PGE2-driven Expression of c-Myc and OncomiR-17-92 Contributes to Apoptosis Resistance in NSCLC (original) (raw)
. Author manuscript; available in PMC: 2015 May 1.
Abstract
Aberrant expression of miRNAs with oncogenic capacities (oncomiRs) has been described for several different malignancies. The first identified oncomiR, miR-17-92, is frequently overexpressed in a variety of cancers and its targets include the tumor suppressor PTEN. The transcription factor c-Myc (MYC) plays a central role in proliferative control and is rapidly up-regulated upon mitogenic stimulation. Expression of c-Myc is frequently deregulated in tumors, facilitating proliferation and inhibiting terminal differentiation. The c-Myc-regulated network comprises a large number of transcripts including those encoding miRNAs. Here prostaglandin E2 (PGE2) exposure rapidly up-regulates expression of the MYC gene followed by the elevation of miR-17-92 levels, which in turn suppresses PTEN expression; thus, enhancing apoptosis resistance in non-small cell lung cancer (NSCLC) cells. Knockdown of MYC expression or the miR-17-92 cluster effectively reverses this outcome. Similarly, miR-17-92 levels are significantly elevated in NSCLC cells ectopically expressing cyclooxygenase-2. Importantly, circulating miR-17-92 was elevated in the blood of lung cancer patients as compared to subjects at risk for developing lung cancer. Furthermore, in patients treated with celecoxib, miR-17-92 levels were significantly reduced. These data demonstrate that PGE2, abundantly produced by NSCLC and inflammatory cells in the tumor microenvironment, is able to stimulate cell proliferation and promote resistance to pharmacologically induced apoptosis in a c-Myc and miR-17-92-dependent manner.
Implications
This study describes a novel mechanism, involving c-Myc and miR-17-92, which integrates cell proliferation and apoptosis resistance.
Keywords: PGE2, c-myc, oncomir, apoptosis resistance, lung cancer
Introduction
Among the fundamental hallmarks of cancer, the capacity to sustain cancer cell proliferation is critical [1]. Chronic proliferation is mediated by several mechanisms, including deregulated growth factor production, abnormal surface receptor expression, loss of negative regulators of cell proliferation due to mutagenesis and transcriptional or epigenetic silencing of tumor suppressors. Importantly, mitogenic signaling by prostaglandin E2 (PGE2)-dependent cross-activation of MAPK/Erk pathway, has been recently shown to promote such hallmarks of cancer as enhanced cell proliferation and apoptosis resistance [2-5]. The c-myc transcription factor plays a central role in control of cellular proliferation [6, 7] and is rapidly upregulated upon mitogenic stimulation [8]. In contrast, anti-proliferative signals have been shown to trigger prompt reduction of c-myc levels due to the transcriptional block and mRNA turnover [9, 10]. CMYC is among the most frequently deregulated protooncogenes in a variety of human malignancies [11]. The causal role of CMYC gene deregulation in cancer development and metastasis has been established in early seminal studies [12, 13]. Recently, c-myc has been identified as a driver of gene signatures associated with poor prognosis in different cancers, including lung cancer, and a necessary component of metastatic cell behavior [14-16]. In keeping with these findings, c-myc overexpression has been reported to be a constituent of a stem cell-like signature in aggressive poorly differentiated tumors [15].
Interestingly, a relationship between c-myc and PTEN (phosphatase and tensin homologue deleted on chromosome 10) has been recently established [17]. PTEN antagonizes PI3-kinase signaling that is necessary for activation of Akt kinase involved in cell survival, and thus increases sensitivity of cells to apoptotic stimuli [18]. Lack of functional redundancy makes PTEN a central (and haploinsufficient) tumor suppressor in various tumor types [19]. PTEN expression is regulated at multiple levels, including transcription, mRNA stability, microRNA targeting, translation and protein stability. PTEN is one the most frequently inactivated tumor suppressor genes [19, 20] and loss of its expression drives tumor development through deregulation of PI3-kinase signaling thus attributing the cells to the second hallmark of cancer, resistance to cell death [1]. Similarly, in murine models inactivation of one allele of PTEN gene increased the incidence of cancer, including lung cancer [21]. Recent studies demonstrate that PTEN is also critical for maintaining the stem cell phenotype and loss of PTEN can contribute to malignant transformation [22, 23].
MicroRNAs (miRNAs) are small regulatory RNAs that control gene expression at the posttranscriptional level and thus serve important roles in a variety of normal and pathologic processes in a wide range of organisms [24, 25]. Recent studies indicate that deregulation of miRNAs is implicated in the pathogenesis of cancer and metastasis [26, 27]. Various miRNAs can function as either tumor suppressors or oncogenes, the latter are often referred to as oncomirs. The first characterized oncomir, miR-17-92 cluster, encodes seven mature microRNAs: miR-17-5p, miR-17-3p, miR-18a, miR-19a, miR-19b, miR-20a and miR-92a, is overexpressed in various cancers and is a direct transcriptional target of c-myc [28, 29]. Recent studies have shown that ectopic expression of miR-17-92 cluster attenuates differentiation and promotes proliferation of lung progenitor cells in transgenic mice [30]. Importantly, PTEN has been identified as one of the main targets of miR-17-92 cluster, specifically, of miR-19 [31]. Studies of the individual contribution of each miRNA to the oncogenic properties of the miR-17-92 cluster identified miR-19, and to a lesser extent miR-18, as the predominant oncogenic miRNA of the cluster [32, 33].
Recent studies demonstrate that miRNA are present not only intracellularly but also in the peripheral blood as circulating miRNA and may reflect those in tumor tissue extracts [34]. MiRNA profiling in both solid tumors and peripheral blood have been used successfully for cancer detection [35, 36]. MiRNAs have been shown to be more robust biomarkers of disease processes than mRNA-based biomarkers [37].
In the current study we describe a novel mechanism of PGE2-dependent apoptosis resistance that includes c-myc-mediated miR-17-92 upregulation and repression of PTEN expression. We report that PGE2 treatment rapidly induces c-myc expression in NSCLC cells. In accord with the previously reported data suggesting that miR-17-92 cluster expression is regulated by c-myc, our studies reveal that this cluster is also rapidly upregulated upon PGE2 treatment in a time-dependent manner. Concomitantly, we discovered that the levels of PTEN expression are reduced and the pattern of this reduction concurs with upregulation of the miR-17-92 cluster. Finally, we demonstrated that the levels of the circulating miR-17-92 were elevated in plasma of lung cancer patients compared to subjects at risk for developing lung cancer. In lung cancer patients treated with the COX-2 inhibitor celecoxib the miR-17-92 levels were significantly reduced following 8 weeks of treatment compared to baseline. Our studies identify a novel mechanism integrating cell proliferation and apoptosis resistance mediated by PGE2 in lung cancer.
Materials and Methods
Reagents
16,16-dimethyl-PGE2 was purchased from Cayman Chemicals (Ann Arbor, MI). Other reagents were purchased from Sigma Chemicals (St. Louis, MO) unless otherwise specified.
Cell culture
The human squamous cell carcinoma NSCLC cell line H157, adenocarcinoma H460 (ATCC, Rockville, MD) and RH2 (previously established in our laboratory [38]), were grown in RPMI supplemented with 10% FBS (Gemini Bio-Products, Calabasas, CA). H157 cells with ectopic expression of COX-2 were generated as previously described [39]. Cells utilized in these studies were passaged no more than eight times. The cells were genotyped regularly (usually, every three months) utilizing the Promega Cell ID system (Promega, Madison, WI). All cell lines were tested and found negative for mycoplasma contamination (MycoAlert Mycoplasma Detection Kit, Lonza, Walkersville, MD).
Western blotting
Cells were washed with cold PBS and lysed with RIPA buffer. Protein concentrations were measured using the BCA Protein Assay Kit (Pierce, Rockford, IL). Western blotting was performed as previously described [40]. Briefly, proteins were separated by SDS-PAGE and transferred to the nitrocellulose membrane (Amersham Biosciences, Piscataway, NJ). Membranes were blocked in 5% Blotto (Santa Cruz Biotechnology, Santa Cruz, CA) in TBS+0.1% Tween-20, incubated with the primary antibodies (Cell Signaling Technology, Danvers, MA) diluted 1:1000 in blocking solution, followed by the incubation with HRP-conjugated secondary antibodies (Santa Cruz, 1:10,000). The blots were developed using the SuperSignal West Pico kit (Pierce).
Human plasma samples
This study utilized plasma samples from the UCLA Lung Cancer SPORE biorepository and samples collected in a randomized Phase II trial evaluating the efficacy of erlotinib plus celecoxib combination in advanced NSCLC [41]. Samples were collected at UCLA or City of Hope respectively in accordance with the institutional IRB requirements. Blood was centrifuged in Vacutainer tubes at 3,000 rpm for 15 minutes within one hour of collection, and plasma was aliquoted and stored at −80°C.
PGE2 assay
PGE2 levels in NSCLC cell culture supernatants were assessed using PGE2 EIA kit (Cayman Chemicals, Ann Arbor, MI) and normalized to the total RNA concentrations.
Circulating miRNA isolation and analysis
Circulating miRNA was isolated from 400 μl of plasma utilizing miRNeasy kit (Qiagen, Valencia, CA). After addition of the lysis reagent, 2.5 fmol of synthetic C. elegans miRNAs (cel-39, cel-54 and cel-238) were added to the samples as spike-in normalization controls as previously described [37]. Circulating miRNA levels were assessed using: a) the miR-ID® platform (Somagenics, Santa Cruz, CA) [42] for the data represented in Figure 3A, or b) TaqMan assays (Life Technologies, Grand Island, NY) in Phase II trial patients. Data was normalized utilizing spike-in controls or RNA concentration (determined using Quant-iT RiboGreen kit, Life Technologies), respectively. Relative expression was calculated utilizing the delta-delta Ct (ΔΔCt) method [43].
Figure 3. Mir-17-92 cluster is upregulated in circulating miRNA from serum of lung cancer patients.
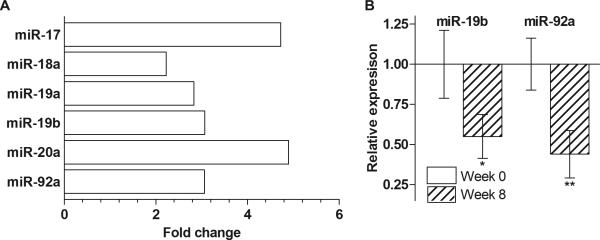
(A) Mir-17-92 is highly elevated in cancer patients compared to subjects at high risk for developing lung cancer. Analysis of mir-17-92 expression in plasma of 30 high-risk controls and 30 advanced stage NSCLC patients by RNA-seq. Results are fold change in advanced stage lung cancer vs. high-risk controls. (B) Celecoxib treatment reduces the levels of the circulating plasma miR-19b and miR-92a in NSCLC patients. Results are mean relative expression ± SEM, p-values computed by a paired samples t-test.
Cellular RNA isolation and analysis
Total RNA was isolated from NSCLC cells utilizing miRNeasy kit. For mRNA, reverse transcription (RT) was performed utilizing iScript kit (Bio-Rad, Hercules, CA) and 2 μl of the cDNA were used for subsequent PCR. For miRNA, RT was performed using the TaqMan MicroRNA RT Kit. Q-RT-PCR was performed using TaqMan assays in the conditions recommended by the manufacturer. Normalization controls: for mRNA — glucuronidase (GUS) or beta-2-microglobulin (B2M); for miRNA — Z30, RNU44 or RNU6B small nucleolar RNAs. All reactions were performed in triplicates. Relative expression was calculated utilizing the ΔΔCt method.
Next generation RNA sequencing (RNA-seq)
RNA-seq was performed on circulating miRNA isolated from pooled 400 μl aliquots of plasma from 30 high-risk control subjects and 30 advanced stage lung cancer patients from the UCLA Lung Cancer SPORE biorepository. The libraries were prepared using Illumina's TruSeq Small RNA Sample Preparation kit as described by the manufacturer. Each sample was sequenced on 1 lane of an Illumina HiSeq instrument to generate approximately 60 million 35 bp reads. Initial data processing was done using Illumina Pipeline SCS 2.9. Adapters were trimmed from the reads using fastx clipper 0.0.13, and reads were aligned to hg19 using Bowtie 0.12.7. BEDTools 2.9.0 was used to obtain expression counts and the data was RPM (reads-per-million) normalized.
C-myc knockdown
Stable c-myc knockdown was performed utilizing Mission® lentiviral shRNA constructs in pLKO.1 vector (Sigma). Three constructs (TRCN0000010389, TRCN0000039640 and TRCN0000174055) targeting different regions of c-myc transcript were utilized. TRC non-hairpin and scrambled shRNA constructs in pLKO.1 vector were used as negative controls. Briefly, the viral particles were produced in genotyped and mycoplasma-negative 293T cells (ATCC) by co-transfection of the shRNA construct, pMD2 viral envelope and pSPAX2 packaging plasmids (Addgene, Cambridge, MA) utilizing the Calcium Phosphate Transfection Kit (Clontech, Mountain View, CA). Next day the medium was replaced with the fresh growth medium and the cells were incubated for another 48 hours before the viral supernatants were collected, filtered through 0.45 μm filters and used for transduction of H157, H460 and RH2 NSCLC cells. The cells were plated at 105 cells/well in 12-well plates. On the following day viral supernatant+10 μg/ml polybrene were added to the cells for 24 h followed by selection with 1 μg/ml puromycin. C-myc knockdown was confirmed by Western blotting (Supplementary Figure 1).
MiRNA inhibition by anti-miR
Anti-miR™ miRNA inhibitors and Anti-miR™—Negative Control #1 were purchased from Life Technologies. Transfection was performed utilizing Lipofectamin 2000 reagent (Invitrogen, Carlsbad, CA). Briefly: a) the day before the transfection, 2×105 cells were plated in 6-well plates, b) on the day of transfection, the medium was changed to 1.6 ml of Opti-MEM, c) 80 pmol of Anti-miR was diluted in 200 μl of Opti-MEM, d) 4 μl of Lipofectamin-2000 was diluted in 200 μl of Opti-MEM, e) Anti-miR and Lipofectamin suspensions were mixed together, incubated for 20 min at room temperature and added to the cells for 24 h. The efficacy of transfection was monitored by the fluorescent microscopy using Cy3 dye-labeled Negative Control #1 (Life Technologies).
Apoptosis assay
Apoptosis was induced by staurosporine treatment. For the apoptosis assays, the cells were plated in T25 flasks at 3.5×105 cells and allowed to adhere overnight. The next day the cells were transfected with Anti-miR™ miRNA inhibitors or Anti-miR™—Negative Control #1 for 4 hours as described above (all volumes were scaled up two-fold). Four hours later PGE2 was added at 5 μg/ml where applicable without changing the medium and the cells were incubated overnight. On the following day apoptosis was induced where applicable by adding 1 μM (H157) or 0.4 μM staurosporine (H460 and RH2 cells) for 60 minutes. The control cells were treated with the equivalent concentration of the diluent (DMSO). Following treatment the cells were washed twice with pre-warmed PBS and the fresh culture medium was added. Cells were incubated overnight and the next morning the apoptosis was assessed utilizing Annexin V/PI kit (BD Biosciences, Franklin Lakes, NJ) according to the manufacturer's instructions. Both the culture medium containing the floating cells and the adherent cells were collected and combined for the apoptosis analysis. The analysis was performed utilizing the FACScan flow cytometer and CellQuest software (BD). All experiments included the non-stained and single stained controls and were performed in triplicates.
Statistical analysis
All statistical analyses were performed using SAS v.9.3 (SAS Institute, Cary, NC) and SPSS v.21 (IBM Corp, Armonk, NY). For circulating miRNA, we tested the celecoxib arm for differences between pre- and post-treatment Ct values for both miR-19b and miR-92a using a paired samples t-test. Differences between groups were compared utilizing a 2-sample t-test. Differences were considered significant for p-values<0.05. Significance values indicated in the figures: * — p<0.05, ** — p<0.01, *** — p<0.001. Graphed data represent mean ± SEM.
Results
PGE2 exposure upregulates c-myc expression in NSCLC cells
To elucidate the molecular mechanisms of PGE2-dependent NSCLC cell proliferation demonstrated in our previous studies [5], we evaluated the expression of several key positive regulators of the cell cycle in a subset of NSCLC cells that were previously shown to be responsive to PGE2 treatment. We found that treatment of H157, H460 and RH2 NSCLC cells with physiologically relevant concentrations of PGE2-induced rapid upregulation of c-myc expression (Figure 1). An increase in c-myc protein was also detected by Western blotting after one hour of treatment, and the elevated protein levels were present throughout the 3-hour exposure (Figure 1A). Similarly, an increase of CMYC gene expression was detected by qPCR 60 minutes following PGE2 exposure and the c-myc mRNA levels remained elevated following 3 hours of treatment (Figure 1B). C-myc is a short-lived protein and our findings indicate that PGE2 induces its expression at the transcriptional level. Among other cell cycle-related proteins we found similar patterns of time-dependent cyclin D1 and c-fos upregulation in response to PGE2 treatment (data not shown). Such upregulation of c-myc, as an immediately early responder to mitogenic signals, as well as other positive regulators of the cell cycle, is consistent with our previously reported findings demonstrating prolonged Erk kinase phosphorylation upon PGE2 treatment [5].
Figure 1. Induction of c-myc expression in NSCLC cells by PGE2.

H157, H460 and RH2 NSCLC cells were serum deprived for 3 hours and treated with 5 μg/ml PGE2 in the fresh serum-free medium for 0, 1, 2 or 3 hours. The cells were then harvested and total RNA and proteins were isolated. Upregulation of c-myc following the PGE2 treatment was detected at transcriptional and protein levels starting at 60 minutes post treatment and for the entire 3 hours of treatment. (A) Proteins were separated by SDS-PAGE and c-myc was detected by immunoblotting. Representative of two independent experiments is shown. (B) C-myc mRNA expression was assessed by TaqMan qRT-PCR. GUS (H157 and H460) or B2M (RH2) expression was used as internal control. Expression was normalized to that observed in the untreated group (0 h).
PGE2 upregulates the miR-17-92 cluster in a c-myc-dependent manner
Given the previously established association between c-myc and multiple miRNAs involved in cell cycle control and apoptosis resistance, we hypothesized that PGE2 may affect miR-17-92 cluster expression. We assessed the expression of several key members of miR-17-92 cluster to determine if the polycistron was upregulated upon PGE2 exposure. Previous studies suggested that the predominant mediators of the oncogenic properties of miR-17-92 are miR-19a and 19b [32, 33] for which sequences differ by 1 nucleotide. Therefore, we assessed the PGE2-dependent expression of miR-19b and another member of the cluster, miR-18a (data not shown), in H157, H460 and RH2 NSCLC cell lines. The expression of both miR-19b and miR-18a followed the upregulation of c-myc and was rapidly elevated following PGE2 exposure in a time-dependent manner (Figure 2A). Because the dynamics of the expression of different cluster members was very similar, in our subsequent studies we evaluated miR-19b as a representative member of the miR-17-92 cluster. To verify that the expression of the miRNA cluster was c-myc dependent, we stably knocked down c-myc expression in NSCLC cells utilizing the shRNA technique. Three different knockdown constructs that expressed shRNAs targeting different regions of c-myc transcript were utilized. Only the constructs that demonstrated a substantial inhibition of c-myc expression were utilized in the subsequent experiments. After the stable c-myc knockdowns were established, we compared their basal levels of miR-18a and miR-19b to the cells transduced with the non-silencing control constructs. We found that both miR-18a and miR-19b basal levels were significantly reduced in c-myc knockdown cells (Figure 2B). Then, we assessed miR-19b expression with and without PGE2 treatment. Knock-down of c-myc expression reversed the PGE2-driven miR-17-92 cluster upregulation (Figure 2C). These data support the notion that the miR-17-92 cluster is a transcriptional target for c-myc and interference with the c-myc expression in NSCLC cells leads to significant reduction in the level of these oncomir.
Figure 2. Expression of miR-19b is upregulated by PGE2.

(A) MiR-19b is upregulated by PGE2 treatment in a time-dependent manner in NSCLC cells. H157, H460 and RH2 cells were serum deprived for 3 hours and treated with 5 μg/ml PGE2 in the fresh serum-free medium for 1, 2 or 3 hours followed by total RNA isolation and TaqMan qRT-PCR for miR-19b and RNU44 (internal control). (B) Basal miR-19b levels are reduced in c-myc knockdown (KD) H157, H460 and RH2 cells compared to non-silencing construct-transduced control cells (NC). (C) C-myc knockdown abolishes PGE2-driven upregulation of miR-19b. C-myc knockdown cells were serum deprived and treated with the diluent control or 5 μg/ml PGE2 in the fresh serum-free medium for 3 hours. (D) PGE2 levels are elevated in H157 cells with ectopic COX-2 expression compared to vector control cells and are significantly reduced by incubation with 2.5 μM celecoxib for 24 or 48 hours. PGE2 data was normalized to the total RNA concentration. (E) Ectopic COX-2 expression leads to the upregulation of the miR-17-92 cluster. Q-RT-PCR was performed on RNA from vector control and COX-2 overexpressing H157 cells. (F) Celecoxib treatment (2.5 μM for 24 or 48 hours) leads to the miR-17-92 downregulation in COX-2 overexpressing cells (COX) compared to vector control (V) as determined by q-RT-PCR.
Ectopic expression of COX-2 leads to an increase in miR-17-92 expression
In order to assess the importance of COX-2 expression and PGE2 production in regulating miR-17-92 expression, we utilized H157 NSCLC cell line ectopically expressing COX-2, which produces nearly 10-fold higher levels of PGE2 than do the vector control cells. Exposure of the H157-COX-2 cells to celecoxib led to a significant reduction in PGE2 production (Figure 2D). We found that compared to the vector control cells, H157-COX-2 cells had significantly elevated levels of miR-19b and miR-92a (Figure 2E). When the cells were cultured in the presence of celecoxib (2.5 μM for 24 or 48 hours), the levels of the miR-17-92 expression were significantly reduced (Figure 2F). These results further corroborate that the miR-17-92 cluster expression is mediated by PGE2 in NSCLC.
The levels of miR-17-92 cluster are elevated in NSCLC patients
Utilizing our own studies and the published work of others, we developed a panel of miRNAs comprising 17 miRNAs highly relevant to lung carcinogenesis, including two members of miR-17-92 cluster, miR-17a and miR-20a. In our initial study utilizing this miRNA panel, we assessed circulating miRNA levels in plasma of five subjects at high-risk for developing lung cancer and five advanced stage lung cancer patients from the UCLA Lung Cancer SPORE biorepository by qRT-PCR. Although the number of samples utilized in the study was insufficient to achieve a significant statistical power, there was an obvious trend towards elevated levels of both miR-17a and miR-20a in the blood of lung cancer patients (data not shown). Then, we performed RNA-seq of circulating miRNA isolated from pooled plasma samples from 30 high-risk control subjects and 30 advanced stage lung cancer patients. RNA-seq yielded a high number of reads with ~87% of reads aligning. Out of the aligned reads, ~29% aligned to miRNA. Our analysis revealed a number of miRNAs that were differentially expressed in cancer compared to high-risk controls. Among these, we found that the expression of the miR-17-92 cluster was elevated by greater than two-fold in the advanced stage cancer sample pool compared to the high-risk control pool (Figure 3A).
Celecoxib reduces circulating miR-17-92 levels in advanced stage NSCLC patients
To further investigate the effect of celecoxib treatment on the levels of circulating miR-19b and miR-92a, we assessed plasma samples collected from a randomized phase II trial that evaluated treatment with erlotinib or erlotinib plus celecoxib in advanced stage NSCLC (Supplementary Table 1) [41]. We measured miR-19b and miR-92a levels by qPCR in plasma collected prior to therapy and following 8 weeks of treatment from 30 patients. We found that celecoxib significantly reduced circulating miR-17-92 expression in plasma (Figure 3B).
PTEN expression is downregulated by PGE2
PTEN functions as a key negative regulator of the PI3-K/Akt/mTOR axis thus affecting cell growth, proliferation and survival. Previous studies in human fibroblast cell culture suggested that PTEN might be a target of miR-19 members of miR-17-92 cluster [32]. However, the causal relationship between PGE2, c-myc and PTEN expression has not yet been established. To define the effect of PGE2 on PTEN levels we treated H157, H460 and RH2 NSCLC cells with PGE2 for up to 3 hours and assessed PTEN mRNA and protein levels. We found that PTEN transcription was sharply reduced upon PGE2 treatment (Figure 4A). This is in accord with our finding of c-myc and miR-17-92 cluster upregulation and supports the previously reported studies suggesting that PTEN is a miR-19b target. We did not detect a reduction of PTEN after 3 hours of PGE2 treatment at the protein level (data not shown), presumably because PTEN is a relatively stable protein. We first compared PTEN levels in the non-silencing construct transduced control cells and c-myc knockdown cells. PTEN levels in the knockdown cells were slightly higher than in the controls (data not shown). We then evaluated the ability of PGE2 to reduce PTEN expression in control and c-myc knockdown cells and found that while in the control cells PGE2 treatment sharply reduced PTEN expression, in c-myc knockdowns this effect was not evident (Figure 4B). These results suggest that in NSCLC cells c-myc contributes to PTEN downregulation upon mitogenic stimuli, whereas the baseline PTEN expression is controlled by the other mechanisms. To elucidate the role of miR-17-92 cluster members in PTEN downregulation we transiently knocked down expression of miR-19b with anti-miR antisense RNA. The control cells were treated with the Anti-miR—Negative Control #1. Then, the controls and cells with knocked down miR-19 expression were treated with PGE2 and the levels of PTEN transcripts were assessed. We found that while in the negative control cells PTEN expression was significantly reduced by PGE2 treatment, knocking down miR-19b expression reversed this effect (Figure 4C). These results clearly indicate that PTEN is a miR-19b target and provided a rationale for the use of miR-19b inhibition in our subsequent functional experiments.
Figure 4. PTEN is negatively regulated by PGE2.

Total RNA was isolated from H157, H460 and RH2 cells and probed for PTEN expression using TaqMan system. Expression was normalized to that observed in the control or untreated groups. (A) PGE2 reduces PTEN mRNA levels. The parental H157, H460 and RH2 cells were serum deprived and treated with the diluent control or 5 μg/ml PGE2 in the fresh serum-free medium for 0, 1, 2 or 3 hours. (B) C-myc knockdown reverses PGE2-driven PTEN downregulation. Non-silencing shRNA construct-transduced control cells and c-myc knockdown cells were plated in 6-well plates and incubated overnight. The cells were then serum deprived and treated with the diluent control (C) or 5 μg/ml PGE2 (PGE2) in the fresh serum-free medium for 3 hours. PGE2 treatment significantly reduced PTEN expression in control cells but not in c-myc knockdown cells. (C) MiR-19b knockdown blocks c-myc-dependent downregulation of PTEN. MiR-19b expression was knocked down with anti-miR antisense RNA in H157, H460 and RH2 cells. The control cells were transfected with the Negative Control anti-miR. Twenty-four hours after the transfection, the cells were serum deprived and treated with the diluent or 5 μg/ml PGE2 for 0, 1, 2 or 3 hours. Negative control- but not anti-miR-19b-transfected cells demonstrated significant downregulation of PTEN by PGE2.
Suppression of miR-19b sensitizes NSCLC cells to apoptosis
Our previous studies demonstrated that PGE2 treatment renders NSCLC cells resistant to pharmacologically induced apoptosis by the modulation of anti-apoptotic proteins levels [4, 40]. Our current findings indicate that PGE2 is also able to modulate PTEN expression through upregulation of the miR-17-92 cluster, suggesting a novel mechanism contributing to apoptosis resistance in lung cancer cells. Therefore we determined if PTEN suppression is functionally relevant in these experimental settings. MiR-19b expression was knocked down by anti-miR oligonucleotides in H157, H460 and RH2 cell lines. Four hours later, the control and miR-19b knockdown cells were treated with PGE2 for 16 hours followed by apoptosis induction where applicable as described in the Materials and Methods section. The apoptosis levels were assessed by flow cytometry utilizing Annexin V/PI method. In accord with our previously published studies, PGE2 treatment rendered cells resistant to apoptosis. Knockdown of miR-19b expression, however, reversed the PGE2 effect and upon the apoptosis induction there was no significant change in cell death levels in PGE2-treated miR-19b knockdowns as compared to the parental cells (Figure 5).
Figure 5. Mir-19b knockdown abolishes the anti-apoptotic effect of PGE2 in NSCLC cells.

PGE2 protects the cells with intact miR-19b expression from apoptosis and this effect is reversed by miR-19 knockdown. The expression of miR-19b was knocked down with anti-miR antisense RNA in H157, H460 and RH2 cells (KD). Negative control anti-miR was used to transfect the control cells (NC). The cells were then pre-treated with 5 μg/ml PGE2 (PG) overnight followed by the induction of apoptosis by staurosporine (St) where applicable. The control cells were treated with the equivalent concentrations of the diluents (diluent control, DC). The cells were allowed to recover overnight (5 μg/ml PGE2 was added to the PGE2 groups) and then were harvested. The apoptosis levels were evaluated by flow cytometry utilizing Annexin V/PI staining. Percent difference (% diff.) indicates the difference in the total number of non-apoptotic cells per 10,000 gated cells (shown in the lower left quadrants) between cells treated with staurosporine alone or in combination with PGE2.
Discussion
Our results demonstrate that PGE2 is able to initiate a sequence of events including c-myc upregulation that induces miR-17-92 cluster expression, which in turn dampens the expression of PTEN ultimately promoting apoptosis resistance in NSCLC cells. Moreover, we found that this PTEN downregulation and augmented apoptosis resistance can be reversed by knocking down c-myc or miR-17-92 cluster expression.
We have previously demonstrated that a subset of approximately 50% NSCLC cell lines respond to PGE2 by the growth factor receptor-independent activation of the MAPK/Erk pathway, induction of cell proliferation and augmented apoptosis resistance [5, 40]. CMYC belongs to a set of immediate early response genes, whose expression is activated early during the transition of cells from a resting to a growing state by a variety of mitogenic stimuli that are usually conveyed through MAPK/Erk cascade activation [8, 44]. Our results suggest that PGE2-driven MAPK/Erk activation is able to induce c-myc expression even in absence of growth factor signaling thus promoting myc-dependent cellular responses. A c-myc-dependent network of genes, including miRNA genes, has been previously described [7, 32, 45]. In accord with these data, we found that the expression of the miR-17-92 cluster was elevated upon PGE2-driven c-myc upregulation in NSCLC cells. Similarly, the miR-17-92 levels in NSCLC cells ectopically expressing COX-2 were significantly elevated compared to the vector control cells and were markedly reduced by the COX-2 inhibitor celecoxib. The miR-17-92 cluster, known as oncomir-1, produces six individual mature miRNAs that are often elevated in various cancers due to the transcriptional activation or genomic amplification [32]. The miR-17-92 cluster has been shown to play a role in embryonic development and to be implicated in tumorigenesis, predominantly through the repression of apoptosis [28]. Subsequent studies revealed that different members of the cluster had differing tumorigenic properties with miR-19 possessing the most potent oncogenic capacities and being absolutely required for the entire cluster activity [28, 31, 33].
The same aforementioned studies that dissected the cluster member's input, identified PTEN as a primary target of miR-19 and a central mediator of miR-17-92 tumorigenicity. Here we report that PGE2-dependent upregulation of c-myc leads to reduction of PTEN levels via the miR-17-92 cluster upregulation leading to apoptosis resistance in NSCLC cells. This finding may partially explain why PTEN loss is frequently found in NSCLC despite somatic PTEN mutations occurring at a low frequency in lung cancer. For example, one study demonstrated that 24% of early stage NSCLC tumors lacked PTEN expression due to promoter methylation [46]. In later reports, PTEN protein expression was reduced or lost in 74% of advanced stage lung cancers [47, 48]. Moreover, the level of PTEN expression is a strong prognostic indicator in lung cancer and may determine treatment outcome. Similarly, PTEN loss has been described as one of the mechanisms of acquired resistance to targeted therapies in lung cancer patients [20]. Thus, our results indicate that PGE2 is able to negatively regulate PTEN via c-myc-mediated expression of the miR-17-92 cluster. This effect contributes to the loss of PTEN expression in mutation-independent manner and increases apoptosis resistance of lung cancer cells. We determined that knocking down miR-19, the most potent suppressor of PTEN expression, restored PTEN levels and susceptibility to apoptosis in PGE2-treated NSCLC cells. Consistent with these data, PGE2-induced miR-17-92 cluster upregulation and PTEN repression were abolished in c-myc knockdown NSCLC cells. Recent studies suggest that circulating miRNAs can serve as robust and tissue-specific non-invasive biomarkers of lung cancer [49]. Utilizing microarrays and next generation sequencing techniques, elevated levels of the miR-17-92 cluster members have been found in the circulating miRNA isolated from serum of patients with colon, gastric, ovarian and prostate cancers [50]. Our results indicate that the miR-17-92 is also highly upregulated in circulating miRNA in lung cancer suggesting that its deregulation may also play a role in the pathogenesis of lung cancer. Furthermore, in lung cancer patients that received celecoxib, the miR-17-92 expression was significantly reduced compared to the baseline. Importantly, the variations in the cluster members’ copy numbers within the samples might reflect the differences in stability of the individual cluster miRNAs or their transfer rate into the bloodstream. This, in turn, may suggest that using some miR-17-92 members as biomarkers might be advantageous compared to others. However, further studies are needed to test this hypothesis. Finally, from the functional point of view, one may speculate that the exosomal transfer of miR-19 from the tumor to the non-involved cells might lead to PTEN downregulation in these cells and thus contribute to their malignant transformation.
In conclusion, our studies integrate elements of the inflammatory tumor microenvironment and promotion of aggressive tumor traits, including lack of response to apoptotic stimuli and enhanced proliferation. These findings identify a novel PGE2-dependent network operative in a subset of NSCLC cells that contributes to apoptosis resistance. Additional lung cancer studies will be required to: a) define the utility of miR-17-92 cluster for early detection, prognosis and response to therapy, b) assess the correlation between the loss of tumor PTEN expression and the levels of circulating miR-19, and c) determine if interference with miR-17-92 cluster expression could be beneficial in clinical settings by helping to restore PTEN expression in NSCLC and to improve response to therapies.
Supplementary Material
1
2
3
Acknowledgements
Flow cytometry was performed in the UCLA Jonsson Comprehensive Cancer Center and Center for AIDS Research Flow Cytometry Core Facility that is supported by NIH awards CA-16042 and AI-28697, and by the JCCC, the UCLA AIDS Institute, and the David Geffen School of Medicine at UCLA. The authors would like to thank Drs. Sergei Kazakov, Pavan Kumar and Brian Johnston (Somagenics, Inc) for helpful discussions.
Financial support: This study was supported by the following grants: NIH/National Center for Advancing Translational Science UCLA CTSI Grant Number UL1TR000124, Early Detection Research Network (EDRN) U01 CA152751, UCLA SPORE in Lung Cancer P50 CA90388, Tobacco Related Disease Program #15RT-0152, American Thoracic Society/LUNGevity Foundation Research Grant LC-06-003, DOD Lung Cancer Research Program LC090615P2 and Veterans Administration — Merit Review 5I01BX000359.
Footnotes
Conflicts of interest: The authors declare no potential conflicts of interests.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2010;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Huang M, Stolina M, Sharma S, Mao JT, Zhu L, Miller PW, et al. Non-small cell lung cancer cyclooxygenase-2-dependent regulation of cytokine balance in lymphocytes and macrophages: up-regulation of interleukin 10 and down-regulation of interleukin 12 production. Cancer Res. 1998;58:1208–16. [PubMed] [Google Scholar]
- 3.Soslow RA, Dannenberg AJ, Rush D, Woerner BM, Khan KN, Masferrer J, et al. COX-2 is expressed in human pulmonary, colonic, and mammary tumors. Cancer. 2000;89:2637–45. doi: 10.1002/1097-0142(20001215)89:12<2637::aid-cncr17>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 4.Krysan K, Dalwadi H, Sharma S, Pold M, Dubinett S. Cyclooxygenase 2-dependent expression of survivin is critical for apoptosis resistance in non-small cell lung cancer. Cancer Res. 2004;64:6359–62. doi: 10.1158/0008-5472.CAN-04-1681. [DOI] [PubMed] [Google Scholar]
- 5.Krysan K, Reckamp KL, Dalwadi H, Sharma S, Rozengurt E, Dohadwala M, et al. Prostaglandin E2 activates mitogen-activated protein kinase/Erk pathway signaling and cell proliferation in non-small cell lung cancer cells in an epidermal growth factor receptor-independent manner. Cancer Res. 2005;65:6275–81. doi: 10.1158/0008-5472.CAN-05-0216. [DOI] [PubMed] [Google Scholar]
- 6.Marcu KB, Bossone SA, Patel AJ. Myc function and regulation. Annu Rev Biochem. 1992;61:809–60. doi: 10.1146/annurev.bi.61.070192.004113. [DOI] [PubMed] [Google Scholar]
- 7.Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol. 2005;6:635–45. doi: 10.1038/nrm1703. [DOI] [PubMed] [Google Scholar]
- 8.Kelly K, Cochran BH, Stiles CD, Leder P. Cell-specific regulation of the c-myc gene by lymphocyte mitogens and platelet-derived growth factor. Cell. 1983;35:603–10. doi: 10.1016/0092-8674(83)90092-2. [DOI] [PubMed] [Google Scholar]
- 9.Lachman HM, Skoultchi AI. Expression of c-myc changes during differentiation of mouse erythroleukaemia cells. Nature. 1984;310:592–4. doi: 10.1038/310592a0. [DOI] [PubMed] [Google Scholar]
- 10.Dean M, Levine RA, Ran W, Kindy MS, Sonenshein GE, Campisi J. Regulation of c-myc transcription and mRNA abundance by serum growth factors and cell contact. J Biol Chem. 1986;261:9161–6. [PubMed] [Google Scholar]
- 11.Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18:3004–16. doi: 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- 12.Hayward WS, Neel BG, Astrin SM. Activation of a cellular onc gene by promoter insertion in ALV-induced lymphoid leukosis. Nature. 1981;290:475–80. doi: 10.1038/290475a0. [DOI] [PubMed] [Google Scholar]
- 13.Payne GS, Bishop JM, Varmus HE. Multiple arrangements of viral DNA and an activated host oncogene in bursal lymphomas. Nature. 1982;295:209–14. doi: 10.1038/295209a0. [DOI] [PubMed] [Google Scholar]
- 14.Wolfer A, Wittner BS, Irimia D, Flavin RJ, Lupien M, Gunawardane RN, et al. MYC regulation of a “poor-prognosis” metastatic cancer cell state. Proc Natl Acad Sci U S A. 2010;107:3698–703. doi: 10.1073/pnas.0914203107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glinsky GV, Berezovska O, Glinskii AB. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Invest. 2005;115:1503–21. doi: 10.1172/JCI23412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Volm M, Koomagi R. Prognostic relevance of c-Myc and caspase-3 for patients with non-small cell lung cancer. Oncol Rep. 2000;7:95–8. doi: 10.3892/or.7.1.95. [DOI] [PubMed] [Google Scholar]
- 17.Vartanian R, Masri J, Martin J, Cloninger C, Holmes B, Artinian N, et al. AP-1 regulates cyclin D1 and c-MYC transcription in an AKT-dependent manner in response to mTOR inhibition: role of AIP4/Itch-mediated JUNB degradation. Mol Cancer Res. 2011;9:115–30. doi: 10.1158/1541-7786.MCR-10-0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simpson L, Parsons R. PTEN: life as a tumor suppressor. Exp Cell Res. 2001;264:29–41. doi: 10.1006/excr.2000.5130. [DOI] [PubMed] [Google Scholar]
- 19.Leslie NR, Downes CP. PTEN function: how normal cells control it and tumour cells lose it. Biochem J. 2004;382:1–11. doi: 10.1042/BJ20040825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer. 2011;11:289–301. doi: 10.1038/nrc3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alimonti A, Carracedo A, Clohessy JG, Trotman LC, Nardella C, Egia A, et al. Subtle variations in PTEN dose determine cancer susceptibility. Nat Genet. 2010;42:454–8. doi: 10.1038/ng.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fournier MV, Fata JE, Martin KJ, Yaswen P, Bissell MJ. Interaction of E-cadherin and PTEN regulates morphogenesis and growth arrest in human mammary epithelial cells. Cancer Res. 2009;69:4545–52. doi: 10.1158/0008-5472.CAN-08-1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hill R, Wu H. PTEN, stem cells, and cancer stem cells. J Biol Chem. 2009;284:11755–9. doi: 10.1074/jbc.R800071200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 25.Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valastyan S, Weinberg RA. MicroRNAs: Crucial multi-tasking components in the complex circuitry of tumor metastasis. Cell Cycle. 2009;8:3506–12. doi: 10.4161/cc.8.21.9802. [DOI] [PubMed] [Google Scholar]
- 27.Ventura A, Jacks T. MicroRNAs and cancer: short RNAs go a long way. Cell. 2009;136:586–91. doi: 10.1016/j.cell.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–33. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–43. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 30.Lu Y, Thomson JM, Wong HY, Hammond SM, Hogan BL. Transgenic over-expression of the microRNA miR-17-92 cluster promotes proliferation and inhibits differentiation of lung epithelial progenitor cells. Dev Biol. 2007;310:442–53. doi: 10.1016/j.ydbio.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mu P, Han YC, Betel D, Yao E, Squatrito M, Ogrodowski P, et al. Genetic dissection of the miR-17~92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev. 2009;23:2806–11. doi: 10.1101/gad.1872909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olive V, Bennett MJ, Walker JC, Ma C, Jiang I, Cordon-Cardo C, et al. miR-19 is a key oncogenic component of mir-17-92. Genes Dev. 2009;23:2839–49. doi: 10.1101/gad.1861409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Haaften G, Agami R. Tumorigenicity of the miR-17-92 cluster distilled. Genes Dev. 2010;24:1–4. doi: 10.1101/gad.1887110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taylor DD, Gercel-Taylor C. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol Oncol. 2008;110:13–21. doi: 10.1016/j.ygyno.2008.04.033. [DOI] [PubMed] [Google Scholar]
- 35.Nygaard S, Jacobsen A, Lindow M, Eriksen J, Balslev E, Flyger H, et al. Identification and analysis of miRNAs in human breast cancer and teratoma samples using deep sequencing. BMC Med Genomics. 2009;2:35. doi: 10.1186/1755-8794-2-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–8. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 37.Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;105:10513–8. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang M, Sharma S, Mao JT, Dubinett SM. Non-small cell lung cancer-derived soluble mediators and prostaglandin E2 enhance peripheral blood lymphocyte IL-10 transcription and protein production. J Immunol. 1996;157:5512–20. [PubMed] [Google Scholar]
- 39.Aljada IS, Ramnath N, Donohue K, Harvey S, Brooks JJ, Wiseman SM, et al. Upregulation of the tissue inhibitor of metalloproteinase-1 protein is associated with progression of human non-small-cell lung cancer. J Clin Oncol. 2004;22:3218–29. doi: 10.1200/JCO.2004.02.110. [DOI] [PubMed] [Google Scholar]
- 40.Krysan K, Merchant FH, Zhu L, Dohadwala M, Luo J, Lin Y, et al. COX-2-dependent stabilization of survivin in non-small cell lung cancer. FASEB J. 2004;18:206–8. doi: 10.1096/fj.03-0369fje. [DOI] [PubMed] [Google Scholar]
- 41.Reckamp K, Cristea M, Dowell J, Gardner B, Milne G, Fouladi Rad S, et al. Randomized, placebo-controlled, phase II trial of EGFR and COX-2 inhibition in advanced non-small cell lung cancer. J Clin Oncol. 2010;28:65s. [Google Scholar]
- 42.Kumar P, Johnston BH, Kazakov SA. miR-ID: a novel, circularization-based platform for detection of microRNAs. RNA. 2011;17:365–80. doi: 10.1261/rna.2490111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 44.Lau LF, Nathans D. Expression of a set of growth-related immediate early genes in BALB/c 3T3 cells: coordinate regulation with c-fos or c-myc. Proc Natl Acad Sci U S A. 1987;84:1182–6. doi: 10.1073/pnas.84.5.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim JW, Mori S, Nevins JR. Myc-induced microRNAs integrate Myc-mediated cell proliferation and cell fate. Cancer Res. 2010;70:4820–8. doi: 10.1158/0008-5472.CAN-10-0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Soria JC, Lee HY, Lee JI, Wang L, Issa JP, Kemp BL, et al. Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin Cancer Res. 2002;8:1178–84. [PubMed] [Google Scholar]
- 47.Marsit CJ, Zheng S, Aldape K, Hinds PW, Nelson HH, Wiencke JK, et al. PTEN expression in non-small-cell lung cancer: evaluating its relation to tumor characteristics, allelic loss, and epigenetic alteration. Hum Pathol. 2005;36:768–76. doi: 10.1016/j.humpath.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 48.Zhang JG, Wang JJ, Zhao F, Liu Q, Jiang K, Yang GH. MicroRNA-21 (miR-21) represses tumor suppressor PTEN and promotes growth and invasion in non-small cell lung cancer (NSCLC). Clin Chim Acta. 411:846–52. doi: 10.1016/j.cca.2010.02.074. [DOI] [PubMed] [Google Scholar]
- 49.Wu X, Piper-Hunter MG, Crawford M, Nuovo GJ, Marsh CB, Otterson GA, et al. MicroRNAs in the pathogenesis of Lung Cancer. J Thorac Oncol. 2009;4:1028–34. doi: 10.1097/JTO.0b013e3181a99c77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lodes MJ, Caraballo M, Suciu D, Munro S, Kumar A, Anderson B. Detection of cancer with serum miRNAs on an oligonucleotide microarray. PLoS One. 2009;4:e6229. doi: 10.1371/journal.pone.0006229. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1
2
3