiu IM et al. Bacteria activate sensory neurons that modulate pain and inflammation. Nature 501, 52–57, doi: 10.1038/nature12479 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Blake KJ et al. Staphylococcus aureus produces pain through pore-forming toxins and neuronal TRPV1 that is silenced by QX-314. Nat Commun 9, 37, doi: 10.1038/s41467-017-02448-6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Diogenes A, Ferraz CC, Akopian AN, Henry MA & Hargreaves KM LPS sensitizes TRPV1 via activation of TLR4 in trigeminal sensory neurons. J Dent Res 90, 759–764, doi: 10.1177/0022034511400225 (2011). [DOI] [PubMed] [Google Scholar]
- 82.Meseguer V et al. TRPA1 channels mediate acute neurogenic inflammation and pain produced by bacterial endotoxins. Nat Commun 5, 3125, doi: 10.1038/ncomms4125 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rudick CN et al. Host-pathogen interactions mediating pain of urinary tract infection. J Infect Dis 201, 1240–1249, doi: 10.1086/651275 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Alpizar YA et al. TRPV4 activation triggers protective responses to bacterial lipopolysaccharides in airway epithelial cells. Nat Commun 8, 1059, doi: 10.1038/s41467-017-01201-3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xu ZZ et al. Inhibition of mechanical allodynia in neuropathic pain by TLR5-mediated A-fiber blockade. Nat Med 21, 1326–1331, doi: 10.1038/nm.3978 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yissachar N et al. An Intestinal Organ Culture System Uncovers a Role for the Nervous System in Microbe-Immune Crosstalk. Cell 168, 1135–1148 e1112, doi: 10.1016/j.cell.2017.02.009 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Maruyama K et al. The ATP Transporter VNUT Mediates Induction of Dectin-1-Triggered Candida Nociception. Science 6, 306–318, doi: 10.1016/j.isci.2018.08.007 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fields HL, Rowbotham M & Baron R Postherpetic neuralgia: irritable nociceptors and deafferentation. Neurobiol Dis 5, 209–227, doi: 10.1006/nbdi.1998.0204 (1998). [DOI] [PubMed] [Google Scholar]
- 89.Steiner I, Kennedy PG & Pachner AR The neurotropic herpes viruses: herpes simplex and varicella-zoster. Lancet Neurol 6, 1015–1028, doi: 10.1016/S1474-4422(07)70267-3 (2007). [DOI] [PubMed] [Google Scholar]
- 90.Marion E et al. Mycobacterial toxin induces analgesia in buruli ulcer by targeting the angiotensin pathways. Cell 157, 1565–1576, doi: 10.1016/j.cell.2014.04.040 (2014). [DOI] [PubMed] [Google Scholar]
- 91.Lotz M, Vaughan JH & Carson DA Effect of neuropeptides on production of inflammatory cytokines by human monocytes. Science 241, 1218–1221 (1988). [DOI] [PubMed] [Google Scholar]
- 92.Sun J, Ramnath RD, Zhi L, Tamizhselvi R & Bhatia M Substance P enhances NF-kappaB transactivation and chemokine response in murine macrophages via ERK1/2 and p38 MAPK signaling pathways. Am J Physiol Cell Physiol 294, C1586–1596, doi: 10.1152/ajpcell.00129.2008 (2008). [DOI] [PubMed] [Google Scholar]
- 93.Lim JE, Chung E & Son Y A neuropeptide, Substance-P, directly induces tissue-repairing M2 like macrophages by activating the PI3K/Akt/mTOR pathway even in the presence of IFNgamma. Sci Rep 7, 9417, doi: 10.1038/s41598-017-09639-7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hong HS & Son Y Substance P ameliorates collagen II-induced arthritis in mice via suppression of the inflammatory response. Biochem Biophys Res Commun 453, 179–184, doi: 10.1016/j.bbrc.2014.09.090 (2014). [DOI] [PubMed] [Google Scholar]
- 95.Baliu-Pique M, Jusek G & Holzmann B Neuroimmunological communication via CGRP promotes the development of a regulatory phenotype in TLR4-stimulated macrophages. Eur J Immunol 44, 3708–3716, doi: 10.1002/eji.201444553 (2014). [DOI] [PubMed] [Google Scholar]
- 96.Nong YH, Titus RG, Ribeiro JM & Remold HG Peptides encoded by the calcitonin gene inhibit macrophage function. J Immunol 143, 45–49 (1989). [PubMed] [Google Scholar]
- 97.Yaraee R, Ebtekar M, Ahmadiani A & Sabahi F Effect of neuropeptides (SP and CGRP) on antigen presentation by macrophages. Immunopharmacol Immunotoxicol 27, 395–404, doi: 10.1080/08923970500240974 (2005). [DOI] [PubMed] [Google Scholar]
- 98.Russell FA, King R, Smillie SJ, Kodji X & Brain SD Calcitonin gene-related peptide: physiology and pathophysiology. Physiol Rev 94, 1099–1142, doi: 10.1152/physrev.00034.2013 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Harzenetter MD et al. Negative regulation of TLR responses by the neuropeptide CGRP is mediated by the transcriptional repressor ICER. J Immunol 179, 607–615 (2007). [DOI] [PubMed] [Google Scholar]
- 100.Gomes RN et al. Calcitonin gene-related peptide inhibits local acute inflammation and protects mice against lethal endotoxemia. Shock 24, 590–594 (2005). [DOI] [PubMed] [Google Scholar]
- 101.Jusek G, Reim D, Tsujikawa K & Holzmann B Deficiency of the CGRP receptor component RAMP1 attenuates immunosuppression during the early phase of septic peritonitis. Immunobiology 217, 761–767, doi: 10.1016/j.imbio.2012.04.009 (2012). [DOI] [PubMed] [Google Scholar]
- 102.Delgado M et al. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit tumor necrosis factor alpha transcriptional activation by regulating nuclear factor-kB and cAMP response element-binding protein/c-Jun. J Biol Chem 273, 31427–31436 (1998). [DOI] [PubMed] [Google Scholar]
- 103.Martinez C et al. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide modulate endotoxin-induced IL-6 production by murine peritoneal macrophages. J Leukoc Biol 63, 591–601 (1998). [DOI] [PubMed] [Google Scholar]
- 104.Fernandes ES et al. TRPV1 deletion enhances local inflammation and accelerates the onset of systemic inflammatory response syndrome. J Immunol 188, 5741–5751, doi: 10.4049/jimmunol.1102147 (2012). [DOI] [PubMed] [Google Scholar]
- 105.Strausbaugh HJ et al. Painful stimulation suppresses joint inflammation by inducing shedding of L-selectin from neutrophils. Nat Med 5, 1057–1061, doi: 10.1038/12497 (1999). [DOI] [PubMed] [Google Scholar]
- 106.Baral P et al. Nociceptor sensory neurons suppress neutrophil and gammadelta T cell responses in bacterial lung infections and lethal pneumonia. Nat Med 24, 417–426, doi: 10.1038/nm.4501 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lei J et al. Transient Receptor Potential Vanilloid Subtype 1 Inhibits Inflammation and Apoptosis via the Release of Calcitonin Gene-Related Peptide in the Heart after Myocardial Infarction. Cardiology 134, 436–443, doi: 10.1159/000444439 (2016). [DOI] [PubMed] [Google Scholar]
- 108.Huang J, Stohl LL, Zhou X, Ding W & Granstein RD Calcitonin gene-related peptide inhibits chemokine production by human dermal microvascular endothelial cells. Brain Behav Immun 25, 787–799, doi: 10.1016/j.bbi.2011.02.007 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zimmerman BJ, Anderson DC & Granger DN Neuropeptides promote neutrophil adherence to endothelial cell monolayers. Am J Physiol 263, G678–682, doi: 10.1152/ajpgi.1992.263.5.G678 (1992). [DOI] [PubMed] [Google Scholar]
- 110.Richter J, Andersson R, Edvinsson L & Gullberg U Calcitonin gene-related peptide (CGRP) activates human neutrophils--inhibition by chemotactic peptide antagonist BOC-MLP. Immunology 77, 416–421 (1992). [PMC free article] [PubMed] [Google Scholar]
- 111.Mulderry PK et al. Differential expression of alpha-CGRP and beta-CGRP by primary sensory neurons and enteric autonomic neurons of the rat. Neuroscience 25, 195–205 (1988). [DOI] [PubMed] [Google Scholar]
- 112.Sui P et al. Pulmonary neuroendocrine cells amplify allergic asthma responses. Science 360, doi: 10.1126/science.aan8546 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Alving K et al. Association between histamine-containing mast cells and sensory nerves in the skin and airways of control and capsaicin-treated pigs. Cell Tissue Res 264, 529–538 (1991). [DOI] [PubMed] [Google Scholar]
- 114.Arizono N et al. Anatomical variation in mast cell nerve associations in the rat small intestine, heart, lung, and skin. Similarities of distances between neural processes and mast cells, eosinophils, or plasma cells in the jejunal lamina propria. Lab Invest 62, 626–634 (1990). [PubMed] [Google Scholar]
- 115.Stead RH et al. Intestinal mucosal mast cells in normal and nematode-infected rat intestines are in intimate contact with peptidergic nerves. Proc Natl Acad Sci U S A 84, 2975–2979 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Furuno T et al. Cell adhesion molecule 1 (CADM1) on mast cells promotes interaction with dorsal root ganglion neurites by heterophilic binding to nectin-3. J Neuroimmunol 250, 50–58, doi: 10.1016/j.jneuroim.2012.05.016 (2012). [DOI] [PubMed] [Google Scholar]
- 117.Jarvikallio A, Harvima IT & Naukkarinen A Mast cells, nerves and neuropeptides in atopic dermatitis and nummular eczema. Arch Dermatol Res 295, 2–7, doi: 10.1007/s00403-002-0378-z (2003). [DOI] [PubMed] [Google Scholar]
- 118.Mollanazar NK, Smith PK & Yosipovitch G Mediators of Chronic Pruritus in Atopic Dermatitis: Getting the Itch Out? Clin Rev Allergy Immunol 51, 263–292, doi: 10.1007/s12016-015-8488-5 (2016). [DOI] [PubMed] [Google Scholar]
- 119.Azimi E et al. Dual action of neurokinin-1 antagonists on Mas-related GPCRs. JCI Insight 1, e89362, doi: 10.1172/jci.insight.89362 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.McNeil BD et al. Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature 519, 237–241, doi: 10.1038/nature14022 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bulut K et al. Sensory neuropeptides and epithelial cell restitution: the relevance of SP- and CGRP-stimulated mast cells. Int J Colorectal Dis 23, 535–541, doi: 10.1007/s00384-008-0447-7 (2008). [DOI] [PubMed] [Google Scholar]
- 122.Kim JH et al. CGRP, a neurotransmitter of enteric sensory neurons, contributes to the development of food allergy due to the augmentation of microtubule reorganization in mucosal mast cells. Biomed Res 35, 285–293 (2014). [DOI] [PubMed] [Google Scholar]
- 123.Russo AF CGRP as a neuropeptide in migraine: lessons from mice. Br J Clin Pharmacol 80, 403–414, doi: 10.1111/bcp.12686 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Eftekhari S, Warfvinge K, Blixt FW & Edvinsson L Differentiation of nerve fibers storing CGRP and CGRP receptors in the peripheral trigeminovascular system. J Pain 14, 1289–1303, doi: 10.1016/j.jpain.2013.03.010 (2013). [DOI] [PubMed] [Google Scholar]
- 125.Mikami N et al. Calcitonin gene-related peptide regulates type IV hypersensitivity through dendritic cell functions. PLoS One 9, e86367, doi: 10.1371/journal.pone.0086367 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ding W, Stohl LL, Wagner JA & Granstein RD Calcitonin gene-related peptide biases Langerhans cells toward Th2-type immunity. J Immunol 181, 6020–6026 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Delgado M, Gonzalez-Rey E & Ganea D VIP/PACAP preferentially attract Th2 effectors through differential regulation of chemokine production by dendritic cells. FASEB J 18, 1453–1455, doi: 10.1096/fj.04-1548fje (2004). [DOI] [PubMed] [Google Scholar]
- 128.Delgado M, Reduta A, Sharma V & Ganea D VIP/PACAP oppositely affects immature and mature dendritic cell expression of CD80/CD86 and the stimulatory activity for CD4(+) T cells. J Leukoc Biol 75, 1122–1130, doi: 10.1189/jlb.1203626 (2004). [DOI] [PubMed] [Google Scholar]
- 129.Ding W et al. Pituitary adenylate cyclase-activating peptide and vasoactive intestinal polypeptide bias Langerhans cell Ag presentation toward Th17 cells. Eur J Immunol 42, 901–911, doi: 10.1002/eji.201141958 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.de Jong PR et al. TRPM8 on mucosal sensory nerves regulates colitogenic responses by innate immune cells via CGRP. Mucosal Immunol 8, 491–504, doi: 10.1038/mi.2014.82 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Voedisch S, Rochlitzer S, Veres TZ, Spies E & Braun A Neuropeptides control the dynamic behavior of airway mucosal dendritic cells. PLoS One 7, e45951, doi: 10.1371/journal.pone.0045951 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wallrapp A et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 549, 351–356, doi: 10.1038/nature24029 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cardoso V et al. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature 549, 277–281, doi: 10.1038/nature23469 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nussbaum JC et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 502, 245–248, doi: 10.1038/nature12526 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mikami N et al. Calcitonin gene-related peptide is an important regulator of cutaneous immunity: effect on dendritic cell and T cell functions. J Immunol 186, 6886–6893, doi: 10.4049/jimmunol.1100028 (2011). [DOI] [PubMed] [Google Scholar]
- 136.Ding W et al. Calcitonin Gene-Related Peptide-Exposed Endothelial Cells Bias Antigen Presentation to CD4+ T Cells toward a Th17 Response. J Immunol 196, 2181–2194, doi: 10.4049/jimmunol.1500303 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Talbot S et al. Silencing Nociceptor Neurons Reduces Allergic Airway Inflammation. Neuron 87, 341–354, doi: 10.1016/j.neuron.2015.06.007 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Delgado M VIP: a very important peptide in T helper differentiation. Trends Immunol 24, 221–224 (2003). [DOI] [PubMed] [Google Scholar]
- 139.Weinstock JV et al. Substance P regulates Th1-type colitis in IL-10 knockout mice. J Immunol 171, 3762–3767 (2003). [DOI] [PubMed] [Google Scholar]
- 140.Cunin P et al. The tachykinins substance P and hemokinin-1 favor the generation of human memory Th17 cells by inducing IL-1beta, IL-23, and TNF-like 1A expression by monocytes. J Immunol 186, 4175–4182, doi: 10.4049/jimmunol.1002535 (2011). [DOI] [PubMed] [Google Scholar]
- 141.Walters N, Trunkle T, Sura M & Pascual DW Enhanced immunoglobulin A response and protection against Salmonella enterica serovar typhimurium in the absence of the substance P receptor. Infect Immun 73, 317–324, doi: 10.1128/IAI.73.1.317-324.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Li WW et al. Neuropeptide regulation of adaptive immunity in the tibia fracture model of complex regional pain syndrome. J Neuroinflammation 15, 105, doi: 10.1186/s12974-018-1145-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.McGillis JP, Humphreys S, Rangnekar V & Ciallella J Modulation of B lymphocyte differentiation by calcitonin gene-related peptide (CGRP). I. Characterization of high-affinity CGRP receptors on murine 70Z/3 cells. Cell Immunol 150, 391–404 (1993). [DOI] [PubMed] [Google Scholar]
- 144.McGillis JP, Humphreys S & Reid S Characterization of functional calcitonin gene-related peptide receptors on rat lymphocytes. J Immunol 147, 3482–3489 (1991). [PubMed] [Google Scholar]
- 145.Payan DG, Brewster DR, Missirian-Bastian A & Goetzl EJ Substance P recognition by a subset of human T lymphocytes. J Clin Invest 74, 1532–1539, doi: 10.1172/JG111567 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Reubi JC, Horisberger U, Kappeler A & Laissue JA Localization of receptors for vasoactive intestinal peptide, somatostatin, and substance P in distinct compartments of human lymphoid organs. Blood 92, 191–197 (1998). [PubMed] [Google Scholar]
- 147.Moran MM & Szallasi A Targeting nociceptive transient receptor potential channels to treat chronic pain: current state of the field. Br J Pharmacol 175, 2185–2203, doi: 10.1111/bph.14044 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Grace PM, Hutchinson MR, Maier SF & Watkins LR Pathological pain and the neuroimmune interface. Nat Rev Immunol 14, 217–231, doi: 10.1038/nri3621 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Qin X, Wan Y & Wang X CCL2 and CXCL1 trigger calcitonin gene-related peptide release by exciting primary nociceptive neurons. J Neurosci Res 82, 51–62, doi: 10.1002/jnr.20612 (2005). [DOI] [PubMed] [Google Scholar]
- 150.Cao DL, Qian B, Zhang ZJ, Gao YJ & Wu XB Chemokine receptor CXCR2 in dorsal root ganglion contributes to the maintenance of inflammatory pain. Brain Res Bull 127, 219–225, doi: 10.1016/j.brainresbull.2016.09.016 (2016). [DOI] [PubMed] [Google Scholar]
- 151.Miller RE et al. CCR2 chemokine receptor signaling mediates pain in experimental osteoarthritis. Proc Natl Acad Sci U S A 109, 20602–20607, doi: 10.1073/pnas.1209294110 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gao YJ et al. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J Neurosci 29, 4096–4108, doi: 10.1523/JNEUROSCI.3623-08.2009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zhang ZJ, Cao DL, Zhang X, Ji RR & Gao YJ Chemokine contribution to neuropathic pain: respective induction of CXCL1 and CXCR2 in spinal cord astrocytes and neurons. Pain 154, 2185–2197, doi: 10.1016/j.pain.2013.07.002 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jiang BC et al. CXCL13 drives spinal astrocyte activation and neuropathic pain via CXCR5. J Clin Invest 126, 745–761, doi: 10.1172/JCI81950 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Biber K et al. Neuronal CCL21 up-regulates microglia P2X4 expression and initiates neuropathic pain development. EMBO J 30, 1864–1873, doi: 10.1038/emboj.2011.89 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Milligan ED et al. Evidence that exogenous and endogenous fractalkine can induce spinal nociceptive facilitation in rats. Eur J Neurosci 20, 2294–2302, doi: 10.1111/j.1460-9568.2004.03709.x (2004). [DOI] [PubMed] [Google Scholar]
- 157.Verge GM et al. Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur J Neurosci 20, 1150–1160, doi: 10.1111/j.1460-9568.2004.03593.x (2004). [DOI] [PubMed] [Google Scholar]