The inflammatory response to cell death (original) (raw)
. Author manuscript; available in PMC: 2011 May 13.
Abstract
When cells die in vivo they trigger an inflammatory response. The ensuing hyperemia, leak of plasma proteins, and recruitment of leukocytes subserve a number of useful functions in host defense and tissue repair. However, this response can also cause tissue damage and in so doing contributes to the pathogenesis of a number of diseases. Given the key role of inflammation in these processes it is important to understand the underlying mechanisms that drive this response. The broad outline of this pathway is understood. Injured cells release “danger signals” that alert the host to cell death. Some of these molecules are recognized by cellular receptors that stimulate the generation of proinflammatory mediators. Other molecules released by dead cells stimulate the generation of mediators from extracellular sources. The resulting mediators then orchestrate the inflammatory response, eliciting its various vascular and cellular components. In addition to stimulating inflammation, dead cells also release danger signals that activate dendritic cells and promote the generation of immune responses to antigens in and around the dying cells. Many of the specific molecules and mechanisms involved in these various processes are still poorly understood. Here we review what is presently known about the sterile inflammatory response and its underlying mechanisms.
Keywords: Necrosis, danger, toll-like receptors, inflammation, cytokines
Introduction
Everyone has experienced injuries and knows from common observation that the injured site becomes inflamed. For example, a burn rapidly becomes red, hot, swollen and painful, a constellation of the four cardinal signs of inflammation that were described over 2,000 years ago by Celsus. It is perhaps not surprising, therefore, that the fact that injury causes inflammation (phlogosis) was recorded in the medical literature at the start of the first millennium AD by Galen. Study of the response to injury also played a key role in the elucidation of the underlying pathophysiologal mechanisms that account for the signs and symptoms recorded by Celsus. Waller (1846) and Cohnheim (1867) first described that injury caused vessels to dilate and leak fluid and stimulated the recruitment of leukocytes. Of course the discovery of infection as the other important trigger of inflammation occurred much later, not in small part because it had to await the discovery of germs and their connection to disease by Pasteur and Koch in the late 1800s.
We now know that there are several mechanisms by which injury can cause inflammation. If microbes are introduced, they can stimulate potent inflammation. However, sterile injury also stimulates inflammation and can do so through a number of different mechanisms. Traumatic stimulation of mast cells and nerves can lead to the release of proinflammatory mediators (1). If trauma causes bleeding, then the hemostatic mechanisms (platelets and clotting) that are triggered can generate inflammatory mediators (2, 3). In addition, cell death is a universal and potent stimulator of sterile inflammation. This latter phenomenon is the focus of this review.
When cells die and undergo necrosis in vivo, the tissue site is rapidly infiltrated with leukocytes, consisting initially of neutrophils followed by accumulations of monocytes. This is seen e.g. in a burn, an infarct (Figure 1) or the experimental implantation of dead tissue or cells (4) (5). This response to injury is so stereotypical that its evolution is used by Pathologists to date the time of tissue injury, e.g. in a myocardial infarct. Although this host response to cell death is universally observed and rather dramatic, its underlying mechanisms are poorly understood. This review explores what is known about this important response.
Figure 1.
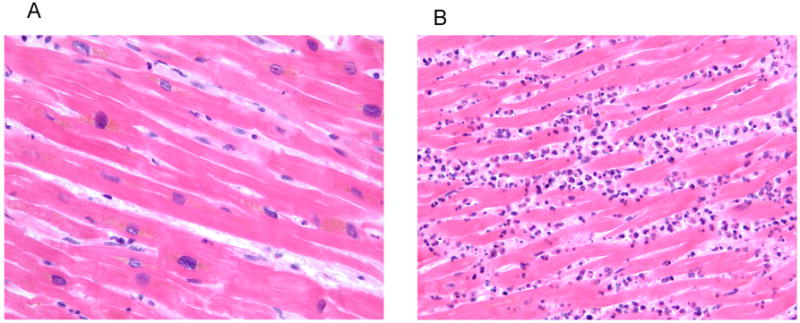
Acute inflammation induced by cell death. (A) Normal myocardium showing the expected absence of leukocytes. (B) Infarcted myocardium showing neutrophils surrounding dead cardiac myocytes. 40X magnification.
Role of the inflammatory response
In an infection, the role of the inflammatory response is clear. Vasodilatation very rapidly increases the delivery of blood borne defenses to the affected site. Increased vascular permeability allows soluble immune proteins such as antibody and complement to leak into the local environment where they can attack the invading microbes. Neoexpression of vascular adhesion molecules and chemokines attract leukocytes that then migrate into the tissue space. Once in the site of infection leukocytes ingest and attempt to destroy the pathogens. Thus, through these mechanisms there is a very rapid delivery of both soluble and cellular immune defenses. The importance of this response is clearly seen in individuals that due to genetic defects or treatments lack key components of the inflammatory response; such individuals have a very high rate of morbidity and mortality (6).
While an inflammatory response is of obvious importance to rapidly combat infection, why does cell death also trigger this response? The answer to this question is less clear, but there are probably several reasons. It has been proposed that the immune system has evolved mechanisms to recognize cell death to alert it to sites of potential danger (7) (see below). Thus, necrotic cell death may be the consequence of an infection, e.g. from a toxin or cytopathic microbe. Even if it is not directly caused by a pathogen, cell death is often occurring at a site where microbes have been introduced as a consequence of an injury, such as a penetrating wound. With a doubling time of 20 minutes, microbes constitute an explosive threat that to be contained requires a very rapid response. By mounting an inflammatory response to dead cells, the host rapidly mobilizes its defenses to a site of potential danger. It should be noted that this notion that dead cells are a “danger signal” was originally conceived to explain the generation of adaptive (T and B cell) immune responses (7). However, we believe the concept is also applicable to the rapid inflammatory (innate immune) response as well.
Nevertheless, in many other situations cells die and provoke inflammation in the absence of microbial infection. What useful role is the inflammatory response playing in these scenarios? In some situations the cells that are dying may still represent a threat to the body (e.g. in an evolving cancer) that may be eliminated by the host immune response (8). In other situations, it is possible that the inflammatory response counteracts certain pathological processes in other ways. E.g. in an ischemic infarct, it is possible that inflammation induced vasodilatation helps perfuse adjacent ischemic areas. In other forms of injury, increased blood flow and fluid leakage may help dilute and drain away soluble injurious agents or the cellular response may encase and wall off offending particulate material (e.g. in a granuloma). Thus, even in these sterile situations, the inflammatory response may still be playing a defensive role and limit further damage to the host.
In addition to containing or neutralizing an injurious process, the inflammatory response is also thought to promote repair at sites of tissue damage. In order to heal an injured site, the dead cells and debris must be cleared and this is accomplished through the action of the recruited phagocytes. To fill in gaps and restore tissue integrity, epithelial cells or fibroblasts must divide and a new blood supply must be established; these processes are stimulated by mediators made by inflammatory cells. In addition, the fibrinogen that leaks from inflamed vessels is converted into fibrin fibrils that provide scaffolding for the growth of many of these cells. Thus, the generation of a rapid inflammatory response subserves a number of different roles in both host defense and tissue repair.
Inflammation to necrosic versus apoptotic cell death
Cells can die through a number of different mechanisms. Two of the major types of cell death are necrosis (oncosis) and apoptosis (9). Necrotic cell death occurs in response to many kinds of insults (e.g. trauma, infarction, toxins, etc.) and therefore is typically the result of a pathological process. Morphologically, it is associated with cell swelling and/or the rapid loss of membrane integrity. In contrast, apoptosis is a process of cellular suicide that can occur as part of physiological events (e.g. loss of trophic factors, elimination of cells during normal development processes, etc.) or in some cases pathological ones (e.g. certain viral infections). Morphologically, apoptotic cells shrink and at least initially maintain integrity of their plasma membrane. It turns out that the host response to these two forms of cell death can be different (Figure 2).
Figure 2.
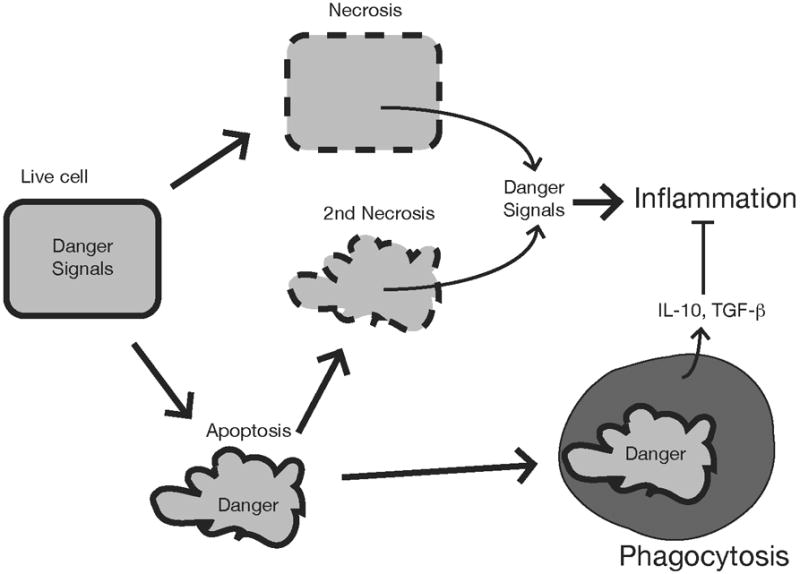
Responses to different forms of cell death. Cells undergoing necrosis lose membrane integrity and leak their intracellular components some of which serve as danger signals that stimulate inflammation. Apoptotic cells may not stimulate inflammation if they are ingested by phagocytes before they release their intracellular contents. Moreover, during this process apoptotic cells can stimulate phagocytes to produce anti-inflammatory cytokines. However, if the apoptotic cells are not cleared rapidly they release danger signals when they proceed into secondary necrosis.
Necrotic cell death stimulates a host inflammatory response (5). In contrast, it is often stated that apoptotic cell death doesn’t provoke inflammation (10). In fact, there are many situations where this is true for apoptotic cells; e.g. there is continuous death of developing T cells in the thymus and this occurs without inducing an inflammatory response. Since apoptotic cells initially maintain membrane integrity, they don’t rapidly release their intracellular contents that, as will be discussed below, contain proinflammatory signals. Instead, before these cells disintegrate, they are rapidly ingested by resident phagocytes. For single apoptotic cells, this clearance process can be remarkably efficient; e.g. despite high levels of apoptosis in the normal thymus, it is actually difficult to see apoptotic T cells in this organ. Moreover, apoptotic cells can stimulate macrophages to generate mediators such as IL-10 or TGF-β that inhibit inflammation inflammation (10, 11). It has been argued on theoretical grounds that this difference in host response to the two different modes of death has evolved because necrotic cell death arises from potentially dangerous situations to the host, while apoptotic death does not (7). According to this idea a rapid inflammatory response is potentially useful in tissues where necrosis occurs but not in sites of apoptosis.
However, the concept that apoptosis is non-inflammatory is not always correct. For example, FAS is a prototypical death receptor that when triggered stimulates cells to undergo apoptosis (12). Injection of an agonistic anti-FAS antibody into mice causes hepatocytes (which express Fas) to undergo apoptosis and this in turn stimulates very strong inflammation (13). If apoptosis is blocked in this situation, then this inflammatory response is inhibited (13). Why then is apoptosis sometimes silent, while at other times inflammatory? While the answer isn’t definitively known, it is likely that a key factor is how rapidly the apoptotic cells are cleared by phagocytes. This is because over time, apoptotic cells undergo a process known as secondary necrosis, in which their membrane becomes permeable to macromolecules (9). If this process occurs before the dead cells are ingested by phagocytes, then their intracellular contents, which contain proinflammatory components, will be released and stimulate a host response (Figure 2, discussed further below). Teleologically, this inflammatory response to apoptotic cells may be useful because there are pathological processes like infections that cause cells to undergo apoptosis (14). Furthermore, if there has been a significant loss of cell mass, the inflammatory response may be useful to promote healing.
Molecular basis of inflammation to infection
Over the last decade there have been major advances in our understanding of the molecular basis of the inflammatory response to microbes. In fact we now understand this process much better that we do for the inflammatory response triggered by tissue injury. Although this review is focused on the inflammatory response to cell death, it is useful to briefly review some of the mechanisms that underlie inflammation to infection. This will set the stage for considering some of the similarities and differences between inflammation to infection and to dying cells. A full review of this subject is beyond the scope of this manuscript and interested readers should consult recent reviews (15, 16). We will focus instead on a few of the inflammatory pathways that are most relevant to issues covered in this review.
The host recognizes the presence of microbes through a number of different mechanisms, some of the most important of which utilize several different families of cellular receptors. These various receptors monitor different subcellular compartments for the presence of microbial molecules. Some, like many of the Toll-like receptors (TLR) (15) and C-type lectins (17) are present on the plasma membrane, where they can detect the presence of microbes that have entered the extracellular fluids. Other receptors, such as a few of the TLR, are present in endocytic vacuoles and monitor these vesicles for microbes that have been ingested by cells (15). Yet other receptors, like the Nod-like receptors (16) and helicases (18), are present in the cytoplasm and detect the presence of pathogens that have invaded this compartment.
Of all of these microbial-sensing receptors, the best understood are the TLR (Figure 3) (15). These are transmembrane proteins with an extracellular antigen recognition domain composed of leucine-rich repeats. There are 11 different TLR each of which recognizes microbial components that are present in large numbers of different kinds of microbes but are chemically distinct from ones normally made by the host (sometimes referred to as PAMPs – pathogen-associated molecular patterns). For example, TLR4 recognizes lipopolysaccarides which are membrane components found in most gram negative bacteria. TLR2 recognizes lipopeptides present in many gram positive organisms. TLR9 recognizes unmethylated C and G-rich DNA sequences that are abundant in most prokaryotic genomes. TLR5 recognizes a protein in flagella and therefore can detect many flagellated microorganisms. TLR 3, 7 and 8 recognize RNA species expressed by many viruses. By focusing on these sets of conserved pathogen-associated molecular patterns the host is able to accomplish two remarkable and important things. It is able to discriminate between self and nonself components, and to do this with a relatively small set of receptors that can nevertheless sense most bacteria, fungi and many viruses, despite the huge diversity among these organisms.
Figure 3.
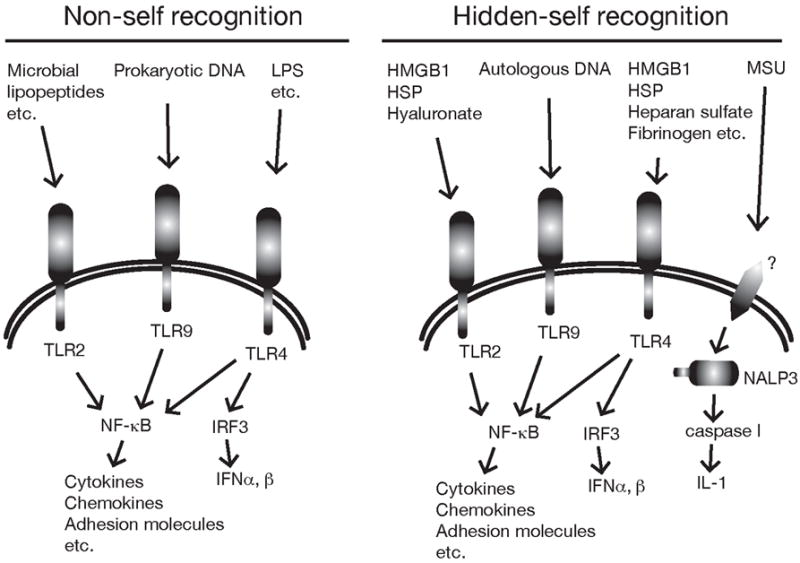
Examples of cellular receptors that sense infection and cell death. TLR detect infections by recognizing microbial molecules that are structurally different from mammalian ones (distinguishing self from nonself). In addition, there is emerging evidence that they can recognize certain autologous molecules that are normally hidden intracellularly (“hidden self”) but are released when cells die. Once stimulated TLR lead to the activation of the transcription factor NFkB and sometimes IRF3 which then turn on many of the key components of the inflammatory response. The danger signal MSU stimulates a pathway that contains the intracellular Nod-like receptor NALP3 and results in the production of the proinflammatory cytokine IL-1.
Once the TLRs bind their ligands, they associate with intracellular adaptor proteins (TIR adaptor molecules), which then initiate an intracellular signaling pathway (19). All TLRs except TLR3 associate with the MyD88 TIR adaptor and this stimulates a kinase cascade that ultimately activates the transcription factor NFkB. In turn, NFkB controls the expression of many of the molecules needed for acute inflammation including proinflamatory cytokines (e.g. IL-1, IL-6, IL-8 and TNFα), chemokines (e.g. MIP-1, MCP1, RANTES, and Eotaxin), and vascular adhesion molecules (e.g. ICAM1, VCAM1, and E-selectin) needed for leukocyte recruitment (20). TLR3 and TLR4 associate with another TIR adaptor protein TRIF which signals to activate type I interferons (19). Through these mechanisms, the host senses and rapidly induces an inflammatory response to infection.
In addition to the various microbial-sensing cellular receptors, there are soluble host proteins that may also be activated by microbes in ways that stimulate or amplify inflammation. The best characterized example of such a soluble system is the complement pathway (21). There are 9 different complement proteins (C1-9) that are constitutively present in the plasma, largely in inactive form. Microbes can activate these proteins in several different ways. The C3 component undergoes spontaneously activation at a low level and is normally rapidly inactivated. However, if it binds to a microbial surface it is stabilized in active form (22). Alternatively, antibodies or mannose-binding lectin that are bound to microbes can activate the formation of C3 convertase complex(23, 24) . Each activated complement component then proteolytically cleaves the next component of the pathway into active form in a cascading and amplifying fashion. Part of each of the cleaved fragments deposit on the surface of the microbe where some promote the binding to and internalization of the microbe by phagocytes (opsinization) and others assemble into a pore that promotes osmotic lysis of the organism (25, 26). Of importance to the inflammatory process, several of the complement proteolytic fragments that don’t bind the microbe remain soluble (e.g. C3a, C4a and C5a) and are potent proinflammatory mediators (27). Therefore, the activation of this set of soluble defense proteins by microbes stimulates an inflammatory response.
Through these mechanisms the TLR and complement systems (and other components not described here) allow the host to rapidly detect pathogens in the extracellular space and respond with a very rapid inflammatory defense. Below, we will consider whether or not these same “sensing mechanisms” contribute to generating inflammation to another extracellular stimulus: Dead cells.
The scent of death: Proinflammatory components from dead cells
The fact that dead cells induce inflammation indicates that the cell corpse must expose or release some sort of proinflammatory signal(s). It is theoretically possible that this signal(s) might be induced while a cell is dying but still metabolically active. However, cells that are instantaneously killed by freeze-thawing ex vivo stimulate robust inflammation when injected into animals (Chen et al. manuscript submitted). In this situation the proinflammatory signal(s) must come from pre-existing cellular components that are released in intact form or are modified by a process that doesn’t require energy. Presumably these components are sequestered inside of live cells but, upon cell death and the subsequent loss of membrane integrity, are passively released into the extracellular environment (Figure 4). Through this mechanism the abnormal release of intracellular components becomes a danger signal that alerts the host to dying cells. Since these components can’t be further synthesized and replenished by dead cells, the proinflammatory activity of dead cells decays over time(5), presumably as the active components are degraded and/or diluted. It should be noted that these overall features are distinct from typical cytokines, which are hormone-like mediators that are synthesized and actively secreted from live cells. While it is possible that in some situations intracellular stores of proinflammatory cytokines (e.g. IL-1 (28)) might be released upon cell disintegration and cause inflammation, most of these mediators have restricted expression in only certain cell types and therefore cannot account for how the majority of cells induce inflammation upon death. We will therefore focus this discussion on cellular proinflammatory molecules that are not classical cytokines.
Figure 4.
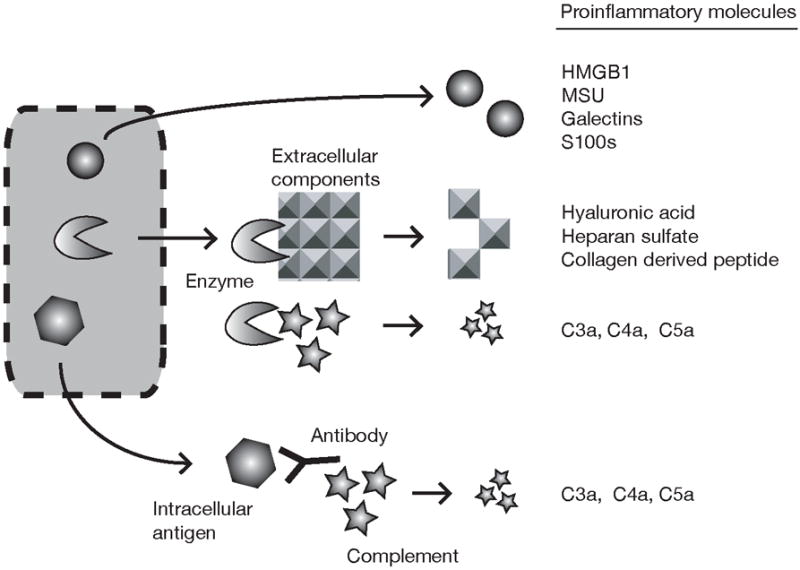
Proinflammatory molecules released from dying cells. Cells contain a number of different danger signals that can potentially stimulate inflammation through different mechanisms. Some of these signal are intracellular molecules that trigger inflammation by directly stimulating cells to make proinflammatory cytokines. Others work by generating proinflammatory mediators from extracellular components such as the extracellular matrix and complement.
The number of different inflammatory triggers released from dying cells is unknown. In fact until recently, the molecular identity of any of these molecules had been unknown. A number of potential candidates have now been identified. Before reviewing these molecules, it is worth considering the criteria for validating a molecule’s bone fide role in inducing an inflammatory response to dead cells. Ideally, multiple criteria should be satisfied: (i) The purified molecule should cause inflammation when injected in vivo; (ii) The purified molecule should also be active in physiological amounts and concentrations; (iii) Microbial contaminants need to be ruled out as the source of the proinflammatory activity. Such molecules (e.g. LPS) are easy to introduce during purification procedures and also contaminate recombinant molecules produced from prokaryotic expression systems. This kind of artifact especially needs to be rigorously excluded for molecules that are found to work through receptors like TLRs, which are known to sense microbial products (see above); (iv) Ideally, elimination or neutralization of the molecule from dead cells should reduce inflammation, although this criterion may be difficult to fulfill, if cells contain multiple danger signals that function redundantly. These criteria have not yet been met in full for any cellular molecule. Nevertheless there are several candidate molecules identified that have various degrees of evidence that support their potential role in triggering the sterile inflammatory response (Figure 4, Table I).
Table 1.
Cellular molecules with intrinsic proinflammatory activity.
| Factor | Cellular distribution | Release from dead cells | Proinflammatory in vitro | Proinflammatory in vivo | Putative receptor | ||
|---|---|---|---|---|---|---|---|
| Purified molecule proinflammatory in vivo | Neutralization reduces inflammation in vivo | ||||||
| HMGB1 | All cells | Necrosis (not apoptosis ) | Chemotaxis(113 ) Cytokine induction (51) Macrophage activation(51) DC activation (108) (114). Endothelial cell activation (115) | Antitumor immunity promotion( 108) | Liver injury( 31) (32) Sepsis (116) endotoxin induced lung injury (117) arthritis (118) | RAGE (82) TLR2&4 (32, 83) | |
| Uric acid (MSU) | All cells | Necrosis & Apoptosis | monocytes/macrophages activation and cytokine induction (93) DC activation (39) | Gout (40) | Adjuvant activity (39) Neutrophil recruitment (41) | ND | TLR2&4 (91) NOT TLR2&4 (92) CD14 (119) |
| nucleosomes | All cells | Secondary necrosis | cytokine induction(47). DC activation (120) Neutrophil activation (121) | lymphocyte necrosis induction (122) and DC maturation (120) | ND | Not specified | |
| Chromatin/DNA | All cells | Necrosis & Apoptosis | DC maturation and macrophage activation (123) B cell and DC activation informs of chromatin-IgG complex(90, 124) | unknown in normals; Yes, in chromati n-autoim mune complexes in SLE patients | Induces DC maturation (123) | ND | TLR9 (with BCR or Fcreceptor) (90, 124) |
| HSP (HSP70, HSP60, gp96) | |||||||
| HSP70 | All cells | Necrosis, some in apoptosis (125) | Nitric oxide production(126) Cytokine induction(127) DC maturation (49) | tumor immunogenicity enhancement(109) | ND | CD14 (127) CD91(128) LOX-1 (129) | |
| HSP60 | All cells | Necrosis, some in apoptosis (125) | Cytokine induction(130) Macrophage and DC activation (49) | ND | ND | TLR4 (84) TLR 2/4 (88) CD14 (131) | |
| gp96 | All cells | Necrosis, some in apoptosis (125) | Nitric oxide production(126) DC maturation (49) | DC maturation and migration(132) | ND | TLR 2/4 (133) CD91(128) SREC-1 (134) Scavenger receptor class A (135) | |
| Galectins | Leukocytes and endothelium (136) | ND | chemotaxis and leukocyte activation(43) DC maturation (137) | Monocyte Recruitment (galectin3)( 44) | ND | CD2 and others with beta-gala ctose (42) | |
| Thioredoxin | All cells | ND | cytokine induction (45) | Chemotaxis on leukocytes (46) | ND | ||
| S100 proteins | Leukocytes, keratinocytes and epithelium (53) | ND | cytokine induction and chemotaxis (51, 52) | ND | ND | RAGE for S100A12 (51) and S100B(138) | |
| Cathelicidins | Variety of cell types, abundant in neutrophils | ND | chemotaxis(54) | ND | ND | formyl peptide receptor-like 1 (54) | |
| Defensins | Leukocytes predominantly in Neutrophils, Paneth cells (human) | ND | chemotaxis(56) DC maturation (110) | ND | ND | CCR6(56 ) TLR4(110) | |
| Adenosine and ATP | all | Necrotic | IL-1 secretion(28) Chemotaxis (139) | ND | ND | P1, P2X and P2Y receptors (140) | |
| Mitochondrial peptides bearing the _N_-formy l group | All | Necrotic | chemotaxis(57) | Neutrophil recruitment (58) | ND | FPR and FPRL1 (141) |
Cellular molecules with intrinsic proinflammatory activity
One interesting molecule that has been implicated in the triggering the inflammatory response to necrotic cells is HMGB1 (29). HMGB1 is a nuclear protein present in all cells where it normally binds to chromatin and plays roles in bending DNA and regulating gene transcription (30). It was found to be passively released from cells after they underwent necrosis but was retained in the nuclei of apoptotic cells (even after they underwent secondary necrosis) (31). Because necrotic but not apoptotic cells were thought to induce inflammation (see section Inflammation to necrosis versus apoptotic cell death), Scaffidi et. al. investigated whether HMGB1 might play a role in inducing inflammation to necrotic cells (31). When tested in vitro, the HMGB1 released from necrotic cells stimulated monocytes to produce TNFα, a potent proinflammatory cytokine. Whether HMGB1 also induced inflammation when injected in vivo has not been reported. However, injecting a neutralizing HMGB1 antibody into animals treated with a hepato-toxic drug, reduced inflammation in the damaged liver (31). Other studies showed that anti-HMGB1 antibodies could also reduce inflammation in livers that had sustained ischemia-reperfusion injury (32). In yet other studies, injection of recombinant HMGB1 into infarcted heart muscle in vivo stimulated regeneration and repair (33). Together, these studies provide evidence that HMGB1 is a cellular factor that plays a role in the inflammatory response to necrotic cells.
In addition to being passively released from necrotic cells, HMGB1 has also recently been found to be released by a non-classical secretion mechanism from a subset of activated leukocytes (monocytes, macrophages, DC and NK cells) (34) (35). This secretion from living cells is stimulated by microbial components or cytokines (35). It is important to note that HMGB1 produced via this pathway would be neutralized when animals are treated with anti-HMGB1 antibodies, which as noted above is one of the key experimental findings that has implicated HMGB1 in the sterile inflammatory response. Therefore, it is possible that HMGB1 might function either as the initial trigger of sterile inflammation or as a later “downstream” factor that amplifies such responses.
It is also unclear presently whether the HMGB1 molecule is actually proinflammatory by itself. In initial studies, purified HMGB1 was reported to activate leukocytes and stimulate the production of proinflamatory mediators in vitro (36, 37). However, more recently highly purified HMGB1 was found to have little proinflammatory activity (38). It was also noted to tightly bind certain microbial or cellular molecules such as phosphatidylserine (38). This raises the possibility that the proinflammatory activity of HMGB1 released from necrotic cells is actually mediated by an associated molecule.
How important is HMGB1 to triggering an inflammatory response to dead cells? Two lines of evidence suggest that it is neither the only factor nor an essential or even dominant one. First, cells undergoing secondary necrosis do not release HMGB1, yet still stimulate inflammation. Therefore, in this setting cells must release other proinflammatory factors. Second, necrotic mutant embryonic fibroblasts that genetically lack HMGB1 stimulate robust inflammation when injected into mice; in fact this response was of similar magnitude to that observed to wild type cells (Chen et al. manuscript submitted). Therefore there must be other factors in cells that play major roles in initiating the sterile inflammatory response. In fact there are several other intracellular molecules that are candidates to participate in this phenomenon.
Another intracellular molecule that might play a role in stimulating inflammation to dying cells is uric acid. Uric acid is the end product of cellular catabolism of purines. It is present at near saturating levels in body fluids and at much higher concentrations in the cytoplasm of cells (39). It was not known to play any physiological role outside of perhaps contributing anti-oxidant activity to body fluids. However, uric acid was identified as a mediator released from necrotic or apoptotic cells that had adjuvant properties (immunostimulatory properties) in vivo (39) (see section Beyond inflammation: cell death and adaptive immune responses). The biologically active form of this molecule is thought to be monosodium urate (MSU) microcrystals that are postulated to form when intracellular uric acid, which is present in the cytosol at concentrations that would be very supersaturated in extracellular fluids, is released and comes in contact with the high levels of free sodium present in the extracellular environment. MSU crystals that form in extracellular sites are known to be highly inflammatory and, when they deposit in the joints of hyperuricemic patients, cause the inflammatory disease of gout (40). Moreover, injection of MSU in vivo stimulates a robust acute inflammatory response (41). Based on these findings it is possible that uric acid released from dying cells might be one of the signals that stimulates sterile inflammation. However, whether and to what extent this plays a role in these responses is not yet known.
Yet other candidates potentially involved in the sterile inflammatory response to cell death are galectins. These molecules are cytosolic lectins that when released can bind to a number of cell surface receptors, including ones known to activate leukocytes (e.g. CD2 and CD3) (42). In vitro they are chemotactic for monocytes and can activate vascular endothelium, mast cells, macrophages and neutrophils (43). Administration of galectin 3 in vivo recruits monocytes to the site of injection (44). Galectins are expressed primarily in certain leukocytes and endothelium (43). Therefore, if these molecules play a role in initiating the sterile inflammatory response to cell death, it will be limited to the restricted set of cells in which they are expressed.
Thioredoxin is a 12 KDa, ubiquitous intracellular enzyme with antioxdant activity. In vitro thioredoxin augments the expression of TNF in monocytes and IL-6 in endothelial cells(45)and in vivo has chemotactic activity for monocytes, PMNs and lymphocytes (46).
There are a number of other intracellular molecules that have been reported to have some type of proinflammatory activity in vitro and which could potentially be released from dying cells to trigger inflammation. These molecules are listed in Table I. They include: (i) DNA-chromatin complexes, which have been shown under some conditions to stimulate proinflammatory cytokine production from splenocytes and endothelial cells in vitro (47); (ii) Heat Shock proteins (HSP), some of which have been shown to stimulate proinflammatory cytokine production from monocytes and macrophages in vitro (48) (49). Here, a note of caution is warranted because in some systems the immune stimulating activity of HSP has been shown to be due to contaminating microbial molecules (50); (iii) Certain of the S100 family of proteins, which stimulate cytokine production and chemotaxis in vitro (51, 52). These proteins are found in granulocytes, monocytes, macrophages, keratinocytes and, in inflammatory conditions, also epithelial cells (53). In some cells these proteins can be actively secreted; (iv) Antimicrobial peptides such as cathelicidins and defensins have chemotactic activity for leukocytes in vitro (54-56); (v) Various purines such as adenosine and ATP. In vitro these molecules can be chemotactic for leukocytes. ATP can bind to cellular receptors on leukocytes that promote the secretion of the proinflammatory cytokine IL-1 (28); (vi) Mitochondrial peptides bearing the _N_-formyl group characteristic of prokaryotic proteins are chemotactic for monocytes and neutrophils in vitro(57) and in vivo (58).Whether any of these various molecules actually stimulate inflammation in vivo and contribute significantly to the sterile inflammatory response to cell death is presently unknown.
As noted above, some putative mediators, like S100s, are not expressed in all cells. Therefore, an unresolved issue is whether the major proinflammatory triggers are the same in all cells or whether different kinds of cells or conditions release different mediators. This is an important question because different kinds of mediators might lead to different kinds of responses and outcomes. To resolve this issue it will be necessary to identify and evaluate the contribution of the various potential mediators in different cell types, e.g. through neutralization or elimination.
Cellular molecules that activate extracellular proinflammatory mediators
In addition to cellular components with intrinsic proinflammatory activity, other kinds of cellular molecules that potentially contribute to the death-induced inflammatory response are ones that are not themselves proinflammatory, but instead work indirectly by activating extracellular inflammatory mediators. Several extracellular proinflammatory pathways have been postulated to play potential roles in this process (Table II). In most of the cases, more is known about the extracellular mediators than of the cellular components that trigger them.
Table II.
Extracellular matrix with proinflammatory activity
| Factor | Proinflammatory in vitro | Proinflammatory in vivo | Putative receptor |
|---|---|---|---|
| Fibrinogen | neutrophil adherance to vascular blood clots (142) cytokine(143) and chemokine(85) expression in mononuclear cells | ND | CD11b/CD18 (142, 143) TLR4(85) |
| Collagen derived peptide | chemotaxis for fibroblasts, neutrophils and monocytes(71) (72, 73) | synthetic N-acetyl-PGP induces PMN infiltration in cornea (144) or lung (72) | CXCR2 (for PGP) (72) |
| Elastin derived peptide | chemotaxis for neutrophil and monocyte(78, 145) | mAb to elastin fragments eliminates cigarette smoke induced emphysema (145) | Not identified |
| fibronectin | Chemotaxis for fibroblasts, endothelial cells, neutrophils and monocytes(74-77) | ND | integrins(146) |
| Hyaluronic acid | Cytokine and chemokine release (67, 89, 147) DC maturation (89, 148) | In vivo administration induces expression of chemokines (67) | CD44 (147) NOT CD44(148) TLR4(67, 86) TLR2 but NOT TLR4(89) TLR2 and 4(149) |
| Heparan sulfate | Cytokine production (68) Macrophage activation (69) DC maturation(70, 87) | ND | TLR4(87) |
| Laminin derived peptide | Induction of MMP-9 production and release in leukocytes and chemotactixis for inflammatory cells (150) | ND | Not identified |
One extracellular proinflammatory pathway that is thought to contribute to sterile inflammation to tissue injury is the complement system. Intrinsic enzymatic activity to cleave C3 fragment was first identified in lysates of rat myocardium (59). It was subsequently found that complement components were deposited on dead cells in ischemic infarcts (60). Since complement was not found on adjacent viable cells, this observation indicated that dead cells were triggering the complement pathway. One of the well known consequences of activating the complement cascade is the generation of complement fragments that are proinflammatory (see section Molecular basis of inflammation to infection). Later experiments found that if the complement pathway was inhibited in vivo, then there was a reduction in the inflammatory response and/or extent of tissue injury in some experimental model(59, 61). Therefore, the complement pathway is triggered by dead cells and this process contributes to the ensuing sterile inflammatory response.
Dead cells may be triggering the complement system through a number of different mechanisms. There is limited evidence that some cellular components can directly activate the complement cascade. Thus, lysates of myocardium were found to cleave the C3 component into active form (59). Presumably, cellular proteases were responsible for this activity but their identity is unknown. In addition, isolated mitochondrial membranes were also shown to activate complement via the C1q component (62). Again the underlying molecular basis for this activity is not known.
More recently it was discovered that certain cellular components could also activate the complement pathway by an indirect mechanism (63). Non-muscle myosin heavy chains (type IIA and C) are released after ischemia-reperfusion injury of the intestine, myocardium and skeletal muscle. By themselves, the myosin molecules are not intrinsically proinflammatory. However, normal animals have circulating “natural” IgM autoantibodies that bind the myosin chains after they are released to form immune complexes (64). The bound autoantibody activates complement that then initiates the inflammatory response (see section Molecular basis of inflammation to infection). Immunodeficient mice that lack antibody fail to develop inflammatory responses in these models (65). However, when an anti-myosin monoclonal antibody is injected into these animals it is sufficient to initiate inflammation at sites of ischemia reperfusion injury (66). Moreover, in normal mice (antibody sufficient) inhibiting the endogenous anti-myosin antibodies by infusion of myosin peptides, reduces the inflammatory response to ischemic injury (64). Therefore, these data identify myosin heavy chain as a cellular trigger of sterile inflammation in vivo and elucidates a novel mechanism by which it generates this response. It is not yet know whether this mechanism plays a role in generating the inflammatory response to cell death in other settings.
Another set of potential extracellular mediators that might be generated by components released from dead cells are ones derived from components of the extracellular matrix (Table II). One set of such matrix components are the glycosaminoglycans, hyaluronic acid and heparin sulfate. Hyaluronic acid fragments have been shown to stimulate the production of proinflammatory mediators from endothelial and dendritic cells in vitro and to stimulate the production of chemokines when injected in vivo (67). Heparan sulfate has also been shown in vitro to activate macrophages and dendritic cells and stimulate the production of proinflammatory cytokines (68-70). There is also limited data that collagen and elastin-derived peptides, laminin and fibronectin fragments can have proinflammatory activites (71)(72, 73) (74-80). The importance of these mediators to the death-induced inflammatory response in vivo is not known.
The release of cellular proteases have also been suggested to trigger other extracellular pathways that generate proinflammatory mediators. These pathways include the clotting, fibrinolytic and kinin cascades (81). Again, the importance of these pathways to the sterile inflammatory response to dead cells hasn’t been determined or quantified.
Sensing death: Receptors that detect cell death and induce inflammation
The proinflammatory signals that are released from dying cells or extracellular matrix are for the most part not ones that had been previously identified to be biologically active mediators. So how do these signals actually stimulate inflammation? This is an issue that is not well understood. One hypothesis is that there are receptors on host cells that sense when molecules that are normally hidden intracellularly (“hidden self”) are released and then trigger the inflammatory process. This would be similar to the process by which the host generates inflammation to extracellular microbes. In fact, there is emerging evidence that sometimes, the exact same mechanisms may be used to sense infection and cell death: both may use TLRs (Figure 3). As we will review next, this appears to be the case for several of the proinflammatory molecules from dead cells (Table I) and the extracellular matrix (Table II). Thus the immune system may use TLR not only to detect molecules that are “non-self” but also ones that are “hidden self”, i.e. molecules that aren’t ordinarily in the extracellular fluids unless cells die.
HMGB1 is bound by the receptor for advanced glycation end products (RAGE) and it was originally suggested that this was responsible for this molecule’s extracellular biological effects (82). However, more recently HMGB1 was reported to stimulate TLR2 and TLR4 (83). In a model of ischemia-reperfusion injury of the liver, essentially all of HMGB1’s effects were found to be mediated by TLR2 and 4 (32). Interestingly, a number of other cellular and extracellular components have also reported to mediate their proinflammatory effects through these same TLR (e.g. via TLR4: HSP(84), fibrin (85), hyaluronic acid (86), and heparan sulfate (87); via TLR2: HSP (88) and hyaluronic acid (89). Other TLR might also play a role in recognizing released cellular components. For example, although TLR9 has specificity for unmethylated C and G-rich DNA sequences that are abundant in prokaryotic DNA, it can also recognize some similar sequences present more rarely in mammalian DNA and this is thought to account for the stimulatory activity of autologous chromatin-DNA complexes (90).
Since TLR are stimulated by microbial components, the possibility that microbial contaminants might account for some examples of endogenous molecules stimulating TLR, especially TLR4, needs to be kept in mind. Uric acid was originally reported to stimulate inflammation through TLR2 and 4 (91), however a subsequent study failed to find any role for TLR2 or 4 in both genetic loss and gain of function experiments (92). Similarly, the stimulatory activity of some HSP has been attributed to microbial contaminants (50). Therefore, while TLRs may indeed recognize some host components, caution is warranted in evaluating such findings.
Is TLR4 or other TLR the principal mechanism by which dead cells stimulate inflammation? While the studies above provide evidence that TLR can recognize cellular components, they don’t quantify how important they are to the sterile inflammatory response. Insight into this issue has come from studies that have injected dead cells into mice that genetically lack various TLR (Chen et al. manuscript submitted). In this situation, inflammation was reduced in mice that were doubly-deficient in TLR2 and TLR4, confirming that these receptors do indeed play a role in the sterile inflammatory response. However, in the absence of these two TLR, the reduction in the inflammatory response was quite modest. This indicates that there must be other receptors involved in this process. There was no statistically significant reduction in the inflammatory response to dead cells in mice deficient in TLR 1, 3, 6, 7, 9, or 11 and mice don’t express TLR10. It remains possible that TLR not examined (TLR5 or 8) play a role in this process or that multiple TLRs are involved and function in a redundant manner (so loss of individual receptors has little impact). However, mice that lack the ability to signal through TLR (MyD88 and TRIF-mutant mice) still generate acute monocytic inflammation to dead cells (Chen et al. manuscript submitted). Therefore, there are almost certainly other as yet unidentified receptors involved in this process. This raises the interesting possibility that the major receptors and pathways that stimulate inflammation to dying cells and microbes could be different. If this is true, it would have important implications for the potential development of therapeutics that might selectively block the sterile inflammatory response.
The other receptors that sense the release of cellular components and trigger inflammation are not yet clear. Recently, an intracellular nod-like receptor, NALP3, has been implicated in the inflammatory response to MSU (93). NALP3 is a component of a molecular complex called the inflammasome that cleaves the IL-1 precursor into its mature form. In some situations, NALP3 is known to function downstream of a surface receptor (e.g. the P2X7 ATP receptor) (94). For MSU it is not known whether NALP3 is similarly needed downstream of a surface MSU-receptor or MSU somehow directly gains access to NALP3 in the cytosol.
Host mediators that drive the inflammatory response to dying cells
After the initial recognition of a dead cell or microbe, the ensuing inflammatory response that evolves is largely orchestrated and amplified by mediators generated by host cells. These factors include cytokines, chemokines, vasoactive amines, and phospholipids metabolites. There are large numbers of such mediators and many have overlapping functions. While many of these mediators are likely to contribute to the inflammatory response to dead cells, the extent of their contributions to this process and whether there are differences in the principal ones that drive inflammation to dying cells versus microbes is not well understood.
Recently, the proinflammatory cytokine IL-1 was unexpectedly found to have a key role in the acute neutrophilic inflammatory response to dead cells (Chen et al. manuscript submitted). When sterile dead cells were injected into mice, wild type animals developed a strong neutrophilic inflammatory response. In contrast this response was markedly suppressed in mice genetically lacking either the IL-1 receptor (IL-1R) or MyD88, the intracellular adaptor that is essential for IL-1R signaling. Similarly, neutrophilic inflammation to hepatocyte death induced by injection of a hepatotoxic drug was markedly reduced in mice lacking the IL-1R-MyD88 pathway (Chen et al. manuscript submitted). Therefore, IL-1 is a particularly important host mediator in the acute neutrophilic inflammatory response to dead cells.
Interestingly, in bone marrow-transplanted (chimeric) mice, the sterile neutrophilic inflammatory response to dead cells required the IL-1R on parenchymal (radioresistant) cells but not ones of bone marrow origin (radiosensitive) (Chen et al. manuscript submitted). Therefore, for this response IL-1 must be principally acting in on cells resident in the tissues rather than on neutrophils or other leukocytes. These data suggest that IL-1 functions to orchestrate the neutrophilic response into tissues at sites of cell death. Whether this is true in all tissues and with all types of dying cells remains to be determined.
Surprisingly, the recruitment of monocytes into sites of cell death was either not reduced or only modestly decreased in IL-1R or MyD88-deficient mice (Chen et al. manuscript submitted). This indicates that after cell death triggers the inflammatory response, the recruitment of monocytes and neutrophils are controlled by different mechanisms. Also surprisingly, neurophilic inflammation to a microbial stimulus, yeast zymosan, was not reduced in mice lacking the IL-1R (Chen et al. manuscript submitted). This indicated that in some situations, the acute inflammatory response to microbial components and dead cells can be controlled by different mediators.
In addition to IL-1, it is likely that other proinflammatory cytokines play important roles in the inflammatory response to dead cells. E.g., TNFα and IL-6 play major roles in many sterile inflammatory responses, e.g. rheumatoid arthritis (95) and systemic-onset juvenile idiopathic arthritis (96), respectively. These and many other cytokines and mediators could well play major roles in death-induced inflammation. However, the role of these various factors in responses to dead cells has not been specifically examined or quantified.
The dark side of sterile inflammation: collateral damage to normal tissues – Therapeutic implications
While the inflammatory response plays important roles in protecting the host and repairing tissues, it can also damage normal tissues. Molecules generated to kill microbes, such as reactive oxygen species and proteases, leak from live and dying leukocytes and kill normal cells. These and other mediators that are generated can also cause the host significant pain and disability. Thus for the host, inflammation is a “double-edged sword”.
The “cost-benefit” ratio of the negative to positive effects of inflammation varies with the situation. In an infection, the collateral damage to normal tissues may be a small price to pay to contain a potentially dangerous microorganism as rapidly as possible. However, in situations where there is sterile tissue injury, inflammation is more costly to the host and in some settings may actually do more harm than good. This is illustrated in experimental models of acute myocardial infarctions and in toxic damage to the liver where tissue damage is substantially reduced by the depletion of neutrophils (97, 98) or blocking the mediators that lead to their accumulation (31, 61). There is similar evidence that wound healing is actually prolonged by neutrophilic inflammation (99). Thus, in a number of acute settings, the neutrophil component of sterile inflammation may be particularly detrimental.
However, tissue damage from inflammation is not only limited to acute responses and neutrophils. Macrophage rich chronic inflammatory infiltrates can also damage normal tissues and this is thought to underlie the pathogenesis of many chronic diseases, e.g. atherosclerosis, autoimmune diseases, chronic obstructive lung diseases and Alzheimer’s disease. There are a number of sterile processes that can stimulate such chronic responses including ones, such as sunburn (in SLE) or cigarette smoke (in COPD), in which cell death causes or contributes to the inflammatory response. Thus the ongoing inflammatory response to sterile cell death may cause disease.
In those settings where inflammation to dying cells does more harm than good it would potentially be useful to block these responses therapeutically. It would be most attractive if this could be done in ways that don’t generally compromise host defense against infections or interfere with the healing process. This might be achieved if it were possible to selectively inhibit inflammation to sterile dead cells. Such selective inhibition might be achieved by neutralizing the cellular triggers of inflammation that are released from dead cells or inhibiting the receptors that sense them. This is one of the reasons why it’s important to elucidate these molecules and identify the most important ones.
Another potential therapeutic strategy might be to inhibit only the neutrophilic component of this response, ideally without blocking effective responses to microbes. This might be achieved by blocking the inflammatory mediators that are needed to recruit neutrophils to the site of injury. That this might be possible and beneficial is shown in experiments where liver damage was induced and the IL-1 receptor pathway was blocked (Chen et al. manuscript submitted). In this situation, neutrophilic inflammation was inhibited and the ensuing liver damage was markedly reduced. Under these same conditions, monocyte recruitment was much less affected and neutrophil recruitment to at lease one microbial stimulus was normal (Chen et al. manuscript submitted). Moreover, blocking IL-1R function in humans has not been associated with infectious complications(100). Thus, it might actually be possible to selectively block neutrophilic inflammation to tissue damage under conditions that preserves host defense functions to microbes.
Beyond inflammation: cell death and adaptive immune responses
In addition to stimulating inflammation (an innate immune response), dying cells may also provide signals that help mobilize adaptive (lymphocyte) immunity to antigens present in the affected tissue. This notion was first conceived on theoretic grounds and termed the “danger hypothesis” (7). This hypothesis proposed that the adaptive immune system evolved to recognize cell death as a potentially dangerous situation and that this recognition would promote immune responses to antigens associated with the site of injury. To understand this idea, it is necessary to first discuss how T cell immune responses are initiated.
The first step in the initiation of a T cell immune response occurs when dendritic cells acquire antigen from the local environment. These cells then hydrolyze the antigen into peptides some of which are displayed on their surface bound to MHC molecules (101). The dendritic cells then migrate into secondary lymphoid organs when they interact with T cells (102). T cells are then stimulated when they recognize their specific antigenic peptide bound to an MHC molecule on the dendritic cell. However this recognition is insufficient to productively activate the T cell and by itself may actually tolerize (inactivate) the lymphocyte (103). To become fully activated the T cell must also receive other co-stimulatory signals from the dendritic cell and these second signals are only provided if the dendritic cells have previously been stimulated in certain ways (104).
One of the important signals that can activate dendritic cells to become immunostimulatory are microbial molecules that engage receptors like the TLRs (105) (19). Through this mechanism, T cell responses will only be generated in response to a potential threat, such as an infection, and not to other innocuous situations. In addition, the danger hypothesis proposed that dying cells also provide immunostimulatory signals (“danger signals”) and that this could explain how T cell responses were generated to cellular components in transplants, cancers and autoimmunity in the absence of infection (7). Subsequent studies showed that dead cells could indeed activate dendritic cells and promote the generation of T cells response to associated antigens (106, 107).
Cells contain multiple danger signals that are released upon death and disintegration of the plasma membrane (7, 39). Whether these molecules are the same ones that stimulate inflammation is not clear but is certainly possible. Uric acid in the form of MSU was shown to be one of the endogenous cellular components capable of activating dendritic cells and promoting CD8 T cell responses to cell-associated antigens (39). In addition, there is also evidence that HMGB1 can also function as a danger signal that could induce DC maturation in vitro and promote antitumor immunity in vivo (108). HSP have also been postulated to have immune stimulating activities (109). It is likely that there are other such molecules e.g. defensins (110) and it will be important to determine whether they have the same or distinct effects on the immune system both with respect to their ability to induce inflammation and to affect adaptive immune responses.
These danger signals when released can help promote the generation of CD4 and CD8 T cell responses to immunogenic antigens that are present in or around the dying cells. This provides an additional surveillance mechanism to detect pathological processes and recruit specialized defenders to reinforce the initial innate defenses of the inflammatory response. Such adaptive responses are important for host defense against microbes and tumors. However, this process also has potential downsides. As with inflammation, the adaptive immune response may cause collateral damage. Moreover, the release of both antigens and danger signals may lead to the initiation of autoimmune disease in those individuals that are not fully tolerant to self antigens (106, 111, 112). Furthermore, this process may also contribute to the initiation of cellular rejection mechanisms against organ transplants (106, 111).
Conclusions
It is clear that when cells die they set in motion a number of important processes. One is the rapid recruitment of innate immune components from the blood as part of a process we recognize as inflammation. Another parallel process is the mobilization of highly specific T and B cell defenses from more distal sites. In this manner this is a rapid but indiscriminate response followed more slowly by one that is much more highly specific. This helps to ensure adequate host defense. These responses induced by cell injury are double-edged swords. On the one hand they protect and help heal injured tissues, while on the other hand they can cause significant damage and disease. The signals that drive these sterile inflammatory and adaptive immune response are normally concealed in the interior of cells and released when cells loses integrity of their plasma membrane. The host then recognizes the release of these normally hidden self molecules and initiates responses. There has been progress in identifying some these signals and their potential modes of action. Nevertheless there still remains much to be learned about the role and mechanism of action of these factors as well as others that are almost certainly waiting to be discovered.
Future Issues.
- How many intracellular molecules have the capacity to trigger inflammation?
- Which of these molecules are the most important?
- Do different cells have different intracellular proinflammatory molecules? Do different conditions lead to the release of different intracellular mediators?
- Do these various molecules all do the same thing or subserve different functions?
- What are the pathways and receptors that are activated by the cellular proinflammatory molecules?
- What are the key mediators that ultimately drive the various components of the sterile inflammatory response?
- Are the “danger signals” that stimulate inflammation the same or different from the ones that stimulate adaptive immune responses?
- Can the inflammatory response to tissue injury be selectively inhibited without compromising host defense and/or healing? What are the molecules and pathways that can be targeted for therapeutic intervention?
Acknowledgments
This work was supported by grants to KLR from the NIH, core resources supported by the Diabetes Endocrinology Research Center grant DK32520, of which KLR is a member, and from Kanae Foundation to HK. We thank Tom Smith for help with photomicroscopy.
Contributor Information
Kenneth L. Rock, Email: kenneth.rock@umassmed.edu.
Hajime Kono, Email: hajime.kono@umassmed.edu.
Literature cited
- 1.Steinhoff M, Vergnolle N, Young SH, Tognetto M, Amadesi S, Ennes HS, Trevisani M, Hollenberg MD, Wallace JL, Caughey GH, Mitchell SE, Williams LM, Geppetti P, Mayer EA, Bunnett NW. Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat Med. 2000;6:151–8. doi: 10.1038/72247. [DOI] [PubMed] [Google Scholar]
- 2.Kaplan AP, Joseph K, Silverberg M. Pathways for bradykinin formation and inflammatory disease. J Allergy Clin Immunol. 2002;109:195–209. doi: 10.1067/mai.2002.121316. [DOI] [PubMed] [Google Scholar]
- 3.Klinger MH. Platelets and inflammation. Anat Embryol (Berl) 1997;196:1–11. doi: 10.1007/s004290050075. [DOI] [PubMed] [Google Scholar]
- 4.Page AR, Good RA. A clinical and experimental study of the function of neutrophils in the inflammatory response. Am J Pathol. 1958;34:645–69. [PMC free article] [PubMed] [Google Scholar]
- 5.Majno G, La Gattuta M, Thompson TE. Cellular death and necrosis: chemical, physical and morphologic changes in rat liver. Virchows Arch Pathol Anat Physiol Klin Med. 1960;333:421–65. doi: 10.1007/BF00955327. [DOI] [PubMed] [Google Scholar]
- 6.Boxer L, Dale DC. Neutropenia: causes and consequences. Semin Hematol. 2002;39:75–81. doi: 10.1053/shem.2002.31911. [DOI] [PubMed] [Google Scholar]
- 7.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 8.Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6:836–48. doi: 10.1038/nri1961. [DOI] [PubMed] [Google Scholar]
- 9.Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol. 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- 10.Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest. 2002;109:41–50. doi: 10.1172/JCI11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chung EY, Kim SJ, Ma XJ. Regulation of cytokine production during phagocytosis of apoptotic cells. Cell Res. 2006;16:154–61. doi: 10.1038/sj.cr.7310021. [DOI] [PubMed] [Google Scholar]
- 12.Nagata S. Apoptosis by death factor. Cell. 1997;88:355–65. doi: 10.1016/s0092-8674(00)81874-7. [DOI] [PubMed] [Google Scholar]
- 13.Faouzi S, Burckhardt BE, Hanson JC, Campe CB, Schrum LW, Rippe RA, Maher JJ. Anti-Fas induces hepatic chemokines and promotes inflammation by an NF-kappa B-independent, caspase-3-dependent pathway. J Biol Chem. 2001;276:49077–82. doi: 10.1074/jbc.M109791200. [DOI] [PubMed] [Google Scholar]
- 14.Fujimoto I, Pan J, Takizawa T, Nakanishi Y. Virus clearance through apoptosis-dependent phagocytosis of influenza A virus-infected cells by macrophages. J Virol. 2000;74:3399–403. doi: 10.1128/jvi.74.7.3399-3403.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 16.Inohara C, McDonald C, Nunez G. NOD-LRR proteins: role in host-microbial interactions and inflammatory disease. Annu Rev Biochem. 2005;74:355–83. doi: 10.1146/annurev.biochem.74.082803.133347. [DOI] [PubMed] [Google Scholar]
- 17.Robinson MJ, Sancho D, Slack EC, Leibundgut-Landmann S, Sousa CR. Myeloid C-type lectins in innate immunity. Nat Immunol. 2006;7:1258–65. doi: 10.1038/ni1417. [DOI] [PubMed] [Google Scholar]
- 18.Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–7. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 19.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 20.Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol. 1994;12:141–79. doi: 10.1146/annurev.iy.12.040194.001041. [DOI] [PubMed] [Google Scholar]
- 21.Kohl J. The role of complement in danger sensing and transmission. Immunol Res. 2006;34:157–76. doi: 10.1385/IR:34:2:157. [DOI] [PubMed] [Google Scholar]
- 22.Volanakis JE. Participation of C3 and its ligands in complement activation. Curr Top Microbiol Immunol. 1990;153:1–21. doi: 10.1007/978-3-642-74977-3_1. [DOI] [PubMed] [Google Scholar]
- 23.Gadjeva M, Takahashi K, Thiel S. Mannan-binding lectin--a soluble pattern recognition molecule. Mol Immunol. 2004;41:113–21. doi: 10.1016/j.molimm.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 24.Cooper NR. The classical complement pathway: activation and regulation of the first complement component. Adv Immunol. 1985;37:151–216. doi: 10.1016/s0065-2776(08)60340-5. [DOI] [PubMed] [Google Scholar]
- 25.Ehlers MR. CR3: a general purpose adhesion-recognition receptor essential for innate immunity. Microbes Infect. 2000;2:289–94. doi: 10.1016/s1286-4579(00)00299-9. [DOI] [PubMed] [Google Scholar]
- 26.Esser AF. Big MAC attack: complement proteins cause leaky patches. Immunol Today. 1991;12:316–8. doi: 10.1016/0167-5699(91)90006-F. discussion 21. [DOI] [PubMed] [Google Scholar]
- 27.Gerard C, Gerard NP. C5A anaphylatoxin and its seven transmembrane-segment receptor. Annu Rev Immunol. 1994;12:775–808. doi: 10.1146/annurev.iy.12.040194.004015. [DOI] [PubMed] [Google Scholar]
- 28.Mariathasan S, Monack DM. Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007;7:31–40. doi: 10.1038/nri1997. [DOI] [PubMed] [Google Scholar]
- 29.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–42. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 30.Landsman D, Bustin M. A signature for the HMG-1 box DNA-binding proteins. Bioessays. 1993;15:539–46. doi: 10.1002/bies.950150807. [DOI] [PubMed] [Google Scholar]
- 31.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–5. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 32.Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA, Billiar TR. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005;201:1135–43. doi: 10.1084/jem.20042614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Limana F, Germani A, Zacheo A, Kajstura J, Di Carlo A, Borsellino G, Leoni O, Palumbo R, Battistini L, Rastaldo R, Müller S, Pompilio G, Anversa P, Bianchi ME, Capogrossi MC. Exogenous high-mobility group box 1 protein induces myocardial regeneration after infarction via enhanced cardiac C-kit+ cell proliferation and differentiation. Circ Res. 2005;97:e73–83. doi: 10.1161/01.RES.0000186276.06104.04. [DOI] [PubMed] [Google Scholar]
- 34.Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME, Rubartelli A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002;3:995–1001. doi: 10.1093/embo-reports/kvf198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang H, Wang H, Czura CJ, Tracey KJ. The cytokine activity of HMGB1. J Leukoc Biol. 2005;78:1–8. doi: 10.1189/jlb.1104648. [DOI] [PubMed] [Google Scholar]
- 36.Li J, Wang H, Mason JM, Levine J, Yu M, Ulloa L, Czura CJ, Tracey KJ, Yang H. Recombinant HMGB1 with cytokine-stimulating activity. J Immunol Methods. 2004;289:211–23. doi: 10.1016/j.jim.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 37.Zimmermann K, Volkel D, Pable S, Lindner T, Kramberger F, Bahrami S, Scheiflinger F. Native versus recombinant high-mobility group B1 proteins: functional activity in vitro. Inflammation. 2004;28:221–9. doi: 10.1023/b:ifla.0000049047.61014.e3. [DOI] [PubMed] [Google Scholar]
- 38.Rouhiainen A, Tumova S, Valmu L, Kalkkinen N, Rauvala H. Analysis of proinflammatory activity of highly purified eukaryotic recombinant HMGB1 (amphoterin) J Leukoc Biol. 2007;81:49–58. doi: 10.1189/jlb.0306200. [DOI] [PubMed] [Google Scholar]
- 39.Shi Y, Evans JE, Rock KL. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425:516–21. doi: 10.1038/nature01991. [DOI] [PubMed] [Google Scholar]
- 40.Terkeltaub R. Pathogenesis and treatment of crystal-induced inflammation. In: Koopman W, Moreland LW, editors. Arthritis and allied conditions. Philadelphia: Lippincott Williams and Wilkins; 2005. p. 2357. [Google Scholar]
- 41.Gordon TP, Kowanko IC, James M, Roberts-Thomson PJ. Monosodium urate crystal-induced prostaglandin synthesis in the rat subcutaneous air pouch. Clin Exp Rheumatol. 1985;3:291–6. [PubMed] [Google Scholar]
- 42.Liu FT, Rabinovich GA. Galectins as modulators of tumour progression. Nat Rev Cancer. 2005;5:29–41. doi: 10.1038/nrc1527. [DOI] [PubMed] [Google Scholar]
- 43.Almkvist J, Karlsson A. Galectins as inflammatory mediators. Glycoconj J. 2004;19:575–81. doi: 10.1023/B:GLYC.0000014088.21242.e0. [DOI] [PubMed] [Google Scholar]
- 44.Sano H, Hsu DK, Yu L, Apgar JR, Kuwabara I, Yamanaka T, Hirashima M, Liu FT. Human galectin-3 is a novel chemoattractant for monocytes and macrophages. J Immunol. 2000;165:2156–64. doi: 10.4049/jimmunol.165.4.2156. [DOI] [PubMed] [Google Scholar]
- 45.Schenk H, Vogt M, Droge W, Schulze-Osthoff K. Thioredoxin as a potent costimulus of cytokine expression. J Immunol. 1996;156:765–71. [PubMed] [Google Scholar]
- 46.Bertini R, Howard OM, Dong HF, Oppenheim JJ, Bizzarri C, Sergi R, Caselli G, Pagliei S, Romines B, Wilshire JA, Mengozzi M, Nakamura H, Yodoi J, Pekkari K, Gurunath R, Holmgren A, Herzenberg LA, Herzenberg LA, Ghezzi P. Thioredoxin, a redox enzyme released in infection and inflammation, is a unique chemoattractant for neutrophils, monocytes, and T cells. J Exp Med. 1999;189:1783–9. doi: 10.1084/jem.189.11.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hefeneider SH, Cornell KA, Brown LE, Bakke AC, McCoy SL, Bennett RM. Nucleosomes and DNA bind to specific cell-surface molecules on murine cells and induce cytokine production. Clin Immunol Immunopathol. 1992;63:245–51. doi: 10.1016/0090-1229(92)90229-h. [DOI] [PubMed] [Google Scholar]
- 48.Tsan MF, Gao B. Cytokine function of heat shock proteins. Am J Physiol Cell Physiol. 2004;286:C739–44. doi: 10.1152/ajpcell.00364.2003. [DOI] [PubMed] [Google Scholar]
- 49.Wallin RP, Lundqvist A, More SH, von Bonin A, Kiessling R, Ljunggren HG. Heat-shock proteins as activators of the innate immune system. Trends Immunol. 2002;23:130–5. doi: 10.1016/s1471-4906(01)02168-8. [DOI] [PubMed] [Google Scholar]
- 50.Bausinger H, Lipsker D, Ziylan U, Manie S, Briand JP, Cazenave JP, Muller S, Haeuw JF, Ravanat C, de la Salle H, Hanau D. Endotoxin-free heat-shock protein 70 fails to induce APC activation. Eur J Immunol. 2002;32:3708–13. doi: 10.1002/1521-4141(200212)32:12<3708::AID-IMMU3708>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 51.Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 52.Ryckman C, Vandal K, Rouleau P, Talbot M, Tessier PA. Proinflammatory activities of S100: proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J Immunol. 2003;170:3233–42. doi: 10.4049/jimmunol.170.6.3233. [DOI] [PubMed] [Google Scholar]
- 53.Foell D, Wittkowski H, Vogl T, Roth J. S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukoc Biol. 2007;81:28–37. doi: 10.1189/jlb.0306170. [DOI] [PubMed] [Google Scholar]
- 54.Zanetti M. Cathelicidins, multifunctional peptides of the innate immunity. J Leukoc Biol. 2004;75:39–48. doi: 10.1189/jlb.0403147. [DOI] [PubMed] [Google Scholar]
- 55.Kurosaka K, Chen Q, Yarovinsky F, Oppenheim JJ, Yang D. Mouse cathelin-related antimicrobial peptide chemoattracts leukocytes using formyl peptide receptor-like 1/mouse formyl peptide receptor-like 2 as the receptor and acts as an immune adjuvant. J Immunol. 2005;174:6257–65. doi: 10.4049/jimmunol.174.10.6257. [DOI] [PubMed] [Google Scholar]
- 56.Yang D, Chertov O, Bykovskaia SN, Chen Q, Buffo MJ, Shogan J, Anderson M, Schroder JM, Wang JM, Howard OM, Oppenheim JJ. Beta-defensins: linking innate and adaptive immunity through dendritic and T cell CCR6. Science. 1999;286:525–8. doi: 10.1126/science.286.5439.525. [DOI] [PubMed] [Google Scholar]
- 57.Carp H. Mitochondrial N-formylmethionyl proteins as chemoattractants for neutrophils. J Exp Med. 1982;155:264–75. doi: 10.1084/jem.155.1.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gawlowski DM, Benoit JN, Granger HJ. Microvascular pressure and albumin extravasation after leukocyte activation in hamster cheek pouch. Am J Physiol. 1993;264:H541–6. doi: 10.1152/ajpheart.1993.264.2.H541. [DOI] [PubMed] [Google Scholar]
- 59.Hill JH, Ward PA. The phlogistic role of C3 leukotactic fragments in myocardial infarcts of rats. J Exp Med. 1971;133:885–900. doi: 10.1084/jem.133.4.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pinckard RN, Olson MS, Giclas PC, Terry R, Boyer JT, O’Rourke RA. Consumption of classical complement components by heart subcellular membranes in vitro and in patients after acute myocardial infarction. J Clin Invest. 1975;56:740–50. doi: 10.1172/JCI108145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weisman HF, Bartow T, Leppo MK, Marsh HC, Jr, Carson GR, Concino MF, Boyle MP, Roux KH, Weisfeldt ML, Fearon DT. Soluble human complement receptor type 1: in vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science. 1990;249:146–51. doi: 10.1126/science.2371562. [DOI] [PubMed] [Google Scholar]
- 62.Pinckard RN, Olson MS, Kelley RE, DeHeer DH, Palmer JD, O’Rourke RA, Goldfein S. Antibody-independent activation of human C1 after interaction with heart subcellular membranes. J Immunol. 1973;110:1376–82. [PubMed] [Google Scholar]
- 63.Carroll MC, Holers VM. Innate autoimmunity. Adv Immunol. 2005;86:137–57. doi: 10.1016/S0065-2776(04)86004-8. [DOI] [PubMed] [Google Scholar]
- 64.Zhang M, Alicot EM, Chiu I, Li J, Verna N, Vorup-Jensen T, Kessler B, Shimaoka M, Chan R, Friend D, Mahmood U, Weissleder R, Moore FD, Carroll MC. Identification of the target self-antigens in reperfusion injury. J Exp Med. 2006;203:141–52. doi: 10.1084/jem.20050390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weiser MR, Williams JP, Moore, K L, M M, H Hb, C Mc. Reperfusion injury of ischemic skeletal muscle is mediated by natural antibody and complement. J Exp Med. 1996;183:2343–8. doi: 10.1084/jem.183.5.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang M, Austen WG, Jr, Chiu I, Alicot EM, Hung R, Ma M, Verna N, Xu M, Hechtman HB, Moore FD, Jr, Carroll MC. Identification of a specific self-reactive IgM antibody that initiates intestinal ischemia/reperfusion injury. Proc Natl Acad Sci U S A. 2004;101:3886–91. doi: 10.1073/pnas.0400347101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Taylor KR, Trowbridge JM, Rudisill JA, Termeer CC, Simon JC, Gallo RL. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J Biol Chem. 2004;279:17079–84. doi: 10.1074/jbc.M310859200. [DOI] [PubMed] [Google Scholar]
- 68.Wrenshall LE, Cerra FB, Carlson A, Bach FH, Platt JL. Regulation of murine splenocyte responses by heparan sulfate. J Immunol. 1991;147:455–9. [PubMed] [Google Scholar]
- 69.Wrenshall LE, Stevens RB, Cerra FB, Platt JL. Modulation of macrophage and B cell function by glycosaminoglycans. J Leukoc Biol. 1999;66:391–400. doi: 10.1002/jlb.66.3.391. [DOI] [PubMed] [Google Scholar]
- 70.Kodaira Y, Nair SK, Wrenshall LE, Gilboa E, Platt JL. Phenotypic and functional maturation of dendritic cells mediated by heparan sulfate. J Immunol. 2000;165:1599–604. doi: 10.4049/jimmunol.165.3.1599. [DOI] [PubMed] [Google Scholar]
- 71.Postlethwaite AE, Kang AH. Collagen-and collagen peptide-induced chemotaxis of human blood monocytes. J Exp Med. 1976;143:1299–307. doi: 10.1084/jem.143.6.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weathington NM, van Houwelingen AH, Noerager BD, Jackson PL, Kraneveld AD, Galin FS, Folkerts G, Nijkamp FP, Blalock JE. A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat Med. 2006;12:317–23. doi: 10.1038/nm1361. [DOI] [PubMed] [Google Scholar]
- 73.Chang C, Houck JC. Demonstration of the chemotactic properties of collagen. Proc Soc Exp Biol Med. 1970;134:22–6. doi: 10.3181/00379727-134-34719. [DOI] [PubMed] [Google Scholar]
- 74.Postlethwaite AE, Keski-Oja J, Balian G, Kang AH. Induction of fibroblast chemotaxis by fibronectin. Localization of the chemotactic region to a 140,000-molecular weight non-gelatin-binding fragment. J Exp Med. 1981;153:494–9. doi: 10.1084/jem.153.2.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tsukamoto Y, Helsel WE, Wahl SM. Macrophage production of fibronectin, a chemoattractant for fibroblasts. J Immunol. 1981;127:673–8. [PubMed] [Google Scholar]
- 76.Bowersox JC, Sorgente N. Chemotaxis of aortic endothelial cells in response to fibronectin. Cancer Res. 1982;42:2547–51. [PubMed] [Google Scholar]
- 77.Norris DA, Clark RA, Swigart LM, Huff JC, Weston WL, Howell SE. Fibronectin fragment(s) are chemotactic for human peripheral blood monocytes. J Immunol. 1982;129:1612–8. [PubMed] [Google Scholar]
- 78.Senior RM, Griffin GL, Mecham RP. Chemotactic activity of elastin-derived peptides. J Clin Invest. 1980;66:859–62. doi: 10.1172/JCI109926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Senior RM, Griffin GL, Mecham RP, Wrenn DS, Prasad KU, Urry DW. Val-Gly-Val-Ala-Pro-Gly, a repeating peptide in elastin, is chemotactic for fibroblasts and monocytes. J Cell Biol. 1984;99:870–4. doi: 10.1083/jcb.99.3.870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kamoun A, Landeau JM, Godeau G, Wallach J, Duchesnay A, Pellat B, Hornebeck W. Growth stimulation of human skin fibroblasts by elastin-derived peptides. Cell Adhes Commun. 1995;3:273–81. doi: 10.3109/15419069509081013. [DOI] [PubMed] [Google Scholar]
- 81.Kaplan AP, Joseph K, Shibayama Y, Reddigari S, Ghebrehiwet B, Silverberg M. The intrinsic coagulation/kinin-forming cascade: assembly in plasma and cell surfaces in inflammation. Adv Immunol. 1997;66:225–72. doi: 10.1016/s0065-2776(08)60599-4. [DOI] [PubMed] [Google Scholar]
- 82.Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, Nagashima M, Lundh ER, Vijay S, Nitecki D, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem. 1995;270:25752–61. doi: 10.1074/jbc.270.43.25752. [DOI] [PubMed] [Google Scholar]
- 83.Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 84.Ohashi K, Burkart V, Flohe S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol. 2000;164:558–61. doi: 10.4049/jimmunol.164.2.558. [DOI] [PubMed] [Google Scholar]
- 85.Smiley ST, King JA, Hancock WW. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J Immunol. 2001;167:2887–94. doi: 10.4049/jimmunol.167.5.2887. [DOI] [PubMed] [Google Scholar]
- 86.Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, Miyake K, Freudenberg M, Galanos C, Simon JC. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. 2002;195:99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Johnson GB, Brunn GJ, Kodaira Y, Platt JL. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J Immunol. 2002;168:5233–9. doi: 10.4049/jimmunol.168.10.5233. [DOI] [PubMed] [Google Scholar]
- 88.Vabulas RM, Ahmad-Nejad P, da Costa C, Miethke T, Kirschning CJ, Hacker H, Wagner H. Endocytosed HSP60s use toll-like receptor 2 (TLR2) and TLR4 to activate the toll/interleukin-1 receptor signaling pathway in innate immune cells. J Biol Chem. 2001;276:31332–9. doi: 10.1074/jbc.M103217200. [DOI] [PubMed] [Google Scholar]
- 89.Scheibner KA, Lutz MA, Boodoo S, Fenton MJ, Powell JD, Horton MR. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. J Immunol. 2006;177:1272–81. doi: 10.4049/jimmunol.177.2.1272. [DOI] [PubMed] [Google Scholar]
- 90.Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:603–7. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 91.Liu-Bryan R, Scott P, Sydlaske A, Rose DM, Terkeltaub R. Innate immunity conferred by Toll-like receptors 2 and 4 and myeloid differentiation factor 88 expression is pivotal to monosodium urate monohydrate crystal-induced inflammation. Arthritis Rheum. 2005;52:2936–46. doi: 10.1002/art.21238. [DOI] [PubMed] [Google Scholar]
- 92.Chen CJ, Shi Y, Hearn A, Fitzgerald K, Golenbock D, Reed G, Akira S, Rock KL. MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest. 2006;116:2262–71. doi: 10.1172/JCI28075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–41. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 94.Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–32. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 95.Maini R, St Clair EW, Breedveld F, Furst D, Kalden J, Weisman M, Smolen J, Emery P, Harriman G, Feldmann M, Lipsky P. Infliximab (chimeric anti-tumour necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised phase III trial. ATTRACT Study Group. Lancet. 1999;354:1932–9. doi: 10.1016/s0140-6736(99)05246-0. [DOI] [PubMed] [Google Scholar]
- 96.Yokota S, Miyamae T, Imagawa T, Iwata N, Katakura S, Mori M, Woo P, Nishimoto N, Yoshizaki K, Kishimoto T. Therapeutic efficacy of humanized recombinant anti-interleukin-6 receptor antibody in children with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2005;52:818–25. doi: 10.1002/art.20944. [DOI] [PubMed] [Google Scholar]
- 97.Romson JL, Hook BG, Kunkel SL, Abrams GD, Schork MA, Lucchesi BR. Reduction of the extent of ischemic myocardial injury by neutrophil depletion in the dog. Circulation. 1983;67:1016–23. doi: 10.1161/01.cir.67.5.1016. [DOI] [PubMed] [Google Scholar]
- 98.Liu ZX, Han D, Gunawan B, Kaplowitz N. Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology. 2006;43:1220–30. doi: 10.1002/hep.21175. [DOI] [PubMed] [Google Scholar]
- 99.Martin P, Leibovich SJ. Inflammatory cells during wound repair: the good, the bad and the ugly. Trends Cell Biol. 2005;15:599–607. doi: 10.1016/j.tcb.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 100.Fleischmann R, Stern R, Iqbal I. Anakinra: an inhibitor of IL-1 for the treatment of rheumatoid arthritis. Expert Opin Biol Ther. 2004;4:1333–44. doi: 10.1517/14712598.4.8.1333. [DOI] [PubMed] [Google Scholar]
- 101.Germain RN. MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell. 1994;76:287–99. doi: 10.1016/0092-8674(94)90336-0. [DOI] [PubMed] [Google Scholar]
- 102.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–52. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 103.Schwartz RH. A cell culture model for T lymphocyte clonal anergy. Science. 1990;248:1349–56. doi: 10.1126/science.2113314. [DOI] [PubMed] [Google Scholar]
- 104.Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol. 2002;2:116–26. doi: 10.1038/nri727. [DOI] [PubMed] [Google Scholar]
- 105.Brightbill HD, Libraty DH, Krutzik SR, Yang RB, Belisle JT, Bleharski JR, Maitland M, Norgard MV, Plevy SE, Smale ST, Brennan PJ, Bloom BR, Godowski PJ, Modlin RL. Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science. 1999;285:732–6. doi: 10.1126/science.285.5428.732. [DOI] [PubMed] [Google Scholar]
- 106.Gallucci S, Lolkema M, Matzinger P. Natural adjuvants: endogenous activators of dendritic cells. Nat Med. 1999;5:1249–55. doi: 10.1038/15200. [DOI] [PubMed] [Google Scholar]
- 107.Shi Y, Zheng W, Rock KL. Cell injury releases endogenous adjuvants that stimulate cytotoxic T cell responses. Proc Natl Acad Sci U S A. 2000;97:14590–5. doi: 10.1073/pnas.260497597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rovere-Querini P, Capobianco A, Scaffidi P, Valentinis B, Catalanotti F, Giazzon M, Dumitriu IE, Muller S, Iannacone M, Traversari C, Bianchi ME, Manfredi AA. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep. 2004;5:825–30. doi: 10.1038/sj.embor.7400205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Melcher A, Todryk S, Hardwick N, Ford M, Jacobson M, Vile RG. Tumor immunogenicity is determined by the mechanism of cell death via induction of heat shock protein expression. Nat Med. 1998;4:581–7. doi: 10.1038/nm0598-581. [DOI] [PubMed] [Google Scholar]
- 110.Biragyn A, Ruffini PA, Leifer CA, Klyushnenkova E, Shakhov A, Chertov O, Shirakawa AK, Farber JM, Segal DM, Oppenheim JJ, Kwak LW. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science. 2002;298:1025–9. doi: 10.1126/science.1075565. [DOI] [PubMed] [Google Scholar]
- 111.Shi Y, Galusha SA, Rock KL. Cutting edge: elimination of an endogenous adjuvant reduces the activation of CD8 T lymphocytes to transplanted cells and in an autoimmune diabetes model. J Immunol. 2006;176:3905–8. doi: 10.4049/jimmunol.176.7.3905. [DOI] [PubMed] [Google Scholar]
- 112.Turley S, Poirot L, Hattori M, Benoist C, Mathis D. Physiological beta cell death triggers priming of self-reactive T cells by dendritic cells in a type-1 diabetes model. J Exp Med. 2003;198:1527–37. doi: 10.1084/jem.20030966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Degryse B, Bonaldi T, Scaffidi P, Muller S, Resnati M, Sanvito F, Arrigoni G, Bianchi ME. The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J Cell Biol. 2001;152:1197–206. doi: 10.1083/jcb.152.6.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Messmer D, Yang H, Telusma G, Knoll F, Li J, Messmer B, Tracey KJ, Chiorazzi N. High mobility group box protein 1: an endogenous signal for dendritic cell maturation and Th1 polarization. J Immunol. 2004;173:307–13. doi: 10.4049/jimmunol.173.1.307. [DOI] [PubMed] [Google Scholar]
- 115.Chavakis E, Hain A, Vinci M, Carmona G, Bianchi ME, Vajkoczy P, Zeiher AM, Chavakis T, Dimmeler S. High-Mobility Group Box 1 Activates Integrin-Dependent Homing of Endothelial Progenitor Cells. Circ Res. 2007 doi: 10.1161/01.RES.0000257774.55970.f4. [DOI] [PubMed] [Google Scholar]
- 116.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–51. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 117.Ueno H, Matsuda T, Hashimoto S, Amaya F, Kitamura Y, Tanaka M, Kobayashi A, Maruyama I, Yamada S, Hasegawa N, Soejima J, Koh H, Ishizaka A. Contributions of high mobility group box protein in experimental and clinical acute lung injury. Am J Respir Crit Care Med. 2004;170:1310–6. doi: 10.1164/rccm.200402-188OC. [DOI] [PubMed] [Google Scholar]
- 118.Kokkola R, Li J, Sundberg E, Aveberger AC, Palmblad K, Yang H, Tracey KJ, Andersson U, Harris HE. Successful treatment of collagen-induced arthritis in mice and rats by targeting extracellular high mobility group box chromosomal protein 1 activity. Arthritis Rheum. 2003;48:2052–8. doi: 10.1002/art.11161. [DOI] [PubMed] [Google Scholar]
- 119.Scott P, Ma H, Viriyakosol S, Terkeltaub R, Liu-Bryan R. Engagement of CD14 Mediates the Inflammatory Potential of Monosodium Urate Crystals. J Immunol. 2006;177:6370–8. doi: 10.4049/jimmunol.177.9.6370. [DOI] [PubMed] [Google Scholar]
- 120.Decker P, Singh-Jasuja H, Haager S, Kötter I, Rammensee HG. Nucleosome, the main autoantigen in systemic lupus erythematosus, induces direct dendritic cell activation via a MyD88-independent pathway: consequences on inflammation. J Immunol. 2005;174:3326–34. doi: 10.4049/jimmunol.174.6.3326. [DOI] [PubMed] [Google Scholar]
- 121.Ronnefarth VM, Erbacher AI, Lamkemeyer T, Madlung J, Nordheim A, Rammensee HG, Decker P. TLR2/TLR4-independent neutrophil activation and recruitment upon endocytosis of nucleosomes reveals a new pathway of innate immunity in systemic lupus erythematosus. J Immunol. 2006;177:7740–9. doi: 10.4049/jimmunol.177.11.7740. [DOI] [PubMed] [Google Scholar]
- 122.Decker P, Wolburg H, Rammensee HG. Nucleosomes induce lymphocyte necrosis. Eur J Immunol. 2003;33:1978–87. doi: 10.1002/eji.200323703. [DOI] [PubMed] [Google Scholar]
- 123.Ishii KJ, Suzuki K, Coban C, Takeshita F, Itoh Y, Matoba H, Kohn LD, Klinman DM. Genomic DNA released by dying cells induces the maturation of APCs. J Immunol. 2001;167:2602–7. doi: 10.4049/jimmunol.167.5.2602. [DOI] [PubMed] [Google Scholar]
- 124.Boule MW, Broughton C, Mackay F, Akira S, Marshak-Rothstein A, Rifkin IR. Toll-like receptor 9-dependent and -independent dendritic cell activation by chromatin-immunoglobulin G complexes. J Exp Med. 2004;199:1631–40. doi: 10.1084/jem.20031942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Basu S, Binder RJ, Suto R, Anderson KM, Srivastava PK. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-kappa B pathway. Int Immunol. 2000;12:1539–46. doi: 10.1093/intimm/12.11.1539. [DOI] [PubMed] [Google Scholar]
- 126.Panjwani NN, Popova L, Srivastava PK. Heat shock proteins gp96 and hsp70 activate the release of nitric oxide by APCs. J Immunol. 2002;168:2997–3003. doi: 10.4049/jimmunol.168.6.2997. [DOI] [PubMed] [Google Scholar]
- 127.Asea A, Kraeft SK, Kurt-Jones EA, Stevenson MA, Chen LB, Finberg RW, Koo GC, Calderwood SK. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat Med. 2000;6:435–42. doi: 10.1038/74697. [DOI] [PubMed] [Google Scholar]
- 128.Basu S, Binder RJ, Ramalingam T, Srivastava PK. CD91 is a common receptor for heat shock proteins gp96, hsp90, hsp70, and calreticulin. Immunity. 2001;14:303–13. doi: 10.1016/s1074-7613(01)00111-x. [DOI] [PubMed] [Google Scholar]
- 129.Delneste Y, Magistrelli G, Gauchat J, Haeuw J, Aubry J, Nakamura K, Kawakami-Honda N, Goetsch L, Sawamura T, Bonnefoy J, Jeannin P. Involvement of LOX-1 in dendritic cell-mediated antigen cross-presentation. Immunity. 2002;17:353–62. doi: 10.1016/s1074-7613(02)00388-6. [DOI] [PubMed] [Google Scholar]
- 130.Chen W, Syldath U, Bellmann K, Burkart V, Kolb H. Human 60-kDa heat-shock protein: a danger signal to the innate immune system. J Immunol. 1999;162:3212–9. [PubMed] [Google Scholar]
- 131.Kol A, Lichtman AH, Finberg RW, Libby P, Kurt-Jones EA. Cutting edge: heat shock protein (HSP) 60 activates the innate immune response: CD14 is an essential receptor for HSP60 activation of mononuclear cells. J Immunol. 2000;164:13–7. doi: 10.4049/jimmunol.164.1.13. [DOI] [PubMed] [Google Scholar]
- 132.Binder RJ, Anderson KM, Basu S, Srivastava PK. Cutting edge: heat shock protein gp96 induces maturation and migration of CD11c+ cells in vivo. J Immunol. 2000;165:6029–35. doi: 10.4049/jimmunol.165.11.6029. [DOI] [PubMed] [Google Scholar]
- 133.Vabulas RM, Braedel S, Hilf N, Singh-Jasuja H, Herter S, Ahmad-Nejad P, Kirschning CJ, Da Costa C, Rammensee HG, Wagner H, Schild H. The endoplasmic reticulum-resident heat shock protein Gp96 activates dendritic cells via the Toll-like receptor 2/4 pathway. J Biol Chem. 2002;277:20847–53. doi: 10.1074/jbc.M200425200. [DOI] [PubMed] [Google Scholar]
- 134.Berwin B, Delneste Y, Lovingood RV, Post SR, Pizzo SV. SREC-I, a type F scavenger receptor, is an endocytic receptor for calreticulin. J Biol Chem. 2004;279:51250–7. doi: 10.1074/jbc.M406202200. [DOI] [PubMed] [Google Scholar]
- 135.Berwin B, Hart JP, Rice S, Gass C, Pizzo SV, Post SR, Nicchitta CV. Scavenger receptor-A mediates gp96/GRP94 and calreticulin internalization by antigen-presenting cells. Embo J. 2003;22:6127–36. doi: 10.1093/emboj/cdg572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Barondes SH, Castronovo V, Cooper DN, Cummings RD, Drickamer K, Feizi T, Gitt MA, Hirabayashi J, Hughes C, Kasai K, et al. Galectins: a family of animal beta-galactoside-binding lectins. Cell. 1994;76:597–8. doi: 10.1016/0092-8674(94)90498-7. [DOI] [PubMed] [Google Scholar]
- 137.Dai SY, Nakagawa R, Itoh A, Murakami H, Kashio Y, Abe H, Katoh S, Kontani K, Kihara M, Zhang SL, Hata T, Nakamura T, Yamauchi A, Hirashima M. Galectin-9 induces maturation of human monocyte-derived dendritic cells. J Immunol. 2005;175:2974–81. doi: 10.4049/jimmunol.175.5.2974. [DOI] [PubMed] [Google Scholar]
- 138.Huttunen HJ, Kuja-Panula J, Sorci G, Agneletti AL, Donato R, Rauvala H. Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. J Biol Chem. 2000;275:40096–105. doi: 10.1074/jbc.M006993200. [DOI] [PubMed] [Google Scholar]
- 139.Cronstein BN, Daguma L, Nichols D, Hutchison AJ, Williams M. The adenosine/neutrophil paradox resolved: human neutrophils possess both A1 and A2 receptors that promote chemotaxis and inhibit O2 generation, respectively. J Clin Invest. 1990;85:1150–7. doi: 10.1172/JCI114547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Burnstock G, Knight GE. Cellular distribution and functions of P2 receptor subtypes in different systems. Int Rev Cytol. 2004;240:31–304. doi: 10.1016/S0074-7696(04)40002-3. [DOI] [PubMed] [Google Scholar]
- 141.Fu H, Karlsson J, Bylund J, Movitz C, Karlsson A, Dahlgren C. Ligand recognition and activation of formyl peptide receptors in neutrophils. J Leukoc Biol. 2006;79:247–56. doi: 10.1189/jlb.0905498. [DOI] [PubMed] [Google Scholar]
- 142.Weber C, Springer TA. Neutrophil accumulation on activated, surface-adherent platelets in flow is mediated by interaction of Mac-1 with fibrinogen bound to alphaIIbbeta3 and stimulated by platelet-activating factor. J Clin Invest. 1997;100:2085–93. doi: 10.1172/JCI119742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Fan ST, Edgington TS. Integrin regulation of leukocyte inflammatory functions. CD11b/CD18 enhancement of the tumor necrosis factor-alpha responses of monocytes. J Immunol. 1993;150:2972–80. [PubMed] [Google Scholar]
- 144.Pfister RR, Haddox JL, Sommers CI. Injection of chemoattractants into normal cornea: a model of inflammation after alkali injury. Invest Ophthalmol Vis Sci. 1998;39:1744–50. [PubMed] [Google Scholar]
- 145.Houghton AM, Quintero PA, Perkins DL, Kobayashi DK, Kelley DG, Marconcini LA, Mecham RP, Senior RM, Shapiro SD. Elastin fragments drive disease progression in a murine model of emphysema. J Clin Invest. 2006;116:753–9. doi: 10.1172/JCI25617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Loftus JC, Smith JW, Ginsberg MH. Integrin-mediated cell adhesion: the extracellular face. J Biol Chem. 1994;269:25235–8. [PubMed] [Google Scholar]
- 147.Kobayashi H, Terao T. Hyaluronic acid-specific regulation of cytokines by human uterine fibroblasts. Am J Physiol. 1997;273:C1151–9. doi: 10.1152/ajpcell.1997.273.4.C1151. [DOI] [PubMed] [Google Scholar]
- 148.Termeer CC, Hennies J, Voith U, Ahrens T, Weiss JM, Prehm P, Simon JC. Oligosaccharides of hyaluronan are potent activators of dendritic cells. J Immunol. 2000;165:1863–70. doi: 10.4049/jimmunol.165.4.1863. [DOI] [PubMed] [Google Scholar]
- 149.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–9. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 150.Adair-Kirk TL, Atkinson JJ, Broekelmann TJ, Doi M, Tryggvason K, Miner JH, Mecham RP, Senior RM. A site on laminin alpha 5, AQARSAASKVKVSMKF, induces inflammatory cell production of matrix metalloproteinase-9 and chemotaxis. J Immunol. 2003;171:398–406. doi: 10.4049/jimmunol.171.1.398. [DOI] [PubMed] [Google Scholar]