Neurovascular Coupling Is Not Mediated by Potassium Siphoning from Glial Cells (original) (raw)
Abstract
Neuronal activity evokes localized changes in blood flow, a response termed neurovascular coupling. One widely recognized hypothesis of neurovascular coupling holds that glial cell depolarization evoked by neuronal activity leads to the release of K+ onto blood vessels (K+ siphoning) and to vessel relaxation. We now present two direct tests of this glial cell-K+ siphoning hypothesis of neurovascular coupling. Potassium efflux was evoked from glial cells in the rat retina by applying depolarizing current pulses to individual cells. Glial depolarizations as large as 100 mV produced no change in the diameter of adjacent arterioles. We also monitored light-evoked vascular responses in Kir4.1 knock-out mice, where functional Kir K+ channels are absent from retinal glial cells. The magnitude of light-evoked vasodilations was identical in Kir4.1 knock-out and wild-type animals. Contrary to the hypothesis, the results demonstrate that glial K+ siphoning in the retina does not contribute significantly to neurovascular coupling.
Keywords: blood flow, glia, astrocytes, Müller cells, Kir4.1, potassium siphoning, retina
Introduction
Neuronal activity within a localized brain region evokes increases in blood flow, a response termed neurovascular coupling. This vascular response is commonly used to monitor brain function using such techniques as functional magnetic resonance imaging and positron emission tomography. Recent work performed both in vivo and ex vivo indicates that glial cells, which contact both neuronal synapses and blood vessels, play an important role in mediating neurovascular coupling.
Neurotransmitters released from active neurons, including glutamate and ATP, stimulate glial cells and evoke glial Ca2+ increases. These Ca2+ increases are thought to trigger the synthesis and release of arachidonic acid metabolites, resulting in either dilation or constriction of neighboring blood vessels. Studies in hippocampal brain slices (Zonta et al., 2003; Mulligan and MacVicar, 2004), the isolated retina (Metea and Newman, 2006), and in the cortex in vivo (Takano et al., 2006) demonstrate that arachidonic acid metabolites such as EETs (epoxyeicosatrienoic acids) and prostaglandins mediate vasodilation, whereas the arachidonic acid metabolite 20-HETE (20-hydroxyeicosatetraenoic acid) mediates vasoconstriction.
A number of years ago, Paulson and Newman (1987) proposed that glial cells could also mediate neurovascular coupling by a directed release of K+ from glial endfeet, a mechanism termed K+ siphoning. In this process, an increase in extracellular K+ ([K+]o) generated by active neurons results in K+ influx into glial cells. This influx depolarizes glial cells and drives out an equal amount of K+ from other cell regions (Newman et al., 1984). Most K+ efflux occurs from glial endfeet, which have a high density of K+ channels (Newman, 1984; Brew et al., 1986). Because glial endfeet terminate on blood vessels, most K+ is siphoned onto the vessels. Inwardly rectifying Kir4.1 K+ channels carry much of the K+ siphoning current in glial cells (Kofuji et al., 2000; Butt and Kamada, 2006). These channels are capable of conducting outward current almost as efficiently as inward current as they are weakly rectifying.
Potassium is a well recognized vasodilator. Artificial increases in [K+]o of several millimolar activate vascular smooth muscle Na+-K+ ATPases. Increased [K+]o also activates smooth muscle Kir channels by increasing channel open probability (Haddy et al., 2006), resulting in raised membrane K+ conductance. The resulting smooth muscle cell hyperpolarization leads to cell relaxation and vasodilation. The hypothesis that neurovascular coupling is mediated by increases in [K+]o generated by glial cell K+ siphoning has gained widespread recognition, appearing in review articles and reference books on neurovascular coupling (Edvinsson et al., 1993; Iadecola, 2004) as well as in neuroscience textbooks (Nicholls et al., 2001). However, the K+ siphoning hypothesis of neurovascular coupling has never been put to a direct test.
We have now performed two tests of the K+ siphoning hypothesis of neurovascular coupling. Experiments were performed in the retina, where the glial cell K+ siphoning process has been demonstrated previously (Newman et al., 1984; Karwoski et al., 1989). The diameter of retinal arterioles was monitored as K+ efflux from glial cell endfeet was evoked by cell depolarization. In addition, neurovascular coupling was assessed in Kir4.1 knock-out (KO) mice, where K+ efflux from glial cells is substantially reduced (Kofuji et al., 2000). The results demonstrate that, contrary to the previous hypothesis, glial K+ siphoning in the retina does not contribute significantly to neurovascular coupling.
Materials and Methods
Animals.
Experiments were performed on acutely isolated whole-mount retinas of male Long–Evans rats (175–300 g; Harlan, Indianapolis, IN) and on postnatal day 15 (P15) to P18 Kir4.1+/+ [wild-type (WT)] and Kir4.1−/− (KO) transgenic mice (Kofuji et al., 2000). Genotyping of the mice was performed as described previously (Kofuji et al., 2000).
Whole-mount retina.
The whole-mount retinal preparation has been described previously (Metea and Newman, 2006). Briefly, retinas were removed from the back half of the eye and the vitreous humor peeled away from retinal pieces. Retinal pieces were superfused in a chamber at 2–3 ml/min with bicarbonate-buffered solution at 34–37°C. The animals used in this study were treated in accordance with the guidelines of the Institutional Animal Care and Use Committee of the University of Minnesota.
Retinal imaging.
Retinas were imaged with a 40× water-immersion objective, infrared-differential interference contrast (IR-DIC) optics, and a cooled CCD camera (CoolSnap ES; Roper Scientific, Duluth, GA) to measure arteriole diameter. As described previously (Metea and Newman, 2006), arterioles were easily distinguished from venules under IR-DIC optics by their morphology. Images were captured and analyzed using MetaMorph image processing software (Molecular Devices, Downingtown, PA). The intraluminal diameter of arterioles, which was clearly demarcated in IR-DIC images, was measured manually from magnified, enhanced retinal images. Some arterioles displayed small luminal oscillations before stimulation. In these cases, the prestimulus baseline diameter was taken as the average diameter of the vessel.
Electrical recording of glial cells.
Retinas were preincubated in collagenase/dispase (2 mg/ml) and DNase (0.1 mg/ml) for 15 min to digest the basal lamina at the vitreal surface of the retina. Whole-cell patch recordings were made from astrocyte somata and Müller cell endfeet at the retinal surface. Patched cells were filled with Lucifer yellow contained in the pipette solution and identified by their morphology. Cell membrane potential and input resistance were monitored using an Axoprobe-1A microelectrode amplifier (Molecular Devices) and custom LabView software. Only cells that had stable membrane potentials more negative than −75 mV were used in experiments. Depolarizing current pulses were injected into cells using the Axoprobe-1A circuitry.
Current–voltage (I–V) relationships of mouse Müller cells were determined using whole-cell patch recordings. Voltage ramps, swept from −180 to +100 mV in 2 s, were applied to cells. For each cell, I–V relationships were determined before and after addition of 100 μm Ba2+. The Ba2+-sensitive component of the current was determined by subtracting the Ba2+ I–V trace from the control trace. The magnitude of the Ba2+-sensitive inward conductance was calculated as the cord conductance measured at −100 mV. Recordings were made in HEPES-buffered solution.
Pressure ejection.
Modified superfusate solutions containing high K+ (3.5–9 mm) were ejected onto blood vessels through pipettes with tip diameters of 2–3 μm. Ejection pressure was low (4–10 psi) to minimize mechanical stimulation of blood vessels, which consistently produced vasoconstriction.
Light stimulation of the retina.
Diffuse flickering white light (100–250 ms flashes repeated every 250–1000 ms) was used to stimulate the retina. The light was focused onto the retina through the microscope objective lens.
Statistics.
Statistical significance was determined by the single tailed Student's t test.
Solutions.
The bicarbonate-buffered superfusate solution contained the following (in mm): 117.0 NaCl, 3.0 KCl, 2.0 CaCl2, 1.0 MgSO4, 0.5 NaH2PO4, 15.0 d-glucose, and 26 NaHCO3, bubbled with 5% CO2 in O2. The HEPES-buffered superfusate solution contained the following (in mm) 135.5, NaCl, 3.0 KCl, 2.0 CaCl2, 1.0 MgSO4, 0.5 NaH2PO4, 15.0 d-glucose, and 10 HEPES, pH 7.44, bubbled with 100% O2. The intracellular pipette solution contained the following (in mm): 5.0 Na-methanesulfonate, 136.0 K-methanesulfonate, 2.0 MgCl2, 2.0 MgATP, 0.2 NaGTP, and 5.0 HEPES, pH 7.4. Lucifer yellow CH (0.1 mg/ml) was added to the pipette solution to visualize glial cell morphology and contacts with blood vessels. In high K+ solutions, K+ was added without reduction of other ions.
Results
Glial cell depolarization
We first determined whether depolarization of glial cells evokes vasomotor responses in adjacent arterioles. Experiments were conducted on the isolated whole-mount retina of the rat. We showed previously that light stimulation evokes arteriole vasodilation in the retina (Metea and Newman, 2006). Before glial cell recordings were made, neurovascular coupling was tested by stimulating the retina with flickering light. Only those preparations where light-evoked vasodilations exceeded 10% (Fig. 1 C) were used in glial cell depolarization experiments.
Figure 1.
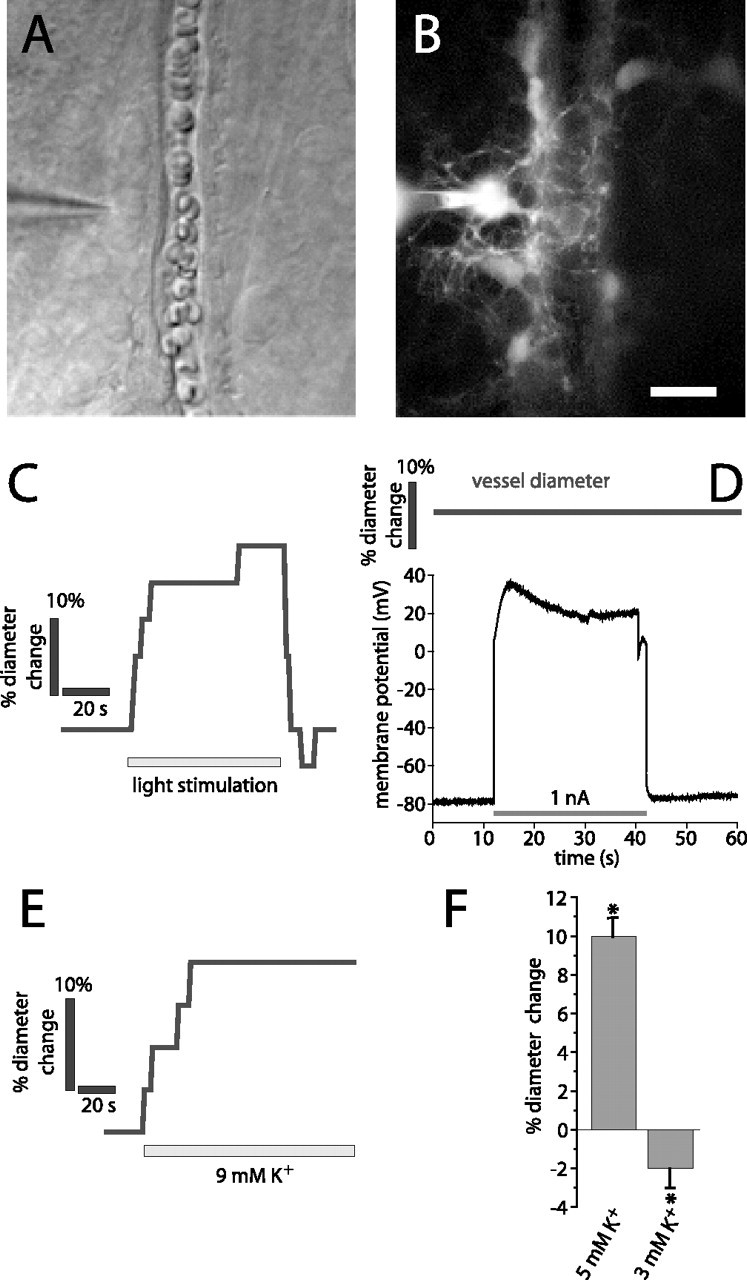
Light stimulation and K+ increase, but not glial cell depolarization, evokes vasodilation. A, B, Micrographs showing a whole-cell-patched astrocyte contacting an arteriole. The patch pipette is seen on the left. A, IR-DIC image. B, Fluorescence image showing the Lucifer-filled astrocyte contacting the arteriole. Additional astrocytes coupled to the patched cell are also seen. Scale bar: (in B) A, B, 10 μm. C–E, Experiments performed on the arteriole and astrocyte in A and B. C, Light stimulation evokes vasodilation. D, The astrocyte is depolarized by injection of 1 nA current. The arteriole diameter does not change during astrocyte depolarization. E, Ejection of 9 mm K+ in the region shown in A evokes vasodilation. F, Ejection of 5 mm K+ solution but not control solution (3 mm) evokes vasodilation in a series of experiments. Asterisks indicate significant change, p < 0.05. Error bars indicate SEM.
Whole-cell patch recordings were made from astrocytes and Müller cells at the vitreal surface of the retina. All recorded cells had processes (visualized with Lucifer yellow) that contacted a nearby arteriole (Fig. 1 A,B).
Constant current pulses of 30 s duration were injected into individual glial cells, producing depolarizations to membrane potentials ranging from −40 to +120 mV (Fig. 1 D, bottom trace). The luminal diameter of the arteriole contacted by the glial cell was monitored during glial cell current injection (19 astrocytes and 34 Müller cells) (Fig. 1 D, top trace). Each cell was depolarized multiple times with the membrane potential recovering between stimulations. There was no change in arteriole diameter during glial depolarization (−1 ± 1%; n = 53; p > 0.05). In additional experiments, no vasomotor responses were seen when glial cells were depolarized to a lesser degree (3–5 mV depolarization). In seven cells, an initial depolarization produced a small increase in vascular diameter (3 ± 1%). However, no vasomotor response was seen when the depolarization was repeated.
The sensitivity of arterioles to externally applied K+ was also tested. Superfusate solution containing elevated K+ was applied to arterioles by pipette ejection (Fig. 1 E). Ejection of 5 mm K+ solution produced vasodilations averaging 10 ± 1% (n = 19 vessels; p < 0.005) (Fig. 1 F). Ejection of control solution (3 mm K+) produced small vasoconstrictions (−2 ± 1%; n = 19 vessels; p < 0.05), most likely attributable to mechanical stimulation of the vessels. Some arterioles did not display K+-induced vasodilations. In these vessels, ejection of solution containing elevated K+ occasionally produced vasoconstrictions. These vasoconstrictions may also have been evoked by mechanical stimulation of the vessels.
Light-evoked vasomotor responses in Kir4.1 knock-out mice
We also tested whether light-evoked vasodilation was compromised in Kir4.1 knock-out animals. Experiments were conducted on wild-type and Kir4.1 KO mice. In wild-type mice, light stimulation evoked vasodilations averaging 13 ± 1% (n = 14) (Fig. 2 A,B,E,G). In Kir4.1 KO mice, light stimulation evoked vasodilations averaging 13 ± 2% (n = 15) (Fig. 2 C,D,F,G). The difference in vasodilation magnitude between the two groups was not significant (p > 0.05). Light-evoked vasoconstriction was occasionally observed in both wild-type mice (vessel diameter reduction of 16 ± 8%; n = 3) and knock-out mice (vessel diameter reduction of 18 ± 2%; n = 4).
Figure 2.
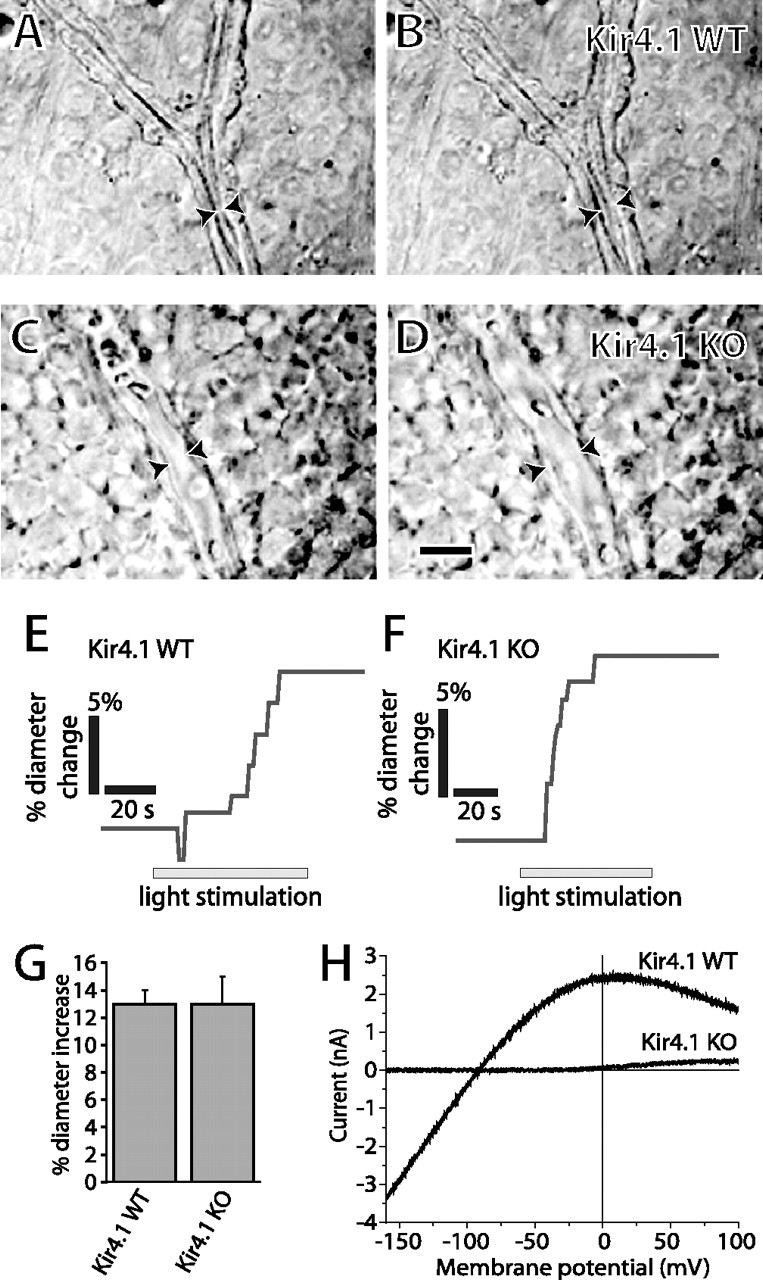
Light-evoked vasodilation is not reduced in Kir4.1 knock-out mice. A–D, IR-DIC micrographs of arterioles before (A, C) and during (B, D) light stimulation. A, B, Wild-type mouse. C, D, Kir4.1 knock-out mouse. Scale bar: (in D) A–D, 10 μm. Arrowheads indicate luminal diameter of arterioles. E–F, Light stimulation evokes vasodilation in wild-type and Kir4.1 knock-out mice. G, The amplitude of light-evoked vasodilation is identical in wild-type and Kir4.1 knock-out mice. H, Ba2+-sensitive current–voltage relationships of Müller cells in Kir4.1 WT and KO mice. Ba2+-sensitive inward current is completely absent in the KO cell. Error bars indicate SEM.
The I–V relationships of Müller cells were determined in Kir4.1 WT and KO mice to confirm that inwardly rectifying channels were absent in the knock-out animals. In WT cells, the Ba2+-sensitive inward conductance at −100 mV averaged 29.4 ± 2.9 nS (12) whereas in KO cells, the Ba2+-sensitive conductance averaged 0.007 ± 0.004 nS (9) (Fig. 2 H), demonstrating that few if any functional Kir channels are present in Kir4.1 KO cells. In addition, the overall conductance of KO cells at −100 mV averaged 0.37 ± 0.07 nS (8), only 1.3% the conductance of WT cells. The sensitivity of arterioles to K+ in Kir4.1 WT and KO mice was also determined. Raising superfusate K+ concentration from 3 to 5 mm evoked similar vasodilatory responses in WT and KO animals.
Discussion
We have performed two tests of the glial cell-K+ siphoning hypothesis of neurovascular coupling. In the first test, we depolarized astrocytes and Müller cells to generate a K+ efflux from cell endfeet onto arterioles. If K+ siphoning contributes to neurovascular coupling, glial cell depolarization should evoke vasodilation. Vessel dilation was not observed after current injection, however, even when glial cells were depolarized by as much as 100 mV.
It could be argued that this experiment is not a valid test of the hypothesis because single glial cells were stimulated in the experiment whereas, in vivo, most glial cells are depolarized by neuronal activity. Although the depolarizing current was applied to single cells, this current will, to some degree, depolarize many neighboring glial cells because glia in the retina are extensively coupled by gap junctions (Zahs and Newman, 1997; Ceelen et al., 2001). Thus, we consider experimental depolarization of glial cells an acceptable test for the widespread glial depolarization that occurs during light-evoked neuronal activity.
Moreover, experimental depolarization should produce greater K+ efflux in stimulated cells than depolarization produced by neuronal activation. Experimental stimulation resulted in glial cell depolarizations of 40–100 mV, whereas neuronal activity evoked by light stimulation depolarizes retinal glial cells by ∼1 mV (Karwoski and Proenza, 1980). Thus, K+ efflux evoked experimentally was many-fold greater than efflux evoked by light stimulation. Even if current injection results in the depolarization of only a single glial cell, the total amount of K+ released onto an arteriole was equivalent to the light-evoked activation of 40–100 glial cells. The sites of glial cell current injection (astrocyte somata and Müller cell endfeet) are closely coupled electrotonically to the cell endfeet contacting vessels (Newman, 1986), ensuring that the experimentally produced depolarizations will evoke significant K+ efflux from the endfeet.
In the second test, we monitored light-evoked vasodilation in animals lacking Kir4.1 channels, the principal K+ channel supporting K+ siphoning current in retinal astrocytes and Müller cells (Kofuji et al., 2000). If K+ siphoning contributes to neurovascular coupling, then light-evoked vasodilations should be substantially reduced in Kir4.1 KO animals. However, light-evoked vasomotor responses were normal in the transgenic animals.
It is possible that K+ channels other than Kir4.1 carry K+ siphoning currents in transgenic animals and that these channels mediate neurovascular coupling. However, voltage-clamp measurements demonstrate that the membrane conductance of Kir4.1 KO cells is just 1.3% the conductance of WT cells. Thus, K+ efflux should be almost completely abolished in KO animals. Yet, the magnitude of light-evoked vasodilations was not reduced in these transgenic animals.
Previously, Filosa et al. (2006) proposed that neurovascular coupling in the brain is mediated, in part, by efflux of K+ through Ca2+-activated K+ (BK) channels in astrocyte endfeet. They suggest that neuronal activity activates glial BK channels, leading to K+ efflux driven by the potential difference between the astrocyte membrane potential and the K+ equilibrium potential. Our results do not rule out such a K+-mediated mechanism of neurovascular coupling. However, it is unlikely that this mechanism contributes to neurovascular coupling in the retina. Although Müller cells express BK channels (Newman, 1985; Bringmann et al., 1997), the channels are not activated at the resting membrane potential, even when intracellular Ca2+ is raised significantly (Newman and Zahs, 1997).
The results of our experiments demonstrate that glial K+ siphoning does not contribute significantly to neurovascular coupling in the retina. Although K+ siphoning is an attractive hypothesis of neurovascular coupling that has gained a degree of acceptance in the field, this mechanism does not appear to be sufficient to initiate activity-induced vasodilation. Note, however, that more subtle contributions of K+ siphoning to vasomotor control, such as the role of glial K+ in setting vascular tone, were not investigated in our experiments. Such a mechanism could influence aspects of neurovascular coupling in vivo.
Our results do not rule out glial cells as mediators of neurovascular coupling. Retinal glial cells mediate light-evoked vasomotor responses by controlling the release of arachidonic acid metabolites (Metea and Newman, 2006). A similar glial mechanism mediates vasomotor responses in the brain (Zonta et al., 2003; Mulligan and MacVicar, 2004; Takano et al., 2006). Neurovascular coupling is undoubtedly mediated by multiple glial and neuronal mechanisms, but glial K+ siphoning does not appear to be a major contributor.
Footnotes
This work was supported by National Institutes of Health Grants EY004077 (E.A.N.) and EY012949 (P.K.) and a National Science Foundation predoctoral fellowship to M.R.M.
References
- Brew H, Gray PTA, Mobbs P, Attwell D. Endfeet of retinal glial cells have higher densities of ion channels that mediate K+ buffering. Nature. 1986;324:466–468. doi: 10.1038/324466a0. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Faude F, Reichenbach A. Mammalian retinal glial (Müller) cells express large-conductance Ca2+-activated K+ channels that are modulated by Mg2+ and pH and activated by protein kinase A. Glia. 1997;19:311–323. doi: 10.1002/(sici)1098-1136(199704)19:4<311::aid-glia4>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Butt AM, Kamada T. Inwardly rectifying potassium channels (Kir) in central nervous system glia: a special role for Kir4.1 in glial functions. J Cell Mol Med. 2006;10:33–44. doi: 10.1111/j.1582-4934.2006.tb00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceelen PW, Lockridge A, Newman EA. Electrical coupling between glial cells in the rat retina. Glia. 2001;35:1–13. doi: 10.1002/glia.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvinsson L, MacKenzie ET, McCulloch J. Cerebral blood flow and metabolism. New York: Raven; 1993. [Google Scholar]
- Filosa JA, Bonev AD, Straub SV, Meredith AL, Wilkerson MK, Aldrich RW, Nelson MT. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat Neurosci. 2006;9:1397–1403. doi: 10.1038/nn1779. [DOI] [PubMed] [Google Scholar]
- Haddy FJ, Vanhoutte PM, Feletou M. Role of potassium in regulating blood flow and blood pressure. Am J Physiol Regul Integr Comp Physiol. 2006;290:R546–R552. doi: 10.1152/ajpregu.00491.2005. [DOI] [PubMed] [Google Scholar]
- Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- Karwoski CJ, Proenza LM. Neurons, potassium, and glia in proximal retina of Necturus. J Gen Physiol. 1980;75:141–162. doi: 10.1085/jgp.75.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karwoski CJ, Lu HK, Newman EA. Spatial buffering of light-evoked potassium increases by retinal Muller (glial) cells. Science. 1989;244:578–580. doi: 10.1126/science.2785716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuji P, Ceelen PW, Zahs KR, Surbeck LW, Lester HA, Newman EA. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J Neurosci. 2000;20:5733–5740. doi: 10.1523/JNEUROSCI.20-15-05733.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metea MR, Newman EA. Glial cells dilate and constrict blood vessels: a mechanism of neurovascular coupling. J Neurosci. 2006;26:2862–2870. doi: 10.1523/JNEUROSCI.4048-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan SJ, MacVicar BA. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature. 2004;431:195–199. doi: 10.1038/nature02827. [DOI] [PubMed] [Google Scholar]
- Newman EA. Regional specialization of retinal glial cell membrane. Nature. 1984;309:155–157. doi: 10.1038/309155a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Voltage-dependent calcium and potassium channels in retinal glial cells. Nature. 1985;317:809–811. doi: 10.1038/317809a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. High potassium conductance in astrocyte endfeet. Science. 1986;233:453–454. doi: 10.1126/science.3726539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA, Zahs KR. Calcium waves in retinal glial cells. Science. 1997;275:844–847. doi: 10.1126/science.275.5301.844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA, Frambach DA, Odette LL. Control of extracellular potassium levels by retinal glial cell K+ siphoning. Science. 1984;225:1174–1175. doi: 10.1126/science.6474173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls JG, Martin AR, Wallace BG, Fuchs PA. From neuron to brain. Sunderland, MA: Sinauer Associates; 2001. [Google Scholar]
- Paulson OB, Newman EA. Does the release of potassium from astrocyte endfeet regulate cerebral blood flow? Science. 1987;237:896–898. doi: 10.1126/science.3616619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, Nedergaard M. Astrocyte-mediated control of cerebral blood flow. Nat Neurosci. 2006;9:260–267. doi: 10.1038/nn1623. [DOI] [PubMed] [Google Scholar]
- Zahs KR, Newman EA. Asymetric gap junctional coupling between glial cells in the rat retina. Glia. 1997;20:10–22. [PubMed] [Google Scholar]
- Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, Carmignoto G. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003;6:43–50. doi: 10.1038/nn980. [DOI] [PubMed] [Google Scholar]