Live-Cell Imaging of Tumor Proteolysis: Impact of Cellular and Non-Cellular Microenvironment (original) (raw)
. Author manuscript; available in PMC: 2013 Jan 1.
Published in final edited form as: Biochim Biophys Acta. 2011 Aug 5;1824(1):123–132. doi: 10.1016/j.bbapap.2011.07.025
Abstract
Our laboratory has had a longstanding interest in how the interactions between tumors and their microenvironment affect malignant progression. Recently, we have focused on defining the proteolytic pathways that function in the transition of breast cancer from the pre-invasive lesions of ductal carcinoma in situ (DCIS) to invasive ductal carcinomas (IDCs). We use live-cell imaging to visualize, localize and quantify proteolysis as it occurs in real-time and thereby have established roles for lysosomal cysteine proteases both pericellularly and intracellularly in tumor proteolysis. To facilitate these studies, we have developed and optimized 3D organotypic co-culture models that recapitulate the in vivo interactions of mammary epithelial cells or tumor cells with stromal and inflammatory cells. Here we will discuss the background that led to our present studies as well as the techniques and models that we employ.
Keywords: Lysosomal proteases, proteolytic networks, functional imaging, 3D culture, organotypic models, tumor microenvironment
1. Introduction
The initial working hypothesis for those studying proteases in cancer was that invasive processes (local and during metastatic spread) required degradation of extracellular matrices by proteases. The roles of proteases in cancer are now known to be much broader, including roles in premalignant lesions, growth, and angiogenesis. Furthermore, any one protease may function differently during the early and late stages of tumor progression (for recent comprehensive reviews on proteases in cancer, see [1]). The proteases themselves derive not only from tumor cells, but also from the multiple cell types present in the tumor stroma, e.g., fibroblasts, macrophages, mast cells, leukocytes, and endothelial cells. An impediment to determining how proteases function in tumors is that a purified protease capable of cleaving a potential substrate in vitro may not be the protease or the single protease responsible for degradation of that substrate in vivo. The in vivo functions of proteases are being studied through the use of transgenic mice deficient in specific proteases. Such studies have been informative; however, relying solely on this approach is problematic for several reasons including: 1) the large number of proteases (~560 in the human genome [2–4]), 2) the interplay between proteases and their endogenous inhibitors and activators, and 3) the redundancy among proteases and the observations of compensatory changes in other proteases. An example of the latter has been found to occur in a transgenic murine model for the development of mammary cancer in which there is altered cell surface expression of the lysosomal cysteine protease cathepsin X in mice deficient in another lysosomal cysteine protease, i.e., cathepsin B [5]. Since proteases of more than one catalytic-type can cleave the same substrate, we would expect that compensatory changes could occur in which deficiency of one class of proteases results in alterations in expression of another class of proteases, e.g., a deficiency in a serine protease might result in alterations in a matrix metalloproteinase (MMP). Such complex interactions will be difficult to detect. Although the biological roles of proteases might be analyzed more successfully in genetically engineered murine cancer models in which the protease gene alleles are latent, conditional or inducible (for review, see [6, 7]), the number of proteases in the cancer degradome will likely restrict the use of such models to only a few proteases.
The complete repertoire of proteases known to be associated with human cancer, i.e., the cancer degradome (for review, see [1]), includes proteases of five catalytic types: aspartic, cysteine, metallo, serine and threonine. Examples of proteases that represent the five catalytic types are: cathepsin D (aspartic), caspases and cathepsin B (cysteine), MMP-9 and ADAM10 (metallo), plasmin and cathepsin G (serine), and the proteasome (threonine). Tumor invasion and metastasis are often assumed to be processes that involve only extracellular soluble proteases and membrane proteases, e.g., MMPs, membrane type-MMPs (MT-MMPs) such as MT1-MMP or MMP-14 and type II transmembrane serine proteases (TTSPs) such as matriptase (for recent reviews, see [8–10]). Indeed, there is extensive and compelling preclinical data implicating MMPs in malignant progression. Nonetheless, the clinical trials on MMP inhibitors (MMPIs) did not fulfill the promise of MMPs as therapeutic targets in cancer. There are several possible explanations for this apparent disconnect between the preclinical and clinical data (for reviews, see [11–14]) and there is still a widespread belief that tumor proteolysis is an extracellular process that involves primarily MMPs.
Intracellular proteases although less commonly implicated in tumor invasion and metastasis mediate invasion in lower organisms such as parasites. Invasion of plasmodium and toxoplasma, the parasites responsible for malaria and toxoplasmosis, respectively, occurs as a result of the secretion of vacuolar cysteine proteases and can be abolished with cysteine protease inhibitors (for reviews, see [15–17]). The working hypothesis in our laboratory has been that the mammalian counterparts of these enzymes, i.e., the lysosomal cysteine cathepsins, play comparable roles in the invasive processes associated with human cancers (for review, see [18]). As discussed below, some of these proteases are secreted from tumor cells and bind to the cell surface. Whether the primary role of cysteine cathepsins in cancer is pericellular, intracellular or both is not yet clear. We have shown that cysteine cathepsins participate in degradation of extracellular matrix proteins intracellularly and pericellularly. Contrary to our expectations, intracellular degradation by cysteine cathepsins occurred in the highly invasive BT549 breast carcinoma cells and pericellular degradation in the less invasive BT20 breast carcinoma cells [19]. Others have confirmed an intracellular role for cysteine cathepsins in degradation of collagens, a role that is causally linked to invasive growth of mammary tumors [20], progression of prostate cancers [21] and migration and invasion of glioma cell lines [22]. The collagens were taken up into the lysosomes via the endocytic collagen receptor uPARAP (urokinase plasminogen activator receptor associated protein)/Endo180. Activity of MT1-MMP, activation of MMP-2 by MT1-MMP and activity of uPA all have been shown to be regulated by uPARAP [23], thus indicating the critical functional interactions among pericellular and intracellular proteases that contribute to collagen degradation specifically and more generally to ‘tumor proteolysis’.
Sylven and colleagues reported an association of lysosomal proteases with cancer in 1957 [24] and later specifically an association of the lysosomal cysteine protease cathepsin B with tumor cell surfaces [25]. Subsequent studies reported secretion of lysosomal proteases: cathepsin B from human breast carcinoma explants [26, 27]; cathepsin L, another lysosomal cysteine protease, from transformed murine fibroblasts [28]; and cathepsin D, a lysosomal aspartic protease, from the human MCF7 breast carcinoma cell line [29]. We have shown that an acidic tumor microenvironment increases secretion of cathepsin B [30] and identified p11 or the light chain of the annexin II heterotetramer as a binding partner that mediates association of cathepsin B with the tumor cell surface [31] and specifically with caveolar lipid rafts [32–34]. In Figure 1A, we illustrate these known interactions of cathepsin B and a number of other proteases (serine and metallo) with caveolae (for reviews, see [18, 35, 36]).
Figure 1. Caveolae (A) and invadopodia (B) cluster proteases in networks at the cell surface.
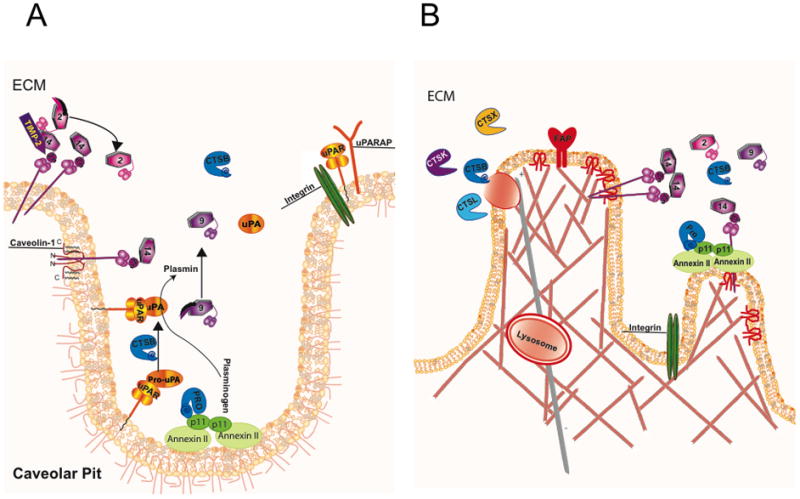
There are structural similarities between caveolae and invadopodia e.g. both require caveolin-1 and lipid rafts for the formation. (A) Caveolae are a subset of lipid rafts in the plasma membrane, which form flask-shaped invaginations as a result of the interactions of dimerized caveolin-1. Many proteases associated with extracellular matrix degradation are localized to caveolae, e.g., plasmin[ogen], MMP-2, -9 and 14 and cathepsin B (CTSB) via binding to receptors, e.g., uPA to uPAR, plasmin[ogen] and CTSB to the annexin II heterotetramer. MMP-14 is a transmembrane MMP, which is also known as MT1-MMP, whereas uPAR is inserted into the membrane by a GPI anchor and is also part of a trimeric complex with β1-integrin and uPARAP, an endocytic receptor associated with collagen uptake. (B) Invadopodia are membrane protrusions associated with filamentous actin and extracellular matrix degradation. MMP-14 and fibroblast activation protein (FAP) or seprase accumulate in the membrane of invadopodia. In addition, there is increased secretion of MMP-2 and lysosomal cysteine cathepsins (e.g., CTSK, CTSL, CTSX) from invadopodia. Depicted here is a lysosome releasing several cysteine cathepsins following trafficking to the membrane on a microtubule (grey). The hexagon shapes represent MMPs with the associated number in the frame; pro-MMPs have an additional swoosh representing their pro-peptide.

2. Proteases and Cancer: Are Proteases Therapeutic Targets?
The failures of MMPIs in clinical trials resulted in allegations that MMPs, and by extrapolation other proteases, are not appropriate therapeutic targets in cancer. Is this the case or might the failure of the MMPI trials reflect problems in clinical trial design for cytostatic agents (see [12, 37, 38] for discussion of clinical trials)? Among the critical questions is whether the MMPIs actually reached and reduced the activity of their target MMPs in vivo. The MMPI trials did not include surrogate endpoints so it is not known whether MMPs were actually inhibited in the test patients [38–40]. Clinical trials without surrogate endpoints to monitor and confirm the efficacy of the therapeutic strategies being tested should not be viewed as definitive [39, 40]. There are other concerns about the MMPI trials. Coussens et al. [12] cite data revealing that the tumors studied in the clinical trials did not necessarily express the particular proteases targeted by the MMPIs. None of the clinical trials with BAY 12-9566 or other MMPIs included patients with breast cancers although this is a cancer for which there is strong preclinical evidence that MMPs impact progression. For example, Sledge and colleagues [41] have demonstrated efficacy for BAY 12-9566 in an orthotopic human breast model. Furthermore, not all MMPs should be inhibited; this is clearly true for MMP-8 (collagenase-2), an MMP expressed by inflammatory neutrophils, which plays a protective role in skin cancer [42]. Similar observations have been made for cathepsin L, a cysteine protease with broad substrate specificity [43]. Cathepsin L plays a protective role in both viral- and carcinogen-induced tumor progression in the mouse epidermis [44, 45]. In mice deficient in cathepsin L, the protection can be restored by transgenic expression of the human orthologue cathepsin V or L2 [44]. Such data on cathepsins L/V, MMP-8 and other MMPs [46, 47] raise the possibility that broad-spectrum inhibitors such as the MMPIs used in clinical trials might have unanticipated side effects and indicate how essential it is to define the intended protease targets before using protease inhibitors for cancer therapy [48]. Unanticipated side effects did occur in the clinical trials, limiting the amount of MMPIs that the patients could take and necessitating MMPI-free holidays in some patients [12]. How endogenous protease inhibitors factor into the equation is also of relevance. Tissue inhibitor of metalloproteinases-1 (TIMP-1) has been shown to promote carcinogenesis of squamous cell carcinoma of the skin, exerting “differential regulation on tissues in a stage-dependent manner” [49]. TIMP-2 binds to MT1-MMP and in so doing acts as a receptor for proMMP-2, thereby activating MMP-2 on the cell surface [50]. TIMPs also can affect tumor proliferation and apoptosis by metalloprotease-independent as well as metalloprotease-dependent mechanisms [51]. In short, we are still far from having a thorough understanding of the roles of proteases in malignant progression: this includes understanding the roles of MMPs (see also [52]), the roles of other catalytic-types of proteases, how those roles may change during the course of malignant progression, whether the critical proteases come from tumor and/or tumor-associated cells, and whether the critical proteases are affected by interactions of the tumor with its microenvironment, etc. Despite ample preclinical evidence that proteases function in the progression of cancer, we do not yet know which protease(s) is the most appropriate target for an anti-protease therapy in a particular cancer or when during the course of cancer development and progression an anti-protease therapy might prove most effective. We might move substantially closer to answers to these questions if we consider the interplay between different proteases within networks; it is becoming quite clear that inhibition of a single protease or a single family of proteases is not sufficient to alter cancer progression.
3. Proteolytic Networks and Tumor Proteolysis
Proteases interact with one another in networks that are generally comprised of more than one catalytic-type of protease (for reviews, see [53–56]). Intracellularly, the colocalization of proteases in structures such as lysosomes and proteasomes or in complexes such as inflammasomes facilitates these networks [57–59]. In a comparable fashion, interactions among proteases at the cell surface may be enhanced by the clustering of proteases within specialized structures, such as caveolar lipid rafts (Figure 1A [60, 61]) and invadopodia (Figure 1B; for review, see [62]). Yamaguchi et al. [9, 63] have linked caveolae to the formation of invadopodia and to degradation of extracellular matrix by breast cancer cells. The cartoons of Figure 1 depict potential interactions among proteases that have been localized to caveolae (Figure 1A) and to invadopodia (Figure 1B). The presence of proteolytic networks in these specialized membrane structures may speak to the critical roles played by protease interactions at the tumor cell surface rather than to the importance of any single protease on the tumor cell surface.
Caveolae are a subset of lipid microdomains within the plasma membrane that are identifiable as flask-shaped invaginations in electron micrographs. The structural protein responsible for the formation of caveolae is caveolin-1 as confirmed by an absence of caveolae in mice deficient in this protein [64]. Increased expression of caveolin-1 has been correlated with a number of cancers: advanced prostate and breast [65], bladder [66], renal [67], lung [68], esophageal [69] and colon [70–72]. In head and neck cancer, caveolin-1 has been reported to function as an oncogene [73]. Others contend that caveolin-1 is a tumor suppressor (for review, see [74]). These contradictory findings may reflect tumor type and stage and the constituents of the caveolae ([32, 33] and for review, see [75]) as well as the putative roles for caveolin-1 in lipid microdomains other than caveolae (for review, see [76]).
Caveolae are abundant in a wide variety of cells. This includes endothelial cells and adipocytes in which caveolin-1 has been shown to play distinct roles [77]. Proteases of the cysteine, serine and metallo catalytic-types have been localized to caveolae, e.g., cathepsin B, plasmin[ogen], urokinase plasminogen activator (uPA), MMPs -2, -9 -11, -13, -14 (for review, see [75]), as have cell surface binding proteins for proteases. These include the annexin II heterotetramer that binds plasmin[ogen] [78, 79] and pro-cathepsin B [80], the urokinase plasminogen activator receptor (uPAR) that binds both inactive and active forms of uPA [81]. Some of the proteases, their binding partners and how they interact are illustrated in Figure 1A. For example, active cathepsin B, shown in the cartoon as a teal structure labeled CTSB, can activate pro-uPA to its active form, which can activate plasminogen to plasmin. MMPs can also be activated in caveolae: pro-MMP-9 by plasmin and MMP-2 by MMP-14 in complex with its inhibitor TIMP-2. We hypothesize that this clustering of proteases leads to activation of proteolytic networks involving the cooperation of several families of proteases.
The colocalization of proteases in caveolae is associated with coordinated endothelial cell migration and formation of tube-like structures [32] and invasion and degradation of type IV collagen by tumor cells [33]. Interfering with this colocalization results in reduced migration and proteolytic activity. For example, in annexin II-null endothelial cells, there is reduced activation of plasmin, MMP-9 and -13 as well as less directed cell migration and vessel sprouting [82]. In colon cancer cells, down-regulation of caveolin-1 decreases the association of cathepsin B and its binding protein p11 and uPA and its receptor uPAR with caveolae fractions, coinciding with decreased degradation of type IV collagen and invasion [33]. The proteases associated with caveolae have been shown to activate downstream proteases and signaling cascades [83, 84]. This clustering of proteases in caveolae is thus the basis for our hypothesis that caveolae enhance focal matrix degradation associated with normal and pathological processes.
Podosomes and invadopodia are other structural elements that cluster proteolytic activity at specific sites. Podosomes are found in cells of myeloid lineage such as dendritic cells, macrophages and osteoclasts [85] and have been proposed to mediate protease-dependent cell migration [86, 87]. Invadopodia are membrane protrusions found in tumor cells through which proteases are delivered to participate in focal degradation and invasion [88, 89] (Figure 1B). Among the proteases that accumulate within invadopodia are MT1-MMP and the serine protease seprase or fibroblast activation protein (FAP) [9, 90, 91]; (Figure 1B). Intriguingly, lipid rafts and caveolin-1 have been linked to formation of invadopodia and extracellular matrix degradation [63, 92]. The relationship between podosomes and invadopodia is not well defined. Yamada and colleagues [93] analyzed this relationship in macrophages, fibroblasts and tumor cells grown on a 2D fibronectin matrix. By live-cell imaging, they observed that tumor cell invadopodia are associated with rapid ruffling of the membrane and formation of invasive filamentous protrusions. This was not observed in either macrophage podosomes or focal adhesions in the fibroblasts. On the other hand, van Goethem et al. [86, 94] have shown that macrophages grown in a 3D gelled collagen I matrix exhibit a mesenchymal migration, similar to that of tumor cells, that is associated with dynamic protrusions and is dependent on proteolytic activity. In this study, an additional level of complexity was apparent as the composition, architecture and stiffness of the matrix were found to affect the type of migration observed as well as whether the migration was protease-dependent. In contrast to tumor cells, in which MT1-MMP has been suggested to be the primary mediator of invasion [95], van Goethem et al. [94] found that MMPIs did not block the protease-dependent 3D mesenchymal migration of macrophages. They propose that lysosomal cysteine proteases such as cathepsin B are responsible, citing a study by Tu et al. [96] in which cathepsin B and lysosomal exocytosis were linked to matrix degradation and invasion by transformed fibroblasts. Another podosome-related degradation process that involves exocytosis of a lysosomal cysteine protease, i.e., cathepsin K, is osteoclastic bone resorption [97]. Mice deficient in cathepsin K exhibit impaired bone resorption, which ultimately results in osteopetrosis [98]. In osteoclasts, cathepsin K is secreted into a resorptive pit formed by a sealing zone of podosome units due to the exocytosis of lysosomes [99]. As a result, lysosomal proton pumps are inserted into the osteoclast membrane, which in turn leads to acidification of the resorptive pit. Consistent with this is that functional proton pumps are present in plasma membranes of invasive tumor cells [100, 101]. Thus, comparable processes may be responsible for the acidification of the resorptive pit between osteoclasts and bone and the microenvironment surrounding solid tumors.
Among the emerging hallmarks added by Weinberg and Hanahan to their original 6 hallmarks of cancer [102] is the reprogramming of energy metabolism [103]. This is often designated as the Warburg effect, named after Otto Warburg who first reported that tumor cells exhibit increased glucose consumption and the ability to survive and progress in acidic environments [104]. A majority of solid tumors have an acidic microenvironment [105] with the extracellular pH (pHe) in tumors ranging from 5.3–7.2 [106, 107]. Acidification can be attributed to a combination of factors including chaotic tumor vasculature, increased aerobic glycolysis, increased export of lactic acid in concert with diminished buffering capacity of tumor interstitial fluids [108] and increased secretion of protons [109, 110]. Whatever the mechanism for acidification the result is a pericellular environment that would favor the activity of proteases such as cysteine cathepsins that have pH optima that are slightly acidic. In addition, an acidic pHe induces a redistribution of lysosomes to the tumor cell surface and secretion of their content of lysosomal proteases via a microtubule dependent process ([111]; Figure 2). Although not experimentally confirmed, the movement of lysosomes to the tumor cell surface should result in insertion of proton pumps into the plasma membrane as has been shown in osteoclasts.
Figure 2. An acidic microenvironment is generated by tumors and enhances invasion and degradation of the extracellular matrix.
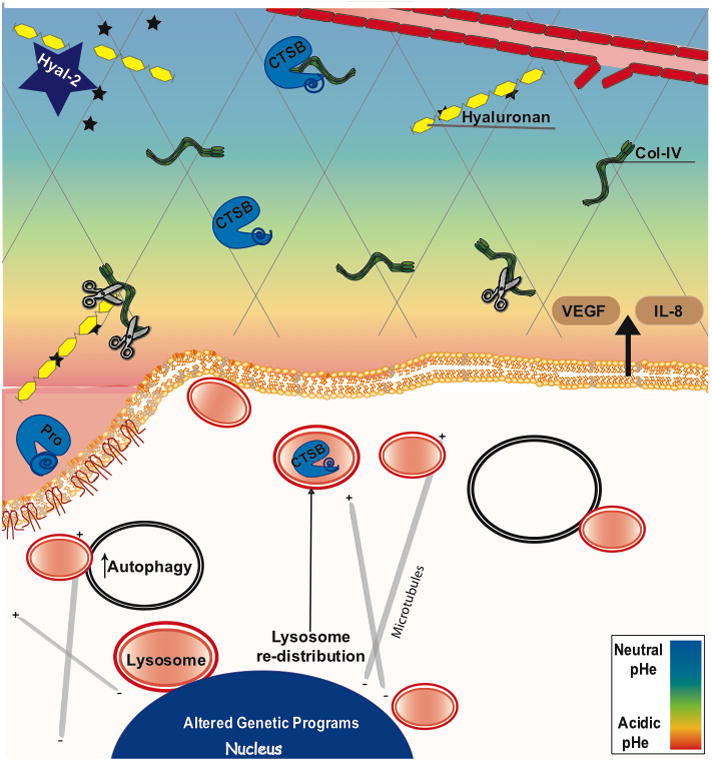
The scissors indicates cleavage of the basement membrane protein collagen IV. In response to the pericellular acidic pH, lysosomes redistribute to the cell periphery via a microtubule-dependent process [111], possibly leading to the observed secretion of both active and pro forms of cathepsin B (Rothberg, Sloane, unpublished data) as well as other lysosomal enzymes from breast cancer cells. This also includes lysosomal glyosidases such as hyaluronase-2 (Hyal-2) [176], which cleaves hyaluronan and release of bioactive molecules bound to this matrix component (stars).
An acidic pericellular pH has several diverse effects that might enhance cancer progression. One could be to help tumors to escape immunosurveillance by inhibiting the immune response [112, 113]. For an in-depth discussion of the effects of an acidic pH on the innate immune system, see review by Lardner [114]. Another is to promote angiogenesis by triggering expression of VEGF and IL-8 by the cancer cells [115–118]. Xu et al. systematically characterized VEGF upregulation following exposure to acidic pH via mechanisms resulting in activation of Ras, Raf, and ERK1/2 MAPK pathways and subsequent down-stream binding of the transcription factor AP-1 to the VEGF promoter [116]. Additional transcription factors responding to acidic pH include NFkB and HIF2α, which also increase angiogenesis [115, 117, 118]. Acidic pH also increases the rupture of tumor-released micro-vesicles that contain angiogenic factors and proteases [119]. Cathepsin B secreted via such micro-vesicles has been suggested to induce activation of MMP-2 and -9 and potentiates endothelial cell invasion in an acidic environment [120]. Human and mouse melanoma cells exposed to an acidic pH before tail vein injection showed a significant increase in experimental lung metastases [121, 122]. Surprisingly, the enhanced metastasis induced by acidic pH can be reduced by putting sodium bicarbonate in the drinking water [123] a non-toxic treatment.
Most of the research in our laboratory has focused on one lysosomal enzyme, the cysteine protease cathepsin B (for reviews, see [60, 124–126]) and a few other papain-family cysteine cathepsins. Analysis of only cysteine cathepsins, however, does not allow us to define the complete repertoire of proteases that contribute to tumor proteolysis. Therefore, we have established a live cell confocal assay to study proteolysis of extracellular matrix proteins that are encountered by invading tumor cells. This assay employs dye-quenched (DQ) derivatives of the extracellular matrix proteins collagens I and IV [127]. This is in contrast to assays measuring degradation of synthetic substrates that are selective for a specific protease or one catalytic-type of protease. We have used the DQ-substrates to analyze proteolysis in vitro by tumor cell lysates and conditioned media [19] and in situ by live tumor cells alone ([19, 128, 129]; Figure 3, also see supplemental movies) or in co-culture with stromal and inflammatory cells [129]. By evaluating the degradation of DQ-collagen IV in the presence and absence of protease inhibitors, we have demonstrated that proteolytic networks are responsible for degradation of collagen IV. For the breast cancer cell lines analyzed, the proteases are cathepsin B, proteases of the plasminogen cascade and MMPs [129]. For the colon cancer cell lines analyzed, they are cathepsin B and MMPs [129]. A caveat to these findings is that as with any model this assay has limitations. The DQ-collagen substrates are heavily labeled with FITC and thus are no longer native proteins. This renders the DQ-collagens more readily cleavable than is endogenous type IV collagen and thus the proteases identified as cleaving these substrates will need to be confirmed by alternative techniques (for further discussion of assay limitations, see [130]). By evaluating the degradation of DQ-collagen IV at neutral and slightly acidic pHe (7.4 vs. 6.8), we have shown that the acidic microenvironment of tumors may substantially enhance tumor proteolysis (Figure 3, also see supplemental movies). Our data indicate that meaningful analyses of tumor proteolysis require assessment of the roles played by multiple proteases, how those proteases interact in networks and knowledge of the properties of the microenvironment in which the proteases are functioning. This includes the non-cellular (e.g., pHe) microenvironment as well as the cellular microenvironment. The incorporation of additional cell types into our analyses will be discussed below.
Figure 3. At pH 6.8, degradation of collagen IV is significantly increased.

After 2 days in culture at either pH 7.4 and 6.8, degradation products (green) accumulate pericellularly around structures formed by MDA-MB-231-RFP human breast carcinoma cells. Fluorescent degradation products are increased at the acidic pH. Red and blue represent expression of RFP in the cytoplasm and nuclear staining, respectively. 3D reconstructions of confocal image stacks were generated with Volocity software (Perkin Elmer). The increase in proteolysis at acidic pH was significant (p ≤ 0.01; x ± SD; n=3) as illustrated by the graph; proteolysis was quantified following our published protocols [127].
4. Contribution of Non-Tumor Cells to Tumor Proteolysis
Liotta and Kohn [131] suggested that cancer therapies should target the stroma or the tumor-stroma interface. The need to target stroma is certainly true when using protease inhibitors, as cells present in the tumor-associated stroma (e.g., fibroblasts, endothelial cells, inflammatory cells, myofibroblasts) are all important sources of proteases (for reviews, see [132–137]). Stromal cells can also impact tumor proteolysis through the expression of endogenous protease inhibitors. Myofibroblasts, for example, are suggested to be the primary source of plasminogen activator inhibitor-1 (PAI-1) in human breast carcinomas [138]. Therefore, we need to study proteolysis in the context of tumor cells interacting with other cells in their microenvironment, interactions that our group has worked to model in vitro in organotypic co-cultures.
In early studies, we had used organotypic co-cultures to establish that stromal cells significantly impact tumor proteolysis [129]. Degradation of DQ-collagen IV is increased as much as 17-fold in live 3D co-cultures of stromal cells (fibroblasts or fibroblasts + macrophages) with breast or colon tumor cells [129]. Furthermore, we demonstrated that serine proteases of the plasminogen cascade degrade DQ-collagen IV in organotypic colon tumor co-cultures, yet not in monotypic colon tumor cultures [129]. These observations could impact whether protease inhibitors are efficacious in vivo. Also relevant to including stromal cells in co-cultures are observations that fibroblasts isolated from invasive ductal breast carcinomas, but not normal breast, can recruit other stromal cells, in this case blood monocytes, into 3D spheroids [139]. Analyzing stromal components is crucial as infiltration of macrophages in vivo potentiates malignant progression of the tumors in a transgenic mouse model for mammary carcinoma [140, 141]. This is also the case for branching morphogenesis of the mammary gland [141, 142], a developmental process already known to be associated with proteolysis [143]. The literature on protease expression by stromal cells and our findings in organotypic co-cultures [129] strongly suggest that delineating the proteases and protease inhibitors that contribute to tumor proteolysis will necessitate evaluating the roles of stromal cells in vitro in organotypic co-cultures, i.e., in systems in which we can dissect out the contribution of individual cell types to tumor proteolysis.
The regulation of protease activity by their endogenous inhibitors and activators, the redundancy among proteases and the complexity of their biological roles suggest that we also should take advantage of cell-based 3D models to analyze the functions of proteases in a more physiological context. Schmeichel and Bissell [144] describe how such models have demonstrated “a role for the stroma as an important regulator of epithelial function and carcinoma progression.” They also predict that, if 3D models were used for high-throughput screening of drugs, they will “yield molecules that will sustain efficacy in clinical trials.” In order to develop a tractable 3D model in which to study breast cancer progression model in vitro, we extended an established model for analyzing morphogenesis and oncogenic transformation of MCF-10A cells in a 3D laminin-rich, reconstituted basement membrane (rBM) overlay culture system [145, 146] to include premalignant and malignant variants of the benign parental MCF-10A cells [147–152]. The MCF-10A cells have previously been characterized in 3D rBM overlay culture as a model for normal mammary epithelia that exhibits apicobasal polarity, cell-cell junctions, strict control of cell proliferation and apoptosis, and formation of functional glandular structures [145, 146, 153]. When isogenic variants of MCF-10A cells are grown in 3D rBM overlay culture, there is increasing derangement of normal acinar structures that parallels progression of the variants to a malignant phenotype. This in vitro MCF-10A variant series includes stages of atypical hyperplasia, dysplasia and carcinoma [154] comparable to those observed in vivo in xenografts [150, 151]. Thus, we have in hand an in vitro model in which the causal roles of proteases and proteolytic pathways in progression to invasive carcinomas can be analyzed.
There is an extensive literature [155–168] on the role of the tumor microenvironment in promoting breast tumor progression. There is also striking evidence from mouse transgenic models of mammary carcinoma that the tumor microenvironment, including normal cellular constituents of mammary tissue or cells that infiltrate as a consequence of angiogenic or inflammatory processes, contributes to progression of these tumors [e.g., see [5, 20, 169–173]. Although increases in expression of proteases have long been associated with tumors, many of those proteases originate from cells that infiltrate the tumors rather than the tumor cells [e.g., see [173, 174]. For example, we have shown that co-culturing macrophages with breast carcinoma cells increases degradation of DQ-collagen IV by ~3-fold, whereas fibroblasts increase degradation ~15-fold [129].
To evaluate temporal, dynamic and reciprocal interactions between the epithelial components of breast tumors and other cell types that infiltrate into tumors, we have established 3D co-culture models that we term MAME for mammary architecture and microenvironment engineering. These complex organotypic 3D culture models represent a new paradigm for in vitro analyses of pathobiological processes for breast cancer, yet possess sufficient flexibility to be applicable to a wide variety of other cancers. Our MAME models: 1) incorporate multiple cell types, 2) allow regulated introduction of soluble factors, 3) allow collection of conditioned media without disturbing cultures, and 4) enable monitoring in real-time of quantitative relevant information. In Figure 4, we illustrate in a schematic form two types of MAME co-cultures: tripartite and mixed. In the tripartite cultures, the first layer is type I collagen in which breast fibroblasts are embedded. The second and third layers are the 3D rBM overlay cultures containing tumor cells such as the MCF-10A variants described above. For the mixed cultures, the two cell types are mixed before plating in rBM overlay cultures. We have maintained MAME cultures up to 24 days and imaged live cultures at intervals over that period. To demonstrate that MAME tripartite and mixed cultures can be used to assess changes in proteolysis, we mix DQ-collagen I with the first layer of type I collagen in the tripartite cultures and DQ-collagen IV with the rBM layer in both tripartite and mixed cultures. In Figure 5 (also see supplemental movies), we show examples of MAME tripartite co-cultures for MCF10.CA1d cells and WS-12Ti human breast tumor-associated fibroblasts over three weeks of growth. The fibroblasts can be observed to infiltrate into the cultures over this time period. A dramatic increase in size of the tumor structure as well as increased invasiveness and proteolysis are observed over the time in culture. In other studies, we have shown that a small molecule MET inhibitor can reduce both invasiveness and proteolysis [175], as can antagonists to a number of chemokines and cytokines (Sameni and Sloane, unpublished data). The fact that we can demonstrate such effects in MAME co-cultures supports their potential utility as a preclinical screening platform that can be used to identify druggable targets as well as test therapies that target tumors in the context of their microenvironment. This includes assessing whether proteolytic networks in the tumor microenvironment are druggable targets.
Figure 4. Schematic diagram illustrating representative components of MAME (mammary architecture and microenvironment engineering) tripartite (A) and mixed (B) co-cultures.
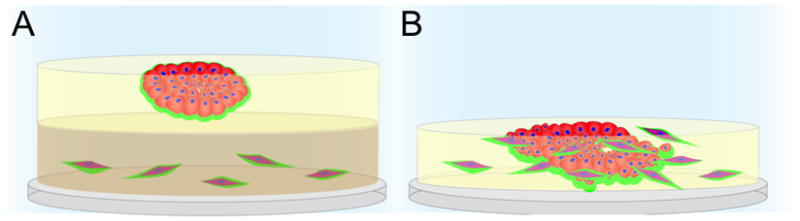
(A) Plastic coverslips are coated with collagen I, containing DQ-collagen I and fibroblasts (red). A 2nd layer of reconstituted basement membrane (rBM) containing DQ-collagen IV is added and tumor cells (red) are plated on top. The cultures are then overlaid with a 3rd layer of 2% rBM, which also is included in subsequent changes of media. (B) Coverslips are coated with rBM containing DQ-collagen IV and a mixture of fibroblasts (red) and tumor cells (red) are plated on top. MAME co-cultures are imaged live using an immersion lens in order to follow changes in morphogenesis and collagen degradation, which is depicted here as green.
Figure 5. Live-cell imaging of tumor proteolysis in 4D.

Extensive proteolysis (green fluorescence) is observed as MAME tripartite co-cultures of MCF10.CA1d human breast carcinoma cells and WS-12Ti human breast carcinoma-associated fibroblasts grow and invade through the surrounding matrix over a 23-day period. Over this time period, fibroblasts become incorporated into the structures. Top (left panels), side (middle panels) and bottom (right panels) views of representative co-cultures reconstructed in 3D are illustrated. Co-cultures were stained with CellTracker Orange to label the carcinoma cells and fibroblasts (red) and then imaged by confocal microscopy at 3 (top row), 16 (middle row) and 23 (bottom row) days. Magnification: top panel, 40X; and middle and bottom panels, 10X.
Conclusions
“Tumor proteolysis” represents a composite of the activity of proteases of multiple catalytic types: metallo, serine, cysteine, aspartic and threonine. Both secreted proteases and intracellular proteases contribute to “tumor proteolysis.” This includes lysosomal proteases, especially cysteine cathepsins, which act both intracellularly within the lysosome and pericellularly in association with specialized regions on the cell surface such as caveolae and invadopodia. These specialized regions cluster proteases in networks that enhance degradation of the extracellular matrix and invasiveness. “Tumor proteolysis” is affected by interactions of tumor cells with the cellular (fibroblasts, macrophages) and non-cellular (acidic pHe) components of the tumor microenvironment. We have taken advantage of live-cell imaging technology to characterize and localize the proteolytic activity of tumor cells and tumor-associated cells within a supportive 3D scaffold that recapitulates the architecture of developing tumors in vivo. This technology has allowed us to analyze and quantify the contributions of various cell types and the non-cellular microenvironment to the composite tumor proteolysis and to establish a role for lysosomal cysteine cathepsins in tumor proteolysis.
Supplementary Material
01
02
03. Supplemental movies – Figure 3.
Animation of 3D reconstruction of confocal images depicted in Figure 3.
04
05. Supplemental movies – Figure 5.
Animation of 3D reconstruction of confocal images depicted in Figure 5.
Highlights of the review.
- Lysosomal proteases contribute to intracellular and pericellular tumor proteolysis.
- Pericellular tumor proteolysis occurs at focal sites where proteolytic networks are present on the cell surface.
- Pericellular proteolysis is increased by an acidic tumor microenvironment.
- Both tumor and tumor-associated cells mediate proteolysis in the tumor microenvironment.
- Tumor proteolysis can be localized in real-time by live-cell imaging techniques.
Acknowledgments
This work was supported by U.S. Public Health Service Grants: R01 CA56586, R01 CA131990, R01 CA131990S and U54 RR022241. The Microscopy, Imaging and Cytometry Resources core is supported by National Institutes of Health Center Grant P30 CA22453.
Abbreviations
3D/4D
three/four dimensional
DCIS
ductal carcinoma in situ
DQ
dye-quenched
FAP
fibroblast activation protein
GPI
glycosylphosphatidylinositol
IDC
invasive ductal carcinoma
MAME
mammary architecture and microenvironment engineering
MMP
matrix metalloproteinase
MMPIs
matrix metalloproteinase inhibitors
MT-MMP
membrane type-MMP
PAI-1
plasminogen activator inhibitor-1
pHe
extracellular pH
rBM
reconstituted basement membrane
TIMP
tissue inhibitors of metalloproteinases
TTSPs
type II transmembrane serine proteases
uPA
urokinase plasminogen activator
uPAR
urokinase plasminogen activator receptor
uPARAP
urokinase plasminogen activator receptor associated protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Edwards DR, Hoyer-Hansen G, Blasi F, Sloane BF, editors. The Cancer Degradome--Proteases and Cancer Biology. Springer; New York: 2008. [Google Scholar]
- 2.Cal S, Moncada-Pazos A, Lopez-Otin C. Expanding the complexity of the human degradome: polyserases and their tandem serine protease domains. Front Biosci. 2007;12:4661–4669. doi: 10.2741/2415. [DOI] [PubMed] [Google Scholar]
- 3.Rawlings N, Barrett AJ. MEROPS: The peptidease database. [ http://merops.sanger.ac.uk/]
- 4.Puente XS, Sanchez LM, Overall CM, Lopez-Otin C. Human and mouse proteases: a comparative genomic approach. Nat Rev Genet. 2003;4:544–558. doi: 10.1038/nrg1111. [DOI] [PubMed] [Google Scholar]
- 5.Vasiljeva O, Papazoglou A, Kruger A, Brodoefel H, Korovin M, Deussing J, Augustin N, Nielsen BS, Almholt K, Bogyo M, Peters C, Reinheckel T. Tumor cell-derived and macrophage-derived cathepsin B promotes progression and lung metastasis of mammary cancer. Cancer Res. 2006;66:5242–5250. doi: 10.1158/0008-5472.CAN-05-4463. [DOI] [PubMed] [Google Scholar]
- 6.Van Dyke T, Jacks T. Cancer modeling in the modern era: progress and challenges. Cell. 2002;108:135–144. doi: 10.1016/s0092-8674(02)00621-9. [DOI] [PubMed] [Google Scholar]
- 7.Tuveson DA, Jacks T. Technologically advanced cancer modeling in mice. Curr Opin Genet Dev. 2002;12:105–110. doi: 10.1016/s0959-437x(01)00272-6. [DOI] [PubMed] [Google Scholar]
- 8.Gialeli C, Theocharis AD, Karamanos NK. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011;278:16–27. doi: 10.1111/j.1742-4658.2010.07919.x. [DOI] [PubMed] [Google Scholar]
- 9.Poincloux R, Lizarraga F, Chavrier P. Matrix invasion by tumour cells: a focus on MT1-MMP trafficking to invadopodia. J Cell Sci. 2009;122:3015–3024. doi: 10.1242/jcs.034561. [DOI] [PubMed] [Google Scholar]
- 10.Webb SL, Sanders AJ, Mason MD, Jiang WG. Type II transmembrane serine protease (TTSP) deregulation in cancer. Front Biosci. 2011;16:539–552. doi: 10.2741/3704. [DOI] [PubMed] [Google Scholar]
- 11.Zucker S, Cao J, Chen WT. Critical appraisal of the use of matrix metalloproteinase inhibitors in cancer treatment. Oncogene. 2000;19:6642–6650. doi: 10.1038/sj.onc.1204097. [DOI] [PubMed] [Google Scholar]
- 12.Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 13.Zucker S, Cao J. Selective matrix metalloproteinase (MMP) inhibitors in cancer therapy: ready for prime time? Cancer Biol Ther. 2009;8:2371–2373. doi: 10.4161/cbt.8.24.10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Overall CM, Kleifeld O. Tumour microenvironment - opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat Rev Cancer. 2006;6:227–239. doi: 10.1038/nrc1821. [DOI] [PubMed] [Google Scholar]
- 15.Binder EM, Kim K. Location, location, location: trafficking and function of secreted proteases of Toxoplasma and Plasmodium. Traffic. 2004;5:914–924. doi: 10.1111/j.1600-0854.2004.00244.x. [DOI] [PubMed] [Google Scholar]
- 16.Rosenthal PJ. Cysteine proteases of malaria parasites. Int J Parasitol. 2004;34:1489–1499. doi: 10.1016/j.ijpara.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 17.McKerrow JH, Caffrey C, Kelly B, Loke P, Sajid M. Proteases in parasitic diseases. Annu Rev Pathol. 2006;1:497–536. doi: 10.1146/annurev.pathol.1.110304.100151. [DOI] [PubMed] [Google Scholar]
- 18.Mohamed MM, Sloane BF. Cysteine cathepsins: multifunctional enzymes in cancer. Nat Rev Cancer. 2006;6:764–775. doi: 10.1038/nrc1949. [DOI] [PubMed] [Google Scholar]
- 19.Sameni M, Moin K, Sloane BF. Imaging proteolysis by living human breast cancer cells. Neoplasia. 2000;2:496–504. doi: 10.1038/sj.neo.7900116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Curino AC, Engelholm LH, Yamada SS, Holmbeck K, Lund LR, Molinolo AA, Behrendt N, Nielsen BS, Bugge TH. Intracellular collagen degradation mediated by uPARAP/Endo180 is a major pathway of extracellular matrix turnover during malignancy. J Cell Biol. 2005;169:977–985. doi: 10.1083/jcb.200411153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kogianni G, Walker MM, Waxman J, Sturge J. Endo180 expression with cofunctional partners MT1-MMP and uPAR-uPA is correlated with prostate cancer progression. Eur J Cancer. 2009;45:685–693. doi: 10.1016/j.ejca.2008.11.023. [DOI] [PubMed] [Google Scholar]
- 22.Takahashi S, Yamada-Okabe H, Hamada K, Ohta S, Kawase T, Yoshida K, Toda M. Downregulation of uPARAP mediates cytoskeletal rearrangements and decreases invasion and migration properties in glioma cells. J Neurooncol. 2010 doi: 10.1007/s11060-010-0398-z. [DOI] [PubMed] [Google Scholar]
- 23.Messaritou G, East L, Roghi C, Isacke CM, Yarwood H. Membrane type-1 matrix metalloproteinase activity is regulated by the endocytic collagen receptor Endo180. J Cell Sci. 2009;122:4042–4048. doi: 10.1242/jcs.044305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sylven B, Malmgren H. The histological distribution of proteinase and peptidase activity in solid tumor transplants; a histochemical study on the enzymic characteristics of the different tumor cell types. Acta Radiol Suppl. 1957:1–124. [PubMed] [Google Scholar]
- 25.Sylven B, Snellman O, Strauli P. Immunofluorescent studies on the occurrence of cathepsin B1 at tumor cell surfaces. Virchows Arch B Cell Pathol. 1974;17:97–112. doi: 10.1007/BF02912840. [DOI] [PubMed] [Google Scholar]
- 26.Poole AR, Tiltman KJ, Recklies AD, Stoker TA. Differences in secretion of the proteinase cathepsin B at the edges of human breast carcinomas and fibroadenomas. Nature. 1978;273:545–547. doi: 10.1038/273545a0. [DOI] [PubMed] [Google Scholar]
- 27.Recklies AD, Tiltman KJ, Stoker TA, Poole AR. Secretion of proteinases from malignant and nonmalignant human breast tissue. Cancer Res. 1980;40:550–556. [PubMed] [Google Scholar]
- 28.Gal S, Gottesman MM. The major excreted protein of transformed fibroblasts is an activable acid-protease. J Biol Chem. 1986;261:1760–1765. [PubMed] [Google Scholar]
- 29.Morisset M, Capony F, Rochefort H. The 52-kDa estrogen-induced protein secreted by MCF7 cells is a lysosomal acidic protease. Biochem Biophys Res Commun. 1986;138:102–109. doi: 10.1016/0006-291x(86)90252-4. [DOI] [PubMed] [Google Scholar]
- 30.Rozhin J, Robinson D, Stevens MA, Lah TT, Honn KV, Ryan RE, Sloane BF. Properties of a plasma membrane-associated cathepsin B-like cysteine proteinase in metastatic B16 melanoma variants. Cancer Res. 1987;47:6620–6628. [PubMed] [Google Scholar]
- 31.Mai J, Finley RL, Jr, Waisman DM, Sloane BF. Human procathepsin B interacts with the annexin II tetramer on the surface of tumor cells. J Biol Chem. 2000;275:12806–12812. doi: 10.1074/jbc.275.17.12806. [DOI] [PubMed] [Google Scholar]
- 32.Cavallo-Medved D, Rudy D, Blum G, Bogyo M, Caglic D, Sloane BF. Live-cell imaging demonstrates extracellular matrix degradation in association with active cathepsin B in caveolae of endothelial cells during tube formation. Exp Cell Res. 2009;315:1234–1246. doi: 10.1016/j.yexcr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cavallo-Medved D, Mai J, Dosescu J, Sameni M, Sloane BF. Caveolin-1 mediates the expression and localization of cathepsin B, pro-urokinase plasminogen activator and their cell-surface receptors in human colorectal carcinoma cells. J Cell Sci. 2005;118:1493–1503. doi: 10.1242/jcs.02278. [DOI] [PubMed] [Google Scholar]
- 34.Victor BC, Anbalagan A, Mohamed MM, Sloane BF, Cavallo-Medved D. Inhibition of cathepsin B activity attenuates extracellular matrix degradation and inflammatory breast cancer invasion. Breast Cancer Research. 2011 doi: 10.1186/bcr3058. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Victor BC, Sloane BF. Cysteine cathepsin non-inhibitory binding partners: modulating intracellular trafficking and function. Biol Chem. 2007;388:1131–1140. doi: 10.1515/BC.2007.150. [DOI] [PubMed] [Google Scholar]
- 36.Cavallo-Medved D, Moin K, Sloane BF. UCSD-Nature Signaling Gateway Molecule Pages. Cathepsin B. (In Press) [PMC free article] [PubMed] [Google Scholar]
- 37.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 38.Chau I, Rigg A, Cunningham D. Matrix metalloproteinase inhibitors--an emphasis on gastrointestinal malignancies. Crit Rev Oncol Hematol. 2003;45:151–176. doi: 10.1016/s1040-8428(02)00015-x. [DOI] [PubMed] [Google Scholar]
- 39.McIntyre JO, Matrisian LM. Molecular imaging of proteolytic activity in cancer. J Cell Biochem. 2003;90:1087–1097. doi: 10.1002/jcb.10713. [DOI] [PubMed] [Google Scholar]
- 40.Li WP, Anderson CJ. Imaging matrix metalloproteinase expression in tumors. Q J Nucl Med. 2003;47:201–208. [PubMed] [Google Scholar]
- 41.Nozaki S, Sissons S, Chien DS, Sledge GW., Jr Activity of biphenyl matrix metalloproteinase inhibitor BAY 12-9566 in a human breast cancer orthotopic model. Clin Exp Metastasis. 2003;20:407–412. doi: 10.1023/a:1025473709656. [DOI] [PubMed] [Google Scholar]
- 42.Balbin M, Fueyo A, Tester AM, Pendas AM, Pitiot AS, Astudillo A, Overall CM, Shapiro SD, Lopez-Otin C. Loss of collagenase-2 confers increased skin tumor susceptibility to male mice. Nat Genet. 2003;35:252–257. doi: 10.1038/ng1249. [DOI] [PubMed] [Google Scholar]
- 43.Turk B, Turk V, Turk D. Structural and functional aspects of papain-like cysteine proteinases and their protein inhibitors. Biol Chem. 1997;378:141–150. [PubMed] [Google Scholar]
- 44.Dennemarker J, Lohmuller T, Mayerle J, Tacke M, Lerch MM, Coussens LM, Peters C, Reinheckel T. Deficiency for the cysteine protease cathepsin L promotes tumor progression in mouse epidermis. Oncogene. 2010;29:1611–1621. doi: 10.1038/onc.2009.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benavides F, Perez C, Blando J, Contreras O, Shen J, Coussens LM, Fischer SM, Kusewitt DF, Digiovanni J, Conti CJ. Protective role of cathepsin L in mouse skin carcinogenesis. Mol Carcinog. 2011 doi: 10.1002/mc.20792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martin MD, Matrisian LM. The other side of MMPs: protective roles in tumor progression. Cancer Metastasis Rev. 2007;26:717–724. doi: 10.1007/s10555-007-9089-4. [DOI] [PubMed] [Google Scholar]
- 47.Lopez-Otin C, Matrisian LM. Emerging roles of proteases in tumour suppression. Nat Rev Cancer. 2007;7:800–808. doi: 10.1038/nrc2228. [DOI] [PubMed] [Google Scholar]
- 48.Kruger A, Kates RE, Edwards DR. Avoiding spam in the proteolytic internet: future strategies for anti-metastatic MMP inhibition. Biochim Biophys Acta. 2010;1803:95–102. doi: 10.1016/j.bbamcr.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 49.Rhee JS, Diaz R, Korets L, Hodgson JG, Coussens LM. TIMP-1 alters susceptibility to carcinogenesis. Cancer Res. 2004;64:952–961. doi: 10.1158/0008-5472.can-03-2445. [DOI] [PubMed] [Google Scholar]
- 50.Murphy G, Knauper V, Lee MH, Amour A, Worley JR, Hutton M, Atkinson S, Rapti M, Williamson R. Role of TIMPs (tissue inhibitors of metalloproteinases) in pericellular proteolysis: the specificity is in the detail. Biochem Soc Symp. 2003:65–80. doi: 10.1042/bss0700065. [DOI] [PubMed] [Google Scholar]
- 51.Cruz-Munoz W, Khokha R. The role of tissue inhibitors of metalloproteinases in tumorigenesis and metastasis. Crit Rev Clin Lab Sci. 2008;45:291–338. doi: 10.1080/10408360801973244. [DOI] [PubMed] [Google Scholar]
- 52.Rodriguez D, Morrison CJ, Overall CM. Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim Biophys Acta. 2010;1803:39–54. doi: 10.1016/j.bbamcr.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 53.Keller KE, Aga M, Bradley JM, Kelley MJ, Acott TS. Extracellular matrix turnover and outflow resistance. Exp Eye Res. 2009;88:676–682. doi: 10.1016/j.exer.2008.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Igarashi Y, Heureux E, Doctor KS, Talwar P, Gramatikova S, Gramatikoff K, Zhang Y, Blinov M, Ibragimova SS, Boyd S, Ratnikov B, Cieplak P, Godzik A, Smith JW, Osterman AL, Eroshkin AM. PMAP: databases for analyzing proteolytic events and pathways. Nucleic Acids Res. 2009;37:D611–618. doi: 10.1093/nar/gkn683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beaufort N, Plaza K, Utzschneider D, Schwarz A, Burkhart JM, Creutzburg S, Debela M, Schmitt M, Ries C, Magdolen V. Interdependence of kallikrein-related peptidases in proteolytic networks. Biol Chem. 2010;391:581–587. doi: 10.1515/BC.2010.055. [DOI] [PubMed] [Google Scholar]
- 56.Mason SD, Joyce JA. Proteolytic networks in cancer. Trends Cell Biol. 2011 doi: 10.1016/j.tcb.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saftig P, Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol. 2009;10:623–635. doi: 10.1038/nrm2745. [DOI] [PubMed] [Google Scholar]
- 58.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 59.Yu HB, Finlay BB. The caspase-1 inflammasome: a pilot of innate immune responses. Cell Host Microbe. 2008;4:198–208. doi: 10.1016/j.chom.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 60.Roshy S, Sloane BF, Moin K. Pericellular cathepsin B and malignant progression. Cancer Metastasis Rev. 2003;22:271–286. doi: 10.1023/a:1023007717757. [DOI] [PubMed] [Google Scholar]
- 61.Kiyan J, Smith G, Haller H, Dumler I. Urokinase-receptor-mediated phenotypic changes in vascular smooth muscle cells require the involvement of membrane rafts. Biochem J. 2009;423:343–351. doi: 10.1042/BJ20090447. [DOI] [PubMed] [Google Scholar]
- 62.Stylli SS, Kaye AH, Lock P. Invadopodia: at the cutting edge of tumour invasion. J Clin Neurosci. 2008;15:725–737. doi: 10.1016/j.jocn.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 63.Yamaguchi H, Takeo Y, Yoshida S, Kouchi Z, Nakamura Y, Fukami K. Lipid rafts and caveolin-1 are required for invadopodia formation and extracellular matrix degradation by human breast cancer cells. Cancer Res. 2009;69:8594–8602. doi: 10.1158/0008-5472.CAN-09-2305. [DOI] [PubMed] [Google Scholar]
- 64.Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, Schedl A, Haller H, Kurzchalia TV. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 2001;293:2449–2452. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- 65.Yang G, Truong LD, Timme TL, Ren C, Wheeler TM, Park SH, Nasu Y, Bangma CH, Kattan MW, Scardino PT, Thompson TC. Elevated expression of caveolin is associated with prostate and breast cancer. Clin Cancer Res. 1998;4:1873–1880. [PubMed] [Google Scholar]
- 66.Rajjayabun PH, Garg S, Durkan GC, Charlton R, Robinson MC, Mellon JK. Caveolin-1 expression is associated with high-grade bladder cancer. Urology. 2001;58:811–814. doi: 10.1016/s0090-4295(01)01337-1. [DOI] [PubMed] [Google Scholar]
- 67.Joo HJ, Oh DK, Kim YS, Lee KB, Kim SJ. Increased expression of caveolin-1 and microvessel density correlates with metastasis and poor prognosis in clear cell renal cell carcinoma. BJU Int. 2004;93:291–296. doi: 10.1111/j.1464-410x.2004.04604.x. [DOI] [PubMed] [Google Scholar]
- 68.Ho CC, Huang PH, Huang HY, Chen YH, Yang PC, Hsu SM. Up-regulated caveolin-1 accentuates the metastasis capability of lung adenocarcinoma by inducing filopodia formation. Am J Pathol. 2002;161:1647–1656. doi: 10.1016/S0002-9440(10)64442-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kato K, Hida Y, Miyamoto M, Hashida H, Shinohara T, Itoh T, Okushiba S, Kondo S, Katoh H. Overexpression of caveolin-1 in esophageal squamous cell carcinoma correlates with lymph node metastasis and pathologic stage. Cancer. 2002;94:929–933. [PubMed] [Google Scholar]
- 70.Fine SW, Lisanti MP, Galbiati F, Li M. Elevated expression of caveolin-1 in adenocarcinoma of the colon. Am J Clin Pathol. 2001;115:719–724. doi: 10.1309/YL54-CCU7-4V0P-FDUT. [DOI] [PubMed] [Google Scholar]
- 71.Cavallo-Medved D, Dosescu J, Linebaugh BE, Sameni M, Rudy D, Sloane BF. Mutant K-ras regulates cathepsin B localization on the surface of human colorectal carcinoma cells. Neoplasia. 2003;5:507–519. doi: 10.1016/s1476-5586(03)80035-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Patlolla JM, Swamy MV, Raju J, Rao CV. Overexpression of caveolin-1 in experimental colon adenocarcinomas and human colon cancer cell lines. Oncol Rep. 2004;11:957–963. [PubMed] [Google Scholar]
- 73.Nohata N, Hanazawa T, Kikkawa N, Mutallip M, Fujimura L, Yoshino H, Kawakami K, Chiyomaru T, Enokida H, Nakagawa M, Okamoto Y, Seki N. Caveolin-1 mediates tumor cell migration and invasion and its regulation by miR-133a in head and neck squamous cell carcinoma. Int J Oncol. 2011;38:209–217. [PubMed] [Google Scholar]
- 74.Razani B, Schlegel A, Liu J, Lisanti MP. Caveolin-1, a putative tumour suppressor gene. Biochem Soc Trans. 2001;29:494–499. doi: 10.1042/bst0290494. [DOI] [PubMed] [Google Scholar]
- 75.Burgermeister E, Liscovitch M, Rocken C, Schmid RM, Ebert MP. Caveats of caveolin-1 in cancer progression. Cancer Lett. 2008;268:187–201. doi: 10.1016/j.canlet.2008.03.055. [DOI] [PubMed] [Google Scholar]
- 76.Goetz JG, Lajoie P, Wiseman SM, Nabi IR. Caveolin-1 in tumor progression: the good, the bad and the ugly. Cancer Metastasis Rev. 2008;27:715–735. doi: 10.1007/s10555-008-9160-9. [DOI] [PubMed] [Google Scholar]
- 77.Briand N, Le Lay S, Sessa WC, Ferre P, Dugail I. Distinct roles of endothelial and adipocyte caveolin-1 in macrophage infiltration and adipose tissue metabolic activity. Diabetes. 2011;60:448–453. doi: 10.2337/db10-0856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kwon M, MacLeod TJ, Zhang Y, Waisman DM. S100A10, annexin A2, and annexin a2 heterotetramer as candidate plasminogen receptors. Front Biosci. 2005;10:300–325. doi: 10.2741/1529. [DOI] [PubMed] [Google Scholar]
- 79.Kim J, Hajjar KA. Annexin II: a plasminogen-plasminogen activator co-receptor. Front Biosci. 2002;7:d341–348. doi: 10.2741/kim. [DOI] [PubMed] [Google Scholar]
- 80.Mai J, Waisman DM, Sloane BF. Cell surface complex of cathepsin B/annexin II tetramer in malignant progression. Biochim Biophys Acta. 2000;1477:215–230. doi: 10.1016/s0167-4838(99)00274-5. [DOI] [PubMed] [Google Scholar]
- 81.Blasi F, Sidenius N. The urokinase receptor: focused cell surface proteolysis, cell adhesion and signaling. FEBS Lett. 2010;584:1923–1930. doi: 10.1016/j.febslet.2009.12.039. [DOI] [PubMed] [Google Scholar]
- 82.Ling Q, Jacovina AT, Deora A, Febbraio M, Simantov R, Silverstein RL, Hempstead B, Mark WH, Hajjar KA. Annexin II regulates fibrin homeostasis and neoangiogenesis in vivo. J Clin Invest. 2004;113:38–48. doi: 10.1172/JCI200419684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kobayashi H, Moniwa N, Sugimura M, Shinohara H, Ohi H, Terao T. Effects of membrane-associated cathepsin B on the activation of receptor-bound prourokinase and subsequent invasion of reconstituted basement membranes. Biochim Biophys Acta. 1993;1178:55–62. doi: 10.1016/0167-4889(93)90109-3. [DOI] [PubMed] [Google Scholar]
- 84.Guo M, Mathieu PA, Linebaugh B, Sloane BF, Reiners JJ., Jr Phorbol ester activation of a proteolytic cascade capable of activating latent transforming growth factor-betaL a process initiated by the exocytosis of cathepsin B. J Biol Chem. 2002;277:14829–14837. doi: 10.1074/jbc.M108180200. [DOI] [PubMed] [Google Scholar]
- 85.Buccione R, Orth JD, McNiven MA. Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat Rev Mol Cell Biol. 2004;5:647–657. doi: 10.1038/nrm1436. [DOI] [PubMed] [Google Scholar]
- 86.Van Goethem E, Guiet R, Balor S, Charriere GM, Poincloux R, Labrousse A, Maridonneau-Parini I, Le Cabec V. Macrophage podosomes go 3D. Eur J Cell Biol. 2011;90:224–236. doi: 10.1016/j.ejcb.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 87.Wolf K, Alexander S, Schacht V, Coussens LM, von Andrian UH, van Rheenen J, Deryugina E, Friedl P. Collagen-based cell migration models in vitro and in vivo. Semin Cell Dev Biol. 2009;20:931–941. doi: 10.1016/j.semcdb.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yamaguchi H, Oikawa T. Membrane lipids in invadopodia and podosomes: Key structures for cancer invasion and metastasis. Oncotarget. 2010;1:320–328. doi: 10.18632/oncotarget.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Caldieri G, Buccione R. Aiming for invadopodia: organizing polarized delivery at sites of invasion. Trends Cell Biol. 2010;20:64–70. doi: 10.1016/j.tcb.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 90.Monsky WL, Lin CY, Aoyama A, Kelly T, Akiyama SK, Mueller SC, Chen WT. A potential marker protease of invasiveness, seprase, is localized on invadopodia of human malignant melanoma cells. Cancer Res. 1994;54:5702–5710. [PubMed] [Google Scholar]
- 91.O’Brien P, O’Connor BF. Seprase: an overview of an important matrix serine protease. Biochim Biophys Acta. 2008;1784:1130–1145. doi: 10.1016/j.bbapap.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 92.Caldieri G, Giacchetti G, Beznoussenko G, Attanasio F, Ayala I, Buccione R. Invadopodia biogenesis is regulated by caveolin-mediated modulation of membrane cholesterol levels. J Cell Mol Med. 2009;13:1728–1740. doi: 10.1111/j.1582-4934.2008.00568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Artym VV, Matsumoto K, Mueller SC, Yamada KM. Dynamic membrane remodeling at invadopodia differentiates invadopodia from podosomes. Eur J Cell Biol. 2011;90:172–180. doi: 10.1016/j.ejcb.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Van Goethem E, Poincloux R, Gauffre F, Maridonneau-Parini I, Le Cabec V. Matrix architecture dictates three-dimensional migration modes of human macrophages: differential involvement of proteases and podosome-like structures. J Immunol. 2010;184:1049–1061. doi: 10.4049/jimmunol.0902223. [DOI] [PubMed] [Google Scholar]
- 95.Sabeh F, Shimizu-Hirota R, Weiss SJ. Protease-dependent versus -independent cancer cell invasion programs: three-dimensional amoeboid movement revisited. J Cell Biol. 2009;185:11–19. doi: 10.1083/jcb.200807195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tu C, Ortega-Cava CF, Chen G, Fernandes ND, Cavallo-Medved D, Sloane BF, Band V, Band H. Lysosomal cathepsin B participates in the podosome-mediated extracellular matrix degradation and invasion via secreted lysosomes in v-Src fibroblasts. Cancer Res. 2008;68:9147–9156. doi: 10.1158/0008-5472.CAN-07-5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Saltel F, Chabadel A, Bonnelye E, Jurdic P. Actin cytoskeletal organisation in osteoclasts: a model to decipher transmigration and matrix degradation. Eur J Cell Biol. 2008;87:459–468. doi: 10.1016/j.ejcb.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 98.Saftig P, Hunziker E, Wehmeyer O, Jones S, Boyde A, Rommerskirch W, Moritz JD, Schu P, von Figura K. Impaired osteoclastic bone resorption leads to osteopetrosis in cathepsin-K-deficient mice. Proc Natl Acad Sci U S A. 1998;95:13453–13458. doi: 10.1073/pnas.95.23.13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stenbeck G. Formation and function of the ruffled border in osteoclasts. Semin Cell Dev Biol. 2002;13:285–292. doi: 10.1016/s1084952102000587. [DOI] [PubMed] [Google Scholar]
- 100.Martinez-Zaguilan R, Lynch RM, Martinez GM, Gillies RJ. Vacuolar-type H(+)-ATPases are functionally expressed in plasma membranes of human tumor cells. Am J Physiol. 1993;265:C1015–1029. doi: 10.1152/ajpcell.1993.265.4.C1015. [DOI] [PubMed] [Google Scholar]
- 101.Sennoune SR, Bakunts K, Martinez GM, Chua-Tuan JL, Kebir Y, Attaya MN, Martinez-Zaguilan R. Vacuolar H+-ATPase in human breast cancer cells with distinct metastatic potential: distribution and functional activity. Am J Physiol Cell Physiol. 2004;286:C1443–1452. doi: 10.1152/ajpcell.00407.2003. [DOI] [PubMed] [Google Scholar]
- 102.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 103.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 104.Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. J Gen Physiol. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wike-Hooley JL, Haveman J, Reinhold HS. The relevance of tumour pH to the treatment of malignant disease. Radiother Oncol. 1984;2:343–366. doi: 10.1016/s0167-8140(84)80077-8. [DOI] [PubMed] [Google Scholar]
- 106.Gillies RJ, Raghunand N, Karczmar GS, Bhujwalla ZM. MRI of the tumor microenvironment. J Magn Reson Imaging. 2002;16:430–450. doi: 10.1002/jmri.10181. [DOI] [PubMed] [Google Scholar]
- 107.van Sluis R, Bhujwalla ZM, Raghunand N, Ballesteros P, Alvarez J, Cerdan S, Galons JP, Gillies RJ. In vivo imaging of extracellular pH using 1H MRSI. Magn Reson Med. 1999;41:743–750. doi: 10.1002/(sici)1522-2594(199904)41:4<743::aid-mrm13>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 108.Stubbs M, Rodrigues L, Howe FA, Wang J, Jeong KS, Veech RL, Griffiths JR. Metabolic consequences of a reversed pH gradient in rat tumors. Cancer Res. 1994;54:4011–4016. [PubMed] [Google Scholar]
- 109.Schornack PA, Gillies RJ. Contributions of cell metabolism and H+ diffusion to the acidic pH of tumors. Neoplasia. 2003;5:135–145. doi: 10.1016/s1476-5586(03)80005-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gillies RJ, Martinez-Zaguilan R, Martinez GM, Serrano R, Perona R. Tumorigenic 3T3 cells maintain an alkaline intracellular pH under physiological conditions. Proc Natl Acad Sci U S A. 1990;87:7414–7418. doi: 10.1073/pnas.87.19.7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rozhin J, Sameni M, Ziegler G, Sloane BF. Pericellular pH affects distribution and secretion of cathepsin B in malignant cells. Cancer Res. 1994;54:6517–6525. [PubMed] [Google Scholar]
- 112.Kraus M, Wolf B. Implications of acidic tumor microenvironment for neoplastic growth and cancer treatment: a computer analysis. Tumour Biol. 1996;17:133–154. doi: 10.1159/000217977. [DOI] [PubMed] [Google Scholar]
- 113.Helmlinger G, Yuan F, Dellian M, Jain RK. Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med. 1997;3:177–182. doi: 10.1038/nm0297-177. [DOI] [PubMed] [Google Scholar]
- 114.Lardner A. The effects of extracellular pH on immune function. J Leukoc Biol. 2001;69:522–530. [PubMed] [Google Scholar]
- 115.Xu L, Fidler IJ. Acidic pH-induced elevation in interleukin 8 expression by human ovarian carcinoma cells. Cancer Res. 2000;60:4610–4616. [PubMed] [Google Scholar]
- 116.Xu L, Fukumura D, Jain RK. Acidic extracellular pH induces vascular endothelial growth factor (VEGF) in human glioblastoma cells via ERK1/2 MAPK signaling pathway: mechanism of low pH-induced VEGF. J Biol Chem. 2002;277:11368–11374. doi: 10.1074/jbc.M108347200. [DOI] [PubMed] [Google Scholar]
- 117.Shi Q, Le X, Wang B, Abbruzzese JL, Xiong Q, He Y, Xie K. Regulation of vascular endothelial growth factor expression by acidosis in human cancer cells. Oncogene. 2001;20:3751–3756. doi: 10.1038/sj.onc.1204500. [DOI] [PubMed] [Google Scholar]
- 118.Hjelmeland AB, Wu Q, Heddleston JM, Choudhary GS, Macswords J, Lathia JD, McLendon R, Lindner D, Sloan A, Rich JN. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 2010 doi: 10.1038/cdd.2010.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Taraboletti G, D’Ascenzo S, Giusti I, Marchetti D, Borsotti P, Millimaggi D, Giavazzi R, Pavan A, Dolo V. Bioavailability of VEGF in tumor-shed vesicles depends on vesicle burst induced by acidic pH. Neoplasia. 2006;8:96–103. doi: 10.1593/neo.05583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Giusti I, D’Ascenzo S, Millimaggi D, Taraboletti G, Carta G, Franceschini N, Pavan A, Dolo V. Cathepsin B mediates the pH-dependent proinvasive activity of tumor-shed microvesicles. Neoplasia. 2008;10:481–488. doi: 10.1593/neo.08178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rofstad EK, Mathiesen B, Kindem K, Galappathi K. Acidic extracellular pH promotes experimental metastasis of human melanoma cells in athymic nude mice. Cancer Res. 2006;66:6699–6707. doi: 10.1158/0008-5472.CAN-06-0983. [DOI] [PubMed] [Google Scholar]
- 122.Schlappack OK, Zimmermann A, Hill RP. Glucose starvation and acidosis: effect on experimental metastatic potential, DNA content and MTX resistance of murine tumour cells. Br J Cancer. 1991;64:663–670. doi: 10.1038/bjc.1991.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Robey IF, Baggett BK, Kirkpatrick ND, Roe DJ, Dosescu J, Sloane BF, Hashim AI, Morse DL, Raghunand N, Gatenby RA, Gillies RJ. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res. 2009;69:2260–2268. doi: 10.1158/0008-5472.CAN-07-5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yan S, Sloane BF. Molecular regulation of human cathepsin B: implication in pathologies. Biol Chem. 2003;384:845–854. doi: 10.1515/BC.2003.095. [DOI] [PubMed] [Google Scholar]
- 125.Cavallo-Medved D, Sloane BF. Cell-surface cathepsin B: understanding its functional significance. Curr Top Dev Biol. 2003;54:313–341. doi: 10.1016/s0070-2153(03)54013-3. [DOI] [PubMed] [Google Scholar]
- 126.Podgorski I, Sloane BF. Cathepsin B and its role(s) in cancer progression. Biochem Soc Symp. 2003:263–276. doi: 10.1042/bss0700263. [DOI] [PubMed] [Google Scholar]
- 127.Jedeszko C, Sameni M, Olive MB, Moin K, Sloane BF. Visualizing protease activity in living cells: from two dimensions to four dimensions. Curr Protoc Cell Biol. 2008;39:20.21–15. doi: 10.1002/0471143030.cb0420s39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sameni M, Dosescu J, Sloane BF. Imaging proteolysis by living human glioma cells. Biol Chem. 2001;382:785–788. doi: 10.1515/BC.2001.094. [DOI] [PubMed] [Google Scholar]
- 129.Sameni M, Dosescu J, Moin K, Sloane BF. Functional imaging of proteolysis: stromal and inflammatory cells increase tumor proteolysis. Mol Imaging. 2003;2:159–175. doi: 10.1162/15353500200303136. [DOI] [PubMed] [Google Scholar]
- 130.Moin K, Sameni M, Victor BC, Rothberg JM, Mattingly RR, Sloane BF. 3D/4D Functional Imaging of Tumor-Associated Proteolysis: Impact of Microenvironment. Meth Enzymol. 2001 doi: 10.1016/B978-0-12-391856-7.00034-2. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. 2001;411:375–379. doi: 10.1038/35077241. [DOI] [PubMed] [Google Scholar]
- 132.DeClerck YA. Interactions between tumour cells and stromal cells and proteolytic modification of the extracellular matrix by metalloproteinases in cancer. Eur J Cancer. 2000;36:1258–1268. doi: 10.1016/s0959-8049(00)00094-0. [DOI] [PubMed] [Google Scholar]
- 133.Johnsen M, Lund LR, Romer J, Almholt K, Dano K. Cancer invasion and tissue remodeling: common themes in proteolytic matrix degradation. Curr Opin Cell Biol. 1998;10:667–671. doi: 10.1016/s0955-0674(98)80044-6. [DOI] [PubMed] [Google Scholar]
- 134.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bogenrieder T, Herlyn M. Axis of evil: molecular mechanisms of cancer metastasis. Oncogene. 2003;22:6524–6536. doi: 10.1038/sj.onc.1206757. [DOI] [PubMed] [Google Scholar]
- 136.van Kempen LC, Coussens LM. MMP9 potentiates pulmonary metastasis formation. Cancer Cell. 2002;2:251–252. doi: 10.1016/s1535-6108(02)00157-5. [DOI] [PubMed] [Google Scholar]
- 137.Almholt K, Johnsen M. Stromal cell involvement in cancer. Recent Results Cancer Res. 2003;162:31–42. doi: 10.1007/978-3-642-59349-9_3. [DOI] [PubMed] [Google Scholar]
- 138.Offersen BV, Nielsen BS, Hoyer-Hansen G, Rank F, Hamilton-Dutoit S, Overgaard J, Andreasen PA. The myofibroblast is the predominant plasminogen activator inhibitor-1-expressing cell type in human breast carcinomas. Am J Pathol. 2003;163:1887–1899. doi: 10.1016/S0002-9440(10)63547-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Silzle T, Kreutz M, Dobler MA, Brockhoff G, Knuechel R, Kunz-Schughart LA. Tumor-associated fibroblasts recruit blood monocytes into tumor tissue. Eur J Immunol. 2003;33:1311–1320. doi: 10.1002/eji.200323057. [DOI] [PubMed] [Google Scholar]
- 140.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193:727–740. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gouon-Evans V, Lin EY, Pollard JW. Requirement of macrophages and eosinophils and their cytokines/chemokines for mammary gland development. Breast Cancer Res. 2002;4:155–164. doi: 10.1186/bcr441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Van Nguyen A, Pollard JW. Colony stimulating factor-1 is required to recruit macrophages into the mammary gland to facilitate mammary ductal outgrowth. Dev Biol. 2002;247:11–25. doi: 10.1006/dbio.2002.0669. [DOI] [PubMed] [Google Scholar]
- 143.Fata JE, Werb Z, Bissell MJ. Regulation of mammary gland branching morphogenesis by the extracellular matrix and its remodeling enzymes. Breast Cancer Res. 2004;6:1–11. doi: 10.1186/bcr634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Schmeichel KL, Bissell MJ. Modeling tissue-specific signaling and organ function in three dimensions. J Cell Sci. 2003;116:2377–2388. doi: 10.1242/jcs.00503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 146.Debnath J, Brugge JS. Modelling glandular epithelial cancers in three-dimensional cultures. Nat Rev Cancer. 2005;5:675–688. doi: 10.1038/nrc1695. [DOI] [PubMed] [Google Scholar]
- 147.Soule HD, Maloney TM, Wolman SR, Peterson WD, Jr, Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, Brooks SC. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50:6075–6086. [PubMed] [Google Scholar]
- 148.Basolo F, Elliott J, Tait L, Chen XQ, Maloney T, Russo IH, Pauley R, Momiki S, Caamano J, Klein-Szanto AJ, et al. Transformation of human breast epithelial cells by c-Ha-ras oncogene. Mol Carcinog. 1991;4:25–35. doi: 10.1002/mc.2940040106. [DOI] [PubMed] [Google Scholar]
- 149.Dawson PJ, Wolman SR, Tait L, Heppner GH, Miller FR. MCF10AT: a model for the evolution of cancer from proliferative breast disease. Am J Pathol. 1996;148:313–319. [PMC free article] [PubMed] [Google Scholar]
- 150.Miller FR, Santner SJ, Tait L, Dawson PJ. MCF10DCIS.com xenograft model of human comedo ductal carcinoma in situ. J Natl Cancer Inst. 2000;92:1185–1186. doi: 10.1093/jnci/92.14.1185a. [DOI] [PubMed] [Google Scholar]
- 151.Miller FR. Xenograft models of premalignant breast disease. J Mammary Gland Biol Neoplasia. 2000;5:379–391. doi: 10.1023/a:1009577811584. [DOI] [PubMed] [Google Scholar]
- 152.Santner SJ, Dawson PJ, Tait L, Soule HD, Eliason J, Mohamed AN, Wolman SR, Heppner GH, Miller FR. Malignant MCF10CA1 cell lines derived from premalignant human breast epithelial MCF10AT cells. Breast Cancer Res Treat. 2001;65:101–110. doi: 10.1023/a:1006461422273. [DOI] [PubMed] [Google Scholar]
- 153.Debnath J, Mills KR, Collins NL, Reginato MJ, Muthuswamy SK, Brugge JS. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell. 2002;111:29–40. doi: 10.1016/s0092-8674(02)01001-2. [DOI] [PubMed] [Google Scholar]
- 154.Li Q, Mullins SR, Sloane BF, Mattingly RR. p21-Activated kinase 1 coordinates aberrant cell survival and pericellular proteolysis in a three-dimensional culture model for premalignant progression of human breast cancer. Neoplasia. 2008;10:314–329. doi: 10.1593/neo.07970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Elenbaas B, Weinberg RA. Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp Cell Res. 2001;264:169–184. doi: 10.1006/excr.2000.5133. [DOI] [PubMed] [Google Scholar]
- 156.Roskelley CD, Bissell MJ. The dominance of the microenvironment in breast and ovarian cancer. Semin Cancer Biol. 2002;12:97–104. doi: 10.1006/scbi.2001.0417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wiseman BS, Werb Z. Stromal effects on mammary gland development and breast cancer. Science. 2002;296:1046–1049. doi: 10.1126/science.1067431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Erickson AC, Barcellos-Hoff MH. The not-so innocent bystander: the microenvironment as a therapeutic target in cancer. Expert Opin Ther Targets. 2003;7:71–88. doi: 10.1517/14728222.7.1.71. [DOI] [PubMed] [Google Scholar]
- 159.Ben-Baruch A. Host microenvironment in breast cancer development: inflammatory cells, cytokines and chemokines in breast cancer progression: reciprocal tumor-microenvironment interactions. Breast Cancer Res. 2003;5:31–36. doi: 10.1186/bcr554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Boudreau N, Myers C. Breast cancer-induced angiogenesis: multiple mechanisms and the role of the microenvironment. Breast Cancer Res. 2003;5:140–146. doi: 10.1186/bcr589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Polyak K, Hu M. Do myoepithelial cells hold the key for breast tumor progression? J Mammary Gland Biol Neoplasia. 2005;10:231–247. doi: 10.1007/s10911-005-9584-6. [DOI] [PubMed] [Google Scholar]
- 162.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 163.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66:605–612. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- 164.Nelson CM, Bissell MJ. Modeling dynamic reciprocity: engineering three-dimensional culture models of breast architecture, function, and neoplastic transformation. Semin Cancer Biol. 2005;15:342–352. doi: 10.1016/j.semcancer.2005.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Schedin P, Elias A. Multistep tumorigenesis and the microenvironment. Breast Cancer Res. 2004;6:93–101. doi: 10.1186/bcr772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Shekhar MP, Pauley R, Heppner G. Host microenvironment in breast cancer development: extracellular matrix-stromal cell contribution to neoplastic phenotype of epithelial cells in the breast. Breast Cancer Res. 2003;5:130–135. doi: 10.1186/bcr580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Vincent-Salomon A, Thiery JP. Host microenvironment in breast cancer development: epithelial-mesenchymal transition in breast cancer development. Breast Cancer Res. 2003;5:101–106. doi: 10.1186/bcr578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Yu JL, Rak JW. Host microenvironment in breast cancer development: inflammatory and immune cells in tumour angiogenesis and arteriogenesis. Breast Cancer Res. 2003;5:83–88. doi: 10.1186/bcr573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Tynan JA, Wen F, Muller WJ, Oshima RG. Ets2-dependent microenvironmental support of mouse mammary tumors. Oncogene. 2005;24:6870–6876. doi: 10.1038/sj.onc.1208856. [DOI] [PubMed] [Google Scholar]
- 170.Iyengar P, Espina V, Williams TW, Lin Y, Berry D, Jelicks LA, Lee H, Temple K, Graves R, Pollard J, Chopra N, Russell RG, Sasisekharan R, Trock BJ, Lippman M, Calvert VS, Petricoin EF, 3rd, Liotta L, Dadachova E, Pestell RG, Lisanti MP, Bonaldo P, Scherer PE. Adipocyte-derived collagen VI affects early mammary tumor progression in vivo, demonstrating a critical interaction in the tumor/stroma microenvironment. J Clin Invest. 2005;115:1163–1176. doi: 10.1172/JCI23424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, Qian H, Xue XN, Pollard JW. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006;66:11238–11246. doi: 10.1158/0008-5472.CAN-06-1278. [DOI] [PubMed] [Google Scholar]
- 172.Duelli D, Lazebnik Y. Cell fusion: a hidden enemy? Cancer Cell. 2003;3:445–448. doi: 10.1016/s1535-6108(03)00114-4. [DOI] [PubMed] [Google Scholar]
- 173.Almholt K, Green KA, Juncker-Jensen A, Nielsen BS, Lund LR, Romer J. Extracellular proteolysis in transgenic mouse models of breast cancer. J Mammary Gland Biol Neoplasia. 2007;12:83–97. doi: 10.1007/s10911-007-9040-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Nielsen BS, Rank F, Illemann M, Lund LR, Dano K. Stromal cells associated with early invasive foci in human mammary ductal carcinoma in situ coexpress urokinase and urokinase receptor. Int J Cancer. 2007;120:2086–2095. doi: 10.1002/ijc.22340. [DOI] [PubMed] [Google Scholar]
- 175.Jedeszko C, Victor BC, Podgorski I, Sloane BF. Fibroblast hepatocyte growth factor promotes invasion of human mammary ductal carcinoma in situ. Cancer Res. 2009;69:9148–9155. doi: 10.1158/0008-5472.CAN-09-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bourguignon LY, Singleton PA, Diedrich F, Stern R, Gilad E. CD44 interaction with Na+-H+ exchanger (NHE1) creates acidic microenvironments leading to hyaluronidase-2 and cathepsin B activation and breast tumor cell invasion. J Biol Chem. 2004;279:26991–27007. doi: 10.1074/jbc.M311838200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01
02
03. Supplemental movies – Figure 3.
Animation of 3D reconstruction of confocal images depicted in Figure 3.
04
05. Supplemental movies – Figure 5.
Animation of 3D reconstruction of confocal images depicted in Figure 5.