Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection (original) (raw)
Abstract
T cell expansion and memory formation are generally more effective when elicited by live organisms than by inactivated vaccines. Elucidation of the underlying mechanisms is important for vaccination and therapeutic strategies. We show that the massive expansion of antigen-specific CD8 T cells that occurs in response to viral infection is critically dependent on the direct action of type I interferons (IFN-Is) on CD8 T cells. By examining the response to infection with lymphocytic choriomeningitis virus using IFN-I receptor–deficient (IFN-IR0) and –sufficient CD8 T cells adoptively transferred into normal IFN-IR wild-type hosts, we show that the lack of direct CD8 T cell contact with IFN-I causes >99% reduction in their capacity to expand and generate memory cells. The diminished expansion of IFN-IR0 CD8 T cells was not caused by a defect in proliferation but by poor survival during the antigen-driven proliferation phase. Thus, IFN-IR signaling in CD8 T cells is critical for the generation of effector and memory cells in response to viral infection.
Antigen-specific CD8 T cells can undergo massive clonal expansion during the acute phase of viral infections. For example, in mice infected with lymphocytic choriomeningitis virus (LCMV), antigen-specific CD8 T cells expand nearly 10,000-fold during week 1 of infection (1). Robust levels of CD8 T cell expansion have been demonstrated in response to several other acute viral and intracellular bacterial infections in both mice and humans (2). The T cell expansion that occurs during the acute phase of infection serves to generate a large number of effector cells that contribute to the clearance of the pathogen and to memory cells that confer increased levels of protection on reexposure. A majority of the effector T cells generated during the acute phase die, but ∼5–10% of these cells differentiate into long-lived memory cells. Despite the death of the majority of the effector cells, the size of the resulting memory T cell pool is substantially higher than the size of the naive precursor pool because of the clonal expansion that takes place during the antigen-driven proliferation phase.
Typical infections with live pathogens provide high initial levels of antigen and lead to activation of APCs via pattern recognition receptors, resulting in the production of inflammatory cytokines that in turn cause efficient activation of APCs. Production of inflammatory cytokines is complex and is dependent on the type of infectious agent, the host, and their reciprocal interaction (3, 4). Type I IFNs (IFN-Is), unlike type II IFN (IFN-γ), are a multigene family of antiviral cytokines produced in high amounts early after viral infection (5). In mice, IFN-Is constitute 13 IFN-Iα subtypes, one IFN-β, and a recently discovered cytokine called limitin. All the IFN-I species are known to act via at least one known heterodimeric receptor complex that is composed of two distinct chains, IFNAR1 and IFNAR2, encoded by separate genes. Although some evidence suggested that distinct IFN-I subtypes might use different IFN-IAR subunits (5), both IFNAR1 and IFNAR2 chains are required to mediate the antiviral functions of IFN-I. All infected cells are capable of making IFN-I, but there are some specialized cell types called plasmacytoid DCs that are equipped with the capacity to produce massive amounts of IFN-I under certain conditions.
In addition to directly interfering with viral replication, IFN-I contributes to adaptive immune responses. IFN-Is are known to induce activation of APCs, notably DCs (6–9). IFN-I also promotes Th1 cytokine production, effector differentiation, proliferation, and survival of activated T cells, at least in vitro (10–15). Mice deficient in the functional IFN-I receptor (IFN-IR0 mice) were previously generated by genetic disruption of IFNAR1 (16). The IFN-IR0 mice are highly susceptible to viral infections because of the absence of the direct viral control effects of IFN-I and show modest levels of CD8 T cell responses to viral infections, and most of the expanding CD8 T cells undergo functional exhaustion or physical deletion (3, 16–17). Whether the effects of IFN-I in vivo on antigen-specific CD8 T cell responses are caused in part by a direct action on the T cells is difficult to address because the IFN-IR0 mice also have aberrant innate immune responses (3, 8), deregulated regulatory cell functions, and control viral infections poorly (16–18). We have addressed this limitation by examining the response of IFN-IR0 and WT CD8 T cells to LCMV infection after adoptive transfer into WT hosts. We find that (a) the expansion of the antigen-specific CD8 T cells in response to LCMV is dependent on IFN-IR expression on these cells, (b) IFN-Is mediate the sustained expansion of CD8 T cells by extending their survival during the antigen-driven proliferation phase, and (c) the IFN-I–mediated survival of effector CD8 T cells during the antigen-driven proliferation phase is critically important for generation of optimal numbers of memory cells.
Results
Direct action of IFN-I on antigen-specific CD8 T cells is critical for their expansion after viral infection
To directly address the role of IFN-IR in responding CD8 T cells, we adoptively transferred WT versus IFN-IR0 TCR transgenic CD8 T cells into WT mice and examined their responses to LCMV infection. IFN-IR0 LCMV GP33-41 epitope–specific P14 TCR transgenic CD8 T cells were generated by backcrossing 129/SvEv IFN-IR0 mice (16) to B6 mice 14 times, followed by intercrossing to P14 TCR transgenic mice (19), which were already backcrossed onto the B6 background (20). We first ensured that the IFN-IR0 P14 CD8 T cells were similar in their phenotype and function to those of WT P14 CD8 T cells. Both were positive for MHC tetramer Db complexed with GP33-41 peptide, and both were CD44low and L-selectinhigh (Fig. 1 A). Likewise, their functional properties were similar; neither population produced detectable levels of IFN-γ after 5 h of stimulation in vitro with GP33-41 peptide, and a fraction of both populations produced equivalently low levels of TNF-α after peptide stimulation (Fig. 1 B). In addition, both WT and IFN-IR0 P14 CD8 T cells were able to undergo several rounds of proliferation after 3-d in vitro cultures in the presence of peptide (Fig. 1 C). Thus, the IFN-IR0 P14 CD8 T cells were not intrinsically defective in their in vitro response.
Figure 1.

IFN-IR0 P14 CD8 T cells respond normally in vitro. (A) Phenotypic analysis of spleen cells derived from WT P14 TCR transgenic mice and IFN-IR0 P14 TCR transgenic mice. Numbers represent the percentage of cells in the indicated quadrant. (B) Spleen cells derived from WT and IFN-IR0 P14 TCR transgenic mice were cultured in vitro for 5 h either in the absence or presence of GP33-41 peptide and stained for cytokines. Data are gated on CD8 T cells. Numbers represent the percentage of cells in the indicated quadrant. (C) CFSE-labeled WT and IFN-IR0 P14 spleen cells cultured for 3 d with (bottom) and without (top) peptide stimulus. Data are gated on CD8 T cells.
To measure response in vivo, purified WT (Ly5.2) and IFN-IR0 (Ly5.2) P14 CD8 T cells from littermate mice were transferred (20,000 cells/mouse) separately into naive C57BL/6 Ly5.1 WT mice. Spleen cell analysis after infection showed that, as expected, the WT donor cells underwent massive clonal expansion by day 5 and continued to expand until day 8 (Fig. 2, A and B, top). In marked contrast to the ∼2,500-fold expansion exhibited by the WT donor cells, the IFN-IR0 donor cells displayed remarkably (>99%) diminished expansion (Fig. 2, A and B, bottom). The endogenous polyclonal (Ly5.1+ve) CD8 T cell response to GP33-41 epitope (Fig. 2 A, left quadrants) and the endogenous CD8 and CD4 T cell responses to other known epitopes of LCMV (not depicted) were similar in both the groups and led to clearance of the virus by day 7 after infection. However, there were marginal differences in the viral titer on day 2 after infection (0.9 ± 0.75 × 107 and 2.8 ± 1.3 × 107 PFU/g of spleen in WT P14 CD8 T cell recipients and IFN-I R0 P14 CD8 T cell recipients, respectively).
Figure 2.

Action of IFN-I on virus-specific CD8 T cells is critical for their clonal expansion. (A) 20,000 purified WT (Ly5.2) or IFN-IR0 (Ly 5.2) P14 CD8 T cells were transferred into individual C57BL/6 (Ly5.1) mice that were infected with LCMV strain Armstrong the next day. Donor cells were analyzed on gated CD8+ cells. Data are representative of at least five mice at each time point from three separate experiments. Numbers represent the percentage of cells in the indicated quadrant. (B) Number of donor cells recovered per spleen on different days after infection. Each point represents the mean ± SD of at least five mice pooled from three experiments. The level of detection was 104 cells/spleen.
Comparison of WT P14 and IFN-IR0 P14 CD8 T cell responses in the same host
To address whether differences in antigenic load or other environmental factors contributed to the observed effect on expansion, WT and IFN-IR0 P14 CD8 T cells bearing different congenic markers (WT = Thy1.2, Ly5.1 and IFN-IR0 = Thy1.2, Ly5.2) were cotransferred into the same host (Thy1.1, C57BL/6) in equal numbers and analyzed after infection (Fig. 3 A). We observed similar differences in expansion in this setting. This finding indicated that differences in the host environment were not responsible for the diminished expansion of IFN-IR0 cells.
Figure 3.

IFN-I–unresponsive CD8 T cells exhibit highly diminished expansion compared with IFN-I–responsive CD8 T cells of identical specificity in the same infected host. 60,000 each of WT (Thy1.2, Ly5.1) plus IFN-IR0 (Thy1.2, Ly5.2) P14 CD8 T cells were cotransferred into a congenic host (Thy1.1, B6 Ly5.2) and infected with LCMV the next day. Donor cells were analyzed on gated CD8+ cells in the spleen (A) and in other compartments (B) on day 8 after infection. Data are representative of two experiments with four mice in each experiment. Numbers represent the percentage of cells in the indicated quadrant.
Reduced expansion of IFN-IR0 P14 CD8 T cells was not caused by preferential sequestration in other tissues
We next addressed whether the diminished expansion observed in the spleen was caused by the preferential sequestration of IFN-IR0 P14 CD8 T cells in another compartment. Fig. 3 B shows that the expansion of IFN-IR0 P14 donor CD8 T cells was also diminished in several other lymphoid and extralymphoid compartments, indicating that the reduced expansion observed in the spleen was not caused by preferential sequestration in other tissues.
Effector cells derived from IFN-IR0 P14 donor CD8 T cells have diminished levels of granzyme B but similar IFN-γ and TNF-α responses after peptide stimulation
Despite poor expansion, the small number of IFN-IR0 P14 donor CD8 T cells recovered after LCMV infection proved capable of producing IFN-γ and TNF-α at levels and frequencies similar to WT donor cells on day 7 after infection (Fig. 4 A). However, the frequency of cells expressing granzyme B directly ex vivo was reduced in IFN-IR0 P14 effector CD8 T cells compared with the WT P14 effector CD8 cells (Fig. 4, B and C). Neither WT nor IFN-IR0 effector CD8 T cells produced the Th2 cytokines IL-4 or IL-10 upon peptide stimulation in vitro (unpublished data). Thus, the direct action of IFN-I on virus-specific CD8 T cells is not only important for their expansion but also has some role in the expression of the cytotoxic effector function–associated molecule granzyme B.
Figure 4.

IFN-I action on virus-specific CD8 T cells affects granzyme B expression but not cytokine effector functions. (A) Spleen cells from the mice indicated in Fig. 3 were stimulated in vitro for 5 h with LCMV GP33-41 peptide in the presence of brefeldin A. IFN-γ or TNF-α production was determined by intracellular cytokine staining in gated donor (Thy1.2+ CD8+) T cells. Ly5.2+ cells are IFN-IR0. Ly5.2− cells were confirmed to be WT donors by positive Ly5.1 staining done separately. Numbers represent the percentage of cells in the indicated quadrant. (B) Granzyme B staining was done ex vivo. These experiments were repeated at least three times with similar results. Numbers represent the percentage of cells in the indicated quadrant. (C) Mean values of percent donor cells expressing granzyme B in three different experiments comprising 11 mice at day 8 and 9 mice at day 6 after infection. Values shown are the mean ± SD.
Contraction, memory generation, and maintenance of IFN-I–unresponsive effector cells
After peak expansion at day 8, the expanded WT effector CD8 T cells, as expected, underwent marked contraction (∼10-fold) and those that survived differentiated into long-lasting memory cells (Fig. 5 A, open circles). In contrast, the small number of IFN-IR0 effector P14 CD8 T cells that were present by day 8 generated a memory pool that was ∼100-fold smaller in size compared with the memory pool generated from WT effector P14 CD8 T cells. The memory cells generated from WT and IFN-IR0 P14 CD8 T cells were both stably maintained for an extended period, suggesting that the action of IFN-I on antigen-specific CD8 T cells is critical during the antigen-driven proliferation/clonal expansion phase but is not necessarily required to maintain resting memory cells. Both WT and IFN-IR0 memory P14 CD8 T cells were capable of producing IFN-γ after in vitro stimulation with cognate peptide, indicating that the cytokine effector function of the IFN-IR0 memory cells was not compromised (Fig. 5 B). Thus, IFN-I action on virus-specific CD8 T cells is critically required for their expansion during the antigen-driven proliferation phase, and the IFN-I–mediated expansion has an important role in the generation of optimal numbers of memory cells.
Figure 5.

IFN-I–unresponsive effector CD8 T cells, although generated in low numbers, differentiate into memory cells that maintain stably thereafter. (A) 20,000 purified WT (Ly5.2) or IFN-IR0 (Ly 5.2) P14 CD8 T cells were transferred into individual C57BL/6 (Ly5.1) mice that were infected with LCMV the next day. The number of donor cells recovered per spleen on different days after infection was calculated by Ly 5.2 and tetramer staining. The level of detection was <104 cells/spleen. Each point represents an individual mouse. The average recovery of WT donor CD8 T cells was 1.10 ± 0.07 × 107 and 0.92 ± 0.2 × 106 at days 8 and 15, respectively, whereas the recovery of IFN-IR0 donor cells was 3.20 ± 0.4 × 104 and 1.70 ± 0.31 × 104 at days 8 and 15, respectively. (B) Spleen cells from 200 d after LCMV infection were stimulated in vitro for 5 h with LCMV GP33-41 peptide in the presence of brefeldin A. IFN-γ or TNF-α production was determined by intracellular cytokine staining, and the data are presented on gated CD8+ T cells. Numbers represent the percentage of cells in the indicated quadrant. Ly5.2−, recipient CD8 T cells; Ly5.2+, donor WT or IFN-IR0 P14 CD8 T cells.
The diminished expansion of IFN-IR0 CD8 T cells was not related to clonal specificity or differences in precursor frequency, virus-mediated cell death, or rejection
It is surprising that the IFN-IR0 P14 CD8 T cells exhibited a >99% reduction in their expansion in vivo (Figs. 2 and 3) despite normal proliferative responses during short-term in vitro cultures (Fig. 1). Additionally, the fact that IFN-IR0 mice, in which neither APCs nor T cells can respond to IFN-I, elicit modest levels of virus-specific CD8 T cell expansion, at least in the initial phase of infection (3, 16–17), prompted us to carefully evaluate several possibilities, including precursor frequency, clonal specificity, rejection by the host, and virus-mediated killing.
The diminished expansion of IFN-IR0 donor P14 CD8 T cells after infection did not reflect differences in initial splenic engraftment because in other experiments, in which higher numbers (107) of cells were transferred, the splenic engraftment was very similar (∼10% of the transferred cell numbers between days 1 and 2 after transfer) for both WT and IFN-IR0 P14 CD8 T cell donors. These donor IFN-IR0 naive P14 cells persisted in uninfected recipient mice for >1 month at levels similar to the WT cells (unpublished data). Moreover, the memory cells generated from IFN-IR0 donor CD8 T cells after the resolution of infection were maintained stably for extended periods (Fig. 5). Additionally, neither naive mice (Fig. 6 A) nor the immune mice that previously received a mixture of WT plus IFN-IR0 P14 cells and LCMV infection (Fig. 6 B) rejected freshly transferred WT or IFN-IR0 P14 CD8 T cells. Also, the preferential expansion of WT P14 CD8 T cells as observed in Fig. 2 was similarly seen in littermate IFN-IR+/+ and IFN-IR+/− hosts (the latter [heterozygous] mice have a mutated IFN-IR allele encoding a G418 resistance gene; Fig. 7 A, middle and top; reference 16). Moreover, the WT P14 CD8 T cells underwent preferential, albeit low, expansion in infected IFN-IR0 hosts (Fig. 7 A, bottom) in which virus was not cleared (Fig. 7 B, bottom), and production of IFN-I was not compromised (reference 3; unpublished data). Collectively, these results demonstrate that the diminished expansion seen in the IFN-IR0 donor cells was not related to rejection but was because of the inability to respond to IFN-I during the antigen-driven proliferation phase.
Figure 6.

Adoptively transferred WT and IFN-IR0 P14 CD8 T cells were maintained at an equal ratio either in naive or in LCMV immune hosts. 0.8 × 106 each of WT (Thy1.2, Ly5.1) and IFN-IR0 (Thy1.2, Ly5.2) P14 CD8 T cells were transferred into (A) naive B6 Thy1.1 mice or into (B) LCMV-immune Thy1.1 mice that had previously received equal numbers of WT plus IFN-IR0 CD8 T cells before LCMV infection. WT/IFN-IR0 donor CD8 T cell ratios were analyzed at days 1 and 8 after transfer. The donor cells were identified based on positive staining for Thy1.2 as well as CFSE (R1 gate). In B, freshly transferred Thy1.2 donor CD8 T cells were distinguished (R1 gate) from previously primed Thy1.2 memory CD8 cells (R2 gate) based on the CFSE label. The numbers in parenthesis indicate the average recovery of donor cells per spleen (n = 3) in each group. Numbers represent the percentage of cells in the indicated quadrant.
Figure 7.

The difference in the expansion of WT and IFN-IR0 CD8 T cells in response to viral infection was observed in IFN-IR0 and heterozygous hosts. 35,000 each of WT (Ly5.1) and IFN-IR0 P14 CD8 T cells (Ly5.2) were cotransferred into littermate IFN-R+/+, IFN-IR+/−, or IFN-IR−/− hosts (Ly5.2) and assessed for their expansion in response to LCMV infection. (A) The ratio of WT/IFN-IR0 P14 donor CD8 T cells was analyzed by gating on MHC tetramer–positive CD8 T cells and assessing Ly5.1 staining among V β8 TCR-positive cells (P14 CD8 T cells express V β8 TCR). The average recovery of IFN-R+/− and IFN-IR−/− P14 donor CD8 T cells per spleen is indicated in parenthesis (n = 3 per group). (B) Spleen viral titers at different times after infection. Each dot represents an individual mouse. IFN-IR expression heterozygosity was confirmed by PCR.
The diminished expansion of IFN-IR0 donor P14 CD8 T cells was unlikely to be caused by their preferential lysis by direct virus infection by itself because LCMV Armstrong is neither known to infect CD8 T cells nor to cause the death of infected cells in mice (21–24). Nevertheless, we tested this possibility by transferring a mixture of bystander ovalbumin-specific WT OT-1 and IFN-IR0 OT-1 TCR transgenic CD8 T cells into WT mice, followed by analyzing their ratio before and after LCMV infection. If LCMV preferentially kills the IFN-IR0 CD8 T cells, the IFN-IR0 OT-1 CD8 T cells should decline more than the WT OT-1 CD8 T cells after infection. The ratio of the IFN-IR0 versus WT OT-1 CD8 T cells (none LCMV specific) did not preferentially decrease after LCMV infection (Fig. 8 A, No infection vs. LCMV only). We repeated these experiments by separately transferring CFSE-labeled WT and IFN-IR0 OT-1 CD8 T cells into WT hosts to assess whether the IFN-IR0 OT-1 cells were undergoing proliferation in a bystander fashion. Neither of them proliferated by day 5 after LCMV infection, suggesting that the IFN-IR0 CD8 T cells were not undergoing any preferential bystander proliferation after infection (unpublished data). These experiments, though they do not test whether LCMV infects CD8 T cells or not, confirm that the IFN-IR0 bystander naive CD8 T cells were not undergoing preferential decline more than the WT bystander CD8 T cells under the conditions of LCMV infection (25). Cross-priming by ovalbumin during LCMV infection (8), however, led to a drastic decrease in the ratio of IFN-IR0 to WT OT-1 CD8 T cells (Fig. 8 A, LCMV + Ova). This was because of a preferential expansion of the WT OT-1 donor CD8 T cells over the IFN-IR0 OT-1 donor cells. (Fig. 8 B). Thus, the striking effect of IFN-I on clonal expansion during LCMV infection applies not only to virus-specific CD8 T cells but also to CD8 T cells responding to a different antigen, ovalbumin.
Figure 8.
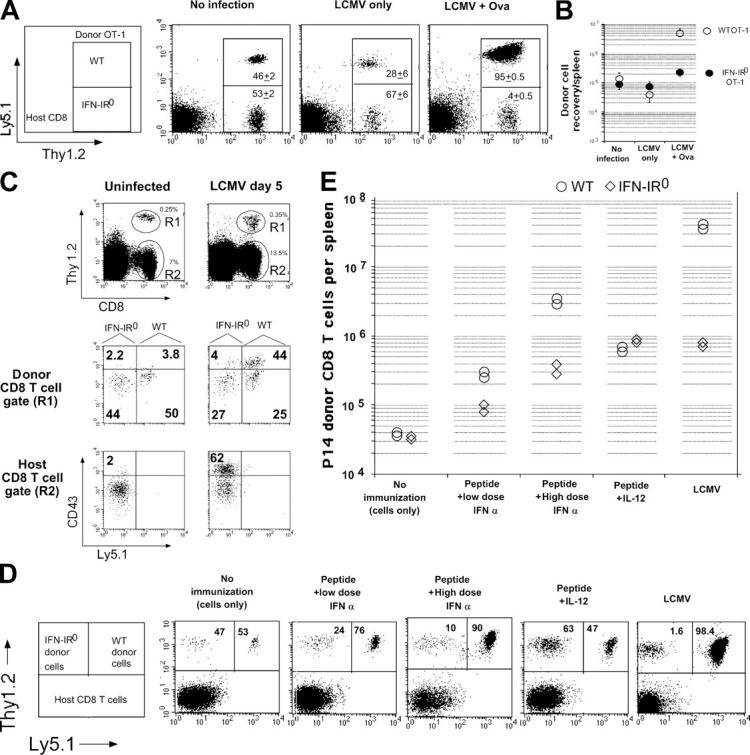
IFN-I action on a variety of CD8 T cell clonal specificities is required for their expansion in response to viral infection, cross priming, and vaccination in the presence of adjuvants that mimic viral infection–induced cytokine profiles. (A) 1.3 ×106 each of WT (Thy1.2, Ly5.1) and IFN-IR0 (Thy 1.2, Ly5.2) OT-1 CD8 T cells were transferred into C57BL/6 Thy1.1 mice. The mice were analyzed the next day (No infection), 5 d after LCMV infection alone (LCMV only), or 5 d after LCMV + ovalbumin immunization (LCMV + Ova). Data are gated on CD8 T cells. Numbers inside the box indicate the average ratio of IFN-IR0 (Thy1.2+ Ly5.1low) to WT (Thy1.2+ Ly5.1+) OT-1 CD8 T cells (n = 5 per group combined from two experiments). Similar trends were seen in a different experiment in which 4 × 106 each of the donor cells were transferred. (B) Average recovery of WT and IFN-IR0 OT-1 donor CD8 T cells per spleen in the experiments indicated in A. Values shown are the mean ± SD. (C) 3.4 × 106 each of enriched polyclonal CD8 T cells derived from age-matched C57BL/6 WT (Thy1.2, Ly5.1) and IFN-IR0 (Thy1.2, Ly5.2) mice were transferred into C57BL/6 Thy1.1 recipients and then infected with LCMV the next day. Mice were analyzed on day 5 after infection (day 6 after cell transfer in the case of uninfected recipients). Similar trends were observed in a different experiment in which mice were analyzed on day 6 after infection. (D) 500,000 each of WT (Thy1.2, Ly5.1) and IFN-IR0 (Thy1.2, Ly5.2) P14 CD8 T cells were transferred into B6 Thy1.1 hosts and left untreated, immunized with 25 μg GP33-41 peptide mixed with low (105 units) or high (106 units) dose universal IFN-I (IFN-α), immunized with peptide + 1 μg recombinant IL-12, or infected with LCMV. Dot plots show the average ratio of the IFN-IR0 versus WT donor P14 cells among the gated donor CD8 T cell population. (E) Recovery of IFN-IR0 and WT donor P14 CD8 T cells per spleen in the experiment described in D. Each point represents an individual mouse.
To assess the role of IFN-I on polyclonal naive CD8 T cell precursors, we purified polyclonal IFN-IR0 (Thy1.2, Ly5.2) and WT (Thy1.2, Ly5.1) CD8 T cells, cotransferred these cells into intact (unirradiated) WT mice (Thy1.1), and examined their activation/expansion in response to LCMV infection. Because of the low frequency of precursor cells in polyclonal populations, we used CD43 up-regulation as a marker to identify virus-specific CD8 T cells (26). Fig. 8 C shows that the donor polyclonal WT CD8 T cells generated far higher numbers of activated CD8 T cells (CD43high) in response to infection than the polyclonal IFN-IR0 donor CD8 T cells present in the same host (Fig. 8 C, middle). Thus, the IFN-I–mediated direct effect on T cell expansion was not restricted to P14 or OT-1 transgenic CD8 T cells but was also seen on naive CD8 T cell precursors that exist in extremely small numbers in a polyclonal population. Importantly, the ratio of IFN-IR0 to WT naive (CD43low) CD8 T cells recovered from the polyclonal donor populations was similar after infection (Fig. 8 B, middle), reaffirming that the IFN-IR0 donor CD8 T cells were neither rejected nor preferentially killed by virus infection.
We next examined the expansion of P14 WT and IFN-IR0 CD8 T cells in response to peptide immunization in normal mice; i.e., mice that were not infected with LCMV using IFN-I as an adjuvant (to mimic virally induced IFN-I). The ratio of the recovered IFN-IR0 versus WT donor P14 CD8 T cells in unimmunized mice was roughly similar (Fig. 8 D, No immunization). After peptide immunization, both IFN-IR0 and WT donor CD8 T cells expanded (Fig. 8 E), but the expansion of WT P14 CD8 T cells was higher, especially with high-dose IFN-I administration. As a result, the ratio of IFN-IR0 to WT cells decreased (Fig. 8 D, Peptide + High dose IFN-α). In contrast, when IL-12 was used as the adjuvant, the IFN-IR0 CD8 T cells expanded at least as well as WT P14 donor cells (Fig. 8, E and D), indicating that priming in the presence of IL-12 can overcome the defect caused by IFN-IR deficiency on CD8 T cells. Collectively, these results show that the diminished expansion of IFN-IR0 P14 CD8 T cells was caused by their specific inability to respond to IFN-I and did not result from host-mediated rejection or viral infection of the cells.
IFN-Is promote clonal expansion by enhancing T cell survival
IFN-Is might control the clonal expansion of CD8 T cells by increasing T cell activation, by increasing the number of naive precursors recruited into the response, by enhancing the rate or duration of their proliferation, and/or by augmenting the survival of daughter cells in vivo. To assess these possibilities, we cotransferred large numbers of WT and IFN-IR0 P14 CD8 T cells into naive congenic mice and analyzed activation early after infection. Fig. 9 A shows that both WT and IFN-IR0 cells up-regulated CD69 and CD25, down-regulated L-selectin, and began to up-regulate CD44 as early as 24 h after infection. These findings suggested that WT and IFN-IR0 P14 CD8 T cells both responded early after infection. Analysis of the recovered cells soon after infection showed that both WT and IFN-IR0 P14 CD8 T cells were maintained at a nearly equal ratio without expansion before 48 h after infection (Fig. 9 B). By 72 h, however, the WT P14 CD8 T cells showed a marked increase over the IFN-IR0 P14 CD8 T cells (Fig. 9 B). Thus, the differences observed in the expansion of IFN-IR0 and WT CD8 T cells were not attributable to differences in early T cell activation or to differences in spontaneous death soon after antigen encounter.
Figure 9.

IFN-IR0 CD8 T cells undergo activation and survive in the early phase of infection. 106 each of WT (Thy1.2, Ly5.1) and IFN-IR0 (Thy1.2, Ly5.2) P14 CD8 T cells were cotransferred into WT (Thy1.1, Ly5.2) mice that were infected with LCMV. (A) Both WT and IFN-IR0 cells underwent activation in response to infection within 24 h as seen by activation markers. Data are gated on donor CD8 T cell populations. Ly5.1+ events are WT, and Ly5.1− events are IFN-IR0 donors. (B) Ratio of WT/IFN-IR0 donors P14 CD8 T cells at various times after infection.
To seek direct information on proliferation during the expansion phase, we transferred CFSE-labeled WT or IFN-IR0 P14 CD8 T cells into naive congenic mice and analyzed cell recruitment, proliferation, and recovery after LCMV infection. Both WT and IFN-IR0 CD8 T cells remained CFSE high in uninfected mice, indicating no spontaneous cell division (Fig. 10 A, left). After LCMV infection, both WT and IFN-IR0 P14 CD8 T cells were recruited into extensive proliferation at 66 h after infection (Fig. 10 A, middle). Surprisingly, the IFN-IR0 cells appeared to proliferate at a faster rate than WT P14 CD8 cells (Fig. 10 A, middle), perhaps because of the lack of antiproliferative effect that IFN-I is known to exert on T cells (see Discussion). By 86 h, both populations were CFSE low, which is indicative of multiple cycles of proliferation (Fig. 10 A, right). To examine T cell proliferation at later stages, we assessed the proportion of actively cycling cells in vivo by giving mice an injection of bromodeoxy-uridine (BrdU) at various days after infection. Staining cells for BrdU incorporation at 12 h after BrdU injection showed that cycling of WT and IFN-IR0 donor cells was very similar on day 5 after infection (Fig. 10 B). An extensive analysis of several mice in three different experiments (Fig. 10 C) showed that (a) both IFN-IR0 and WT cells continued to cycle until day 8 after infection, (b) the proportion of cycling cells progressively decreased from day 3 to 8 after infection among both IFN-IR0 and WT cells, and (c) the IFN-IR0 cells were cycling at a slightly higher rate compared with WT cells at the early phase of response. Despite similar levels of recruitment and a higher initial rate of proliferation, the expansion of IFN-IR0 P14 CD8 T cells was greatly reduced, indicating that most of the daughter cells generated from IFN-IR0 P14 CD8 T cells failed to survive.
Figure 10.
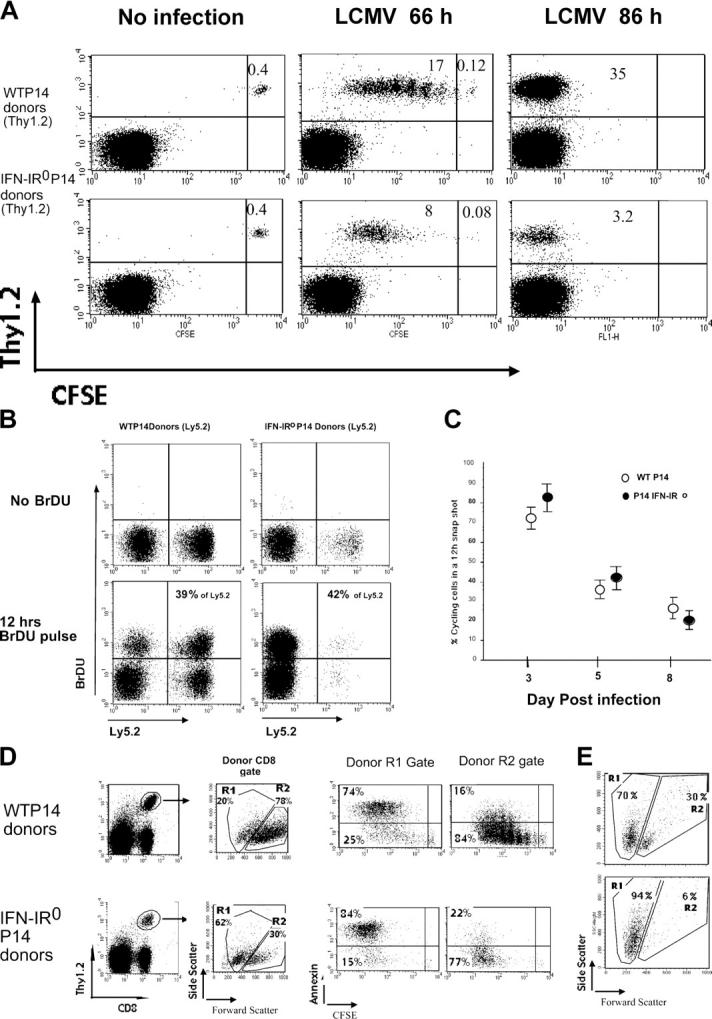
IFN-IR0 CD8 T cells were not defective in proliferation but survived poorly. (A) 400,000 each of purified, CFSE-labeled WT P14 or IFN-IR0 P14 naive CD8 T cells (Thy1.2, Ly5.2) were transferred into congenic naive C57BL/6 (Thy1.1) mice and left uninfected or infected the next day. Gated CD8+ T cells from the spleen were analyzed at 66 or 86 h after infection for division and frequencies of donor Thy1.2+ cells. Data are representative of five mice that received WT cells and seven mice that received IFN-IR0 donors at each time point in two individual experiments. Note that the IFN-IR0 P14 CD8 T cells proliferate but exhibit diminished accumulation. Numbers represent the percentage of cells in the indicated quadrant. (B) IFN-IR0 CD8 T cells continue to proliferate at later times. 400,000 cells each of purified WT P14 naive CD8 T cell (Ly5.2) or IFN-IR0 P14 naive CD8 T cell (Ly5.2) donors were transferred into congenic naive C57BL/6 (Ly5.1) mice. Mice were infected with LCMV the next day and were injected with 0.2 mg BrdU i.p on day 5 after infection. Gated CD8+ spleen cells were analyzed for BrdU incorporation in donor (Ly5.2+) and endogenous (Ly5.2−) CD8 T cells 12 h after BrdU injection. Numbers indicate the percentage of BrdU-positive cells among the donor Ly5.2+ cells. Data are representative of two experiments and a total of four mice in each group. (C) The BrdU pulse was done at days 3, 5, and 7 after infection, and mice were analyzed at 12 h after BrdU injection. Data are pooled from five experiments. Values shown are the mean ± SD. (D) CFSE-labeled donor P14 CD8 cells from LCMV-infected mice indicated in the panels were analyzed after 66 h for scatter property and ex vivo annexin V staining (representative of four mice in each group). (E) Spleen cells at day 5 after infection were cultured in vitro for 24 h without peptide stimulation or cytokines. After 24 h, gated WT or IFN-IR0 donor P14 CD8 T cells were analyzed for scatter properties. Data are an example of two mice per group in one experiment and one mouse per group in a different experiment.
Ex vivo assessment of side scatter at 66 h after infection (high side scatter, dying, or dead cells) confirmed that a greater proportion of IFN-IR0 P14 CD8 T cells were undergoing death (Fig. 10D). This finding was further confirmed by demonstrating that a majority of cells with high side scatter were annexin V positive (Fig. 10 D, R1 gate). In vitro culture of WT and IFN-IR0 P14 effector CD8 T cells from day 5–infected mice showed that both populations were undergoing spontaneous death, but the death was vastly increased in IFN-IR0 cells (Fig. 10 E). Thus, IFN-I action on CD8 T cells is critical for keeping the progeny of the effector CD8 T cells alive. In the absence of IFN-I action, the antigen-experienced effector CD8 T cells proliferate, but a majority of the progeny derived from them undergoes rapid death.
Discussion
Our study shows that the massive expansion of virus-specific CD8 T cells seen in the acute phase of live viral infection is critically dependent on survival signals provided by the direct action of IFN-I on responding T cells. The naive CD8 T cell precursors recruited into response through appropriate APCs proliferate rapidly, but the decision of whether their progeny should live or die is dependent on their exposure to IFN-I, a set of innate cytokines produced after infection. Previous studies suggested that antigen-experienced CD8 T cells undergo programmed expansion (which is a cumulative outcome of proliferation plus survival of the daughter cells) for several days after infection of mice with Listeria monocytogenes, even if the bacterium was eliminated by antibiotic treatment within the first 24–48 h (27–29). Whether this expansion was programmed in a cell-intrinsic way or whether cell-extrinsic factors play a role remained unclear. Our studies highlight that T cell proliferation and survival are interdependently regulated by separate stimuli (i.e., proliferation by cognate TCR interaction and survival by inflammatory stimuli) and may be differentially programmed under the conditions of viral infection. Whether a brief exposure to IFN-I is sufficient for maintaining the effector T cell survival or whether the effector cells need continuous exposure to IFN-I in order to remain alive requires further investigation.
It is generally viewed that the effects of IFN-I on clonal expansion in vivo are related largely via their actions on APCs and to some extent via skewing activated T cells toward a type I cytokine response (6–15). Our studies highlight a previously unappreciated role for IFN-I in causing clonal expansion via direct action on antigen-specific CD8 T cells. We think this effect was not appreciated in previous experiments that analyzed T cell responses directly in virally infected IFN-IR0 mice (16), in which neither T cells nor APCs can respond to IFN-I, possibly because (a) the infected IFN-IR0 mice may elicit altered innate responses and (b) the IFN-IR0 mice have a high persistent viral load that makes it difficult to dissociate effects because of the absence of IFN-I action from effects related to overwhelming viral loads (16, 17). For example, IFN-IR0 mice are known to elicit elevated levels of IL-12 (which may overcome the defect caused by IFN-IR0 deficiency on CD8 T cells; Fig. 8) because of the absence of an IFN-I–mediated IL-12 feedback inhibition pathway (3). Additionally, other arms of the immune system may also be affected in the IFN-IR0 host, depending on the context, thus making it difficult to assess what might be the effect that IFN-Is have on T cell response under normal physiological conditions. A recent study reported that IFN-IR0 mice can even elicit more enhanced levels of CD8 T cell responses than WT mice during DNA immunization, possibly because of deregulated control of IL-10–producing suppressor cells (18). Our results suggesting that IL-12 might contribute to overcoming the defect caused by IFN-IR deficiency on CD8 T cells is further supported by a recent study showing that purified naive CD8 T cells stimulated in vitro with microspheres bearing MHC class I/peptide Ag and B7-1 ligand expand and differentiate into effectors in the presence of exogenously added IFN-I and that exogenously added IL-12 can substitute for IFN-I in mediating this effect (15). Based on these results, together with the observation that LCMV induces almost undetectable levels of IL-12 (3), we would predict that the responding CD8 T cells become less dependent on IFN-I–mediated signals under the conditions that induce more IL-12 and less IFN-I. Consistent with this prediction, ongoing studies from our lab indicate that in response to infection with recombinant vaccinia virus, as well as _L. monocytogenes_–expressing LCMV GP33-41 epitope, the IFN-IR0 P14 CD8 T cells exhibit ∼80% reduced expansion compared with the expansion of WT P14 CD8 T cells in a WT host (unpublished data). This suggests that the extent to which IFN-I contributes to T cell expansion may be dependent on the type of pathogen and the context of immunization. More comprehensive studies are warranted to assess which factors contribute to T cell expansion in different types of infection.
Another striking observation of our study is the effect that the IFN-I have on the generation of a memory pool. The size of the memory pool generated from the IFN-IR0 effector CD8 T cells was ∼100-fold smaller than the memory CD8 T cell pool generated from WT P14 cells (Fig. 5). The mechanisms that contribute to the generation of memory cells are poorly understood. Previous studies suggested that antigen-experienced CD8 T cells undergo programmed expansion for about 1 wk after infection and then undergo programmed cell death irrespective of whether the antigen persists or not (27–29). A proportion of effector CD8 T cells that were able to up-regulate molecules such as IL-7Rα chain, Spi2A, and CD8αα homodimer (30–32) were shown to have a better capacity to escape death and become memory precursor cells that will differentiate further in a progressive manner in the absence of additional antigenic stimulus into functional memory cells (33). Our results show that a majority of the daughter cells derived from antigen-experienced CD8 T cells undergo death in parallel to proliferation during the acute phase of viral infection. Direct IFN-I action rescues them from this death, thereby tilting the balance effectively toward clonal expansion. The IFN-I–mediated rescue from death during the antigen-driven proliferation is critical not only for the expansion of effector cells, but also for the expansion of memory precursors.
Why do the majority of IFN-I–responsive CD8 cells that have received IFN-I–mediated signals and survived until day 8 still undergo massive contraction afterwards? One possibility is that these cells begin death because the IFN-I–mediated survival signals become limited by this time. Indeed, the serum IFN-I levels usually peak by day 3 and reach baseline levels by about day 7 (34). An alternative possibility is that the effector T cells may become progressively refractory to IFN-I–mediated signals, especially after the withdrawal of antigenic exposure and they may now need survival signals provided by other factors such as IL-7 and IL-15. Exposure to IFN-I is shown to cause down-regulation of IFN-IR α chain in primary human CD34+ bone marrow cells and in several human cell lines of diverse origin (35–36). Whether similar receptor downmodulation/desensitization occurs in activated effector CD8 T cells requires further investigation. Alternatively, it is possible that the precipitous death of the CD8 T cells after the peak expansion may be caused by a completely unrelated event, including homeostatic compensatory mechanisms (37). Indeed, we were unable to prevent WT P14 effector CD8 T cell death by exogenous IFN-I treatment on day 8 after infection (unpublished data; Shaulov, A., personal communication), strengthening the latter possibilities. The memory cells generated from the IFN-I–unresponsive effector CD8 T cells, albeit low in number, were maintained stably for extended periods, indicating that the IFN-I–mediated signals required for survival during the antigen-driven proliferation become dispensable once the antigen–driven proliferation ceases and the cells differentiate into resting memory cells.
Why is IFN-I action on T cells so important when there are several other cytokines available in vivo? Although various inflammatory cytokines have been historically used to increase effector T cell survival in vitro and in vivo by exogenous administration, their direct role on T cells under the normal physiological conditions of an immune response to infection is becoming apparent only from recent experiments. Naive CD8 T cells stimulated in vitro produce IL-2, which is known to provide antiapoptotic signals to the T cells. However, IL-2 is produced only transiently in vivo. Recent studies show that T cells that cannot respond to IL-2 exhibit only modestly diminished expansion under the conditions of viral infections (38, 39). A recent study suggested that CD8 T cells that lack IFN-γ receptor exhibit nearly 2–4-fold decreased expansion compared with WT cells in response to LCMV infection (40). Surprisingly, in our study, the IFN-IR0 CD8 T cells, despite having an intact IFN-γ receptor, exhibit strikingly diminished expansion, suggesting that IFN-γ signals alone cannot rescue the deficiency caused by IFN-IR deficiency. This may be related to the fact that prior IFN-I exposure contributes to optimal IFN-γ action, as proposed in previous experiments (41).
Another factor that may influence the dependence on IFN-I is the type of cell that displays antigen. Under the conditions of viral infection, a majority of antigen-specific CD8 T cells that were recruited into response via activated professional APCs may subsequently encounter several infected cell types that are not necessarily professional APCs. Many of these infected targets, although deficient in the expression of the costimulatory molecules/inflammatory cytokines typical of professional APCs, are capable of producing IFN-I. We predict that repetitive exposure of the antigen-experienced T cells to these “nonprofessional” targets may lead to activation-induced cell death, but the IFN-I produced in response to infection may help in tilting the balance from cell death to survival/expansion. This may ensure continued expansion of antigen-experienced T cells at the time when the availability of professional APCs becomes limited. The frequency and duration of antigen-experienced T cell encounters with such infected nonprofessional targets is likely to be higher under the conditions of infection with noncytopathic viruses such as LCMV.
Continued production of such inflammatory signals under the conditions of persistent infection is likely to be deleterious to the host, because this may allow uncontrollable expansion of effector T cell progeny that may result in immune-mediated pathology. Consistent with this, IFN-Is are induced in a large burst in the acute phase of viral infection but do not continue to be produced in such large amounts at later phases if the virus establishes persistent infection (42). This raises the question of whether the activation-induced cell death and functional exhaustion of T cells that is typically seen under the conditions of persisting viral load (43–45) is related, at least in part, to the diminished availability of IFN-I–mediated survival signals and warrants further investigations.
The level of IFN-I produced and the expansion of CD8 T cells in response to LCMV infection are considerably higher compared with the levels produced in response to other viral and intracellular bacterial infections (e.g., vaccinia virus, vesicular stomatitis virus, and _L. monocytogen_es) (1, 3, 8, 28). Based on the remarkable effect that IFN-I has on CD8 T cell expansion, we predict that the robustness of T cell responses elicited in response to various infections may be related, at least in part, to the level of IFN-I produced.
How do IFN-Is prevent T cell death? IFN-Is were originally discovered as antiviral and antiproliferative cytokines and are shown to exert opposing functions on cell survival or death, depending on the context of its action, the type of cell, and the stage of the cell (5, 12, 25, 46–47). IFN-Is are also known to delay cell cycle progression from G0/G1 to S phase (48). Previous experiments showed that activated T cells, when cultured in vitro in the presence of IFN-I, survive better and that this increased survival was not associated with elevated levels of Bcl-2 or Bcl-XL (12). This suggests that IFN-I may confer survival signals via a pathway that is distinct from that induced by members of the IL-2 family or CD28 engagement. Consistent with this, we find that the IFN-IR0 CD8 T cells expressed at least as much bcl-2 and IL-7Rα chain as WT CD8 T cells at various time points after infection (Fig. S1, A and B, available at http://www.jem.org/cgi/content/full/jem.20050821/DC1). It is also possible that IFN-I might enhance T cell survival via induction/up-regulation of receptors for other cytokine families. For example, previous experiments indicated that IFN-I enhances memory CD8 T cell proliferation via induction of IL-15, a cytokine that is shown to maintain proliferative renewal of memory CD8 T cells (49). We think the IFN-I–mediated survival of the effector cells is unlikely to be mediated via the IL-15 pathway because previous studies show that IL-15R α chain–deficient and IL-15–deficient mice elicit potent CD8 T cell expansion after LCMV infection (49–50).
Interestingly, the antiproliferative signaling pathway activated by IFN-I has some level of cross talk with TCR signaling via the P53 pathway (51–52). Our observation that IFN-IR0 P14 CD8 cells recovered at 66 h after infection display more advanced division history than WT P14 CD8 T cells (Fig. 7 A) indicates that IFN-I may be applying brakes on T cell proliferation, at least in the beginning of response. Rapid division usually accompanies DNA duplication errors. DNA repair mechanisms are generally more efficient during the G1/S phase. It was proposed that continuous division with shorter intermitotic times might reduce the efficiency by which DNA repairs occur (53). This raises the question of whether IFN-I increases cell survival by applying brakes to uncontrollable cell proliferation and warrants further investigation.
In summary, our study shows that IFN-Is, a set of innate cytokines produced in response to viral infection, act directly on antigen-specific CD8 T cells to allow their clonal expansion. IFN-Is mediate this process by prolonging the survival of effector CD8 T cells. The IFN-I–mediated survival during the antigen-driven proliferation phase is critical for generation of optimal numbers of memory CD8 T cells. This knowledge will have broad implications for further understanding the role of IFN-Is in sustaining T cell survival during vaccination/therapeutic strategies.
Materials and Methods
Mice.
IFN-IR–deficient mice on a 129/SvEv background (16) were backcrossed to B6 for 14 generations. P14 and OT-1 TCR transgenic mice that were on a B6 background were bred to B6 Ly5.1 mice to generate mice cells with a congenic marker. C57BL/6 (Thy1.2) and C57BL/6 (Thy1.1) mice were purchased from the Jackson Laboratory. Male or female mice at 4–12 wk of age were used for experiments. All mice were maintained under specific pathogen-free conditions at the University of Washington animal care facility under the guidelines of the Institutional Animal Care and Use Committee.
Antibodies and staining.
All antibodies, except anti–granzyme B, were purchased from either BD Biosciences or eBioscience. Anti–human granzyme B was purchased from Caltag. Intracellular staining was done by culturing the cells for 5 h in a CO2 incubator at 37°C in the presence of brefeldin A, followed by surface and intracellular staining using a Cytofix/Cytoperm kit (BD Biosciences) as previously described (1). BrdU staining was done using a BrdU flow kit (BD Biosciences). Mice were injected with 200 μg BrdU 12 h before analysis.
Purification of CD8 T cells and adoptive transfers.
CD8 T cells were purified by negative sorting using a cell sorter (FACSVantage; Beckton Dickinson) after staining with a mixture of antibodies against CD4, B220, CD11c, and CD19. All adoptive cell transfers were done i.v.
CFSE labeling.
Cells were washed two times with serum-free medium, followed by incubation with 5 μM CFSE for 7 min, quenching with 20% FCS, and washing with RPMI 1640 with 10% FCS.
Virus and immunizations.
LCMV strain Armstrong was plaque purified, grown in baby hamster kidney cells, and titered on Vero cells. 2 × 105 PFU LCMV was injected into mice 24–48 h after cell transfer. Doses of universal IFN-I (IFN-α; PBL Biomedical Laboratories) or murine recombinant IL-12 (R&D Systems) were mixed with peptide in a volume of 200 μl PBS and injected i.p and s.c on days 0 and 2, as indicated in the figures.
Online supplemental material.
Fig. S1 shows that IFN-IR0 CD8 T cells expressed at least as much bcl-2 and IL-7Rα chain as did WT CD8 T cells at various time points after infection. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20050821/DC1.
Acknowledgments
We thank C.B. Wilson, M. Bevan, A. Rudensy, P. Fink, E. Clark, P. Greenburg, R. Ahmed, and members of the Kaja laboratory for helpful discussions, as well as K. Madhavi for technical assistance.
This work was supported in part by National Institutes of Health grants 1R01AI053146 and R21AI051386 (to K. Murali-Krishna) and by funds from the Washington National Primate Center and the University of Washington Department of Immunology.
The authors have no conflicting financial interests.
Abbreviations used: BrdU, bromodeoxy-uridine; IFN-I, type I IFN; LCMV, lymphocytic choriomeningitis virus.
References
- 1.Murali-Krishna, K., J.D. Altman, M. Suresh, D.J. Sourdive, A.J. Zajac, J. Miller, J. Slansky, and R. Ahmed. 1998. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. 8:177–187. [DOI] [PubMed] [Google Scholar]
- 2.Kaech, S.M., E.J. Wherry, and R. Ahmed. 2002. Effector and memory T-cell differentiation: implications for vaccine development. Nat. Rev. Immunol. 2:251–262. [DOI] [PubMed] [Google Scholar]
- 3.Cousens, L.P., R. Peterson, S. Hsu, A. Dorner, J.D. Altman, R. Ahmed, and C.A. Biron. 1999. Two roads diverged: interferon α/β– and interleukin 12–mediated pathways in promoting T cell interferon γ responses during viral infection. J. Exp. Med. 189:1315–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hochrein, H., K. Shortman, D. Vremec, B. Scott, P. Hertzog, and M. O'Keeffe. 2001. Differential production of IL-12, IFN-α, and IFN-γ by mouse dendritic cell subsets. J. Immunol. 166:5448–5456. [DOI] [PubMed] [Google Scholar]
- 5.Stark, G.R., I.M. Kerr, B.R. Williams, R.H. Silverman, and R.D. Schreiber. 1998. How cells respond to interferons. Annu. Rev. Biochem. 67:227–264. [DOI] [PubMed] [Google Scholar]
- 6.Luft, T., K.C. Pang, E. Thomas, P. Hertzog, D.N. Hart, J. Trapani, and J. Cebon. 1998. Type I IFNs enhance the terminal differentiation of dendritic cells. J. Immunol. 161:1947–1956. [PubMed] [Google Scholar]
- 7.Biron, C.A. 2001. Interferons alpha and beta as immune regulators–a new look. Immunity. 14:661–664. [DOI] [PubMed] [Google Scholar]
- 8.Le Bon, A., N. Etchart, C. Rossmann, M. Ashton, S. Hou, D. Gewert, P. Borrow, and D.F. Tough. 2003. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat. Immunol. 4:1009–1015. [DOI] [PubMed] [Google Scholar]
- 9.Krug, A., R. Veeraswamy, A. Pekosz, O. Kanagawa, E.R. Unanue, M. Colonna, and M. Cella. 2003. Interferon-producing cells fail to induce proliferation of naive T cells but can promote expansion and T helper 1 differentiation of antigen-experienced unpolarized T cells. J. Exp. Med. 197:899–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parronchi, P., M. De Carli, R. Manetti, C. Simonelli, S. Sampognaro, M.P. Piccinni, D. Macchia, E. Maggi, G. Del Prete, and S. Romagnani. 1992. IL-4 and IFN (alpha and gamma) exert opposite regulatory effects on the development of cytolytic potential by Th1 or Th2 human T cell clones. J. Immunol. 149:2977–2983. [PubMed] [Google Scholar]
- 11.Wenner, C.A., M.L. Guler, S.E. Macatonia, A. O'Garra, and K.M. Murphy. 1996. Roles of IFN-gamma and IFN-alpha in IL-12-induced T helper cell-1 development. J. Immunol. 156:1442–1447. [PubMed] [Google Scholar]
- 12.Marrack, P., J. Kappler, and T. Mitchell. 1999. Type I interferons keep activated T cells alive. J. Exp. Med. 189:521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nguyen, K.B., W.T. Watford, R. Salomon, S.R. Hofmann, G.C. Pien, A. Morinobu, M. Gadina, J.J. O'Shea, and C.A. Biron. 2002. Critical role for STAT4 activation by type 1 interferons in the interferon-gamma response to viral infection. Science. 297:2063–2066. [DOI] [PubMed] [Google Scholar]
- 14.Marrack, P., and J. Kappler. 2004. Control of T cell viability. Annu. Rev. Immunol. 22:765–787. [DOI] [PubMed] [Google Scholar]
- 15.Curtsinger, J.M., J.O. Valenzuela, P. Agarwal, D. Lins, and M.F. Mescher. 2005. Cutting edge: type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J. Immunol. 174:4465–4469. [DOI] [PubMed] [Google Scholar]
- 16.Muller, U., U. Steinhoff, L.F. Reis, S. Hemmi, J. Pavlovic, R.M. Zinkernagel, and M. Aguet. 1994. Functional role of type I and type II interferons in antiviral defense. Science. 264:1918–1921. [DOI] [PubMed] [Google Scholar]
- 17.Ou, R., S. Zhou, L. Huang, and D. Moskophidis. 2001. Critical role for alpha/beta and gamma interferons in persistence of lymphocytic choriomeningitis virus by clonal exhaustion of cytotoxic T cells. J. Virol. 75:8407–8423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dikopoulos, N., A. Bertoletti, A. Kroger, H. Hauser, R. Schirmbeck, and J. Reimann. 2005. Type I IFN negatively regulates CD8+ T cell responses through IL-10-producing CD4+ T regulatory 1 cells. J. Immunol. 174:99–109. [DOI] [PubMed] [Google Scholar]
- 19.Zimmerman, C., K. Brduscha-Riem, C. Blaser, R.M. Zinkernagel, and H. Pircher. 1996. Visualization, characterization, and turnover of CD8+ memory T cells in virus-infected hosts. J. Exp. Med. 183:1367–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murali-Krishna, K., L.L. Lau, S. Sambhara, F. Lemonnier, J. Altman, and R. Ahmed. 1999. Persistence of memory CD8 T cells in MHC class I-deficient mice. Science. 286:1377–1381. [DOI] [PubMed] [Google Scholar]
- 21.Homann, D., D.B. McGavern, and M.B. Oldstone. 2004. Visualizing the viral burden: phenotypic and functional alterations of T cells and APCs during persistent infection. J. Immunol. 172:6239–6250. [DOI] [PubMed] [Google Scholar]
- 22.Rambukkana, A., S. Kunz, J. Min, K.P. Campbell, and M.B. Oldstone. 2003. Targeting Schwann cells by nonlytic arenaviral infection selectively inhibits myelination. Proc. Natl. Acad. Sci. USA. 100:16071–16076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kagi, D., and H. Hengartner. 1996. Different roles for cytotoxic T cells in the control of infections with cytopathic versus noncytopathic viruses. Curr. Opin. Immunol. 8:472–477. [DOI] [PubMed] [Google Scholar]
- 24.Binder, D., M.F. van den Broek, D. Kagi, H. Bluethmann, J. Fehr, H. Hengartner, and R.M. Zinkernagel. 1998. Aplastic anemia rescued by exhaustion of cytokine-secreting CD8+ T cells in persistent infection with lymphocytic choriomeningitis virus. J. Exp. Med. 187:1903–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McNally, J.M., and R.M. Welsh. 2002. Bystander T cell activation and attrition. Curr. Top. Microbiol. Immunol. 263:29–41. [DOI] [PubMed] [Google Scholar]
- 26.Harrington, L.E., M. Galvan, L.G. Baum, J.D. Altman, and R. Ahmed. 2000. Differentiating between memory and effector CD8 T cells by altered expression of cell surface O-glycans. J. Exp. Med. 191:1241–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaech, S.M., and R. Ahmed. 2001. Memory CD8+ T cell differentiation: initial antigen encounter triggers a developmental program in naive cells. Nat. Immunol. 2:415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Badovinac, V.P., B.B. Porter, and J.T. Harty. 2002. Programmed contraction of CD8(+) T cells after infection. Nat. Immunol. 3:619–626. [DOI] [PubMed] [Google Scholar]
- 29.van Stipdonk, M.J., G. Hardenberg, M.S. Bijker, E.E. Lemmens, N.M. Droin, D.R. Green, and S.P. Schoenberger. 2003. Dynamic programming of CD8+ T lymphocyte responses. Nat. Immunol. 4:361–365. [DOI] [PubMed] [Google Scholar]
- 30.Kaech, S.M., J.T. Tan, E.J. Wherry, B.T. Konieczny, C.D. Surh, and R. Ahmed. 2003. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 4:1191–1198. [DOI] [PubMed] [Google Scholar]
- 31.Liu, N., T. Phillips, M. Zhang, Y. Wang, J.T. Opferman, R. Shah, and P.G. Ashton-Rickardt. 2004. Serine protease inhibitor 2A is a protective factor for memory T cell development. Nat. Immunol. 5:919–926. [DOI] [PubMed] [Google Scholar]
- 32.Madakamutil, L.T., U. Christen, C.J. Lena, Y. Wang-Zhu, A. Attinger, M. Sundarrajan, W. Ellmeier, M.G. von Herrath, P. Jensen, D.R. Littman, and H. Cheroutre. 2004. CD8 alpha alpha-mediated survival and differentiation of CD8 memory T cell precursors. Science. 304:590–593. [DOI] [PubMed] [Google Scholar]
- 33.Kaech, S.M., S. Hemb, E. Kersh, and R. Ahmed. 2002. Molecular and functional profiling of memory CD8 T cell differentiation. Cell. 111:837–851. [DOI] [PubMed] [Google Scholar]
- 34.Biron, C.A., K.B. Nguyen, and G.C. Pien. 2002. Innate immune responses to LCMV infections: natural killer cells and cytokines. Curr. Top. Microbiol. Immunol. 263:7–27. [DOI] [PubMed] [Google Scholar]
- 35.Constantinescu, S.N., E. Croze, A. Murti, C. Wang, L. Basu, D. Hollander, D. Russell-Harde, M. Betts, V. Garcia-Martinez, and J.E. Mullersman. 1995. Expression and signaling specificity of the IFNAR chain of the type I interferon receptor complex. Proc. Natl. Acad. Sci. USA. 92:10487–10491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ito, K., H. Tanaka, T. Ito, T.A. Sultana, T. Kyo, F. Imanaka, Y. Ohmoto, and A. Kimura. 2004. Initial expression of interferon alpha receptor 2 (IFNAR2) on CD34-positive cells and its down-regulation correlate with clinical response to interferon therapy in chronic myelogenous leukemia. Eur. J. Haematol. 73:191–205. [DOI] [PubMed] [Google Scholar]
- 37.Welsh, R.M., L.K. Selin, and E. Szomolanyi-Tsuda. 2004. Immunological memory to viral infections. Annu. Rev. Immunol. 22:711–743. [DOI] [PubMed] [Google Scholar]
- 38.D'Souza, W.N., and L. Lefrancois. 2003. IL-2 is not required for the initiation of CD8 T cell cycling but sustains expansion. J. Immunol. 171:5727–5735. [DOI] [PubMed] [Google Scholar]
- 39.Wong, P., and E.G.P. Am. 2004. Disparate in vitro and in vivo requirements for IL-2 during antigen-independent CD8 T cell expansion. J. Immunol. 172:2171–2176. [DOI] [PubMed] [Google Scholar]
- 40.Whitmire, J.K., J.T. Tan, and J.L. Whitton. 2005. Interferon-γ acts directly on CD8+ T cells to increase their abundance during virus infection. J. Exp. Med. 201:1053–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takaoka, A., Y. Mitani, H. Suemori, M. Sato, Y. Yokochi, S. Noguchi, N. Tanaka, and T. Taniguchi. 2000. Cross talk between interferon-gamma and -alpha/beta signaling components in caveolar membrane domains. Science. 288:2357–2360. [DOI] [PubMed] [Google Scholar]
- 42.Bukowski, J.F., C.A. Biron, and R.M. Welsh. 1983. Elevated natural killer cell-mediated cytotoxicity, plasma interferon, and tumor cell rejection in mice persistently infected with lymphocytic choriomeningitis virus. J. Immunol. 131:991–996. [PubMed] [Google Scholar]
- 43.Zajac, A.J., J.N. Blattman, K. Murali-Krishna, D.J. Sourdive, M. Suresh, J.D. Altman, and R. Ahmed. 1998. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 188:2205–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gallimore, A., A. Glithero, A. Godkin, A.C. Tissot, A. Pluckthun, T. Elliott, H. Hengartner, and R. Zinkernagel. 1998. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I–peptide complexes. J. Exp. Med. 187:1383–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Missale, G., E. Cariani, and C. Ferrari. 2004. Role of viral and host factors in HCV persistence: which lesson for therapeutic and preventive strategies? Dig. Liver Dis. 36:703–711. [DOI] [PubMed] [Google Scholar]
- 46.Dondi, E., L. Rogge, G. Lutfalla, G. Uze, G, and S. Pellegrini. 2003. Down-modulation of responses to type I IFN upon T cell activation. J. Immunol. 170:749–756. [DOI] [PubMed] [Google Scholar]
- 47.Tanabe, Y., T. Nishibori, L. Su, R.M. Arduini, D.P. Baker, and M. David. 2005. Cutting edge: role of STAT1, STAT3, and STAT5 in IFN-alpha beta responses in T lymphocytes. J. Immunol. 174:609–613. [DOI] [PubMed] [Google Scholar]
- 48.Sangfelt, O., S. Erickson, and D. Grander. 2000. Mechanisms of interferon-induced cell cycle arrest. Front Biosci. 5:D479–D487. [DOI] [PubMed] [Google Scholar]
- 49.Schluns, K.S., and L. Lefrancois. 2003. Cytokine control of memory T-cell development and survival. Nat. Rev. Immunol. 3:269–279. [DOI] [PubMed] [Google Scholar]
- 50.Becker, T.C., E.J. Wherry, D. Boone, K. Murali-Krishna, R. Antia, A. Ma, and R. Ahmed. 2002. Interleukin 15 is required for proliferative renewal of virus-specific memory CD8 T cells. J. Exp. Med. 195:1541–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takaoka, A., S. Hayakawa, H. Yanai, D. Stoiber, H. Negishi, H. Kikuchi, S. Sasaki, K. Imai, T. Shibue, K. Honda, and T. Taniguchi. 2003. Integration of interferon-alpha/beta signaling to p53 responses in tumour suppression and antiviral defence. Nature. 424:516–523. [DOI] [PubMed] [Google Scholar]
- 52.Petricoin, E.F., III, S. Ito, B.L. Williams, S. Audet, L.F. Stancato, A. Gamero, K. Clouse, P. Grimley, A. Weiss, J. Beeler, et al. 1997. Antiproliferative action of interferon-alpha requires components of T-cell-receptor signalling. Nature. 390:629–632. [DOI] [PubMed] [Google Scholar]
- 53.Liu, L., H. Parekh-Olmedo, and E.B. Kmiec. 2003. The development and regulation gene repair. Nat. Rev. Genet. 4:679–689. [DOI] [PubMed] [Google Scholar]