Behaviour of NMDA and AMPA receptor-mediated miniature EPSCs at rat cortical neuron synapses identified by calcium imaging (original) (raw)
Abstract
- Simultaneous recording of intracellular calcium concentration at a synapse and synaptic currents from the cell body allows mapping of miniature excitatory postsynaptic currents (mEPSCs) to single synapses.
- In the absence of extracellular Mg2+, 77% of synapses had mEPSCs with fast and slow components, attributed to AMPA- and NMDA-type glutamate receptors, respectively. The remainder of synapses (23%) had mEPSCs that lacked a fast component; these responses were attributed to NMDA receptors.
- A strong positive correlation between the amplitude of the calcium transient and the NMDA receptor-mediated mEPSC was observed, indicating that the mEPSCs originate from an identified synapse.
- At synapses that had both mEPSC components, the AMPA receptor component was positively correlated with charge influx mediated by NMDA receptors during repeated synaptic events. No periodic failure in the AMPA receptor mEPSC was observed at synapses expressing both receptor components.
- A significant positive correlation between the mean amplitudes of NMDA and AMPA receptor components of mEPSCs is observed across different synapses.
- We suggest that factors effecting both receptor classes, such as the amount of transmitter in synaptic vesicles, might contribute to the variation in mEPSC amplitude during repeated miniature events at a single synapse. Although the average postsynaptic response at different synapses can vary in amplitude, there appears to be a mechanism to keep the ratio of each receptor subtype within a narrow range.
Fast excitatory synaptic transmission in the central nervous system is primarily mediated by two major classes of ionotropic glutamate receptors, AMPA receptors (AMPARs) and NMDA receptors (NMDARs) (Bekkers & Stevens, 1989; Jonas & Spruston, 1994). It has been shown that both receptor subtypes can be co-localized at synapses, but at some synapses NMDARs dominate over AMPARs, and other synapses exist that lack AMPARs altogether (Bekkers & Stevens, 1989; Silver et al. 1992; Wu et al. 1996; Malenka & Nicoll, 1997; O'Brien et al. 1998; Petralia et al. 1999). Processes that regulate the distribution of functional AMPARs have been proposed as important regulators of activity-dependent synaptic modulation and development (Wu et al. 1996; Malenka & Nicoll, 1997; Kamboj & Huganir, 1998; O'Brien et al. 1998; Petralia et al. 1999). Regarding the mechanism of differential receptor distribution, it has been shown that anchoring proteins are required for the clustering of AMPARs and NMDARs (Ehlers et al. 1996; Muller et al. 1996; Dong et al. 1997; O'Brien et al. 1998).
A number of studies suggest that the amplitude of AMPAR-mediated mEPSCs varies during repeated events at single synapses (Liu & Tsien, 1995; Forti et al. 1997; Liu et al. 1999). If AMPARs are not saturated by glutamate from single synaptic vesicles, presynaptic factors, such as the amount of neurotransmitter in a synaptic vesicle, would contribute to the response amplitude variability (Liu & Tsien, 1995; Forti et al. 1997; Liu et al. 1999). In the case of NMDARs, as they have a high affinity for glutamate, it was suggested that they may be saturated by glutamate from single synaptic vesicles (Patneau & Mayer, 1990) but more recent evidence suggests that NMDARs are not saturated by the quantal transmitter release and that presynaptic factors are thus capable of controlling synaptic responses mediated by NMDARs (Min et al. 1998; Mainen et al. 1999).
The mechanism of quantal response variation has been intensely studied, since it may provide insight into how synaptic efficacy is changed during neuronal plasticity (Bekkers & Stevens, 1989; Liu & Tsien, 1995; Murphy et al. 1995; Forti et al. 1997; Gomperts et al. 1998). To determine the variability in synaptic events at single synapses and between synapses, it is necessary to map events to a single synapse because neurons receive inputs from multiple synapses. Calcium imaging techniques take advantage of the high Ca2+ permeability of NMDARs to identify synaptic events at a single synapse (Muller & Connor, 1991; Malinow et al. 1994; Murphy et al. 1994, 1995; Schiller et al. 1998). Here, we have recorded mEPSCs from identified synapses of cultured cortical neurons by simultaneously recording synaptic currents from the cell body and intracellular calcium concentration at the synapse (Murphy et al. 1994, 1995). At synapses that expressed both NMDAR and AMPAR components we found significant positive correlation between the amplitude of AMPAR- and NMDAR-mediated mEPSCs during repeated synaptic events. This correlation suggests that presynaptic factors could contribute to the variation in mEPSC amplitude. In addition, we found significant positive correlation between the synaptic responses mediated by AMPARs and NMDARs across synapses. It is suggested that synapses have regulatory mechanisms to maintain a relatively constant ratio of each receptor type.
METHODS
Primary cultures of cortical neurons were prepared as described previously (Murphy et al. 1995). Briefly, neurons and glia were dissociated from postnatal day 1 rat pups killed by decapitation under the guidelines of the Tohoku University animal care committee. The neurons were plated at a concentration of 105 per 35 mm dish, and maintained for at least 4 weeks to mature. Neurons were viewed with an inverted microscope equipped with a × 100 objective lens (Olympus, Hachioji Japan). Recordings were made in voltage clamp mode (_V_m = −65 mV) using the whole-cell patch clamp technique at room temperature by an Axopatch 200B amplifier (Axon Instruments). The calcium sensitive dyes Fluo-3 (1 mM) and Mag-fura-2 (0·5 mM, Molecular Probes) were included in the pipette solution. The pipette solution contained (mM): 130 KMeSO4, 4 NaCl, 20 Hepes, 2 MgCl2, 3 MgATP, 1 Fluo-3 and 0·5 Mag-fura-2 (adjusted to pH 7·2 with KOH), and the pipette resistance was ∼3 MΩ. The bathing solution for the recording contained (mM): 137 NaCl, 5 KCl, 10 Hepes, 4 CaCl2, 0·001 TTX and 0·01 bicuculline (adjusted to pH 7·4 with NaOH). Our medium was not supplemented with glycine. We have recorded NMDAR-mediated mEPSCs in the presence of glycine (10 μM), and found that the amplitude and decay time constants were not significantly different in the absence of glycine (P > 0·41 and P > 0·23, respectively, n = 6). Therefore, under the conditions used in this study it is likely that the glycine concentration in the synaptic cleft is high enough to effectively activate NMDARs. Currents were filtered at 2 kHz, sampled at 5 kHz by the Digidata 1200A A/D board (Axon Instruments) and Acquire software (Bruxton, Seattle, WA, USA) and stored on a PC. The access resistance was monitored throughout the experiment.
Fluorescence images were recorded using an intensified CCD camera (XRGenIII+; Solamere, Salt Lake City, UT, USA) at video rate (30 frames s−1), captured to a PC using a video frame grabber (XCIP; EPIX, Buffalo Grove), and stored on a hard disk. We used 380 nm light for excitation of Mag-fura-2, a low affinity [Ca2+]i probe. Excitation of Mag-fura-2 enabled us to identify the dendrites of the neuron and map their structure, since Fluo-3 fluorescence is low at the resting [Ca2+]i. Excitation of Fluo-3 by 490/25 nm light was used to measure [Ca2+]i. After the establishment of the whole-cell configuration, ∼15 min was required to allow the dyes to fill the dendrites. We examined several sites to find synapses on the dendrites with a reasonable frequency of synaptic activity. To avoid photobleaching of calcium sensitive dyes and cell damage, 15 s of optical recording was followed by at least a 30 s break. Analysis of baseline data indicated that little or no bleaching of Mag-fura-2 or Fluo-3 fluorescence occurred; in further support of this no time-dependent run-down of miniature postsynaptic calcium transient (MSCT) amplitude at single dendrite sites was observed. The mean image acquisition time for single synapses was 6 min (over a 20 min total recording period). Recordings were stopped if the basal fluorescence level of Fluo-3 increased by more than 20% of Mag-fura-2 fluorescence. Changes in [Ca2+]i were estimated by taking the ratio of fluorescence of Fluo-3 to that of Mag-fura-2. Images and currents were analysed off-line using programs written by the IDL (Research System, Boulder, CO, USA). The correlation coefficient (r), was quantified with Pearson's correlation test using Prism statistics software (GraphPad, San Diego, CA, USA).
To measure calcium transients, we placed a 7 × 5 pixel box on the dendrite (long axis is placed parallel to the dendrite; pixel size, 0·2 μm2) and the fluorescence intensity of Fluo-3 in the box was averaged and normalized to that of Mag-fura-2. The baseline was defined as the mean of six consecutive images and the onset of MSCTs was defined as the first point that exceeded 1 s.d. from the baseline. The amplitude of the calcium transient was measured as a peak of a smoothed fluorescence record (moving average of 5 points; 167 ms) within 500 ms after the onset of the MSCT. Our experiments indicate that the noise levels of fluorescence based recordings of MSCTs at single synapses are small ∼6% (c.v.) and therefore would contribute only about 3% to the observed variance in the MSCT.
RESULTS
In the whole-cell recording configuration, using a patch pipette containing calcium-sensitive dyes, we simultaneously recorded mEPSCs from the soma of a cortical neuron and miniature postsynaptic calcium transients (MSCTs) at synapses (Murphy et al. 1994, 1995). TTX was added to the bathing solution to suppress spontaneous excitation and to allow isolation of synaptic events induced by the spontaneous quantal transmitter release. Magnesium was eliminated from the bathing solution to promote calcium influx at negative holding potentials. Previous work has shown that the MSCT is largely mediated by calcium influx through NMDARs (Murphy et al. 1994, 1995). To isolate individual mEPSCs, we used low-density cultures to decrease the mEPSC frequency by reducing the number of synaptic inputs onto a single neuron (Fig. 1_A_ and B). Figure 1_A_ and D shows examples of how mEPSCs are mapped to an identified synapse. Changes in intracellular calcium concentration were estimated by normalizing changes in Fluo-3 fluorescence to Mag-fura-2 fluorescence (Fig. 1). The origin of calcium transients was identified as the earliest point of the onset on the line-scan image of the dendrite, and we regarded events that originated within a 1 μm range of a dendrite as events from the same synapse. MSCT onset was defined as a 1 s.d. increase in intracellular calcium concentration from the baseline that persisted for six measurements. As the sampling frequency of video images was 30 Hz, delays of up to 33 ms were expected when the onset of MSCTs and mEPSCs were compared (Figs 1_C_, 1_D_ and 2_A_). Events with multiple overlapping mEPSCs were eliminated from the analysis. The frequency of mEPSCs recorded from the soma was 6·9 ± 1·4 Hz (mean ±s.e.m.) (n = 14 neurons). The mean frequency of synaptic events at single synapses was 0·04 ± 0·01 Hz and the mean recording time was 327 ± 36 s (10 ± 1·6 mEPSCs at 36 synapses on 14 neurons).
Figure 1. Recording of mEPSCs from an identified synapse.
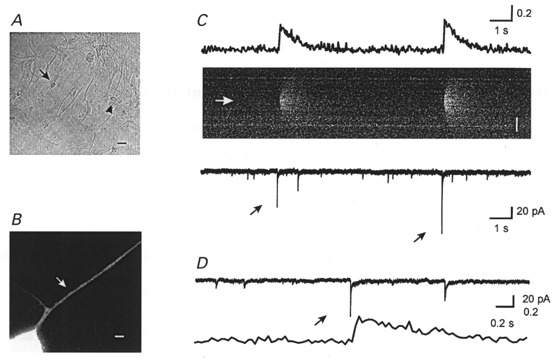
A, bright field image of neurons. The arrow indicates the neuron that was recorded from and the arrowhead indicates an adjacent neuron. Scale bar, 20 μm. B, higher power Mag-fura-2 image of the neuron of interest. The arrow indicates the putative synapse. Scale bar, 2 μm. C, time course of MSCTs (top trace) and mEPSCs (bottom trace). Two MSCTs were observed within the 15 s of data shown. The time course of intracellular calcium concentration is estimated by normalizing the fluorescence intensity of Fluo-3 to Mag-fura-2 fluorescence. The middle panel shows the line-scan image of Fluo-3 fluorescence in the dendrite. The white arrow indicates the synapse location. Scale bar, 5 μm. Black arrows indicate mEPSCs associated with the MSCT on the line-scan image. The first mEPSC, along with the associated MSCT, is shown on a higher time scale in D (top trace, mEPSCs; bottom trace, MSCTs).
Figure 2. mEPSCs identified at a single synapse by correlated calcium imaging.
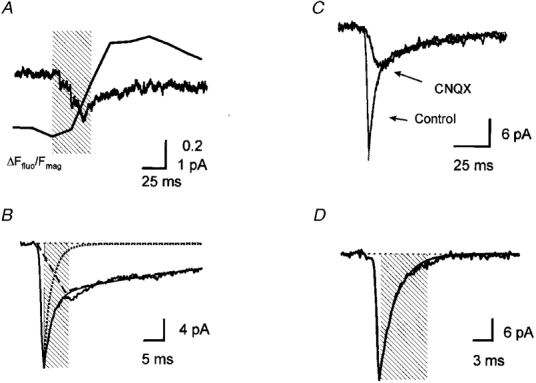
A, time course of the mean MSCT and mean mEPSC from 40 synaptic events. The mEPSCs were aligned by the first point of MSCTs (alignment subject to ± 33 ms timing jitter). The shaded area indicates a 60 ms period of two video frames at the MSCT onset and the previous frame. B, onset and decay time course of the mean mEPSC at an identified synapse. The decay phase of the mean mEPSC was fitted to a sum of two exponential components (thin line). The dotted line is the exponential decay at a time constant of 2·0 ms from the peak of the mean trace. The dashed line is the linear activation and the exponential decay of the slow component (time to peak, 7 ms; decay time constant, 53 ms). The shaded area indicates a 6 ms period following the peak that was excluded for the quantification of the amplitude of NMDAR component (measured between 6–56 ms). A and B are averages of recordings from the same neuron. C, time course of mEPSCs in the presence and absence of CNQX (5 μM). Each current trace is the mean of 14 mEPSCs. The decay time course of the current was fitted to a sum of two exponential components with time constants of 3·0 and 40 ms. D, time course of a mean AMPA receptor-mediated mEPSC in the presence of AP5 (50 μM). The current trace is the mean of 10 events. The decay time course of the current was fitted to a single exponential component with a time constant of 2·5 ms (continuous line). The shaded area indicates the 7·5 ms duration that was excluded from the measurement of the charge mediated by NMDARs.
The decay of mEPSCs that were coincident with MSCTs occurring at an identified synapse could be described by fast and slow components. Figure 2_A_ shows the time course of the mean mEPSC at an identified synapse. The decay phase of the mEPSC was described by a sum of two exponential components with time constants of 2·0 and 53·2 ms (Fig. 2_A_). The decay time constants of the fast and slow components were 1·8 ± 0·1 ms (28 synapses) and 51·0 ± 1·6 ms (36 synapses), respectively. It is likely that the fast and the slow components are mediated by AMPARs and NMDARs, respectively, because the time constants agree well with previous work in the hippocampal neurons and cerebellar granule cells (Silver et al. 1992; Spruston et al. 1995; Gomperts et al. 1998). Consistent with the work of others (Bekkers & Stevens, 1989; Silver et al. 1992; Gomperts et al. 1998), the fast component was reduced to non-detectable levels by CNQX (5 μM), a kainate and AMPAR antagonist (Wilding & Huettner, 1996) (Fig. 2_A_), while addition of an NMDAR antagonist, D-aminophosphonovalerate (AP5, 50 μM) blocked the slow component of the mEPSC (n = 5, Fig. 2_A_). No mEPSCs were detected in the presence of CNQX or AP5 (data not shown). The charge mediated by the NMDAR component was calculated by integrating 50 ms of the mEPSC record, beginning at a point 3 times the decay time constant of the fast component after the mEPSC peak (6 ms at the synapse shown in 2_B_). During this time period, the contribution of the fast component should be negligible (averages ∼1% over the 50 ms measurement period, Fig. 2_A_ and D). The mean charge mediated by the slow component was 0·28 ± 0·02 pC and the c.v. was 0·42 ± 0·05 at 36 synapses (Fig. 3_A_).
Figure 3. Behaviour of repeated mEPSCs at a single synapse.
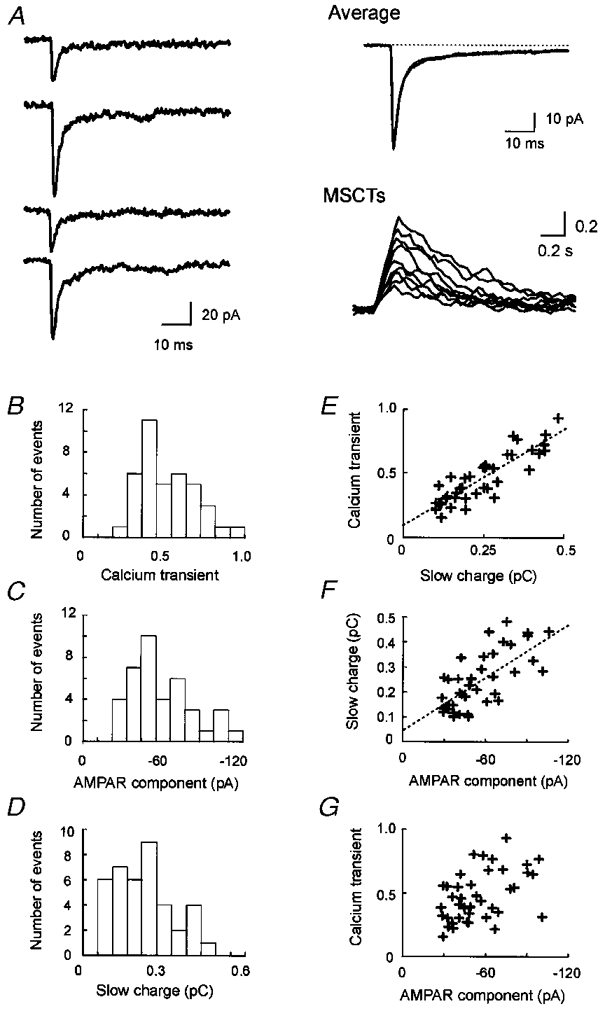
A, individual mEPSC traces and the mean mEPSC at a synapse identified by correlated calcium imaging. The individual traces are aligned at the onset. At this synapse, 40 mEPSCs were observed. The mean trace is fitted to sum of two exponential components with decay time constants of 2·3 and 52 ms (continuous line). The time course (moving average of 5 points) of nine superimposed MSCTs is shown in the right lower panel. B, amplitude histogram of MSCTs. C, amplitude histogram of the AMPAR components of mEPSCs. D, amplitude histogram of the charge mediated by the slow component, calculated as an integral of the current amplitude between 6·9 and 56·9 ms after the peak. E, scatter plot of charge influx mediated by the slow component versus the MSCT amplitude. F, scatter plot of the amplitude of the AMPAR component versus charge influx mediated by the slow component. G, scatter plot of the amplitude of the AMPAR component versus the MSCT amplitude. r values for the above relationships were 0·87, 0·70 and 0·51, respectively.
As in previous reports (Murphy et al. 1995), we found a strong correlation between the charge influx mediated by the slow component of mEPSCs and the MSCT amplitude at this synapse (Fig. 3_A_); the mean r value was 0·87 ± 0·02 at eighteen synapses (P < 10−12). This strong correlation indicates that we have recorded currents from a dendritic region of interest that is probably attributed to a single synapse. Using electron microscopic reconstruction we have shown that in the majority of cases (> 75%) MSCT signals were associated with a single synapse (Mackenzie et al. 1999).
In cultured hippocampal neurons, the amplitude of the AMPAR component of the mEPSC is positively correlated with the amplitude of NMDAR component recorded from the cell body (Gomperts et al. 1998). To determine whether the correlation is due to event-to-event variation of mEPSCs at single synapses, we examined the relationship between the amplitude of NMDAR and AMPAR components during repeated mEPSCs at nine synapses at which we observed more than nine mEPSCs. Synapses were considered to be AMPAR dominant if the fast component was at least twice the amplitude of the slow component and the rise time of the mean mEPSC was < 1·4 ms (Fig. 4_A_ and D). In our recordings, the mean rise time (from 20 to 80% of the peak) of mEPSCs at AMPAR dominant synapses was 0·71 ± 0·11 ms (n = 9) and that at NMDAR dominant synapses was 4·4 ± 0·4 ms (n = 8) (see below). To estimate the contribution of the NMDAR component activated at the peak of mEPSCs (predominantly AMPAR-mediated for dual component mEPSCs), we assumed that the time course of NMDAR activation is linear. As the decay of the slow component is described by a single exponential function (time constant, ∼50 ms), we could extrapolate the peak amplitude of the NMDAR component (Fig. 2_A_). Using this assumption the average current amplitude of the NMDAR component activated at the peak of the mEPSC would be ∼17% of the peak NMDAR component (Fig. 2_A_). This extrapolated peak amplitude was estimated to be 1·5 times the mean current amplitude during the 50 ms measurement period shown in Fig. 2_B_. Therefore, the amplitude of the NMDAR component activated at the peak of the mEPSC should be given by 0·17 × 1·5 of the mean amplitude during the 50 ms measurement period. For each of the mEPSCs, we corrected the amplitude of the AMPAR component (mEPSC peak amplitude) by subtracting the amplitude of the NMDAR component activated at the peak of the mEPSC. The amplitude of AMPAR component was −54·0 ± 3·2 pA (39 mEPSCs) and the c.v. was 0·37 at the synapse shown in Fig. 3, and the mean AMPAR component and c.v. at nine synapses were 42·6 ± 6·8 pA and 0·31 ± 0·03, respectively (Fig. 3_A_). At the synapse shown in Fig. 3, the AMPAR component amplitude was correlated to the charge influx mediated by the NMDAR component (r = 0·70, P < 10−6; Fig. 3_A_). We found a significant correlation between the NMDAR and the AMPAR components at all synapses examined (mean r = 0·69± 0·03, P < 0·05). To determine whether the correlation between the mEPSC mediated by AMPARs and the charge associated with the slow component was due to random factors, such as baseline variation, we examined baseline noise 50 ms before the onset of the fast mEPSC current. No significant correlation was observed between baseline noise and the peak mEPSC amplitude (r = 0·05± 0·02, n = 4).
Figure 4. Relationship between peak mEPSC amplitude and slow mEPSC amplitude across different synapses.
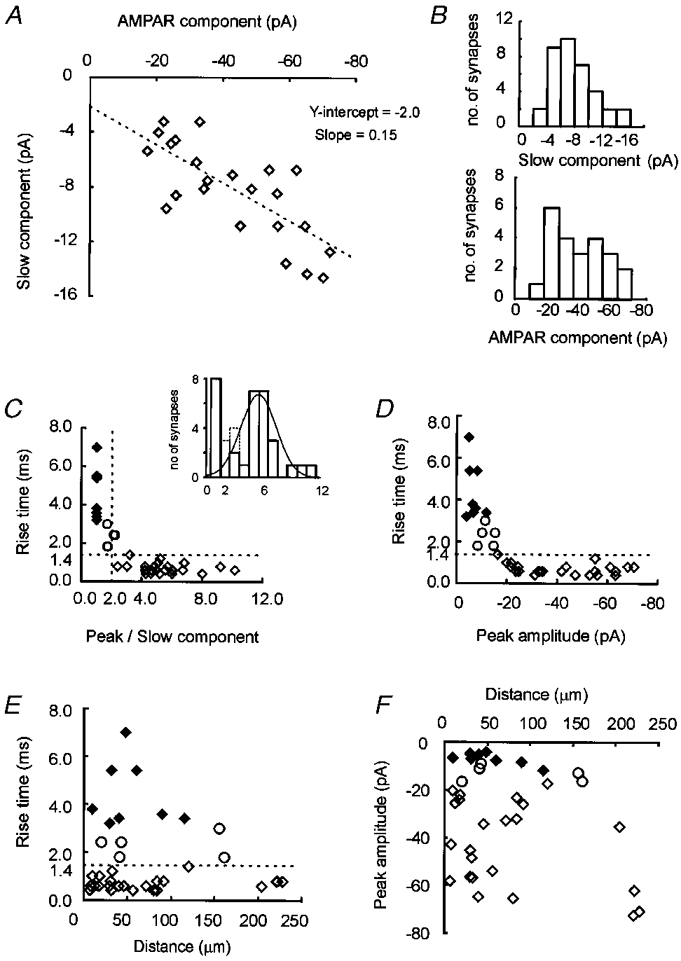
A, scatter plot of the amplitude of the AMPAR and NMDAR components of averaged mEPSCs at single synapses (r = 0·75, 23 synapses, 11 cells). B, amplitude histogram of the AMPAR and NMDAR components. C, scatter plot of the ratio of the fast component (peak) amplitude and the slow component amplitude versus the rise time. The dashed lines indicate the criteria of AMPAR dominant synapses; the rise time of 1·4 ms and a peak/slow ratio of two. The inset is a histogram of the ratios of the peak amplitude of mEPSCs to slow component amplitude. The dotted line shows the distribution of synapses that did not fall in the two groups. The smooth line shows the normal distribution of AMPAR dominant synapses calculated from the mean and the standard deviation. D, scatter plot of the peak amplitude of mEPSCs versus the rise time. The dashed line indicates a rise time of 1·4 ms. E, scatter plot of the distance of a synapse from the cell body versus the rise time. The dashed line indicates a rise time of 1·4 ms. F, scatter plot of the distance of the synapse from the cell body versus the peak amplitude of mEPSCs. ♦, NMDAR dominant synapses; ⋄, AMPAR dominant synapses; ○, synapses that did not fall within the two categories.
Recently, hypotheses concerning the mechanism of long term potentiation (LTP) and synaptic development have focused on the selective recruitment of AMPARs at synapses that contain only NMDARs (Isaac et al. 1995; Liao et al. 1995; Malenka & Nicoll, 1997; Petralia et al. 1999). An alternative explanation for the selective failure of AMPAR-mediated responses would be that some synaptic vesicles contain only enough glutamate to activate NMDARs but not AMPARs, since the affinity of NMDARs for glutamate is higher than that of AMPARs (Patneau & Mayer, 1990; Gomperts et al. 1998). We have tested this hypothesis by examining AMPAR-mediated mEPSCs at synapses identified by correlated calcium imaging. At nine synapses that expressed both components, we observed that no mEPSCs exhibited selective failures of the AMPAR component (see Fig. 3_A_ and D). Thus, it is unlikely that the failure of AMPAR-mediated EPSCs is due to underfilling of glutamate vesicles or inaccessibility of receptors.
In cultured hippocampal neurons, the amplitude of the AMPAR component of the mEPSC is positively correlated to the amplitude of the NMDAR component (Gomperts et al. 1998). The positive correlation suggests that each receptor subtype is distributed in the same relative proportions at synapses expressing both components. To determine if the correlation originates from the distribution of functional AMPARs and NMDARs at single synapses, the relative distribution of each receptor subtype was estimated from the mean mEPSCs observed at 23 synapses which displayed both components. The mean mEPSC at each synapse was fitted to the sum of two exponential functions and the amplitude of the NMDAR component was estimated from the slow component. We estimated the amplitude of the AMPAR component from the peak amplitude of mEPSCs that were corrected for contamination by the NMDAR component (see above). The mean amplitude of the AMPAR component was 41·9 ± 3·7 pA and that of the NMDAR component was 8·3 ± 0·73 pA at synapses that displayed both components (n = 23) (Fig. 4_A_). We found a significant positive correlation between AMPAR and NMDAR components across synapses (r = 0·75, P < 5·0× 10−5; Fig. 4_A_). Analysis of the peak to slow component ratios indicated that synapses with a rapid mEPSC rise time formed a unique group that also had relatively higher peak mEPSC amplitude (Fig. 4_A_). At synapses which displayed both components the mEPSC rise times were less than 1·4 ms (mean, 0·7 ± 0·06 ms) and were not inversely correlated with peak amplitude suggesting that the effect of dendritic filtering is small (_r_ = 0·003, _P_ > 0·15) (Fig. 4_D_; Bekkers & Stevens, 1996). In further support of the relatively minor effect of dendritic filtering, we found no significant correlation between the mEPSC rise time and the distance from the soma (r = 0·001, P > 0·5; mean distance, 73 ± 16 μm) (Fig. 4_A_), or between the peak mEPSC amplitude and the distance from the soma (r = 0·05, P > 0·10) (Fig. 4_A_). Also, no correlation was observed between the decay time constant of the fast component and the distance (r = 0·002, P > 0·25) (data not shown). The inability to readily explain these results by dendritic filtering suggests that there is a mechanism to keep the relative number of AMPARs and NMDARs at single synapses within a certain range.
At eight of 36 synapses, the decay of the mean mEPSC was described by a single exponential function with a slow time constant (49·8 ± 3·8 ms), similar to that of the slow component of synapses with biphasic kinetics (P > 0·7). The mean amplitude of mEPSCs at these synapses was −6·9 ± 0·9 pA and was not different from that at synapses with two components (P > 0·1). The rise time of mEPSCs at these synapses was significantly slower than that observed at synapses expressing mEPSCs with two components (4·4 ± 0·4 ms versus 0·7 ± 0·06 ms; P < 10−7) (Fig. 4_D, E_ and F). The lack of a rapidly rising and/or decaying component suggested that synapses with slow monophasic mEPSCs have little contribution from AMPARs (Fig. 4_A_ and 5_A_; Gomperts et al. 1998). On the other hand, the lack of a rapid rise time or decay time constant could be the result of other factors that include, the electrotonic filtering properties of the dendrite, the time course of transmitter in the synaptic cleft, and the deactivation of receptors (Jonas & Spruston, 1994; Bekkers & Stevens, 1996). It is unlikely that dendritic filtering selectively eliminates the fast components of these mEPSCs, since the distance between NMDAR dominant synapses (slow decay only) and the cell body (44 ± 9 μm) was not significantly different from that observed at synapses with fast and slow components (_P_ > 0·25) (Fig. 4_A_ and F). In addition, AMPAR-positive synapses were observed at sites (on the same dendrite) that were further from the soma than NMDAR dominant synapses (4 synapses on 4 neurons) (Fig. 5_A_). We also tested the contribution of AMPARs to the slow mEPSC pharmacologically. We found that CNQX (5 μM) was without effect on slow mEPSCs occurring at apparently NMDAR dominant synapses. This result indicated that neither AMPARs nor kainate receptors mediated the slow mEPSCs (P > 0·4 at four synapses on three neurons; Fig. 5_A_). The lack of CNQX effect also suggested that the slow mEPSCs were not attributed to AMPARs even if the clearance of glutamate from the synaptic cleft were delayed. Slow clearance of glutamate is also unlikely because the decay time constant of slow mEPSCs was longer than the time course of the desensitization of AMPARs (Jonas & Sakmann, 1992). Therefore, it is likely that the contribution of AMPARs to mEPSCs at NMDAR dominant synapses is small and the majority of the current is attributed to NMDARs.
Figure 5. NMDA receptor dominant synapse.
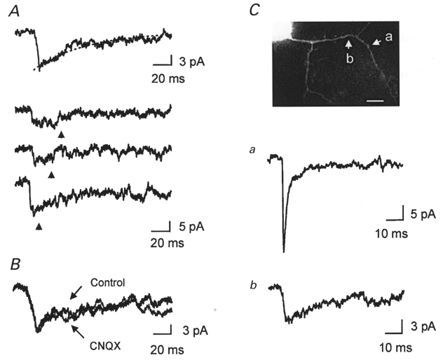
A, mEPSCs at NMDAR dominant synapses. The mean mEPSC (n = 15) from a synapse identified by correlated calcium imaging that lacked a fast component of the mEPSC. Top trace, the mean mEPSC was fitted to a single exponential function with a time constant of 45 ms. Bottom traces, three examples of individual mEPSCs (raw data). Arrows indicate the beginning of the video frame of MSCT onset. B, effect of CNQX (5 μM) on mEPSCs at the NMDAR dominant synapse. The mean mEPSCs in the absence and presence of CNQX are superimposed. The charges mediated by mEPSCs in the absence and presence of CNQX (5 μM) were 0·47 ± 0·06 pC (n = 15 in 225 s) and 0·45 ± 0·09 pC (n = 8 in 120 s) (P > 0·8), respectively. C, a NMDAR dominant synapse and a synapse with biphasic mEPSCs (both NMDAR and AMPAR components) on the same dendrite at a more distal site. Traces are the means of 11 and 6 mEPSCs, for a and b, respectively. Scale bar, 10 μm.3
DISCUSSION
We have recorded mEPSCs at identified synapses using simultaneous recording of electrical signals and intracellular calcium concentration at synapses. Using this approach we show a positive correlation between the AMPAR- and the NMDAR-mediated component of mEPSCs during repeated events at single synapses. This correlation suggests a contribution of presynaptic factors (explaining the effect on both receptor classes) in controlling variability of AMPAR mEPSCs. These results are consistent with the recent findings of Liu et al. (1999) in which local perfusion was used to elicit mEPSCs at single synapses. It is somewhat surprising that a presynaptic factor would contribute to the control of mEPSC amplitude mediated by NMDARs since NMDARs have high affinity for glutamate (Patneau & Mayer, 1990). However, in hippocampal neurons the lack of the saturation of NMDARs by the quantal transmitter release and an apparent presynaptic control of NMDAR-mediated responses is also observed (Min et al. 1998; Mainen et al. 1999). One possible explanation of the lack of saturation would be that glutamate released from single synaptic vesicles could diffuse out from the synaptic cleft and activate extrasynaptic NMDARs (Asztely et al. 1997; Min et al. 1998). The other possibility is that the clearance of glutamate was too fast to saturate all receptors (Kullmann, 1999).
Based on the mean amplitude of the AMPAR-mediated mEPSC (43 pA), and the single channel conductance of AMPARs (Spruston et al. 1995) we estimated that the number of AMPARs activated by the quantal transmitter release is ∼70. The observed variability of synaptic responses mediated by AMPA receptors (31% c.v.) was larger than that expected from the random opening of agonist bound receptors (∼15% c.v.) (Faber et al. 1992). The number of activated AMPARs could be underestimated, since we have not accounted for an apparent reduction in receptor number due to dendritic filtering. In any case, the variability of AMPAR components is larger than that expected from the random opening of this number of receptors. The large trial to trial variability of AMPAR components further supports our hypothesis that AMPARs are not saturated by glutamate from single synaptic vesicles (Liu & Tsien, 1995; Forti et al. 1997; Min et al. 1998). If AMPARs are not saturated large variability in postsynaptic response could be expected to arise from differences in the amount of transmitter loaded in single vesicles. In other systems such as the amacrine cell dinapse preparation co-variance between pre- and postsynaptic GABAergic responses has been used to argue for presynaptic control of miniature amplitude variability (Frerking et al. 1995). These authors propose that differences in synaptic vesicle diameters (10% c.v.) account for a large part of the IPSC variability because the amount of transmitter in single vesicles would vary by the 3rd power of the vesicle diameter (Bekkers et al. 1990; Frerking et al. 1995). In addition, the binding properties of glutamate receptors would compound this variance, as multiple glutamate molecules must bind to the receptor resulting in a power dependence with glutamate concentration (Clements & Westbrook, 1991).
Although both AMPA and NMDA receptor-mediated components of mEPSCs vary in amplitude, we did not observe selective failure of the AMPAR component across repeated trials at synapses that express both responses. This result suggests that during miniature transmission sufficient glutamate is released to activate both receptor classes despite the relatively low affinity of glutamate for AMPARs (Patneau & Mayer, 1990). From these observations we conclude that selective failure of AMPAR-mediated EPSCs during plasticity experiments is due to inactive receptors and not failure of transmitters to reach AMPARs (Isaac, 1995; Liao et al. 1995; Durand et al. 1996; Malenka & Nicoll, 1997).
Current hypotheses suggest that there are synapses that express only functional NMDARs and that enhancement of synaptic efficacy during long term potentiation or development involves the induction of functional AMPARs (Isaac et al. 1995; Liao et al. 1995; Durand et al. 1996; Malenka & Nicoll, 1997; Petralia et al. 1999). We have tested this hypothesis and provide additional evidence that a subset of synapses may not express functional AMPARs. In these experiments we cannot exclude the possibility that NMDARs, but not AMPARs, are activated by spillover of glutamate from neighbouring synapses (Min et al. 1998). However, under our recording conditions in which only asynchronous transmitter release occurs, the source of glutamate spillover would have to be a synapse onto a dendrite of a neighbouring neuron. We believe that this is unlikely since we have recorded from low density cultures of cortical neurons to reduce the contribution of transmitter spillover from synapses onto nearby neurons (Fig. 1). As MSCTs in most cases are associated with the activation of a single synapse, the probability of glutamate spillover would be expected to be low. Thus, it is unlikely that the exclusive activation of NMDARs by glutamate spillover occurs at a relatively high proportion of synapses (∼20% with apparently only NMDARs). The model that is most consistent with our data is one in which some synapses express functional AMPARs at low levels relative to the expression of NMDARs.
Our functional imaging experiments are consistent with recent immunocytochemical data showing that some developing synapses lack AMPARs (Gomperts et al. 1998; Nusser et al. 1998; Rao et al. 1998; Liao et al. 1999; Petralia et al. 1999). Our experiments indicate, consistent with the previous findings, that when synapses express both classes of receptor, each type is expressed in relatively similar proportions (Gomperts et al. 1998). We extend the findings of Gomperts et al. (1998) by demonstrating that identified synapses can express mEPSCs of different mean amplitude. Our combined imaging/electrophysiology experiments provide an important distinction in that they differentiate between variation in miniature amplitude due to trial to trial fluctuation within synapses and differences in mean amplitude between synapses. It has been shown that the clustering of receptors at synapses require anchoring proteins (Ehlers et al. 1996; O'Brien et al. 1998). Since the interaction between anchoring proteins and receptors are very specific, the distribution of each receptor subtype may be controlled in a framework of anchoring molecules that could maintain the ratio of receptor subtypes within a certain range. In this scenario, synapses express NMDARs first and then incorporate AMPARs during the development and the synaptic modulation (Bear & Malenka, 1994; Malenka, 1994; Wu et al. 1996; Wu & Cline, 1998).
Acknowledgments
We would thank Drs T. Kitamoto, H. Sakagami and H. Yawo of Tohoku University, and Drs L. A. Raymond and P. J. Mackenzie of the University of British Columbia. M.U. is supported by the Ministry of Education, Science, Sports and Culture of Japan. T.H.M. is supported by an operating grant from the Medical Research Council of Canada and is a MRC Scientist.
References
- Asztely F, Erdemli G, Kullmann DM. Extrasynaptic glutamate spillover in the hippocampus: dependence on temperature and the role of active glutamate uptake. Neuron. 1997;18:281–293. doi: 10.1016/s0896-6273(00)80268-8. [DOI] [PubMed] [Google Scholar]
- Bear MF, Malenka RC. Synaptic plasticity: LTP and LTD. Current Opinion in Neurobiology. 1994;4:389–399. doi: 10.1016/0959-4388(94)90101-5. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Richerson GB, Stevens CF. Origin of variability in quantal size in cultured hippocampal neurons and hippocampal slices. Proceedings of the National Academy of Sciences of the USA. 1990;87:5359–5362. doi: 10.1073/pnas.87.14.5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. NMDA and non-NMDA receptors are co-localized at individual excitatory synapses in cultured rat hippocampus. Nature. 1989;341:230–233. doi: 10.1038/341230a0. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Cable properties of cultured hippocampal neurons determined from sucrose-evoked miniature EPSCs. Journal of Neurophysiology. 1996;75:1250–1255. doi: 10.1152/jn.1996.75.3.1250. [DOI] [PubMed] [Google Scholar]
- Clements JD, Westbrook GL. Activation kinetics reveal the number of glutamate and glycine binding sites on the N-methyl-D-aspartate receptor. Neuron. 1991;7:605–613. doi: 10.1016/0896-6273(91)90373-8. [DOI] [PubMed] [Google Scholar]
- Dong H, O'Brien RJ, Fung ET, Lanahan AA, Worley PF, Huganir RL. GRIP: a synaptic PDZ domain-containing protein that interacts with AMPA receptors. Nature. 1997;386:279–284. doi: 10.1038/386279a0. [DOI] [PubMed] [Google Scholar]
- Durand GM, Kovalchuk Y, Konnerth A. Long-term potentiation and functional synapse induction in developing hippocampus. Nature. 1996;381:71–75. doi: 10.1038/381071a0. [DOI] [PubMed] [Google Scholar]
- Ehlers MD, Mammen AL, Lau LF, Huganir RL. Synaptic targeting of glutamate receptors. Current Opinion in Cell Biology. 1996;8:484–489. doi: 10.1016/s0955-0674(96)80024-x. [DOI] [PubMed] [Google Scholar]
- Faber DS, Young WS, Legendre P, Korn H. Intrinsic quantal variability due to stochastic properties of receptor-transmitter interactions. Science. 1992;258:1494–1498. doi: 10.1126/science.1279813. [DOI] [PubMed] [Google Scholar]
- Forti L, Bossi M, Bergamaschi A, Villa A, Malgaroli A. Loose-patch recordings of single quanta at individual hippocampal synapses. Nature. 1997;388:874–878. doi: 10.1038/42251. [DOI] [PubMed] [Google Scholar]
- Frerking M, Borges S, Wilson M. Variation in GABA mini amplitude is the consequence of variation in transmitter concentration. Neuron. 1995;15:885–895. doi: 10.1016/0896-6273(95)90179-5. [DOI] [PubMed] [Google Scholar]
- Gomperts SN, Rao A, Craig AM, Malenka RC, Nicoll RA. Postsynaptically silent synapses in single neuron cultures. Neuron. 1998;21:1443–1451. doi: 10.1016/s0896-6273(00)80662-5. [DOI] [PubMed] [Google Scholar]
- Hessler NA, Shirke AM, Malinow R. The probability of transmitter release at a mammalian central synapse. Nature. 1993;366:569–572. doi: 10.1038/366569a0. [DOI] [PubMed] [Google Scholar]
- Isaac JT, Nicoll RA, Malenka RC. Evidence for silent synapses: implications for the expression of LTP. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- Jonas P, Sakmann B. Glutamate receptor channels in isolated patches from CA1 and CA3 pyramidal cells of rat hippocampal slices. The Journal of Physiology. 1992;455:143–171. doi: 10.1113/jphysiol.1992.sp019294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas P, Spruston N. Mechanisms shaping glutamate-mediated excitatory postsynaptic currents in the CNS. Current Opinion in Neurobiology. 1994;4:366–372. doi: 10.1016/0959-4388(94)90098-1. [DOI] [PubMed] [Google Scholar]
- Kamboj S, Huganir RL. Receptor clustering - activate to accumulate. Current Biology. 1998;8:R719–721. doi: 10.1016/s0960-9822(98)70457-9. [DOI] [PubMed] [Google Scholar]
- Kullmann DM. Excitatory synapses. Neither too loud nor too quiet. Nature. 1999;399:111–112. doi: 10.1038/20089. [DOI] [PubMed] [Google Scholar]
- Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- Liao D, Zhang X, O'Brien R, Ehlers MD, Huganir RL. Regulation of morphological postsynaptic silent synapses in developing hippocampal neurons. Nature Neuroscience. 1999;2:37–43. doi: 10.1038/4540. [DOI] [PubMed] [Google Scholar]
- Liu G, Choi S, Tsien RW. Variability of neurotransmitter concentration and nonsaturation of postsynaptic AMPA receptors at synapses in hippocampal cultures and slices. Neuron. 1999;22:395–409. doi: 10.1016/s0896-6273(00)81099-5. [DOI] [PubMed] [Google Scholar]
- Liu G, Tsien RW. Properties of synaptic transmission at single hippocampal synaptic boutons. Nature. 1995;375:404–408. doi: 10.1038/375404a0. [DOI] [PubMed] [Google Scholar]
- Mackenzie PJ, Kenner GS, Prange O, Shayan H, Umemiya M, Murphy TH. Ultrastructural correlates of quantal synaptic function at single CNS synapses. Journal of Neuroscience. 1999;19:RC13. doi: 10.1523/JNEUROSCI.19-12-j0003.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainen ZF, Malinow R, Svoboda K. Synaptic calcium transients in single spines indicate that NMDA receptors are not saturated. Nature. 1999;399:151–155. doi: 10.1038/20187. [DOI] [PubMed] [Google Scholar]
- Malenka RC. Synaptic plasticity in the hippocampus: LTP and LTD. Cell. 1994;78:535–538. doi: 10.1016/0092-8674(94)90517-7. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Silent synapses speak up. Neuron. 1997;19:473–476. doi: 10.1016/s0896-6273(00)80362-1. [DOI] [PubMed] [Google Scholar]
- Malinow R, Otmakhov N, Blum KI, Lisman J. Visualizing hippocampal synaptic function by optical detection of Ca2+ entry through the N-methyl-D-aspartate channel. Proceedings of the National Academy of Sciences of the USA. 1994;91:8170–8174. doi: 10.1073/pnas.91.17.8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min MY, Rusakov DA, Kullmann DM. Activation of AMPA, kainate, and metabotropic receptors at hippocampal mossy fiber synapses: role of glutamate diffusion. Neuron. 1998;21:561–570. doi: 10.1016/s0896-6273(00)80566-8. [DOI] [PubMed] [Google Scholar]
- Muller BM, Kistner U, Kindler S, Chung WJ, Kuhlendahl S, Fenster SD, Lau LF, Veh RW, Huganir RL, Gundelfinger ED, Garner CC. SAP102, a novel postsynaptic protein that interacts with NMDA receptor complexes in vivo. Neuron. 1996;17:255–265. doi: 10.1016/s0896-6273(00)80157-9. [DOI] [PubMed] [Google Scholar]
- Muller W, Connor JA. Dendritic spines as individual neuronal compartments for synaptic Ca2+ responses. Nature. 1991;354:73–76. doi: 10.1038/354073a0. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Baraban JM, Wier WG. Mapping miniature synaptic currents to single synapses using calcium imaging reveals heterogeneity in postsynaptic output. Neuron. 1995;15:159–168. doi: 10.1016/0896-6273(95)90073-x. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Baraban JM, Wier WG, Blatter LA. Visualization of quantal synaptic transmission by dendritic calcium imaging. Science. 1994;263:529–532. doi: 10.1126/science.7904774. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Lujan R, Laube G, Roberts JD, Molnar E, Somogyi P. Cell type and pathway dependence of synaptic AMPA receptor number and variability in the hippocampus. Neuron. 1998;21:545–559. doi: 10.1016/s0896-6273(00)80565-6. [DOI] [PubMed] [Google Scholar]
- O'Brien RJ, Lau LF, Huganir RL. Molecular mechanisms of glutamate receptor clustering at excitatory synapses. Current Opinion in Neurobiology. 1998;8:364–369. doi: 10.1016/s0959-4388(98)80062-7. [DOI] [PubMed] [Google Scholar]
- Patneau DK, Mayer ML. Structure-activity relationships for amino acid transmitter candidates acting at N-methyl-D-aspartate and quisqualate receptors. Journal of Neuroscience. 1990;10:2385–2399. doi: 10.1523/JNEUROSCI.10-07-02385.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralia RS, Esteban JA, Wang YX, Partridge JG, Zhao H-M, Wenthold RJ, Malinow R. Selective acquisition of AMPA receptors over postnatal development suggests a molecular basis for silent synapses. Nature Neuroscience. 1999;2:31–36. doi: 10.1038/4532. [DOI] [PubMed] [Google Scholar]
- Rao A, Kim E, Sheng M, Craig AM. Heterogeneity in the molecular composition of excitatory postsynaptic sites during development of hippocampal neurons in culture. Journal of Neuroscience. 1998;18:1217–1229. doi: 10.1523/JNEUROSCI.18-04-01217.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Clements JD, Westbrook GL. Nonuniform probability of glutamate release at a hippocampal synapse. Science. 1993;262:754–757. doi: 10.1126/science.7901909. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Westbrook GL. Calcium-induced actin depolymerization reduces NMDA channel activity. Neuron. 1993;10:805–814. doi: 10.1016/0896-6273(93)90197-y. [DOI] [PubMed] [Google Scholar]
- Schiller J, Schiller Y, Clapham D. NMDA receptors amplify calcium influx into dendritic spines during associative pre- and postsynaptic activation. Nature Neuroscience. 1998;1:114–118. doi: 10.1038/363. [DOI] [PubMed] [Google Scholar]
- Silver RA, Traynelis SF, Cull Candy SG. Rapid-time-course miniature and evoked excitatory currents at cerebellar synapses in situ. Nature. 1992;355:163–166. doi: 10.1038/355163a0. [DOI] [PubMed] [Google Scholar]
- Spruston N, Jonas P, Sakmann B. Dendritic glutamate receptor channels in rat hippocampal CA3 and CA1 pyramidal neurons. The Journal of Physiology. 1995;482:325–352. doi: 10.1113/jphysiol.1995.sp020521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong G, Shepherd D, Jahr CE. Synaptic desensitization of NMDA receptors by calcineurin. Science. 1995;267:1510–1512. doi: 10.1126/science.7878472. [DOI] [PubMed] [Google Scholar]
- Wilding TJ, Huettner JE. Antagonist pharmacology of kainate- and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-preferring receptors. Molecular Pharmacology. 1996;49:540–546. [PubMed] [Google Scholar]
- Wu G, Malinow R, Cline HT. Maturation of a central glutamatergic synapse. Science. 1996;274:972–976. doi: 10.1126/science.274.5289.972. [DOI] [PubMed] [Google Scholar]
- Wu GY, Cline HT. Stabilization of dendritic arbor structure in vivo by CaMKII. Science. 1998;279:222–226. doi: 10.1126/science.279.5348.222. [DOI] [PubMed] [Google Scholar]
- Zhang S, Ehlers MD, Bernhardt JP, Su CT, Huganir RL. Calmodulin mediates calcium-dependent inactivation of N-methyl-D-aspartate receptors. Neuron. 1998;21:443–453. doi: 10.1016/s0896-6273(00)80553-x. [DOI] [PubMed] [Google Scholar]