Regulation of HCN Channel Surface Expression by a Novel C-Terminal Protein-Protein Interaction (original) (raw)
Abstract
Hyperpolarization-activated cation currents (_I_h) are carried by channels encoded by a family of four genes (HCN1-4) that are differentially expressed within the brain in specific cellular and subcellular compartments. HCN1 shows a high level of expression in apical dendrites of cortical pyramidal neurons and in presynaptic terminals of cerebellar basket cells, structures with a high density of _I_h. Expression of _I_h is also regulated by neuronal activity. To isolate proteins that may control HCN channel expression or function, we performed yeast two-hybrid screens using the C-terminal cytoplasmic tails of the HCN proteins as bait. We identified a brain-specific protein, which has been previously termed TRIP8b (for TPR-containing Rab8b interacting protein) and PEX5Rp (for Pex5p-related protein), that specifically interacts with all four HCN channels through a conserved sequence in their C-terminal tails. In situ hybridization and immunohistochemistry show that TRIP8b and HCN1 are colocalized, particularly within dendritic arbors of hippocampal CA1 and neocortical layer V pyramidal neurons. The dendritic expression of TRIP8b in layer V pyramidal neurons is disrupted after deletion of HCN1 through homologous recombination, demonstrating a key in vivo interaction between HCN1 and TRIP8b. TRIP8b dramatically alters the trafficking of HCN channels heterologously expressed in Xenopus oocytes and human embryonic kidney 293 cells, causing a specific decrease in surface expression of HCN protein and _I_h density, with a pronounced intracellular accumulation of HCN protein that is colocalized in discrete cytoplasmic clusters with TRIP8b. Finally, TRIP8b expression in cultured pyramidal neurons markedly decreases native _I_h density. These data suggest a possible role for TRIP8b in regulating HCN channel density in the plasma membrane.
Keywords: hyperpolarization-activated channel, _I_h, dendrite, hippocampus, endosome, trafficking
Introduction
Ion channel expression is regulated through various cellular mechanisms that fine tune electrical activity over a range of time scales (Moss and Henley, 2002). One important means of controlling channel function and expression is through direct interactions with accessory proteins (Sheng, 2001; Arikkath and Campbell, 2003; Kass et al., 2003). Some of these proteins, such as β subunits, form an integral, fixed component of the channel. Other proteins associate with channels transiently to dynamically regulate channel assembly and trafficking.
Here we address the importance of protein-protein interactions for the control of expression and function of the hyperpolarization-activated, cyclic nucleotide-regulated (HCN) channels, which generate the hyperpolarization-activated cation current _I_h (Robinson and Siegelbaum, 2003). The biophysical properties of _I_h vary among different cells, reflecting the diverse functions of these currents, including determination of the resting membrane potential, dendritic integration, pacemaker activity, and modulation of synaptic transmission. The heterogeneity of _I_h is attributable to the presence of four closely related HCN genes (HCN1-4) that generate _I_h channels with distinct biophysical properties and tissue distribution (Santoro et al., 1998, 2000; Moosmang et al., 2001). Recently, the integral membrane protein MiRP1 (Mink-related protein 1) has been found to serve as a β subunit for HCN channels (Yu et al., 2001; Qu et al., 2004).
The distribution of HCN1 has been resolved in fine detail (Santoro et al., 1997; Lorincz et al., 2002; Notomi and Shigemoto, 2004), which has revealed differential protein localization within specific neurons and subcellular compartments that is dependent on cell type. Thus, inhibitory basket cells express the highest density of HCN1 protein within their presynaptic terminals, whereas neocortical and hippocampal pyramidal neurons show a distinct somatodendritic gradient of HCN1 protein density, with expression in apical dendrites increasing as a function of distance from the soma.
Recent studies indicate that levels of _I_h expression are regulated during development (Bender et al., 2001; Vasilyev and Barish, 2002), by abnormal patterns of neuronal activity associated with seizures (K. Chen et al., 2001; Brewster et al., 2002; Santoro and Baram, 2003) and neuropathic pain (Chaplan et al., 2003), and by physiological patterns of activity during induction of long-term synaptic plasticity (Wang et al., 2003; Van Welie et al., 2004). We have addressed possible molecular mechanisms that contribute to the spatial and temporal control of _I_h expression by identifying a protein expressed in the nervous system that specifically interacts with HCN channels to regulate their expression.
We describe a novel protein-protein interaction between the C-terminal cytoplasmic tails of HCN channel proteins and a brain-specific protein, which has been variously termed TPR-containing Rab8b interacting protein (TRIP8b), Pex5p-related protein (PEX5Rp), and H-channel interacting protein 1 (HIP1) (Amery et al., 2001; S. Chen et al., 2001; Santoro et al., 2002). We refer to this protein here as TRIP8b, adopting the nomenclature of S. Chen et al. (2001), who identified this protein on the basis of its interaction with Rab8b, a member of the Rab family of small GTPase proteins important for vesicle trafficking. Binding of TRIP8b to the HCN channels leads to a drastic reduction in their surface expression, both in heterologous expression systems and in neurons. Because TRIP8b is expressed at very high levels in the apical dendrites of pyramidal neurons, where it colocalizes strongly with the HCN1 channel protein, we suggest a possible role for this interaction in the regulation of local _I_h density.
Materials and Methods
Yeast two-hybrid screen
A yeast two-hybrid screen was performed using the Grow'N′Glow Two-Hybrid kit (Bio101, Vista, CA) and yeast strain EGY48. A mouse brain cDNA library was constructed in the vector pJG4-5 using random hexamer priming (Santoro et al., 1997). The C-terminal bait constructs were ligated in vector pEG202 and contained the following HCN channel sequences: mHCN1, amino acids 777-910; mHCN2, amino acids 758-863; mHCN3, amino acids 586-779; mHCN4, amino acids 1035-1201; mHCN1ΔC, amino acids 777-867. Colonies visible after 3-5 d of growth in galactose +-Leu - selective medium were screened for green fluorescent protein (GFP) expression under a UV light. Plasmid DNA was isolated from GFP-positive colonies and tested for specificity after retransformation into an independent yeast strain in the presence of the pEG202 vector that lacked or contained the indicated bait sequences.
Glutathione S_-transferase pull-down assay_
The p-glutathione _S_-transferase (pGST)-HCN1 plasmid was obtained by subcloning amino acids 777-910 (stop) of HCN1 into pGEX-1λT (Amersham Biosciences, Arlington Heights, IL). Bacterially expressed GST-HCN1 fusion proteins were prepared as described previously (Santoro et al., 1997), except that proteins were not eluted from glutathione-agarose beads. In vitro translation of TRIP8b was performed using the Rabbit Reticulocyte Lysate System (Promega, Madison, WI) and TRIP8b mRNA transcribed from plasmid pGH-TRIP8b (see below), in the presence of [35S]methionine. Binding reactions were performed by adding the in vitro translation reaction mix (diluted 1:20 in PBS/0.1% Triton X-100) to 20 μl of GST control beads or GST-HCN1 beads for 1 hr at room temperature. Beads were washed extensively in PBS/0.1% Triton X-100, and proteins were eluted directly in Laemmli buffer for SDS-PAGE electrophoresis.
Northern blot and in situ hybridization
A PCR-amplified cDNA fragment corresponding to amino acids 467-600 of TRIP8b was used to probe a mouse Multiple Tissue Northern Blot (Clontech, Cambridge, UK) using Express Hyb (Clontech), according to the manufacturer's instructions. In situ hybridization was performed essentially as described previously (Wisden and Morris, 1994) using oligonucleotide probes labeled with [33P]ATP and terminal transferase. Hybridizations were performed at 42°C (50% formamide, 4× SSC, 10% dextran sulfate) and washes at 60°C in 1× SSC. Probes corresponded to amino acids 648-657 of HCN1 and amino acids 531-546 of TRIP8b.
Immunohistochemistry
Immunohistochemistry was performed on free-floating sections (40 μm) from adult mouse brain (age, 8-12 weeks), cut after perfusion with 4% PFA/PBS. Slices were washed in PBS plus 0.1% Triton X-100 and then blocked with PBS plus 3% goat serum. Primary antibody labeling was performed overnight at 4°C in blocking solution. The following antibodies were used: affinity-purified rabbit polyclonal antibody against HCN1 (Alomone Labs, Jerusalem, Israel; dilution 1:100), rat monoclonal antibody against HCN1 (generous gift from F. Mueller and B. Kaupp, Forschungszentrum Julich, Germany; dilution 1:25), and affinity-purified rabbit polyclonal antibody against TRIP8b (generous gift from S. Chen and A. Ting, Institute of Molecular and Cell Biology, Singapore; dilution 1:50), as indicated in figure legends. Secondary antibody labeling was performed for 2-4 hr at room temperature in blocking solution. The following antibodies were used: rhodamine-red-X-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA; dilution 1:200) and FITC-conjugated goat anti-rat IgG (Jackson ImmunoResearch; dilution 1:200), as appropriate. Double labeling was performed in the presence of the rat monoclonal antibody against HCN1 and the rabbit polyclonal antibody against TRIP8b. Negative controls for double-labeling immunofluorescence included omitting the primary antibody against TRIP8b (all mouse lines) or omitting the primary antibody against HCN1 (wild-type, HCN1f/f and HCN1f/f,cre lines); in all cases, no cross-labeling or signal spillover was observed. Labeling was detected using confocal microscope imaging (see figure legends).
The anti-TRIP8b polyclonal antibody (S. Chen et al., 2001) was raised against the N-terminal 190 amino acids of the TRIP8b sequence reported in GenBank accession number AF324454, which is not the same splicing variant used in the present study; however, this polyclonal antibody is expected to recognize all splice variants of TRIP8b (see Results), which differ only in amino acid sequence N-terminal to position 56 in the mouse protein described here (which corresponds to position 43 in the protein of GenBank accession number AF324454). The finding of S. Chen et al. (2001) that Western blot analysis of native tissue reveals only a single band (which we confirmed here; data not shown) could be attributed to the fact that the size difference between the splicing variants may be too small to resolve (exons 1-4 each encode only 7-35 amino acids) or that one splice variant is predominantly expressed in vivo (a quantitative difference that would be obscured by the nonquantitative RT-PCR approach used here). Despite this uncertainty, we assume that our immunohistochemistry results reflect the distribution of all splicing isoforms of TRIP8b.
Plasmids and constructs for heterologous expression
Oocyte expression. Mouse HCN1 and HCN2 cDNAs were subcloned into pGH19 and pGHE expression vectors, respectively (Santoro et al., 2000). Deletion constructs were generated essentially as described previously (Wainger et al., 2001) by introducing a premature stop codon and an overlapping _Xba_I site using the PCR. Truncation mutants were HCN2ΔC-X (Q665stop) and HCN1ΔSNL (S908stop). The mouse TRIP8b cDNA (same as GenBank accession AB032591, except for P110T) was amplified by RT-PCR from mouse brain poly(A+) RNA (Clontech) and subcloned into vector pGHE. The enhanced GFP cDNA, between sites _Age_I and _Eco_RI in the pEGFP-C1 vector (Clontech), was inserted into plasmid pGH-TRIP8b to obtain a GFP-TRIP8b fusion product and into plasmid pGH-HCN2 to obtain a GFP-HCN2 fusion product. Both fusion proteins contain the GFP moiety N-terminal to the channel protein. All cRNAs were transcribed from _Nhe_I-linearized (pGH-HCN1 constructs, pGH-TRIP8b constructs) or _Sph_I-linearized (pGH-HCN2 constructs) DNA using T7 RNA polymerase (Message Machine, Ambion, Austin, TX). Plasmids pCEM1 (GFP cDNA in pGHE vector) and pREG1 (GFP-CNGA1 fusion product in pGHE vector) were generous gifts from E. Young (Columbia University, New York). KAT1 was kindly provided by R. Gaber (Northwestern University, Evanston, IL). The plasmid encoding the dominant-negative subunit, HCN1-AAA, was kindly provided by R. Li (Johns Hopkins University, Baltimore, MD) (Xue et al., 2002).
Mammalian expression. For human embryonic kidney (HEK) 293 cell expression studies, the mouse TRIP8b cDNA was subcloned into the vector pCI (Promega) to obtain the plasmid pCI-TRIP8b (expression without GFP labeling) and into vectors pEGFP-C1 and pDsRed2-C1 (Clontech) to obtain plasmids expressing a GFP-TRIP8b and a DsRed-TRIP8b fusion protein, respectively. The mouse HCN1 and HCN2 cDNAs were subcloned into vector pEGFP-C1 (Clontech) to obtain pECHCN1 and pEC-HCN2, which express a GFP-HCN1 and a GFP-HCN2 fusion protein, respectively. All fusion proteins contain the GFP moiety N-terminal to the channel protein. For the construction of plasmid pECHCN2[hemagglutinin (HA)], a fragment between restriction sites _Nhe_I and _Pfl_MI from plasmid ntHCN2-HA (generous gift from M. Sanguinetti, University of Utah, Salt Lake City, UT) (Chen et al., 2000) was inserted in a corresponding position after partial digestion with _Nhe_I and _Pfl_MI of plasmid pEC-HCN2. The resulting construct contained the full-length mouse HCN2 sequence in frame with GFP, including an extracellular HA tag sequence inserted between transmembrane segments S3 and S4.
Oocyte injection and electrophysiology
Xenopus laevis oocytes were prepared and injected with 50 nl of cRNA solution each as described previously (Wainger et al., 2001), with RNA concentration kept at 0.5 μg/μl for HCN channel constructs and 0.2 μg/μl for GFP or GFP-TRIP8b. Experiments were performed using pairs of oocytes from the same frog a fixed number of days after the injection of HCN cRNA and either GFP cRNA (control) or a given experimental cRNA. Cell-attached patch recordings were obtained 4-6 d after cRNA injection so that control current levels would be consistently high. Data were digitized and acquired using an ITC-16 interface (InstruTech, Port Washington, NY) and acquired and analyzed with Pulse and PulseFit software (HEKA Electronik, Lambrecht/Pfalz, Germany). Data were filtered at 1 kHz and sampled at 2 kHz. The pipette solution contained (in mm): 107 KCl, 5 NaCl, 10 HEPES, and 2 MgCl2, pH 7.4 with KOH; the bath solution contained 107 KCl, 5 NaCl, 10 HEPES, 1 MgCl2, and 1 EGTA, pH 7.4 with KOH. The Ag-AgCl ground wire was connected to the bath solution by a 3 m KCl agar bridge electrode. Two microelectrode voltage-clamp recordings were obtained 3 d after cRNA injection (unless noted otherwise) using a Warner Instruments (Hamden, CT) OC-725C amplifier. Data were acquired, filtered, and analyzed as described above. The extracellular recording solution contained (in mm): 96 KCl, 2 NaCl, 10 HEPES, 2 MgCl2. The intracellular electrodes were filled with 3 m KCl. All recordings were obtained at room temperature (22-24°C).
Tail-current amplitudes were measured at -40 mV after the decay of capacitive transients. Tail current _I_-V curves were fitted by the following Boltzmann equation: I(V) = A1 + A2/{1 + exp[(V - _V_1/2)/_s_]}, in which A1 is the offset caused by holding current, A2 is the maximal tail-current amplitude, V is the test pulse voltage, _V_1/2 is the midpoint voltage of activation, and s is the slope (in millivolts). HCN channel current magnitude was determined from A2 rather than tail-current amplitude at a given voltage, because the latter may be affected by shifts in voltage dependence.
Oocytes were imaged using a Bio-Rad (Hercules, CA) MRC-1000 confocal microscope. cRNA concentrations were the same as used for patch-clamp recordings. Basic biophysical properties of the GFP-labeled channels were unaffected by fusion of GFP (data not shown).
HEK293 cell immunocytochemistry
HEK293 cells were transfected using Effectene (Qiagen, Hilden, Germany). Data were collected 24-48 hr after transfection. For immunocytochemistry, cells were washed in PBS, fixed in 3.7% formaldehyde for 25 min at 4°C, washed in PBS, and blocked with PBS plus 10% goat serum plus 0.25% Triton X-100, followed by antibody labeling in PBS plus 3% goat serum plus 0.25% Triton X-100 for 1 hr at room temperature. For nonpermeabilizing conditions, Triton X-100 was omitted from all solutions. Primary antibodies used were anti-HA (rat monoclonal 3F10; Roche, Hertfordshire, UK; 1:250); anti-calnexin (CNX) (mouse monoclonal AF18; Affinity BioReagents; 1:500); anti-early endosome marker protein (EEA1) (mouse monoclonal; BD Transduction Labs; 1:500). Secondary antibody labeling was performed using rhodamine-red-X-conjugated goat anti-rat IgG (Jackson ImmunoResearch; 1:200) or Cy3-conjugated goat anti-mouse IgG, and labeling was detected using a confocal microscope.
Sindbis virus construction
_Mlu_I and _Sph_I restriction sites were introduced by PCR to flank the GFP-TRIP8b fusion cDNA (see above), and this fragment was subcloned into the pSinRep vector (Invitrogen, Gaithersburg, MD) The resulting plasmid was linearized using _Not_I and in vitro transcribed using the SP6 promoter (Machine Message, Ambion). Baby hamster kidney cells were cotransfected with the GFP-TRIP8b and DH-BB helper virus cRNAs using the TransMessenger Transfection Reagent kit (Qiagen). Virus was harvested as described in the manufacturer's protocol (Invitrogen K750-01). GFP control Sindbis virus was kindly provided by A. Morozov (Unit on Behavioral Genetics, National Institute of Mental Health, Bethesda, MD). Neuronal cultures were infected with 5-20 μl of virus per 5 cm dish.
Neuronal culture and electrophysiology
Dissociated cultures were made from 2-d-old postnatal Sprague Dawley rats following the general method described in O'Dell et al. (1991), with modifications introduced by Evans et al. (1998). All material preparations and dissections were performed in a laminar flow culture hood. Cultures were grown on plastic dishes coated with poly-d-lysine and laminin in a 37°C, 5% CO2 incubator for 18-20 d before use. Neurons were visualized with phase-contrast microscopy using a Nikon inverted microscope equipped with an Intracellular Imaging 300W Xenon light source for fluorescence detection. Whole-cell patch recordings were obtained from pyramidal-shaped neurons using 3-5 MΩ borosilicate glass (KG-33, Garner Glass Company, Claremont, CA) pipettes. Data were digitized and acquired as above for patch experiments. Data were filtered at 1 kHz and sampled at 2 kHz (10 kHz filtering and 33 kHz sampling to monitor series resistance). The internal solution contained (in mm): 120 potassium methylsulfate, 20 KCl, 10 HEPES, 2 MgCl2, 0.1 EGTA, 4 Na2ATP, 0.3 Na2GTP, 7.5 phosphocreatine (pH adjusted to 7.30 with KOH). The external bath solution contained (in mm): 145 NaCl, 3 KCl, 10 HEPES, 3 CaCl2, 8 glucose, 2 MgCl2 (pH adjusted to 7.30 with NaOH). To eliminate spontaneous activity, 2 μm tetrodotoxin (TTX) (Calbiochem, La Jolla, CA) was added to the bath solution. The bath was per-fused in between recordings. Series resistance was monitored before and after recording protocols. Series resistance was not compensated and ranged from 10 to 20 MΩ.
All data were obtained in pairs, with one GFP-expressing neuron matched with each GFP-TRIP8b neuron. In recordings without TTX, all cells displayed firing patterns consistent with those expected for pyramidal neurons in that they showed rapid accommodation and low maximum firing rates. Cell health was confirmed by three criteria: visual inspection to note an intact membrane without blebbing or inclusion bodies, an input resistance >100 MΩ, and a resting potential more negative than -50 mV. Note that the resting potential for cultured hippocampal neurons has been found to be more depolarized than for pyramidal neurons from acute slice recordings (Evans et al., 1998). Cells were held at a potential of -55 mV and then stepped to a range of test potentials before tail currents were measured at -115 mV.
Results
A conserved C-terminal tripeptide sequence mediates the interaction of HCN channels with TRIP8b
We analyzed ∼106 clones from a mouse brain cDNA library in a yeast two-hybrid screen using the C-terminal 130 amino acids of the mouse HCN1 protein as a bait (Santoro et al., 1997). This region, although highly variable among the four HCN channel proteins, contains two highly conserved regions. The first is an 11 amino acid cassette, located ∼100 residues before the C terminus (Fig. 1_A,B_) (labeled “SAS”). The second is a potential class I PDZ (postsynaptic density-95/Discs large/zona occludens-1) domain-binding sequence corresponding to the terminal three amino acids (Fig. 1_A,B_) (“SNL” in HCN1, HCN2, and HCN4, and “ANM” in HCN3). Clones positive for interaction with the HCN1 bait were subsequently isolated and tested for interaction with baits derived from the corresponding region of HCN2, HCN3, and HCN4 (Fig. 1_A_).
Figure 1.
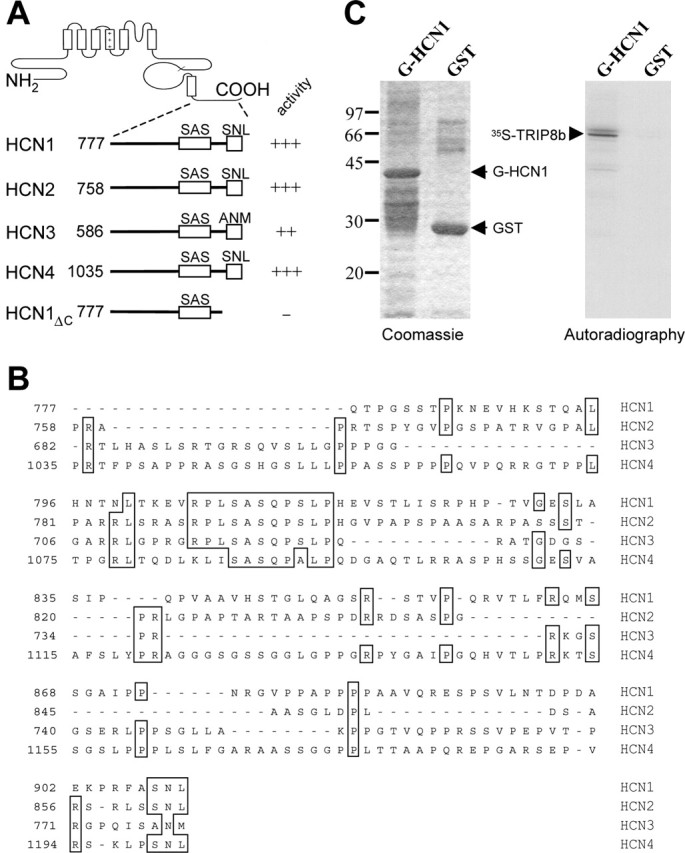
HCN channel proteins interact with TRIP8b through their conserved C-terminal end. A, Schematic representation of HCN baits used in the yeast two-hybrid screen and their respective activation of a GFP reporter gene in the presence of the TRIP8b target construct. Numbers on the left indicate the N-terminal starting position of bait construct in the HCN protein sequence. B, Alignment of the regions corresponding to the HCN baits. Numbers on the left indicate starting position (the HCN3 bait construct extends to position 586). C, GST pull-down assay. The panel on the left shows a Coomassie stain of protein product eluted from glutathione-agarose beads after incubation with in vitro translated TRIP8b; beads carried either a GST-HCN1 fusion protein (G-HCN1, amino acids 777-910) or a control GST protein (GST). Corresponding bands are indicated (arrows), demonstrating equal loading of beads. The panel on the right shows an autoradiogram from the same gel, with the 35S-signal corresponding to the full-length TRIP8b protein. Protein molecular weight markers are indicated on the left (in kilodaltons).
One target clone (corresponding to a partial mouse cDNA for TRIP8b) (S. Chen et al., 2001) displayed a strong, specific, and positive reaction with all four HCN-derived baits, suggesting that binding might be mediated by one of the conserved C-terminal cassettes. To identify the sequence, an additional bait was constructed that retained the first (SAS) but lacked the second (SNL) conserved cassette (Fig. 1_A_) (HCN1ΔC). The observed loss of positive signal from this bait suggests a specific involvement of the extreme C-terminal sequence, SNL, in binding the target protein. Finally, to rule out the possibility that binding might be mediated (i.e., bridged) by an intervening endogenous yeast protein, an in vitro binding assay was performed using a GST-HCN1 fusion protein (HCN1 bait) and an in vitro translated 35S-labeled TRIP8b target protein. The specific retention of the 35S-labeled TRIP8b protein on the column expressing the GST-HCN1 fusion product indicates that direct binding indeed occurs between the two proteins (Fig. 1_C_).
Sequence analysis of the cDNA revealed this protein to be the mouse ortholog of rat TRIP8b (S. Chen et al., 2001). The mouse TRIP8b is a 615 amino acid protein that contains an N-terminal domain with no sequence homology to other known proteins and a C-terminal domain containing six tetratricopeptide repeats (TPRs), a well characterized 34 amino acid protein binding motif (for review, see Blatch and Lassle, 1999). The human ortholog of TRIP8b has also been characterized and termed PEX5Rp because of the homology of its C-terminal TPR domain with that of the peroxisomal import protein PEX5p (Amery et al., 2001). Both the rat and human orthologs of TRIP8b were previously found to interact specifically with C-terminal tripeptide sequences closely related to SNL (Amery et al., 2001; S. Chen et al., 2001). In both cases, the binding was mediated by the C-terminal TPR domain of the protein (Amery et al., 2001; S. Chen et al., 2001).
A comparison of the various GenBank entries for TRIP8b orthologs (accession numbers AB032591, AF324454, AB032592, and AB032593) suggests that the N-terminal domain of the protein might be subject to alternative splicing. Based on our analysis of the intron-exon structure of the TRIP8b gene, as inferred by comparison of deposited cDNA and genomic sequences, we designed a series of oligonucleotides suitable for the specific amplification of individual splice forms. RT-PCR analysis of mouse brain cDNA demonstrated the presence of a number of alternatively spliced isoforms of TRIP8b (data not shown). All variants included one of two alternative starting exons and different combinations of exons 2-4; no variations were found in the splicing of exons 5-16, which comprise the bulk of the protein, including the C-terminal TPR region (Fig. 2_A_). Northern blot analysis confirmed that the expression of mouse TRIP8b is essentially restricted to the brain (Fig. 2_B_), as was shown for the rat and human TRIP8b/PEX5Rp orthologs (Amery et al., 2001; S. Chen et al., 2001), suggesting a primary role for this protein in nervous system function.
Figure 2.
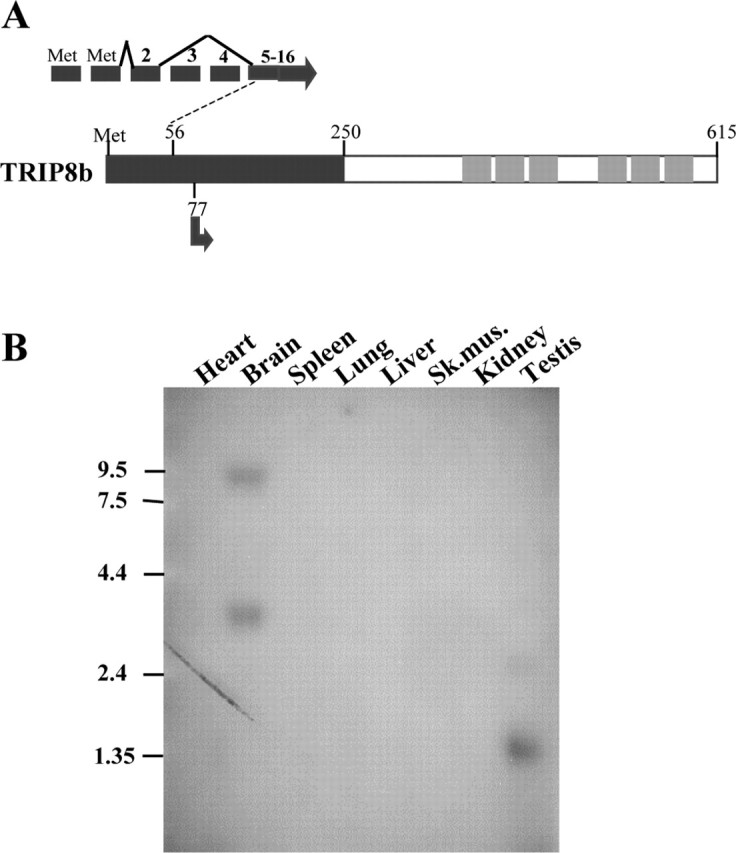
Domain structure and tissue distribution of TRIP8b. A, Schematic representation of the TRIP8b protein. The N-terminal domain, having no known homology to other proteins, is represented by a dark gray box; the C-terminal domain contains six TPRs represented by light grayboxes. Numbers indicate positions in the protein sequence. The starting point of the TRIP8b partial clone isolated in the yeast two-hybrid screen is indicated (arrow at position 77; clone extends through the C-terminal end of protein). Inset on the top illustrates the intron-exon structure of the TRIP8b gene and the splicing isoform used in this study. Exons are labeled by numbers; starting methionines are labeled “Met.” No alternative splicing was found after exon 5 through exon 16, which contains the stop codon. B, Northern blot analysis of TRIP8b mRNA expression in mouse tissues. The probe, corresponding to amino acids 467-600, recognizes all alternatively spliced isoforms. RNA molecular weight markers are indicated on the left (in kilobases). The significance of the shorter transcript in testis is unclear, because its size is insufficient to encode a full-length protein.
TRIP8b is strongly expressed in pyramidal neurons and specifically colocalizes with HCN1 in the apical dendrites of cortical layer V pyramidal neurons
Because previous studies on TRIP8b and PEX5Rp did not investigate the distribution of these gene products within distinct brain regions and cell types, we proceeded to characterize the pattern of both TRIP8b mRNA and protein expression within the mouse brain.
Analysis of TRIP8b mRNA expression
In situ hybridization, performed using radiolabeled oligonucleotides directed to a nonvariant region of the TRIP8b mRNA (exon 15), demonstrated a distinct pattern of TRIP8b distribution within the brain, with high levels of expression within the CA1 pyramidal cell layer of the hippocampus and subiculum and in layer V pyramidal neurons of the neocortex (Fig. 3_B,D_). Relatively high levels of expression were also observed in one layer of the superior colliculus and in the inferior colliculus (Fig. 3_D_). This pattern of distribution is strikingly similar to the distribution of HCN1 mRNA, which is also highest in the CA1, subiculum, and layer V pyramidal neurons, as well as in neurons within the superior and inferior colliculus (Santoro et al., 1997, 2000) (for reference, serial sections radiolabeled with an HCN1-specific probe are presented in Fig. 3_A,C_). The overlap in expression is only partial, however, because other classes of neurons that strongly express HCN1 mRNA, such as cerebellar Purkinje cells and brainstem motoneurons, do not display similarly high levels of TRIP8b expression (Fig. 3, compare A,C and B,D). These findings indicate that, first and foremost, TRIP8b is indeed coexpressed with HCN channels in distinct cell types in vivo; second, TRIP8b might be relevant to the specific function of HCN1 in some neurons but not in others.
Figure 3.

In situ hybridization analysis of TRIP8b mRNA expression in the mouse brain. A, C, Sections hybridized with an HCN1 probe. B,D, Sections hybridized with a TRIP8b probe. Sections in A and B are consecutive sagittal sections; sections in C and D are also consecutive but collected in a more medial position.
Analysis of TRIP8b protein expression
The subcellular distribution of the HCN1 protein within pyramidal neurons has been characterized in great detail (Santoro et al., 1997; Lorincz et al., 2002; Notomi and Shigemoto, 2004). A graded increase in HCN1 immunoreactivity is found as a function of increasing distance from the soma along the apical dendrites of pyramidal neurons from the CA1 region of the hippocampus, subiculum, and layer V of the neocortex. This distribution is paralleled by a similar somatodendritic gradient of increasing _I_h current density (Magee, 1998; Berger et al., 2001). Given the high levels of TRIP8b mRNA expression in these same neurons, we investigated the subcellular localization of the TRIP8b protein within these cells.
Immunohistochemistry performed on brain sections from adult mice showed striking similarities in the distribution of the HCN1 and TRIP8b proteins; their pattern of expression was closely matched in the hippocampus (Fig. 4) and neocortex (Figs. 5, 6) as well as subiculum (including presubiculum and parasubiculum), entorhinal cortex, and superior colliculus (data not shown). In the hippocampus, TRIP8b expression was strongest in stratum lacunosum moleculare, suggesting colocalization with HCN1 in the apical dendrites of CA1 pyramidal neurons (Fig. 4). Dendritic localization was even more evident in neocortical layer V neurons, where apical dendrites were distinctly labeled by both HCN1 (Fig. 5_A_) and TRIP8b antibodies (Fig. 5_B_); however, as described above for mRNA distribution (Fig. 3), the pattern of protein distribution was not identical. Thus, there was a clear expression of TRIP8b protein in neuronal somata (Fig. 4_B_, pyramidal cell layer; Fig. 5_B_, arrowhead), where HCN1 protein was essentially absent. Furthermore, TRIP8b was present in stratum lucidum of CA3 (mossy fiber pathway), whereas HCN1 was missing from this region (Fig. 4, compare A, B). Although these differences imply that proteins other than HCN1 might also interact with TRIP8b in distinct cell types and subcellular compartments, including other HCN isoforms expressed in these cells, the striking localization of HCN1 and TRIP8b within the distal ends of apical dendrites from pyramidal neurons suggests strongly that they do indeed interact in vivo.
Figure 4.

Immunohistochemical analysis of TRIP8b protein expression in mouse hippocampus. A, Anti-HCN1 antibody staining obtained by immunofluorescence on a coronal brain section (40 μm) from a wild-type mouse. B, Anti-TRIP8b antibody staining obtained by immunofluorescence on a serial coronal brain section (40 μm) from a wild-type mouse. Imaging was performed by confocal microscopy (10× objective); all images represent a single focal plane and were collected at the point of maximum labeling intensity for each section.
Figure 5.

Immunohistochemical analysis of TRIP8b protein expression in neocortex of a wild-type mouse and a mouse line with an unrestricted deletion of HCN1. A, C, Anti-HCN1 antibody staining obtained by immunofluorescence on a coronal brain section (40 μm) from a wild-type (A) or an HCN1-/- (C) mouse. B, D, Anti-TRIP8b antibody staining obtained by immunofluorescence on a serial coronal brain section (40 μm) from a wild-type (B) or HCN1-/- (D) mouse. Arrowheads show the location of somata, and arrows indicate the location of apical dendrites from layer V pyramidal neurons. Imaging was performed by confocal microscopy (10× objective); all images represent a single focal plane and were collected at the point of maximum labeling intensity for each section, using identical settings for samples in A and C and in B and D, respectively.
Figure 6.

Double-immunofluorescence labeling of TRIP8b and HCN1 protein expression in the neocortex of mice with a forebrain-restricted deletion of HCN1 (HCN1f/f,cre) and matched littermates with normal HCN1 expression (HCN1f/f). A,C, Confocal image (40× objective) of FITC signal as a marker of anti-HCN1 antibody staining on a coronal brain section (40 μm) from an HCN1f/f (A) or an HCN1f/f,cre (C) mouse. B, D, Corresponding confocal image of rhodamine-red signal as a marker of anti-TRIP8b antibody staining in the same area, from an HCN1f/f (B) or an HCN1f/f,cre (D) mouse. All images represent a single focal plane. Images in A and C were taken under identical conditions and illustrate the dramatic reduction of HCN1 labeling in the apical dendrites from layer V neurons of the HCN1f/f,cre mice. Images in B and D were also taken under identical conditions and illustrate the correspondingly strong reduction of TRIP8b labeling in the apical dendrites from the HCN1f/f,cre mouse. E-G, Colocalization of HCN1 and TRIP8b protein in the apical dendrites of layer V pyramidal neurons. Confocal images (40× objective) of FITC labeling for HCN1 (E) and rhodamine-red labeling for TRIP8b (F) on the same brain section from an HCN1f/f,cre mouse, together with a merged image (G) demonstrating colabeling (yellow) of the same dendritic processes by both antibodies. Note that illumination settings were greatly increased with respect to images in _A_-D to enhance detection of residual dendritic label.
TRIP8b protein distribution is altered in the brain of HCN1 knock-out mice
To further investigate the hypothesis of a functional interaction between TRIP8b and HCN1 in vivo, we examined the effect of genetic deletion of HCN1 through homologous recombination on the pattern of TRIP8b protein expression [for a detailed description of the mouse lines used, see Nolan et al. (2003, 2004)]. The first line of mice that we examined, HCN1-/-, carries an unrestricted knock-out of the HCN1 gene (Nolan et al., 2003) (Fig. 5 A, C). Immunohistochemical analysis of brain sections isolated from the HCN1-/- mice revealed a striking redistribution of TRIP8b protein expression (Fig. 5, compare B, D). The most apparent change was a drastic reduction of TRIP8b labeling in the superficial layers of the cortex (Fig. 5_D_, arrow), with a clear loss of normal expression in the apical dendrites of layer V pyramidal neurons. The reduction in TRIP8b expression was particularly evident in layer I, which contains the terminal plexus of the apical dendrites from layer V pyramidal neurons. In contrast, TRIP8b signal was retained in the somata of these neurons (Fig. 5_D_, arrowhead).
To rule out the possibility that the loss of TRIP8b represents a nonspecific effect of the HCN1 deletion, for example because of developmental abnormalities or a deleterious effect on the health of the mice, we examined a second line of mice, HCN1f/f,cre, in which deletion of the HCN1 gene product is limited to the forebrain (Nolan et al., 2003, 2004). These mice contain two floxed alleles of the HCN1 gene (HCN1f/f) and express Cre recombinase driven by the forebrain-specific CaMKIIα promoter (HCN1f/f,cre). In this line of mice, HCN1 is deleted from the vast majority of cortical neurons, although expression is preserved in a small fraction of pyramidal cells because of the mosaic expression of the CaMKIIα promoter (Nolan et al., 2004) (Fig. 6_A,C_). Because expression through the CaMKIIα promoter occurs relatively late in development, the forebrain-specific knock-out of HCN1 occurs only after postnatal day 10, thus minimizing any potential developmental abnormalities.
We observed a similar pattern of reduced TRIP8b staining in apical dendrites in the HCN1f/f,cre forebrain-restricted HCN1 knock-out mice as was seen in the general knock-out. In these experiments, we used double-immunofluorescence labeling to compare the pattern of HCN1 and TRIP8b expression in the cortex of HCN1f/f,cre mice and sex-matched HCN1-expressing littermates (HCN1f/f), minimizing any difference attributable to genetic background. The knock-out mice showed a strong reduction in dendritic TRIP8b labeling (Fig. 6, compare B, D) that closely matched the reduction of HCN1-labeled dendrites (Fig. 6, compare A, C). The double labeling also clearly demonstrated that TRIP8b dendritic staining was present and highly colocalized with HCN1 in those neurons that still expressed HCN1 (Fig. 6_E_-G).
The latter finding both confirms the colocalization of both proteins within the same subcellular structure and strengthens the idea that the proper dendritic targeting of TRIP8b is specifically dependent on the presence of HCN1. As an additional control for specificity, we demonstrated that HCN1 deletion had no effect on the dendritic localization of a different protein, the TrkB receptor (Yan et al., 1997; Drake et al., 1999), in either line of knock-out mice (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
In contrast to the changes described above in neocortical areas, rather minor changes in TRIP8b expression were observed in the hippocampus, subiculum, entorhinal cortex, and surrounding areas (data not shown). In particular, expression remained generally high in stratum lacunosum moleculare, suggesting that other factors contribute to maintain high TRIP8b protein expression in the apical dendrites of CA1 pyramidal cells (see Discussion). Small changes in absolute protein levels may not be detected by our immunohistochemical analysis and could still occur in these regions.
In conclusion, the discrete changes in TRIP8b protein localization that we observed in response to the loss of functional HCN1 channel subunits confirm the existence of a link between the two proteins in vivo, most likely mediated by a direct protein-protein interaction.
TRIP8b drastically reduces HCN channel current expression in Xenopus oocytes
In light of the direct binding between the TRIP8b and the HCN channel proteins observed in vitro and the strong colocalization of these proteins within specific sub-cellular compartments in the brain, we sought to determine the effects of TRIP8b coexpression on HCN channel function. HCN channels were expressed in Xenopus oocytes after injection of HCN cRNA, either with or without coinjection of TRIP8b cRNA. To control for potential artifacts caused by the introduction of a second cRNA species (that could conceivably impair the translation efficiency of the first cRNA species), baseline values of HCN channel expression in the absence of TRIP8b were always obtained using coinjection of an equal amount of GFP cRNA. Furthermore, in some experiments, a GFPTRIP8b fusion construct (GFP fused to the N terminus of TRIP8b) was used to monitor the expression levels of the TRIP8b product; no differences were observed between experiments using the TRIP8b or GFP-TRIP8b constructs, suggesting that these proteins are functionally equivalent.
As shown in Figure 7, A and B, coexpression of TRIP8b with HCN2 caused a dramatic reduction in the amplitude of _I_h measured in cell-attached patch recordings. In contrast, coexpression of TRIP8b had no effect on the current magnitude generated by a truncated HCN2 channel protein, HCN2ΔC-X, which lacks the C-terminal 200 amino acids, and thus the SNL tripeptide sequence (Wainger et al., 2001) (Fig. 7_C,D_). The group data (Fig. 7_E_) demonstrate an ∼20-fold reduction in tail-current amplitude when the wild-type channel was expressed in the presence of TRIP8b (HCN2 plus GFP, 154 ± 24 pA; HCN2 plus GFP-TRIP8b, 7 ± 2 pA; mean ± SE; t test; p < 10-5). In contrast, no significant current reduction was observed when TRIP8b was coexpressed with the truncated HCN2 channel (HCN2ΔC-X plus GFP, 118 ± 27 pA; HCN2ΔC-X plus GFP-TRIP8b, 64 ± 14 pA; p = 0.10). This finding is consistent with the idea that the SNL peptide mediates, at least in part, the binding of the channel to TRIP8b. Moreover, it argues that the effect of TRIP8b to inhibit _I_h is not caused by a nonspecific effect on protein expression.
Figure 7.
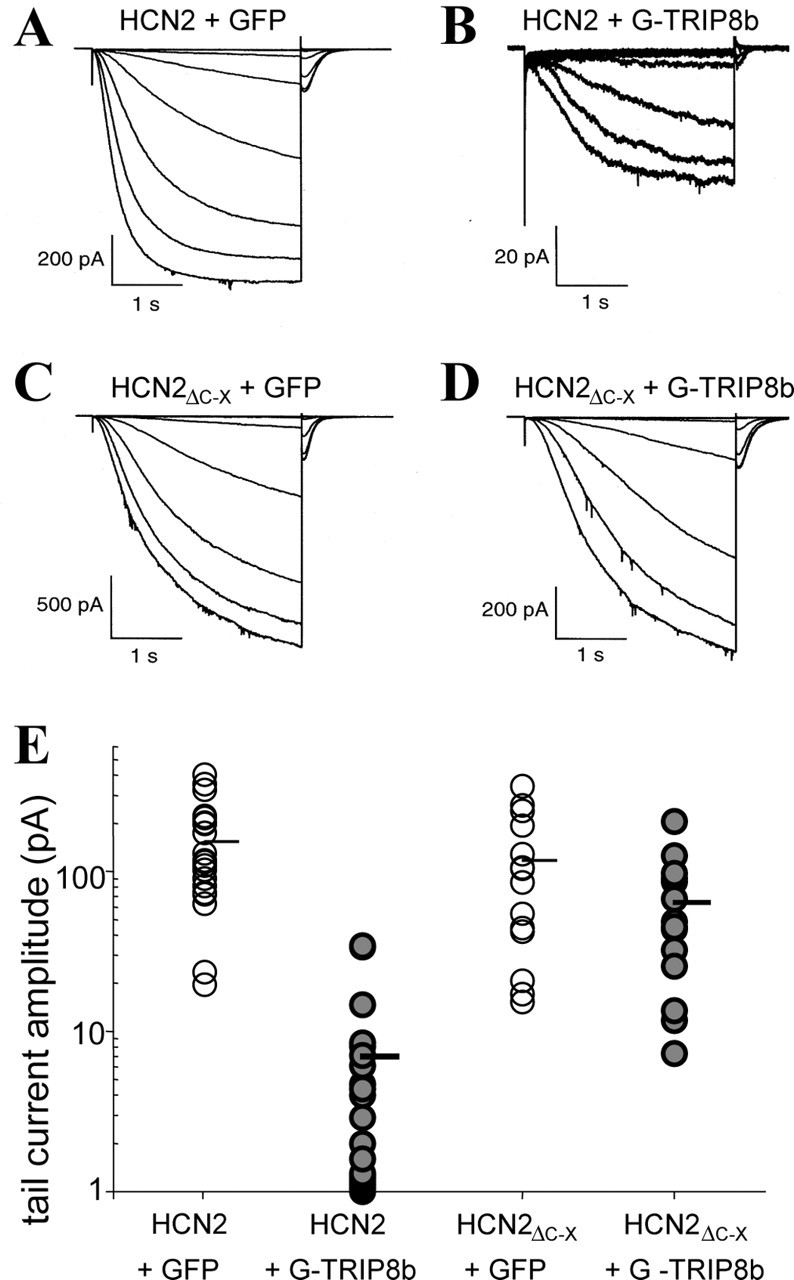
TRIP8b expression strongly reduces HCN2 currents in cell-attached patches. A-D, Sample current traces from Xenopus oocytes coinjected with cRNA encoding HCN2 and GFP (A), HCN2 and GFP-TRIP8b (B), HCN2ΔC-X and GFP (C), HCN2ΔC-X and GFP-TRIP8b (D). Currents elicited by 3 sec hyperpolarizations from a holding potential of -30 mV to a range of test potentials between -75 and -145 mV, in increments of 10 mV. Note differences in current scale bars. E, Plot of maximal tail-current amplitude (measured at -40 mV). For each patch, tail-current activation curves were fit with a Boltzmann equation (see Materials and Methods) to obtain the tail-current amplitude. Each point corresponds to a single patch recording, and only one patch was taken from each oocyte. Note logarithmic _y_-axis scale. Horizontal bar shows mean current under each condition.
The reduction in _I_h in cell-attached patches might reflect an action of TRIP8b to cluster HCN channels in a limited number of dense patches, which could have caused us to underestimate total levels of HCN2 surface expression resulting from limited sampling. To explore this possibility, we repeated the coexpression experiments using a two-microelectrode voltage clamp, which reports the current through all channels in the surface membrane (Fig. 8). In these experiments we used HCN1 cRNA, rather than HCN2, because the former typically expresses at a lower level and so does not saturate the recording amplifier, unlike the high-expressing HCN2 subunits. We observed a 100-fold reduction in the whole-oocyte tail-current amplitude after coexpression of TRIP8b with HCN1, indicating a true decrease in total surface _I_h current density (HCN1 plus GFP, 8728 ± 1168 nA; HCN1 plus GFP-TRIP8b, 79 ± 22 nA; p < 10-5).
Figure 8.

TRIP8b specifically suppresses HCN1 currents in whole-cell recordings. A, Data obtained from oocytes injected with wild-type HCN1 and coinjected with either GFP (left) or GFP-TRIP8b fusion protein cRNAs (right). B, Data from oocytes injected with HCN1ΔSNL truncation mutant and coinjected with either GFP (left) or GFP-TRIP8b cRNAs (right). C, Data from oocytes injected with KAT1 and coinjected with either GFP (left) or GFP-TRIP8b cRNAs (right). Top, Plots of maximal tail-current amplitude obtained as in Figure 7_E_ for HCN constructs. For KAT1, tail-current amplitude was measured at -40 mV after a step to -115 mV. Each point represents data from a single oocyte. Bottom, Sample current traces from two-microelectrode voltage-clamp recordings from Xenopus oocytes coinjected with cRNAs encoding the indicated gene products. Currents were elicited by 3 sec hyperpolarizations from a holding potential of -30mV to a range of test potentials between -35 and -105mV, in increments of 10 mV.
The loss of HCN1 current was entirely dependent on the presence of the C-terminal SNL tripeptide sequence. Thus, the amplitude of whole-oocyte tail currents generated by expression of a mutant HCN1 construct lacking only its terminal tripeptide (HCN1ΔSNL) showed no significant change after coexpression with TRIP8b (HCN1ΔSNL plus GFP, 10530 ± 1230 nA; HCN1ΔSNL plus GFP-TRIP8b, 13425 ± 1278 nA; p = 0.10). As a final control, we coexpressed TRIP8b with KAT1, a hyperpolarization-activated K+ channel from Arabidopsis that, like HCN channels, is a member of the six-transmembrane segment K+ channel superfamily; however, KAT1 terminates in a distinct tripeptide sequence (SSN). Coexpression of TRIP8b had no significant effect on the amplitude of KAT1 tail currents measured in two-microelectrode voltage clamp (KAT1 plus GFP, 8380 ± 2156 nA; KAT1 plus GFP-TRIP8b, 5791 + 545 nA; p = 0.25). Hence, TRIP8b caused a reduction in current amplitude that is specific to HCN channels and dependent on the presence of an intact C-terminal SNL tripeptide sequence.
TRIP8b alters the intracellular trafficking of HCN channels
The observed decrease in HCN current amplitude as a result of TRIP8b protein binding could be explained by either an inhibitory action on channel function (an effect on gating or permeability) or a decrease in the number of channels present in the plasma membrane (an effect on trafficking). Although an accurate measure of the kinetics of HCN channel activation in the presence of TRIP8b could not be obtained [because of the small size of the residual _I_h and the confounding effect of the endogenous chloride currents of the oocytes (Kowdley et al., 1994)], we did not observe any effect of TRIP8b on the voltage dependence of HCN channel activation, suggesting a limited effect on the biophysical properties of the channels (in cell-attached patch recordings, _V_1/2 for HCN2 plus GFP was -120 ± 6 mV, n = 10; _V_1/2 for HCN2 plus TRIP8b was -124 ± 6 mV, n = 10; mean ± SD; t test; p > 0.05). Thus, we proceeded to examine whether TRIP8b altered surface expression of HCN channels.
Subcellular distribution of HCN proteins in oocytes
To explore the hypothesis that the reduced current density might be caused by altered trafficking of the channel protein, we visualized with confocal microscopy the subcellular localization of HCN protein in oocytes using fusion constructs in which the GFP protein was attached to the N terminus of HCN, thus leaving the SNL C-terminal end free. Both the GFP-HCN1 and GFP-HCN2 fusion proteins showed normal electrophysiological properties (data not shown). In the absence of TRIP8b, the GFP-HCN fluorescence signal was localized to a rim corresponding to the surface membrane, with little intracellular signal (Fig. 9). Coexpression of TRIP8b dramatically increased the HCN signal detected in the intracellular portion of the cell (Fig. 9_A_). In contrast, TRIP8b coexpression did not affect the distribution of a fusion protein in which the GFP moiety was attached to the N terminus of the rod photoreceptor cyclic nucleotide-gated channel CNGA1 subunit (Fig. 9_B_, GFP-CNGA1), again indicating that the action of TRIP8b is specific to the HCN family of channels. These results suggest that TRIP8b binding does indeed specifically alter HCN channel trafficking, which could contribute to the observed reduction in whole-oocyte current density.
Figure 9.

Trafficking of HCN channels is altered in oocytes coexpressing TRIP8b. The orientation of each oocyte is schematically shown at the side of each panel (black, animal pole; white, vegetal pole). A, Confocal image of oocyte injected with cRNA encoding GFP-HCN2 fusion protein, in the presence (right) or absence (left) of TRIP8b. Note substantial fluorescence in vegetal pole after coexpression with TRIP8b. Only protein in the vegetal pole of the oocyte is visible, because of the dark pigmentation of the Xenopus oocyte animal pole. B, Confocal image of oocyte injected with cRNA encoding GFP-CNGA1 fusion protein, in the presence (right) or absence (left) of TRIP8b. All images are single planes taken approximately perpendicular to the equatorial line dividing the animal from vegetal pole.
Subcellular distribution of HCN proteins in HEK293 cells
Because the resolution of imaging in the oocyte is limited, we proceeded to characterize the surface expression of HCN channels after transfection in the HEK293 mammalian cell line. Furthermore, to unambiguously determine levels of HCN surface expression, we also generated a GFP-HCN2 protein construct in which an extracellular HA “tag” sequence was inserted between transmembrane segments S3 and S4 (Chen et al., 2000). This permits the selective detection of channel protein present in the plasma membrane by specific labeling of the externally accessible epitope using anti-HA antibodies under nonpermeabilizing conditions. When expressed alone, GFP-HCN2 fusion proteins were found distributed primarily either in the plasma membrane, appearing as a rim of fluorescence around the surface of the cell, or in large intracellular structures near the nucleus, presumably the endoplasmic reticulum or Golgi apparatus (Fig. 10_A,B_).
Figure 10.
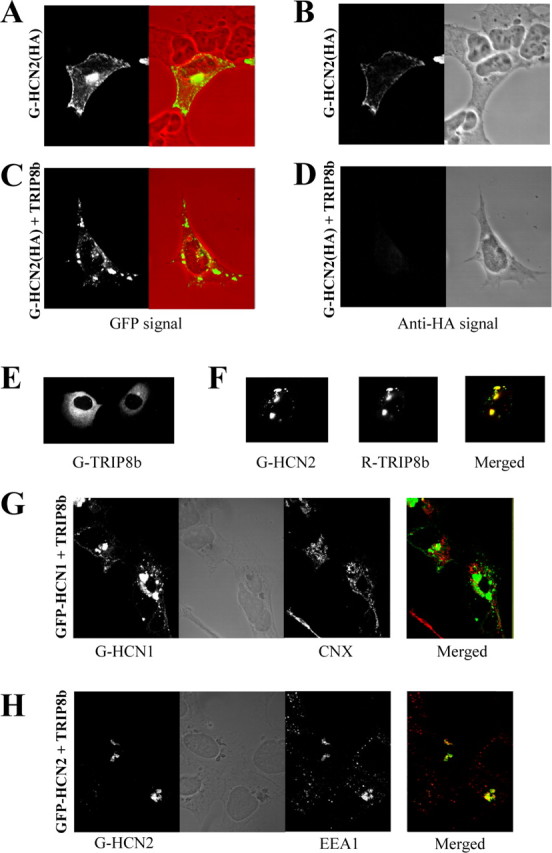
Coexpression of TRIP8b results in the intracellular accumulation of HCN channels. HEK293 cells were transfected with constructs indicated on the left of each panel, or as specified. A, C, Confocal images of GFP signal as marker of total GFP-HCN2 fusion protein expression. Images on left show GFP fluorescence alone. Images on right (red background) represent merged transmitted light image, with cell outlines in red and GFP fluorescence image in green. B, D, Confocal images of surface expression of HA epitope-tagged GFP-HCN2 protein. Fluorescence images on the left show staining after intact cells were labeled with an anti-HA antibody followed by detection with a rhodamine-conjugated secondary antibody. Images on the right show transmitted light. Note complete loss of surface staining after coexpression with TRIP8b. E, Confocal image of GFP fluorescence from cell transfected with GFP-TRIP8b protein alone. Note diffuse cytoplasmic staining. F, Colocalization of HCN protein and TRIP8b protein in HEK293 cells cotransfected with GFP-HCN2 and DsRed-TRIP8b. Left, Confocal image of GFP fluorescence indicating localization of HCN2. Middle, Image of DsRed fluorescence representing TRIP8b. Right, Merged image of GFP and DsRed fluorescence (with yellow representing overlapping green and red signals). G, H, Colocalization of internalized HCN protein and internal membrane compartments after coexpression of GFP-HCN1 or GFP-HCN2 with TRIP8b. HEK293 cells were cotransfected with HCN protein and TRIP8b as indicated, permeabilized, and stained using either anti-CNX antibodies to label the endoplasmic reticulum (G) or antibodies to EEA1 to label early endosomes (H). Confocal image comparing the GFP signal, representing HCN1 (G) or HCN2 (H) channel expression and the rhodamine-red signal, representing internal membrane markers. Middle images show transmitted light. Merged images of GFP and rhodamine signals are shown on the extreme right.
Cotransfection of GFP-HCN2 with TRIP8b resulted in a striking redistribution of channel protein, with the disappearance of label from the cell surface and the appearance of prominent staining in dense puncta scattered throughout the cytoplasm, distinct from the internal staining pattern in the absence of TRIP8b (Fig. 10_C,D_). Interestingly, when transfected alone, the TRIP8b protein yielded a diffuse pattern of cytoplasmic staining (Fig. 10_E_) (S. Chen et al., 2001; Amery et al., 2001). After coexpression with HCN protein, however, TRIP8b was colocalized with HCN channels in the dense intracellular puncta (Fig. 10_F_). Furthermore, TRIP8b coexpression caused a complete disappearance of HCN1 surface expression, as determined by the anti-HA antibody (Fig. 10_D_). Thus all of the dense clusters appeared to be confined intracellularly. This result further suggests that an alteration in channel trafficking is responsible, in large part, for the observed decrease in HCN current density.
To determine whether the intracellular accumulation of HCN-TRIP8b proteins occurs at a specific step in the biosynthetic pathway, we tested for colabeling of the protein clusters with markers of internal membrane compartments. No overlap was observed between the HCN-TRIP8b channel clusters and the endoplasmic reticulum (ER) marker CNX (Fig. 10_G_); however, a strong association was found with the early endosome marker EEA1 (Fig. 10_H_). The distribution of EEA1 was also dramatically altered in HCN-TRIP8b cotransfected cells, with the normal scattered punctate staining replaced by the staining of large intracellular structures that colocalized with the HCN protein signal. Although not all of the intracellular HCN clusters were labeled by EEA1, a large proportion of HCN protein was confined in early endosomes, suggesting strongly that TRIP8b might promote the internalization of HCN channels from the plasma membrane.
TRIP8b decreases the half-life of HCN channels expressed on the cell surface
The reduced surface expression of HCN channels in the presence of TRIP8b could be the result of impaired delivery of protein to the plasma membrane, or enhanced internalization, as suggested by the above experiments. To distinguish further between these two possibilities, we again took advantage of the Xenopus oocyte expression system to perform pulse-chase experiments for functional HCN channel expression.
To study the effect of TRIP8b on the half-life of HCN channels expressed on the plasma membrane, we first determined the stability of the channel in the absence of TRIP8b using a double cRNA injection procedure (Fig. 11_A_, inset). The strategy used a dominant-negative HCN1 channel subunit, in which the GYG selectivity filter was mutated to the sequence AAA (Xue et al., 2002). Incorporation of a single mutant subunit into a tetrameric channel abrogates its function, even in the presence of wild-type subunits, as demonstrated by the complete lack of detectable currents up to 48 hr after coinjection of HCN1 with HCN1AAA (data not shown). Wild-type HCN1 cRNA was injected at time 0, and 24 hr later current amplitudes were determined to verify proper expression of the channel. At this time, half of the HCN1-expressing oocytes were then injected with an excess amount of HCN1AAA cRNA. Because of the high HCN1AAA/HCN1 cRNA ratio, few functional channels should assemble after this time because of the dominant-negative action of the mutant subunits. Thus, currents measured on days 2 and 3 in the doubly injected oocytes should reflect primarily channels that were assembled and delivered to the surface during the first 24 hr after the initial HCN1 cRNA injection.
Figure 11.
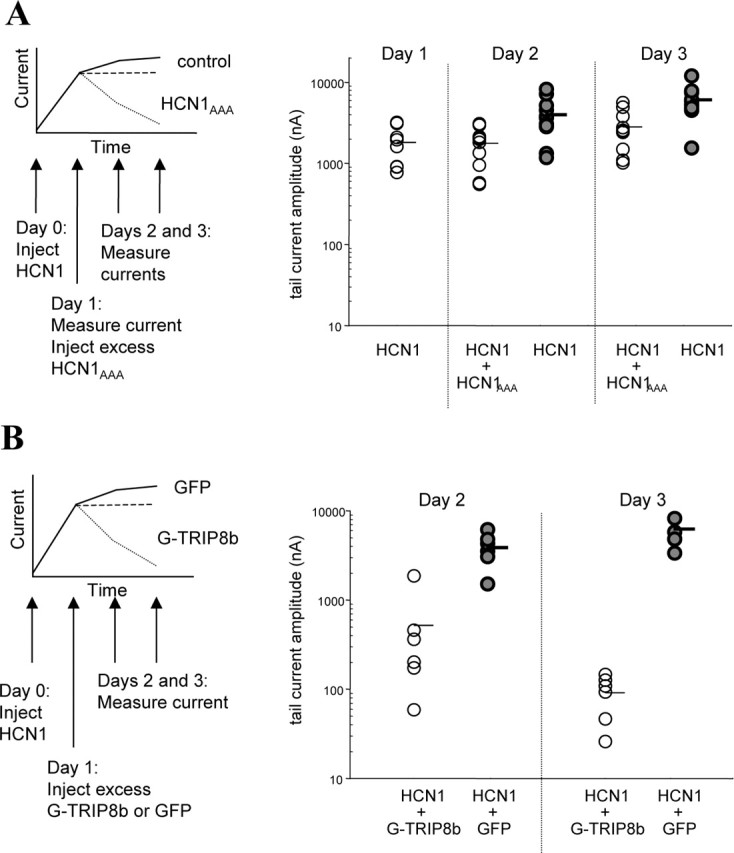
TRIP8b expression decreases the half-life of HCN channels expressed on the plasma membrane of Xenopus oocytes. _A,_Determination of HCN current stability after block of new functional channel synthesis. Schematic of experiment is shown on the left (see Results for explanation). On day 0, oocytes were injected with an HCN1 cRNA. Twenty-four hours later on day 1, oocytes either were injected with cRNA for an HCN1AAA dominant-negative constructor received no injection. Solid line indicates predicted continuous increase in total current after HCN1 cRNA injection on day 0 for oocytes that were not coinjected with HCN1AAA. The dashed line shows predicted stable current level for oocytes injected with HCN1AAA on day 1 if rate of membrane turnover is slow. Dotted line shows predicted decrease in HCN1 current after injection of HCN1AAA if membrane turnover is fast. Experimental data are shown on the right. Plot of maximal tail-current amplitudes was obtained as in Figure 7_E_ from two-electrode recordings. Note relative stability of HCN current after injection of HCN1AAA, indicating slow rate of membrane turnover. B, Rate of HCN channel turnover increased after expression of TRIP8b. Schematic of experiment is shown on the left (see Results for explanation). Experiment is similar to that shown in A, except either GFP-TRIP8b or GFP is injected on day 1, instead of HCN1AAA. Solid line indicates prediction for GFP-injected control oocytes; broken lines indicates two possible outcomes after TRIP8b injection. If TRIP8b only blocks insertion of newly synthesized protein, current should resemble that seen using HCN1AAA (dashed line). If TRIP8b enhances channel internalization, then we predict an accelerated loss of current (dotted line). Experimental tail-current data are shown on the right. Note greatly increased loss of HCN current after injection of TRIP8b, compared with injection of GFP alone or HCN1AAA (A).
As shown in Figure 11_A_, although control oocytes showed a sizeable increase in tail-current amplitude between day 1 (1821 ± 340 nA) and day 2 (3995 + 664 nA) or day 3 (6085 ± 856 nA; t test day 1 to day 2, p < 0.02; day 1 to day 3, _p_ < 0.001), no significant change in _I_h magnitude was observed in the oocytes injected with HCN1AAA cRNA (day 1, 1821 ± 340 nA; day 2, 1774 ± 257 nA; day 3, 2824 ± 491; _t_ test day 1 to day 2, _p_ > 0.9; day 1 to day 3, p > 0.1). This result confirms the idea that the dominant-negative subunit effectively interrupts the synthesis of new functional channels and demonstrates that channels delivered to the plasma membrane before the mutant subunit injection are indeed stable for at least 2 d.
Next, we used a similar double-injection procedure to determine whether coexpression of TRIP8b would reduce the stability of the surface pool of channels (Fig. 11_B_). Wild-type HCN1 cRNA was again injected at time 0, and current amplitudes were evaluated 24 hr later. One-half of the HCN1-expressing oocytes were then injected with an excess amount of GFP-encoding cRNA and the other half with GFP-TRIP8b-encoding cRNA. Expression of GFP-TRIP8b resulted in a striking downregulation in HCN1 tail-current amplitudes compared with current levels in control oocytes expressing GFP cRNA. On day 2, the mean tail-current amplitude in control GFP-expressing oocytes was 3890 + 650 nA, whereas in oocytes expressing GFPTRIP8b the tail-current amplitude was only 522 + 276 nA (p < 0.001). The difference was even more striking on day 3; the mean current amplitude in oocytes coinjected with GFP was 6267 + 1087 nA, whereas coinjection with GFP-TRIP8b reduced the current amplitude to 91 + 19 nA (_p_ < 0.0005). Because the half-life of HCN channels expressed on the oocyte plasma membrane is >2 d in the absence of TRIP8b (Fig. 11_A_), the large reduction in current amplitude observed in these experiments supports the idea that TRIP8b promotes channel internalization.
TRIP8b reduces endogenous _I_h currents in cultured hippocampal neurons
Does TRIP8b also reduce levels of native _I_h currents in neurons? Hippocampal pyramidal neurons in dissociated cell culture for 17-21 d reliably expressed _I_h currents and showed positive labeling with anti-HCN1 antibodies (data not shown). To determine the action of TRIP8b on these currents, we infected the neuronal cultures with Sindbis virus vectors that expressed either GFP or GFP-TRIP8b. Twenty-four hours after infection, we recorded _I_h using whole-cell patch-clamp recording (Fig. 12). Neurons infected with the virus expressing GFP-TRIP8b showed a more than fivefold reduction in _I_h tail-current amplitude compared with neurons infected with the virus that expressed GFP alone (GFP, 162 ± 30 pA; GFP-TRIP8b, 30 ± 11 pA; p < 0.0005). We thus conclude that the TRIP8b protein reduces the surface expression of native HCN channels in neurons as well as in heterologous systems.
Figure 12.
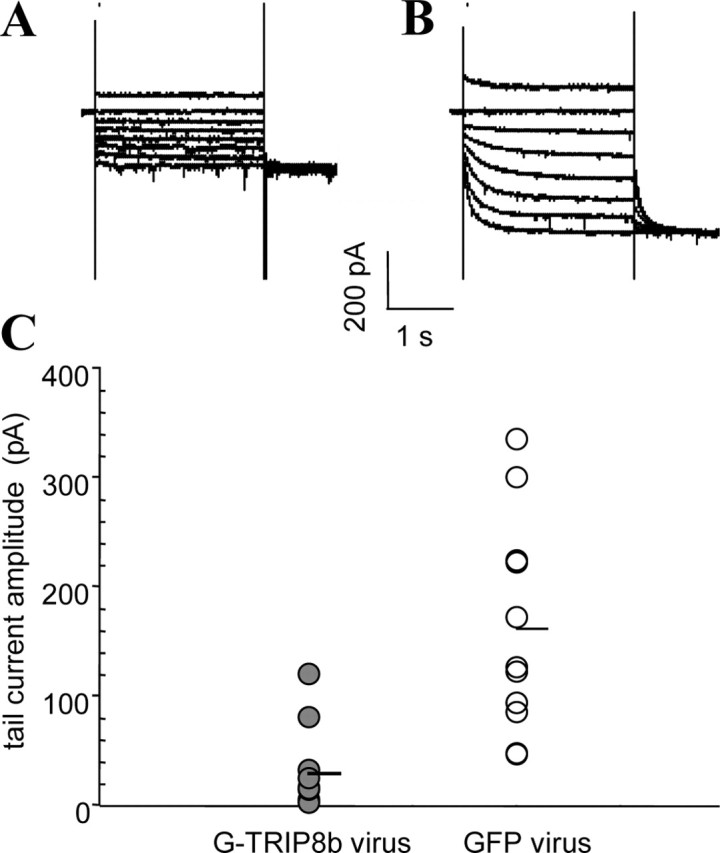
TRIP8b suppresses native _I_h currents in hippocampal neurons. A, B, Sample whole-cell patch recordings from cultured hippocampal pyramidal neurons, obtained 24 hr after infection with Sindbis virus expressing either GFP-TRIP8b (A) or GFP alone (B). Cells were held at -55 mV, and currents were elicited by voltage steps from -45 to -115 mV, in increments of 10 mV. C, Plot of tail-current amplitude of Boltzmann fits (one point per cell, as before) after tail current measurement at -115 mV.
Discussion
In this study we have demonstrated an interaction between the conserved C-terminal tripeptide sequence of HCN channel proteins with a TPR-containing protein, which has previously been termed TRIP8b (S. Chen et al., 2001) or PEX5Rp (Amery et al., 2001) by others and HIP1 by us (Santoro et al., 2002). Furthermore, we have shown that this interaction is likely to occur in the dendrites of cortical and hippocampal pyramidal cells. Finally, functional coexpression of TRIP8b with HCN protein, either in native cells or heterologous systems, results in a strong downregulation of HCN channel in the plasma membrane.
Functional characterization of TRIP8b-PEX5Rp
PEX5Rp, the human ortholog of TRIP8b, was initially isolated in a homology screen aimed at identifying proteins related to the peroxin 5 protein PEX5 (hence the name Pex5p-related protein) (Amery et al., 2001). PEX5 is a ubiquitous protein, conserved from yeast to mammals, responsible for the recognition and import of peroxisomal matrix proteins from the cytosol into the peroxisome. Interestingly, the recognition of peroxisomal matrix proteins is based on the presence at their C terminus of a conserved tripeptide, known as a peroxisome-targeting signal 1 (PTS1), the consensus sequence of which is “SKL.” PEX5 binds to PTS1 through a series of TPRs that are present in its C-terminal half. This TPR domain is highly conserved between PEX5 and TRIP8b-PEX5Rp (57% identity).
An x-ray crystal structure of the TPR domain of PEX5 in complex with the PTS1 peptide (Gatto et al., 2000) provides insight into the interaction between the homologous TPR domain of TRIP8b and the SNL tripeptide sequence that it recognizes. Thus, in PEX5 a glutamic acid at residue 379 contacts the lysine at the -2 position in the SKL sequence. In TRIP8b, the residue at the corresponding position is a polar uncharged amino acid, threonine 396. This provides a potential explanation for why this latter TPR domain favors interactions with the SNL sequence, in which the threonine could form a favorable hydrogen bond with the asparagine at the -2 position. Furthermore, there is a common degeneracy in the requirement for the terminal tripeptide sequence between PEX5 and TRIP8b. Thus, although the PEX5 consensus binding sequence is SKL, this protein also recognizes target proteins in which the last three amino acids consist of “ARM,” where only the basic nature of the -2 position is conserved. Similarly, we find that TRIP8b not only recognizes the SNL sequence of HCN1, HCN2, and HCN4, it also recognizes the “ANM” sequence of HCN3, indicating a similar tolerance of the binding domain for the “SXL” or “AXM” sequence, as long as the -2 position is conserved.
Although the C-terminal portions containing the TPR domains of TRIP8b and PEX5 are very similar and functionally equivalent in that they both mediate the recognition of a C-terminal tripeptide target sequence, TRIP8b does not appear to be important for peroxisomal import (Amery et al., 2001). This finding is not unexpected, given the lack of homology between the N-terminal halves of the two proteins (sequence identity is only 13%). Importantly, the six copies of a WXXXF/Y pentapeptide sequence present in the N terminus of PEX5, necessary for the docking of the PEX5-PTS1 complex to the peroxisome membrane, are missing from TRIP8b (Amery et al., 2001). Thus, it appears that although PEX5 and TRIP8b recognize their target proteins through an evolutionary conserved C-terminal protein-protein interaction module (the TPR domain), these protein-protein interactions subserve very different functions, which are determined by their unique N termini.
An additional clue to the function of TRIP8b is provided by the study that identified TRIP8b in a yeast two-hybrid screen using the small GTP-binding protein, Rab8b, as a bait (hence the name, TPR-containing Rab8b interacting protein) (S. Chen et al., 2001). Although the precise function of Rab8b is unknown, the large Rab family is thought to play an important role in the trafficking of vesicles to their proper targets (Zerial and McBride, 2001). Consistent with a functional role for this protein-protein interaction, S. Chen et al. (2001) showed that TRIP8b overexpression caused a dramatic increase in induced hormone release in a pituitary cell line. The finding that TRIP8b interacts with both Rab8b and the HCN proteins is consistent with our observation that overexpression of the TPR protein causes a marked change in trafficking of HCN channels.
Role of TRIP8b in the regulation of HCN channel expression
Both TRIP8b mRNA and protein are predominantly expressed in the brain (Fig. 2) (Amery et al., 2001; S. Chen et al., 2001). Within the brain, the highest expression levels are found in two specific cell types: pyramidal neurons in layer V of the neocortex and pyramidal neurons in area CA1 of the hippocampus (Figs. 3, 4, 5). Intriguingly, in both cells there is a striking somatodendritic gradient of increasing levels of expression of HCN1 (Santoro et al., 1997; Lorincz et al., 2002) and _I_h (Magee, 1998; Berger et al., 2001) as a function of distance from the soma. Our immunohistochemical analysis indicates a similar gradient of TRIP8b protein. Importantly, the TRIP8b dendritic gradient in layer V neurons is dependent on the presence of HCN1 protein, as demonstrated by the disruption of TRIP8b dendritic expression in HCN1 knock-out mice (Figs. 5, 6). In contrast, we found relatively little change in the distribution of TRIP8b in other brain regions of the knock-out mice, including the CA1 region of the hippocampus. It is possible then that factors in addition to HCN1 may help target TRIP8b to its proper location. Such factors may comprise HCN2 subunits, which are strongly expressed in CA1 pyramidal neurons, or other dendritic ion channels, which might also interact with TRIP8b [e.g., Kv4.2, which terminates in the related tripeptide sequence “SAL” and the expression of which has been suggested to be homeostatically linked to that of HCN channels (MacLean et al., 2003)].
On the basis of the above evidence, we suggest two hypotheses for the physiological role of TRIP8b in HCN channel function. One hypothesis is suggested by the interaction between TRIP8b and Rab8b (S. Chen et al., 2001). Although the specific function of Rab8b is not known, the closest relative of Rab8b, Rab8, is involved in polarized transport of vesicles to the plasma membrane of hippocampal neuron dendrites (Huber et al., 1993; Peranen et al., 1996). If this function were conserved in Rab8b, TRIP8b could participate in the transport of HCN channel proteins to the dendrites of pyramidal neurons, by mediating their association with Rab8b-linked vesicles. Because TRIP8b is not transported to dendrites in the absence of HCN1 (Figs. 5, 6), any targeting or “anchoring” function for TRIP8b would have to be conditional on the previous formation of an HCN1-TRIP8b complex. Furthermore, given our finding that TRIP8b overexpression results in internalization of HCN surface channels, we must postulate the necessity for the association of some additional protein, such as Rab8b, to form a ternary forward trafficking complex; however, in preliminary experiments, in which Rab8b was cotransfected in HEK293 cells expressing HCN channels and TRIP8b protein, we did not observe an increased mobilization of channel clusters to the cell surface. Rather, the Rab8b protein was also found to colocalize within HCN-TRIP8b-labeled internal puncta (data not shown).
A second hypothesis, consistent with the results from the HCN1 knock-out mice and our coexpression studies, is that TRIP8b is a negative regulator of HCN1 expression that is localized to dendrites by its interaction with HCN1 channels. According to this view, the somatodendritic gradient of HCN1 expression drives the parallel gradient of TRIP8b expression. To explain why the high dendritic levels of TRIP8b do not constitutively downregulate native HCN channels, we postulate that this negative effect of TRIP8b on HCN channel expression levels is itself dynamically regulated in vivo by a specific factor or signal. Evidence for the existence of an equilibrium between the amount of HCN channel inserted in the plasma membrane or retained intracellularly comes from electron microscopy immunolocalization studies performed in different hippocampal cell types (Bräuer et al., 2001; Lorincz et al., 2002). In particular, Lorincz et al. (2002) determined that in the soma of hippocampal CA1 pyramidal neurons ∼91% of the total HCN1 channel protein is found within the cytoplasm; in contrast, in distal dendrites the cytoplasmic pool represents only ∼29% of the total channel. Furthermore, the levels of expression of _I_h and HCN protein have recently been shown to be regulated by neuronal activity. Thus, _I_h current density is altered in hippocampal pyramidal neurons after induction of long-term potentiation (Wang et al., 2003) and seizures (K. Chen et al., 2001; Brewster et al., 2002; Van Welie et al., 2004). These observations raise the intriguing possibility that the functional interaction among TRIP8b, HCN channels, and perhaps other scaffolding proteins might be regulated by the action of protein kinases or phosphatases, after the activation of specific signal transduction cascades. Further elucidation of the function of the interaction between HCN channels and TRIP8b may allow us to define this new mechanism in the regulation of HCN channels and explore its possible role in neuronal plasticity and disease.
Footnotes
This research was partially supported by National Institutes of Health Grant NS36658 to S.A.S. We thank Stanislav Zakharenko, Anthony DeCostanzo, and Lei Zhou for their helpful contributions throughout the course of this study. We thank Huan Yao, John Riley, and Natasha Krougliak for expert technical assistance. We are particularly grateful to Alexei Morozov, Matt Nolan, and Eric Kandel for providing the HCN1 knock-out mice and Shan Chen, Anthony Ting, and Johnny Ngsee for providing the anti-TRIP8b antibodies. We also thank Susan Patterson and Stu Feinstein for sharing the TrkB23-36 antibody, Ronald Li for sharing the HCN1AAA construct, and Mike Sanguinetti for sharing the ntHCN2-HA construct.
Correspondence should be addressed to Dr. Bina Santoro, Center for Neurobiology and Behavior, Columbia University, 1051 Riverside Drive, New York, NY 10032. E-mail: bs73@columbia.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/2410750-13$15.00/0
References
- Amery L, Sano H, Mannaerts GP, Snider J, Van Looy J, Fransen M, Van Veldhoven PP (2001) Identification of PEX5p-related novel peroxisome-targeting signal 1 (PTS1)-binding proteins in mammals. Biochem J 357: 635-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arikkath J, Campbell KP (2003) Auxiliary subunits: essential components of the voltage-gated calcium channel complex. Curr Opin Neurobiol 13: 298-307. [DOI] [PubMed] [Google Scholar]
- Bender RA, Brewster A, Santoro B, Ludwig A, Hofmann F, Biel M, Baram TZ (2001) Differential and age-dependent expression of hyperpolarization-activated, cyclic nucleotide-gated cation channel isoforms 1-4 suggests evolving roles in the developing rat hippocampus. Neuroscience 106: 689-698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger T, Larkum ME, Luscher HR (2001) High I(h) channel density in the distal apical dendrite of layer V pyramidal cells increases bidirectional attenuation of EPSPs. J Neurophysiol 85: 855-868. [DOI] [PubMed] [Google Scholar]
- Blatch GL, Lassle M (1999) The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. BioEssays 21: 932-939. [DOI] [PubMed] [Google Scholar]
- Bräuer AU, Savaskan NE, Kole MH, Plaschke M, Monteggia LM, Nestler EJ, Simburger E, Deisz RA, Ninnemann O, Nitsch R (2001) Molecular and functional analysis of hyperpolarization-activated pacemaker channels in the hippocampus after entorhinal cortex lesion. FASEB J 14: 2689-2701. [DOI] [PubMed] [Google Scholar]
- Brewster A, Bender RA, Chen Y, Dube C, Eghbal-Ahmadi M, Baram TZ (2002) Developmental febrile seizures modulate hippocampal gene expression of hyperpolarization-activated channels in an isoform- and cell-specific manner. J Neurosci 22: 4591-4599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Guo HQ, Lee DH, Luo L, Liu C, Kuei C, Velumian AA, Butler MP, Brown SM, Dubin AE (2003) Neuronal hyperpolarization-activated pacemaker channels drive neuropathic pain. J Neurosci 23: 1169-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Mitcheson JS, Lin M, Sanguinetti MC (2000) Functional roles of charged residues in the putative voltage sensor of the HCN2 pacemaker channel. J Biol Chem 275: 36465-36471. [DOI] [PubMed] [Google Scholar]
- Chen K, Aradi I, Thon N, Eghbal-Ahmadi M, Baram TZ, Soltesz I (2001) Persistently modified h-channels after complex febrile seizures convert the seizure-induced enhancement of inhibition to hyperexcitability. Nat Med 7: 331-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Liang MC, Chia JN, Ngsee JK, Ting AE (2001) Rab8b and its interacting partner TRIP8b are involved in regulated secretion in AtT20 cells. J Biol Chem 276: 13209-13216. [DOI] [PubMed] [Google Scholar]
- Drake CT, Milner TA, Patterson SL (1999) Ultrastructural localization of full-length trkB immunoreactivity in rat hippocampus suggests multiple roles in modulating activity-dependent synaptic plasticity. J Neurosci 19: 8009-8026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MS, Collings MA, Brewer GJ (1998) Electrophysiology of embryonic, adult and aged rat hippocampal neurons in serum-free culture. J Neurosci Methods 79: 37-46. [DOI] [PubMed] [Google Scholar]
- Gatto Jr GJ, Geisbrecht BV, Gould SJ, Berg JM (2000) Peroxisomal targeting signal-1 recognition by the TPR domains of human PEX5. Nat Struct Biol 7: 1091-1095. [DOI] [PubMed] [Google Scholar]
- Huber LA, de Hoop MJ, Dupree P, Zerial M, Simons K, Dotti C (1993) Protein transport to the dendritic plasma membrane of cultured neurons is regulated by rab8p. J Cell Biol 123: 47-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass RS, Kurokawa J, Marx SO, Marks AR (2003) Leucine/isoleucine zipper coordination of ion channel macromolecular signaling complexes in the heart. Roles in inherited arrhythmias. Trends Cardiovasc Med 13: 52-56. [DOI] [PubMed] [Google Scholar]
- Kowdley GC, Ackerman SJ, John III JE, Jones LR, Moorman JR (1994) Hyperpolarization-activated chloride currents in Xenopus oocytes. J Gen Physiol 103: 217-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz A, Notomi T, Tamas G, Shigemoto R, Nusser Z (2002) Polarized and compartment-dependent distribution of HCN1 in pyramidal cell dendrites. Nat Neurosci 5: 1185-1193. [DOI] [PubMed] [Google Scholar]
- MacLean JN, Zhang Y, Johnson BR, Harris-Warrick RM (2003) Activity-independent homeostasis in rhythmically active neurons. Neuron 37: 109-120. [DOI] [PubMed] [Google Scholar]
- Magee JC (1998) Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J Neurosci 18: 7613-7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moosmang S, Stieber J, Zong X, Biel M, Hofmann F, Ludwig A (2001) Cellular expression and functional characterization of four hyperpolarization-activated pacemaker channels in cardiac and neuronal tissues. Eur J Biochem 268: 1646-1652. [DOI] [PubMed] [Google Scholar]
- Moss SJ, Henley J (2002) Receptor and ion-channel trafficking. New York: Oxford UP.
- Nolan MF, Malleret G, Lee KH, Gibbs E, Dudman JT, Santoro B, Yin D, Thompson RF, Siegelbaum SA, Kandel ER, Morozov A (2003) The hyperpolarization-activated HCN1 channel is important for motor learning and neuronal integration by cerebellar Purkinje cells. Cell 115: 551-564. [DOI] [PubMed] [Google Scholar]
- Nolan MF, Malleret G, Dudman JT, Buhl DL, Santoro B, Gibbs E, Vronskaya S, Buzsáki G, Siegelbaum SA, Kandel ER, Morozov A (2004) A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell 119: 719-732. [DOI] [PubMed] [Google Scholar]
- Notomi T, Shigemoto R (2004) Immunohistochemical localizaiton of Ih channel subunits, HCN1-4, in rat brain. J Comp Neurol 471: 241-276. [DOI] [PubMed] [Google Scholar]
- O'Dell TJ, Hawkins RD, Kandel ER, Arancio O (1991) Tests of the roles of two diffusible substances in long-term potentiation: evidence for nitric oxide as a possible early retrograde messenger. Proc Natl Acad Sci USA 88: 11285-11289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peranen J, Auvinen P, Virta H, Wepf R, Simons K (1996) Rab8 promotes polarized membrane transport through reorganization of actin and microtubules in fibroblasts. J Cell Biol 135: 153-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu J, Kryukova Y, Potapova IA, Doronin SV, Larsen M, Krishnamurthy G, Cohen IS, Robinson RB (2004) MiRP1 modulates HCN2 channel expression and gating in cardiac myocytes. J Biol Chem 279: 43497-43502. [DOI] [PubMed] [Google Scholar]
- Robinson RB, Siegelbaum SA (2003) Hyperpolarization-activated cation currents: from molecules to physiological function. Annu Rev Physiol 65: 453-480. [DOI] [PubMed] [Google Scholar]
- Santoro B, Baram TZ (2003) The multiple personalities of h-channels. Trends Neurosci 26: 550-554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Grant SG, Bartsch D, Kandel ER (1997) Interactive cloning with the SH3 domain of N-src identifies a new brain specific ion channel protein, with homology to eag and cyclic nucleotide-gated channels. Proc Natl Acad Sci USA 94: 14815-14820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Liu DT, Yao H, Bartsch D, Kandel ER, Siegelbaum SA, Tibbs GR (1998) Identification of a gene encoding a hyperpolarization-activated pacemaker channel of brain. Cell 93: 717-729. [DOI] [PubMed] [Google Scholar]
- Santoro B, Chen S, Luthi A, Pavlidis P, Shumyatsky GP, Tibbs GR, Siegelbaum SA (2000) Molecular and functional heterogeneity of hyperpolarization-activated pacemaker channels in the mouse CNS. J Neurosci 20: 5264-5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Wainger BJ, Siegelbaum SA (2002) Regulation of HCN channel surface expression by a novel C-terminal protein-protein interaction. Soc Neurosci Abstr 28: 744.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M (2001) Molecular organization of the postsynaptic specialization. Proc Natl Acad Sci USA 98: 7058-7061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Welie I, Van Hooft JA, Wadman WJ (2004) Homeostatic scaling of neuronal excitability by synaptic modulation of somatic hyperpolarization-activated Ih channels. Proc Natl Acad Sci USA 101: 5123-5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilyev DV, Barish ME (2002) Postnatal development of the hyperpolarization-activated excitatory current _I_h in mouse hippocampal pyramidal neurons. J Neurosci 22: 8992-9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainger BJ, DeGennaro M, Santoro B, Siegelbaum SA, Tibbs GR (2001) Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature 411: 805-810. [DOI] [PubMed] [Google Scholar]
- Wang Z, Xu NL, Wu CP, Duan S, Poo MM (2003) Bidirectional changes in spatial dendritic integration accompanying long-term synaptic modifications. Neuron 37: 463-472. [DOI] [PubMed] [Google Scholar]
- Wisden W, Morris BJ (1994) In situ hybridization with synthetic oligonucleotide probes. In: In situ hybridization protocols for the brain (Wisden W, Morris BJ, eds), pp 9-26. New York: Academic.
- Xue T, Marban E, Li RA (2002) Dominant-negative suppression of HCN1- and HCN2-encoded pacemaker currents by an engineered HCN1 construct: insights into structure-function relationships and multimerization. Circ Res 90: 1267-1273. [DOI] [PubMed] [Google Scholar]
- Yan Q, Radeke MJ, Matheson CR, Talvenheimo J, Welcher AA, Feinstein SC (1997) Immunocytochemical localization of TrkB in the central nervous system of the adult rat. J Comp Neurol 378: 135-157. [PubMed] [Google Scholar]
- Yu H, Wu J, Potapova I, Wymore RT, Holmes B, Zuckerman J, Pan Z, Wang H, Shi W, Robinson RB, El-Maghrabi MR, Benjamin W, Dixon J, McKinnon D, Cohen IS, Wymore R (2001) MinK-related peptide 1: a beta subunit for the HCN ion channel subunit family enhances expression and speeds activation. Circ Res 88: E84-E87. [DOI] [PubMed] [Google Scholar]
- Zerial M, McBride H (2001) Rab proteins as membrane organizers. Nat Rev Mol Cell Biol 2: 107-117. [DOI] [PubMed] [Google Scholar]