Checkpoint Blockade in Cancer Immunotherapy (original) (raw)
. Author manuscript; available in PMC: 2007 Aug 24.
Abstract
The progression of a productive immune response requires that a number of immunological checkpoints be passed. Passage may require the presence of excitatory costimulatory signals or the avoidance of negative or coinhibitory signals, which act to dampen or terminate immune activity. The immunoglobulin superfamily occupies a central importance in this coordination of immune responses, and the CD28/cytotoxic T-lymphocyte antigen-4 (CTLA-4):B7.1/B7.2 receptor/ligand grouping represents the archetypal example of these immune regulators. In part the role of these checkpoints is to guard against the possibility of unwanted and harmful self-directed activities. While this is a necessary function, aiding in the prevention of autoimmunity, it may act as a barrier to successful immunotherapies aimed at targeting malignant self-cells that largely display the same array of surface molecules as the cells from which they derive. Therapies aimed at overcoming these mechanisms of peripheral tolerance, in particular by blocking the inhibitory checkpoints, offer the potential to generate antitumor activity, either as monotherapies or in synergism with other therapies that directly or indirectly enhance presentation of tumor epitopes to the immune system. Such immunological molecular adjuvants are showing promise in early clinical trials. This review focuses on the results of the archetypal example of checkpoint blockade, anti-CTLA-4, in preclinical tumor models and clinical trials, while also highlighting other possible targets for immunological checkpoint blockade.
1. Introduction
Progress in antitumor immunotherapy has been aided by advances in the understanding of antigen presentation and the rules governing polarization of subsequent immune responses toward CD4+ or CD8+ compartments and Th1/Th2 or Tc1/Tc2 phenotypes. A number of approaches aimed at enhancing tumor-specific activities have provided important proofs of principle in both murine models and early clinical trials in humans. However, while many methodologies aimed at enhancing these earliest of events in the immune response (such as peptide or protein vaccines, dendritic cell vaccines loaded with peptides or modified to express tumor antigens, DNA vaccines with or without modifications to enhance CD8+ T-cell responses, and cytokine-secreting cellular vaccines derived from primary tumor) have provided encouraging results in specific preclinical models, or have been demonstrated to enable the generation of measurable antitumor activity based on sensitive laboratory read-outs of immunological reactivity, the generation of prolonged, objectively quantifiable and clinically meaningful responses in patients has proven more difficult than initially envisaged. Of course, part of the difficulty arises from the fact that the tumors are host-derived and express mostly the same array of self-antigens as the cell types from which they arise. Many of the molecules identified as potentially therapeutic targets in human cancers are self or “altered self” antigens, either aberrantly expressed or overexpressed on malignant cells. Overcoming multiple mechanisms of peripheral tolerance to these tumor-associated targets may prove crucial for effective recruitment of the immune effectors required for successful immune-based therapies. Just as our knowledge of the sentinel role of dendritic cells (T-cell extrinsic elements) in directing the outcome of early events in immune responses has expanded, we have also become increasingly aware of the roles of both T-cell intrinsic cell-autonomous regulatory elements, and of T-cell intrinsic non cell-autonomous mediators (regulatory T cells) in the induction of peripheral tolerance. And as we have learned more about the rules governing the progression of productive immune responses, we have discovered an extended network of immunological checkpoints that need to be passed in order for these responses to proceed. Attention to these immunological bottlenecks may prove critical for us to fully harness the therapeutic potential of immunotherapy.
Given the latent destructive capacity inherent in the mammalian adaptive immune system, it is perhaps no surprise that multiple immunological checkpoints are in place to prevent inappropriate activation events such as those targeted toward self-antigens. However, the true complexity of these pathways has only relatively recently become apparent, and continues to be unraveled with the discovery of new molecules whose physiological significance remains uncertain. At a basic level these checkpoints may be viewed as those that are required to provide additional excitatory costimulatory activity for progression of immune priming or activation, initiation of cell division, or development of particular effector phenotypes following T-cell receptor (TCR) ligation, and those that provide “coinhibitory” influences and which may be more important both for the prevention of the initiation of inappropriately directed responses and for limiting the size, duration, or premature focusing of immune responses once initiated. As a group these molecules allow fine-tuning of the response to TCR ligation by cognate antigen. Each feeds into overlapping or identical downstream signaling pathways and by virtue of the contribution of multiple costimulatory signals with overlapping but nonredundant function acts as a rheostat for T-cell activation, survival, and function.
The initial foundation of self-tolerance is a fundamental function of the central tolerance established through positive and negative selection in the thymus. Self-proteins are processed and presented in association with self-major histocompatability complex (MHC) molecules on the surface of thymic antigen presenting cells (APCs). The subsequent outcome of interactions with T cells depends on the avidity between TCRs and self-peptide-MHC complexes. Interactions of very low-avidity result in T-cell deletion by apoptotic death by neglect, while high-avidity interactions result in similar termination of T cells by apoptotic negative selection. Intermediate-avidity binding provides positive selection with further T-cell differentiation and establishment of a T-cell repertoire characterized by a population of relatively weakly autoreactive T cells. Subthreshold recognition of self-antigens may thus be a prerequisite for the generation and survival of regulatory T cells and both naïve and memory T cells. This ontogeny has important implications for tumor immunotherapy in a system now established to include rheostatic mechanisms for resetting the threshold for T-cell activation events. If T cells capable of responding to tumor epitopes are present in the host, therapeutic manipulation of activation thresholds could recruit these cells, or enhance their functional capabilities sufficiently to effect meaningful antitumor activity. Evidence suggesting the existence of such tumor-reactive T cells can be derived from correlative studies in a number of human cancers demonstrating prolonged survival and/or reduced metastases in those patients who have greater levels of intratumor infiltration with T cells (Marrogi et al., 1997; Naito et al., 1998; Nakano et al., 2001; Vesalainen et al., 1994; Zhang et al., 2003a).
It is also apparent that the evolution of the malignant phenotype of tumor cells could be characterized by adaptations involving these regulatory molecules, which could enhance evasion of the immune responses that target aberrant cell outgrowth. For example, reduced expression of costimulatory ligands could render the malignant cells “invisible” to the immune system, while overexpression of coinhibitory ligands could effectively dampen or terminate active antitumor immunity (Iwai et al., 2002; Townsend and Allison, 1993). Subjugation of regulatory T-cell populations could also affect a similar outcome (Curiel et al., 2004; Liyanage et al., 2002; Viguier et al., 2004; Woo et al., 2001, 2002). Examples of all of these mechanisms have now been described in a variety of murine tumor models and human cancers, providing further impetus for attempts to manipulate these pathways for therapeutic benefit. Blockade of any of the inhibitory checkpoints could potentially enhance any preexistent antitumor immunity, and synergize with other therapies that either directly or indirectly augment such activities.
One further general point needs to be addressed when considering the potential of immune checkpoint blockade as a therapeutic modality. These checkpoints have a vital physiological role in limiting the potential damage that can be caused by an auto-reactive T-cell repertoire. Blockade might theoretically result in uncontrolled auto-reactivity and significant toxicity. Adverse immune events have been noted in some of the early clinical trials of cytotoxic T-lymphocyte antigen-4 (CTLA-4) blockade (Attia et al., 2005; Phan et al., 2003b; Ribas et al., 2005b; Sanderson et al., 2005). A question of vital importance is whether such adverse events are an inherent part of effective checkpoint blockade, or whether they can be dissociated by manipulation of dose scheduling, targeting immunological rather than clinical endpoints as a primary objective, or by combinatorial approaches involving strategies that will enhance presentation of tumor-selective antigens to the immune system over-and-above those of normal tissues. This review focuses on CTLA-4 blockade in tumor immunotherapy as the prototypical example of checkpoint blockade in order to address these issues, and highlight other potential targets warranting further exploration.
2. The Extended CD28:B7 Immunoglobulin Superfamily
Since the initial description of the costimulatory receptor ligand pair of CD28: B7, an extended family of structurally and functionally related molecules have been described, which due to their commonality with the immunoglobulin variable-like (IgV) and constant (IgC) domains of the immunoglobulins have become known collectively as the CD28:B7 immunoglobulin superfamily. The CD28 family is composed of CD28, CTLA-4, inducible T-cell costimulator (ICOS), programmed death-1 (PD-1), and B-and T-lymphocyte attenuator (BTLA). CD28 and ICOS typify costimulatory family members, whereas CTLA-4, PD-1, and BTLA appear to be coinhibitory. Both PD-1 and BTLA possess immunoreceptor tyrosine-inhibitory plus/minus tyrosine-switch motifs (ITIMs and ITSMs) and, like CTLA-4, are able to recruit phosphatases such as Src-homology protein tyrosine phosphatase-2 (SHP-2). Each immunoglobulin superfamily member, except BTLA, is characterized by a single extracellular IgV domain followed by a short cytoplasmic tail (although the major human form of B7-H3 uniquely contains two extracellular tandem IgV-IgC domains (Sun et al., 2002)). The crystal structure of BTLA, in contrast, suggests it to be a member of the I-set of Ig domains (Compaan et al., 2005). CD28, CTLA-4, and ICOS have an unpaired cysteine that allows them to homodimerize on the cell surface, while neither PD-1 nor BTLA has an unpaired cysteine and both likely exist as monomers on the cell surface (Compaan et al., 2005; Zhang et al., 2004). The seven known members of the B7 family (B7.1, B7.2, ICOS-L, PD-L1, PD-L2, B7-H3, and B7x/B7-H4) are characterized by extracellular IgV-like and IgC-like domains and short intracellular cytoplasmic domains (19–62 amino acids). The intracellular domains contain serine and threonine residues, which may have significance as potential phosphorylation sites given the evidence that B7-family members can signal into the cell on which they are expressed. They are predicted to form homodimers at the cell surface (as shown for B7.1), although B7.2 likely exists as a monomer (Schwartz et al., 2001; Zhang et al., 2003b). The ligand for BTLA has been identified as the tumor-necrosis factor (TNF) receptor family member herpesvirus entry mediator (HVEM) (Sedy et al., 2005), providing a previously unknown structural interaction allowing reverse signaling from a TNF receptor family member toward lymphocytes that may be a barrier to T-cell activation. This, and the I-set structure, suggests that BTLA may be evolutionarily the most distantly related of the CD28 family.
2.1. CD28/CTLA-4:B7.1/B7.2: The Archetypal Immune Regulators
2.1.1. CD28: The Original Second Signal
The ability of CD28-dependent signaling to rescue T-cell clones from TCR-mediated anergy was recognized in the early 1990s (Harding et al., 1992), and the critical role of engagement by either of the structurally-homologous B7.1 (CD80) and B7.2 (CD86) ligand pair for costimulatory activity also established (Hathcock et al., 1994; Linsley et al., 1991). Blocking antibodies to B7.1 or B7.2 diminished, and a combination of both or a CTLA-4-Ig fusion protein effectively eliminated T-cell responses to a variety of in vitro and in vivo stimuli (Lenschow and Bluestone, 1993). Meanwhile, CD28−/− knockout mice showed severe diminution of most T-cell responses (although the ability to reject some viruses was retained, and subsequent studies suggested that the duration of TCR stimulation may determine the costimulatory requirement of certain T-cell responses) (Kundig et al., 1996; Shahinian et al., 1993), and B7.1/B7.2 double knockout mice demonstrated a virtual absence of T-cell responses (Borriello et al., 1997). Dendritic cells (DCs) expressed the highest levels of these costimulatory ligands, particularly after activating stimuli, and also demonstrated an enhanced ability to sample, process, and present exogenous antigen back to the adaptive immune system via the intermediary of MHC molecules. They became drawn as the sentinels of the immune system.
CD28 is constitutively expressed by most mouse T cells, 90% of human CD4+ T cells and 50% of human CD8+ T cells. The restricted localization of B7.1 and B7.2 expression to lymphoid and antigen-presenting cells (such as DCs, macrophages, and activated B cells) provides a compartmentalization that may function as a means by which early events in T-cell activation are directed to maintain peripheral tolerance by restricting T-cell activation to areas of inflammation or injury. B7.2 is expressed at a low level in nonactivated DCs and can be rapidly upregulated by a variety of activating stimuli (infection, tissue injury, inflammatory cytokines, and interaction of DCs with activated T cells). B7.1 is virtually absent from nonactivated DCs, is upregulated by similar stimuli but is expressed on the cell surface later than the peak of B7.2 expression.
There are no clearly defined differences in the downstream biochemical effects of ligation of CD28 by B7.1 or B7.2 respectively, and mice that are deficient for either B7.1 or B7.2 show that they have partially overlapping functions. The functional differences that can be observed may be more a result of the differences in expression kinetics of the two ligands than of any unique biochemical properties of the signals they induce. Provision of signaling via both TCR and CD28 results in dramatic changes in numerous aspects of T-cell function, the most immediate of which include rearrangement of plasma-membrane and cytoskeletal components, reorientation of the microtubule organizing center (MTOC), accelerated intracellular vesicular trafficking, and both general and locus-specific chromatin changes that are concomitant with nuclear localization and the activation of transcription factors (reviewed in Acuto and Michel, 2003). While the majority of these changes can be directed by strong and long-lasting supra-physiological stimulation via the TCR alone, the TCR ligation conditions that occur in vivo under normal circumstances are likely to generate only short-lasting and incomplete activation events that are insufficient to cause cell proliferation and differentiation, and which more likely result in the induction of anergy or cell death. In addition, some biological outcomes of TCR ligation do appear to have a mandatory requirement for CD28 signaling. Isolated CD28 ligation results in transient expression of a restricted number of the same genes induced by TCR ligation with no discernable dedicated signaling element, but engagement in concert with TCR ligation strongly amplifies weak TCR signals and modifies the gene regulation induced by TCR stimulation (Diehn et al., 2002; Riley et al., 2002). Thus, ligation of CD28 decreases the number of ligated TCRs that are required for a given biological response (Viola and Lanzavecchia, 1996), resulting in an apparent dependency on a qualitative second signal when TCR occupancy is low.
The recognition that B7 expression could enhance T-cell responses led to studies in murine models confirming that transfection-induced expression of B7.1/B7.2 on many immunogenic tumors resulted in T-cell mediated rejection in both a prophylactic and treatment setting (Chen et al., 1992, 1994; Townsend and Allison, 1993). The efficacy of antitumor activity depended on inherent tumor immunogenicity (Chen et al., 1994). Primary rejection of tumor cells was frequently documented to result in long-term protection against a secondary challenge, even with the B7− parental tumor, as well as eradication of preestablished micro-metastases. Protection was demonstrated to be due to CD8+ T cells by depletion studies, and it was initially envisaged that CD8+ cytotoxic T cells (CTL) were being primed directly by transfected tumor cells. Bone marrow chimera experiments in which the haplotype of the reconstituted APC population was distinct from that of the tumor, however, demonstrated that while B7-transfected tumor could directly serve as APC for the priming of naïve CTL in vivo, the efficiency of this process was far less than that of CTL priming by the host’s APC, requiring multiple immunizations to establish detectable responses as compared to the single immunization required for responses restricted to the haplotype of the bone marrow (Huang et al., 1996). These findings suggested that B7 expression might enhance an initial wave of direct tumor lysis, but that it was the efficient cross-presentation of tumor debris via DCs that resulted in the majority of the enhanced immunogenicity seen with B7+ tumors, consistent with the earlier finding that generation of effective systemic immunity was dependent on inherent tumor immunogenicity and that immunity to the parental tumor could be established. Nonimmunogenic tumors might be targeted by the initial wave of direct B7-directed killing of transfected cells but would not then generate potent systemic immunity.
2.1.2. CTLA-4 as a T Cell Intrinsic Cell-Autonomous Negative Regulator
CTLA-4 was recognized as a closely related homologue of CD28 capable of binding both of the B7.1/B7.2 ligand pair well before its true function became apparent. Following its establishment as a key negative regulator of immune responses these four molecules became acknowledged as perhaps the most important cofactors functioning at the tip of an immunological cascade. Together they act as the arbiters of initial immunological “stop–go” decisions, and emerged as the archetypal example of interdependent antagonistic costimulatory/inhibitory pathways. It has subsequently become apparent that their role may not be as straight forward as the simple licensing of “stop–go” signaling, but that they may be more subtly involved in the shaping of the immune repertoire and that their interdependence, coupled with the tight control of the temporal and spatial kinetics of their expression, might enable a mechanism to escape the restraints imposed by system based purely on Boolean logic. CTLA-4 has significantly higher affinities for both B7 ligands than does CD28. The interaction of CTLA-4 with B7.1 is of higher affinity than that with B7.2, whereas CD28 is predicted to bind to B7.2 more effectively than B7.1 (Collins et al., 2002). Ligand binding is important for the accumulation of both CD28 and CTLA-4 at the synapse. While CD28 is recruited to the synapse in the absence of B7.1 and B7.2 binding, it is not effectively stabilized there, and its localization can be disrupted by CTLA-4. The latter is more critically dependent on ligand binding for concentration at the synapse (Pentcheva-Hoang et al., 2004). The structure of cocrystals of CTLA-4 and B7.1 suggests that these molecules may form extended lattice-like networks, enabled by the distal positioning of CTLA-4 binding sites from the B7.1 dimer interface (Stamper et al., 2001; van der Merwe and Davis, 2003). This contrasts with a likely monovalent interaction between CD28 and B7.2 (Collins et al., 2002). The function of CTLA-4 as a negative regulator of CD28-dependent T-cell responses is perhaps most strikingly demonstrated by the phenotype of CTLA-4 knockout mice, which succumb to a rapidly lethal polyclonal CD4-dependent lympho-proliferation within 3–4 weeks of birth (Chambers et al., 1997; Tivol et al., 1995; Waterhouse et al., 1995).
It is likely that the temporal and spatial separation of expression of CD28 and CTLA-4 are critical for regulation of immune responses. CTLA-4 expression is difficult to detect on resting T cells, but it is recognized to influence some of the earliest events in T-cell activation (Blair et al., 1998; Brunner et al., 1999). It undergoes complex intracellular trafficking mediated by its binding to the clathrin-adaptor molecules AP-1 and AP-2 (Chuang et al., 1997; Shiratori et al., 1997; Zhang and Allison, 1997), and is mobilized from intracellular vesicles in the proximity of the MTOC to the synaptic site of TCR engagement rapidly after TCR engagement (Egen and Allison, 2002). Strong TCR agonists are more efficient at inducing translocation of CTLA-4 to the immunological synapse, suggesting that TCR signal strength itself may inversely influence subsequent signaling elements. In contrast to CD28, CTLA-4 has a short cell-surface half-life in activated T cells, and surface expression is thus tightly linked to gene transcription and/or translation. In the unphosphorylated state, an intracellular localization motif mediates rapid binding to AP-2, endocytosis, and lysosomal targeting (Shiratori et al., 1997). Its ability to form a lattice structure of alternating CTLA-4 and B7.1 homodimers coupled to the 500–2500-fold higher affinities for both ligands than those of CD28 (Greene et al., 1996) provides one possible physical mechanism for its role as a negative regulator of CD28 signaling. It may exclude CD28 from the immunological synapse and out-compete it for the shared ligands. Transfection of T cells with CTLA-4 expressing mutations or truncations provide confirmatory evidence for the hypothesis that the CTLA-4 cytoplasmic tail is not always necessary for inhibitory function and that competition for shared ligands may be an important mechanism of inhibition (Carreno et al., 2000; Nakaseko et al., 1999). Expression of a mutant CTLA-4 molecule without a functional intracellular domain blocked the massive lethal lymphoproliferation that characterizes CTLA-4−/− mice completely (Takahashi et al., 2005), or partially (Chikuma et al., 2005; Masteller et al., 2000) depending on the surface expression level, presumably as a consequence of competition for CD28/B7 ligation or indo-leamine 2,3-dioxygenase (IDO) induction (Fallarino et al., 2003). However, although such competition may be most effective when B7 levels are limiting, direct signaling through the tail appears to be necessary if B7 levels are high (Carreno et al., 2000). In addition, an alternatively spliced ligand independent form of CTLA-4 (liCTLA-4) lacking the B7.1/B7.2 ligand-binding domain has been reported to be expressed on resting T cells, particularly on CD4+CD45Rblo cells, to be rapidly downregulated following T-cell activation, and to inhibit both T-cell proliferation and cytokine production (Vijayakrishnan et al., 2004). It has been speculated that this isoform controls survival and/or homeostasis of naïve T-cell subsets. Taken together, the results suggest that inhibitory signals are transduced via both ligand-independent and B7 ligation-dependent mechanisms to maximize negative signaling. One important aspect of the mechanism of inhibition is that a direct physical competition with CD28 for ligand binding may be negated by antibodies that block the ligand interaction, resulting in a more effective blockade than agents that specifically target the CTLA-4 signaling pathway. While the precise mechanisms by which CTLA-4 inhibits T-cell responses remain unclear, the importance of the codependence of its activity on CD28-mediated signaling is emphasized by gene expression analyses demonstrating that CTLA-4 engagement selectively blocked augmentation of gene regulations by CD28-mediated costimulation but did not ablate gene regulation induced by TCR triggering alone (Riley et al., 2002).
Two distinct, but not mutually exclusive, models that attempt to integrate the effects of CTLA-4 engagement within the framework of TCR and CD28 signaling and subsequent biological responses have been proposed. They have been termed the threshold and attenuation models (Chambers et al., 2001). In the threshold model CTLA-4 increases the TCR and/or CD28 signal strength required for activation of a naïve T cell, and effectively prevents those cells receiving low amounts of stimulation from proceeding to a state of full activation. This could be affected by competition with CD28 for B7.1/B7.2 on APCs under conditions when CD28-B7.1/B7.2 signaling is limiting for T-cell activation. The lymphoproliferation in CTLA-4−/− mice is dependent on both CD28 costimulation and TCR signals generated from low-affinity peptide-MHC interactions. The absence of B7.1 and B7.2 signals ablates the autoimmune phenotype in triple-mutant mice deficient in B7.1, B7.2, and CTLA-4 (Mandelbrot et al., 1999). CTLA-4 could set the threshold for activation above that delivered by the weak TCR signals that will be an inevitable function of thymic positive selection. When CD28-B7.1/B7.2 signaling is not limiting, CTLA-4 could restrict cell division following T-cell activation (Brunner et al., 1999; Doyle et al., 2001; Krummel and Allison, 1996), and hence attenuate rather than prevent T-cell responses. If this inhibition of expansive capacity were differentially regulated as a function of TCR signal strength (Chambers et al., 1999; Egen and Allison, 2002), the antigen-driven selection of T cells bearing high-affinity TCRs that occurs during T-cell responses might be decreased allowing the maintenance of a broader diversity of clonal responses early during antigen encounter. Attenuation of T-cell expansion could also influence the induction of T-cell anergy, as T cells can be rendered anergic if they fail to progress through enough rounds of division (Vanasek et al., 2001).
TCR transgenic T cells from CTLA-4-deficient mice allowed the study of the role of CTLA-4 in the regulation of peptide specific responses. These studies indicated that the inhibitory effect of CTLA-4 was more pronounced during secondary rather than primary responses, and indicated a role in both CD4+ and CD8+ T-cell responses (Chambers et al., 1998, 1999; Luhder et al., 2000). Together the data are consistent with a model in which chronic T-cell stimulation results in persistently higher levels of CTLA-4 expression. Thus, CTLA-4 blockade offered a further potential mechanism by which to try to enhance the antitumor responses of preexistent chronically stimulated tumor-reactive T cells by a combination of reducing the threshold for activation of weakly reacting clones and removal of the attenuation of subsequent T-cell proliferation, the effect of which might be accentuated by prior upregulation of CTLA-4 in the higher affinity population (Fig. 1). Blockade might result in the promotion of higher affinity clones that would otherwise be more restricted by CTLA-4 signaling.
Figure 1.
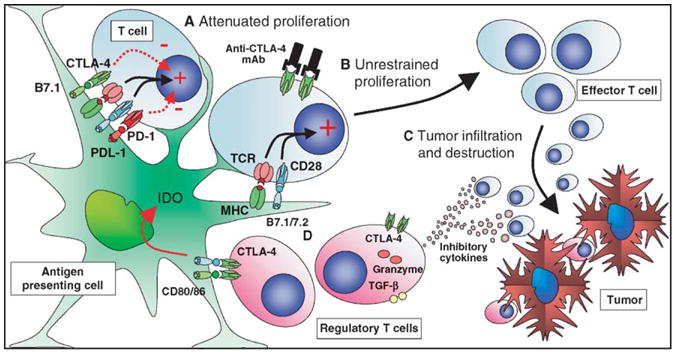
CTLA-4 blockade in cancer immunotherapy. Dendritic cells can sample tumor antigens and present them to T cells. (A) Although activation of dendritic cells may result in upregulation of B7.1/B7.2, the potentially responsive T cells expressing reactive TCR may be inhibited from effector function by inhibitory signaling via CTLA-4 and PD-1. (B) Blockade of CTLA-4 signaling may allow unopposed CD28 costimulation, resulting in recruitment of these T cells as antitumor effectors, either directly or as helpers of CD8-mediated T cells responses. (C) Activated cytotoxic T cells can then affect antitumor responses. (D) CTLA-4 expressing regulatory T cell populations may still be locally active in suppressing antitumor responses. Their activity could also be directly downregulated by CTLA-4 blockade, although the relative importance of CTLA-4 expression to their function remains controversial.
2.1.3. CTLA-4 as a Non Cell-Autonomous Regulator: Regulatory T Cells and Dendritic Cells
The function of CTLA-4 has so far been discussed with respect to a cell-autonomous mode of action that is T-cell intrinsic (i.e., a direct action mediated via a receptor located on the helper or effector T cell). Adoptive transfer experiments demonstrate that CTLA-4-deficient T cells show enhanced responsiveness to antigenic stimulation compared to wild-type cells, confirming the importance of a cell-autonomous mechanism of CTLA-4-mediated inhibition (Greenwald et al., 2001). However, negative regulatory mechanisms within the immune cascade include those that invoke additional subsets of cells, including regulatory T cells (in which CTLA-4 could potentially fulfill a T-cell intrinsic, non cell-autonomous regulatory role) and dendritic cells (T-cell extrinsic regulators). The role of immunoglobulin coinhibitory family members in the function of both continues to be elucidated. Both are important targets in their own right for manipulation in the field of immunotherapy. Data has demonstrated the presence of infiltrating CD4+CD25+Foxp3+ regulatory T cells in human ovarian, breast, pancreatic, and lung carcinoma, and metastatic melanoma (Curiel et al., 2004; Liyanage et al., 2002; Viguier et al., 2004; Woo et al., 2001). A number of T-cell subtypes with regulatory or suppressive activity are now recognized. They fall broadly into one of two categories: those which are continuously produced by the thymus, express CD4, CD25, and GITR, and appear crucially dependent on the expression of the X-linked forkhead/winged helix transcription factor, Foxp3, for their development [so-called “naturally occurring” regulatory T (Treg) cells]; and those which arise as a result of “tolerogenic” encounters in the periphery [interleukin (IL)-10-producing, Foxp3-negative Tr1 cells (Groux et al., 1997; Levings et al., 2001a) and TGFβ-producing Th3 cells (Weiner, 2001) being the best characterized of these “inducible” or “adaptive” regulatory T cells]. In addition, CD4+CD25−Foxp3+ T cells with regulatory capabilities have also been described (Zhou et al., 2005). CD4+CD25+ regulatory T cells are selected by high-affinity interactions in the thymus (Caton et al., 2004; Jordan et al., 2001), although the factors dictating the choice between Treg development and T cell negative selection remain unclear. Interactions between CD28 and B7.1/B7.2 are implicated in the selection process as it is defective in mice lacking these molecules, helping to explain the unexpected finding of increased severity and accelerated onset of diabetes that develops in CD28−/− and B7.1/B7.2−/− NOD mice (Salomon et al., 2000). The finding that the CD4+CD25+ Treg subset constitutively expresses CTLA-4 at high levels (Salomon et al., 2000) led to the suggestion that it may be important in the function of these cells and that “blockade” with monoclonal antibodies may suppress the function of or mediate the depletion of these cells. An alternative explanation for the high levels of CTLA-4 that does not require the invocation of additional mechanisms of function is that its role in CD4+CD25+ Treg is the same as in other T cells. That is, it works in a cell-autonomous fashion to restrict proliferation of a population of T cells with higher affinities for self than those of nonregulatory populations, and that the higher levels of CTLA-4 are a manifestation of chronic stimulation by self-antigen in vivo.
Precise details of the mechanisms involved in effecting suppression in different immune responses are not yet entirely clear. CD4+CD25+ Treg are able to suppress the proliferation of other T cells in an antigen-nonspecific manner, although Treg activation is antigen specific. Activity in vitro appears to be cytokine-independent and to involve direct interactions between T cells (Gondek et al., 2005; Thornton and Shevach, 2000), while in many in vivo experimental systems suppression requires TGF-β or IL-10 (Fahlen et al., 2005). CTLA-4 has been implicated in CD4+CD25+ Treg function in some, but not all, studies. Inhibition of the CTLA-4 pathway by using antibodies or CTLA-4 deficient regulatory cells reduces suppressor function in some experimental systems (Read et al., 2000; Takahashi et al., 2000). Normal mice treated with high doses of anti-CTLA-4 or a mixture of anti-CTLA-4 and anti-CD25 develop autoimmune gastritis, and the administration of anti-CTLA-4 reverses the Treg-mediated inhibition of CD25− T cell induced colitis in vivo (Read et al., 2000). It is of course difficult to exclude the possibility of a T cell intrinsic cell-autonomous mechanism of action in these studies via the direct blockade of inhibitory signals via CTLA-4 on effector populations. The find-ing that bone marrow chimeras generated from CTLA-4−/− and wild-type CTLA-4+/+ donors are protected from the lethal lymphoproliferative disorder that characterizes the CTLA-4−/− mice (Bachmann et al., 1999) has also been used to suggest an important non cell-autonomous role for CTLA-4 in negative regulation of T-cell responses. Wild-type CTLA-4 expressing regulatory T cells are obvious candidate populations to mediate these effects. In two alternate, but not mutually exclusive, models, CTLA-4 could be required for the normal development or maintenance of a regulatory population, or could be key to the inhibitory function of these cells. In this respect, the finding that CTLA-4 binding to B7.1/B7.2 appears to result in backward or “outside-in” signaling, resulting in induction of IDO in dendritic cells, which in turn leads to immune suppression as a consequence of tryptophan depletion and production of pro-apoptotic metabolites, may have relevance (Fallarino et al., 2003).
Further evidence supports the possibility of a similar direct effect on T cells (a truly T-cell intrinsic, non cell-autonomous activity). B7.2, but not B7.1, is constitutively expressed on some resting T cells, and both can be upregulated. CD4+CD25− effector T cells from B7.1/B7.2−/− mice are resistant to suppression by CD4+CD25+ Treg compared to wild-type CD4+CD25− effector Tcells in vitro, and these cells provoke a lethal wasting disease in lymphopaenic mice despite the presence of regulatory T cells (Paust et al., 2004). Susceptibility of B7.1/B7.2-deficient cells to suppression could be restored by lentiviral-based expression of full-length B7.1 or B7.2, but not of truncated molecules lacking the transmembrane/cytoplasmic domain, despite restoration of CD28-dependent costimulatory activity by these truncation mutants. In addition, T cells that constitutively overexpress B7.2 exhibit reduced alloreactivity and graft-versus-host disease mortality in murine transplantation models (Taylor et al., 2004). Conversely, T cells from B7.1/B7.2−/− mice effect accelerated alloresponses and increased graft-versus-host disease-related mortality. The downregulation of responsiveness mediated via B7.1/B7.2 appeared to be dependent on interaction with T cell-associated CTLA-4. Collectively these data suggest that bi-directional signaling may also be important in T-cell regulation by B7.1/B7.2-CTLA-4 interactions.
Anti-CTLA-4 does not, however, successfully reverse in vitro suppression in all studies (Chai et al., 2002; Thornton and Shevach, 1998). Studies with human cultured cells have suggested that CTLA-4 blockade has only a moderate or no effect on the suppressive function of CD4+CD25+ regulatory T cells (Annunziato et al., 2002; Levings et al., 2001b). In addition, CTLA-4 expression does not appear to be critical for the development of CD4+CD25+ T cells within the thymus of CTLA-4−/− mice (Kataoka et al., 2005), and CD4+CD25+CD62L+ T cells from CTLA-4-deficient mice with the CTLA-4Ig transgene express levels of Foxp3 that are similarly high to those of wild-type Treg by Western blot analysis, suggesting that they are of the same lineage (Tang et al., 2004). Both of these CTLA-4−/− Treg populations exhibit regulatory function in in vitro assays that is equivalent to that of wild-type Treg cells, although the mechanism of suppression may differ as there is some evidence of compensatory changes in CTLA-4−/− Treg that enable them to suppress in a partially TGF-β-dependent fashion in in vitro assays, unlike their wild-type counterparts (Tang et al., 2004). Thus, data suggest that the role of CTLA-4 in both the development and function of CD4+CD25+ Treg is, at least, partially redundant.
3. Preclinical Models of Checkpoint Blockade as Tumor Immunotherapy
The demonstration that blockade of CTLA-4/B7 interactions with anti-CTLA-4 monotherapy was able to induce rejection of several types of established transplantable tumors in mice, including colon carcinoma, fibrosarcoma, prostatic carcinoma, lymphoma, and renal carcinoma was an important proof of principle establishing checkpoint blockade as a potentially viable therapeutic modality (Kwon et al., 1997; Leach et al., 1996; Shrikant et al., 1999; Sotomayor et al., 1999; Yang et al., 1997). Antitumor activity appears dependent on inherent tumor immunogenicity and response longevity, in terms of ability to reject subsequent tumor challenge, is variable. The failure of anti-CTLA-4 monotherapy in the less immunogenic tumors (e.g., B16 melanoma) led to the exploration of its combination with vaccination approaches. Irradiated tumor vaccines that are engineered to produce granulocyte-macrophage colony stimulating factor (GMCSF) are highly effective as prophylaxis in tumor challenge experiments but poorly effective in treatment models. Combination with CTLA-4 blockade in the B16 melanoma model results in synergism of activity, which can cause tumor rejection if initiated 4 days after tumor implantation, and significant retardation in tumor growth if delayed by up to 1 week (van Elsas et al., 1999). A similar effect was observed for the poorly immunogenic SM1 mammary carcinoma line (Hurwitz et al., 1998) and in a transgenic model of prostate carcinoma (Hurwitz et al., 2000). Analogous synergism is observed with DNA rather than cellular vaccines. Xenogeneic or heteroclitic DNA vaccines have shown some promise in preclinical murine models, and combination with CTLA-4 blockade enhanced T-cell responses to melanoma differentiation antigens (tyrosinase-related protein 2 and gp100) and enhanced B16 tumor rejection (Gregor et al., 2004). The effect was stronger when anti-CTLA-4 was administered with booster vaccinations with the suggestion that the sequence and schedule of administration of CTLA-4 blockade may be an important variable to consider in clinical studies. CTLA-4 blockade also increased the T-cell responses to prostate-specific membrane antigen (PSMA) when given with the booster rather than the priming vaccinations.
In the B16 melanoma vaccination/CTLA-4 blockade models, rejection is accompanied by depigmentation, reminiscent of that seen in patients with melanoma who respond to immunotherapy, and suggesting that the immune targets for these responses may be normally expressed differentiation antigens. Depigmentation is uncommon following vaccination alone, suggesting that CTLA-4 blockade is important in breaking peripheral tolerance in this system. Importantly it is not seen with anti-CTLA-4 monotherapy. An additional manipulation that may enhance therapeutic efficacy in murine models is depletion of CD25+ Treg cells. Depletion prior to combination therapy results in induction of increased numbers of TRP-2180–188-specific T cells and an enhanced ability to reject larger tumor burdens (Sutmuller et al., 2001). Efficacy of the antitumor therapy correlated with the extent of autoimmune skin depigmentation as well as with the frequency of TRP-2180–188-specific cytotoxic T cells detected in the periphery.
Combination of CTLA-4 blockade with a number of other therapeutic modalities has confirmed the versatility of a therapy which can enhance immune responses induced by a variety of interventions. Multiple mechanisms may account for this effect, such as reducing tumor burden, increasing the available pool of tumor antigen, or upregulating costimulatory molecules. Ionizing radiation is an important component in the management of breast cancer, but although the primary tumor can be successfully treated by surgery and radiotherapy, metastatic breast cancer remains a therapeutic challenge. CTLA-4 blockade has no effect on primary tumor growth or survival in the poorly immunogenic metastatic mouse mammary carcinoma 4T1 model when used as a monotherapy (Demaria et al., 2005). Radiotherapy delays the growth of the primary tumor, but in the absence of CTLA-4 blockade survival is not improved compared to control mice. In contrast, mice treated with radiotherapy and CTLA-4 blockade demonstrate a statistically significant survival advantage, correlating with an inhibition of the formation of metastatic lesions within the lung. This activity required CD8+ T cells, while CD4+ T cells were dispensable (Demaria et al., 2005) as in earlier studies of CTLA-4 blockade as a therapeutic rather than prophylactic modality (van Elsas et al., 1999). Chemotherapy is another mainstay for the treatment of many human malignancies with which CTLA-4 blockade may synergize. Anti-CTLA-4 monotherapy is ineffective in retarding tumor growth in the murine MOPC-315 tumor system, but demonstrates significant therapeutic benefits when combined with a sub-therapeutic dose of the chemotherapeutic agent melphalan (Mokyr et al., 1998).
Finally, combination of anti-CTLA-4 with both peptide vaccination and CpG, which acts as an immunological adjuvant by activating the innate immunity via Toll-like receptor (TLR)-9 signaling, has also been shown to enhance antitumor immunity in the B16 melanoma model (Davila et al., 2003). The antitumor effect of this combination immunotherapy required both CD4+ and CD8+ T lymphocytes.
3.1. CTLA-4 Blockade in Tumor Immunotherapy: A Cell-Autonomous or Noncell-Autonomous Mode of Action?
There are no data that definitively address the issue of the role of CTLA-4 blockade on Treg cells as opposed to helper/effector cells during antitumor therapy. Studies documenting the possibility of Treg depletion have given contradictory results. Depletion was not seen in the lymph nodes of BALB/c mice following administration of blocking antibody despite the induction of autoimmunity (Takahashi et al., 2000) and did not occur following therapy in nonhuman primates (Keler et al., 2003). However, blood samples from human subjects receiving anti-CTLA antibody indicate that there may be a reduction in the percentage of CD4+CD25+ T cells that has been suggested to imply a depletion of the Treg compartment (Attia et al., 2005). Depletion of CD25+ cells prior to tumor challenge significantly increases the effectiveness of combination therapy with CTLA-4 blockade and cellular vaccines (Sutmuller et al., 2001). If the efficacy of CTLA-4 blockade was dependent on inhibition of CD4+CD25+ Treg activity, one might predict that depletion of CD4+CD25+ Treg prior to blockade would abrogate its activity to some degree. The synergism in the effects of CTLA-4 blockade and depletion of CD4+CD25+ Treg cells suggests that CD25+ regulatory T cells and CTLA-4 signaling represent two alternative pathways for suppression of autoreactive T-cell immunity, although it does not formally exclude a direct effect on Treg function, particularly if this were effected through the CD4+CD25−Foxp3+ population, which is not depleted by CD25-depleting antibodies.
Studies in our laboratory favor a T cell intrinsic cell-autonomous mode of action of anti-CTLA-4 on both the effector and regulatory compartments (Quezada et al., manuscript submitted). Anti-CTLA-4 increased proliferation of the purified CD4+CD25− effector population and was unable to inhibit the suppressive capacity of CD4+CD25+ regulatory T cells at a 1:1 suppressor: effector ratio in standard in vitro suppressor assays. In addition, chronic anti-CTLA-4 therapy in vivo (over a period of 2 weeks) induced expansion of intranodal Treg rather than depletion, suggesting a direct cell-autonomous effect of blockade of CTLA-4-mediated inhibitory signaling on the Treg. In a mouse model of the poorly immunogenic B16 melanoma tumor, tumor growth induced the expansion and accumulation of Treg in the lymph nodes in absence of any therapy. Anti-CTLA-4 monotherapy resulted in a further accumulation of Treg within the lymph nodes, presumably driven by self-antigen (Fig. 2A), but did not result in increased intratumoral T-cell infiltration. Administration of a GM-CSF-expressing B16 cellular vaccine facilitated efficient priming of tumor-specific effector T cells. As a monotherapy this enhanced tumor infiltration by CD8+ T cells. However, intratumoral proliferation was still presumably under the restraints imposed by CTLA-4 mediated signaling and tumor growth was not arrested. Combination with CTLA-4 blockade resulted in maximal effects on nonregulatory T cell numbers by allowing unrestrained proliferation driven by tumor antigens, resulting in the inversion of the ratio of effectors to regulators (Fig. 2B). The data suggest that with combination therapy the priming induced by the cellular vaccine results in a massive increase in the effector compartment within the tumor and once the inhibitory cellular restraints imposed by CTLA-4 signaling are removed the inhibitory activities of the Treg are overwhelmed, resulting in tumor rejection. In the absence of the cellular vaccine, insufficient effector T-cells infiltrate the tumor and the outcome of CTLA-4 blockade still favors Treg over the effector populations resulting in continued tumor growth. Hence, the overall outcome will depend on the priming history of the T-cell populations and the local antigenic milieu. It remains difficult to completely exclude an additional non cell-autonomous effect mediated via inhibition of Treg function and further studies addressing these issues are clearly warranted.
Figure 2.
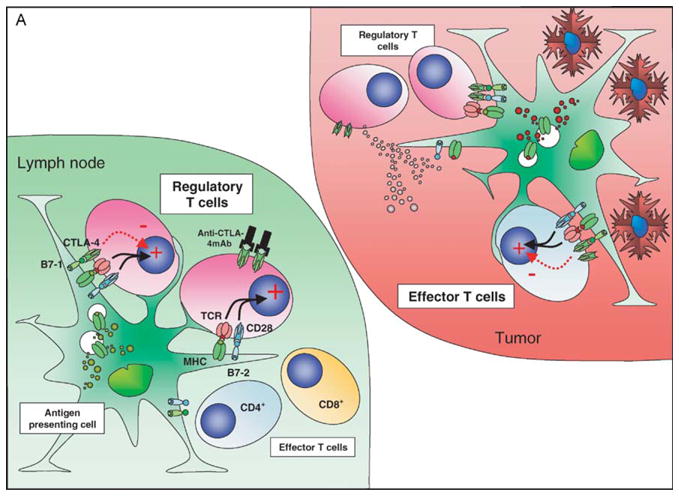
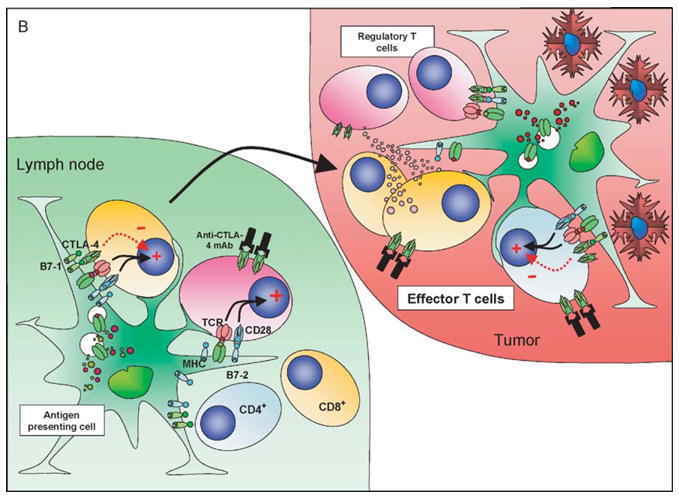
CTLA-4 blockade and cellular vaccines in poorly immunogenic tumors: the effect of CTLA-4 blockade and vaccination with a GM-CSF expressing cellular vaccine for poorly immuno-genic tumors may depend on the local antigenic milieu and priming history of the T cells. In the nonvaccinated host (A) a direct effect on Treg may predominate due to higher TCR affinities, lower requirements for costimulation and predominance of self-antigen presentation, resulting in a relative expansion of Treg compared to other T-cell populations. The tumor infiltrate is characterized by a relatively high proportion of Treg and very low levels of CD8+ T cells. CTLA-4 blockade under these conditions still favors Treg over effector T cells. In the vaccinated host (B) the preferential presentation of tumor-associated antigens in association with high levels of costimulatory molecules allows expansion of effector T-cell populations that can traffic to the tumor, although these cells will now upregulate CTLA-4 expression. Vaccination may be insufficient on its own to eradicate tumor due to the presence of both cell-autonomous and non cell-autonomous inhibitory circuits. The addition of CTLA-4 blockade now tips the balance in favor of the effector population, perhaps aided by the numerical superiority of these cells over tumor-resident Treg resulting in tumor eradication.
3.2. Adverse Immune Manifestations of CTLA-4 Blockade in Preclinical Models
The ability of anti-CTLA-4 to exacerbate autoimmunity in experimental models is well established (Hurwitz et al., 1997). In general, autoimmunity has been amplified when mice are vaccinated with self-antigens in combination with CTLA-4 blockade (Karandikar et al., 1996; Luhder et al., 1998; Perrin et al., 1996; Wang et al., 2001). While preclinical studies of antitumor immunity demonstrated the potential for CTLA-4 blockade in tumor immunotherapy, they also illustrated the possibility of induction of autoimmunity. However, toxicities were limited to predicted “target” tissues sharing the same epitopic topiary as the cellular vaccines (depigmentation in the melanoma model (van Elsas et al., 1999), prostatitis in the prostate cancer model (Hurwitz et al., 2000)). More serious systemic toxicities were not documented, perhaps in part due to the shorter duration of therapy, and shorter half life of the original hamster antimouse antibody (clone 9H10) used in these studies as compared to that of the fully human antibody used for subsequent clinical studies. Nonhuman primates treated with CTLA-4 blockade and a human melanoma whole cell vaccine showed enhanced development of antibodies to some self-antigens, in particular to those present in lysates prepared from their melanocyte rich iris tissue (Keler et al., 2003). Continued administration of anti-CTLA-4 in high doses for up to 6 months, however, did not result in any clinically or pathologically detectable end organ damage, even in those animals with detectable humoral anti-self responses.
4. Clinical Trials of CTLA-4 Blockade: Overview
Inhibition of the activity of CTLA-4 has begun to be tested in humans through the use of human antibodies developed using mice that are transgenic for human immunoglobulin loci. MDX-010 is a human IgG1 antibody that binds human and rhesus CTLA-4 and inhibits the binding of CTLA-4 to B7.1 and B7.2. In preclinical studies in cynomolgus monkeys, the antibody was shown to augment antibody responses to a Hepatitis B surface antigen vaccine and a human melanoma cell line, SK-MEL (Keler et al., 2003). These studies also revealed that multiple dosing at 10 mg/kg resulted in no serious adverse events. CP-675, 206, another human antibody to human CTLA-4 in development, is an IgG2 isotype and blocks B7 ligand binding (Ribas et al., 2005a). MDX-010 has a reported half life of 12–14 days (Davis et al., 2002), while CP-675, 206 has been reported as 22.1 days (Ribas et al., 2005a). This difference may be due to the different isotypes of the antibodies. These antibodies have entered human clinical trials and have been tested in multiple therapeutic regimens.
CTLA-4 blockade has been predominantly tested in metastatic melanoma and in renal cancer, those cancers in which immunological responses are thought to be most relevant. In these settings, treatment by anti-CTLA-4 antibodies is capable of inducing objective tumor responses as monotherapy. These responses can be durable in nature with several ongoing after several years. The response rate to monotherapy is variable (from 5 to 13%). Dosing has ranged from single doses of 0.01–15 mg/kg and repeated dosing from 3 to 15 mg/kg at various frequencies of administration. Redosing or maintenance dosing in those patients showing clinical benefit have also been explored. It remains unclear what the optimal scheduling is for CTLA-4 blockade either in monotherapy or concurrently with vaccines. Other cancers in which CTLA-4 blockade has been tested include prostate, ovarian, breast, and colon carcinomas.
In studies in which CTLA-4 blockade is combined with gp100 melanoma peptides, objective durable responses have also been noted (Attia et al., 2005; Phan et al., 2003b). These have involved multiple subcutaneous skin lesions, as well as multiple visceral sites in the lung as well as brain metastases. CTLA-4 blockade did not result in increased numbers of antigen specific T cells when the antipeptide responses were measured in peripheral blood relative to historical controls. The lack of augmented antigen specific responses using CTLA-4 blockade has also been observed using other peptides as well (Sanderson et al., 2005). Vaccination with peptide vaccines may indirectly result in the activation and increase in numbers of tumor-specific T cells with specificity distinct from the tumor antigen immunogen as shown by the work of Lurquin et al. (2005).
It is likely that monotherapy is able to activate preexisting antitumor responses. However, it is also possible that de novo antitumor responses are also generated. T-cell subset analysis in patients treated with MDX-010 revealed that HLA-DR+ T cells were increased as well as frequent increases in CD45RO T cells (Attia et al., 2005; Maker et al., 2005a; Phan et al., 2003b). These cells may include those effector T cells or their precursors that are responsible for the antitumor effects as well as the adverse events observed with anti-CTLA-4 treatment. In addition, CTLA-4 blockade is unlikely to function through effects on Treg cells, since patients treated with anti-CTLA-4 do not show demonstrable changes in Treg activity (Maker et al., 2005a).
4.1. Clinical Trials of CTLA-4 Blockade: Adverse Events
Treatment with anti-CTLA-4 antibodies is frequently accompanied by a wide variety of adverse events. The most common events involve the skin (rash and pruritis) and the gastrointestinal tract (diarrhea and colitis). In a small number of patients, these side effects can be of a serious nature (Grade III/IV). In a collection of 198 patients with metastatic melanoma and renal cancer that were treated with MDX-010 in a variety of regimens, Grade III/IV colitis was observed in 21% of the patients (Beck et al., 2005). These adverse events are due to inflammatory T-cell infiltrates where studied. Infiltrates of CD4 and CD8 T cells can be observed in skin and colon biopsies. Other serious adverse events include hypophysitis, uveitis, and hepatitis (Blansfield et al., 2005; Phan et al., 2003b; Ribas et al., 2005a; Robinson et al., 2004); it is presumed that these events are also the consequence of inflammatory T-cell infiltration. Colectomy has been necessary in some cases of colitis and two deaths were observed in a combination trial with DTIC, although these may be ascribed to causes other than anti-CTLA-4 (Fischkoff et al., 2005; Korman et al., 2005). The adverse events can appear at various times after anti-CTLA-4 treatment. Management of these adverse events includes cessation of drug and treatment with high-dose steroids. Steroid use can temper the inflammation in most cases and paradoxically antitumor responses have been observed to be maintained after steroid use (Attia et al., 2005). While glucocorticoids have multiple activities and are thought to impair T-cell responses, their use may not impair activated T-cells responses as shown in a murine model in which dexa-methasone treatment did not alter the effectiveness of adoptive cell therapy (Hinrichs et al., 2005). Tumor-necrosis factor blockade has also been used in the management of colitis in a small number of patients (Beck et al., 2005). In some trials, no treatment of the adverse events was attempted and resolution of many of the side effects occurred over time after cessation of antibody treatment (Ribas et al., 2005b).
One of the observations from these studies is that serious adverse events (Grade III/IV) may be correlated with effective antitumor responses (Attia et al., 2005; Beck et al., 2005; Phan et al., 2003b). For example, in patients with enterocolitis, Beck et al. (2005) report that 45% and 46% of patients with metastatic melanoma and renal cell cancer, respectively showed objective tumor responses. This suggests that a component of T-cell activation is directed at tumor antigens as well as putative self-antigens and that CTLA-4 blockade may overcome tolerance to self-antigens. However, in no instance has specificity of the T cells mediating the adverse events been determined. It is most likely that nonantigen specific expansion/infiltration of T cells catalyzes these adverse events. In this respect, these side events are suggestive of the phenotype of the CTLA-4 knockout mouse. While the adverse event profile of CTLA-4 blockade has frequently been referred to as autoimmune in nature, it should be noted that the phenomenon of uncontrolled T-cell proliferation in the murine system has not been shown to be directed at specific self-antigens.
4.2. Clinical Trials of CTLA-4 Blockade: Specific Trials
4.2.1. Monotherapy for Malignant Melanoma
MDX-010 was initially given to 17 patients in a phase I clinical trial at a single dose of 3 mg/kg. At this dose level the drug was well tolerated and side effects of rash were noted. There were two partial responses. Many of the patients in this trial received vaccines at various times prior to treatment with MDX-010. It was observed that prior treatment with GM-CSF modified vaccines engendered a greater frequency of immune cell infiltrates into the tumors and tumor necrosis, although objective responses were not noted in these patients (Hodi et al., 2003).
Dose escalation studies have been carried out for CP-675, 206 as a single agent in 39 patients that included 34 patients with melanoma using from doses of 0.01–15 mg/kg (Ribas et al., 2005a). Dose limiting toxicities were observed at 15 mg/kg and a maximum tolerated dose was considered to be 10 mg/kg. Two complete and 2 partial responses were noted out of 34 melanoma patients (response rate of 12%) with responses continuing from 24+ to 35+ months. Further studies with CP-675, 206 have explored multiple dosing regimens at high-antibody levels in malignant melanoma (16 patients at 10 mg/kg and 10 patients at 15 mg/kg every 3 months) (Reuben et al., 2005). Five objective responses were noted. In addition, monthly dosing of CP-675, 206 from 3 to 10 mg/kg has been examined in 14 patients (Ribas et al., 2005a). Adverse events included Grade III diarrhea and were similar to those adverse events previously observed. One patient had an ongoing response while receiving 10 doses at 10 mg/kg with the sole adverse event of autoimmune thyroiditis. These studies suggest that prolonged CTLA-4 blockade may be tolerated in some individuals.
4.2.2. Monotherapy for Renal Cell Carcinoma
MDX-010 has been administered to renal cancer patients at a dose of 3 mg/kg every 3 weeks (n = 38 or 3 mg/kg followed by 1 mg/mg every 3 weeks (n = 21) (Yang et al., 2005). In the 3 mg/kg cohort, 5 responses of partial duration were noted with 3 ongoing (8+, 10+, and 12+); 1 partial response was noted in the 3/1 cohort. All the responders were among the group with serious adverse events. A similar profile of adverse events was described (enteritis, colitis, hypophysitis, and rash) that are similar to that observed in malignant melanoma. The type of adverse events does not appear to correlate with the type of cancer. The contribution of prior IL-2 treatment in these patients remains to be analyzed since the majority of these patients were previously treated IL-2 nonresponders.
4.3. CTLA-4 Blockade and Vaccines
After initial single dose trials of MDX-010, multiple dosing trials were initiated together with peptide vaccines in patients with Stage 4 nonresectable meta-static melanoma (Attia et al., 2005; Phan et al., 2003b). This vaccine consists of two modified peptides from the melanocyte antigen gp100 that are restricted by HLA-A2 administered with the adjuvant Montanide. A total of 56 patients were treated: one cohort of 29 patients were treated with 3 mg/kg of MDX-010 every 3 weeks together with peptide vaccinations (for 1–12 cycles) and a second cohort of 27 were treated with an initial dose of 3 mg/kg and peptide vaccination followed by 1 mg/kg of MDX-010 and peptide vaccinations every 3 weeks (2–12 cycles completed). Four patients out of 29 (14%) patients in the high-dose group had objective clinical responses with 2 complete and 1 partial response (continuing for 2–3 years). The response rate in the second cohort was 3/27 (11%) with 2 partial responses continuing greater than 2 years. There appeared to be no significant differences either in efficacy between the two cohorts or the frequency of induction of adverse events. Treatment with MDX-010 was associated with significant immune related adverse events and these adverse events were associated with antitumor activity. Of 14 patients with grade III/IV toxicities, 5 tumor responses were noted, while 2 responses were noted among 42 patients without grade III/IV toxicity.
It is unclear from the above studies whether there is an effect of the vaccine and/or adjuvant in the clinical responses. Vaccination with gp100 peptides had previously been shown to induce antigen specific T-cell responses but few significant antitumor responses (Phan et al., 2003a). The gp100 vaccine together with MDX-010 generates no greater T-cell responses than that previously described for the peptides alone; however, these responses are monitored in peripheral blood and may not reflect the number or activity of antigen specific T-cells at effector sites. Thus it is possible that the effects observed are a consequence of MDX-010 alone. A phase III trial comparing response rates among groups treated with vaccine/adjuvant alone, MDX-010 alone, and MDX-010 together with vaccine/adjuvant will address this question directly.
CTLA-4 blockade may reactivate antitumor responses as seen in patients in which administration of anti-CTLA-4 has been given after prior vaccination with various types of vaccines. In one study of nine previously vaccinated patients with advanced cancer there was clear evidence of antitumor activity. In patients who had been previously vaccinated with autologous GM-CSF-secreting tumor cell vaccines, anti-CTLA-4 induced extensive tumor necrosis along with lymphocytic and granulocytic infiltration of tumors in each of three patients with metastatic melanoma and CA-125 levels were stabilized or decreased in two of two patients with metatastic ovarian carcinoma (Hodi et al., 2003). In another example, a complete response (CR) was obtained after treatment with anti-CTLA-4 antibody after tumor progression occurred despite multiple DC/Mart-1 peptide vaccinations (Ribas et al., 2004). In addition, T cell reactivity to additional tumor antigens as well as infectious agents were augmented in this patient following CTLA-4 blockade. These observations, albeit limited in number, suggest that anti-CTLA-4 was able to reactivate latent antitumor responses and prolong objective antitumor responses.
4.4. Anti-CTLA-4 and Chemotherapy
MDX-010 has been combined with the chemotherapeutic, dacarbazine (DTIC) in patients with unresectable metastatic melanoma (Fischkoff et al., 2005). MDX-010 (3 mg/kg) was administered monthly for 4 cycles together with DTIC (250 mg/m2 for 5 consecutive days for up to 6 cycles); in addition, there was a second arm where MDX-010 (3 mg/kg for 4 cycles) alone was administered. In the combination arm 6/35 responses were noted for a response rate of 17.1%. This included 2 complete responses (ongoing at 17+ and 20+ months) and 4 partial responses (one ongoing at 21+ months). Two partial responses were noted in the 35 patients in the monotherapy arm (both ongoing at 16 and 18 months). In addition, a significant increase in overall survival (14.2 months for MDX-010 and DTIC and 11.2 months for MDX-010) as well as progression-free survival was observed in the MDX-010 groups relative to historical values for DTIC (Fischkoff et al., 2005). Adverse events, such as colitis, were also observed in this trial including two deaths.
4.5. Anti-CTLA-4 and IL-2
High-dose bolus IL-2 is an approved treatment for metastatic melanoma that results in response rates of ~15% of which a high percentage can be durable (Atkins et al., 1999; Rosenberg, 2000). Thirty-six patients have been treated with a combination of MDX-010 and high-dose bolus IL-2 in a dose escalation trial of MDX-010 (Maker et al., 2005b). Twelve patients were treated with MDX-010 at 0.1, 0.3, 1, and 2 mg/kg (3 patients per group) while 24 patients were treated at 3 mg/kg of MDX-010. The response rate in all groups was 22.2% (3 complete and 5 partial responses) with ongoing responses in 6 patients of 13–19+ month duration. Given the response rate to MDX-010 together with gp100 peptides described above, it appears that the combination resulted in an additive effect. Five of 36 patients experienced Grade III/IV adverse events, but the association of inflammatory adverse events with outcome was not observed in this study. This may be explained by responses that are due to IL-2 treatment alone, which is not expected to result in an adverse event profile like that for anti-CTLA-4. An alternative explanation is that IL-2 promotes the activity of regulatory T cells, which may limit the inflammatory reactions of those T cells that are otherwise activated by anti-CTLA-4; however, this must be accomplished while retaining the activity of those T-effector cells that mediate antitumor activity.
5. Other Potential Coinhibitory Targets for Checkpoint Blockade
Given its earlier identification compared to other members of the immuno-globulin superfamily and its position at the tip of the regulatory cascade, CTLA-4 blockade was a natural first choice for investigation as a therapeutic intervention in immune-based therapies. However, the identification of other inhibitory checkpoints has highlighted other potential targets for therapeutic blockade (Fig. 3). These include PD-1 and its ligands PD-L1 and PD-L2, and possibly B7-H3, B7x/B7-H4, and BTLA.
Figure 3.
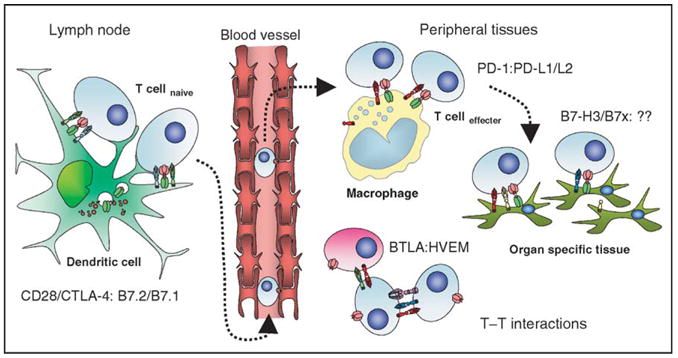
Immunological inhibitory checkpoints: potential therapeutic targets. All of the members of the immunoglobulin superfamily that act as inhibitory checkpoints are potential targets for manipulation in immunotherapies. CD28/CTLA-4:B7.1/B7.2 are centrally important for the initial activation of naïve T cells and regulation of the clonal composition of the responding repertoire following migration of activated dendritic cells to lymphoid organs. As activated effectors traffic back into peripheral tissues they come under the influence of PD1:PD-L1/L2 mediated signaling, both as a result of interactions with tissue macrophages and with ligands expressed on malignant cells. B7-H3 and B7x could be poised to act as the final arbiters of the fate of T effector interactions with nonlymphoid target tissues, and could potentially protect any tumor cells expressing them from cytotoxic T cell mediated killing. The potential for cross-talk between T-cell populations via many of these pathways is complex, particularly as activated T cells can upregulate receptors and/or ligands, which can potentially signal bidirectionally. Blockade of BTLA may remove inhibitory restraints imposed by HVEM-expressing cells, but effects on T–T interactions mediated by blockade of CTLA-4, PD-1/PD-L1, or B7-H3 are also possible.
PD-1 is more broadly expressed than CD28/CTLA-4. It is expressed on activated, but not resting, CD4+ and CD8+ T cells, B cells, monocytes, and at low levels on NKT cells. It shares the same pattern as CTLA-4 of dual ligands (PD-L1 (B7-H1) and PD-L2 (B7-DC)). PD-1 displays a higher affinity for PD-L2 than for PD-L1 (Youngnak et al., 2003). The ligands exhibit distinct expression profiles (reviewed in Greenwald et al., 2005). PD-L1 is expressed on resting and upregulated on activated B, T, myeloid, and dendritic cells, and on CD4+CD25+ Treg cells. It is also expressed on nonhematopoietic cells including microvascular endothelial cells and in nonlymphoid organs including heart, lung, pancreas, muscle, and placenta. This distribution suggests that interactions of ligands and receptors may be important in regulating effector T-cell responses in the peripheral tissues by “professional” APCs, such as DCs, macrophages, and also endothelial cells. PD-L2 is induced by cytokines on macrophages and DCs. The widespread expression of PD-L1 and expression of PD-1 on T cells, B cells, and macrophages presents ample opportunities for bidirectional signaling, further complicated by evidence of “outside-in” signaling (Dong et al., 2003; Nguyen et al., 2002; Radhakrishnan et al., 2004).
In a situation somewhat reminiscent of the early studies of CTLA-4 function, controversy exists over the function of PD-1. Studies using antibodies specific for PD-1 or the natural ligands demonstrate that engagement of PD-1 is able to inhibit T-cell proliferation under suboptimal CD3 stimulation (Carter et al., 2002; Freeman et al., 2000; Latchman et al., 2001). At high-antigen concentrations the TCR/CD28 signal predominates over PD-L1 or PD-L2 transduced signals, although cytokine production may be diminished. Colocalization of PD-1 with TCR/CD28 is necessary for this inhibitory activity. The phenotype of PD-1−/− mice also provides evidence for an inhibitory role of the receptor (Nishimura et al., 1999, 2001). These mice develop an autoimmune phenotype, delayed in onset compared to CTLA-4−/− mice, and characterized by high titers of autoantibodies in keeping with a negative regulatory effect on T and/or B cells. The phenotype manifested depends on the genetic background of the mouse. PD-1 deficiency on the C57BL/6 background results in development of a late-onset progressive arthritis and lupus-like glomerulone-phritis (Nishimura et al., 1999), while on the BALB/c background it results in development of a lethal dilated cardiomyopathy that shows incomplete penetrance, with concomitant evidence of autoantibodies to troponin-1 (Nishimura et al., 2001). PD-L1−/− mice accumulate CD8+ T cells in the liver that do not mediate spontaneous hepatic disease but which can enhance autoimmune hepatitis when experimentally challenged (Dong et al., 2004). Numerous models of autoimmune disease, and transplantation and graft-versus-host disease provide additional evidence that inhibition of PD-1/PD-L1 signaling results in enhanced severity and/or accelerated onset of disease, or that signaling induced by PD-L1Ig can synergize with conventional immunosuppressive drugs to enhance graft survival. However, other evidence points to a possible costimulatory role of PD-1 engagement (Dong et al., 1999; Tamura et al., 2001; Tseng et al., 2001; Wang et al., 2003), and PD-L2−/− DCs have a reduced capacity to stimulate CD4+ T cells (Shin et al., 2003). Site-directed mutagenesis studies in both PD-L1 and PD-L2 demonstrate mutations that can abrogate binding to PD-1 but which retain or enhance costimulatory activity (Wang et al., 2003). The possibility that an undiscovered second stimulatory receptor exists that interacts with the PD-L1/L2 ligands would add further symmetry to the similarities with the CD28/CTLA-4:CD80/86 pathway, and remains an intriguing possibility. Finally, the likelihood that this pathway has functional importance in the induction or function of regulatory T cell or DC populations appears increasingly probable (Aramaki et al., 2004; Baecher-Allan et al., 2001).
The expression of PD-L1 and PD-L2 by a range of murine and human tumors has led to the hypothesis that this may be a mechanism by which they evade immune surveillance. PD-L1 is expressed on many human carcinomas (mammary, cervical, lung, ovarian, colonic, renal), as well as melanoma, glio-blastoma, some primary T-cell lymphomas, and most thymic epithelial tumors including thymomas and thymic carcinoma (Blank et al., 2004; Brown et al., 2003; Dong et al., 2002; Thompson et al., 2004). In renal cancer, PD-L1 expression on tumors and on infiltrating T cells is associated with poor prognosis (Thompson et al., 2004). By contrast, PD-L2 is highly expressed on Hodgkin’s lymphoma cell lines and is the best genomic discriminator between primary mediastinal B-cell lymphoma and other less favorable diffuse large B-cell lymphomas (Rosenwald et al., 2003). PD-L1 and PD-L2 expression has been observed alone and together in esophageal cancer and poor survival is associated with either PD-L1 or PD-L2 expression (Ohigashi et al., 2005).
Transfection of murine tumors with PD-L1 rendered them less susceptible to the specific T-cell antigen receptor-mediated lysis by cytotoxic T cells in vitro, and markedly enhanced tumor growth and invasiveness in vivo (Iwai et al., 2002). Both effects could be reversed by blockade by anti-PD-L1 antibody (Iwai et al., 2002; Strome et al., 2003). Transfection with PD-L1 was able to negate the enhanced immunogenicity conferred by transfection of P815 mastocytoma cells with CD80 (Dong et al., 2002). The 4T1 mammary cell carcinoma is PD-L1 negative in culture but expresses PD-L1 in vivo (or can be induced to express PD-L1 in culture by γ-IFN) (Hirano et al., 2005). This tumor is refractory to tumor rejection mediated by an agonistic anti-41BB antibody, an activating receptor that is a member of the TNF family of receptors. While anti-41BB shows a modest decrease in tumor growth, treatment with anti-41BB together with anti-PD-L1 results in dramatic tumor rejection. Murine myeloma cell lines naturally express PD-L1, and their growth in vivo was also inhibited significantly, but transiently, by the administration of anti-PD-L1 antibody, although a direct effect of the antibody on the growth of the tumor (by other mechanisms such as ADCC) was not excluded. Their growth was suppressed completely in syngeneic PD-1-deficient mice (Iwai et al., 2002). In addition, PD-1−/− CD8+ TCR transgenic T cells caused tumor rejection in an adoptive transfer model in which wild-type and CTLA-4 −/− T cells failed to mediate rejection (Blank et al., 2004). These results are consistent with a model in which CTLA-4 is more vital for regulation of CD4+ T-cell responses, particularly early at the APC interface, whereas PD-1 has a relatively minor role at this stage (when CD28 costimulation can overcome its inhibitory effects (Carter et al., 2002)) but a more critical role in suppressing the execution of T-cell effector function against cells that do not express CD80/86 (Fig. 3). Intriguingly, PD-L2 expression on J558 plasmacytoma cells actually enhanced CD8-mediated immunity, tumor rejection, and establishment of immunological memory to subsequent rechallenge (Liu et al., 2003). The effect was evident for PD-1−/− as well as wild-type-T cells, suggesting that it may be mediated by interaction with a receptor other than PD-1.
Very few studies have examined the ability of anti-PD-1 antibodies to promote antitumor responses directly. Two metastatic models have been shown to be sensitive to PD-1 blockade (Iwai et al., 2005). Utilizing CT26, a colon carcinoma that metastasizes to the lung after intravenous injection, tumor growth was inhibited by 50% after treatment with anti-PD-1 antibody. This study also reported that B16 melanoma metastasis to the liver after intrasplenic injection of tumor cells, in which PD-L1 expression was found to be upregulated in vivo, could be inhibited by anti-PD1 treatment. In studies using the immunogenic fibrosarcoma line, Sa1N, PD-1 blockade was able to prevent the outgrowth of tumor in unstaged tumors and showed a partial effect on staged tumors. Sa1N cells were unable to express PD-L1. This suggests that host PD-L1 expression in dendritic cells and/or APCs play a role in limiting antitumor responses (unpublished data).
Other possible targets for checkpoint blockade are currently somewhat more speculative. B7-H3 (B7-RP2) and B7x (B7-H4, B7-S1) are the most recently described B7 family members, which currently remain orphan ligands. They display greater similarity to each other than other family members. Both B7-H3 and B7x/B7-H4 appear to bind to a receptor(s) expressed on activated but not naïve T cells, but the identity of the receptor(s) remains unclear. The broad tissue distribution and inducible nature of both ligands has led to the suggestion that they downregulate immune responses in the periphery and play a role in regulating T-cell tolerance. In addition, the presence in tumor cells suggests a possible role in immune evasion as suggested for PD-L1, and another potential target for immune manipulation in tumor therapy.
B7-H3 is not constitutively expressed but can be induced, on T and B cells, NK cells, and monocytes. It is expressed on immature and mature myeloid DC. It has very general mRNA expression in nonlymphoid tissues (e.g., heart, kidney, testes, and nasal epithelium). It has been reported to possess both stimulatory and inhibitory functions. These contradictory findings are yet to be fully reconciled but might be explained by the existence of two receptors with opposing function, or the effects of signaling mediated by different cell types. The phenotype of B7-H3-deficient mice supports an inhibitory role, with enhanced severity of pathology under Th1-polarizing conditions in airway inflammation experiments coupled with increased T-cell responses and accelerated development of experimental autoimmune encephalomyelitis (Suh et al., 2003). However, expression of B7-H3 by transfection of the mouse P815 tumor line enhanced its immunogenicity, leading to the regression of tumors and amplification of tumor-specific CD8+ CTL responses (Luo et al., 2004). In addition, intratumoral injection of an expression plasmid encoding B7-H3 led to complete regression of 50% EL4 tumors. Mice whose tumors completely regressed resisted a challenge with parental tumor cells, indicating systemic immunity had been generated. This immunity was mediated by CD8+ T and NK cells. These tumor models suggest a positive costimulatory function for B7-H3, implying that blocking antibodies may actually reduce rather than enhance antitumor activity.
B7x/B7-H4 appears to be a further negative regulator of T-cell responses, and to inhibit proliferation and cytokine production in both CD4+ and CD8+ T cells (Prasad et al., 2003; Sica et al., 2003; Zang et al., 2003). It has a similarly broad mRNA expression pattern to B7-H3 in both humans and mice, where it is expressed in lymphoid (spleen, and thymus) and nonlymphoid (lung, liver, small intestine, pancreas, placenta, ovary, testis, and skeletal muscle) tissues (Sica et al., 2003; Zang et al., 2003). Protein expression studies suggest that it is absent from freshly isolated human T and B cells, DCs, and monocytes but that it can be induced on these cells following stimulation. Immunohistochemical analyses demonstrate that it is not expressed on the majority of normal human tissues (Choi et al., 2003), although it has been reported to be expressed by some tissues, such as normal breast, Fallopian tubal epithelium, endometrial and endocervical glands, and pancreas (Tringler et al., 2005). It is, however, expressed on several tumor lines (prostate, lung, and colon) as demonstrated by quantitative real-time polymerase chain reaction analyses, and on human cancers (ovarian and lung) at higher levels than in the normal tissue counterparts as demonstrated by immunohistochemical analyses (Choi et al., 2003; Salceda et al., 2005; Tringler et al., 2005). Expression levels on lymphoid and antigen-presenting cells are currently controversial.
It is unclear whether the differences in mRNA expression patterns and immunohistochemistry relate to low-level protein expression, technical issues related to the antibodies employed, or translational regulation of protein levels. Administration of anti-B7x/B7-H4 antibodies to mice exacerbates EAE with concomitant increases in the numbers of CD8+ and CD4+ T cells infiltrating the brain (Prasad et al., 2003), and B7x/B7-H4Ig both reduces T-cell proliferation and cytotoxic T-cell activity, and extends survival in a murine model of graft-versus-host disease (Sica et al., 2003). The apparently inhibitory function, coupled to the demonstration of enhanced expression on human tumors, indicate that it is another potentially useful target in tumor immunotherapy.
Finally, BTLA is expressed on activated T and B cells, shows high expression by resting B cells, is induced on anergic CD4+ T cells and has lower expression on macrophages, DCs, and NK cells (Han et al., 2004; Hurchla et al., 2005; Watanabe et al., 2003). It remains expressed on Th1 but not Th2 cells, suggesting that it may specifically downregulate Th1-mediated inflammatory responses. Its ligand has recently been identified as HVEM (Sedy et al., 2005). HVEM is constitutively expressed by naïve T cells, is downregulated after activation, and then reexpressed as the T cell returns toward a resting (memory) state. Lymphotoxins, inducible, competes with herpes simplex virus glycoprotein D for HVEM, expressed by T cells (LIGHT) can costimulate T cells through HVEM activation, but it remains unclear whether BTLA activates HVEM or what effects BTLA might exert on LIGHT or lymphotoxin (LT) α interactions with HVEM. BTLA binding does not occlude the TNF family binding site, which is on the opposite face of HVEM, suggesting that HVEM may form stable ternary complexes with BTLA and either LIGHT or LTα (Compaan et al., 2005). BTLA exerts inhibitory effects on both B and T cells (Watanabe et al., 2003). Both cell types show moderately enhanced responsiveness in BTLA−/− mice. The phenotype of these mice is more similar to that of PD-1 knockout than to that of CTLA-4 knockout mice (Han et al., 2004; Watanabe et al., 2003). They have an increased susceptibility to EAE suggesting that the function of BTLA is not entirely redundant, although no propensity to spontaneous autoimmunity with age has yet been documented. Thus, the negative regulation induced by BTLA ligation on B and T cells can potently regulate the strength of the immune response and alter the balance governing immune tolerance. The consequences of immunological blockade in tumor models await further study.
6. Conclusions
The potential for therapeutic immunological checkpoint blockade has been amply demonstrated in preclinical murine models of a variety of cancers and in combination with a variety of other therapeutic interventions. Clinical studies remain in their infancy but have demonstrated exciting initial responses in heavily pretreated patients with advanced stage disease. The association of clinical responses with immune related adverse events is perhaps not surprising given the mode of action of these therapies, but whether such adverse events are an inherent part of effective checkpoint blockade, or whether they can be dissociated remains an area for future study. These reactions have been organ-specific and no evidence of generalized systemic autoimmunity has been documented to date. Evidence for the antigen specificity of the clones causing toxicity is lacking but seems likely to differ from those of the clones mediating the therapeutic benefits. The majority resolves with systemic immune suppression without long-term sequelae, and antitumor responses do not appear to be compromised. This has led to current exploration of dose escalation as one potential way to improve response rates. While CTLA-4 blockade remains the archetypal example of these approaches, other immunological checkpoints offer further targets for intervention. Manipulation of any of these pathways will likely be most effective when combined with other therapeutic strategies, particularly as this may offer the best balance of benefits and toxicities. In addition, combined or sequential manipulation of multiple members of the costimulatory family may ultimately prove more effective in generation of sustained responses and immunological memory. It is anticipated that the results from preclinical studies of combinatorial approaches will help to inform the rational evolution of clinical strategies in the coming years.
References
- Acuto O, Michel F. CD28-mediated costimulation: A quantitative support for TCR signalling. Nat Rev Immunol. 2003;3:939–951. doi: 10.1038/nri1248. [DOI] [PubMed] [Google Scholar]
- Annunziato F, Cosmi L, Liotta F, Lazzeri E, Manetti R, Vanini V, Romagnani P, Maggi E, Romagnani S. Phenotype, localization, and mechanism of suppression of CD4(+) CD25(+) human thymocytes. J Exp Med. 2002;196:379–387. doi: 10.1084/jem.20020110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aramaki O, Shirasugi N, Takayama T, Shimazu M, Kitajima M, Ikeda Y, Azuma M, Okumura K, Yagita H, Niimi M. Programmed death-1-programmed death-L1 interaction is essential for induction of regulatory cells by intratracheal delivery of alloantigen. Transplantation. 2004;77:6–12. doi: 10.1097/01.TP.0000108637.65091.4B. [DOI] [PubMed] [Google Scholar]
- Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, Abrams J, Sznol M, Parkinson D, Hawkins M, Paradise C, Kunkel L, Rosenberg SA. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: Analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105–2116. doi: 10.1200/JCO.1999.17.7.2105. [DOI] [PubMed] [Google Scholar]
- Attia P, Phan GQ, Maker AV, Robinson MR, Quezado MM, Yang JC, Sherry RM, Topalian SL, Kammula US, Royal RE, Restifo NP, Haworth LR, et al. Autoimmunity correlates with tumor regression in patients with metastatic melanoma treated with anti-cytotoxic T-lymphocyte antigen-4. J Clin Oncol. 2005;23:6043–6053. doi: 10.1200/JCO.2005.06.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann MF, Kohler G, Ecabert B, Mak TW, Kopf M. Cutting edge: Lymphoproliferative disease in the absence of CTLA-4 is not T cell autonomous. J Immunol. 1999;163:1128–1131. [PubMed] [Google Scholar]
- Baecher-Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+CD25high regulatory cells in human peripheral blood. J Immunol. 2001;167:1245–1253. doi: 10.4049/jimmunol.167.3.1245. [DOI] [PubMed] [Google Scholar]
- Beck KE, Blansfield JA, Tran KQ, Hughes SM, Royal RE, Kammula US, Topalian SL, Sherry RM, Rosenberg SA, Yang JC. Enterocolitis in patients after antibody blockade of CTLA-4. J Immunother. 2005;28:647. [Google Scholar]
- Blair PJ, Riley JL, Levine BL, Lee KP, Craighead N, Francomano T, Perfetto SJ, Gray GS, Carreno BM, June CH. CTLA-4 ligation delivers a unique signal to resting human CD4 T cells that inhibits interleukin-2 secretion but allows Bcl-X(L) induction. J Immunol. 1998;160:12–15. [PubMed] [Google Scholar]
- Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, Gajewski TF. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004;64:1140–1145. doi: 10.1158/0008-5472.can-03-3259. [DOI] [PubMed] [Google Scholar]
- Blansfield JA, Beck KE, Tran K, Yang JC, Hughes MS, Kammula US, Royal RE, Topalian SL, Haworth LR, Levy C, Rosenberg SA, Sherry RM. Cytotoxic T-lymphocyte-associated antigen-4 blockage can induce autoimmune hypophysitis in patients with metastatic melanoma and renal cancer. J Immunother. 2005;28:593–598. doi: 10.1097/01.cji.0000178913.41256.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borriello F, Sethna MP, Boyd SD, Schweitzer AN, Tivol EA, Jacoby D, Strom TB, Simpson EM, Freeman GJ, Sharpe AH. B7-1 and B7-2 have overlapping, critical roles in immunoglobulin class switching and germinal center formation. Immunity. 1997;6:303–313. doi: 10.1016/s1074-7613(00)80333-7. [DOI] [PubMed] [Google Scholar]
- Brown JA, Dorfman DM, Ma FR, Sullivan EL, Munoz O, Wood CR, Greenfield EA, Freeman GJ. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol. 2003;170:1257–1266. doi: 10.4049/jimmunol.170.3.1257. [DOI] [PubMed] [Google Scholar]
- Brunner MC, Chambers CA, Chan FK, Hanke J, Winoto A, Allison JP. CTLA-4-Mediated inhibition of early events of T cell proliferation. J Immunol. 1999;162:5813–5820. [PubMed] [Google Scholar]
- Carreno BM, Bennett F, Chau TA, Ling V, Luxenberg D, Jussif J, Baroja ML, Madrenas J. CTLA-4 (CD152) can inhibit T cell activation by two different mechanisms depending on its level of cell surface expression. J Immunol. 2000;165:1352–1356. doi: 10.4049/jimmunol.165.3.1352. [DOI] [PubMed] [Google Scholar]
- Carter L, Fouser LA, Jussif J, Fitz L, Deng B, Wood CR, Collins M, Honjo T, Freeman GJ, Carreno BM. PD-1:PD-L inhibitory pathway affects both CD4 (+) and CD8(+) T cells and is overcome by IL-2. Eur J Immunol. 2002;32:634–643. doi: 10.1002/1521-4141(200203)32:3<634::AID-IMMU634>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Caton AJ, Cozzo C, Larkin J, III, Lerman MA, Boesteanu A, Jordan MS. CD4+ CD25+ regulatory T cell selection. Ann NY Acad Sci. 2004;1029:101–114. doi: 10.1196/annals.1309.028. [DOI] [PubMed] [Google Scholar]
- Chai JG, Tsang JY, Lechler R, Simpson E, Dyson J, Scott D. CD4+CD25+ T cells as immunoregulatory T cells in vitro. Eur J Immunol. 2002;32:2365–2375. doi: 10.1002/1521-4141(200208)32:8<2365::AID-IMMU2365>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Chambers CA, Kuhns MS, Allison JP. Cytotoxic T lymphocyte antigen-4 (CTLA-4) regulates primary and secondary peptide-specific CD4(+) T cell responses. Proc Natl Acad Sci USA. 1999;96:8603–8608. doi: 10.1073/pnas.96.15.8603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers CA, Kuhns MS, Egen JG, Allison JP. CTLA-4-mediated inhibition in regulation of T cell responses: Mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol. 2001;19:565–594. doi: 10.1146/annurev.immunol.19.1.565. [DOI] [PubMed] [Google Scholar]
- Chambers CA, Sullivan TJ, Allison JP. Lymphoproliferation in CTLA-4-deficient mice is mediated by costimulation-dependent activation of CD4+ T cells. Immunity. 1997;7:885–895. doi: 10.1016/s1074-7613(00)80406-9. [DOI] [PubMed] [Google Scholar]
- Chambers CA, Sullivan TJ, Truong T, Allison JP. Secondary but not primary Tcell responses are enhanced in CTLA-4-deficient CD8+ T cells. Eur J Immunol. 1998;28:3137–3143. doi: 10.1002/(SICI)1521-4141(199810)28:10<3137::AID-IMMU3137>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Chen L, Ashe S, Brady WA, Hellstrom I, Hellstrom KE, Ledbetter JA, McGowan P, Linsley PS. Costimulation of antitumor immunity by the B7 counterreceptor for the T lymphocyte molecules CD28 and CTLA-4. Cell. 1992;71:1093–1102. doi: 10.1016/s0092-8674(05)80059-5. [DOI] [PubMed] [Google Scholar]
- Chen L, McGowan P, Ashe S, Johnston J, Li Y, Hellstrom I, Hellstrom KE. Tumor immunogenicity determines the effect of B7 costimulation on T cell-mediated tumor immunity. J Exp Med. 1994;179:523–532. doi: 10.1084/jem.179.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikuma S, Abbas AK, Bluestone JA. B7-independent inhibition of T cells by CTLA-4. J Immunol. 2005;175:177–181. doi: 10.4049/jimmunol.175.1.177. [DOI] [PubMed] [Google Scholar]
- Choi IH, Zhu G, Sica GL, Strome SE, Cheville JC, Lau JS, Zhu Y, Flies DB, Tamada K, Chen L. Genomic organization and expression analysis of B7-H4, an immune inhibitory molecule of the B7 family. J Immunol. 2003;171:4650–4654. doi: 10.4049/jimmunol.171.9.4650. [DOI] [PubMed] [Google Scholar]
- Chuang E, Alegre ML, Duckett CS, Noel PJ, Vander Heiden MG, Thompson CB. Interaction of CTLA-4 with the clathrin-associated protein AP50 results in ligand-independent endocytosis that limits cell surface expression. J Immunol. 1997;159:144–151. [PubMed] [Google Scholar]
- Collins AV, Brodie DW, Gilbert RJ, Iaboni A, Manso-Sancho R, Walse B, Stuart DI, van der Merwe PA, Davis SJ. The interaction properties of costimulatory molecules revisited. Immunity. 2002;17:201–210. doi: 10.1016/s1074-7613(02)00362-x. [DOI] [PubMed] [Google Scholar]
- Compaan DM, Gonzalez LC, Tom I, Loyet KM, Eaton D, Hymowitz SG. Attenuating lymphocyte activity: The crystal structure of the BTLA-HVEM complex. J Biol Chem. 2005;280:39553–39561. doi: 10.1074/jbc.M507629200. [DOI] [PubMed] [Google Scholar]
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- Davila E, Kennedy R, Celis E. Generation of antitumor immunity by cytotoxic T lymphocyte epitope peptide vaccination, CpG-oligodeoxynucleotide adjuvant, and CTLA-4 blockade. Cancer Res. 2003;63:3281–3288. [PubMed] [Google Scholar]
- Davis TA, Tchekmedyian S, Korman A, Keler T, Deo Y, Small EJ. MDX-010 (human anti-CTLA4): A phase 1 trial in hormone refractory prostate carcinoma (HRPC) ASCO Annual Meeting. 2002;Abstract 74 [Google Scholar]
- Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, Formenti SC. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res. 2005;11:728–734. [PubMed] [Google Scholar]
- Diehn M, Alizadeh AA, Rando OJ, Liu CL, Stankunas K, Botstein D, Crabtree GR, Brown PO. Genomic expression programs and the integration of the CD28 costimulatory signal in T cell activation. Proc Natl Acad Sci USA. 2002;99:11796–11801. doi: 10.1073/pnas.092284399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Strome SE, Matteson EL, Moder KG, Flies DB, Zhu G, Tamura H, Driscoll CL, Chen L. Costimulating aberrant T cell responses by B7-H1 autoantibodies in rheumatoid arthritis. J Clin Invest. 2003;111:363–370. doi: 10.1172/JCI16015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, Lennon VA, Celis E, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- Dong H, Zhu G, Tamada K, Flies DB, van Deursen JM, Chen L. B7-H1 determines accumulation and deletion of intrahepatic CD8(+) T lymphocytes. Immunity. 2004;20:327–336. doi: 10.1016/s1074-7613(04)00050-0. [DOI] [PubMed] [Google Scholar]
- Doyle AM, Mullen AC, Villarino AV, Hutchins AS, High FA, Lee HW, Thompson CB, Reiner SL. Induction of cytotoxic T lymphocyte antigen 4 (CTLA-4) restricts clonal expansion of helper T cells. J Exp Med. 2001;194:893–902. doi: 10.1084/jem.194.7.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egen JG, Allison JP. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity. 2002;16:23–35. doi: 10.1016/s1074-7613(01)00259-x. [DOI] [PubMed] [Google Scholar]
- Fahlen L, Read S, Gorelik L, Hurst SD, Coffman RL, Flavell RA, Powrie F. T cells that cannot respond to TGF-{beta} escape control by CD4+CD25+ regulatory T cells. J Exp Med. 2005;201:737–746. doi: 10.1084/jem.20040685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, Belladonna ML, Fioretti MC, Alegre ML, Puccetti P. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol. 2003;4:1206–1212. doi: 10.1038/ni1003. [DOI] [PubMed] [Google Scholar]
- Fischkoff SA, Hersh E, Weber J, Powderly J, Khan K, Pavlick A, Samlowski W, O’Day S, Nichol G, Yellin M. Durable responses and long-term progression-free survival observed in a Phase II study of MDX-010 alone or in combination with dacarbazine (DTIC) in metastatic melanoma. ASCO Annual Meeting. 2005;Abstract 7525 [Google Scholar]
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, Horton HF, Fouser L, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: Contact-mediated suppression by CD4+CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol. 2005;174:1783–1786. doi: 10.4049/jimmunol.174.4.1783. [DOI] [PubMed] [Google Scholar]
- Greene JL, Leytze GM, Emswiler J, Peach R, Bajorath J, Cosand W, Linsley PS. Covalent dimerization of CD28/CTLA-4 and oligomerization of CD80/CD86 regulate T cell costimulatory interactions. J Biol Chem. 1996;271:26762–26771. doi: 10.1074/jbc.271.43.26762. [DOI] [PubMed] [Google Scholar]
- Greenwald RJ, Boussiotis VA, Lorsbach RB, Abbas AK, Sharpe AH. CTLA-4 regulates induction of anergy in vivo. Immunity. 2001;14:145–155. doi: 10.1016/s1074-7613(01)00097-8. [DOI] [PubMed] [Google Scholar]
- Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–548. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- Gregor PD, Wolchok JD, Ferrone CR, Buchinshky H, Guevara-Patino JA, Perales MA, Mortazavi F, Bacich D, Heston W, Latouche JB, Sadelain M, Allison JP, Scher HI, Houghton AN. CTLA-4 blockade in combination with xenogeneic DNA vaccines enhances T-cell responses, tumor immunity and autoimmunity to self antigens in animal and cellular model systems. Vaccine. 2004;22:1700–1708. doi: 10.1016/j.vaccine.2003.10.048. [DOI] [PubMed] [Google Scholar]
- Groux H, O’Garra A, Bigler M, Rouleau M, Antonenko S, de Vries JE, Roncarolo MG. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389:737–742. doi: 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- Han P, Goularte OD, Rufner K, Wilkinson B, Kaye J. An inhibitory Ig superfamily protein expressed by lymphocytes and APCs is also an early marker of thymocyte positive selection. J Immunol. 2004;172:5931–5939. doi: 10.4049/jimmunol.172.10.5931. [DOI] [PubMed] [Google Scholar]
- Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature. 1992;356:607–609. doi: 10.1038/356607a0. [DOI] [PubMed] [Google Scholar]
- Hathcock KS, Laszlo G, Pucillo C, Linsley P, Hodes RJ. Comparative analysis of B7–1 and B7–2 costimulatory ligands: Expression and function. J Exp Med. 1994;180:631–640. doi: 10.1084/jem.180.2.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichs CS, Palmer DC, Rosenberg SA, Restifo NP. Glucocorticoids do not inhibit antitumor activity of activated CD8+ T cells. J Immunother. 2005;28:517. doi: 10.1097/01.cji.0000177999.95831.7b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, Rietz C, Flies DB, Lau JS, Zhu G, Tamada K, Chen L. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65:1089–1096. [PubMed] [Google Scholar]
- Hodi FS, Mihm MC, Soiffer RJ, Haluska FG, Butler M, Seiden MV, Davis T, Henry-Spires R, MacRae S, Willman A, Padera R, Jaklitsch MT, et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated meta-static melanoma and ovarian carcinoma patients. Proc Natl Acad Sci USA. 2003;100:4712–4717. doi: 10.1073/pnas.0830997100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang AY, Bruce AT, Pardoll DM, Levitsky HI. Does B7–1 expression confer antigen-presenting cell capacity to tumors in vivo? J Exp Med. 1996;183:769–776. doi: 10.1084/jem.183.3.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurchla MA, Sedy JR, Gavrielli M, Drake CG, Murphy TL, Murphy KM. B and T lymphocyte attenuator exhibits structural and expression polymorphisms and is highly Induced in anergic CD4+ T cells. J Immunol. 2005;174:3377–3385. doi: 10.4049/jimmunol.174.6.3377. [DOI] [PubMed] [Google Scholar]
- Hurwitz AA, Foster BA, Kwon ED, Truong T, Choi EM, Greenberg NM, Burg MB, Allison JP. Combination immunotherapy of primary prostate cancer in a transgenic mouse model using CTLA-4 blockade. Cancer Res. 2000;60:2444–2448. [PubMed] [Google Scholar]
- Hurwitz AA, Sullivan TJ, Krummel MF, Sobel RA, Allison JP. Specific blockade of CTLA-4/B7 interactions results in exacerbated clinical and histologic disease in an actively-induced model of experimental allergic encephalomyelitis. J Neuroimmunol. 1997;73:57–62. doi: 10.1016/s0165-5728(96)00168-3. [DOI] [PubMed] [Google Scholar]
- Hurwitz AA, Yu TF, Leach DR, Allison JP. CTLA-4 blockade synergizes with tumor-derived granulocyte-macrophage colony-stimulating factor for treatment of an experimental mammary carcinoma. Proc Natl Acad Sci USA. 1998;95:10067–10071. doi: 10.1073/pnas.95.17.10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci USA. 2002;99:12293–12297. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai Y, Terawaki S, Honjo T. PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int Immunol. 2005;17:133–144. doi: 10.1093/intimm/dxh194. [DOI] [PubMed] [Google Scholar]
- Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, Naji A, Caton AJ. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol. 2001;2:301–306. doi: 10.1038/86302. [DOI] [PubMed] [Google Scholar]
- Karandikar NJ, Vanderlugt CL, Walunas TL, Miller SD, Bluestone JA. CTLA-4: A negative regulator of autoimmune disease. J Exp Med. 1996;184:783–788. doi: 10.1084/jem.184.2.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka H, Takahashi S, Takase K, Yamasaki S, Yokosuka T, Koike T, Saito T. CD25+CD4+ regulatory T cells exert in vitro suppressive activity independent of CTLA-4. Int Immunol. 2005;17:421–427. doi: 10.1093/intimm/dxh221. [DOI] [PubMed] [Google Scholar]
- Keler T, Halk E, Vitale L, O’Neill T, Blanset D, Lee S, Srinivasan M, Graziano RF, Davis T, Lonberg N, Korman A. Activity and safety of CTLA-4 blockade combined with vaccines in cynomolgus macaques. J Immunol. 2003;171:6251–6259. doi: 10.4049/jimmunol.171.11.6251. [DOI] [PubMed] [Google Scholar]
- Korman A, Yellin M, Keler T. Tumor immunotherapy: Preclinical and clinical activity of anti-CTLA4 antibodies. Curr Opin Invest Drugs. 2005;6:582–591. [PubMed] [Google Scholar]
- Krummel MF, Allison JP. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J Exp Med. 1996;183:2533–2540. doi: 10.1084/jem.183.6.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundig TM, Shahinian A, Kawai K, Mittrucker HW, Sebzda E, Bachmann MF, Mak TW, Ohashi PS. Duration of TCR stimulation determines costimulatory requirement of T cells. Immunity. 1996;5:41–52. doi: 10.1016/s1074-7613(00)80308-8. [DOI] [PubMed] [Google Scholar]
- Kwon ED, Hurwitz AA, Foster BA, Madias C, Feldhaus AL, Greenberg NM, Burg MB, Allison JP. Manipulation of T cell costimulatory and inhibitory signals for immunotherapy of prostate cancer. Proc Natl Acad Sci USA. 1997;94:8099–8103. doi: 10.1073/pnas.94.15.8099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R, Greenfield EA, Bourque K, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- Lenschow DJ, Bluestone JA. T cell co-stimulation and in vivo tolerance. Curr Opin Immunol. 1993;5:747–752. doi: 10.1016/0952-7915(93)90132-c. [DOI] [PubMed] [Google Scholar]
- Levings MK, Sangregorio R, Galbiati F, Squadrone S, de Waal MR, Roncarolo MG. IFN-alpha and IL-10 induce the differentiation of human type 1 T regulatory cells. J Immunol. 2001a;166:5530–5539. doi: 10.4049/jimmunol.166.9.5530. [DOI] [PubMed] [Google Scholar]
- Levings MK, Sangregorio R, Roncarolo MG. Human cd25(+)cd4(+) T regulatory cells suppress naive and memory T cell proliferation and can be expanded in vitro without loss of function. J Exp Med. 2001b;193:1295–1302. doi: 10.1084/jem.193.11.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsley PS, Brady W, Grosmaire L, Aruffo A, Damle NK, Ledbetter JA. Binding of the B cell activation antigen B7 to CD28 costimulates T cell proliferation and interleukin 2 mRNA accumulation. J Exp Med. 1991;173:721–730. doi: 10.1084/jem.173.3.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Gao JX, Wen J, Yin L, Li O, Zuo T, Gajewski TF, Fu YX, Zheng P, Liu Y. B7DC/PDL2 promotes tumor immunity by a PD-1-independent mechanism. J Exp Med. 2003;197:1721–1730. doi: 10.1084/jem.20022089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liyanage UK, Moore TT, Joo HG, Tanaka Y, Herrmann V, Doherty G, Drebin JA, Strasberg SM, Eberlein TJ, Goedegebuure PS, Linehan DC. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002;169:2756–2761. doi: 10.4049/jimmunol.169.5.2756. [DOI] [PubMed] [Google Scholar]
- Luhder F, Chambers C, Allison JP, Benoist C, Mathis D. Pinpointing when T cell costimulatory receptor CTLA-4 must be engaged to dampen diabetogenic T cells. Proc Natl Acad Sci USA. 2000;97:12204–12209. doi: 10.1073/pnas.200348397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhder F, Hoglund P, Allison JP, Benoist C, Mathis D. Cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) regulates the unfolding of autoimmune diabetes. J Exp Med. 1998;187:427–432. doi: 10.1084/jem.187.3.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Chapoval AI, Flies DB, Zhu G, Hirano F, Wang S, Lau JS, Dong H, Tamada K, Flies AS, Liu Y, Chen L. B7-H3 enhances tumor immunity in vivo by costimulating rapid clonal expansion of antigen-specific CD8+ cytolytic T cells. J Immunol. 2004;173:5445–5450. doi: 10.4049/jimmunol.173.9.5445. [DOI] [PubMed] [Google Scholar]
- Lurquin C, Lethe B, De Plaen E, Corbiere V, Theate I, van Baren N, Coulie PG, Boon T. Contrasting frequencies of antitumor and anti-vaccine T cells in metastases of a melanoma patient vaccinated with a MAGE tumor antigen. J Exp Med. 2005;201:249–257. doi: 10.1084/jem.20041378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maker AV, Attia P, Rosenberg SA. Analysis of the cellular mechanism of antitumor responses and autoimmunity in patients treated with CTLA-4 blockade. J Immunol. 2005a;175:7746–7754. doi: 10.4049/jimmunol.175.11.7746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maker AV, Phan GQ, Attia P, Yang JC, Sherry RM, Topalian SL, Kammula US, Royal RE, Haworth LR, Levy C, Kleiner D, Mavroukakis SA, Yellin M, Rosenberg SA. Tumor Regression and Autoimmunity in Patients Treated With Cytotoxic T Lymphocyte-Associated Antigen 4 Blockade and Interleukin 2: A Phase I/II Study. Ann Surg Oncol. 2005b;12:1005–1016. doi: 10.1245/ASO.2005.03.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelbrot DA, McAdam AJ, Sharpe AH. B7–1 or B7–2 is required to produce the lymphoproliferative phenotype in mice lacking cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) J Exp Med. 1999;189:435–440. doi: 10.1084/jem.189.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrogi AJ, Munshi A, Merogi AJ, Ohadike Y, El Habashi A, Marrogi OL, Freeman SM. Study of tumor infiltrating lymphocytes and transforming growth factor-beta as prognostic factors in breast carcinoma. Int J Cancer. 1997;74:492–501. doi: 10.1002/(sici)1097-0215(19971021)74:5<492::aid-ijc3>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Masteller EL, Chuang E, Mullen AC, Reiner SL, Thompson CB. Structural analysis of CTLA-4 function in vivo. J Immunol. 2000;164:5319–5327. doi: 10.4049/jimmunol.164.10.5319. [DOI] [PubMed] [Google Scholar]
- Mokyr MB, Kalinichenko T, Gorelik L, Bluestone JA. Realization of the therapeutic potential of CTLA-4 blockade in low-dose chemotherapy-treated tumor-bearing mice. Cancer Res. 1998;58:5301–5304. [PubMed] [Google Scholar]
- Naito Y, Saito K, Shiiba K, Ohuchi A, Saigenji K, Nagura H, Ohtani H. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 1998;58:3491–3494. [PubMed] [Google Scholar]
- Nakano O, Sato M, Naito Y, Suzuki K, Orikasa S, Aizawa M, Suzuki Y, Shintaku I, Nagura H, Ohtani H. Proliferative activity of intratumoral CD8(+) T-lymphocytes as a prognostic factor in human renal cell carcinoma: Clinicopathologic demonstration of antitumor immunity. Cancer Res. 2001;61:5132–5136. [PubMed] [Google Scholar]
- Nakaseko C, Miyatake S, Iida T, Hara S, Abe R, Ohno H, Saito Y, Saito T. Cytotoxic T lymphocyte antigen 4 (CTLA-4) engagement delivers an inhibitory signal through the membrane-proximal region in the absence of the tyrosine motif in the cytoplasmic tail. J Exp Med. 1999;190:765–774. doi: 10.1084/jem.190.6.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen LT, Radhakrishnan S, Ciric B, Tamada K, Shin T, Pardoll DM, Chen L, Rodriguez M, Pease LR. Cross-linking the B7 family molecule B7-DC directly activates immune functions of dendritic cells. J Exp Med. 2002;196:1393–1398. doi: 10.1084/jem.20021466. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–151. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N, Honjo T. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–322. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- Ohigashi Y, Sho M, Yamada Y, Tsurui Y, Hamada K, Ikeda N, Mizuno T, Yoriki R, Kashizuka H, Yane K, Tsushima F, Otsuki N, et al. Clinical significance of programmed death-1 ligand-1 and programmed death-1 ligand-2 expression in human esophageal cancer. Clin Cancer Res. 2005;11:2947–2953. doi: 10.1158/1078-0432.CCR-04-1469. [DOI] [PubMed] [Google Scholar]
- Paust S, Lu L, McCarty N, Cantor H. Engagement of B7 on effector T cells by regulatory T cells prevents autoimmune disease. Proc Natl Acad Sci USA. 2004;101:10398–10403. doi: 10.1073/pnas.0403342101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentcheva-Hoang T, Egen JG, Wojnoonski K, Allison JP. B7-1 and B7-2 selectively recruit CTLA-4 and CD28 to the immunological synapse. Immunity. 2004;21:401–413. doi: 10.1016/j.immuni.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Perrin PJ, Maldonado JH, Davis TA, June CH, Racke MK. CTLA-4 blockade enhances clinical disease and cytokine production during experimental allergic encephalomyelitis. J Immunol. 1996;157:1333–1336. [PubMed] [Google Scholar]
- Phan GQ, Touloukian CE, Yang JC, Restifo NP, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ, Seipp CA, Freezer LJ, Morton KE, Mavroukakis SA, et al. Immunization of patients with metastatic melanoma using both class I-and class II-restricted peptides from melanoma-associated antigens. J Immunother. 2003a;26:349–356. doi: 10.1097/00002371-200307000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan GQ, Yang JC, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ, Restifo NP, Haworth LR, Seipp CA, Freezer LJ, Morton KE, Mavroukakis SA, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci USA. 2003b;100:8372–8377. doi: 10.1073/pnas.1533209100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad DV, Richards S, Mai XM, Dong C. B7S1, a novel B7 family member that negatively regulates T cell activation. Immunity. 2003;18:863–873. doi: 10.1016/s1074-7613(03)00147-x. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan S, Nguyen LT, Ciric B, Flies D, Van Keulen VP, Tamada K, Chen L, Rodriguez M, Pease LR. Immunotherapeutic potential of B7-DC (PD-L2) cross-linking antibody in conferring antitumor immunity. Cancer Res. 2004;64:4965–4972. doi: 10.1158/0008-5472.CAN-03-3025. [DOI] [PubMed] [Google Scholar]
- Read S, Malmstrom V, Powrie F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J Exp Med. 2000;192:295–302. doi: 10.1084/jem.192.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuben JM, Lee BN, Shen DY, Gutierrez C, Hernandez I, Parker CA, Bozon VA, Gomez-Navarro J, Lopez-Berestein G, Camacho LH. Therapy with human monoclonal anti-CTLA-4 antibody, CP-675, 206, reduces regulatory T cells and IL-10 production in patients with advanced malignant melanoma (MM) ASCO Annual Meeting. 2005;Abstract 7505 [Google Scholar]
- Ribas A, Glaspy JA, Lee Y, Dissette VB, Seja E, Vu HT, Tchekmedylan NS, Oseguera D, Comin-Anduix B, Wargo JA, Amornani SN, McBride WH, et al. Role of dendritic cell phenotype, determine spreading, and negative costimulatory blockade in dendritic cell-based melanoma immunotherapy. J Immunother. 2004;27:354–367. doi: 10.1097/00002371-200409000-00004. [DOI] [PubMed] [Google Scholar]
- Ribas A, Bozon VA, Lopez-Berestein G, Pavlov D, Reuben JM, Parker CA, Seja E, Glaspy JA, Gomez-Navarro J, Camacho LH. Phase 1 Trial of Monthly Doses of the Human Anti-CTLA4 Monoclonal Antibody CP-675, 206 in Patients with Advanced Melanoma. ASCO Annual Meeting. 2005b;Abstract 7524 [Google Scholar]
- Ribas A, Camacho LH, Lopez-Berestein G, Pavlov D, Bulanhagui CA, Millham R, Comin-Anduix B, Reuben JM, Seja E, Parker CA, Sharma A, Glaspy JA, et al. Antitumor activity in melanoma and anti-self responses in a phase I trial with the anti-cytotoxic T lymphocyte-associated antigen 4 monoclonal antibody CP-675, 206. J Clin Oncol. 2005a;23:8968–8977. doi: 10.1200/JCO.2005.01.109. [DOI] [PubMed] [Google Scholar]
- Riley JL, Mao M, Kobayashi S, Biery M, Burchard J, Cavet G, Gregson BP, June CH, Linsley PS. Modulation of TCR-induced transcriptional profiles by ligation of CD28, ICOS, and CTLA-4 receptors. Proc Natl Acad Sci USA. 2002;99:11790–11795. doi: 10.1073/pnas.162359999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MR, Chan CC, Yang JC, Rubin BI, Gracia GJ, Sen HN, Csaky KG, Rosenberg SA. Cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma: A new cause of uveitis. J Immunother. 2004;27:478–479. doi: 10.1097/00002371-200411000-00008. [DOI] [PubMed] [Google Scholar]
- Rosenberg SA. Interleukin-2 and the development of immunotherapy for the treatment of patients with cancer. Cancer J Sci Am. 2000;6(Suppl 1):S2–S7. [PubMed] [Google Scholar]
- Rosenwald A, Wright G, Leroy K, Yu X, Gaulard P, Gascoyne RD, Chan WC, Zhao T, Haioun C, Greiner TC, Weisenburger DD, Lynch JC, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med. 2003;198:851–862. doi: 10.1084/jem.20031074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salceda S, Tang T, Kmet M, Munteanu A, Ghosh M, Macina R, Liu W, Pilkington G, Papkoff J. The immunomodulatory protein B7-H4 is overexpressed in breast and ovarian cancers and promotes epithelial cell transformation. Exp Cell Res. 2005;306:128–141. doi: 10.1016/j.yexcr.2005.01.018. [DOI] [PubMed] [Google Scholar]
- Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- Sanderson K, Scotland R, Lee P, Liu D, Groshen S, Snively J, Sian S, Nichol G, Davis T, Keler T, Yellin M, Weber J. Autoimmunity in a phase I trial of a fully human anti-cytotoxic T-lymphocyte antigen-4 monoclonal antibody with multiple melanoma peptides and Montanide ISA 51 for patients with resected stages III and IV melanoma. J Clin Oncol. 2005;23:741–750. doi: 10.1200/JCO.2005.01.128. [DOI] [PubMed] [Google Scholar]
- Schwartz JC, Zhang X, Fedorov AA, Nathenson SG, Almo SC. Structural basis for co-stimulation by the human CTLA-4/B7–2 complex. Nature. 2001;410:604–608. doi: 10.1038/35069112. [DOI] [PubMed] [Google Scholar]
- Sedy JR, Gavrieli M, Potter KG, Hurchla MA, Lindsley RC, Hildner K, Scheu S, Pfeffer K, Ware CF, Murphy TL, Murphy KM. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol. 2005;6:90–98. doi: 10.1038/ni1144. [DOI] [PubMed] [Google Scholar]
- Shahinian A, Pfeffer K, Lee KP, Kundig TM, Kishihara K, Wakeham A, Kawai K, Ohashi PS, Thompson CB, Mak TW. Differential T cell costimulatory requirements in CD28-deficient mice. Science. 1993;261:609–612. doi: 10.1126/science.7688139. [DOI] [PubMed] [Google Scholar]
- Shin T, Kennedy G, Gorski K, Tsuchiya H, Koseki H, Azuma M, Yagita H, Chen L, Powell J, Pardoll D, Housseau F. Cooperative B7–1/2 (CD80/CD86) and B7-DC costimulation of CD4+ T cells independent of the PD-1 receptor. J Exp Med. 2003;198:31–38. doi: 10.1084/jem.20030242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiratori T, Miyatake S, Ohno H, Nakaseko C, Isono K, Bonifacino JS, Saito T. Tyrosine phosphorylation controls internalization of CTLA-4 by regulating its interaction with clathrin-associated adaptor complex AP-2. Immunity. 1997;6:583–589. doi: 10.1016/s1074-7613(00)80346-5. [DOI] [PubMed] [Google Scholar]
- Shrikant P, Khoruts A, Mescher MF. CTLA-4 blockade reverses CD8+ T cell tolerance to tumor by a CD4+ T cell-and IL-2-dependent mechanism. Immunity. 1999;11:483–493. doi: 10.1016/s1074-7613(00)80123-5. [DOI] [PubMed] [Google Scholar]
- Sica GL, Choi IH, Zhu G, Tamada K, Wang SD, Tamura H, Chapoval AI, Flies DB, Bajorath J, Chen L. B7-H4, a molecule of the B7 family, negatively regulates T cell immunity. Immunity. 2003;18:849–861. doi: 10.1016/s1074-7613(03)00152-3. [DOI] [PubMed] [Google Scholar]
- Sotomayor EM, Borrello I, Tubb E, Allison JP, Levitsky HI. In vivo blockade of CTLA-4 enhances the priming of responsive T cells but fails to prevent the induction of tumor antigen-specific tolerance. Proc Natl Acad Sci USA. 1999;96:11476–11481. doi: 10.1073/pnas.96.20.11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamper CC, Zhang Y, Tobin JF, Erbe DV, Ikemizu S, Davis SJ, Stahl ML, Seehra J, Somers WS, Mosyak L. Crystal structure of the B7–1/CTLA-4 complex that inhibits human immune responses. Nature. 2001;410:608–611. doi: 10.1038/35069118. [DOI] [PubMed] [Google Scholar]
- Strome SE, Dong H, Tamura H, Voss SG, Flies DB, Tamada K, Salomao D, Cheville J, Hirano F, Lin W, Kasperbauer JL, Ballman KV, Chen L. B7-H1 blockade augments adoptive T-cell immunotherapy for squamous cell carcinoma. Cancer Res. 2003;63:6501–6505. [PubMed] [Google Scholar]
- Suh WK, Gajewska BU, Okada H, Gronski MA, Bertram EM, Dawicki W, Duncan GS, Bukczynski J, Plyte S, Elia A, Wakeham A, Itie A, et al. The B7 family member B7-H3 preferentially down-regulates T helper type 1-mediated immune responses. Nat Immunol. 2003;4:899–906. doi: 10.1038/ni967. [DOI] [PubMed] [Google Scholar]
- Sun M, Richards S, Prasad DV, Mai XM, Rudensky A, Dong C. Characterization of mouse and human B7-H3 genes. J Immunol. 2002;168:6294–6297. doi: 10.4049/jimmunol.168.12.6294. [DOI] [PubMed] [Google Scholar]
- Sutmuller RP, van Duivenvoorde LM, van Elsas A, Schumacher TN, Wildenberg ME, Allison JP, Toes RE, Offringa R, Melief CJ. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25(+) regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J Exp Med. 2001;194:823–832. doi: 10.1084/jem.194.6.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Kataoka H, Hara S, Yokosuka T, Takase K, Yamasaki S, Kobayashi W, Saito Y, Saito T. In vivo overexpression of CTLA-4 suppresses lymphoproliferative diseases and thymic negative selection. Eur J Immunol. 2005;35:399–407. doi: 10.1002/eji.200324746. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Tagami T, Yamazaki S, Uede T, Shimizu J, Sakaguchi N, Mak TW, Sakaguchi S. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303–310. doi: 10.1084/jem.192.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura H, Dong H, Zhu G, Sica GL, Flies DB, Tamada K, Chen L. B7-H1 costimulation preferentially enhances CD28-independent T-helper cell function. Blood. 2001;97:1809–1816. doi: 10.1182/blood.v97.6.1809. [DOI] [PubMed] [Google Scholar]
- Tang Q, Boden EK, Henriksen KJ, Bour-Jordan H, Bi M, Bluestone JA. Distinct roles of CTLA-4 and TGF-beta in CD4+CD25+ regulatory T cell function. Eur J Immunol. 2004;34:2996–3005. doi: 10.1002/eji.200425143. [DOI] [PubMed] [Google Scholar]
- Taylor PA, Lees CJ, Fournier S, Allison JP, Sharpe AH, Blazar BR. B7 expression on T cells down-regulates immune responses through CTLA-4 ligation via T-T interactions. J Immunol. 2004;172:34–39. doi: 10.4049/jimmunol.172.1.34. [DOI] [PubMed] [Google Scholar]
- Thompson RH, Gillett MD, Cheville JC, Lohse CM, Dong H, Webster WS, Krejci KG, Lobo JR, Sengupta S, Chen L, Zincke H, Blute, et al. Costimulatory B7-H1 in renal cell carcinoma patients: Indicator of tumor aggressiveness and potential therapeutic target. Proc Natl Acad Sci USA. 2004;101:17174–17179. doi: 10.1073/pnas.0406351101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. 1998;188:287–296. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton AM, Shevach EM. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol. 2000;164:183–190. doi: 10.4049/jimmunol.164.1.183. [DOI] [PubMed] [Google Scholar]
- Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- Townsend SE, Allison JP. Tumor rejection after direct costimulation of CD8+ T cells by B7-transfected melanoma cells. Science. 1993;259:368–370. doi: 10.1126/science.7678351. [DOI] [PubMed] [Google Scholar]
- Tringler B, Zhuo S, Pilkington G, Torkko KC, Singh M, Lucia MS, Heinz DE, Papkoff J, Shroyer KR. B7-h4 is highly expressed in ductal and lobular breast cancer. Clin Cancer Res. 2005;11:1842–1848. doi: 10.1158/1078-0432.CCR-04-1658. [DOI] [PubMed] [Google Scholar]
- Tseng SY, Otsuji M, Gorski K, Huang X, Slansky JE, Pai SI, Shalabi A, Shin T, Pardoll DM, Tsuchiya H. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med. 2001;193:839–846. doi: 10.1084/jem.193.7.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Merwe PA, Davis SJ. Molecular interactions mediating T cell antigen recognition. Annu Rev Immunol. 2003;21:659–684. doi: 10.1146/annurev.immunol.21.120601.141036. [DOI] [PubMed] [Google Scholar]
- van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. 1999;190:355–366. doi: 10.1084/jem.190.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanasek TL, Khoruts A, Zell T, Mueller DL. Antagonistic roles for CTLA-4 and the mammalian target of rapamycin in the regulation of clonal anergy: Enhanced cell cycle progression promotes recall antigen responsiveness. J Immunol. 2001;167:5636–5644. doi: 10.4049/jimmunol.167.10.5636. [DOI] [PubMed] [Google Scholar]
- Vesalainen S, Lipponen P, Talja M, Syrjanen K. Histological grade, perineural infiltration, tumour-infiltrating lymphocytes and apoptosis as determinants of long-term prognosis in prostatic adenocarcinoma. Eur J Cancer. 1994;30A:1797–1803. doi: 10.1016/0959-8049(94)e0159-2. [DOI] [PubMed] [Google Scholar]
- Viguier M, Lemaitre F, Verola O, Cho MS, Gorochov G, Dubertret L, Bachelez H, Kourilsky P, Ferradini L. Foxp3 expressing CD4+CD25(high) regulatory T cells are overrepresented in human metastatic melanoma lymph nodes and inhibit the function of infiltrating T cells. J Immunol. 2004;173:1444–1453. doi: 10.4049/jimmunol.173.2.1444. [DOI] [PubMed] [Google Scholar]
- Vijayakrishnan L, Slavik JM, Illes Z, Greenwald RJ, Rainbow D, Greve B, Peterson LB, Hafler DA, Freeman GJ, Sharpe AH, Wicker LS, Kuchroo VK. An autoimmune disease-associated CTLA-4 splice variant lacking the B7 binding domain signals negatively in T cells. Immunity. 2004;20:563–575. doi: 10.1016/s1074-7613(04)00110-4. [DOI] [PubMed] [Google Scholar]
- Viola A, Lanzavecchia A. T cell activation determined by T cell receptor number and tunable thresholds. Science. 1996;273:104–106. doi: 10.1126/science.273.5271.104. [DOI] [PubMed] [Google Scholar]
- Wang HB, Shi FD, Li H, Chambers BJ, Link H, Ljunggren HG. Anti-CTLA-4 antibody treatment triggers determinant spreading and enhances murine myasthenia gravis. J Immunol. 2001;166:6430–6436. doi: 10.4049/jimmunol.166.10.6430. [DOI] [PubMed] [Google Scholar]
- Wang S, Bajorath J, Flies DB, Dong H, Honjo T, Chen L. Molecular modeling and functional mapping of B7-H1 and B7-DC uncouple costimulatory function from PD-1 interaction. J Exp Med. 2003;197:1083–1091. doi: 10.1084/jem.20021752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, Hurchla MA, Zimmerman N, Sim J, Zang X, Murphy TL, Russell JH, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol. 2003;4:670–679. doi: 10.1038/ni944. [DOI] [PubMed] [Google Scholar]
- Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW. Lymphoproliferative disorders with early lethality in mice deficient in CTLA-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- Weiner HL. Induction and mechanism of action of transforming growth factor-beta-secreting Th3 regulatory cells. Immunol Rev. 2001;182:207–214. doi: 10.1034/j.1600-065x.2001.1820117.x. [DOI] [PubMed] [Google Scholar]
- Woo EY, Chu CS, Goletz TJ, Schlienger K, Yeh H, Coukos G, Rubin SC, Kaiser LR, June CH. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 2001;61:4766–4772. [PubMed] [Google Scholar]
- Woo EY, Yeh H, Chu CS, Schlienger K, Carroll RG, Riley JL, Kaiser LR, June CH. Cutting edge: Regulatory T cells from lung cancer patients directly inhibit autologous T cell proliferation. J Immunol. 2002;168:4272–4276. doi: 10.4049/jimmunol.168.9.4272. [DOI] [PubMed] [Google Scholar]
- Yang JC, Beck KE, Blansfield JA, Tran K, Lowy I, Rosenberg SA. Tumor regression in patients with metastatic renal cancer treated with a monoclonal antibody to CTLA4 (MDX-010) ASCO Annual Meeting. 2005;Abstract 2501 [Google Scholar]
- Yang YF, Zou JP, Mu J, Wijesuriya R, Ono S, Walunas T, Bluestone J, Fujiwara H, Hamaoka T. Enhanced induction of antitumor T-cell responses by cytotoxic T lymphocyte-associated molecule-4 blockade: The effect is manifested only at the restricted tumor-bearing stages. Cancer Res. 1997;57:4036–4041. [PubMed] [Google Scholar]
- Youngnak P, Kozono Y, Kozono H, Iwai H, Otsuki N, Jin H, Omura K, Yagita H, Pardoll DM, Chen L, Azuma M. Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem Biophys Res Commun. 2003;307:672–677. doi: 10.1016/s0006-291x(03)01257-9. [DOI] [PubMed] [Google Scholar]
- Zang X, Loke P, Kim J, Murphy K, Waitz R, Allison JP. B7x: A widely expressed B7 family member that inhibits T cell activation. Proc Natl Acad Sci USA. 2003;100:10388–10392. doi: 10.1073/pnas.1434299100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN, Rubin SC, Coukos G. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348:203–213. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- Zhang X, Schwartz JC, Almo SC, Nathenson SG. Crystal structure of the receptor-binding domain of human B7–2: Insights into organization and signaling. Proc Natl Acad Sci USA. 2003;100:2586–2591. doi: 10.1073/pnas.252771499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Schwartz JC, Guo X, Bhatia S, Cao E, Lorenz M, Cammer M, Chen L, Zhang ZY, Edidin MA, Nathenson SG, Almo SC. Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity. 2004;20:337–347. doi: 10.1016/s1074-7613(04)00051-2. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Allison JP. Interaction of CTLA-4 with AP50, a clathrin-coated pit adaptor protein. Proc Natl Acad Sci USA. 1997;94:9273–9278. doi: 10.1073/pnas.94.17.9273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Drake CG, Levitsky HI. Amplification of tumor-specific regulatory T cells following therapeutic cancer vaccines. Blood. 2005;107:628–636. doi: 10.1182/blood-2005-07-2737. [DOI] [PMC free article] [PubMed] [Google Scholar]