Fusions with histone H3 result in highly specific alteration of gene expression (original) (raw)
Abstract
Hap1 is a yeast transcriptional activator which controls expression of genes such as CYC1 and CYC7. Our results show that Hap1 activity is dependent on a functional chromatin remodeling complex SWI/SNF. Using a modified two-hybrid screen with Hap1 as bait, we recovered expression vectors encoding the Gal4 activation domain fused to histone H3 [Gal4(AD)–H3]. Hap1 activity at CYC1 or CYC7 was increased by Gal4(AD)–H3 and the effect was dependent on the presence of the activation domain of Hap1 and a functional SWI complex. Importantly, overexpression of H3 alone had no effect on Hap1 activity. Analysis of Gal4(AD)–H3 revealed that the fusion is not incorporated into the nucleosome while a functional Gal4 activation domain is dispensable. Activity of many other transcriptional activators was unchanged or slightly affected in the presence of Gal4(AD)–H3. Thus, our results identify a new class of histone H3 variants that cause highly specific alteration of gene expression. Hap1 may interact directly with H3 favoring chromatin remodeling by the SWI/SNF complex.
INTRODUCTION
In the nucleus, DNA is associated with histones (two molecules of each of the core histones H2A, H2B, H3 and H4) to form a nucleosome core, the basic repeating unit of chromatin (1,2). In the absence of DNA, H2A and H2B form a heterodimer while H3 and H4 form a heterotetramer. Histones H3 and H4 show the strongest evolutionary conservation. A central tetramer (H3–H4)2 is flanked by a H2A–H2B dimer on both sides (1). Core histones have a long central α-helix flanked by a loop segment that bridges smaller α-helices (3). The strongly basic N-termini of H3 and H4 extend outside the core nucleosome and are subject to post-transcriptional modifications, including acetylation of lysines (see below).
Numerous studies have shown the importance of histones in regulating gene expression. Changes in histone gene dosage alter yeast gene transcription (4–7). Similarly, deletions of the N-termini of histones H3 and H4 result in altered gene activation (8,9) and repression (10–13). In addition, mutations in the dimer–tetramer interfaces of H4 and H2A–H2B affect expression of cell cycle genes (14).
A number of complexes have been shown to remodel chromatin structure (reviewed in 15–18). Yeast Gcn5 and its Tetrahymena homolog were shown to possess histone acetyltransferase activity (19,20). However, acetylation by Gcn5 is not observed when histones are present in nucleosomes (21). Such an enzymatic activity is seen only when Gcn5 is part of the Ada or other related complexes such as SAGA (Spt–Ada–Gcn5–acetyltransferase) (21,22). One model stipulates that the Ada complex is recruited by transcriptional activators allowing acetylation of the neighboring histones, which would result in local remodeling of the chromatin structure (23). In agreement with this model, a strong correlation between Gcn5 enzymatic activity and VP16- or Gcn4-mediated transcriptional activation has been observed (24) and a direct link between acetylated histones at specific genes and transcription was recently reported (25). Finally, the SAGA complex was shown to increase VP16- and Gcn4-mediated transcriptional activity in vitro by recruiting the SAGA complex to nucleosomes (26,27). Other gene products like CBP (CREB-binding protein), P/CAF (P300/CBP-associated factor), TAF250 and the nuclear receptor co-activators ACTR and SRC possess histone acetyltransferase activity (reviewed in 15).
Yeast genetic screens have led to the identification of many genes whose products form a complex called SWI/SNF (referred to as SWI hereafter). This complex has 11 subunits including the SWI1, SWI2, SWI3, SWP73, SNF6, ARP7 and ARP9 gene products and potentiates the activity of many transcriptional activators (reviewed in 18,28). Drosophila and human homologs have also been isolated recently (18,29 and references therein). Evidence for SWI acting at chromatin remodeling was provided by the fact that many yeast suppressors of swi– mutants are mutated components of chromatin, such as histones H2A, H2B, H3 and H4 (see 30 for references). Furthermore, SWI is involved in the formation of DNase I hypersensitive sites in vitro (31). In addition, the nucleosome arrangement at the promoter of the SUC2 gene (encoding sucrose invertase) is altered in a swi– strain as shown by in vivo studies (32). The yeast and human SWI complexes enhance binding in vitro on nucleosomal DNA of proteins containing the DNA-binding domain of the transcriptional activator Gal4 (33,34) and yeast SWI is required for binding of Gal4 to target sites in vivo (35). The SWI complex stably alters nucleosome structure and does not need the continuous presence of ATP (31,36–38). Thus, it seems plausible that SWI alters the chromatin structure to facilitate access of transcriptional activators to target DNA sequences, although other models have been proposed (see for example 39).
These data are in agreement with the SWI complex functioning to remodel chromatin. The SWI complex binds preferentially to four-way junction DNA, a structure that resembles DNA found at the entry and exit points of a nucleosome (40). However, these observations do not show how SWI is recruited to specific promoters. One mechanism of action may involve specialized SWI complexes, as recently described for the PYR complex, which is related to the SWI complex (41). The PYR complex is associated with fetal-to-adult globin gene switching. Interestingly, the SWI complex was shown to be associated with the RNA polymerase II holoenzyme (42). However, this observation is controversial (43). More recent observations show that the SWI complex interacts directly with transcriptional activators (82–84).
Yeast transcriptional activator Hap1
Many transcriptional regulators contain a zinc finger known as a binuclear zinc cluster. Two well-characterized members of this family are Gal4 (44), a positive regulator of the expression of genes involved in galactose metabolism, and Hap1 (45), which controls expression of genes required for cellular respiration. Hap1 possesses an acidic activation domain at its C-terminus (residues 1307–1483) and an N-terminal DNA-binding domain (residues 1–174).
Hap1 interacts with specific DNA sites, such as the upstream activating sequences (UASs) identified in the CYC1 and CYC7 genes, which encode isoforms of cytochrome c (46 and references therein). These sites share only modest homology and their dissimilarity results in different levels of Hap1-dependent activation. Activity at the CYC1 promoter is higher than at CYC7, even if the UASs are placed in an identical promoter context, and is not a result of different affinity of Hap1 for CYC1 and CYC7 (46,47). Furthermore, a single amino acid change in Hap1 of Ser63→Arg (mutant Hap1-18), immediately N-terminal to the first cysteine of the zinc cluster, prevents binding to the UAS of CYC1 and results in greatly increased activation (10- to 100-fold) at CYC7, even though Hap1-18 has a similar affinity for CYC7 to wild-type Hap1. Other Hap1 mutations that flank the zinc cluster, termed positive control (PC) mutants, show wild-type binding and activation at CYC1 but are transcriptionally inactive at CYC7, in spite of wild-type or increased binding at that site (48).
Thus, there are several classes of mutations in the DNA-binding domain of Hap1 that do not affect the in vitro affinity of Hap1 for DNA but dramatically alter its ability to activate transcription. The crystal structure of Hap1 and Hap1-18 bound to CYC7 revealed alternate protein–DNA interactions induced by the Ser63→Arg mutation (49,50). How these structural changes result in altered transcriptional activity is not clear. It has been proposed that Hap1–DNA interactions have allosteric effects on the activator (50). Similarly, it was hypothesized that the PC mutations alter the Hap1–DNA interface resulting in altered transcriptional activity of the PC mutants at CYC7 (50). In spite of the fact that the DNA-binding domain of Hap1 has no transcriptional activity on its own, the results clearly show that mutations in that region can affect the transcriptional activity of the entire protein. These observations suggest that the lack of correlation between in vitro binding of Hap1 on naked DNA and in vivo activity is due to chromatin effects.
In this report, we show that Hap1 activity is strongly dependent on the SWI complex and that fusions with histone H3 result in hyperactivation of Hap1. The H3 fusion has no effect on many other transcriptional activators. Thus, our results identify a new class of histone H3 variants that cause highly specific alteration of gene expression.
MATERIALS AND METHODS
Yeast strains
The yeast strains used were: YPH499 (MATa ura3-52 lys2-801 _ade2-101 his3-Δ_2000 leu2-Δ_1; 51); THl (MATa ura3-52 his4-119 ade1-100 Δ_hap1::LEU2; 46); YA1 (MATa ura3-52 lys2-801 ade2-101 trp1-Δ_63 Δ_hap1::_HIS3 leu2-Δ_1; this study), derived from YPH499; CY337 (MATa ura3-52 lys2-801 _ade2-101 his3-Δ_200 leu2-Δ_1; C. L. Peterson); CY407 (MATa ura3-52 lys2-801 ade2-101 leu2-Δ_1 HIS3::swi2; C. L. Peterson), derived from CY337; Y190 (MATa ura3-52 leu2-3,112 trp1-901 his3-200 ade2-101 Δ_gal4 Δ_gal80 URA3::GAL1-lacZ LYS2::GAL-HIS3 cyhr; 80); MX4-22A [MAT**a** Δ(_HHT1 HHF1_) Δ(_HHT2 HHF2_) _leu2-3,112_ _lys2_Δ_201 ura3-52_ pMS329 (_URA3 HHT1_ _HHF1_); 14]. For YA1, most of the Hap1 coding sequences were deleted using the method of Baudin et al. (52). The HIS3 gene was amplified using plasmid pMHIS3 (53) and oligos AACACCAGCTCTAACTCTCCACCCTTGCACATGTCTTCAGATTCAGGGTTTTCCCAGTCA and TCAATT-TCAACATTATCAACGGGTAGAAAATCAACCGCGTC-ATGCGGATAACAATTTCAC.
Plasmids
Gal4(AD)–H3 deletion mutants. pGal4(AD)-H3, isolated from a two-hybrid screen (see Results) was cut with _Hin_dIII and the fragment containing the coding sequences of Gal4(AD) and H3 was subcloned into the _Hin_dIII site of pZERO-2 (Invitrogen) to give pZERO-H3. The resulting plasmid was transformed into Escherichia coli strain CJ236 for single-stranded DNA isolation and used as a template for site-directed mutagenesis (54,55). The following oligos were used to perform site-directed mutagenesis (or deletions): Gal4(AD)–H3(1–120), AA-GCGTGTCACTATCTAGGCCTAGGATATCAAGTTGGC; Gal4(AD)–H3(1–100), CTGTCGAAGCCTACTAGGCCT-CTTTATTTGAAGAT; Gal4(AD)–H3(1–65), CTGTTGATC-AGAAAGTAGGCCTTCCAAAGATTGGT; Gal4(AD)–H3(1–36), TCTACCGGTGGTGTTTAGGCCTCTCACAGATATAAG; Gal4(AD)–H3(Δ4–20), CGAGCAAACAATGGC-CAGATTAGCTTCTAAGGCTGCCAG; Gal4(AD)–H3(Δ1–50), ATTCGGCACGAGCAAACAGAAATCAGAAGATTCCAA; Gal4(AD)–H3(Δ1–80), ATTCGGCACGAGCAAACAACC-GACTTGAGATTTCAA; Gal4–mutant-H3 (double mutant), TTGATGATGGTAATAATCAAAACCACTGTCACC and GAAGATACCCCACCAAGACCCAAAAAAAGAGATC. The _Hin_dIII fragment containing the desired mutations (or deletions) was then subcloned back into pGal4(AD)–H3.
Other histone plasmids. Plasmid pH3 (a 2 µ overexpression vector for H3 under control of the ADH1 promoter) was constructed by deleting Gal4 sequences of pZERO-H3 by cutting with _Eco_RI, treating with Klenow and inserting a _Hin_dIII linker by religation of the backbone vector. The _Hin_dIII–_Xho_I fragment containing H3 sequences was then inserted into pVP16 (56) cut with the same enzymes. The resulting plasmid was then cut with _Xho_I and _Not_I, treated with Klenow and religated. Plasmid pVP16-H3 was made by inserting a _Bgl_II fragment (containing HA and H3 sequences) from pZERO-H3 into the _Bam_HI site of pVP16 (56). Plasmid pGal4(AD)-H4 was obtained by amplifying the H4 open reading frame YBR009C with primers GGAATTCGGATGTCCGGTAGAGGTAAAGG and TAACTCGAGTTAACCACCGAAACCGTAT and yeast genomic DNA isolated from strain YPH499. The PCR product was cut with _Eco_RI and _Xho_I and subcloned into pACT2 cut with the same restriction enzymes. A LYS2 marked expression vector for histone H4 (pH4-LYS2) was obtained by amplifying the HHF1 gene using plasmid pMS329 (14) as a template with oligos CCCAAGCTTAGCCGTGGAGGGTACCAAA and AACTGCAGTCTCC-TAAACCCGCTATAA. The PCR product was then cut with _Hin_dIII and _Pst_I and subcloned into pRS317 (57) cut with the same enzymes.
Other plasmids. For plasmid p424ERα the cDNA encoding estrogen receptor α was obtained by cutting hER0 (58) with _Eco_RI and subcloning into the _Eco_RI site of plasmid p424MET25 (59). Plasmid pCYC7-5-HIS4 (48) was cut with _Eae_I and _Bst_YI and the fragment containing two copies of the UAS of CYC7 was subcloned into pRS315HIS (60) cut with _Not_I and _Bam_HI to give pCYC7-HIS3. A 2 µ TRP1 derivative was constructed by subcloning an _Apa_I–_Sac_I fragment (containing CYC7, GAL1 and HIS3 sequences) into pRS424 (61) to give p424CYC7-HIS3. A LEU2 version of the CYC7 reporter pCYC7-5-HIS4 (48) was obtained by removing URA3 sequences by cutting with _Not_I and inserting a _Not_I fragment containing the LEU2 gene to give pCYC7-5lacZLEU2. The CYC1 version UAS1lacZLEU2 was obtained by exchanging _Dra_III fragments between pLGΔ312ΔAX-HIS (48) and pCYC7-5lacZLEU2. Plasmid pGPD-Hap1, a Hap1 expression vector under control of the GPD promoter, was made by introducing a _Nhe_I linker into the _Sma_I site of SD5-HAP1 (48) to give SD5-HAP1-N. GPD sequences were amplified with oligos CCGCTCGAGTCGAGTTTATCATTATCA and CG-GCTAGCTCGAAACTAAGTTCTTGG using pG-1 (62) as template. The PCR product was then cut with _Xho_I and _Nhe_I and subcloned into SD5-HAP1-N cut with the same enzymes. Expression vectors for Gal4–VP16 and mutants were obtained by digesting pVS1.VP16C (and mutants) (85) with _Alw_NI and _Sac_I and subcloning into plasmid pRS314 cut with the same enzymes. The resulting TRP-marked low copy plasmid expresses Gal4(1–147)–VP16C (or mutants) (85) from the ADH1 promoter. All constructs derived from mutagenesis or PCR products were verified by DNA sequencing.
β-Galactosidase assays
Strains were transformed with plasmids and grown on selective plates and resultant colonies were grown overnight in YPD (63). Cells were then diluted and grown for 12–18 h (OD600 0.6–1.0) in synthetic drop-out medium with 2% glucose and assayed for β-galactosidase activity with permeabilized cells (64). Results are an average of at least two experiments performed in duplicate with a variation of usually <25%. For Figure 1, β-galactosidase activity was measured in extracts and normalized to protein concentration.
Figure 1.

Hap1 activity is dependent on the SWI complex. (a) Reporters for CYC1 (pUAS1LacZLEU2) or CYC7 (pCYC7-5lacZLEU2) were transformed into a wild-type strain (CY337), a swi2– strain (CY407) or a Δhap1 strain (YA1) and their activity assayed as described in Materials and Methods. GPD–HAP1 is a Hap1 expression vector under the control of the glyceraldehyde 3-phosphate dehydrogenase (GPD) promoter transformed along with the CYC1 reporter. (b) The lacZ reporters ‘CYC1’ and ‘CYC7’ are shown schematically.
Two-hybrid screen
Strain YPH499 containing reporter plasmids pCYC7-HIS3 and pCYC7-lacZ (plasmid containing one copy of the UAS of CYC7 inserted in front of a minimal CYC1 promoter; 46) was transformed with a yeast cDNA library (obtained from S. J. Elledge) constructed in vector pACT2 (80,81). Following heat shock, cells were grown in liquid in the absence of histidine for 2 h and spread on plates lacking histidine and containing 5 mM 3-aminotriazole. Colonies were replica-plated on identical plates and then assayed for β-galactosidase activity. Representative clones were shuttled to E.coli (strain KC8; Clontech) for further analysis.
Northern blot analysis
RNA was isolated using the hot phenol method (54). Electrophoresis, transfer and hybridization were performed as described (86). Probes were obtained by amplifying open reading frames YPR060C (ARO7), YMR303C (ADH2), YOL110W (SHR5) and YKL095W (YJU2) using primers from Research Genetics. The amplification products were subcloned into plasmid pCR2.1. Probes were generated by PCR amplification with the primers used for the initial amplification and plasmids containing open reading frames described above as template. PCR products were then randomly labeled (54).
RESULTS
The SWI complex mediates Hap1 transcriptional activity
The chromatin remodeling complex SWI has been shown to modulate the activity of many transcriptional activators. We tested whether the activity of the transcriptional activator Hap1 is also controlled by the SWI complex using reporter plasmids containing two UASs of CYC1 or CYC7 in front of a minimal CYC1 promoter driving lacZ transcription. Reporter plasmids were introduced into a wild type, an isogenic strain carrying a deletion of the SWI2 gene (which encodes a subunit of the SWI complex) and a Δhap1 strain. The activity of Hap1 at CYC1 or CYC7 was greatly reduced (5- to 20-fold) in a strain carrying a non-functional SWI2 gene (Fig. 1, lanes 1–6). We compared the Hap1 levels in SWI2+ and swi2– strains using electrophoretic mobility shift assays with a CYC1 probe and found that Hap1 levels were reduced <2-fold with extracts prepared from a swi2– strain (data not shown). These results are in agreement with the fact that RNA levels of Hap1 are not significantly affected in a swi2– background (65). In addition, we expressed Hap1 from the glyceraldehyde 3-phosphate dehydrogenase (GPD) promoter, known to be more active in a swi2– strain than in a SWI2+ strain (66), and found that activity of Hap1 is reduced 12-fold when assayed at CYC1 (Fig. 1, lanes 7 and 8). Similar results were obtained with a CYC7 reporter (data not shown). Thus, reduced transcriptional activity of Hap1 in a swi– strain is not due to decreased levels of the activator. In conclusion, Hap1 activity is strongly dependent on the SWI complex.
Identification of potential Hap1-interacting proteins
We wished to identify the proteins that interact with Hap1 and may be responsible for its transcriptional activation using a modified two-hybrid screen (67). As a bait we used the native Hap1 activator and as reporters plasmids containing one or two copies of the UAS of CYC7 inserted in front of the minimal GAL1 and CYC1 promoters driving HIS3 or lacZ transcription, respectively. The UAS of CYC7 was chosen because activation by Hap1 at this site is relatively weak. Indeed, this kind of screen probably would not work with strong activators, since the activity of the reporter would be very high even in the absence of an expression vector for the prey (from the library). Preliminary experiments have shown that activity of wild-type Hap1 at a CYC7–HIS3 reporter is not strong enough to allow growth of a Δhis3 yeast strain on plates lacking histidine and containing an inhibitor of the HIS3 gene product, 3-aminotriazole. However, normal growth is observed when using the hyperactive mutant Hap1-18 (data not shown).
We screened a library carrying sequences encoding the Gal4 activation domain fused to cDNAs as described in Materials and Methods. About 1 × 107 transformants of the cDNA library were obtained and screened for growth on selective plates lacking histidine. Colonies were then assayed for β-galactosidase activity. About 60 colonies showed a strong β-galactosidase activity and their cDNA inserts were amplified by PCR of yeast colonies for restriction enzyme analysis. Three different groups of clones were identified and representative members were transformed into E.coli for plasmid isolation and retransformation into yeast. All clones allowed growth on selective plates and showed increased β-galactosidase activity.
One type of clone had DNA sequences of unknown origin fused to a fragment of chromosome V and was not further analyzed. Interestingly, the two other groups (Fig. 2) correspond to expression vectors encoding the Gal4 activation domain fused to the entire open reading frames of the HHT1 or the HHT2 genes, which both encode histone H3 [fusions referred to as Gal4(AD)–H3 hereafter]. Expression of either fusion protein increased Hap1 activity ~8- to 10-fold at CYC1 or CYC7 (with the HAP1 gene in its normal chromosomal location; Fig. 3, lanes 1–6 and data not shown). Similar results were obtained when the effect of Gal4(AD)–H3 on Hap1 was assayed with a CYC7 reporter stably incorporated into the yeast genome (data not shown). Our results are reminiscent of those of Alevizopoulos et al. (68). Indeed, using a fusion of the DNA-binding domain of Gal4 and the activation domain of the proline-rich mammalian activator CTF-1 (68) as bait in a yeast two-hybrid screen, a clone encoding a fusion of the activation domain of VP16 and H3 was isolated. Histone H3 was further shown to interact with the activation domain of CTF-1 in vitro and in mammalian cells (68).
Figure 2.

Sequences of the junction of the fusion proteins recovered from the ‘two-hybrid’ screen. Amino acid and DNA sequences of the junction of Gal4(AD) and histone H3 are shown. Open reading frames YBR010W and YNL031C correspond to the HHT1 and HHT2 genes, respectively, and both encode histone H3.
Figure 3.

Effect of Gal4(AD)–H3 on activity of Hap1 activity and derivatives. Activity of Hap1 was assayed at CYC1 or CYC7 using lacZ reporters. Reporters contain one copy of the UAS of CYC1 or CYC7 inserted upstream of the minimal CYC1 promoter driving lacZ transcription (46). Hap1 and None refer to use of the wild-type strain YPH499 and the Δhap1 strain YA1, respectively. Empty vector, plasmid pACT2 (80) used for construction of the cDNA library and expressing the activation domain of Gal4.
Since both types of H3 fusions isolated from our screen have an identical amino acid sequence [with the exception of a few amino acids at the junction between Gal4(AD) and H3; Fig. 2], we arbitrarily concentrated our analysis on the plasmid carrying the HHT1 gene (encoding histone H3). No effect on CYC1 or CYC7 activity was observed when Gal4(AD)–H3 was expressed in the absence of Hap1 (Fig. 3, lanes 7 and 8). Similarly, co-expression of Gal4(AD)–H3 and a truncated form of Hap1 lacking the activation domain (but not defective in DNA binding; 48) gave background transcriptional activity (data not shown). Thus, these results indicate that Gal4(AD)–H3 has no effect on its own.
Altered gene expression has been associated with changes in gene dosage of histones (4–7). However, overexpression of H3 had no effect on Hap1 activity, indicating that Gal4 sequences are required for increased Hap1 activity (Fig. 3, lanes 9 and 10). Moreover, co-expression of wild-type Hap1 and Gal4(AD)–H3 in a swi2– strain gave background activity (Fig. 3, lanes 11 and 12). Thus, increased Hap1 activity induced by Gal4(AD)–H3 is dependent on: (i) the activation domain of Hap1; (ii) the Gal4(AD) sequences present in the fusion; (iii) a functional SWI complex. Moreover, the increased activity of Hap1 induced by Gal4(AD)–H3 is not related to the basic CYC1 promoter since the initial ‘two-hybrid’ selection relied on the UAS of CYC7 linked to the minimal GAL1 promoter. Finally, overexpression of Hap1 from a high copy number (2 µ) plasmid also increased activity with Gal4(AD)–H3, but to a lesser extent (4- to 5-fold) than chromosomal Hap1 (data not shown).
Thus, our results show that the activity of CYC1 and CYC7 reporters is increased by expression of Gal4(AD)–H3. However, this increased activity may be indirect and related to increased transcription of the HAP1 promoter resulting in higher levels of the activator Hap1. To rule out this possibility, we expressed Hap1 from a promoter under the control of Gal4. The activity of the Gal4-responsive promoter is minimally affected (2-fold reduction) by expression of Gal4(AD)–H3 (see Fig. 6). In this assay, activity of a CYC7 reporter was increased ~20-fold with co-expression of Gal4(AD)–H3. Thus, our results strongly suggest that Gal4(AD)–H3 (along with Hap1) directly affects activity of the CYC1 and CYC7 reporters.
Figure 6.

Effect of Gal4(AD)–H3 and derivatives on various activators. Activity of various transcriptional activators was assayed with lacZ reporters. Results are an average of two to three independent transformations. Uga3 and Leu3 reporters contain one copy (20mer) of the UAS of UGA1 and LEU2, respectively, inserted upstream of the minimal CYC1 promoter (53,73). The Gal4 reporter contains four copies of UASGAL4 found in the GAL1-10 intergenic region (pLGSD5) (74). The ER reporter contains three estrogen-responsive elements inserted upstream of the minimal GAL1 gene (75). The Gcn4 reporter contains binding sites for Bas1, Bas2 and Gcn4 (76). The Hap2/3/4/5 reporter contains CYC1 sequences up to –265 bp (77). Uga3, a yeast activator inducible by γ-aminobutyrate (GABA) (78), was assayed in the presence (0.1%) or absence of GABA. ER, human estrogen receptor α expressed in yeast (see Materials and Methods). Activity of ER was induced with 500 nM estradiol (+hormone). Uga3, Leu3, Gal4, Gcn4 and Hap2/3/4/5 were expressed from their natural chromosomal locations. Empty vector, plasmid pACT2 (80) used for construction of the cDNA library and expressing the activation domain of Gal4.
Several mechanisms may explain the stimulation of Hap1-dependent transcription by Gal4(AD)–H3: this enhancement may simply result from a direct interaction between H3 and Hap1, tethering the Gal4 activation domain to the promoter, a two-hybrid effect (Fig. 9). On the other hand, expression of Gal4(AD)–H3 in yeast cells may interfere with formation of nucleosomes, resulting in a more open chromatin structure and facilitated access of Hap1 or other factors to promoters (Fig. 9).
Figure 9.

Models for the mode of action of Gal4(AD)–H3. Models (a–d) for the mechanism of action of the H3 fusion protein Gal4(AD)–H3 are schematically shown. The exact position of the nucleosomes on the minimal CYC1 promoter is not known and they were drawn at arbitrary locations. TFIID is apparently constitutively bound to CYC1 (79). (a) Normal situation: Hap1 binding to target sites is favored by the SWI complex allowing interaction of Hap1 with the basic transcriptional machinery and initiation of transcription. (b) Gal4(AD)–H3 is incorporated into the nucleosome resulting in a less stable nucleosomal structure that can be more easily remodeled by the SWI complex resulting in increased activity by Hap1. (c) Gal4(AD)–H3 is not incorporated into the nucleosome and the H3 moiety of the fusion protein interacts with Hap1 allowing a classical ‘two-hybrid effect’. (d) Gal4(AD)–H3 forms a complex with Hap1 favoring chromatin remodeling by the SWI complex resulting in increased transcription.
Gal4(AD)–H3 is not incorporated into the nucleosome
We performed an analysis of the sequences of Gal4(AD)–H3 required for hyperactivation of Hap1. We constructed a series of Gal4(AD)–H3 deletion mutants and tested their activity in a strain containing a CYC7 reporter with a single UAS (Fig. 4). Interestingly, deletion of 15 amino acids at the C-terminus of H3 had no effect on the activity of Gal4(AD)–H3 (Fig. 4, lanes 2 and 3). In vivo studies in Xenopus have shown that the 10 amino acids at the C-terminus of histone H3 are essential for assembly of H3 into the nucleosome (87). Since Gal4(AD)–H3(1–120) is functional, this strongly suggests that the fusion is functioning outside the nucleosome. Larger truncations at the C-terminus of H3 abolished the hyperactivation of Hap1 (Fig. 4, lanes 4–6). Similarly, deletion of H3 amino acids 4–20 at the N-terminus of Gal4(AD)–H3 resulted in only a slight decrease in reporter activity (Fig. 4, lane 7). Larger deletions (amino acids 1–50 and 1–80) abolished hyperactivation of Hap1 (Fig. 4, lanes 8 and 9). H3 amino acids 1–63 were shown to be not essential for assembly into the nucleosome or heterodimerization with histone H4 (87). Thus, it conceivable that Hap1 interacts with the N-terminus of histone H3. In summary, our deletion analysis strongly suggests that Gal4(AD)–H3 functions outside the nucleosomal context.
Figure 4.

Domains of Gal4(AD)–H3 required for hyperactivation of Hap1. A lacZ reporter containing one UAS of CYC7 (46) was introduced into the HAP1+ strain YPH499 along with an expression vector for Gal4(AD)–H3 (or derivatives) and its activity assayed. Expression vectors (see Materials and Methods) are shown schematically on the left and their effect on Hap1 activity on the right.
A functional Gal4 activation domain is not required for Hap1 hyperactivation
Our next analysis focused on the Gal4 moiety of the Gal4(AD)–H3 fusion. Previous results showed that overexpression of histone H3 alone has no effect on Hap1 activity (Figs 3 and 4, lane 10), suggesting a requirement for Gal4 sequences. In order to test whether a functional activation domain of Gal4 is required for hyperactivation of Hap1, we introduced one frameshift mutation in Gal4(AD) so that critical amino acids for activation at the C-terminus of Gal4 were totally changed. The second frameshift allowed translation back to the normal reading frame and, hence, translation of H3, resulting in a fusion protein of the same bulk as ‘wild-type’ Gal4(AD)–H3.
Expression of this mutant protein resulted in reduced but significant (~3-fold) hyperactivation by Hap1 (Fig. 4, lane 11). As controls, we fused the wild-type or the mutated activation domain of Gal4 to its DNA-binding domain (amino acids 1–147) and assayed their transcriptional activity with a Gal4-responsive reporter. As expected, increased reporter activity was observed with a fusion containing the wild-type activation domain of Gal4 but not with a fusion containing the mutated activation domain (data not shown). Thus, our results strongly suggest that a functional activation domain in Gal4(AD)–H3 is not necessary for hyperactivation by Hap1. In agreement with these results, a fusion of the strong activation domain of VP16 and H3 did not affect Hap1 activity (Fig. 4, lane 12). Although the initial strategy that allowed recovery of Gal4(AD)–H3 was based on a modified two-hybrid screen, our data suggest that the mode of action of Gal4(AD)–H3 does not involve a ‘two-hybrid effect’. Further evidence for specificity of action of Gal4(AD)–H3 was provided by expressing histone H4 (which heterodimerizes with H3) fused to the activation domain of Gal4. The Gal4(AD)–H4 fusion slightly reduced the doubling time of cells (unpublished results) but had no effect on Hap1 activity (Fig. 4, lane 13).
Our analysis of Gal4(AD)–H3 mutants has provided strong evidence that the fusion is not incorporated into the nucleosome. Thus, the Gal4(AD)–H3 fusion should not functionally replace wild-type H3. We used a strain deleted of the HHT1 and HHT2 genes (encoding histone H3) and carrying the essential wild-type H3 gene on a URA3 plasmid. H3 variants on a LEU2 plasmid were introduced into the strain. We then tested whether the H3 gene on the URA3 plasmid can be lost by FOA selection for ura3– cells. As expected, a LEU2 plasmid overexpressing histone H3 allowed growth on selective plates but not when H3 sequences were lacking (Fig. 5). No growth was observed when expressing Gal4(AD)–H3 or a Gal4–mutant H3 carrying a non-functional activation domain (Fig. 5). The results show that Gal4(AD)–H3 (or mutant) cannot substitute for wild-type H3. We cannot formally rule out the possibility that incorporation of Gal4(AD)–H3 into the nucleosome results in toxic effects to the cells. However, deletion analysis of histone H3 rather suggests that Gal4(AD)–H3 is not incorporated into the nucleosome and, hence, cannot substitute for histone H3.
Figure 5.

Gal4(AD)–H3 cannot substitute for histone H3. Yeast strain MX4-22A (14) carrying deletions of all H3 and H4 genes and containing plasmid pMS329 (URA3; HHT1 HHF1, coding for H3 and H4) was transformed with a LYS2 plasmid carrying a wild-type H4 gene along with expression vectors for H3 or variants on a LEU2 plasmid. Transformants were then grown overnight in YPD and streaked on FOA plates to test whether the URA3 plasmid can be lost. WILD TYPE H3, plasmid pH3; NO H3, pACT2 (80).
The effect of Gal4(AD)–H3 is restricted to a subset of transcriptional activators
We tested if Gal4(AD)–H3 has broad effects on transcriptional activity or is restricted to a subset of transcriptional activators. Activity of the yeast transcriptional activator Uga3 or the human estrogen receptor α was unchanged in the presence of Gal4(AD)–H3 (Fig. 6, lanes 3, 4, 7 and 8). Moreover, basal activity of the uninduced activators was also not affected by Gal4(AD)–H3 (Fig. 6, lanes 1, 2, 5 and 6). Activity of Leu3 was induced ~2 times in the presence of Gal4(AD)–H3 (lanes 9 and 10) while it was slightly reduced for Gal4 (lanes 11 and 12). Other activators tested (Gcn4 and Hap2/3/4/5) were minimally affected by expression of Gal4(AD)–H3 (Fig. 6, lanes 13–16). Note that activity of Leu3 and Uga3 was assayed with reporters almost identical to CYC7–lacZ, the only difference being the UAS inserted upstream of the minimal CYC1 gene. Thus, the effect of Gal4(AD)–H3 is restricted to a subset of transcriptional activators (Hap1 and, to a lesser extent, Leu3).
Hyperactivation by Gal4(AD)–H3 does not correlate with the strength of the activator
The results presented above indicate that hyperactivation by Gal4(AD)–H3 is restricted to Hap1 and, to a lesser extent, Leu3. However, activation by Hap1 is relatively weak when compared with the other activators tested above. Thus, the action of Gal4(AD)–H3 may be restricted to weak activators. To test this possibility, we expressed the chimeric activator Gal4(1–147)–VP16 (and mutants that show reduced transcriptional activity) along with Gal4(AD)–H3 or Gal4(AD) in a yeast strain containing an integrated GAL1–lacZ reporter. Expression of Gal4(AD)–H3 had no effect on ‘wild-type’ Gal4(1–147)–VP16 activity (Fig. 7, lanes 1 and 2). Similarly, a mutant with greatly reduced activity was insensitive to Gal4(AD)–H3 (mutant F475A; Fig. 7, lanes 3 and 4). Some mutants showed increased activity in the presence of Gal4(AD)–H3 but the effect was 2-fold or less (Fig. 7, lanes 5–8), as opposed to many-fold for Hap1. In addition, no effect was observed for mutants that are greatly impaired (or inactive) for transcriptional activation (Fig. 7, lanes 9–12). As observed for Hap1 reporters, no significant increase in reporter activity was observed in the absence of an activator (Fig. 7, lanes 13 and 14). Thus, our results show that there is no correlation between the strength of an activator and the response to Gal4(AD)–H3.
Figure 7.

Hyperactivation by Gal4(AD)–H3 is not dependent on the strength of the activator. Expression vectors for Gal4(1–147)–VP16C (or mutants) along with an expression vector for Gal4(AD)–H3 (or the empty vector) were transformed into yeast strain Y190, which contains an integrated GAL1–lacZ reporter, and β-galactosidase activity assayed. Amino acid changes in the Gal4(1–147)–VP16C mutants (85) are given on the left. + refers to the presence of the expression vector for Gal4(AD)–H3, – to the empty vector pACT2 (80).
Effect of Gal4(AD)–H3 on the expression of endogenous genes
We further extended our analysis of the specificity of Gal4(AD)–H3 by monitoring mRNA levels expressed from endogenous genes by northern blot analysis (Fig. 8). RNA levels for ARO7 and YJU2 were not significantly affected by Gal4(AD)–H3 while they were slightly reduced for SHR5 (Fig. 8). Interestingly, a drastic reduction in ADH2 mRNA was observed in the presence of Gal4(AD)–H3. Thus, the results show that Gal4(AD)–H3 can have both positive and negative effects on gene expression.
Figure 8.
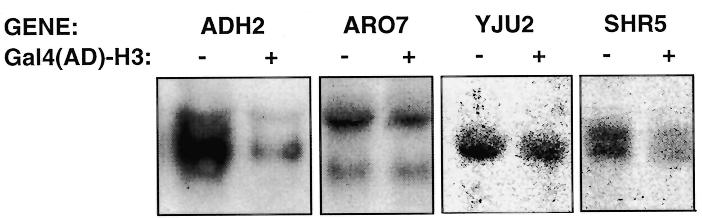
Effect of Gal4(AD)–H3 on the expression of endogenous genes. Northern blot analysis of endogenous genes. Yeast strain T17 was transformed with an expression vector for Gal4(AD)–H3 or the parental vector pACT2 (80) and RNA isolated for northern blot analysis (see Materials and Methods). Twenty micrograms of total RNA were loaded per lane. GENE, probe used for northern blot analysis; +, presence of the expression vector for Gal4(AD)–H3; –, empty vector pACT2 (80).
DISCUSSION
In this report, we have shown that activity of the yeast transcriptional activator Hap1 is strongly dependent on a functional SWI complex, known to be involved in chromatin remodeling. In order to identify proteins that interact with Hap1 and may be responsible for its activation properties, we set up a modified two-hybrid screen where wild-type Hap1 was used as bait. Strikingly, plasmids encoding fusion of the Gal4 activation domain to histone H3 were isolated. It is noteworthy that fusions encoded by both H3 yeast genes were recovered from the screen. The fusions were shown to increase Hap1 activity by 8- to 10-fold at target sites on both episomal and integrated reporters. Characterization of the effect of Gal4(AD)–H3 fusions has shown that a functional Hap1 activator is required for hyperactivation as well as a functional SWI complex. Importantly, overexpression of histone H3 had no effect on Hap1 activity, showing that the effect of Gal4(AD)–H3 does not involve a histone imbalance as observed for other transcription factors (4–7).
Figure 9 shows models that could account for the hyperactivation of Hap1 by Gal4(AD)–H3. One possibility is that the H3 fusion is incorporated within the nucleosome. This would result in the formation of less stable nucleosomes and, hence, more facilitated access of Hap1 (or other transcription factors) to target DNA sites (Fig. 9b). However, results from a deletion analysis of Gal4(AD)–H3 argue against this model. In vivo studies in Xenopus have shown that the last 10 amino acids of histone H3 are essential for incorporation of H3 into the nucleosome (87). Interestingly, deletion of 15 amino acids at the C-terminus of Gal4(AD)–H3 does not impair hyperactivation of Hap1 by the fusion protein (Fig. 4). Thus our results imply that Gal4(AD)–H3 must be acting out of the nucleosome. In agreement with these results, Gal4(AD)–H3 cannot substitute for histone H3 (Fig. 5). Taken together, these results strongly suggest that Gal4(AD)–H3 acts outside the nucleosomal context.
Analysis of the Gal4 sequences of the fusion protein revealed that a H3 fusion carrying a non-functional activation domain of Gal4 hyperactivates Hap1, although to a lesser extent than Gal4(AD)–H3 (Fig. 4). Moreover, a fusion of the strong activating domain of VP16 with H3 does not affect Hap1 activity (Fig. 4). This is in contrast to another study where fusion of VP16 and H3 has been shown to increase activity of the proline-rich CTF transcription factor (68,88). Our results suggest that hyperactivation of Hap1 requires a critical size of the fusion protein. Importantly, the results strongly suggest that Gal4(AD)–H3 does not act by a classical ‘two-hybrid’ effect (Fig. 9c), even though the initial strategy that allowed recovery of Gal4(AD)–H3 was based on such a screen.
Many of the activators tested are minimally or not responsive to the action of Gal4(AD)–H3 (Figs 6–8). Moreover, there is no correlation between the strength of the activator and the response to Gal4(AD)–H3 (Figs 6 and 7). Although the activity of Hap1, Leu3 and Uga3 is strongly dependent on the SWI complex (Fig. 1 and data not shown), Gal4(AD)–H3 shows the strongest effect on Hap1 (8-fold), a moderate effect on Leu3 (2-fold) and no effect on Uga3. Thus, there is no correlation between dependence on the SWI complex and effect of Gal(AD)–H3. Although a functional Gal4 activation domain is not required for hyperactivation, the results do not rule out the possibility that Hap1 interacts with histone H3. Other proteins have been shown to interact with H3, including Spt6, a yeast protein involved in controlling transcription elongation (69), the yeast repressor Tup1 (70) and the silent information regulators Sir3 and Sir4, involved in transcriptional repression of the yeast silent mating loci and telomeric regions (71). For Tup1, Sir3 and Sir4, interaction was shown to occur with the N-terminal tail of histone H3. In contrast, our deletion analysis of Gal4(AD)–H3 has shown that amino acids 20–50 of histone H3 are required for hyperactivation of Hap1. These amino acids have been shown not to be essential for assembly within the nucleosome or heterodimerization with histone H4 (87). Thus, Hap1 may interact with the N-terminus of histone H3 (amino acids 20–50).
How can we explain the hyperactivation of Hap1 by Gal4(AD)–H3? Since Gal4(AD)–H3 is not incorporated into the nucleosome, it is possible that Hap1 forms, in the absence of DNA, a complex with Gal4(AD)–H3 (or histone H3 in a natural context) and other factors. Interestingly, histone H3 (and H4) have been shown to be part of the upstream activation factor, a transcription complex involved in high level transcription by RNA polymerase I (72). Overexpression of Gal4(AD)–H3 may favor formation of a complex with Hap1, unlike wild-type H3, which is more readily incorporated into chromatin (Fig. 9d). Interestingly, our unpublished results show that high levels of the H3 fusion protein are required since no effect on Hap1 activity is observed when Gal4(AD)–H3 is expressed from a low copy number plasmid. Our data suggest that, to some extent, activation of transcription by Hap1 differs from other transcriptional activators tested in this study. Activators have been shown to interact directly with the SWI complex (82–84). Thus, Hap1 may interact with both the SWI complex and histone H3. Formation of a complex between Hap1 and H3 may therefore favor chromatin remodeling by the SWI complex, resulting in increased transcription. In summary, we have isolated a novel histone H3 variant whose expression results in highly specific alteration of gene expression.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Drs F. Winston (Harvard), X.-J. Yang (McGill), S. Berger (Wistar Institute) and M. Featherstone (McGill) for comments and suggestions on this work. We also thank members of the Laboratory of Molecular Endocrinology for stimulating discussions. We thank Drs M. Smith, S. Berger, C. L. Peterson, S. Elledge, S. J. Triezenberg and K. R. Yamamoto for providing reagents (yeast strains, plasmids and two-hybrid library). We also thank Drs M. Raymond and S. Roudani for advice on northern blot analysis. This work was supported by grants from the Natural Sciences and Engineering Research Council of Canada and the Canadian Genome and Analysis Technology Program (Canada). B.T. is a scholar from the Fonds de la recherche en santé du Québec (FRSQ). N.H. was supported by a studentship from FRSQ.
REFERENCES
- 1.Wolffe A. (1998) Chromatin, Structure and Function. Academic Press, London, UK.
- 2.Wolffe A.P. and Hayes,J.J. (1999) Nucleic Acids Res., 27, 711–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luger K., Mader,A.W., Richmond,R.K., Sargent,D.F. and Richmond,T.J. (1997) Nature, 389, 251–260. [DOI] [PubMed] [Google Scholar]
- 4.Clark-Adams C.D., Norris,D., Osley,M.A., Fassler,J.S. and Winston,F. (1988) Genes Dev., 2, 150–159. [DOI] [PubMed] [Google Scholar]
- 5.Han M. and Grunstein,M. (1988) Cell, 55, 1137–1145. [DOI] [PubMed] [Google Scholar]
- 6.Kim U.-J., Han,M., Kayne,P. and Grunstein,M. (1988) EMBO J., 7, 2211–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han M., Kim,U.-J., Kayne,P. and Grunstein,M. (1988) EMBO J., 7, 2221–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Durrin L.K., Mann,R.K., Kayne,P.S. and Grunstein,M. (1991) Cell, 65, 1023–1031. [DOI] [PubMed] [Google Scholar]
- 9.Mann R.K. and Grunstein,M. (1992) EMBO J., 11, 3297–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lenfant F., Mann,R.K., Thomsen,B., Ling,X. and Grunstein,M. (1996) EMBO J., 15, 3974–3985. [PMC free article] [PubMed] [Google Scholar]
- 11.Kayne P.S., Kim,U.-J., Han,M., Mullen,J.R., Yoshizaki,F. and Grunstein,M. (1988) Cell, 55, 27–39. [DOI] [PubMed] [Google Scholar]
- 12.Thompson J.S., Ling,X. and Grunstein,M. (1994) Nature, 369, 245–247. [DOI] [PubMed] [Google Scholar]
- 13.Huang L., Zhang,W. and Roth,S.Y. (1997) Mol. Cell. Biol., 17, 6555–6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Santisteban M.S., Arents,G., Moudrianakis,E.N. and Smith,M.M. (1997) EMBO J., 16, 2493–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Workman J.L. and Kingston,R.E. (1998) Annu. Rev. Biochem., 67, 545–579. [DOI] [PubMed] [Google Scholar]
- 16.Struhl K. (1998) Genes Dev., 12, 599–606. [DOI] [PubMed] [Google Scholar]
- 17.Gregory P.D. and Horz,W. (1998) Eur. J. Biochem., 251, 9–18. [DOI] [PubMed] [Google Scholar]
- 18.Travers A. (1999) Cell, 96, 311–314. [DOI] [PubMed] [Google Scholar]
- 19.Brownell J.E., Zhou,J., Ranalli,T., Kobayashi,R., Edmondson,D.G., Roth,S.Y. and Allis,C.D. (1996) Cell, 84, 843–851. [DOI] [PubMed] [Google Scholar]
- 20.Candau R., Zhou,J.X., Allis,C.D. and Berger,S.L. (1997) EMBO J., 16, 555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grant P.A., Duggan,L., Côté,J., Roberts,S.M., Brownell,J.E., Candau,R., Ohba,R., Owenhughes,T., Allis,C.D., Winston,F., Berger,S.L. and Workman,J. (1997) Genes Dev., 11, 1640–1650. [DOI] [PubMed] [Google Scholar]
- 22.Sterner D.E., Grant,P.A., Roberts,S.M., Duggan,L.J., Belotserkovskaya,R., Pacella,L.A., Winston,F., Workman,J.L. and Berger,S.L. (1999) Mol. Cell. Biol., 19, 86–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Struhl K. and Moqtaderi,Z. (1998) Cell, 94, 1–4. [DOI] [PubMed] [Google Scholar]
- 24.Wang L., Liu,L. and Berger,S.L. (1998) Genes Dev., 12, 640–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuo M.H., Brownell,J.E., Sobel,R.E., Ranalli,T.A., Cook,R.G., Edmondson,D.G., Roth,S.Y. and Allis,C.D. (1996) Nature, 383, 269–272. [DOI] [PubMed] [Google Scholar]
- 26.Utley R.T., Ikeda,K., Grant,P.A., Côté,J., Steger,D.J., Eberharter,A., John,S. and Workman,J.L. (1998) Nature, 394, 498–502. [DOI] [PubMed] [Google Scholar]
- 27.Ikeda K., Steger,D.J., Eberharter,A. and Workman,J.L. (1999) Mol. Cell. Biol., 19, 855–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kadonaga J.T. (1998) Cell, 92, 307–313. [DOI] [PubMed] [Google Scholar]
- 29.Wang W., Xue,Y., Zhou,S., Kuo,A., Cairns,B.R. and Crabtree,G.R. (1996) Genes Dev., 10, 2117–2130. [DOI] [PubMed] [Google Scholar]
- 30.Kingston R.E., Bunker,C.A. and Imbalzano,A.N. (1996) Genes Dev., 10, 905–920. [DOI] [PubMed] [Google Scholar]
- 31.Owen-Hughes T., Utley,R.T., Côté,J., Peterson,C.L. and Workman,J.L. (1996) Science, 273, 513–516. [DOI] [PubMed] [Google Scholar]
- 32.Hirschhorn J.N., Brown,S.A., Clark,C.D. and Winston,F. (1992) Genes Dev., 6, 2288–2298. [DOI] [PubMed] [Google Scholar]
- 33.Côté J., Quinn,J., Workman,J.L. and Peterson,C.L. (1994) Science, 265, 53–60. [DOI] [PubMed] [Google Scholar]
- 34.Kwon H., Imbalzano,A.N., Khavari,P.A., Kingston,R.E. and Green,M.R. (1994) Nature, 370, 477–481. [DOI] [PubMed] [Google Scholar]
- 35.Burns L.G. and Peterson,C.L. (1997) Mol. Cell. Biol., 17, 4811–4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Imbalzano A.N., Schnitzler,G.R. and Kingston,R.E. (1996) J. Biol. Chem., 271, 20726–20733. [DOI] [PubMed] [Google Scholar]
- 37.Schnitzler G., Sif,S. and Kingston,R.E. (1998) Cell, 94, 17–27. [DOI] [PubMed] [Google Scholar]
- 38.Côté J., Peterson,C.L. and Workman,J.L. (1998) Proc. Natl Acad. Sci. USA, 95, 4947–4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ryan M.P., Jones,R. and Morse,R.H. (1998) Mol. Cell. Biol., 18, 1774–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Quinn J., Fyrberg,A.M., Ganster,R.W., Schmidt,M.C. and Peterson,C.L. (1996) Nature, 379, 844–847. [DOI] [PubMed] [Google Scholar]
- 41.O’Neill D., Yang,J., Erdjument-Bromage,H., Bornschlegel,K., Tempst,P. and Bank,A. (1999) Proc. Natl Acad. Sci. USA, 96, 349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilson C.J., Chao,D.M., Imbalzano,A.N., Schnitzler,G.R., Kingston,R.E. and Young,R.A. (1996) Cell, 84, 235–244. [DOI] [PubMed] [Google Scholar]
- 43.Cairns B.R., Lorch,Y., Li,Y., Zhang,M., Lacomis,L., Erdjument-Bromage,H., Tempst,P., Du,J., Laurent,B. and Kornberg,R.D. (1996) Cell, 87, 1249–1260. [DOI] [PubMed] [Google Scholar]
- 44.Lohr D., Venkov,P. and Zlatanova,J. (1995) FASEB J., 9, 777–787. [DOI] [PubMed] [Google Scholar]
- 45.Pfeifer K., Kim,K.S., Kogan,S. and Guarente,L. (1989) Cell, 56, 291–301. [DOI] [PubMed] [Google Scholar]
- 46.Ha N., Hellauer,K. and Turcotte,B. (1996) Nucleic Acids Res., 24, 1453–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim K.S. and Guarente,L. (1989) Nature, 342, 200–203. [DOI] [PubMed] [Google Scholar]
- 48.Turcotte B. and Guarente,L. (1992) Genes Dev., 6, 2001–2009. [DOI] [PubMed] [Google Scholar]
- 49.King D.A., Zhang,L., Guarente,L. and Marmorstein,R. (1999) Nature Struct. Biol., 6, 22–27. [DOI] [PubMed] [Google Scholar]
- 50.King D.A., Zhang,L., Guarente,L. and Marmorstein,R. (1999) Nature Struct. Biol., 6, 64–71. [DOI] [PubMed] [Google Scholar]
- 51.Sikorski R.S. and Hieter,P. (1989) Genetics, 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baudin A., Ozier-Kalogeropoulos,O., Denouel,A., Lacroute,F. and Cullin,C. (1993) Nucleic Acids Res., 21, 3329–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Noël J. and Turcotte,B. (1998) J. Biol. Chem., 273, 17463–17468. [DOI] [PubMed] [Google Scholar]
- 54.Ausubel F.M., Brent,R., Kingston,R.E., Moore,D.D., Seidman,J.G., Smith,J.A. and Struhl,K. (1989) Current Protocols in Molecular Biology. Greene Publishing Associates and Wiley-Interscience, New York, NY.
- 55.Kunkel T.A. (1985) Proc. Natl Acad. Sci. USA, 82, 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vojtek A.B., Hollenberg,S.M. and Cooper,J.A. (1993) Cell, 74, 205–214. [DOI] [PubMed] [Google Scholar]
- 57.Sikorski R.S. and Boeke,J.D. (1991) Methods Enzymol., 194, 302–318. [DOI] [PubMed] [Google Scholar]
- 58.Green S., Walter,P., Kumar,V., Krust,A., Bornert,J.M., Argos,P. and Chambon,P. (1986) Nature, 320, 134–139. [DOI] [PubMed] [Google Scholar]
- 59.Mumberg D., Muller,R. and Funk,M. (1994) Nucleic Acids Res., 22, 5767–5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chevray P.M. and Nathans,D. (1992) Proc. Natl Acad. Sci. USA, 89, 5789–5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Christianson T.W., Sikorski,R.S., Dante,M., Shero,J.H. and Hieter,P. (1992) Gene, 110, 119–122. [DOI] [PubMed] [Google Scholar]
- 62.Schena M. and Yamamoto,K.R. (1988) Science, 241, 965–967. [DOI] [PubMed] [Google Scholar]
- 63.Sherman F., Fink,G.R. and Hicks,J.B. (1986) Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 64.Guarente L. and Mason,T. (1983) Cell, 32, 1279–1286. [DOI] [PubMed] [Google Scholar]
- 65.Holstege F.C.P., Jennings,E.G., Wyrick,J.J., Lee,T.I., Hengartner,C.J., Green,M.R., Golub,T.R., Lander,E.S. and Young,R.A. (1998) Cell, 95, 717–728. [DOI] [PubMed] [Google Scholar]
- 66.Yoshinaga S.K., Peterson,C.L., Herskowitz,I. and Yamamoto,K.R. (1992) Science, 258, 1598–1604. [DOI] [PubMed] [Google Scholar]
- 67.Fields S. and Song,O. (1989) Nature, 340, 245–246. [DOI] [PubMed] [Google Scholar]
- 68.Alevizopoulos A., Dusserre,Y., Tsai-Pflugfelder,M., von der Weid,T., Wahli,W. and Mermod,N. (1995) Genes Dev., 9, 3051–3066. [DOI] [PubMed] [Google Scholar]
- 69.Hartzog G.A.,Wada,T., Handa,H. and Winston,F. (1998) Genes Dev., 12, 357–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Edmondson D.G., Smith,M.M. and Roth,S.Y. (1996) Genes Dev., 10, 1247–1259. [DOI] [PubMed] [Google Scholar]
- 71.Hecht A., Strahl-Bolsinger,S. and Grunstein,M. (1996) Nature, 383, 92–96. [DOI] [PubMed] [Google Scholar]
- 72.Keener J., Dodd,J.A., Lalo,D. and Nomura,M. (1997) Proc. Natl Acad. Sci. USA, 94, 13458–13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hellauer K., Rochon,M.-H. and Turcotte,B. (1996) Mol. Cell. Biol., 16, 6096–6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guarente L., Yocum,R.R. and Gifford,P. (1982) Proc. Natl Acad. Sci. USA, 79, 7410–7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Metzger D., Losson,R., Bornert,J.M., Lemoine,Y. and Chambon,P. (1992) Nucleic Acids Res., 20, 2813–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Arndt K.T., Styles,C. and Fink,G.R. (1987) Science, 237, 874–880. [DOI] [PubMed] [Google Scholar]
- 77.Guarente L., Lalonde,B., Gifford,P. and Alani,E. (1984) Cell, 36, 503–511. [DOI] [PubMed] [Google Scholar]
- 78.André B. (1990) Mol. Gen. Genet., 220, 269–76. [DOI] [PubMed] [Google Scholar]
- 79.Chen J., Ding,M. and Pederson,D.S. (1994) Proc. Natl Acad. Sci. USA, 91, 11909–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bai C. and Elledge,S.J. (1997) Methods Enzymol., 283, 141–156. [DOI] [PubMed] [Google Scholar]
- 81.Durfee T., Becherer,K., Chen,P.L., Yeh,S.H., Yang,Y., Kilburn,A.E., Lee,W.H. and Elledge,S.J. (1993) Genes Dev., 7, 555–569. [DOI] [PubMed] [Google Scholar]
- 82.Natarajan K., Jackson,B.M., Zhou,H., Winston,F. and Hinnebusch,A.G. (1999) Mol. Cell, 4, 657–664. [DOI] [PubMed] [Google Scholar]
- 83.Neely K.E., Hassan,A.H., Wallberg,A.E., Steger,D.J., Cairns,B.R., Wright,A.P.H. and Workman,J.L. (1999) Mol. Cell, 4, 649–655. [DOI] [PubMed] [Google Scholar]
- 84.Yudkovsky N., Logie,C., Hahn,S. and Peterson,C.L. (1999) Genes Dev., 13, 2369–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sullivan S.M., Horn,P.J., Olson,V.A., Koop,A.H., Niu,W., Ebright,R.H. and Triezenberg,S.J. (1998) Nucleic Acids Res., 26, 4487–4496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Alarco A.M., Balan,I., Talibi,D., Mainville,N. and Raymond,M. (1997) J. Biol. Chem., 272, 19304–19313. [DOI] [PubMed] [Google Scholar]
- 87.Freeman L., Kurumizaka,H. and Wolffe,A.P. (1996) Proc. Natl Acad. Sci. USA, 93, 12780–12785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wolffe A.P. and Pruss,D. (1996) Curr. Biol., 6, 234–237. [DOI] [PubMed] [Google Scholar]