Pheochromocytoma and Paraganglioma: Diagnosis, Genetics, Management, and Treatment (original) (raw)
. Author manuscript; available in PMC: 2015 Jan 15.
Introduction
Pheochromocytomas (PHEOs) are rare neuroendocrine tumors that arise from the chromaffin cells of the adrenal glands. Paragangliomas (PGLs), the extra-adrenal counterparts of PHEOs, arise from ganglia along the sympathetic and parasympathetic chain. Although these tumors have been recognized since the early 20th century, many recent advances in the field of PHEO/PGL have fundamentally changed our understanding of these tumors, leading to better diagnostic evaluation, more appropriate patient-specific treatment strategies, and improved patient outcomes. However, there is still no cure for these tumors or successful long-term treatment for patients with metastatic disease; therefore a great deal of research still needs to be done. This extensive review will focus on the most updated information about the diagnosis, genetics, and management of patients with PHEO/PGL and conclude with some perspectives on future treatment strategies and continuing research.
Incidence of PHEO/PGL
PHEO/PGL are rare tumors, affecting about 1 in 2500-6500 individuals, with 500-1600 cases diagnosed annually in the United States.1 However, their true incidence may be higher due to lack of diagnosis until after death; a review of autopsy cases in Australia found that 0.05% had undiagnosed PHEO/PGL.2 They are a rare cause of secondary hypertension, with an incidence in hypertensive patients of only about 0.3-0.5%.3,4 Although adrenal PHEOs, the more common of the two, account for about 80-85% of these tumors,5 only approximately 5-7% of adrenal incidentalomas are PHEOs.6,7 The mean age at diagnosis is approximately 43 years of age, but 10-20% of PHEO/PGL are identified in children, commonly associated with underlying genetic conditions.8–10
Genetics of PHEO/PGL
Underlying germline mutations in one of 17 susceptibility genes have been associated with approximately 35% of PHEO/PGL. An additional 15% of tumors are associated with somatic mutations in these same genes.11–14 In children, this rate is even higher, with 69% of pediatric PHEO/PGL cases in a Spanish study8 and 87.5% of metastatic PHEO/PGL patients who developed their first tumor in childhood9 linked to underlying germline mutations. This high percentage of genetically linked tumors underlines the need for appropriate genetic testing as part of the work-up for most patients with PHEO/PGL. A summary of the clinical characteristics of patients with each genetic mutation is presented in Table 1.
Table 1.
Clinical characteristics of genetic mutations associated with PHEO/PGL.
| GENE | PROTEIN FUNCTION | SYNDROME | GERMLINE/SOMATIC | PHEO/PGL Penetrance | DE NOVO MUTATIONS | MEAN AGE | BIOCHEMICAL PHENOTYPE | COMMON PHEO/PGL SITES | BILATERAL PHEO | MALIGNANCY | OTHER ASSOCIATED CLINICAL CHARACTERISTICS/TUMORS |
|---|---|---|---|---|---|---|---|---|---|---|---|
| VHL | E3 ubiquitin ligase | VHL | Both reported | 10-20% | 20% | 30 | Noradrenergic | Adrenal PHEOs (rarely sympathetic or head and neck PGLs) | 50% | <5% | Hemangioblastomas Renal cell carcinoma Islet cell tumors of pancreas |
| RET | Receptor tyrosine kinase | MEN2 | Both reported | 50% | 5% (MEN2A), 50% (MEN2B) | 30-40 | Adrenergic | Adrenal PHEOs | 50-80% | Rare | Medullary thyroid carcinoma (95% MEN2A, 100% MEN2B) MEN2A: hyperparathyroidism (15-30%) MEN2B: marphanoid habitus; mucosal ganglioneuromas |
| NF1 | GTPase activating protein | NF1 | Both reported | <6% | 50% | 42 | Adrenergic | Adrenal PHEOs (rarely sympathetic PGLs) | 16% | ∼12% | Café-au-lait spots Neurofibromas Freckles Benign iris hamartomas Optic-nerve gliomas Sphenoid bone dysplasia/pseudoarthritis |
| SDHB | Catalytic subunit of succinate dehydrogenase (mitochondrial complex II) | PGL4 | Both reported | 30-100% | Further study needed | 30 | Noradrenergic and/or dopaminergic (Rarely biochemically silent) | Sympathetic PGLs (rarely adrenal PHEOs and head and neck PGLs) | Rare | 31-71% | Renal cell carcinoma Gastrointestinal stromal tumors Pituitary adenomas Possibly breast carcinoma Possibly papillary thyroid carcinoma |
| SDHD | Membrane anchoring subunit of succinate dehydrogenase | PGL1 | Both reported | 73-90% (paternal transmission) | Further study needed | 35 | Noradrenergic, dopaminergic, or silent | Head and neck PGLs, commonly multiple (rarely extra-adrenal abdominal PGLs or adrenal PHEOs) | Rare | <5% | Renal cell carcinoma Gastrointestinal stromal tumors Pituitary adenomas |
| SDHC | Membrane anchoring subunit of succinate dehydrogenase | PGL3 | Germline | Further study needed | Further study needed | 40-50 | Noradrenergic, dopaminergic, or silent | Head and neck PGLs, sometimes multiple (rarely sympathetic PGLs or adrenal PHEOs) | Further study needed | Rare | Renal cell carcinoma Gastrointestinal stromal tumors Pituitary adenomas |
| SDHA | Catalytic subunit of succinate dehydrogenase (mitochondrial complex II) | Germline | Further study needed | Further study needed | 40 | Further study needed | Adrenal PHEOs or extra-adrenal PGLs | Further study needed | 0-14% | Homozygous patients: Leigh syndrome Renal cell carcinoma Gastrointestinal stromal tumors Pituitary adenomas | |
| SDHAF2 | Assembly factor for succinate dehydrogenase (mitochondrial complex II) | PGL2 | Germline | 100% (paternal transmission) | Further study needed | 30-40 | Further study needed | Head and neck PGLs, sometimes multiple | No known cases of PHEO | Further study needed | |
| MAX | Transcription factor | Both reported | Further study needed (paternal transmission likely) | Further study needed | 32 | Adrenergic and noradrenergic | Adrenal | 67% | 20-25% | ||
| TMEM127 | Further study needed | Germline | Further study needed | Further study needed | 43 | Adrenergic and noradrenergic | Adrenal | 33% | <5% | Possibly linked to breast carcinoma Possibly linked to papillary thyroid carcinoma | |
| HIF2A | Hypoxia-inducible factor | Pacak-Zhuang | Mostly somatic; germline reported | N/A | Somatic mutations | Noradrenergic | Extra-adrenal PGLs, usually multiple or PHEOs, possibly bilateral | Further study needed | None reported | Multiple somatostatinomas Polycythemia | |
| IDH | Metabolic enzyme in Krebs cycle | Somatic (one case) | N/A | N/A | N/A | Not specified | Carotid PGL | No known cases of PHEO | None reported | Glioblastoma multiforme | |
| FH | Metabolic enzyme in Krebs cycle | Germline (one case) | N/A | N/A | N/A | Noradrenergic | Adrenal PHEO | None reported | Patient had metastatic tumor | Leiomyomatosis Renal cell carcinoma | |
| KIF1Bβ | Kinesin family protein (transport protein) | Germline | N/A | N/A | N/A | Further study needed | Further study needed | Further study needed | None reported | ||
| PHD2 | Prolyl hydroxylase | Germline (one case) | N/A | N/A | N/A | Not specified | Multiple PGLs | No known cases of PHEO | None reported | Erythrocytosis | |
| H-RAS | GTPase | Somatic | N/A | Somatic mutations | N/A | Adrenergic and/or noradrenergic | Both PHEO and PGL | None reported | None reported |
Multiple endocrine neoplasia type 2
Multiple endocrine neoplasia type 2 (MEN2) is associated with underlying mutations in the rearranged during transfection (RET) protooncogene. The RET protein is a receptor tyrosine kinase that regulates cellular proliferation and apoptosis. Patients with MEN2 are usually first diagnosed with medullary thyroid cancer (MTC), which is the most common condition in these patients. MEN2 is divided into three subclassifications: MEN2A, MEN2B, and familial MTC. Patients with MEN2A have a 95% chance of developing MTC, a 50% chance of PHEO, and a 15-30% chance of hyperparathyroidism; this form of MEN2 is the most common, accounting for approximately 90% of cases. Patients with MEN2B have a 100% chance of developing MTC and a 50% chance of PHEO, but also typically present with a marphanoid body habitus and mucosal ganglioneuromas. Patients with familial MTC do not have a risk of developing PHEO.15,16
Patients who do develop PHEOs usually present with epinephrine/metanephrine-secreting tumors in the adrenal gland, with approximately half presenting with bilateral tumors. However, malignancy is rare, though higher in patients with the more aggressive MEN2B. The age of onset for PHEO is typically between 30 and 40. Although approximately 5% of MEN2A and 50% of MEN2B patients present with de novo mutations, the majority have a strong family history, so carriers are typically identified early in life. Thus, early and regular screening usually catches tumors while they are still small, keeping the rate of metastasis low for patients with MEN2.15,16
Von Hippel-Lindau
Mutations in the von Hippel-Lindau (VHL) gene cause VHL syndrome.17 The VHL protein regulates the activity of hypoxia-inducible factor alpha (HIFα) and regulates cellular processes, including angiogenesis. As with MEN2, VHL is characterized by a predisposition to multiple tumor types and can be divided into subclassifications based on the risk of PHEO/PGL. Patients with VHL type 1, the more common form, develop retinal angiomas, central nervous system hemangioblastomas, renal carcinomas, islet cell tumors of the pancreas, endolymphatic sac tumors, or cysts and cystadenomas of the kidney, pancreas, epididymis, or broad ligament, but do not develop PHEO/PGL. Patients with VHL type 2 are at risk of PHEO/PGL and are further divided into type 2A (without renal carcinomas and infrequent type 1 tumors), type 2B (with renal cell carcinoma or any type 1 tumors), and type 2C (only PHEO/PGL, without any type 1 tumors).15,16
VHL patients usually develop norepinephrine/normetanephrine-secreting adrenal PHEOs, with a high rate of bilateral tumors. The age of onset of PHEO/PGL with VHL is approximately 30 years, though patients as young as 5 have been reported. In addition, there is a relatively high rate of de novo mutations (approximately 20%). Metastases are infrequent in patients with VHL, though recurrent and multiple primary tumors can occur.15,16
Neurofibromatosis type 1
Neurofibromatosis type 1 (NF1) has multiple manifestations that can include PHEO/PGL, in addition to MTC, carcinoid tumors, parathyroid tumors, peripheral nerve sheath tumors, and chronic myeloid leukemia. However, the rate of PHEO/PGL development in NF1 is significantly lower than in VHL or MEN2. Although NF1 is caused by germline mutations in the NF1 gene,18 which encodes a GTPase activating protein involved in multiple signaling cascades important to cellular growth and differentiation, genetic testing is rarely performed due to the large size of the gene. Instead, diagnosis is usually based on clinical criteria, often at a young age due to the frequent presence of characteristic café au lait spots from birth. Family history, while a factor in diagnosis, is not necessary, as 50% of cases result from de novo mutations. PHEO/PGL are relatively infrequent in NF1 patients, and therefore screening is not usually performed as regularly as with other symptoms. PHEO/PGL tumors usually appear at the same age as sporadic tumors, with a mean age at diagnosis of 42. Epinephrine/metanephrine-secreting adrenal PHEOs are more common than PGLs, and bilaterality is infrequent. However, the metastatic rate for NF1 tumors, approximately 12%, is higher than MEN2 or VHL.15,16
Recently, somatic NF1 mutations have been linked to the pathogenesis of apparently sporadic PHEO/PGL. In a study of 53 sporadic tumors, 41% were found to have inactivating somatic NF1 mutations, suggesting that these events are a relatively common cause of PHEO.19
Succinate dehydrogenase mutations
For many years, additional familial syndromes associated with PHEO/PGL development were recognized clinically, but the mechanism of inheritance was unexplained. It was only with the identification of succinate dehydrogenase subunit D (SDHD) mutations in families with familial PGL in 2000 that these syndromes began to be explained molecularly.20 Further study in the following years identified two other subunits, SDHB21 and SDHC,22 as heritability genes for PHEO/PGL. The final subunit in the complex, SDHA, was initially only linked to a rare early onset encephalopathy, Leigh syndrome, found in homozygous carriers; however, very recently, heterozygous SDHA mutation carriers with PHEO/PGL have been identified.23 Mutations in a complex assembly factor, SDH assembly factor 2 (SDHAF2, also known as SDH5 in yeast), were also linked to familial PHEO/PGL.24 Because of its role as mitochondrial complex II in both the Krebs cycle and the electron transport chain, SDH mutations severely disrupt cellular metabolism. Studies have shown that mutated SDH proteins are recognized by cellular protein degradation machinery and have shorter half-lives than wild-type SDHB.25 This results in insufficient levels of the SDH complex within cells, increasing the accumulation of succinate and causing a state of pseudohypoxia.
Although mutations in the SDH genes all affect the same complex, their clinical presentations can vary greatly. _SDH_-related tumors are typically extra-adrenal, although some cases of adrenal PHEOs have also been reported. Tumors related to these mutations usually have noradrenergic or dopaminergic phenotypes, though biochemically silent tumors have also been more frequently associated with _SDH_-related PGLs.15,16,26,27 PGLs in the mediastinum and organ of Zuckerkandl are frequently related to underlying SDHB or SDHD mutations.28,29 SDHD mutations are also common in head and neck PGLs, the majority of which are biochemically silent.30 However, approximately 20% secrete dopamine and/or its metabolite methoxytyramine, which can be useful for monitoring these patients.31 Of note, SDHD undergoes maternal imprinting, and therefore PHEO/PGL only arise in patients with affected fathers.15,16,32 Multiple tumors are common with SDHD, but metastases are rare.15,16,30,33
SDHB mutations, which are the most common gene mutations in PHEO/PGL, tend to be linked more frequently to abdominal or thoracic extra-adrenal PGLs.30 Multiple tumors are identified in many SDHB carriers. SDHB mutations are also associated with more aggressive tumors, with younger ages at presentation and higher rates of metastases.15,16,30,33,34 The explanation for this is unclear, but may be due to lower catecholamine activity in _SDHB_-related tumors, leading to later presentations. In addition, the penetrance of these mutations is lower than with other clinical syndromes, so mutation carriers are often not identified until after they present with a tumor.
SDHC, SDHAF2, and SDHA mutations are rare, so clinical information is limited. SDHC mutations are most frequently associated with multiple head and neck tumors, with a mean age of onset similar to that of sporadic patients. Some extra-adrenal abdominal/thoracic PGLs and adrenal PHEOs have also been found in SDHC carriers.15,16 SDHAF2 also appears to be associated with the development of multiple head and neck tumors, often in young patients. Like SDHD, SDHAF2 also appears to undergo maternal imprinting.15,16,32 The rate of penetrance of SDHAF2 mutations appears to be high.15,16 SDHA mutations have been found in patients with PHEOs and PGLs, but these cases have been isolated, so no larger conclusions can be drawn regarding the importance of SDHA testing or the clinical presentation of these carriers.15,16
Although the SDH genes were initially thought to be linked exclusively to PHEO/PGL, additional tumor types linked to these mutations have been discovered. Renal cell carcinoma has been found in a fraction of SDH carriers, particularly those with SDHB, SDHC, and SDHD mutations, with an estimated 14% of SDHB carriers developing this tumor type.35–37 Some cases of rare tumor syndromes, Carney-Stratakis dyad and Carney triad have also been linked to mutations in SDH genes.38,39 Carney-Stratakis dyad consists of gastrointestinal stromal tumors (GIST) and PHEO/PGL, while Carney triad also includes pulmonary chondromas in addition to GIST and PHEO/PGL. A recent link between SDH gene mutations and pituitary adenomas has also been identified, with SDHA, SDHB, SDHC, and SDHD mutations all linked to these tumors.35,40,41 SDH mutations have also been identified in patients with neuroblastoma.42–44 Finally, there is also an unclear association between SDH mutations and breast cancer development45,46; papillary thyroid carcinomas have also been reported in SDH mutation carriers.45,47 Taken together, these data suggest that SDH mutations may represent a metabolic tumor syndrome.
SDH mutations are often found in the absence of family history. However, this is not due to a high rate of de novo mutations, but rather a low rate of penetrance. Maternal imprinting in patients with SDHD and SDHAF2 mutations can mask familial inheritance. However, SDH carriers who lack a family history of PHEO/PGL may have a family history of other conditions, such as renal cell carcinoma or pituitary adenomas, which could be related to the SDH mutation. Studies of the penetrance of SDH mutations have established various penetrances. A report by Benn et al. determined a 29% and 45% penetrance of SDHB mutations by ages 30 and 40, respectively, and a 48% and 73% penetrance of SDHD mutations by ages 30 and 40, respectively (in patients who inherited the mutation from their fathers).30 Similar penetrance values have been determined by Ricketts et al., who calculated a 52% penetrance in SDHB carriers by age 60 and a 71% penetrance for SDHD carriers.37 However, Schiavi et al. estimated a much lower penetrance of only 30% by age 80 for SDHB carriers.48 Further study on large patient cohorts will need to be performed to resolve this debate, but the rarity of these mutations makes these studies difficult.
There is a great deal of interest in understanding the mechanism by which SDH mutations lead to tumor formation. Loss of heterozygosity resulting in the loss of the wild-type allele has been observed in tumor tissue from affected patients, consistent with Knudsen's two-hit hypothesis.13,49 The tumorigenic properties of SDH mutations are not believed to be due to dysfunction of the SDH protein, but rather to increased mutant protein degradation. In fact, studies of mutant SDHB proteins have found a markedly reduced protein half-life, but intact protein localization and SDH complex formation.25 However, this increased degradation, and the loss of heterozygosity in tumors, results in decreased activity of complex II (SDH), which also results in decreased ATP production and an increase in succinate accumulation.50 A recent study has also suggested that increased succinate may lead to increased methylation that affects gene expression, leading to tumorigenesis.51
Although mutation analysis is the recommended procedure for diagnosing SDH mutations, immunohistochemistry can be used on resected tumors to indicate patients with these mutations. Previous reports have shown that SDHB immunostaining accurately detects the presence or absence of the SDHB protein.52–54 In cases in which SDHB immunostaining is weak, a somatic or germline SDH mutation is highly probable. This procedure has a sensitivity of 100% and a specificity ranging from 84-94%.52,54 It has also been suggested that the intensity of staining can prioritize certain testing; absent staining is more suggestive of an SDHB mutation, while weak staining may be indicative of an SDHD mutation.53 Furthermore, patients with SDHA mutations, which can sometimes be hard to identify by traditional sequencing due to gene structure, can be identified by performing a second immunostain for the SDHA protein. While all tumors with SDHA, B, C, and D mutations will show negative staining for SDHB, only tumors with SDHA mutations will have negative SDHA immunostaining.23,55
MAX
In recent years, the rate of gene discovery in PHEO/PGL has accelerated rapidly, due in large part to advances in genetic research techniques and the broader availability and lower costs of performing genetic analysis. Such studies have identified several genes that are minor contributors to the spectrum of heritable PHEO/PGL through whole-genome analyses. One such gene is myc-associated factor X (MAX), which encodes a transcription factor that acts as part of the MYC/MAX/MXD1 network in regulating the myc oncoprotein and interacting with the mTOR pathway to control cellular differentiation, growth, and apoptosis.15,16,56 This rare gene mutation, reported in 1.12% of presumed sporadic PHEO/PGL, is predominantly associated with adrenal PHEOs, though some extra-adrenal tumors have been identified.15,16,57,58 Bilateral adrenal PHEOs are common.15,16,57 Higher rates of metastases have also been reported in patients with MAX mutations.15,16,56,57 Data has suggested that these mutations may, like SDHD and SDHAF2, be paternally transmitted.15,16,56
TMEM127
Another gene associated with PHEO/PGL development is transmembrane protein 127 (TMEM127).59 This gene is linked to the mTOR pathway, though its exact function is unknown. A suggested role in protein trafficking within the endomembrane system has been proposed.60 TMEM127 mutations are a rare cause of PHEO/PGL.15,16,58–60 Patients typically present at a mean age of 43, usually with benign, unilateral or bilateral adrenal PHEOs that secrete both norepinephrine/normetanephrine and epinephrine/metanephrine.15,16,60 Very rare extra-adrenal tumors have been associated with TMEM127 carriers. In addition, carriers with breast cancer and papillary thyroid cancer have been identified, but whether these cancers are linked to TMEM127 mutations is unclear at the present time.15,16
Hypoxia-inducible factor 2-alpha
One of the most recently discovered genes in PHEO/PGL pathogenesis is hypoxia-inducible factor 2-alpha (HIF2A). It was first identified as somatic mutations in tumor tissue from patients who presented with multiple PGLs and polycythemia, one of whom also had multiple somatostatinomas.61 After two additional patients with multiple PGLs, somatostatinomas, and polycythemia were described, a new syndrome, Pacak-Zhuang syndrome, was proposed.62 Additional studies confirmed these findings63 and extended this syndrome to adrenal PHEOs.64 The mutations in HIF2A affected the hydroxylation site, preventing recognition by VHL and decreasing the degradation rate.61,65 Interestingly, all patients identified with this syndrome have been female. Germline mutations of HIF2A have also been identified, including in one male patient with PGL and polycythemia.66
After the initial discovery of HIF2A mutations in PHEO/PGL, additional studies were performed to determine whether these mutations underlie other cases of PHEO/PGL previously identified as sporadic. A study of 41 PHEO/PGL with no known mutations identified 7 patients with somatic HIF2A mutations, 3 of whom had multiple PHEO/PGLs and polycythemia. The other 4, however, had no known polycythemia, suggesting that HIF2A mutations can be factors in the development of PHEO/PGL even in the absence of polycythemia, most likely due to differences in the timing of the occurrence of the mutation.67
Other rare genes
Because of the frequency of isocitrate dehydrogenase (IDH) mutations in glioblastoma multiforme, a screening of 365 PHEO/PGL tumors was performed to determine if these mutations also contribute to PHEO/PGL. Only one somatic mutation was discovered in a patient with no underlying germline mutation.68 Four additional genes, KIF1Bβ, PHD2 (also known as EGLN1), fumarate hydratase (FH), and BRCA-1 associated protein-1 (BAP1), have been identified in isolated cases of familial PHEO/PGL.15,16 KIF1Bβ is a gene involved in the regulation of apoptosis. Rare cases of patients with PHEOs and neuroblastomas have been reported in association with KIF1Bβ mutations.69 PHD2 is a member of the prolyl hydroxylase family and is involved in interactions with HIFα. A PHD2 mutation has been identified in one family with multiple PGLs and congenital erythrocytosis.70 A germline FH mutation was identified in one patient with an adrenal PHEO.51 FH mutations have previously been identified in patients with leiomyomatosis and renal cell carcinoma,71 but so far no widespread evidence has been found for their involvement in PHEO/PGL.51 After previous reports identifying BAP1 mutations in melanoma, meningioma, and mesothelioma, one family with a BAP1 mutation was found, with one carrier in the family manifesting with a PGL with confirmed loss of the wild-type allele in the PGL tumor; the significance of this finding is currently unclear.72
Somatic mutations in H-RAS have also been identified in limited cases of PHEO/PGL. Although mutations in RAS have been previously identified in other types of cancer, no definitive evidence of these mutations had ever been found in PHEO/PGL until recently. In a screening of 58 tumors with no previously identified mutations, 6.9% had somatic H-RAS mutations. These mutations led to the constitutive activation of the GTPase domain of RAS, increasing cell proliferation through activated RAS/RAF/ERK and PI3K/AKT/mTOR pathways.73
Algorithm and procedures for genetic testing
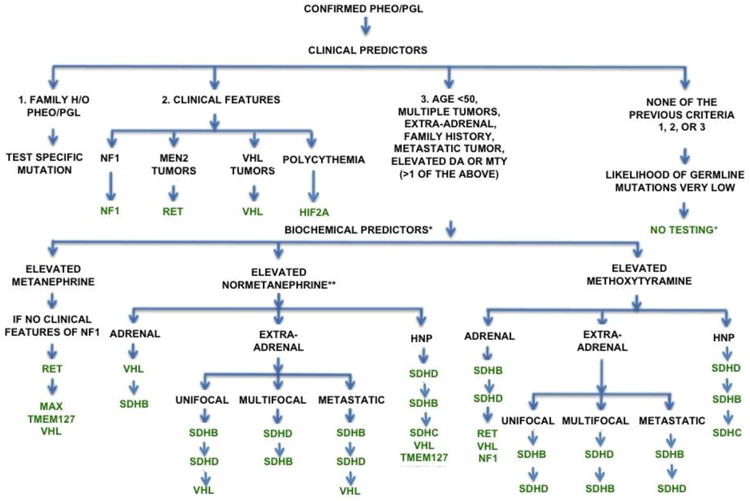
Although genetic testing is a critical component of the clinical evaluation of patients with PHEO/PGL, testing can be costly. Therefore, determining an algorithm for prioritizing gene testing can help reduce costs while maintaining high accuracy.1,15 Family history can eliminate the need for such an algorithm, as only the known mutation needs to be analyzed. In patients lacking family history, the biochemical and clinical profile can be combined to determine a cost-effective strategy.15,74 The current recommended algorithm is detailed in Figure 1.
Figure 1.
Recommended genetic testing algorithm for patients with PHEO/PGL. (Adapted from Karasek et al. 2013)
*If both normetanephrine and methoxytyramine are elevated, follow the algorithm for methoxytyramine. If both normetanephrine and metanephrine are elevated, follow the algorithm for metanephrine.
**In patients with elevated normetanephrine with clinical features that do not clearly indicate which gene to test, perform immunohistochemistry for SDHB and SDHA.
+If tumor is adrenal, TMEM127 testing may be considered.
Abbreviations: DA: dopamine; HIF2A: hypoxia-inducible factor 2-alpha; HNP: head and neck PGL; h/o: history of; MAX: myc-associated factor X; MEN2: multiple endocrine neoplasia type 2; MTY: methoxytyramine; NF1: neurofibromatosis type 1; RET: rearranged during transfection; SDHA: succinate dehydrogenase subunit A; SDHB: succinate dehydrogenase subunit B; SDHC: succinate dehydrogenase subunit C; SDHD: succinate dehydrogenase subunit D; TMEM127: transmembrane protein 127; VHL: von Hippel-Lindau
As more genes become identified in the pathogenesis of PHEO/PGL, the need for more effective and less expensive genetic testing strategies is becoming evident. Although algorithmic testing based on clinical presentation can reduce costs and remain effective, it is a timely process, as each gene must be ruled out individually before testing can proceed. The use of next-generation sequencing techniques has been proposed as a method for more rapidly analyzing multiple genes in at-risk patients at a reduced cost. This method was preliminarily tested in a study of 205 patients, 85 of whom had known mutations detected by traditional sequencing. Nine of the most common genes (SDHA, SDHB, SDHC, SDHD, SDHAF2, MAX, RET, TMEM127, and VHL) were sequenced simultaneously, with a 98.7% sensitivity for detecting mutations.75 Whole exome sequencing has also recently been introduced as a possible technique for both rapidly sequencing known PHEO/PGL susceptibility genes and also potentially identifying new genes. In a study of whole exome sequencing in PHEO/PGL, the authors were able to effectively identify mutations in SDHB, SDHC, SDHD, RET, and VHL in a small sample of patients when appropriate techniques were used, at a much lower cost than traditional sequencing.76
Beyond genetic testing
A great deal of interest has been given to determining underlying similarities in various forms of hereditary PHEO/PGL, in the hopes of determining broader mechanisms and pathways for pathogenesis. Microarray studies determining the expression profiles of hereditary PHEO/PGL have broadly classified these tumors into two clusters. Cluster 1 tumors, which include VHL, PHD2, and SDH tumors, as well as presumably FH and IDH mutations, have increased hypoxia and angiogenesis signatures.77–79 Changes in oxidation and reduction enzyme levels have also been described in these tumors.77 Overexpressed genes include the glucose transporter, vascular endothelial growth factor (VEGF), and genes known to be involved in angiogenesis.78 Tumors in this cluster also appear to have reduced SDHB protein levels, regardless of the underlying genetic mutation, representing a broader oxidoreductase signature caused by mitochondrial dysfunction.77 Although both HIF-1α and HIF-2α overexpression have been linked to Cluster 1 tumors, their exact roles remain unclear.77,78,80,81 One study reported that HIF-1α was particularly overexpressed in VHL tumors compared to SDH tumors81; however, other reports have described overexpression of HIF-2α in VHL tumors and increased HIF-1α in SDH tumors.82 Cluster 2 tumors, including MEN2, NF1, KIF1Bβ, MAX, TMEM127, and presumably H-RAS tumors, are associated with disruptions in kinase signaling.59,60,77 Sporadic tumors are almost equally distributed between the two clusters.60
In addition to the well-established clustering of PHEO/PGL, other microarray analysis studies have proposed other clustering methods. Recent microarray data comparing SDHB, SDHD head and neck, SDHD abdominal and thoracic, and VHL tumors revealed two distinct clusters based on expression profiles: SDHB and SDHD abdominal and thoracic tumors in one cluster and SDHD head and neck and VHL tumors in the other. This suggests that SDHD tumors of the parasympathetic and sympathetic nervous system, though related to the same underlying pathogenic mutation, develop by different mechanisms. Whether this is true for all PHEO/PGL of the parasympathetic and sympathetic nervous system remains to be seen.83
MicroRNAs (miRNAs) have become an area of interest in many cancers, due to their ability to regulate mRNA expression through degradation. miRNA profiling has been done in several series of PHEO/PGL. These expression profiles appear to vary based on genetic background, with unique signatures that broadly cluster similar to the previously described mRNA profile clusters. Certain miRNAs have been suggested to contribute to the development of PHEO/PGL tumors by interfering with cellular differentiation, but further studies are needed.84 In addition, increased expression of specific miRNAs, particularly those associated with IGF2, was found to be more frequently associated with malignant PHEO/PGLs and could represent a novel marker.85,86 Differences in miRNA expression between different hereditary forms of PHEO and between recurrent, metastatic, and primary tumors have also been described.87
Unifying pathways and mechanisms linking multiple underlying germline mutations across clusters are also being explored. One pathway linking these is the egl nine homolog 3 (EGLN3)/c-Jun/JunB apoptotic pathway. Mutant VHL proteins demonstrated a failure to downregulate JunB; higher quantities of JunB lead to increased antagonization of c-Jun and therefore inhibition of apoptosis. The accumulation of succinate due to dysfunctional SDH protein blocks EGLN3 activity, which is necessary for Jun-induced apoptosis. NF1 and RET mutations have been found to work upstream of JunB, affecting a neuronal growth factor receptor and thereby preventing apoptosis.88
Another unifying paradigm is the link between hereditary forms of PHEO/PGL and HIFs. A link between hypoxia and PHEO/PGL is well-established, demonstrated for example by the high rates of PGL in Peruvian patients living at high altitudes in the Andes.89 As previously mentioned, Cluster 1 tumors have a hypoxic signature. The VHL protein directly binds to HIFs and targets them for hydroxylation and subsequent degradation in oxygen-rich conditions; this degradation cannot occur in the absence of a functional VHL protein. This hydroxylation is carried out by PHDs, such as PHD2; therefore mutations in PHD2 also prevent HIF degradation. The accumulation of succinate due to dysfunction of the SDH protein inhibit PHD activity, also leading to overexpression of HIFs. As additional enzymes involved in the Krebs cycle, IDH and FH mutations also result in the accumulation of metabolic intermediates that prevent HIF hydroxylation by PHDs.80,90 In tumors with RET, NF1, and H-RAS mutations, activation of the Ras/MAPK pathway leads to upregulation of HIF. The NF1 protein directly activates Ras, which in turn activates the MAPK, PI3K, and mTOR pathways, all of which may increase HIF levels. RET mutations lead to increased activation of the Ras/MAPK pathways and PI3K/AKT pathways, also leading to HIF upregulation. H-RAS mutations also affect the Ras/MAPK pathway and lead to increased HIF signaling. TMEM127 and MAX appear to upregulate HIF through the mTOR pathway; TMEM127 is directly involved in negatively regulating the mTOR signaling pathway, so mutations prevent this inactivation and lead to increased mTOR signaling. The MAX protein is involved in c-Myc signaling, which affects both the PI3K/mTOR pathway and directly regulates HIF-1alpha. KIF1Bβ is a downstream target of PHD3 and therefore may be involved in HIF signaling as well, though current evidence is lacking.90
Diagnosis of PHEO/PGL
Symptoms
One of the most challenging aspects of diagnosing PHEO/PGL can be identifying the signs and symptoms of a tumor. In fact, many tumors are missed and are not discovered until autopsy.2,5,7 Patients can present with a variety of non-specific symptoms that can mimic many other conditions. These can vary greatly from one patient to another, even within the same family. The frequency of various symptoms is summarized in Table 2. The classic triad of PHEO/PGL symptoms is headaches, sweating, and palpitations.5 Many patients also present with hypertension, which may be sustained or paroxysmal.5,91 Other symptoms may include pallor, feelings of anxiety or panic, fever, or nausea and vomiting.5,91 Nausea and vomiting may specifically be exercise-induced, which is particularly common in children.92 Another rare sign is the onset of diabetes, particularly in younger patients without typical risk factors for diabetes.93 Hypertensive crises caused by catecholamine surges after accidental tumor manipulation or anesthesia administration may also indicate the presence of a PHEO/PGL.5 In addition, patients with resistant hypertension should be considered for evaluation of PHEO/PGL.5 Patients with a family history of PHEO/PGL who begin exhibiting suspicious symptoms or patients with incidentally discovered adrenal masses, even in the absence of symptoms, should also undergo evaluation to rule out PHEO/PGL.5–7 Recognizing the signs and symptoms of PHEO/PGL and making the appropriate diagnosis is critical, as patients who are undiagnosed or misdiagnosed can suffer severe consequences of hypertensive crises, including heart attacks, strokes, and even death. A recent review of published cases in the literature found 106 cases of patients who experienced hypertensive emergencies as a result of PHEO/PGL, with 15% resulting in death.94
Table 2.
Frequency of signs and symptoms in patients with PHEO/PGL.
| Signs | Symptoms | ||
|---|---|---|---|
| Hypertension | ++++ | Headaches | ++++ |
| Sustained hypertension | ++ | Palpitations | ++++ |
| Paroxysmal hypertension | ++ | Anxiety/nervousness | +++ |
| Postural hypotension | + | Tremulousness | ++ |
| Tachycardia or reflex bradycardia | ++ + | Weakness, fatigue | ++ |
| Excessive sweating | ++++ | Nausea/vomiting | + |
| Pallor | ++ | Pain in chest/abdomen | + |
| Flushing | + | Dizziness or faintness | + |
| Weight loss | + | Paresthesias | + |
| Fasting hyperglycemia | ++ | Constipation (rarely diarrhea) | + |
| Decreased gastrointestinal motility | + | Visual disturbances | + |
| Increased respiratory rate | + |
Biochemistry
PHEO/PGL tumors produce, store, synthesize, and metabolize catecholamines. Although previous methods of diagnosis relied on the measurement of catecholamines in the plasma or urine, these are not always the most effective measurements. Many tumors have fluctuating levels of catecholamine release,5 which can lead to false-negatives during periods of low catecholamine release. Instead, the measurement of plasma or urine metanephrines, the metabolites of catecholamines, are the most accurate test currently available.1,5,95 Although catecholamine release fluctuates, their metabolism remains fairly constant, leading to a steady release of metanephrines.1,5 Therefore, these are consistently elevated in patients with biochemically active PHEO/PGL. At the present time, there is no clear evidence favoring plasma or urine metanephrines.
In addition to the measurement of metanephrines, recent studies have demonstrated the utility of plasma methoxytyramine in diagnosing PHEO/PGL. Measurements of this biomarker can be valuable for detecting exclusively dopamine-secreting tumors, which are rare but easily overlooked by traditional measurements of metanephrines.96,97 Methoxytyramine, a metabolite of dopamine, also appears to serve as a predictor of malignancy.98
Chromogranin A (CgA) is often commonly measured in patients with PHEO/PGL. CgA is a polypeptide that is commonly secreted by chromaffin cells, typically with catecholamines.99 Elevated CgA is found in 91% of PHEO/PGL patients.100 Although it is a nonspecific marker of neuroendocrine tumors, in some patients CgA can be a valuable marker for monitoring disease.101 When combined with catecholamine measurements, the sensitivity for diagnosing PHEO/PGL can be close to 100%.100 CgA has been found to be significantly higher in patients with certain hereditary syndromes than in other patients, suggesting possible differences in vesicle formation and catecholamine secretion rates between different hereditary forms of PHEO/PGL.102
In most patients, especially those presenting with signs and symptoms of PHEO/PGL, catecholamines and metanephrines will be elevated to diagnostic levels, defined as levels greater than four times the upper reference limit. In these patients, diagnostic workup can immediately move forward to anatomical and functional imaging. However, some patients will have equivocal test results, with elevations between the upper reference limit and the diagnostic level. In these patients, several steps should be initiated. First, medication interferences should be ruled out. Anti-depressants, some anti-hypertensives, and other common medications can cause false-positive elevations. Patients on these medications should, if possible, be taken off them or switched to other medications before testing is repeated; a list of contraindicated medications for patients with known or suspected PHEO/PGL is listed in Table 3.5,103–106 In addition, several foods, such as caffeine, can cause elevations in catecholamines and metanephrines and should be avoided before repeat testing.5,103,104 Caution should also be exercised when evaluating patients with chronic kidney disease, particularly those on dialysis, as elevated plasma metanephrines are common in this population, even in the absence of PHEO/PGL.107
Table 3. Medications contraindicated in patients with known or suspected PHEO/PGL.
| Drug class | Relevant clinical uses |
|---|---|
| β-Adrenergic blockers1 | May be used to treat conditions that result from catecholamine excess (e.g. hypertension, cardiomyopathy, heart failure, panic attacks, migraine, tachycardia and cardiac dysrhythmias) |
| Dopamine D2 receptor antagonists | Control of nausea, vomiting, psychosis, hot flashes and for tranquilizing effect |
| Tricyclic antidepressants | Treatment of insomnia, neuropathic pain, nocturnal enuresis in children, headaches, depression (rarely) |
| Other antidepressants (serotonin and NE reuptake inhibitors) | Depression, anxiety, panic attacks, antiobesity agents |
| Monoamine oxidase inhibitors | Non-selective agents rarely used as antidepressants (due to “cheese effect”). |
| Sympathomimetics1 | Control of low blood pressure during surgical anesthesia; decongestants; antiobesity agents |
| Chemotherapeutic agents1 | Antineoplastic actions; treatment of malignant pheochromocytoma |
| Opiate analgesics1 | Induction of surgical anesthesia |
| Neuromuscular blocking agents1 | Induction of surgical anesthesia |
| Peptide and steroid hormones1 | Diagnostic testing |
If interfering drugs cannot be discontinued or if medication interferences have been ruled out, a clonidine suppression test should be performed. This can only be done for patients with elevated norepinephrine or normetanephrine. The clonidine test is most sensitive when performed with plasma normetanephrine as the biomarker.5,103 If levels of plasma normetanephrine fail to suppress below the upper reference limit or by 40% of the initial value even after the addition of clonidine, further workup for suspected PHEO/PGL should be performed.5 Previously, clinicians used a similar strategy with the glucagon stimulation test, but this test is no longer recommended due to its low sensitivity and high risk of complications such as hypertensive crises.108
While most PHEO/PGL are biochemically active, a small percentage have no abnormal hormonal activity. These tumors are deemed biochemically silent and are often associated with underlying SDH mutations.26,27 In other rare cases, PHEO/PGL can co-secrete other hormones, such as cortisol or ACTH. These patients often present with Cushing's disease in addition to PHEO/PGL.109–111
Appropriate biochemical testing is critical both for the diagnosis and management of PHEO/PGL. Determining a patient's biochemical phenotype (adrenergic for patients with predominantly epinephrine/metanephrine secretion, noradrenergic for patients with predominantly norepinephrine/normetanephrine secretion, and dopaminergic for patients with predominantly dopamine/methoxytyramine secretion) can help guide genetic testing in the absence of family history. Biochemical levels can also serve as important markers for monitoring the efficacy and response to treatment. Therefore, obtaining accurate measurements is critical. Plasma catecholamine and metanephrine levels should be drawn through an in-dwelling catheter after the patient has rested supine for at least 20 minutes in a dark, quiet room, to remove any environmental impacts on stress levels; failure to obtain blood tests under these conditions can result in false-positive elevations relative to supine reference ranges.112 Patients should have fasted overnight before the blood draw.112 The use of appropriate age-adjusted reference ranges is critical; a recent study showed an increase in the sensitivity of plasma metanephrine and normetanephrine from 88.3% to 96.0% when reference intervals based on patient age were used.113 Urine measurements should be done over a 24-hour period. Interfering medications should be discontinued or avoided, if possible, and foods that can elevate catecholamines or metanephrines should be avoided from at least 24 hours prior to testing until testing is complete.
Imaging
In addition to biochemical testing, imaging plays an important role in the diagnosis of PHEO/PGL. If the biochemical testing has been completed and is positive for elevated metanephrine/epinephrine, imaging can be focused on the adrenal gland, since the majority of tumors that secrete epinephrine are found in the adrenal gland. Computed tomography (CT) or magnetic resonance imaging (MRI) should be sufficient to detect such a tumor.5 If the PHEO is less than 3 cm and the patient is under 40 years of age and has no family history of PHEO, no further imaging workup needs to be performed.114 If adrenal imaging is negative, imaging of additional areas of the body should be performed. Imaging should be completed of the abdomen, followed by the pelvis, chest, and neck.
With regards to sensitivity, CT and MRI have similar success in detecting PHEO/PGL.5 However, MRI may be slightly favored in patients with extra-adrenal tumors. MRI is also preferred in patients with CT-contrast allergies, in pregnant or pediatric patients, and in patients in whom radiation exposure should be limited.5 On CT, PHEO/PGL typically have a heterogeneous appearance, often with some cystic areas.115–118 They typically have attenuation values greater than 10 Hounsfield units, though some PHEOs with fatty components may have appearances more consistent with adenomas.117 Calcifications or hemorrhage may also be present.117 On dual-phase contrast-enhanced CT, PHEOs can also be distinguished from other adrenal masses due to higher intensity during the arterial phase, with enhancement levels greater than 110 Hounsfield units.118 On MRI, PHEO/PGL typically appear as T2-bright lesions, although cystic or necrotic components may affect this classic appearance.115–117 On T1 imaging, PHEO/PGL enhance about equally to muscle and are less intense than the liver.117 PHEO/PGL also typically enhance with gadolinium contrast agents, though cystic or necrotic areas can reduce this enhancement.117
Ultrasound has also been used in PHEO/PGL, but its utility is limited. However, it can be valuable in evaluating metastatic liver lesions as well as tumors in the urinary bladder. On ultrasound, PHEO/PGL can have varied appearances; some appear cystic, while others may be solid, and still others may be somewhere between the two extremes. Necrotic areas or hemorrhages can be present and may appear echogenic.117
In most cases, functional imaging also plays an important part in the work-up of PHEO/PGL. Functional imaging may help detect primary or metastatic tumors that could be missed on CT/MRI. It can also help characterize tumors in terms of their metabolic activity in vivo. Historically, functional imaging has been performed with 123I- or 131I-metaiodobenzylguanidine (MIBG) scintigraphy. MIBG has a structure that resembles norepinephrine and enters cells through norepinephrine transporters. While both 123I- and 131I-MIBG have been used in imaging, 123I-MIBG has been found to be more sensitive and clinically useful, due to its better detection rate, higher possible doses, and shorter intervals between injection and image acquisition.114,117,119,120 Both CT and MRI have been used in combination with single-photon emission computed tomography (SPECT) imaging for added colocalization. A recent comparison study of these techniques within the same patients have found that 123I-MIBG SPECT/MRI has the highest sensitivity for adrenal PHEOs. Both adrenal MRI and SPECT/CT were found to be equally sensitive in diagnosing adrenal lesions and inferior to SPECT/MRI, but they had the advantage of offering better diagnostic imaging in patients in whom PHEO was ruled out.121 However, it is important to note that this study did not evaluate the use of these techniques on extra-adrenal or metastatic tumors, for which MIBG scintigraphy has been found to be less sensitive.122 A less common form of MIBG, 124I-MIBG, has been infrequently used in positron emission tomography (PET) imaging for neuroendocrine tumors. This technique offers the advantage of PET scanning, which provides higher quality images than traditional scintigraphy. Available studies are limited, but a recent case report using 124I-MIBG PET/MRI in a patient with metastatic PHEO found that this technique allowed for more accurate tumor volume determination and therefore better dose-planning for 131I-MIBG therapy.123 It has also been suggested that the intensity of MIBG scintigraphy uptake could be used to distinguish benign from malignant disease, as a study of 9 patients with benign PHEO and 9 patients with malignant disease found more intense MIBG uptake in the metastatic patients.124 However, further validation of these findings must be done in a larger series.
In addition to improvements in diagnostic accuracy, important limitations of MIBG scintigraphy have also been discovered in recent years. False-negative results occur more commonly with extra-adrenal tumors or tumors associated with succinate dehydrogenase subunit B (SDHB) mutations.114,125 MIBG scintigraphy may also miss metastatic disease.114 Certain medications, such as opioids, tricyclic antidepressants, and antihypertensives like labetalol, can also affect MIBG uptake, leading to less intense or false-negative scans.114,126 MIBG scintigraphy is also suboptimal for head and neck PGLs.114 Therefore, additional functional imaging techniques may be warranted or preferred.
PET has become more widely available and more widely used in the field of PHEO/PGL in recent years, due to its increased sensitivity, shorter acquisition times, and higher image resolution compared to SPECT.114,123 PET also offers the advantage of standard uptake values (SUV), which can quantify tracer uptake and therefore some aspect of tumor metabolism.114 Often, PET scans are performed with a corresponding CT for attenuation purposes to increase their sensitivity. Multiple tracers have been studied in patients with PHEO/PGL. The most widely available is 18F-fluorodeoxyglucose (FDG), an analog of glucose that is taken up by the glucose transporter.117,119 FDG PET scanning can be a valuable technique, particularly for patients with SDHB mutations or metastatic disease.127–129 Higher standard uptake values on FDG PET have also been suggested as possible indications of malignant disease, though further validation is needed.127 However, FDG is not specific to PHEO/PGL, so caution should be exercised when interpreting scan results, as other tumor types may also be identified by this technique.114
More specific tracers have been developed, but these are less widely available. The first is 18F-fluorodopa (FDOPA), an amino acid analog and catecholamine precursor that is taken up by the amino acid transporter.117,119 FDOPA is specific to neuroendocrine tumors, increasing the likelihood that findings represent true PHEO/PGL, though other neuroendocrine tumors may also be identified by this modality.114 However, false-positives are rare.130 Pretreatment with carbidopa, which inhibits DOPA decarboxylase, enhances tumor uptake and improves its sensitivity.131 FDOPA PET is extremely sensitive for patients with head/neck PGLs, sometimes identifying small tumors missed by all other imaging techniques.132,133 This technique also appears to be particularly effective for patients with SDH mutations and/or biochemically silent PHEO/PGL and may be valuable as a screening technique, particularly for patients with SDHD mutations.133,134 In a recent large study focusing on tumors missed by FDOPA PET, a high rate of SDH mutations were found, suggesting that patients with false-negative FDOPA PET scans should be tested for these mutations.135
The second PHEO/PGL-specific tracer is 18F-fluorodopamine, which is similar to dopamine and taken up by norepinephrine transporters, though with higher affinity than MIBG.117,119 Unfortunately, this technique is only available at limited institutions worldwide, but studies have shown its value in identifying PHEO/PGL, particularly for primary tumors in the abdomen.136–139 It has been found to be more sensitive than MIBG or Octreoscan.136,138,139 FDA PET also appears to be a valuable modality for patients with metastatic tumors.129,138,139 Increased availability of FDOPA and FDA PET in the future will significantly improve diagnosis; at the present time, patients with more challenging cases of PHEO/PGL who would benefit from these imaging modalities should be referred to tertiary care centers that can offer these techniques.
Newer PET scanning tracers are also being explored in PHEO/PGL, though clinical experience remains limited. One such tracer is 18F-fluorothymidine (FLT), which has been used previously in multiple cancers to detect rapidly proliferating tumor cells. Studies on this imaging modality in PHEO/PGL are currently being performed. However, one published report on a patient with metastatic PGL imaged with FLT PET showed no uptake in any tumors; the only uptake was a bright rim around metastatic bone lesions, where proliferating bone cells took up the tracer.140 While this provides interesting insights into the in vivo activity of PHEO/PGL tumors, this imaging modality does not appear to have any utility for patient diagnosis.
More promising results have been found with radiolabeled DOTA peptides (DOTATATE, DOTATOC, and DOTANOC), which target somatostatin receptors on the cell membrane. Recent studies of 68Ga-labeled DOTA peptides on patients with neuroendocrine tumors, including PHEO/PGL, have found high sensitivities of these modalities, even for small tumors and head and neck tumors.141–150 These DOTA peptides may also help distinguish adrenocortical adenomas from PHEOs; in a recent series, 10 patients with adrenal lesions were identified with FDG PET, but only the 2 patients with PHEO had positive uptake on DOTA imaging.148 Their superiority to MIBG scintigraphy for metastatic tumors and possibly for primary tumors as well has also been demonstrated.142,145–147,150 68Ga-DOTATOC PET/CT was also found to be superior to FDOPA PET/CT in the diagnosis of metastatic tumors.144 However, these results have been limited, and ongoing research at limited centers is being performed to try to expand these findings to larger cohorts of PHEO/PGL patients.
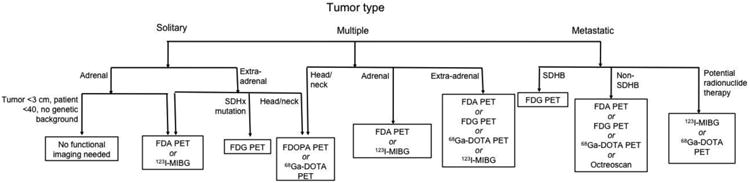
Other functional imaging techniques have also been used in PHEO/PGL. One such technique is Octreoscan, or 111In-pentetreotide scintigraphy. This modality exploits the somatostatin receptors often expressed on the cell membranes of PHEO/PGL by introducing radioactively labeled octreotide to bind to these receptors.114 However, the expression of these receptors can vary between PHEO/PGL patients, with certain subtypes less expressed or even absent in certain tumor specimens, affecting the sensitivity of this technique.151 Several efforts have been made to incorporate this technique more broadly into the algorithm for PHEO/PGL diagnosis, but suboptimal sensitivities have precluded more widespread use. Small or metastatic tumors, as well as PGLs in the head and neck or abdomen, are more frequently missed on Octreoscan.114 False-positives have also been reported in patients with renal cysts, abdominal hernias, accessory spleens, inflammatory diseases, and other neuroendocrine tumors.114 However, Octreoscan can be of value in patients with metastatic disease.136 In addition, a recent study of SDHB mutation carriers found that adding Octreoscan to the routine screening of carriers increased diagnostic sensitivity.152 As Octreoscan is more widely available than FDOPA/FDA PET, these findings may benefit a great deal of patients. In addition to Octreoscan, bone scans are sometimes performed in patients with bony metastases. These scans are not commonly used, but can be of value for patients with osseous metastases.129 Figure 2 shows the current recommended functional imaging algorithm for patients with PHEO/PGL, and Table 4 summarizes sensitivities of imaging modalities in different PHEO/PGL types.
Figure 2.
Recommended functional imaging algorithm for patients with PHEO/PGL.
Abbreviations: FDA: 18F-fluorodopamine; FDG: 18F-fluorodeoxyglucose; FDOPA: 18F-fluorodopa; 68Ga-DOTA: 68Ga-DOTA-peptides; 123I-MIBG: 123I-metaiodobenzylguanidine; PET: positron emission tomography;; SDHB: succinate dehydrogenase subunit B; SDHx: succinate dehydrogenase mutation
Table 4. Sensitivities and specificities of imaging modalities.
| Imaging modality | Primary (non-metastatic) | Adrenal PHEO | Extra-adrenal PGL | Head/neck PGL | SDHx carriers | Metastatic | SDHB metastatic | Non-SDHB metastatic | Bone metastases | |
|---|---|---|---|---|---|---|---|---|---|---|
| Sensitivity | Specificity | |||||||||
| CT/MRI | 66-100% | 40-90% | --- | --- | 80-92% | 85.7-87.5% | 45-100% | 78-96% | 71% | 37.8-96% |
| FDA PET | 77-88% | 90% | --- | --- | 40-46% | --- | 76-97% | 76-88% | 76% | 79-100% |
| FDG PET | 58-88% | 90% | --- | --- | 69-80% | --- | 74-91.4% | 74-100% | 62-67.3% | 76-93.7% |
| FDOPA PET | 67-93% | 95-100% | 93.9% | 47.1-90% | 96.5-100% | --- | 45-100% | 20-45% | 93% | --- |
| MIBG | 52-87% | 75-100% | 85-87% | 58-67% | 18-50% | 42.70% | 38-92.4% | 44-80% | 59-66% | 20.75-76% |
| Octreoscan | 25-54% | 75% | --- | --- | 64-100% | 69.50% | 68.5-88.9% | 59-81% | --- | --- |
| 68Ga-DOTA peptides | 80-100% | 85.7% | --- | --- | 100% | 60% | 91.70% | --- | --- | 100% |
Imaging is also an important component of the screening process for patients with genetic predispositions to PHEO/PGL development and of follow-up for patients with a history of PHEO/PGL. For carrier screening, a CT or MRI is often recommended every few years, in conjunction with annual biochemical testing, to detect potential tumor growth. Adding whole-body imaging is particularly important for SDH mutation carriers, as these patients more frequently have normal biochemistry, so tumors can be missed by only biochemical evaluations.153 In patients with specific genetic backgrounds, particularly those with SDHB mutations or a family history of biochemically silent tumors, occasional functional imaging may be a valuable addition to the regular screening evaluation. A similar strategy should be employed for patients after PHEO/PGL removal. Biochemical testing 6-8 weeks after the procedure can be used to determine the success of the surgical resection, with additional biochemical testing and imaging studies on approximately 6-month intervals. Once the likelihood of recurrence has decreased based on patient risk factors such as age, genetic background, and tumor size and location, follow-up intervals can be extended to one to two years.
Metastatic PHEO/PGL
One of the largest challenges in PHEO/PGL management is the inability to predict which patients may develop metastatic disease. As previously mentioned, there are no clear features that distinguish benign from malignant primary PHEO/PGL. A scoring system, deemed the “pheochromocytoma of the adrenal gland scaled score” (PASS), was proposed,154 but a large retrospective analysis found no significant correlation between PASS score and future malignancy.155 While the Ki-67 index is often used as a marker of proliferation in other cancers, there is no clear value to this marker in PHEO/PGL. One study has suggested that the Ki-67 index, in addition to pS100 staining and the presence of tumor necrosis, may be a predictor of malignancy;156 another study reported Ki-67 and c-erbB-2 staining was higher in malignant versus primary tumors.157 However, studies of the Ki-67 index in relation to imaging findings, particularly standard uptake values on FDG PET or lesion intensity on MIBG, have found no correlation.158 A large-scale microarray analysis of benign versus malignant tumors identified a large cohort of genes that were underexpressed in malignant tumors, suggesting that malignant tumors may develop due to dedifferentiated gene expression. Further analysis of this dataset may reveal genes that could be predictive markers for metastatic PHEO/PGL development.159 Recently, expression of heat shock protein 90 (Hsp90) and activator of transcription 3 (STAT3) have been proposed as potential markers for distinguishing between benign and malignant tumors, as malignant tumors were more likely to stain positively for these proteins on immunohistochemistry. However, 22.37% and 26.32% of benign tumors also stained positively for Hsp90 and STAT3, respectively, showing that these technique may not be sufficient for distinguishing between these two types of tumors.160 High telomerase activity has also been proposed as a marker of malignancy in PHEO/PGL, as this was more frequently associated with malignant PHEOs in one study, but its predictive value is unclear.161 Increased expression of angiogenesis genes has also been reported in malignant PHEO/PGL compared to benign tumors, but one study also found overexpression of these genes in 30% of benign tumors as well.162,163 A recent study has also found that high copy numbers of an N-terminal truncated splice isoform of carboxypeptidase E accurately predicted future recurrence or metastases in PHEO/PGL, but further study is needed to validate these findings in larger patient populations.164 While these features may indicate a possibility for malignancy, more accurate and comprehensive predictive tools that can be used in routine diagnosis still need to be developed.
Several independent risk factors for metastases have been established. The first is the presence of an SDHB mutation.98,165,166 As previously discussed, SDHB tumors are more frequently associated with metastases. SDHB mutations have also been independently linked to higher rates of mortality in patients with PHEO/PGL.165 One study found that approximately half of patients with metastatic PGL had SDHB mutations.34 Extra-adrenal location has also been independently associated with an increased risk of malignancy and a decreased rate of survival.98,166,167 Primary tumors in the mediastinum and organ of Zuckerkandl had particularly high metastatic rates in one study of 371 patients with metastatic PHEO/PGL.167 The size of the primary tumor is a third risk factor for metastases.98,166–169 Tumor sizes over 5 cm have been associated with increased risk of metastatic disease development and shorter overall survival.98,166 The age at primary tumor diagnosis is also associated with increased risk of metastatic disease development, with patients who develop metastatic disease presenting at a statistically significantly younger age, a mean of 41 years vs. 50 years for patients without metastases.169 Finally, increased levels of plasma methoxytyramine, even when not associated with extra-adrenal or _SDHB_-related tumors, have been established as an indication of metastatic disease risk.98
With regards to the clinical characteristics of metastatic PHEO/PGL patients, a recent large retrospective analysis of adults with metastatic PHEO/PGL has been performed, which identified 287 patients with metastatic PHEO and 221 patients with metastatic PGL. Similar numbers of males and females were identified, suggesting there is no gender difference in the development of metastases. The mean age at diagnosis was in the sixth decade of life. Survival was significantly better for patients with metastatic PGL than for those with metastatic PHEO. The majority of patients underwent surgery, typically for primary tumors, though some patients were not identified until metastases were present, in which case debulking procedures were performed. Patients who did not undergo surgery for PHEO or who had metastases at presentation for PGL were at the highest risk of death from disease. As previously discovered, the metastatic patients included in the study typically had large primary tumor sizes (with mean sizes >5 cm). However, no improvement in survival rates were noted over the two decades encompassed by the patients in this study, highlighting the need for improved treatment strategies.170
Typical sites of PHEO/PGL metastases include the lungs, liver, bones, and lymph nodes.166,171 Patients with PHEO/PGL, particularly metastatic disease, suffer diminished quality of life due to pain caused by tumor effects, side effects from treatments, and consequences of elevated catecholamines.166,172 Patients with bone metastases frequently report bone pain.173 Other skeletal complications include spinal cord compression, bone fractures, and/or hypercalcemia.173 In patients with bone metastases, skeletal events were reduced if patients responded to therapy.173 Bone metastases appear to be less aggressive than other forms of metastatic tumors; in one study of patients with metastatic PHEO/PGL, patients with bone metastases only had an average survival of 12 years, compared to 7.5 years for patients without skeletal metastases and 5 years for patients with both skeletal and non-skeletal metastases.173 However, the overall 5-year survival rate for patients with metastatic PHEO/PGL is less than 60%.171,174
While treatment of patients with metastatic PHEO/PGL can have some benefit, the limited number of available therapeutic options warrants careful consideration of available options before treatment is initiated. Therapy should only be considered in patients with disease that is clearly progressing. Patients with metastatic PHEO/PGL may often have stable disease, even in the absence of therapy, and therefore should undergo close clinical monitoring without treatment.174,175 For instance, a recent retrospective study of 90 metastatic PHEO patients who were monitored but never treated at several French institutions found that half of patients had stable disease after 1 year even in the absence of any therapeutic intervention.175
Management of PHEO/PGL
Blockade
Patients with biochemically active PHEO/PGL should immediately be placed on anti-hypertensive medications to control symptoms and reduce the risk of hypertensive crises. A summary of the available drugs and suggested doses is listed in Table 5. Alpha blockade should always be initiated first, followed by beta blockade, if necessary. If beta blockade is initiated first, unopposed stimulation of alpha-adrenoceptors due to beta-adrenoceptor vasodilation can result in hypertensive crises.5,91,106 Several alpha-adrenoceptor blockers are available. Phenoxybenzamine is a long-lasting alpha blocker that is commonly used in patients with PHEO/PGL.91,106 However, phenoxybenzamine is not widely available, especially outside the United States.91 Short-acting alpha blockers can be used as alternatives to phenoxybenzamine, either when phenoxybenzamine is not available or when a patient's hypertension is not severe enough to warrant the use of a long-acting alpha blocker. These include prazosin, terazosin, and doxazosin. These medications should be started at bedtime, as they can cause orthostatic hypotension after the first dose.91,106 Doses should be adjusted until normotension or even mild hypotension is achieved.106
Table 5.
Medications used for symptom management and pre-procedural blockade.
| Drug | Classifications | Doses | Recommended Use |
|---|---|---|---|
| Alpha blockers | First choice for alpha-adrenoceptor blockade | ||
| Phenoxybenzamine (Dibenzyline) | long-lasting; irreversible; noncompetitive | 10 mg 1-3 times daily | When phenoxybenzamine is not available For patients who cannot tolerate phenoxybenzamine For patients with mild hypertension |
| Prazosin (Minipress) | short-acting; specific; competitive | 2-5 mg 2-3 times daily | |
| Terazosin (Hytrin) | short-acting; specific; competitive | 2-5 mg per day | |
| Doxazosin (Cardura) | short-acting; specific; competitive | 2-8 mg per day | |
| Beta blockers | |||
| Atenolol (Tenormin) | cardioselective | 12.5-25 mg 2-3 times daily | |
| Metoprolol (Lopressor) | cardioselective | 25-50 mg 3-4 times daily | |
| Propranol (Inderal) | non-selective | 20-80 mg 1-3 times daily | To control tachyarrythmia caused by catecholamines or alpha blockade |
| Calcium channel blockers | |||
| Amlodopine (Norvasc) | 10-20 mg per day | To provide additional blood pressure control for patients on alpha blockers For patients who cannot tolerate alpha blockers For patients with intermittent hypertension | |
| Nicardipine (Cardene) | 60-90 mg per day | ||
| Nifedipine (Adalat) | extended-release action | 30-90 mg per day | |
| Verapamil (Covera-HS, Calan-SR) | extended-release action | 180-540 mg per day | |
| Catecholamine synthesis inhibitors | |||
| Metyrosine (Demser) | 250 mg every 8-12 h for a total dose of 1.5-2 g per day | To provide additional blood pressure control for patients on adrenoceptor blockade |
After alpha blockade has been established, beta blockers may need to be introduced to address additional symptoms, such as tachyarrythmia. Cardioselective beta blockers are frequently preferred in the management of patients with PHEO/PGL and include metoprolol and atenolol. However, the nonselective beta blocker propranolol may also be used.91
While combined alpha- and beta-adrenoceptor antagonists such as labetalol may seem ideally suited for patients with PHEO/PGL, these agents are not recommended. The beta-blocking activity of labetalol far outweighs its alpha-adrenoceptor activity, which could result in hypertensive crises.91,106 The lower alpha-blocking ability of labetalol also results in inadequate blood pressure control in most patients.91,106 Finally, labetalol can interfere with MIBG uptake, which can affect both scanning and treatment.106,114
Calcium channel blockers can also be used for additional blood pressure and symptom control. Patients with persistent hypertension after alpha blockers may benefit from the addition of a calcium channel blocker rather than increased doses of alpha blocker. In addition, some patients may be unable to tolerate alpha blockers, in which case calcium channel blockers should be used.106 Calcium channel blockers can also be valuable in the management of patients with very mild hypertension, in whom alpha blockade would cause hypotension.91,106 Amlodipine, nicardapine, nifedipine, and verapamil are all commonly used for pre-operative blockade.91,106
In patients who do not achieve adequate hypertensive or symptom control with alpha and beta blockers, metyrosine (Demser) can be added to prevent catecholamine synthesis. Metyrosine is a competitive inhibitor of tyrosine hydroxylase, a critical enzyme in catecholamine synthesis. Metyrosine can significantly decrease levels of catecholamines and provide additional blood pressure control for patients with biochemically active PHEO/PGL. However, its availability is limited. In addition, its ability to cross the blood-brain barrier and deplete catecholamine levels in the brain can lead to side effects such as depression, anxiety and sleepiness, so patients should be carefully monitored.106
Although blockade may not eliminate the possibility of intra-procedural hypertensive crises, it can dramatically reduce their severity and improve the ease of management.176 In one study, intraoperative complications were present in only 3% of patients who received appropriate pre-surgical blockade, compared to 69% of patients without blockers.177 Patients who are candidates for surgical resection or any treatment that could induce the release of catecholamines should be initiated on adequate alpha and beta blockade at least 2 weeks before the procedure or treatment. This includes patients who are normotensive, as unanticipated catecholamine release by the tumor during surgery or other procedures may lead to hypertensive crises. Mild alpha blockade or calcium channel inhibitors may be most appropriate for these patients. The only patients who may not require pre-procedural blockade are patients with non-secreting head and neck tumors.106
Surgery
Currently, surgical resection remains the only curative treatment option for patients with PHEO/PGL. Laparascopic surgery has been successfully performed in patients with both adrenal PHEO and extra-adrenal PGLs with outcomes similar to open surgery and is the preferred technique when feasible.5,176,178–185 For larger tumors over 6 cm, laparascopy may still be used, though these are frequently converted to open procedures intraoperatively.176,184 Multiple, recurrent, or metastatic tumors can also be approached laparascopically if performed by experienced surgeons, although open resection may be preferable to ensure complete removal of tumors suspected to be metastatic.171,185 Robotic assistance or robotic procedures can be used with similar success rates, with the added advantages of lower morbidity, less postoperative pain, and shorter postoperative hospital stays.186 For patients with adrenal PHEOs, full adrenalectomies should be performed in the absence of a genetic background, in patients with a low risk of bilateral disease, or in patients with larger tumors. However, in patients with bilateral tumors or a high risk of bilateral tumors (such as in patients with VHL or MEN2), cortex-sparing surgery may be sufficient if the tumor is small enough, thereby eliminating the need for steroid replacement.5,187–193 The risk of recurrence with cortical-sparing adrenalectomies is small (approximately 7%) as long as the whole tumor is removed,189 but repeat subtotal adrenalectomies in these patients may be successfully performed if tumors recur.187,188 The risks of operative mortality are extremely low if performed by an experienced surgical team, including a skilled anesthesiologist to monitor for intra-operative hypertensive crises.5 In the immediate post-operative period, patient should undergo fluid replacement to mitigate post-operative hypotension caused by the sudden drop in the amount of circulating catecholamines.5
For patients with small tumors, surgical resection can be curative, although hypertension may persist.5,194 In the absence of genetic background, with complete tumor removal, rates of metastases and recurrence can be very low. In a large study of 114 patients who underwent successful removals of PHEO/PGL, only 16 (14%) later developed recurrent or metastatic disease.194 Unfortunately, as there is no clear method for distinguishing benign from malignant tumors pathologically, patients should undergo close clinical follow-up after surgery, typically at least annually for ten years,5,194 regardless of pathological features of the tumor. Surgery can be used as a curative treatment for primary, recurrent, or limited metastatic tumors; it can also be used as a debulking technique for patients with extensive metastatic disease, to reduce symptoms and imminent complications from tumor size. However, the long-term benefits of debulking procedures for patients with metastatic disease may be limited.1,174,195 A recent study found that only 8.3% of patients who underwent a non-curative debulking procedure were able to cease antihypertensive medications for more than 6 months. In addition, only one patient out of 30 had a biochemical response to surgery that lasted for 12 months.195 In this same study, though, patients who underwent aggressive surgical intervention with the goal of complete resection had very successful outcomes.195 In addition, surgery may be the only option, though not curative, if tumors may pose an immediate risk to vital processes or are affecting critical organ structures.1 The reduction of tumor burden through surgical debulking may also increase the efficacy of post-surgical therapies.1,174
Radiofrequency ablation
In some patients for whom surgery may not be the best option, tumors in accessible locations can be addressed by radiofrequency ablation (RFA). RFA has been successfully used on osseous and liver metastases.196–199 One study of 10 patients who received RFA found that 56% had successful ablations without recurrence; in 2 of the patients, all identified metastatic lesions were ablated.197 Another study of 6 patients with 7 metastatic lesions demonstrated complete ablation in 6 out of 7 lesions, with no serious adverse events reported.199 Due to catecholamine release by the tumor during the procedure, experienced radiologists, in conjunction with experienced anesthesiologists, should perform the technique while monitoring blood pressure to reduce risks of intra-procedural catecholamine-induced hypertensive crises.196
External radiation
In some cases, external beam radiation has been used for inoperable tumors or for symptom palliation. This is particularly popular for the treatment of bone lesions.171,174 The outcomes of radiation therapy on metastatic PHEO/PGL are unclear.171 One study of 17 patients who underwent external beam radiation for non-head and neck metastases reported local control and/or symptom relief in 76% of patients, all of which lasted at least 1 year or until death, suggesting an important palliative role for this therapy. Of note, 5 of these 17 patients also received systemic therapy with 131I-MIBG.200 It is important to exercise restraint when using external beam radiation on metastatic PHEO/PGL, as patients may have multiple bone lesions that require treatment and doses should be limited to avoid further radiation-related complications.171
External beam radiation is also a common treatment modality for non-resectable head and neck PGLs. In a retrospective analysis of 31 patients with head and neck PGLs, 14 of whom had previously undergone partial resections, long-term local control was observed, with limited toxicity. Five-year, 10-year, and 15-year local control rates were 96%, 90%, and 90%, respectively.201 Glomus jugulare PGLs are particularly popular candidates for external beam radiation, with high success rates and limited toxicities.202 More recently, radiosurgery using gamma knife, LINAC, or CyberKnife have begun to replace traditional external beam radiation for glomus jugulare tumors, due to their more precise targeting of radiation and increased dose capability.202 High success rates, determined by local control through stable or decreased tumor sizes, have been reported with all three techniques, up to 100%, with limited complications.202–205
Radiotherapy
For patients with positive MIBG scintigraphy, MIBG therapy can be a valuable treatment modality. When considering patients for treatment with radiolabeled MIBG, it is critical to obtain a recent MIBG scan to determine tumor uptake. Before the scan and for the duration of therapy, the patient should be taken off medications that can block MIBG uptake such as labetalol, tricyclic antidepressants, and certain calcium antagonists.5,114,126,206,207 The therapy is based on the emission of beta particles once the radioactive compound has been taken up into tumor cells, leading to their destruction.206 Broadly, two strategies are being evaluated with regards to MIBG therapy. In some trials and studies, 131I-MIBG therapy is given in small doses over a longer period. Doses usually range from 100-300 milliCuries (mCi) and are given once every 3-4 months for up to an approximately 1000 mCi dose total, although there are no strictly established guidelines on maximum cumulative dosage or repeated doses.206 A retrospective analysis of 116 patients who received mean single doses of 158 mCi for a range of 1-11 treatments, resulting in cumulative doses ranging from 96-2322 mCi, reported improved symptoms in 76% of patients, biochemical responses in 45%, and tumor responses in 30%; 5 patients reported complete tumor and biochemical responses for 16-58 months. Side effects were generally mild and reported in 41% of patients; only one patient suffered extreme toxicities and died from bone marrow asplasia.208 However, some centers have experimented with giving extremely large one-time doses of MIBG, followed by stem cell infusion to replace bone marrow.206 In one such study, 49 patients were treated with single doses of 492-1160 mCi, with 15 patients receiving multiple doses, for total doses of 492-3191 mCi. Fifty-seven percent of patients had a complete, partial, or minor response, and an additional 8% achieved stable disease for at least one year. However, severe toxicities were observed, including 2 patient deaths from myelodysplastic syndrome, acute respiratory distress in 3 patients, leading to an additional patient death, as well as high rates of grades 3-4 neutropenia (87%) and thrombocytopenia (83%).209 While there is no proven benefit to one method over the other, the significantly reduced toxicity of the first approach suggests that this may be a favorable treatment strategy, and this method is used far more frequently.206 Repeat staging is usually done 3-6 months after treatment to assess response and determine whether another dose is warranted.206
Complete response is rare from MIBG treatment, regardless of the technique, with the highest reported rates only around 15%.206 A systematic review of published studies on 131I-MIBG treatment found that of 243 patients treated with this modality, only 3% had a complete response; 27% had a partial response and 52% had stable disease.210 However, many patients benefit from partial responses, reduced symptoms, and/or lower biochemical levels.171,206 In addition to clinical benefits to the patient with regards to symptoms, the ease of this therapy, which can be performed as an outpatient treatment, and the relative lack of side effects compared to other available treatments makes MIBG the preferred first treatment for patients with moderately progressing MIBG-avid metastatic PHEO/PGL.174,206
More recently, therapies targeting somatostatin receptors have been introduced for patients with PHEO/PGL. Using DOTA peptides (DOTATATE, DOTATOC, and DOTANOC) radiolabeled with lutetium (177Lu), yttrium (90Y), or indium (111In), researchers have been able to give doses to patients with positive Octreoscan or Ga-DOTA-peptide imaging. Although data on this treatment technique are limited, preliminary reports have shown this to be a promising strategy. For example, a report on pediatric patients with neuroendocrine tumors, including 3 with PGL, used 90Y-DOTATOC; 2 patients had stable disease and 1 had a minor response, with all 3 reporting symptomatic relief.211 Another study on 4 patients with non-resectable, non-metastatic _SDHD_-related PGL found stable disease or partial responses in all 4 patients.212 In a larger study by van Essen et al., 12 patients with PGL were treated with 177Lu DOTATATE. Six patients had stable disease on follow-up, and two had a minor or partial response.213 A retrospective analysis of 28 patients treated with 90Y-DOTATOC found partial responses in 2 patients, minor responses in 5, mixed responses in 2, and stable disease in 13; responses were sustained from 6-44 months.214
Chemotherapy
For patients with metastatic disease, chemotherapy may be valuable to palliate patient symptoms, reduce or stop the rate of tumor growth, and in some cases, shrink tumors. Although no chemotherapeutic treatment has been discovered with long-term efficacy, some chemotherapy regimens can maintain disease status for several years, prolonging patient survival and improving patient quality of life. Traditional chemotherapy with cyclophosphamide, vincristine, and dacarbazine (CVD) has been used most extensively with PHEO/PGL and still remains one of the most effective treatments for widespread metastatic disease.171,174 An early study on 14 patients with metastatic PHEO/PGL found at least partial tumor responses in 57% of patients, with 79% showing complete or partial biochemical responses, with response durations from 5 to more than 35 months.215 A single institution study of patients receiving CVD or similar chemotherapy regimens found a 33% rate of response (measured in terms of symptom relief) and prolonged overall survival.216 In a more recent study of 17 metastatic PHEO/PGL patients from Japan who were followed between 12 and 192 months after initiating CVD, 47.1% showed partial responses in tumor size and/or biochemical levels; stable disease without significant response was observed in another 23.5%. Although no patients in the study showed a complete tumor response, progression-free survival ranged from 31 to 60 months (with a mean of 40 months) in patients with partial responses.217 However, in a study that followed 18 patients with a 22-year follow-up, while a complete response was seen in 11% and a partial in 44% of patients, no significant difference was noted in overall survival between patients who responded to CVD and those who did not. The main benefit of CVD, therefore, is symptom improvement.218 CVD chemotherapy has been found to be particularly effective for patients with SDHB mutations (unpublished observations). CVD chemotherapy is usually well-tolerated for long periods, with side effects being relatively minor, such as nausea, vomiting, hair loss, thromobocytopenia, and paresthesia.171,174,215 Patients exhibiting toxicities can be offered reduced doses or prolonged intervals between cycles.171
There is limited experience with other chemotherapeutic agents in PHEO/PGL. Other chemotherapeutic combinations have been tried in limited cases. These include temozolomide; streptozotocin with other agents; ifosfamide; cyclophosphamide and methotrexate; etoposide, carboplatin, vincristine, cyclophosphamide, and doxorubicin; and cisplatin and 5-fluorouracil.166,171,174 The experience with most of these combinations is anecdotal. For example, a case report of a metastatic PHEO patient treated with streptozotocin reported tumor shrinkage, decreased biochemical levels, and reduced symptoms.219 An isolated case report of cisplatin and 5-fluorouracil reported a minor tumor response and the loss of a need for antihypertensive medications followed by stable disease for 2 years after only 3 treatments with this regimen.220 Other therapies have been tried on small cohorts of neuroendocrine tumor patients. A retrospective analysis of patients with neuroendocrine tumors treated with temozolomide included 1 patient with PGL, who had stable disease after treatment.221 Temozolomide has also been used in conjuction with thalidomide, which is thought to inhibit angiogenesis. One study of patients with neuroendocrine tumors, including 3 patients with PHEO/PGL, found a 33% partial response rate with limited mild toxicities.222 Due to the isolated nature of these treatment options, no conclusions or recommendations about their use can be made for patients with PHEO/PGL.
Molecular targeted therapies
One drug that has begun to receive a great deal of attention is sunitinib (sutent), which was developed as a treatment for renal cell carcinoma. Sunitinib is a tyrosine kinase inhibitor that prevents angiogenesis through the targeting of vascular endothelial growth factor receptors (VEGFR) and other angiogenic processes. Conflicting reports have found varying effects of sunitinib on PHEO/PGL. In vitro studies have found that sunitinib appears to induce apoptosis in rat PHEO cells.223 Further studies have also suggested that sunitinib directly inhibits catecholamine synthesis by reducing the activity of tyrosine hydroxylase, a critical enzyme in catecholamine synthesis.224 While some studies have found partial or even complete responses in terms of tumor size, symptoms, catecholamine secretion, and metabolic activity as measured by PET scanning in small patient cohorts,225–227 other studies have not reported much success. For example, a case report of a patient with metastatic PGL reported a partial response to sunitinib initially, but disease progression was observed after 6 months of treatment.228 It has also been suggested that patients with SDHB mutations may respond less frequently, perhaps due to a lack of the necessary receptors for sunitinib targeting (unpublished observations).
Other more experimental regimens have been evaluated in limited cases of PHEO/PGL. Everolimus has been used in limited cases of PHEO/PGL to target the mTOR pathway, with disappointing results. In a study of 4 patients treated with everolimus, progressive disease was noted in all 4 after relatively short treatment periods of 3-6 months; one patient died of metastatic disease while on the therapy.229 A phase II study of everolimus on patients with neuroendocrine tumors, including 7 patients with metastatic PHEO/PGL, reported stable disease in 5 and progressive disease in 2, with only a mean 3.8 month progression-free survival time.230
Future directions
The field of PHEO/PGL is rapidly expanding. As diagnostic techniques improve and the understanding and access to genetic testing in these tumors identifies more carriers, more patients are identified, leading to increased sample sizes for clinical trials and expanding the available knowledge database. However, there are still many aspects of PHEO/PGL that remain to be understood. The underlying pathogenetic mechanisms, particularly those that govern the transformation to malignancy, are not well understood. This is partially due to the lack of a human cell line. While several established mouse and rat cell lines have been used extensively in research, the development of a human cell line would improve in vitro experimentation and accelerate research to an unprecedented degree.
In addition, therapeutic options are still relatively limited for patients with metastatic PHEO/PGL. While some strategies can provide symptom relief and extend progression-free survival for some time, only surgery can cure patients long-term. Discovering new treatment targets is essential for providing additional treatment options. In addition, developing drugs to target potentially relevant targets needs to be accelerated. A link between PHEO/PGL development and the HIF genes has been explored in a recent review article, suggesting this as a potential treatment target.90 However, there is no clinically approved drug to directly target the HIF protein.231 Although HIF1-targeting drugs have been tested, compensatory mechanisms in PHEO/PGL tumors make these drugs ineffective in vitro. Therefore, a broader HIF1/2-targeting drug, or a HIF2-targeting drug that can be used in combination, is necessary and could prove extremely valuable in the treatment of PHEO/PGL, regardless of genetic background.
Another potential target is the mTOR pathway. The mTOR protein, a serine/threonine protein kinase, is regulated by two major mTOR complexes (mTORCs), mTORC1 and mTORC2. Cell proliferation and migration were reduced in vitro through the use of an ATP-competitive inhibitors that target both major mTORCs, cell proliferation and migration was reduced in vitro.232 Furthermore, the introduction of a dual mTORC inhibitor to metastatic PHEO/PGL mouse models significantly reduced tumor burden.232 Although previous results with everolimus, an mTORC1 inhibitor, have been disappointing, dual mTORC1/2 inhibitors could overcome compensatory mechanisms that may be activated upon reduction in one mTORC due to targeted treatment.231
Heat shock proteins, molecular chaperones thought to play a role in protein folding and degradation, have been investigated as potential treatment targets in various tumor types. Preliminarily promising results of inhibitors of a specific protein, Hsp90, have been seen in multiple cancers, including melanoma, leukemia, prostate cancer, lung cancer, multiple myeloma, and breast cancer.233 Hsp90 is of particular interest due to its overexpression in metastatic PHEO/PGL.160 A study of two Hsp90 inhibitors, 17-AAG and ganetespib, found that cell proliferation and migration were reduced in vitro with Hsp90 inhibitors in available mouse and rat PHEO cell lines and in primary human PHEO/PGL tissue cultures. In addition, metastatic tumor burden was reduced in a mouse model of metastatic PHEO/PGL after treatment with Hsp90 inhibitors.234 A second study on rat PHEO cell lines also confirmed reduced cell proliferation after 17-AAG treatment and also reported increased apoptosis.235
Other potential targets for therapy will be discovered as more insight is gained into PHEO/PGL pathogenesis. A recent study on insulin-like growth factor 1 (IGF1) found a link between circulating levels of IGF1 and PHEO/PGL tumor development and progression in mice, suggesting this as a possible therapeutic target.236 An in vitro study of NVP-AEW541, an IGF1 receptor antagonist, found significantly decreased cell viability in mouse PHEO/PGL cell lines with high doses, although compensatory upregulation of other cellular pathways was also observed.237 Drugs targeting RET, which have been successfully used in MTC, may be of value in PHEO/PGL, particularly Cluster 2 tumors.231 Another receptor tyrosine kinase, v-erb-b2 erythroblastic leukemia viral oncogene homolog 2 (ERBB2, Her2/Neu), has been found to lead to PHEO formation in mice when overexpressed238; high levels have also been found in metastatic human PHEO/PGL.239 Although no studies of this target have been done, several existing drugs target this protein and could be introduced into clinical trial for patients with metastatic PHEO/PGL.231
Fatty acids have also been suggested to play a role in apoptosis induction in several cancers. One such fatty acid, eicosapentaenoic acid (EPA), has been found to induce apoptosis in rat PHEO cells. This is thought to result at least partially from lipid peroxidation.240 Whether this could be used as a treatment modality for PHEO/PGL remains to be seen, but its limited side effects make this an appealing possibility.231
Another potential target is the previously mentioned N-terminal truncated splice isoform of carboxypeptidase E, which has been found to be present in high copy numbers in metastatic PHEO/PGL.164 The protein product, CPE-deltaN, of this truncated mRNA was found to upregulate the expression of genes associated with metastases formation in melanoma.164 Using siRNA targeted to this splice isoform reduced tumor growth and invasion in mice injected with highly metastatic human liver cells.164 As previously mentioned, carboxypeptidase E copy numbers are also thought to be a potential biomarker of malignant potential in benign tumors, though further investigation is needed.164,231
Another mechanism for treatment involves increasing apoptosis in tumor cells. Several drugs have been found to have this ability in vitro and in animal models, but are still awaiting introduction into clinical trials. One such treatment is the use of histone deacetylase (HDAC) inhibitors. Two HDAC inhibitors were tested in mouse PHEO cell lines, and both showed inhibition of cell proliferation. However, perhaps more interesting, both also increased the uptake of 131I-MIBG into metastatic PHEOs in a mouse model, suggesting a possible role for HDAC inhibitors as a pretreatment enhancer for patients undergoing MIBG therapy.241 The use of HDAC inhibitors has also been proposed to prevent the degradation of mutant SDHB protein, allowing it to be transported to the mitochondria.25 Similarly, a nuclear factor kappa B (NFκB) inhibitor was found to induce apoptosis in mouse and rat PHEO cell lines, reduce metastases in a mouse model, and increase the levels of the norepinephrine transporter system, which thereby increases the available entrance sites for 131I-MIBG treatment.242 Topoisomerase inhibitors have also been proposed as treatment targets in cancer, due to their role in unraveling DNA supercoiling and in apoptosis.243
Immunotherapy is a novel but increasingly popular potential treatment modality in many cancers. Through the use of vaccines targeted to specific cancer molecules, the patient's immune system can recognize and attack these molecules.244 The challenge is identifying molecules that would be specific to the tumor cells and still be capable of recognition. One proposed target in PHEO/PGL is CgA. A study in mice with vaccines targeted to CgA found that cytotoxic T-cells were successfully produced in response to the vaccines and were capable of recognizing CgA and inducing lysis in PHEO cells. Vaccinated mice also exhibited less tumor growth in the liver.245 This preliminary study suggests that exploring immunotherapy as a treatment option in PHEO/PGL, with CgA or other potential target molecules, could be an effective therapy.
The use of MIBG in combination with other therapies, such as chemotherapy or DOTA-peptide radionuclides, has been proposed, but so far these techniques have not been reported in the literature.171,206,246 Combinations of multiple targeted molecular therapies directed toward multiple pathways may overcome compensatory mechanisms of PHEO/PGL cells, reduce necessary doses and therefore reduce the risk of resistance development, and overall lead to greater therapeutic success.171,231 Combined treatment with NVP-BEZ235, a dual PI3K/mTORC1/2 inhibitor, and lovastatin, a drug known to reduce ERK signaling, showed a significant additive effect leading to reduced cell viability in mouse PHEO/PGL cells, supporting the use of combination therapies in overcoming compensatory upregulation of other pathways and increasing treatment efficacy.237
Conclusions
The application of novel techniques and improved understanding of PHEO/PGL pathogenesis have led to a great deal of progress in this field in recent years. However, successful long-term treatments for patients who develop metastatic disease are still lacking. Several promising options have been identified and need to be introduced into clinical trials. These targeted treatment options will not only provide insight into the molecular mechanisms of PHEO/PGL pathogenesis, but also improve the quality of life for patients who suffer from this devastating disease.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chen H, Sippel RS, O'Dorisio MS, et al. The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas. 2010;39:775–783. doi: 10.1097/MPA.0b013e3181ebb4f0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McNeil AR, Blok BH, Koelmeyer TD, et al. Phaeochromocytomas discovered during coronial autopsies in Sydney, Melbourne and Auckland. Aust N Z J Med. 2000;30:648–652. doi: 10.1111/j.1445-5994.2000.tb04358.x. [DOI] [PubMed] [Google Scholar]
- 3.Anderson GH, Blakeman N, Streeten DH. The effect of age on prevalence of secondary forms of hypertension in 4429 consecutively referred patients. J Hypertens. 1994;12:609–615. doi: 10.1097/00004872-199405000-00015. [DOI] [PubMed] [Google Scholar]
- 4.Omura M, Saito J, Yamaguchi K, et al. Prospective study on the prevalence of secondary hypertension among hypertensive patients visiting a general outpatient clinic in Japan. Hypertens Res. 2004;27:193–202. doi: 10.1291/hypres.27.193. [DOI] [PubMed] [Google Scholar]
- 5.Lenders JWM, Eisenhofer G, Mannelli M, et al. Phaeochromocytoma. Lancet. 2005;366:665–675. doi: 10.1016/S0140-6736(05)67139-5. [DOI] [PubMed] [Google Scholar]
- 6.Arnaldi G, Boscaro M. Adrenal incidentaloma. Best Pract Res Clin Endocrinol Metab. 2012;26:405–419. doi: 10.1016/j.beem.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 7.Mannelli M, Lenders JWM, Pacak K, et al. Subclinical phaeochromocytoma. Best Pract Res Clin Endocrinol Metab. 2012;26:507–515. doi: 10.1016/j.beem.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cascón A, Inglada-Pérez L, Comino-Méndez I, et al. Genetics of pheochromocytoma and paraganglioma in Spanish pediatric patients. Endocr Relat Cancer. 2013;20:L1–6. doi: 10.1530/ERC-12-0339. [DOI] [PubMed] [Google Scholar]
- 9.King KS, Prodanov T, Kantorovich V, et al. Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: significant link to SDHB mutations. J Clin Oncol. 2011;29:4137–4142. doi: 10.1200/JCO.2011.34.6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waguespack SG, Rich T, Grubbs E, et al. A current review of the etiology, diagnosis, and treatment of pediatric pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. 2010;95:2023–2037. doi: 10.1210/jc.2009-2830. [DOI] [PubMed] [Google Scholar]
- 11.Burnichon N, Vescovo L, Amar L, et al. Integrative genomic analysis reveals somatic mutations in pheochromocytoma and paraganglioma. Hum Mol Genet. 2011;20:3974–3985. doi: 10.1093/hmg/ddr324. [DOI] [PubMed] [Google Scholar]
- 12.Dannenberg H, de Krijger RR, van der Harst E, et al. Von Hippel-Lindau gene alterations in sporadic benign and malignant pheochromocytomas. Int J Cancer. 2003;105:190–195. doi: 10.1002/ijc.11060. [DOI] [PubMed] [Google Scholar]
- 13.Gimm O, Armanios M, Dziema H, et al. Somatic and occult germ-line mutations in SDHD, a mitochondrial complex II gene, in nonfamilial pheochromocytoma. Cancer Res. 2000;60:6822–6825. [PubMed] [Google Scholar]
- 14.van Nederveen FH, Korpershoek E, Lenders JWM, et al. Somatic SDHB mutation in an extraadrenal pheochromocytoma. N Engl J Med. 2007;357:306–308. doi: 10.1056/NEJMc070010. [DOI] [PubMed] [Google Scholar]
- 15.Karasek D, Shah U, Frysak Z, et al. An update on the genetics of pheochromocytoma. J Hum Hypertens. 2013;27:141–147. doi: 10.1038/jhh.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gimenez-Roqueplo AP, Dahia PL, Robledo M. An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Horm Metab Res. 2012;44:328–333. doi: 10.1055/s-0031-1301302. [DOI] [PubMed] [Google Scholar]
- 17.Latif F, Tory K, Gnarra J, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317–1320. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 18.Wallace MR, Marchuk DA, Andersen LB, et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science. 1990;249:181–186. doi: 10.1126/science.2134734. [DOI] [PubMed] [Google Scholar]
- 19.Burnichon N, Buffet A, Parfait B, et al. Somatic NF1 inactivation is a frequent event in sporadic pheochromocytoma. Hum Mol Genet. 2012;21:5397–5405. doi: 10.1093/hmg/dds374. [DOI] [PubMed] [Google Scholar]
- 20.Baysal BE, Ferrell RE, Willett-Brozick JE, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 21.Astuti D, Latif F, Dallol A, et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001;69:49–54. doi: 10.1086/321282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niemann S, Müller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. 2000;26:268–270. doi: 10.1038/81551. [DOI] [PubMed] [Google Scholar]
- 23.Burnichon N, Briere JJ, Libé R, et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. 2010;19:3011–3020. doi: 10.1093/hmg/ddq206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hao HX, Khalimonchuk O, Schraders M, et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009;325:1139–1142. doi: 10.1126/science.1175689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang C, Matro JC, Huntoon KM, et al. Missense mutations in the human SDHB gene increase protein degradation without altering intrinsic enzymatic function. FASEB J. 2012;26:4506–4516. doi: 10.1096/fj.12-210146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Timmers HJLM, Pacak K, Huynh TT, et al. Biochemically silent abdominal paragangliomas in patients with mutations in the succinate dehydrogenase subunit B gene. J Clin Endocrinol Metab. 2008;93:4826–4832. doi: 10.1210/jc.2008-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Timmers HJLM, Gimenez-Roqueplo AP, Mannelli M, et al. Clinical aspects of SDHx-related pheochromocytoma and paraganglioma. Endocr Relat Cancer. 2009;16:391–400. doi: 10.1677/ERC-08-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghayee HK, Havekes B, Corssmit EPM, et al. Mediastinal paragangliomas: association with mutations in the succinate dehydrogenase genes and aggressive behavior. Endocr Relat Cancer. 2009;16:291–299. doi: 10.1677/ERC-08-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lodish MB, Adams KT, Huynh TT, et al. Succinate dehydrogenase gene mutations are strongly associated with paraganglioma of the organ of Zuckerkandl. Endocr Relat Cancer. 2010;17:581–588. doi: 10.1677/ERC-10-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Benn DE, Gimenez-Roqueplo AP, Reilly JR, et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab. 2006;91:827–836. doi: 10.1210/jc.2005-1862. [DOI] [PubMed] [Google Scholar]
- 31.van Duinen N, Corssmit EPM, de Jong WHA, et al. Plasma levels of free metanephrines and 3-methoxytyramine indicate a higher number of biochemically active HNPGL than 24-h urinary excretion rates of catecholamines and metabolites. Eur J Endocrinol. 2013;169:377–382. doi: 10.1530/EJE-13-0529. [DOI] [PubMed] [Google Scholar]
- 32.Baysal BE. Mitochondrial complex II and genomic imprinting in inheritance of paraganglioma tumors. Biochim Biophys Acta. 2013;1827:573–577. doi: 10.1016/j.bbabio.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 33.van Hulsteijn LT, Dekkers OM, Hes FJ, et al. Risk of malignant paraganglioma in SDHB-mutation and SDHD-mutation carriers: a systematic review and meta-analysis. J Med Genet. 2012;49:768–776. doi: 10.1136/jmedgenet-2012-101192. [DOI] [PubMed] [Google Scholar]
- 34.Brouwers FM, Eisenhofer G, Tao JJ, et al. High frequency of SDHB germline mutations in patients with malignant catecholamine-producing paragangliomas: implications for genetic testing. J Clin Endocrinol Metab. 2006;91:4505–4509. doi: 10.1210/jc.2006-0423. [DOI] [PubMed] [Google Scholar]
- 35.Papathomas T, Gaal J, Corssmit EPM, et al. Non-pheochromocytoma/paraganglioma tumors in patients with succinate dehydrogenase-related pheochromocytoma-paraganglioma syndromes: a clinicopathologic and molecular analysis. Eur J Endocrinol. 2013 doi: 10.1530/EJE-13-0623. [DOI] [PubMed] [Google Scholar]
- 36.Ricketts C, Woodward ER, Killick P, et al. Germline SDHB mutations and familial renal cell carcinoma. J Natl Cancer Inst. 2008;100:1260–1262. doi: 10.1093/jnci/djn254. [DOI] [PubMed] [Google Scholar]
- 37.Ricketts CJ, Forman JR, Rattenberry E, et al. Tumor risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum Mutat. 2010;31:41–51. doi: 10.1002/humu.21136. [DOI] [PubMed] [Google Scholar]
- 38.Ayala-Ramirez M, Callender GG, Kupferman ME, et al. Paraganglioma syndrome type 1 in a patient with Carney-Stratakis syndrome. Nat Rev Endocrinol. 2010;6:110–115. doi: 10.1038/nrendo.2009.250. [DOI] [PubMed] [Google Scholar]
- 39.Pasini B, McWhinney SR, Bei T, et al. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet. 2008;16:79–88. doi: 10.1038/sj.ejhg.5201904. [DOI] [PubMed] [Google Scholar]
- 40.Dwight T, Mann K, Benn DE, et al. Familial SDHA mutation associated with pituitary adenoma and pheochromocytoma/paraganglioma. J Clin Endocrinol Metab. 2013;98:E1103–1108. doi: 10.1210/jc.2013-1400. [DOI] [PubMed] [Google Scholar]
- 41.Xekouki P, Stratakis CA. Succinate dehydrogenase (SDHx) mutations in pituitary tumors: could this be a new role for mitochondrial complex II and/or Krebs cycle defects? Endocr Relat Cancer. 2012;19:C33–40. doi: 10.1530/ERC-12-0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schimke RN, Collins DL, Stolle CA. Paraganglioma, neuroblastoma, and a SDHB mutation: Resolution of a 30-year-old mystery. Am J Med Genet A. 2010;152A:1531–1535. doi: 10.1002/ajmg.a.33384. [DOI] [PubMed] [Google Scholar]
- 43.Armstrong R, Greenhalgh KL, Rattenberry E, et al. Succinate dehydrogenase subunit B (SDHB) gene deletion associated with a composite paraganglioma/neuroblastoma. J Med Genet. 2009;46:215–216. doi: 10.1136/jmg.2008.060749. [DOI] [PubMed] [Google Scholar]
- 44.Cascón A, Landa I, López-Jiménez E, et al. Molecular characterisation of a common SDHB deletion in paraganglioma patients. J Med Genet. 2008;45:233–238. doi: 10.1136/jmg.2007.054965. [DOI] [PubMed] [Google Scholar]
- 45.Ni Y, Zbuk KM, Sadler T, et al. Germline mutations and variants in the succinate dehydrogenase genes in Cowden and Cowden-like syndromes. Am J Hum Genet. 2008;83:261–268. doi: 10.1016/j.ajhg.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim S, Kim DH, Jung WH, et al. Succinate dehydrogenase expression in breast cancer. Springerplus. 2013;2:299. doi: 10.1186/2193-1801-2-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neumann HPH, Pawlu C, Peczkowska M, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 2004;292:943–951. doi: 10.1001/jama.292.8.943. [DOI] [PubMed] [Google Scholar]
- 48.Schiavi F, Milne RL, Anda E, et al. Are we overestimating the penetrance of mutations in SDHB? Hum Mutat. 2010;31:761–762. doi: 10.1002/humu.21269. [DOI] [PubMed] [Google Scholar]
- 49.Gimenez-Roqueplo AP, Favier J, Rustin P, et al. Functional consequences of a SDHB gene mutation in an apparently sporadic pheochromocytoma. J Clin Endocrinol Metab. 2002;87:4771–4774. doi: 10.1210/jc.2002-020525. [DOI] [PubMed] [Google Scholar]
- 50.Rao JU, Engelke UFH, Rodenburg RJT, et al. Genotype-specific abnormalities in mitochondrial function associate with distinct profiles of energy metabolism and catecholamine content in pheochromocytoma and paraganglioma. Clin Cancer Res. 2013;19:3787–3795. doi: 10.1158/1078-0432.CCR-12-3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Letouzé E, Martinelli C, Loriot C, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23:739–752. doi: 10.1016/j.ccr.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 52.Castelblanco E, Santacana M, Valls J, et al. Usefulness of Negative and Weak-Diffuse Pattern of SDHB Immunostaining in Assessment of SDH Mutations in Paragangliomas and Pheochromocytomas. Endocr Pathol. 2013 doi: 10.1007/s12022-013-9269-4. [DOI] [PubMed] [Google Scholar]
- 53.Gill AJ, Benn DE, Chou A, et al. Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC, and SDHD in paraganglioma-pheochromocytoma syndromes. Hum Pathol. 2010;41:805–814. doi: 10.1016/j.humpath.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 54.van Nederveen FH, Gaal J, Favier J, et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncol. 2009;10:764–771. doi: 10.1016/S1470-2045(09)70164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Korpershoek E, Favier J, Gaal J, et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J Clin Endocrinol Metab. 2011;96:E1472–1476. doi: 10.1210/jc.2011-1043. [DOI] [PubMed] [Google Scholar]
- 56.Comino-Méndez I, Gracia-Aznárez F, Schiavi F, et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet. 2011;43:663–667. doi: 10.1038/ng.861. [DOI] [PubMed] [Google Scholar]
- 57.Burnichon N, Cascón A, Schiavi F, et al. MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin Cancer Res. 2012;18:2828–2837. doi: 10.1158/1078-0432.CCR-12-0160. [DOI] [PubMed] [Google Scholar]
- 58.Pęczkowska M, Kowalska A, Sygut J, et al. Testing new susceptibility genes in the cohort of apparently sporadic phaeochromocytoma/paraganglioma patients with clinical characteristics of hereditary syndromes. Clin Endocrinol (Oxf) 2013 doi: 10.1111/cen.12218. [DOI] [PubMed] [Google Scholar]
- 59.Qin Y, Yao L, King EE, et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet. 2010;42:229–233. doi: 10.1038/ng.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jiang S, Dahia PLM. Minireview: the busy road to pheochromocytomas and paragangliomas has a new member, TMEM127. Endocrinology. 2011;152:2133–2140. doi: 10.1210/en.2011-0052. [DOI] [PubMed] [Google Scholar]
- 61.Zhuang Z, Yang C, Lorenzo F, et al. Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N Engl J Med. 2012;367:922–930. doi: 10.1056/NEJMoa1205119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pacak K, Jochmanova I, Prodanov T, et al. New syndrome of paraganglioma and somatostatinoma associated with polycythemia. J Clin Oncol. 2013;31:1690–1698. doi: 10.1200/JCO.2012.47.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Toledo RA, Qin Y, Srikantan S, et al. In vivo and in vitro oncogenic effects of HIF2A mutations in pheochromocytomas and paragangliomas. Endocr Relat Cancer. 2013;20:349–359. doi: 10.1530/ERC-13-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Taïeb D, Yang C, Delenne B, et al. First report of bilateral pheochromocytoma in the clinical spectrum of HIF2A-related polycythemia-paraganglioma syndrome. J Clin Endocrinol Metab. 2013;98:E908–913. doi: 10.1210/jc.2013-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang C, Sun MG, Matro J, et al. Novel HIF2A mutations disrupt oxygen sensing, leading to polycythemia, paragangliomas, and somatostatinomas. Blood. 2013;121:2563–2566. doi: 10.1182/blood-2012-10-460972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lorenzo FR, Yang C, Ng Tang Fui M, et al. A novel EPAS1/HIF2A germline mutation in a congenital polycythemia with paraganglioma. J Mol Med (Berl) 2013;91:507–512. doi: 10.1007/s00109-012-0967-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Comino-Méndez I, de Cubas AA, Bernal C, et al. Tumoral EPAS1 (HIF2A) mutations explain sporadic pheochromocytoma and paraganglioma in the absence of erythrocytosis. Hum Mol Genet. 2013;22:2169–2176. doi: 10.1093/hmg/ddt069. [DOI] [PubMed] [Google Scholar]
- 68.Gaal J, Burnichon N, Korpershoek E, et al. Isocitrate dehydrogenase mutations are rare in pheochromocytomas and paragangliomas. J Clin Endocrinol Metab. 2010;95:1274–1278. doi: 10.1210/jc.2009-2170. [DOI] [PubMed] [Google Scholar]
- 69.Schlisio S, Kenchappa RS, Vredeveld LCW, et al. The kinesin KIF1Bbeta acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor. Genes Dev. 2008;22:884–893. doi: 10.1101/gad.1648608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ladroue C, Carcenac R, Leporrier M, et al. PHD2 mutation and congenital erythrocytosis with paraganglioma. N Engl J Med. 2008;359:2685–2692. doi: 10.1056/NEJMoa0806277. [DOI] [PubMed] [Google Scholar]
- 71.Tomlinson IPM, Alam NA, Rowan AJ, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30:406–410. doi: 10.1038/ng849. [DOI] [PubMed] [Google Scholar]
- 72.Wadt K, Cho J, Chung JY, et al. A cryptic BAP1 splice mutation in a family with uveal and cutaneous melanoma, and paraganglioma. Pigment Cell Melanoma Res. 2012;25:815–818. doi: 10.1111/pcmr.12006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Crona J, Delgado Verdugo A, Maharjan R, et al. Somatic Mutations in H-RAS in Sporadic Pheochromocytoma and Paraganglioma Identified by Exome Sequencing. J Clin Endocrinol Metab. 2013;98:E1266–E1271. doi: 10.1210/jc.2012-4257. [DOI] [PubMed] [Google Scholar]
- 74.Eisenhofer G, Lenders JWM, Timmers H, et al. Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin Chem. 2011;57:411–420. doi: 10.1373/clinchem.2010.153320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rattenberry E, Vialard L, Yeung A, et al. A comprehensive next generation sequencing-based genetic testing strategy to improve diagnosis of inherited pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. 2013;98:E1248–1256. doi: 10.1210/jc.2013-1319. [DOI] [PubMed] [Google Scholar]
- 76.McInerney-Leo AM, Marshall MS, Gardiner B, et al. Whole Exome Sequencing is an Efficient and Sensitive Method for Detection of Germline Mutations in Patients with Phaeochromocytomas and Paragangliomas. Clin Endocrinol (Oxf) 2013 doi: 10.1111/cen.12331. [DOI] [PubMed] [Google Scholar]
- 77.Dahia PLM, Ross KN, Wright ME, et al. A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet. 2005;1:72–80. doi: 10.1371/journal.pgen.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Eisenhofer G, Huynh TT, Pacak K, et al. Distinct gene expression profiles in norepinephrine- and epinephrine-producing hereditary and sporadic pheochromocytomas: activation of hypoxia-driven angiogenic pathways in von Hippel-Lindau syndrome. Endocr Relat Cancer. 2004;11:897–911. doi: 10.1677/erc.1.00838. [DOI] [PubMed] [Google Scholar]
- 79.Favier J, Briere JJ, Burnichon N, et al. The Warburg effect is genetically determined in inherited pheochromocytomas. PloS One. 2009;4:e7094. doi: 10.1371/journal.pone.0007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Favier J, Gimenez-Roqueplo AP. Pheochromocytomas: the (pseudo)-hypoxia hypothesis. Best Pract Res Clin Endocrinol Metab. 2010;24:957–968. doi: 10.1016/j.beem.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 81.López-Jiménez E, Gómez-López G, Leandro-García LJ, et al. Research resource: Transcriptional profiling reveals different pseudohypoxic signatures in SDHB and VHL-related pheochromocytomas. Mol Endocrinol. 2010;24:2382–2391. doi: 10.1210/me.2010-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pollard PJ, El-Bahrawy M, Poulsom R, et al. Expression of HIF-1alpha, HIF-2alpha (EPAS1), and their target genes in paraganglioma and pheochromocytoma with VHL and SDH mutations. J Clin Endocrinol Metab. 2006;91:4593–4598. doi: 10.1210/jc.2006-0920. [DOI] [PubMed] [Google Scholar]
- 83.Shankavaram U, Fliedner SMJ, Elkahloun AG, et al. Genotype and tumor locus determine expression profile of pseudohypoxic pheochromocytomas and paragangliomas. Neoplasia. 2013;15:435–447. doi: 10.1593/neo.122132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.de Cubas AA, Leandro-García LJ, Schiavi F, et al. Integrative analysis of miRNA and mRNA expression profiles in pheochromocytoma and paraganglioma identifies genotype-specific markers and potentially regulated pathways. Endocr Relat Cancer. 2013;20:477–493. doi: 10.1530/ERC-12-0183. [DOI] [PubMed] [Google Scholar]
- 85.Meyer-Rochow GY, Jackson NE, Conaglen JV, et al. MicroRNA profiling of benign and malignant pheochromocytomas identifies novel diagnostic and therapeutic targets. Endocr Relat Cancer. 2010;17:835–846. doi: 10.1677/ERC-10-0142. [DOI] [PubMed] [Google Scholar]
- 86.Patterson E, Webb R, Weisbrod A, et al. The microRNA expression changes associated with malignancy and SDHB mutation in pheochromocytoma. Endocr Relat Cancer. 2012;19:157–166. doi: 10.1530/ERC-11-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tömböl Z, Eder K, Kovács A, et al. MicroRNA expression profiling in benign (sporadic and hereditary) and recurring adrenal pheochromocytomas. Mod Pathol. 2010;23:1583–1595. doi: 10.1038/modpathol.2010.164. [DOI] [PubMed] [Google Scholar]
- 88.Lee S, Nakamura E, Yang H, et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell. 2005;8:155–167. doi: 10.1016/j.ccr.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 89.Saldana MJ, Salem LE, Travezan R. High altitude hypoxia and chemodectomas. Hum Pathol. 1973;4:251–263. doi: 10.1016/s0046-8177(73)80012-7. [DOI] [PubMed] [Google Scholar]
- 90.Jochmanová I, Yang C, Zhuang Z, et al. Hypoxia-inducible factor signaling in pheochromocytoma: turning the rudder in the right direction. J Natl Cancer Inst. 2013;105:1270–1283. doi: 10.1093/jnci/djt201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mazza A, Armigliato M, Marzola MC, et al. Anti-hypertensive treatment in pheochromocytoma and paraganglioma: current management and therapeutic features. Endocrine. 2013 doi: 10.1007/s12020-013-0007-y. [DOI] [PubMed] [Google Scholar]
- 92.King KS, Darmani NA, Hughes MS, et al. Exercise-induced nausea and vomiting: another sign and symptom of pheochromocytoma and paraganglioma. Endocrine. 2010;37:403–407. doi: 10.1007/s12020-010-9319-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.La Batide-Alanore A, Chatellier G, Plouin PF. Diabetes as a marker of pheochromocytoma in hypertensive patients. J Hypertens. 2003;21:1703–1707. doi: 10.1097/00004872-200309000-00020. [DOI] [PubMed] [Google Scholar]
- 94.Whitelaw B, Prague JK, Mustafa O, et al. Pheochromocytoma crisis. Clin Endocrinol (Oxf) 2013 doi: 10.1111/cen.12324. [DOI] [PubMed] [Google Scholar]
- 95.Lenders JWM, Pacak K, Walther MM, et al. Biochemical diagnosis of pheochromocytoma: which test is best? JAMA. 2002;287:1427–1434. doi: 10.1001/jama.287.11.1427. [DOI] [PubMed] [Google Scholar]
- 96.Eisenhofer G, Goldstein DS, Sullivan P, et al. Biochemical and clinical manifestations of dopamine-producing paragangliomas: utility of plasma methoxytyramine. J Clin Endocrinol Metab. 2005;90:2068–2075. doi: 10.1210/jc.2004-2025. [DOI] [PubMed] [Google Scholar]
- 97.Poirier É, Thauvette D, Hogue JC. Management of exclusively dopamine-secreting abdominal pheochromocytomas. J Am Coll Surg. 2013;216:340–346. doi: 10.1016/j.jamcollsurg.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 98.Eisenhofer G, Lenders JWM, Siegert G, et al. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer. 2012;48:1739–1749. doi: 10.1016/j.ejca.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Eiden LE, Iacangelo A, Hsu CM, et al. Chromogranin A synthesis and secretion in chromaffin cells. J Neurochem. 1987;49:65–74. doi: 10.1111/j.1471-4159.1987.tb03395.x. [DOI] [PubMed] [Google Scholar]
- 100.Grossrubatscher E, Dalino P, Vignati F, et al. The role of chromogranin A in the management of patients with phaeochromocytoma. Clin Endocrinol (Oxf) 2006;65:287–293. doi: 10.1111/j.1365-2265.2006.02591.x. [DOI] [PubMed] [Google Scholar]
- 101.d'Herbomez M, Do Cao C, Vezzosi D, et al. Chromogranin A assay in clinical practice. Ann Endocrinol. 2010;71:274–280. doi: 10.1016/j.ando.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 102.Cleary S, Phillips JK, Huynh TT, et al. Chromogranin a expression in phaeochromocytomas associated with von Hippel-Lindau syndrome and multiple endocrine neoplasia type 2. Horm Metab Res. 2007;39:876–883. doi: 10.1055/s-2007-993135. [DOI] [PubMed] [Google Scholar]
- 103.Eisenhofer G, Goldstein DS, Walther MM, et al. Biochemical diagnosis of pheochromocytoma: how to distinguish true- from false-positive test results. J Clin Endocrinol Metab. 2003;88:2656–2666. doi: 10.1210/jc.2002-030005. [DOI] [PubMed] [Google Scholar]
- 104.Eisenhofer G, Siegert G, Kotzerke J, et al. Current progress and future challenges in the biochemical diagnosis and treatment of pheochromocytomas and paragangliomas. Horm Metab Res. 2008;40:329–337. doi: 10.1055/s-2008-1073156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Neary NM, King KS, Pacak K. Drugs and pheochromocytoma--don't be fooled by every elevated metanephrine. N Engl J Med. 2011;364:2268–2270. doi: 10.1056/NEJMc1101502#SA1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pacak K. Preoperative management of the pheochromocytoma patient. J Clin Endocrinol Metab. 2007;92:4069–4079. doi: 10.1210/jc.2007-1720. [DOI] [PubMed] [Google Scholar]
- 107.Niculescu DA, Ismail G, Poiana C. Plasma Free Metanephrine and Normetanephrine Levels are Increased in Patients with Chronic Kidney Disease. Endocr Pr. 2013 doi: 10.4158/EP13251.OR. [DOI] [PubMed] [Google Scholar]
- 108.Lenders JWM, Pacak K, Huynh TT, et al. Low sensitivity of glucagon provocative testing for diagnosis of pheochromocytoma. J Clin Endocrinol Metab. 2010;95:238–245. doi: 10.1210/jc.2009-1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nijhoff MF, Dekkers OM, Vleming LJ, et al. ACTH-producing pheochromocytoma: Clinical considerations and concise review of the literature. Eur J Intern Med. 2009;20:682–685. doi: 10.1016/j.ejim.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 110.Kumar M, Kumar V, Talukdar B, et al. Cushing Syndrome in an Infant Due to Cortisol Secreting Adrenal Pheochromocytoma: A Rare Association. J Pediatr Endocrinol Metab. 2010;23 doi: 10.1515/jpem.2010.102. [DOI] [PubMed] [Google Scholar]
- 111.Berenyi MR, Singh G, Gloster ES, et al. ACTH-producing pheochromocytoma. Arch Pathol Lab Med. 1977;101:31–35. [PubMed] [Google Scholar]
- 112.Därr R, Pamporaki C, Peitzsch M, et al. Biochemical diagnosis of phaeochromocytoma using plasma free normetanephrine, metanephrine and methoxytyramine: Importance of supine sampling under fasting conditions. Clin Endocrinol (Oxf) 2013 doi: 10.1111/cen.12327. [DOI] [PubMed] [Google Scholar]
- 113.Eisenhofer G, Lattke P, Herberg M, et al. Reference intervals for plasma free metanephrines with an age adjustment for normetanephrine for optimized laboratory testing of phaeochromocytoma. Ann Clin Biochem. 2013;50:62–69. doi: 10.1258/acb.2012.012066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Taïeb D, Timmers HJ, Hindié E, et al. EANM 2012 guidelines for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging. 2012;39:1977–1995. doi: 10.1007/s00259-012-2215-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bhatia KSS, Ismail MM, Sahdev A, et al. 123I-metaiodobenzylguanidine (MIBG) scintigraphy for the detection of adrenal and extra-adrenal phaeochromocytomas: CT and MRI correlation. Clin Endocrinol (Oxf) 2008;69:181–188. doi: 10.1111/j.1365-2265.2008.03256.x. [DOI] [PubMed] [Google Scholar]
- 116.Havekes B, King K, Lai EW, et al. New imaging approaches to phaeochromocytomas and paragangliomas. Clin Endocrinol (Oxf) 2010;72:137–145. doi: 10.1111/j.1365-2265.2009.03648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Leung K, Stamm M, Raja A, et al. Pheochromocytoma: the range of appearances on ultrasound, CT, MRI, and functional imaging. AJR Am J Roentgenol. 2013;200:370–378. doi: 10.2214/AJR.12.9126. [DOI] [PubMed] [Google Scholar]
- 118.Northcutt BG, Raman SP, Long C, et al. MDCT of Adrenal Masses: Can Dual-Phase Enhancement Patterns Be Used to Differentiate Adenoma and Pheochromocytoma? AJR Am J Roentgenol. 2013;201:834–839. doi: 10.2214/AJR.12.9753. [DOI] [PubMed] [Google Scholar]
- 119.Chen CC, Carrasquillo JA. Molecular imaging of adrenal neoplasms. J Surg Oncol. 2012;106:532–542. doi: 10.1002/jso.23162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Furuta N, Kiyota H, Yoshigoe F, et al. Diagnosis of pheochromocytoma using [123I]-compared with [131I]-metaiodobenzylguanidine scintigraphy. Int J Urol. 1999;6:119–124. doi: 10.1046/j.1442-2042.1999.06310.x. [DOI] [PubMed] [Google Scholar]
- 121.Derlin T, Busch JD, Wisotzki C, et al. Intraindividual comparison of 123I-mIBG SPECT/MRI, 123I-mIBG SPECT/CT, and MRI for the detection of adrenal pheochromocytoma in patients with elevated urine or plasma catecholamines. Clin Nucl Med. 2013;38:e1–6. doi: 10.1097/RLU.0b013e318263923d. [DOI] [PubMed] [Google Scholar]
- 122.Ilias I, Meristoudis G. Intraindividual Comparison of 123I-mIBG SPECT/MRI, 123I-mIBG SPECT/CT, and MRI for the Detection of Adrenal Pheochromocytoma in Patients with Elevated Urine or Plasma Catecholamines. Clin Nucl Med. 2013;38:810. doi: 10.1097/RLU.0b013e31829f8e7d. [DOI] [PubMed] [Google Scholar]
- 123.Hartung-Knemeyer V, Rosenbaum-Krumme S, Buchbender C, et al. Malignant pheochromocytoma imaging with [124I]mIBG PET/MR. J Clin Endocrinol Metab. 2012;97:3833–3834. doi: 10.1210/jc.2012-1958. [DOI] [PubMed] [Google Scholar]
- 124.Maurea S, Cuocolo A, Imbriaco M, et al. Imaging characterization of benign and malignant pheochromocytoma or paraganglioma: comparison between MIBG uptake and MR signal intensity ratio. Ann Nucl Med. 2012;26:670–675. doi: 10.1007/s12149-012-0624-1. [DOI] [PubMed] [Google Scholar]
- 125.Fonte JS, Robles JF, Chen CC, et al. False-negative 123I-MIBG SPECT is most commonly found in SDHB-related pheochromocytoma or paraganglioma with high frequency to develop metastatic disease. Endocr Relat Cancer. 2012;19:83–93. doi: 10.1530/ERC-11-0243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Apeldoorn L, Voerman HJ, Hoefnagel CA. Interference of MIBG uptake by medication: a case report. Neth J Med. 1995;46:239–243. doi: 10.1016/0300-2977(94)00098-0. [DOI] [PubMed] [Google Scholar]
- 127.Saad FFA, Kroiss A, Ahmad Z, et al. Localization and prediction of malignant potential in recurrent pheochromocytoma/paraganglioma (PCC/PGL) using 18F-FDG PET/CT. Acta Radiol. 2013 doi: 10.1177/0284185113504330. [DOI] [PubMed] [Google Scholar]
- 128.Timmers HJLM, Chen CC, Carrasquillo JA, et al. Staging and functional characterization of pheochromocytoma and paraganglioma by 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography. J Natl Cancer Inst. 2012;104:700–708. doi: 10.1093/jnci/djs188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zelinka T, Timmers HJLM, Kozupa A, et al. Role of positron emission tomography and bone scintigraphy in the evaluation of bone involvement in metastatic pheochromocytoma and paraganglioma: specific implications for succinate dehydrogenase enzyme subunit B gene mutations. Endocr Relat Cancer. 2008;15:311–323. doi: 10.1677/ERC-07-0217. [DOI] [PubMed] [Google Scholar]
- 130.Imani F, Agopian VG, Auerbach MS, et al. 18F-FDOPA PET and PET/CT accurately localize pheochromocytomas. J Nucl Med. 2009;50:513–519. doi: 10.2967/jnumed.108.058396. [DOI] [PubMed] [Google Scholar]
- 131.Timmers HJLM, Hadi M, Carrasquillo JA, et al. The effects of carbidopa on uptake of 6-18F-Fluoro-L-DOPA in PET of pheochromocytoma and extraadrenal abdominal paraganglioma. J Nucl Med. 2007;48:1599–1606. doi: 10.2967/jnumed.107.042721. [DOI] [PubMed] [Google Scholar]
- 132.King KS, Chen CC, Alexopoulos DK, et al. Functional imaging of SDHx-related head and neck paragangliomas: comparison of 18F-fluorodihydroxyphenylalanine, 18F-fluorodopamine, 18F-fluoro-2-deoxy-D-glucose PET, 123I- metaiodobenzylguanidine scintigraphy, and 111In-pentetreotide scintigraphy. J Clin Endocrinol Metab. 2011;96:2779–2785. doi: 10.1210/jc.2011-0333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Marzola MC, Chondrogiannis S, Grassetto G, et al. 18F-DOPA PET/CT in the Evaluation of Hereditary SDH-Deficiency Paraganglioma-Pheochromocytoma Syndromes. Clin Nucl Med. 2013 doi: 10.1097/RLU.0b013e31829aface. [DOI] [PubMed] [Google Scholar]
- 134.Miederer M, Fottner C, Rossmann H, et al. High incidence of extraadrenal paraganglioma in families with SDHx syndromes detected by functional imaging with [18F]fluorodihydroxyphenylalanine PET. Eur J Nucl Med Mol Imaging. 2013;40:889–896. doi: 10.1007/s00259-013-2346-6. [DOI] [PubMed] [Google Scholar]
- 135.Gabriel S, Blanchet EM, Sebag F, et al. Functional characterization of nonmetastatic paraganglioma and pheochromocytoma by (18) F-FDOPA PET: focus on missed lesions. Clin Endocrinol (Oxf) 2013;79:170–177. doi: 10.1111/cen.12126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ilias I, Chen CC, Carrasquillo JA, et al. Comparison of 6-18F-fluorodopamine PET with 123I-metaiodobenzylguanidine and 111in-pentetreotide scintigraphy in localization of nonmetastatic and metastatic pheochromocytoma. J Nucl Med. 2008;49:1613–1619. doi: 10.2967/jnumed.108.052373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Pacak K, Eisenhofer G, Carrasquillo JA, et al. 6-[18F]fluorodopamine positron emission tomographic (PET) scanning for diagnostic localization of pheochromocytoma. Hypertension. 2001;38:6–8. doi: 10.1161/01.hyp.38.1.6. [DOI] [PubMed] [Google Scholar]
- 138.Timmers HJLM, Chen CC, Carrasquillo JA, et al. Comparison of 18F-fluoro-L-DOPA, 18F-fluoro-deoxyglucose, and 18F-fluorodopamine PET and 123I-MIBG scintigraphy in the localization of pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. 2009;94:4757–4767. doi: 10.1210/jc.2009-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Timmers HJLM, Eisenhofer G, Carrasquillo JA, et al. Use of 6-[18F]-fluorodopamine positron emission tomography (PET) as first-line investigation for the diagnosis and localization of non-metastatic and metastatic phaeochromocytoma (PHEO) Clin Endocrinol (Oxf) 2009;71:11–17. doi: 10.1111/j.1365-2265.2008.03496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Blanchet EM, Martucci V, Millo C, et al. Multitracer PET imaging of bone metastases from paraganglioma: peripheral halo of uptake on (18)F-FLT PET mismatching with central uptake of (18)F-FDOPA, (18)F-fluorodopamine, and (18)F-FDG. Eur J Nucl Med Mol Imaging. 2013 doi: 10.1007/s00259-013-2507-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Fanti S, Ambrosini V, Tomassetti P, et al. Evaluation of unusual neuroendocrine tumours by means of 68Ga-DOTA-NOC PET. Biomed Pharmacother. 2008;62:667–671. doi: 10.1016/j.biopha.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 142.Kroiss A, Putzer D, Uprimny C, et al. Functional imaging in phaeochromocytoma and neuroblastoma with 68Ga-DOTA-Tyr 3-octreotide positron emission tomography and 123I-metaiodobenzylguanidine. Eur J Nucl Med Mol Imaging. 2011;38:865–873. doi: 10.1007/s00259-010-1720-x. [DOI] [PubMed] [Google Scholar]
- 143.Kroiss A, Putzer D, Decristoforo C, et al. 68Ga-DOTA-TOC uptake in neuroendocrine tumour and healthy tissue: differentiation of physiological uptake and pathological processes in PET/CT. Eur J Nucl Med Mol Imaging. 2013;40:514–523. doi: 10.1007/s00259-012-2309-3. [DOI] [PubMed] [Google Scholar]
- 144.Kroiss A, Putzer D, Frech A, et al. A retrospective comparison between (68)Ga-DOTA-TOC PET/CT and (18)F-DOPA PET/CT in patients with extra-adrenal paraganglioma. Eur J Nucl Med Mol Imaging. 2013 doi: 10.1007/s00259-013-2548-y. [DOI] [PubMed] [Google Scholar]
- 145.Maurice JB, Troke R, Win Z, et al. A comparison of the performance of 68Ga-DOTATATE PET/CT and 123I-MIBG SPECT in the diagnosis and follow-up of phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging. 2012;39:1266–1270. doi: 10.1007/s00259-012-2119-7. [DOI] [PubMed] [Google Scholar]
- 146.Naji M, Zhao C, Welsh SJ, et al. 68Ga-DOTA-TATE PET vs. 123I-MIBG in identifying malignant neural crest tumours. Mol Imaging Biol. 2011;13:769–775. doi: 10.1007/s11307-010-0396-8. [DOI] [PubMed] [Google Scholar]
- 147.Naswa N, Sharma P, Nazar AH, et al. Prospective evaluation of 68Ga-DOTA-NOC PET-CT in phaeochromocytoma and paraganglioma: preliminary results from a single centre study. Eur Radiol. 2012;22:710–719. doi: 10.1007/s00330-011-2289-x. [DOI] [PubMed] [Google Scholar]
- 148.Naswa N, Sharma P, Soundararajan R, et al. Preoperative characterization of indeterminate large adrenal masses with dual tracer PET-CT using fluorine-18 fluorodeoxyglucose and gallium-68-DOTANOC: initial results. Diagn Interv Radiol. 2013;19:294–298. doi: 10.5152/dir.2013.048. [DOI] [PubMed] [Google Scholar]
- 149.Sharma P, Thakar A, Suman KCS, et al. 68Ga-DOTANOC PET/CT for baseline evaluation of patients with head and neck paraganglioma. J Nucl Med. 2013;54:841–847. doi: 10.2967/jnumed.112.115485. [DOI] [PubMed] [Google Scholar]
- 150.Win Z, Al-Nahhas A, Towey D, et al. 68Ga-DOTATATE PET in neuroectodermal tumours: first experience. Nucl Med Commun. 2007;28:359–363. doi: 10.1097/MNM.0b013e32808ea0b0. [DOI] [PubMed] [Google Scholar]
- 151.Mundschenk J, Unger N, Schulz S, et al. Somatostatin receptor subtypes in human pheochromocytoma: subcellular expression pattern and functional relevance for octreotide scintigraphy. J Clin Endocrinol Metab. 2003;88:5150–5157. doi: 10.1210/jc.2003-030262. [DOI] [PubMed] [Google Scholar]
- 152.Gimenez-Roqueplo AP, Caumont-Prim A, Houzard C, et al. Imaging work-up for screening of paraganglioma and pheochromocytoma in SDHx mutation carriers: a multicenter prospective study from the PGL. EVA Investigators J Clin Endocrinol Metab. 2013;98:E162–173. doi: 10.1210/jc.2012-2975. [DOI] [PubMed] [Google Scholar]
- 153.Jasperson KW, Kohlmann W, Gammon A, et al. Role of rapid sequence whole-body MRI screening in SDH-associated hereditary paraganglioma families. Fam Cancer. 2013 doi: 10.1007/s10689-013-9639-6. [DOI] [PubMed] [Google Scholar]
- 154.Thompson LDR. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol. 2002;26:551–566. doi: 10.1097/00000478-200205000-00002. [DOI] [PubMed] [Google Scholar]
- 155.Agarwal A, Mehrotra PK, Jain M, et al. Size of the tumor and pheochromocytoma of the adrenal gland scaled score (PASS): can they predict malignancy? World J Surg. 2010;34:3022–3028. doi: 10.1007/s00268-010-0744-5. [DOI] [PubMed] [Google Scholar]
- 156.de Wailly P, Oragano L, Radé F, et al. Malignant pheochromocytoma: new malignancy criteria. Langenbecks Arch Surg. 2012;397:239–246. doi: 10.1007/s00423-011-0850-3. [DOI] [PubMed] [Google Scholar]
- 157.Tavangar SM, Shojaee A, Moradi Tabriz H, et al. Immunohistochemical expression of Ki67, c-erbB-2, and c-kit antigens in benign and malignant pheochromocytoma. Pathol Res Pract. 2010;206:305–309. doi: 10.1016/j.prp.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 158.Lin M, Wong V, Yap J, et al. FDG PET in the evaluation of phaeochromocytoma: a correlative study with MIBG scintigraphy and Ki-67 proliferative index. Clin Imaging. 2013 doi: 10.1016/j.clinimag.2013.07.011. [DOI] [PubMed] [Google Scholar]
- 159.Brouwers FM, Elkahloun AG, Munson PJ, et al. Gene expression profiling of benign and malignant pheochromocytoma. Ann NY Acad Sci. 2006;1073:541–556. doi: 10.1196/annals.1353.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Xu Y, Qi Y, Rui W, et al. Expression and diagnostic relevance of heat shock protein 90 and signal transducer and activator of transcription 3 in malignant pheochromocytoma. J Clin Pathol. 2013;66:286–290. doi: 10.1136/jclinpath-2012-201134. [DOI] [PubMed] [Google Scholar]
- 161.Boltze C, Mundschenk J, Unger N, et al. Expression profile of the telomeric complex discriminates between benign and malignant pheochromocytoma. J Clin Endocrinol Metab. 2003;88:4280–4286. doi: 10.1210/jc.2002-021299. [DOI] [PubMed] [Google Scholar]
- 162.Favier J, Plouin PF, Corvol P, et al. Angiogenesis and vascular architecture in pheochromocytomas: distinctive traits in malignant tumors. Am J Pathol. 2002;161:1235–1246. doi: 10.1016/S0002-9440(10)64400-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Salmenkivi K, Heikkilä P, Liu J, et al. VEGF in 105 pheochromocytomas: enhanced expression correlates with malignant outcome. APMIS Acta Pathol Microbiol Immunol. 2003;111:458–464. doi: 10.1034/j.1600-0463.2003.1110402.x. [DOI] [PubMed] [Google Scholar]
- 164.Lee TK, Murthy SRK, Cawley NX, et al. An N-terminal truncated carboxypeptidase E splice isoform induces tumor growth and is a biomarker for predicting future metastasis in human cancers. J Clin Invest. 2011;121:880–892. doi: 10.1172/JCI40433. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 165.Amar L, Baudin E, Burnichon N, et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab. 2007;92:3822–3828. doi: 10.1210/jc.2007-0709. [DOI] [PubMed] [Google Scholar]
- 166.Plouin PF, Fitzgerald P, Rich T, et al. Metastatic pheochromocytoma and paraganglioma: focus on therapeutics. Horm Metab Res. 2012;44:390–399. doi: 10.1055/s-0031-1299707. [DOI] [PubMed] [Google Scholar]
- 167.Ayala-Ramirez M, Feng L, Johnson MM, et al. Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: primary tumor size and primary tumor location as prognostic indicators. J Clin Endocrinol Metab. 2011;96:717–725. doi: 10.1210/jc.2010-1946. [DOI] [PubMed] [Google Scholar]
- 168.Park J, Song C, Park M, et al. Predictive characteristics of malignant pheochromocytoma. Korean J Urol. 2011;52:241–246. doi: 10.4111/kju.2011.52.4.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zelinka T, Musil Z, Duskova J, et al. Metastatic pheochromocytoma: does the size and age matter? Eur J Clin Invest. 2011;41:1121–1128. doi: 10.1111/j.1365-2362.2011.02518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Goffredo P, Sosa JA, Roman SA. Malignant pheochromocytoma and paraganglioma: a population level analysis of long-term survival over two decades. J Surg Oncol. 2013;107:659–664. doi: 10.1002/jso.23297. [DOI] [PubMed] [Google Scholar]
- 171.Jimenez C, Rohren E, Habra MA, et al. Current and future treatments for malignant pheochromocytoma and sympathetic paraganglioma. Curr Oncol Rep. 2013;15:356–371. doi: 10.1007/s11912-013-0320-x. [DOI] [PubMed] [Google Scholar]
- 172.van Hulsteijn LT, Louisse A, Havekes B, et al. Quality of life is decreased in patients with paragangliomas. Eur J Endocrinol. 2013;168:689–697. doi: 10.1530/EJE-12-0968. [DOI] [PubMed] [Google Scholar]
- 173.Ayala-Ramirez M, Palmer JL, Hofmann MC, et al. Bone metastases and skeletal-related events in patients with malignant pheochromocytoma and sympathetic paraganglioma. J Clin Endocrinol Metab. 2013;98:1492–1497. doi: 10.1210/jc.2012-4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Adjallé R, Plouin PF, Pacak K, et al. Treatment of malignant pheochromocytoma. Horm Metab Res. 2009;41:687–696. doi: 10.1055/s-0029-1231025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hescot S, Leboulleux S, Amar L, et al. One-Year Progression-Free Survival of Therapy-Naive Patients With Malignant Pheochromocytoma and Paraganglioma. J Clin Endocrinol Metab. 2013;98:4006–4012. doi: 10.1210/jc.2013-1907. [DOI] [PubMed] [Google Scholar]
- 176.Conzo G, Musella M, Corcione F, et al. Laparoscopic adrenalectomy, a safe procedure for pheochromocytoma. A retrospective review of clinical series. Int J Surg. 2013;11:152–156. doi: 10.1016/j.ijsu.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 177.Goldstein RE, O'Neill JA, Holcomb GW, et al. Clinical experience over 48 years with pheochromocytoma. Ann Surg. 1999;229:755–764. doi: 10.1097/00000658-199906000-00001. discussion 764–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Cheah WK, Clark OH, Horn JK, et al. Laparoscopic Adrenalectomy for Pheochromocytoma. World J Surg. 2002;26:1048–1051. doi: 10.1007/s00268-002-6669-x. [DOI] [PubMed] [Google Scholar]
- 179.Goers TA, Abdo M, Moley JF, et al. Outcomes of resection of extra-adrenal pheochromocytomas/paragangliomas in the laparoscopic era: a comparison with adrenal pheochromocytoma. Surg Endosc. 2013;27:428–433. doi: 10.1007/s00464-012-2451-9. [DOI] [PubMed] [Google Scholar]
- 180.Henry JF, Defechereux T, Raffaelli M, et al. Complications of Laparoscopic Adrenalectomy: Results of 169 Consecutive Procedures. World J Surg. 2000;24:1342–1346. doi: 10.1007/s002680010222. [DOI] [PubMed] [Google Scholar]
- 181.Hwang JJ, Shoaf G, Uchio EM, et al. Laparoscopic Management of Extra-Adrenal Pheochromocytoma. J Urol. 2004;171:72–76. doi: 10.1097/01.ju.0000102081.46348.a4. [DOI] [PubMed] [Google Scholar]
- 182.Janetschek G, Finkenstedt G, Gasser R, et al. Laparascopic Surgery for Pheochromocytoma: Adrenalectomy, Partial Resection, Excision of Paragangliomas. J Urol. 1998;160:330–334. doi: 10.1016/s0022-5347(01)62886-6. [DOI] [PubMed] [Google Scholar]
- 183.Sprung J, O'Hara JF, Gill IS, et al. Anesthetic aspects of laparoscopic and open adrenalectomy for pheochromocytoma. Urology. 2000;55:339–343. doi: 10.1016/s0090-4295(99)00466-5. [DOI] [PubMed] [Google Scholar]
- 184.Vargas HI, Kavoussi LR, Bartlett DL, et al. Laparoscopic adrenalectomy: A new standard of care. Urology. 1997;49:673–678. doi: 10.1016/s0090-4295(97)00083-6. [DOI] [PubMed] [Google Scholar]
- 185.Walz MK, Peitgen K, Neumann HPH, et al. Endoscopic Treatment of Solitary, Bilateral, Multiple, and Recurrent Pheochromocytomas and Paragangliomas. World J Surg. 2002;26:1005–1012. doi: 10.1007/s00268-002-6632-x. [DOI] [PubMed] [Google Scholar]
- 186.Aliyev S, Karabulut K, Agcaoglu O, et al. Robotic Versus Laparoscopic Adrenalectomy for Pheochromocytoma. Ann Surg Oncol. 2013 doi: 10.1245/s10434-013-3134-z. [DOI] [PubMed] [Google Scholar]
- 187.Brauckhoff M, Gimm O, Brauckhoff K, et al. Repeat Adrenocortical-Sparing Adrenalectomy for Recurrent Hereditary Pheochromocytoma. Surg Today. 2004;34:251–255. doi: 10.1007/s00595-003-2690-4. [DOI] [PubMed] [Google Scholar]
- 188.Fallon SC, Feig D, Lopez ME, et al. The utility of cortical-sparing adrenalectomy in pheochromocytomas associated with genetic syndromes. J Pediatr Surg. 2013;48:1422–1425. doi: 10.1016/j.jpedsurg.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 189.Grubbs EG, Rich TA, Ng C, et al. Long-term outcomes of surgical treatment for hereditary pheochromocytoma. J Am Coll Surg. 2013;216:280–289. doi: 10.1016/j.jamcollsurg.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 190.Neumann HPH, Reincke M, Bender BU, et al. Preserved Adrenocortical Function After Laparoscopic Bilateral Adrenal Sparing Surgery for Hereditary Pheochromocytoma. J Clin Endocrinol Metab. 1999;84:2608–2610. doi: 10.1210/jcem.84.8.5872. [DOI] [PubMed] [Google Scholar]
- 191.Volkin D, Yerram N, Ahmed F, et al. Partial adrenalectomy minimizes the need for long-term hormone replacement in pediatric patients with pheochromocytoma and von Hippel-Lindau syndrome. J Pediatr Surg. 2012;47:2077–2082. doi: 10.1016/j.jpedsurg.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Walther MM, Herring J, Choyke PL, et al. Laparascopic Partial Adrenalectomy in Patients With Hereditary Forms of Pheochromocytoma. J Urol. 2000;164:14–17. [PubMed] [Google Scholar]
- 193.Yip L, Lee JE, Shapiro SE, et al. Surgical management of hereditary pheochromocytoma. J Am Coll Surg. 2004;198:525–534. doi: 10.1016/j.jamcollsurg.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 194.Plouin PF, Chatellier G, Fofol I, et al. Tumor Recurrence and Hypertension Persistence After Successful Pheochromocytoma Operation. Hypertension. 1997;29:1133–1139. doi: 10.1161/01.hyp.29.5.1133. [DOI] [PubMed] [Google Scholar]
- 195.Ellis RJ, Patel D, Prodanov T, et al. Response after surgical resection of metastatic pheochromocytoma and paraganglioma: can postoperative biochemical remission be predicted? J Am Coll Surg. 2013;217:489–496. doi: 10.1016/j.jamcollsurg.2013.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Mamlouk MD, van Sonnenberg E, Stringfellow G, et al. Radiofrequency Ablation and Biopsy of Metastatic Pheochromocytoma: Emphasizing Safety Issues and Dangers. J Vasc Interv Radiol. 2009;20:670–673. doi: 10.1016/j.jvir.2009.01.031. [DOI] [PubMed] [Google Scholar]
- 197.McBride JF, Atwell TD, Charboneau WJ, et al. Minimally Invasive Treatment of Metastatic Pheochromocytoma and Paraganglioma: Efficacy and Safety of Radiofrequency Ablation and Cryoablation Therapy. J Vasc Interv Radiol. 2011;22:1263–1270. doi: 10.1016/j.jvir.2011.06.016. [DOI] [PubMed] [Google Scholar]
- 198.Pacak K, Fojo T, Goldstein DS, et al. Radiofrequency ablation: a novel approach for treatment of metastatic pheochromocytoma. J Natl Cancer Inst. 2001;93:648–649. doi: 10.1093/jnci/93.8.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Venkatesan AM, Locklin J, Lai EW, et al. Radiofrequency ablation of metastatic pheochromocytoma. J Vasc Interv Radiol. 2009;20:1483–1490. doi: 10.1016/j.jvir.2009.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Fishbein L, Bonner L, Torigian DA, et al. External beam radiation therapy (EBRT) for patients with malignant pheochromocytoma and non-head and -neck paraganglioma: combination with 131I-MIBG. Horm Metab Res. 2012;44:405–410. doi: 10.1055/s-0032-1308992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Chino JP, Sampson JH, Tucci DL, et al. Paraganglioma of the head and neck: long-term local control with radiotherapy. Am J Clin Oncol. 2009;32:304–307. doi: 10.1097/COC.0b013e318187dd94. [DOI] [PubMed] [Google Scholar]
- 202.Li G, Chang S, Adler JR, et al. Irradiation of glomus jugulare tumors: a historical perspective. Neurosurg Focus. 2007;23:E13. doi: 10.3171/FOC-07/12/E13. [DOI] [PubMed] [Google Scholar]
- 203.Lim M, Bower R, Nangiana JS, et al. Radiosurgery for glomus jugulare tumors. Technol Cancer Res Treat. 2007;6:419–423. doi: 10.1177/153303460700600507. [DOI] [PubMed] [Google Scholar]
- 204.Poznanovic SA, Cass SP, Kavanagh BD. Short-term tumor control and acute toxicity after stereotactic radiosurgery for glomus jugulare tumors. Otolaryngol Head Neck Surg. 2006;134:437–442. doi: 10.1016/j.otohns.2005.10.040. [DOI] [PubMed] [Google Scholar]
- 205.Wegner RE, Rodriguez KD, Heron DE, et al. Linac-based stereotactic body radiation therapy for treatment of glomus jugulare tumors. Radiother Oncol. 2010;97:395–398. doi: 10.1016/j.radonc.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 206.Carrasquillo JA, Pandit-Taskar N, Chen CC. Radionuclide therapy of adrenal tumors. J Surg Oncol. 2012;106:632–642. doi: 10.1002/jso.23196. [DOI] [PubMed] [Google Scholar]
- 207.Giammarile F, Chiti A, Lassmann M, et al. EANM procedure guidelines for 131I-meta-iodobenzylguanidine (131I-mIBG) therapy. Eur J Nucl Med Mol Imaging. 2008;35:1039–1047. doi: 10.1007/s00259-008-0715-3. [DOI] [PubMed] [Google Scholar]
- 208.Loh KC, Fitzgerald PA, Matthay KK, et al. The treatment of malignant pheochromocytoma with iodine-131 metaiodobenzylguanidine (131I-MIBG): a comprehensive review of 116 reported patients. J Endocrinol Invest. 1997;20:648–658. doi: 10.1007/BF03348026. [DOI] [PubMed] [Google Scholar]
- 209.Gonias S, Goldsby R, Matthay KK, et al. Phase II study of high-dose [131I]metaiodobenzylguanidine therapy for patients with metastatic pheochromocytoma and paraganglioma. J Clin Oncol. 2009;27:4162–4168. doi: 10.1200/JCO.2008.21.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Van Hulsteijn LT, Niemeijer ND, Dekkers OM, et al. (131) I-MIBG therapyfor Malignant Paraganglioma and Pheochromocytoma: Systematic Review and Meta-Analysis. Clin Endocrinol (Oxf) 2013 doi: 10.1111/cen.12341. [DOI] [PubMed] [Google Scholar]
- 211.Menda Y, O'Dorisio MS, Kao S, et al. Phase I trial of 90Y-DOTATOC therapy in children and young adults with refractory solid tumors that express somatostatin receptors. J Nucl Med. 2010;51:1524–1531. doi: 10.2967/jnumed.110.075226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Zovato S, Kumanova A, Dematte S, et al. Peptide receptor radionuclide therapy (PRRT) with 177Lu-DOTATATE in individuals with neck or mediastinal paraganglioma (PGL) Horm Metab Res. 2012;44:411–414. doi: 10.1055/s-0032-1311637. [DOI] [PubMed] [Google Scholar]
- 213.van Essen M, Krenning EP, Kooij PP, et al. Effects of therapy with [177Lu-DOTA0, Tyr3]octreotate in patients with paraganglioma, meningioma, small cell lung carcinoma, and melanoma. J Nucl Med. 2006;47:1599–1606. [PubMed] [Google Scholar]
- 214.Forrer F, Riedweg I, Maecke HR, et al. Radiolabeled DOTATOC in patients with advanced paraganglioma and pheochromocytoma. Q J Nucl Med Mol Imaging. 2008;52:334–340. [PubMed] [Google Scholar]
- 215.Averbuch SD, Steakley CS, Young RC, et al. Malignant pheochromocytoma: effective treatment with a combination of cyclophosphamide, vincristine, and dacarbazine. Ann Intern Med. 2008;109:267–273. doi: 10.7326/0003-4819-109-4-267. [DOI] [PubMed] [Google Scholar]
- 216.Ayala-Ramirez M, Feng L, Habra MA, et al. Clinical benefits of systemic chemotherapy for patients with metastatic pheochromocytomas or sympathetic extra-adrenal paragangliomas: insights from the largest single-institutional experience. Cancer. 2012;118:2804–2812. doi: 10.1002/cncr.26577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Tanabe A, Naruse M, Nomura K, et al. Combination chemotherapy with cyclophosphamide, vincristine, and dacarbazine in patients with malignant pheochromocytoma and paraganglioma. Horm Cancer. 2013;4:103–110. doi: 10.1007/s12672-013-0133-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Huang H, Abraham J, Hung E, et al. Treatment of malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: recommendation from a 22-year follow-up of 18 patients. Cancer. 2008;113:2020–2028. doi: 10.1002/cncr.23812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Feldman JM. Treatment of metastatic pheochromocytoma with streptozocin. Arch Intern Med. 1983;143:1799–1800. [PubMed] [Google Scholar]
- 220.Srimuninnimit V, Wampler GL. Case report of metastatic familial pheochromocytoma treated with cisplatin and 5-fluorouracil. Cancer Chemother Pharmacol. 1991;28:217–219. doi: 10.1007/BF00685513. [DOI] [PubMed] [Google Scholar]
- 221.Ekeblad S, Sundin A, Janson ET, et al. Temozolomide as monotherapy is effective in treatment of advanced malignant neuroendocrine tumors. Clin Cancer Res. 2007;13:2986–2991. doi: 10.1158/1078-0432.CCR-06-2053. [DOI] [PubMed] [Google Scholar]
- 222.Kulke MH, Stuart K, Enzinger PC, et al. Phase II study of temozolomide and thalidomide in patients with metastatic neuroendocrine tumors. J Clin Oncol. 2006;24:401–406. doi: 10.1200/JCO.2005.03.6046. [DOI] [PubMed] [Google Scholar]
- 223.Saito Y, Tanaka Y, Aita Y, et al. Sunitinib induces apoptosis in pheochromocytoma tumor cells by inhibiting VEGFR2/Akt/mTOR/S6K1 pathways through modulation of Bcl-2 and BAD. Am J Physiol Endocrinol Metab. 2012;302:E615–625. doi: 10.1152/ajpendo.00035.2011. [DOI] [PubMed] [Google Scholar]
- 224.Aita Y, Ishii K, Saito Y, et al. Sunitinib inhibits catecholamine synthesis and secretion in pheochromocytoma tumor cells by blocking VEGF receptor 2 via PLC-γ-related pathways. Am J Physiol Endocrinol Metab. 2012;303:E1006–1014. doi: 10.1152/ajpendo.00156.2012. [DOI] [PubMed] [Google Scholar]
- 225.Ayala-Ramirez M, Chougnet CN, Habra MA, et al. Treatment with sunitinib for patients with progressive metastatic pheochromocytomas and sympathetic paragangliomas. J Clin Endocrinol Metab. 2012;97:4040–4050. doi: 10.1210/jc.2012-2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Jimenez C, Cabanillas ME, Santarpia L, et al. Use of the tyrosine kinase inhibitor sunitinib in a patient with von Hippel-Lindau disease: targeting angiogenic factors in pheochromocytoma and other von Hippel-Lindau disease-related tumors. J Clin Endocrinol Metab. 2009;94:386–391. doi: 10.1210/jc.2008-1972. [DOI] [PubMed] [Google Scholar]
- 227.Joshua AM, Ezzat S, Asa SL, et al. Rationale and evidence for sunitinib in the treatment of malignant paraganglioma/pheochromocytoma. J Clin Endocrinol Metab. 2009;94:5–9. doi: 10.1210/jc.2008-1836. [DOI] [PubMed] [Google Scholar]
- 228.Prochilo T, Savelli G, Bertocchi P, et al. Targeting VEGF-VEGFR Pathway by Sunitinib in Peripheral Primitive Neuroectodermal Tumor, Paraganglioma and Epithelioid Hemangioendothelioma: Three Case Reports. Case Reports Oncol. 2013;6:90–97. doi: 10.1159/000348429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Druce MR, Kaltsas GA, Fraenkel M, et al. Novel and evolving therapies in the treatment of malignant phaeochromocytoma: experience with the mTOR inhibitor everolimus (RAD001) Horm Metab Res. 2009;41:697–702. doi: 10.1055/s-0029-1220687. [DOI] [PubMed] [Google Scholar]
- 230.Oh DY, Kim TW, Park YS, et al. Phase 2 study of everolimus monotherapy in patients with nonfunctioning neuroendocrine tumors or pheochromocytomas/paragangliomas. Cancer. 2012;118:6162–6170. doi: 10.1002/cncr.27675. [DOI] [PubMed] [Google Scholar]
- 231.Matro J, Giubellino A, Pacak K. Current and future therapeutic approaches for metastatic pheochromocytoma and paraganglioma: focus on SDHB tumors. Horm Metab Res. 2013;45:147–153. doi: 10.1055/s-0032-1331211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Giubellino A, Bullova P, Nolting S, et al. Combined inhibition of mTORC1 and mTORC2 signaling pathways is a promising therapeutic option in inhibiting pheochromocytoma tumor growth: in vitro and in vivo studies in female athymic nude mice. Endocrinology. 2013;154:646–655. doi: 10.1210/en.2012-1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Jhaveri K, Taldone T, Modi S, et al. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta. 2012;1823:742–755. doi: 10.1016/j.bbamcr.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Giubellino A, Sourbier C, Lee MJ, et al. Targeting heat shock protein 90 for the treatment of malignant pheochromocytoma. PloS One. 2013;8:e56083. doi: 10.1371/journal.pone.0056083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Zhang C, Xu Y, Chen D, et al. Effect of HSP90 inhibitor in pheochromocytoma PC12 cells: an experimental investigation. Tumour Biol. 2013 doi: 10.1007/s13277-013-0996-4. [DOI] [PubMed] [Google Scholar]
- 236.Fernández MC, Venara M, Nowicki S, et al. Igf-I regulates pheochromocytoma cell proliferation and survival in vitro and in vivo. Endocrinology. 2012;153:3724–3734. doi: 10.1210/en.2012-1107. [DOI] [PubMed] [Google Scholar]
- 237.Nölting S, Garcia E, Alusi G, et al. Combined blockade of signalling pathways shows marked anti-tumour potential in phaeochromocytoma cell lines. J Mol Endocrinol. 2012;49:79–96. doi: 10.1530/JME-12-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Lai EW, Rodriguez OC, Aventian M, et al. ErbB-2 induces bilateral adrenal pheochromocytoma formation in mice. Cell Cycle. 2007;6:1946–1950. doi: 10.4161/cc.6.15.4521. [DOI] [PubMed] [Google Scholar]
- 239.Yuan W, Wang W, Cui B, et al. Overexpression of ERBB-2 was more frequently detected in malignant than benign pheochromocytomas by multiplex ligation-dependent probe amplification and immunohistochemistry. Endocr Relat Cancer. 2008;15:343–350. doi: 10.1677/ERC-07-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Li M, Kong ZM, Liu ZL. Antioxidant enzyme activities and lipid peroxidation induced by eicosapentaenoic acid (EPA) in PC12 cells. Cell Biol Toxicol. 2006;22:331–337. doi: 10.1007/s10565-006-0060-x. [DOI] [PubMed] [Google Scholar]
- 241.Martiniova L, Perera SM, Brouwers FM, et al. Increased uptake of [123I]meta-iodobenzylguanidine, [18F]fluorodopamine, and [3H]norepinephrine in mouse pheochromocytoma cells and tumors after treatment with the histone deacetylase inhibitors. Endocr Relat Cancer. 2011;18:143–157. doi: 10.1677/ERC-10-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Pacak K, Sirova M, Giubellino A, et al. NF-κB inhibition significantly upregulates the norepinephrine transporter system, causes apoptosis in pheochromocytoma cell lines and prevents metastasis in an animal model. Int J Cancer. 2012;131:2445–2455. doi: 10.1002/ijc.27524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Sordet O, Goldman A, Redon C, et al. Topoisomerase I requirement for death receptor-induced apoptotic nuclear fission. J Biol Chem. 2008;283:23200–23208. doi: 10.1074/jbc.M801146200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Ogi C, Aruga A. Immunological monitoring of anticancer vaccines in clinical trials. Oncoimmunology. 2013;2:e26012. doi: 10.4161/onci.26012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Papewalis C, Kouatchoua C, Ehlers M, et al. Chromogranin A as potential target for immunotherapy of malignant pheochromocytoma. Mol Cell Endocrinol. 2011;335:69–77. doi: 10.1016/j.mce.2010.05.021. [DOI] [PubMed] [Google Scholar]
- 246.Madsen MT, Bushnell DL, Juweid ME, et al. Potential increased tumor-dose delivery with combined 131I-MIBG and 90Y-DOTATOC treatment in neuroendocrine tumors: a theoretic model. J Nucl Med. 2006;47:660–667. [PubMed] [Google Scholar]