Finding distal regulatory elements in the human genome (original) (raw)
. Author manuscript; available in PMC: 2012 Apr 8.
Published in final edited form as: Curr Opin Genet Dev. 2009 Oct 23;19(6):541–549. doi: 10.1016/j.gde.2009.09.006
Abstract
Transcriptional regulation of human genes depends not only on promoters and nearby _cis_-regulatory elements, but also on distal regulatory elements such as enhancers, insulators, locus control regions, and silencing elements, which are often located far away from the genes they control. Our knowledge of human distal regulatory elements is very limited, but the last several years have seen rapid progress in the development of strategies to identify these long-range regulatory sequences throughout the human genome. Here, we review these advances, focusing on two important classes of distal regulatory sequences — enhancers and insulators.
Introduction
Eight years have passed since the human genome sequence was first mapped [1,2], but much of the genome still remains unannotated today. Roughly 1.5% of the genome is devoted to protein coding, and 45% is repetitive DNA — mostly remnants of retrotransposons over eons of evolution. The remaining ~50% of the human genome was sometimes referred to as ‘junk’ sequences in the past, but increasing evidence now suggests that these noncoding sequences contain key regulatory elements responsible for the elaborate expression programs in the diverse cell types of the human body. The critical, unresolved questions are: Where are these regulatory sequences? Which specific genes do they control? What transcriptional programs are they part of? How many of these sequences are there? These questions seemed very daunting only a short time ago, but thanks to recent progress in genomic technologies and new understanding of chromatin landscape in the nuclei, the answers appear to be in sight.
Temporal and tissue-specific gene expression in mammals depends on _cis_-regulatory elements in the genome. These noncoding sequences can be divided into many classes depending on their regulatory functions (Figure 1) [3]. Among the better-characterized elements are promoters, enhancers, silencers, and insulators. Transcription initiates from promoters, which serve as anchor points for the recruitment of the general transcriptional machinery [4,5]. Enhancers act to recruit a complex array of transcription factors and chromatin-modifying activities that facilitate gene transcription [6,7]. Silencing elements, on the other hand, bind proteins and/or modify chromatin structure to inhibit gene transcription [6,8]. Insulator elements provide additional regulation by preventing the spread of heterochromatin and restricting transcriptional enhancers from activating unrelated promoters [9]. Besides these four classes of _cis_-regulatory sequences, there are also locus control regions that facilitate the activation of a cluster of genes through still poorly understood mechanisms. A recent comprehensive survey of 1% of the human genome, using a combination of multiple genomic and computational methods, has identified a large number of transcripts and potential regulatory elements [10•]. The results indicate that the regulatory elements are more abundant in the genome than the genes they control, they are mostly distal to the genes that they regulate, and they undergo more rapid turnover during evolution.
Figure 1.
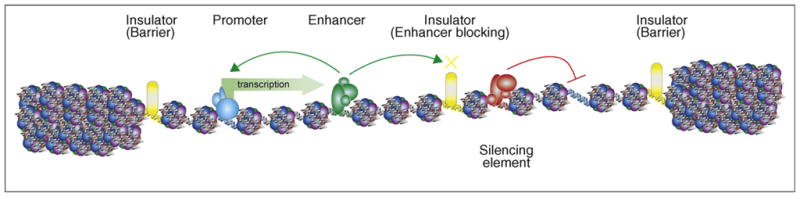
Different classes of _cis_-regulatory elements in a typical human genome. Transcription initiates at promoters (blue DNA), which are further activated by enhancers (green DNA) or repressed by silencers (red DNA). The activity of enhancers and silencers may be confined by insulators (yellow DNA), which also prevent the spreading of repressive condensed chromatin structures (shown at each end of this chromosomal region). This model depicts nucleosomes as DNA (gray helix) wound around histone proteins (various colors), which are less dense at exposed DNA regulatory elements when bound by various transcription factors (blue ovals), activator and repressor proteins (green and red ovals, respectively), and CTCF (yellow oval).
In this review, we focus on two types of distal regulatory elements: insulators and enhancers. Compared to promoters, which have been extensively characterized and amply reviewed elsewhere (see [11] and references therein), the landscape of insulators and enhancers in the human genome and their roles in cell type specific gene expression have only recently become evident. A number of new findings now point to the existence of a remarkably large number of enhancers and insulators in the genome, and indicate that these elements contribute to cell type specific gene expression in distinct ways. It has emerged that enhancers are likely of primary importance in determining cell type specific gene expression. On the other hand, the activities of insulators across the genome are largely cell type invariant. These results point to key regulatory pathways that determine lineage specification and cellular identity, and have broad impacts on understanding mechanisms of human development and molecular basis of many human diseases.
Insulators
Insulator elements affect gene expression by preventing the spread of heterochromatin (barrier function) and/or restricting transcriptional enhancers from activation of unrelated promoters (enhancer blocking) [12]. In vertebrates, enhancer-blocking function of insulators requires association with the CCCTC-binding factor (CTCF), a protein with 11 zinc-finger motifs in its DNA binding domain that is capable of recognizing long and diverse nucleotide sequences [13–15]. To identify CTCF binding sites in the human genome, Kim et al. performed ChIP-chip analysis using genome-tiling microarrays, and determined a total of 13 804 regions bound by CTCF [16•]. They defined a consensus motif that is shared by most of these CTCF binding sequences, which is capable of interacting with CTCF in vitro. Therefore, CTCF’s binding in vivo is to a great extent mediated via this consensus motif. Interestingly, using bioinformatics approaches, Xie et al. independently identified the same motif as one of the most abundant and evolutionarily conserved long motifs in the human genome, suggesting a strong evolutionary pressure to maintain the insulator function and a general role for the CTCF protein in this process [17].
Several additional studies using ChIP-based approaches have identified CTCF binding sites in additional cell types including CD4+ T cells, HeLa cells, and activated T cells [18,19,20••]. Remarkably, the majority of CTCF binding sites identified in the fibroblasts and CD4+ T cells were found to be in common, implying that most insulator elements are not specific to individual cell types. This notion is further supported by several additional studies. Crawford and colleagues used a high throughput method (DNase-chip) to identify sites sensitive to DNaseI treatment in isolated nuclei from several cell types [21]. They showed that the DNaseI hypersensitive sites (DHS) shared among different cell types are highly enriched for the CTCF motif, and overlap with previously identified CTCF binding sites in IMR90 cells. Further functional studies confirmed that many of these cell type invariant DHS are indeed insulators. More recently, Heintzman et al. investigated the binding of CTCF in 1% of the human genome (ENCODE regions) in five diverse cell types [22•]. They found over 600 CTCF binding sites, and demonstrated that most of them are actually associated with CTCF in each of cell types investigated. Therefore, most CTCF binding insulator elements appear to function in a way that is independent of cell type.
How does CTCF protein mediate enhancer-blocking function? Several research groups using a chromatin conformation capture (3C) approach have pointed out a role for CTCF in mediating interchromosomal and intrachromosomal interactions [23–25]. This finding is further supported by recent studies that link CTCF to the cohesin complex ([26] and references therein). Using the ChIP-chip method, Wendt et al. found that over 90% of CTCF binding sites are also occupied by the cohesin complex in a diverse set of human cells [20]. Since the cohesin complex has a well documented role in keeping sister chromatids together during DNA replication and before mitosis, it is conceivable that the cohesin complex may play a similar structural role in stabilizing long-range interactions between distant chromosomal regions or between DNA on two different chromosomes. Indeed, this is shown to be the case for several CTCF binding sites located upstream and downstream of the γ-interferon locus [27].
Enhancers
In order for transcription to start, the transcription machinery must first overcome the negative effects of chromatin, the highly ordered compact form in which native DNA exists inside every cell [28]. The fundamental structural units of chromatin are nucleosomes, which consist of 146 base pairs of DNA wrapped around a single histone octamer composed of two histone H2A–H2B dimers and one H3–H4 histone tetramer [29]. The compaction of DNA into chromatin prevents the protein–DNA interactions required for transcription, unless these chromatin structures are decondensed and altered in ways to make the underlying DNA sequence available to transcription factors and RNA polymerase II (RNAPII).
Enhancer elements can be defined as DNA sequences that serve to recruit transcription factors which promote the decondensation of repressed chromatin and/or facilitate the assembly of the transcription machinery at gene promoters [30]. The human genome encodes approximately 1700–1900 sequence-specific transcription factors [31••]. These proteins usually contain two distinct domains, one responsible for the recognition of specific DNA sequences (DNA-binding domain), the other carrying out a regulatory function (regulatory domain). One primary function of the regulatory domain is to recruit cofactors that carry chromatin-remodeling activities or can directly interact with the RNAPII transcriptional machinery [32,33].
Several classes of protein complexes are recruited to specific enhancer elements to remodel the local chromatin structures [32] (Figure 2). One class of proteins, represented by the SWI/SNF complexes, modifies the chromatin structure noncovalently in an ATP-dependent fashion [34]. These proteins, once recruited to enhancer elements, can reposition specific nucleosomes along the DNA. Consequently, core promoters may be exposed to allow transcription to start [35,36]. Alternatively, key transcription factor target sites may also be exposed to allow the assembly of functional enhancer complexes. Another class of cofactors remodels chromatin structure by introducing covalent modifications to the N-terminal tails of histones [8,37]. One of the well-known modifications involves the acetylation of histones H3 and H4 at the N-terminal domains. Such modifications may directly induce the decondensation of packed nucleosomes, or serve as a platform for the recruitment of additional chromatin-remodeling factors. A number of histone acetyl-transferases (HATs) have been identified that catalyze the acetylation of histones at specific residues. The protein complexes that catalyze histone acetylation include PCAF, CBP, p300, GCN5, TRRAP, and others, which are also known to function as cofactors for many transcriptional activators [38].
Figure 2.

Three mechanisms by which enhancers act to enhance transcription at target promoters. (a) Transcription factors recruit nucleosome-remodeling complexes containing SWI/SNF proteins (green ovals), ATPases that can slide nucleosomes along the DNA in an ATP-dependent fashion. The resulting change of nucleosome positions at the target promoter (blue DNA) facilitates assembly of the transcription machinery (blue ovals) and gene expression (represented by green block arrow). (b) Transcription factors also recruit coactivator complexes (green ovals) with histone acetyltransferase (HAT) activities, which in turn lead to histone acetylation (represented by stars on the nucleosomes). Such chromatin modification may provide binding surfaces for other activator proteins (additional green ovals) to bind, or result in decondensation of chromatin to allow transcription machinery (blue ovals) easier access. (c) Transcription factors may also recruit the mediator complex (purple ovals), which directly interact with RNAPII to activate transcription.
The third class of cofactors that can be recruited to enhancer elements includes so-called mediator complexes [39]. These proteins facilitate transcription by serving as interfaces between sequence-specific transcription factors and the general transcription apparatus in eukaryotes. Transcriptional coactivators in this category, including MED1, p160, Asc2, and others, have been shown to be recruited to specific enhancer sequences to promote the assembly of functional transcription initiation complexes [40,41].
One type of experimental evidence to suggest a DNA sequence as an enhancer element is its association with an activator protein that binds to specific DNA sequences. Although this strategy has been successfully carried out for a number of transcription factors in a variety of cell types (for example, see Refs. [10,42]), the strategy is not really feasible for the determination of all enhancer elements, because of the large number of transcription factors encoded by the human genome and the number of cell types needed. Further, the mere binding of a sequence-specific DNA binding protein could lead to activation, repression, or no transcriptional consequence. Therefore, an alternative approach has been used to determine the binding sites of coactivator proteins, such as p300, binding of which is more closely related to transcriptional activation. Using this strategy, Visel et al. recently determined the p300-binding sites in the mouse genome in forebrain, mid brain, and limb of e11.5 mouse embryos [43••]. Between 500 and 2500 binding sites were identified in each of the embryonic tissues. That these elements function as tissue-specific enhancers was confirmed by mouse transgenic reporter assays, which showed that over 80% of the tested elements drive reporter gene expression in the tissue where p300-binding was detected. Because virtually all known transcription factors function by recruiting transcriptional coregulators, and because the number of chromatin-remodeling complexes or mediators in the genome is much less than the number of sequence-specific transcription factors, the strategy of using cofactors as one ‘marker’ for enhancer elements is a more practical approach to identify all enhancer elements in the genome. The main hurdle, however, is the availability of suitable antibodies against each of the known coactivator proteins.
Another strategy to experimentally determine enhancers stems from the initial observation that distal p300-binding sites are associated with a unique combination of chromatin modifications that involves, among others, the presence of mono-methylated histone H3 lysine 4 (H3K4me1) and the absence of the tri-methylated form of this lysine (H3K4me3) [22,44••] (Figure 3). Indeed, when this pattern of chromatin modification signature was used to search for additional similar genomic regions in 1% sampling of the human genome, approximately 400 putative enhancers were identified that included 85% of the p300-binding sites and ~300 other sequences. Importantly, the majority of these putative enhancers are associated with DNaseI hypersensitivity, bound by coactivators p300 or MED1, and associated with additional ‘active’ chromatin marks such as histone acetylation, making them likely enhancers. When tested in reporter assays, the predicted enhancers can indeed support transcriptional activation, providing preliminary evidence for their function. With the enhancer-specific chromatin signatures, we have generated a list of more than 90 000 potential enhancers in four types of human cells ([22]; Hawkins et al., unpublished data). To date, a total of 26 predicted enhancers have been tested by reporter assay in transient transfection in vitro, and over 80% (21 out of 26) of the tested fragments were shown to possess enhancer activity, supporting the validity of this enhancer-finding method ([22]; Hawkins et al., unpublished data). It is worth noting that although many chromatin modification marks are found at enhancers and can be used to predict such elements in the genome, Heintzman et al. found that with the use of profiles of just two chromatin marks — H3K4me1 and H3K4me3 — one can achieve excellent specificity and sensitivity [22,44••]. Additionally, this minimal chromatin signature has been used to identify enhancers in a variety of different cell types, in both humans and mice (Ren et al., unpublished data).
Figure 3.

A strategy to map enhancers based on their chromatin signatures. (a) Derivation of the average chromatin modification profiles from known enhancers. Enhancers (green DNA) are flanked by nucleosomes containing mono-methylated histone H3 lysine 4 (H3K4me1, represented by single pentagons). Plotting H3K4me1 enrichment relative to chromosomal position for many enhancers yields an average enhancer H3K4me1 profile or ‘chromatin signature.’ (b) Scanning the chromatin signature along the genome (fading green profile) to discover regions with similar chromatin profiles. A new enhancer is identified based on correlation of its H3K4me1 profile to the enhancer chromatin signature (see thick green profile), whereas other regulatory elements lacking this signature (insulators, yellow; promoters, blue) are passed over by the scanning algorithm. Promoters are characterized by tri-methylated and di-methylated H3K4 (represented by triple and double pentagons), and acetylation (stars) may be found at either kind of regulatory element.
Cell type specific activity of enhancers
One of the most prominent features displayed by enhancers, compared to that of promoters and insulator elements, is their cell type specific activities. While previous works on classical enhancers such as those in beta-globin genes have suggested such properties of enhancers, recent genome-wide studies have confirmed this on a global scale. Among the p300-binding sites identified in three embryonic tissues, the majority are occupied by the coactivator in only one of the tissues, and when tested in mouse transgenic assays exhibited tissue-specific enhancer activities [43]. Similarly, p300-binding sites found in three human cell lines demonstrated highly cell type specific occupancy by the factor [22•]. Furthermore, the enhancers identified in different cell types are associated with cell type dependent chromatin modification patterns. The cell type specific presence of chromatin marks, such as H3K4me1, at enhancers is closely correlated with cell type specific expression of the putative targets of these enhancers. These findings indicate that enhancers are more dynamically regulated in different cell types, suggesting that these elements are of primary importance in driving cell type specific gene expression.
Enhancer-specific transcription factors
Computational analysis of the putative enhancers discovered in the human genome has revealed a number of over-represented DNA motifs, with some matching the recognition sites of known transcription factors [22•]. Interestingly, of the 41 motifs identified in these enhancer sequences, over 90% appear to be unique to enhancers, and exhibit no enrichment at promoters, suggesting that some transcription factors may function exclusively through these distal _cis_-regulatory elements. Indeed, recent investigations into the genomic binding sites of 14 sequence-specific transcription factors in the mouse embryonic stem cells revealed two classes of in vivo binding sites by these factors — nearly half of them, including Oct4, Sox2, Nanog, appear to bind more preferentially to distal regulatory sequences, while the rest, including cMyc, prefer to occupy promoters [42].
Target genes of enhancers
One of the challenges in characterizing enhancer function is determining which genes they control. The issue arises because frequently these distal _cis_-regulatory elements are located tens or hundreds of kilobases away from their target genes, and could be located at the gene body of nearby genes. Further complicating the issue, there has also been report that enhancers could activate target genes located on different chromosomes [45].
To resolve the target genes of enhancers, researchers frequently assign the enhancers to the nearest genes as a first order approximation [43••]. While in most cases, such assignment would sufficiently explain cell type specific expression of genes, there has not been any report on the rate of false positives by this strategy. A variation of the above strategy is to assign enhancers to the genes located within the same genomic segments bounded by the enhancer-blocking insulator elements, which can be experimentally determined as CTCF binding sites [22•]. This strategy appears to capture nicely the correlation between chromatin modification patterns at enhancers and the differential gene expression at the presumed target genes. Consistent with this model, upon depletion of CTCF and presumably loss of enhancer-blocking function by the insulator elements, a significant number of genes located near previously shielded enhancers become activated (Figure 4). While this strategy is conceptually simple, the limitation has been a lack of understanding of the functional mechanism for enhancer blocking by insulators. As discussed above, an emerging consensus is that CTCF binding sites act to establish long-range chromosomal interactions that would lead to the formation of local topographical constraints. Depending on the way such topographical constraints are formed, the enhancer/promoter interactions may be restricted in different ways, and therefore different assignments may be made for the enhancers.
Figure 4.

Enhancer activity in the absence of CTCF. The effect of CTCF knockdown in human cells is depicted in this model, illustrating that activation of transcription (green block arrows) occurs due to loss of the enhancer-blocking insulator function, as described in Ref. [22•]. When CTCF (yellow ovals) is knocked down by siRNA (red no symbol), enhancers (green) are free to upregulate promoters (blue) that were previously separated by insulators (yellow DNA) bound by CTCF.
In principle, a more direct approach for assigning enhancers to target genes is to experimentally determine the long-range chromosomal interactions between enhancers and target promoters [46]. This can be accomplished by the Chromosome Conformation Capture (3C) method [47] or its high throughput variations including 4C (circular 3C) [48] or 5C (3C carbon copy) [45,49–51]. This strategy is based on the observations that active enhancers are brought in close proximity to target promoters through DNA looping [47]. In many cases, this method has helped to define unexpected target genes, such as those located in different chromosomes [45]. The future will see more enhancer/target relationships defined using this strategy.
What is next?
Although great strides have been made in recent years in unraveling the distal regulatory sequences in the human genome, it should be recognized that a long journey is still ahead of us. We are only beginning to comprehend the landscape of enhancers in the human genome, and it is almost certain that more enhancers are yet to be identified. Since these elements are active in a cell type specific manner, their identification will require investigation of many more additional cell types than current efforts have engaged. Based on the limited sampling, however, the number of enhancers in the human genome is likely on the order of hundreds of thousands, or even millions, making them the most abundant class of _cis_-regulatory sequences.
With genome-wide identification of enhancer sequences in different cell types, tissues, and developmental stages, the next important question is which transcription factors act through these elements to mediate transcriptional activation of the target genes. Motif analysis can lend insights into potential transcription factors involved. However, a bottleneck has been our limited knowledge of consensus DNA binding sites recognized by the over 1700 transcription factors encoded by the human genome [31••]. So far, such information is available for just a few hundred of transcription factors, described in databases such as TRANSFAC, JASPAR, and UniPROBE [52–54]. Fortunately, investigations using high throughput methods, including Protein Binding Microarrays and one hybrid methods are quickly enriching our knowledge base and will likely provide the full spectrum of DNA binding for most human transcription factors in the near future [55–60].
In parallel, a consortium of investigators is now generating genome-wide data for various transcription factors using ChIP-seq in many different human cell lines [10•]. This will surely one day lead to systematic determination of functional sequences in the human genome, and provide a rich resource for understanding gene regulatory sequences.
A fundamental question remains to be resolved regarding the molecular mechanisms by which long-range elements such as enhancers and insulator elements act to regulate gene expression. A commonly accepted model is that DNA sequences between enhancers and promoters form loops to allow the distal regulatory sequences to interact directly with the promoters [46] (Figure 5). In this model, the function of enhancers is to facilitate the assembly of the RNA polymerase complex at the promoters, or subsequent steps of RNA polymerase transcription cycle. Although this model is supported by strong evidence over the years, important details remain to be elucidated. How does the loop form? What mechanism allows the enhancers to be positioned to the appropriate target promoters? Which step(s) of the RNA polymerase transcription process do enhancers act on?
Figure 5.
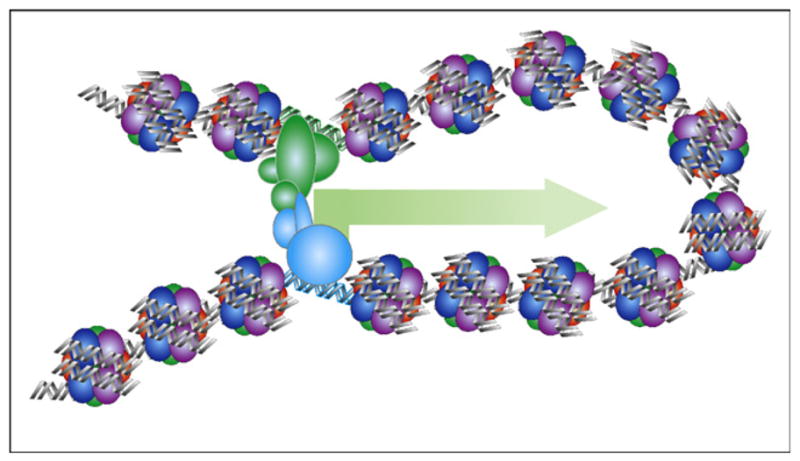
Looping model of enhancer function. During transcriptional activation, an enhancer (green) is positioned in close proximity to the target promoter (blue) via DNA looping.
Another theory, not mutually exclusive to the looping model, is the RNA polymerase factory model [61]. In this model, synthesis of RNA transcripts occurs in nuclear compartments that contain stores of RNA polymerase and accessory factors. Active genes are dynamically shuttled in and out of the ‘factories’ during transcription. Since there are a limited number of such factories, ranging from several hundreds to thousands in a single nucleus, each factory needs to simultaneously transcribe multiple genes. This model therefore predicts colocalization of coordinately regulated genes in the nucleus, and the role of the enhancers, thusly, is to facilitate the movement of genes into the RNA polymerase factories. Indeed, recent studies by Hu and colleagues showed that nuclear receptors induce the rapid movement of enhancer sequences to specific nuclear speckles, and the new interchromosomal interactions formed this way are required for the induction of nuclear receptor target genes [62].
Future work will also help resolve other questions regarding the mechanisms of chromatin signatures at enhancers [44••]. Are the chromatin signatures dependent on transcription factor binding? Which enzymes are responsible for depositing and maintaining these chromatin modifications? Are the modifications necessary for enhancer function? If so, how do they affect transcriptional activation? Answers to these questions promise to shed light on the mysterious mechanisms of enhancer function.
Acknowledgments
We apologize to those whose are not referenced here due to limitation of space. We thank Gary Hon for comments on the manuscript. The work is supported by funds from the Ludwig Institute for Cancer Research, Nation Institute of Health, and the California Institute of Regenerative Medicine.
Footnotes
This review comes from a themed issue on Genomes and evolution
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 2.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. The sequence of the human genome. Science (New York, NY) 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 3.Maston GA, Evans SK, Green MR. Transcriptional regulatory elements in the human genome. Annu Rev Genomics Hum Genet. 2006;7:29–59. doi: 10.1146/annurev.genom.7.080505.115623. [DOI] [PubMed] [Google Scholar]
- 4.Butler JE, Kadonaga JT. The RNA polymerase II core promoter: a key component in the regulation of gene expression. Genes Dev. 2002;16:2583–2592. doi: 10.1101/gad.1026202. [DOI] [PubMed] [Google Scholar]
- 5.Lee TI, Young RA. Transcription of eukaryotic protein-coding genes. Annu Rev Genet. 2000;34:77–137. doi: 10.1146/annurev.genet.34.1.77. [DOI] [PubMed] [Google Scholar]
- 6.Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14:121–141. [PubMed] [Google Scholar]
- 7.Tjian R, Maniatis T. Transcriptional activation: a complex puzzle with few easy pieces. Cell. 1994;77:5–8. doi: 10.1016/0092-8674(94)90227-5. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Y, Reinberg D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 2001;15:2343–2360. doi: 10.1101/gad.927301. [DOI] [PubMed] [Google Scholar]
- 9.West AG, Gaszner M, Felsenfeld G. Insulators: many functions, many mechanisms. Genes Dev. 2002;16:271–288. doi: 10.1101/gad.954702. [DOI] [PubMed] [Google Scholar]
- 10•.Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. This article is a summary of the discoveries made by the international ENCODE consortium to identify functional elements in 1% of the human genome. The main findings, relevant to this review, are that there exist a large number of cis-regulatory elements in the genome and they are frequently not conserved. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heintzman ND, Ren B. The gateway to transcription: identifying, characterizing and understanding promoters in the eukaryotic genome. Cell Mol Life Sci. 2007;64:386–400. doi: 10.1007/s00018-006-6295-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wallace JA, Felsenfeld G. We gather together: insulators and genome organization. Curr Opin Gen Dev. 2007;17:400–407. doi: 10.1016/j.gde.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Felsenfeld G, Burgess-Beusse B, Farrell C, Gaszner M, Ghirlando R, Huang S, Jin C, Litt M, Magdinier F, Mutskov V, et al. Chromatin boundaries and chromatin domains. Cold Spring Harb Symp Quant Biol. 2004;69:245–250. doi: 10.1101/sqb.2004.69.245. [DOI] [PubMed] [Google Scholar]
- 14.Bell AC, West AG, Felsenfeld G. Insulators and boundaries: versatile regulatory elements in the eukaryotic. Science. 2001;291:447–450. doi: 10.1126/science.291.5503.447. [DOI] [PubMed] [Google Scholar]
- 15.Bell AC, West AG, Felsenfeld G. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell. 1999;98:387–396. doi: 10.1016/s0092-8674(00)81967-4. [DOI] [PubMed] [Google Scholar]
- 16•.Kim TH, Abdullayev Z, Smith A, Ching KA, Loukinov D, Green RD, Zhang MQ, Lobanenkov VV, Ren B. Analysis of the vertebrate insulator protein CTCF binding in the human genome. Cell. 2007;128:1231–1245. doi: 10.1016/j.cell.2006.12.048. This article is the first report of CTCF binding sites in the human genome. The authors used ChIP-chip and genome-tiling arrays to identify genomic regions occupied by the insulator binding protein in human fibroblasts. The main finding is that most CTCF binding sites share a consensus motif, and that the distribution of the potential insulator elements correlates well with genes. Additionally, CTCF binds to a similar set of genomic sequences in different cell types. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie X, Mikkelsen TS, Gnirke A, Lindblad-Toh K, Kellis M, Lander ES. Systematic discovery of regulatory motifs in conserved regions of the human genome, including thousands of CTCF insulator sites. Proc Natl Acad Sci U S A. 2007;104:7145–7150. doi: 10.1073/pnas.0701811104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barski A, Cuddapah S, Cui KR, Roh TY, Schones DE, Wang ZB, Wei G, Chepelev I, Zhao KJ. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 19.Cuddapah S, Jothi R, Schones DE, Roh T-Y, Cui K, Zhao K. Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains. Genome Res. 2009;19:24–32. doi: 10.1101/gr.082800.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20••.Wendt KS, Yoshida K, Itoh T, Bando M, Koch B, Schirghuber E, Tsutsumi S, Nagae G, Ishihara K, Mishiro T, et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008;451:796–801. doi: 10.1038/nature06634. This landmark paper, among a series of four concurrent studies, revealed a close association between the insulator binding protein CTCF and the cohesin complex. Both are found together at the insulator elements in the genome, and are necessary for the enhancer-blocking function of the binding sites. The study immediately suggested a model for mechanism of insulator function. [DOI] [PubMed] [Google Scholar]
- 21.Xi H, Shulha HP, Lin JM, Vales TR, Fu Y, Bodine DM, McKay RDG, Chenoweth JG, Tesar PJ, Furey TS, et al. Identification and characterization of cell type-specific and ubiquitous chromatin regulatory structures in the human genome. PLoS Genet. 2007;3:e136. doi: 10.1371/journal.pgen.0030136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22•.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. This study revealed that enhancers are abundant in the human genome, and are associated with chromatin modifications in a cell type dependent way that also correlates with cell type specific gene expression. Additionally, the study found that chromatin modification patterns at promoters, or CTCF binding at insulator elements, is less dynamic and cell type dependent than the chromatin modifications at enhancers, suggesting the importance of enhancers in driving cell specific gene expression programs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ling JQ, Li T, Hu JF, Vu TH, Chen HL, Qiu XW, Cherry AM, Hoffman AR. CTCF mediates interchromosomal colocalization between Igf2/H19 and Wsb1/Nf1. Science (New York, NY) 2006;312:269–272. doi: 10.1126/science.1123191. [DOI] [PubMed] [Google Scholar]
- 24.Kurukuti S, Tiwari VK, Tavoosidana G, Pugacheva E, Murrell A, Zhao Z, Lobanenkov V, Reik W, Ohlsson R. CTCF binding at the H19 imprinting control region mediates maternally inherited higher-order chromatin conformation to restrict enhancer access to Igf2. Proc Natl Acad Sci U S A. 2006;103:10684–10689. doi: 10.1073/pnas.0600326103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hou C, Zhao H, Tanimoto K, Dean A. CTCF-dependent enhancer-blocking by alternative chromatin loop formation. Proc Natl Acad Sci U S A. 2008;105:20398–20403. doi: 10.1073/pnas.0808506106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wendt KS, Peters J-M. How cohesin and CTCF cooperate in regulating gene expression. Chromosome Res. 2009;17:201–214. doi: 10.1007/s10577-008-9017-7. [DOI] [PubMed] [Google Scholar]
- 27.Hadjur S, Williams LM, Ryan NK, Cobb BS, Sexton T, Fraser P, Fisher AG, Merkenschlager M. Cohesins form chromosomal cis-interactions at the developmentally regulated IFNG locus. Nature. 2009;20:20. doi: 10.1038/nature08079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Felsenfeld G, Groudine M. Controlling the double helix. Nature. 2003;421:448–453. doi: 10.1038/nature01411. [DOI] [PubMed] [Google Scholar]
- 29.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A ° resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 30.Maston GA, Evans SK, Green MR. Transcriptional regulatory elements in the human genome. Annu Rev Genomics Hum Genet. 2006;23:23. doi: 10.1146/annurev.genom.7.080505.115623. [DOI] [PubMed] [Google Scholar]
- 31••.Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM. A census of human transcription factors: function, expression and evolution. Nat Rev Genet. 2009;10:252–263. doi: 10.1038/nrg2538. The authors manually annotated 1391 sequence-specific transcription factors in the human genome and presented analysis of the expression patterns, evolutionary conservation, and potential functions of the proteins. This work will lay a foundation for systematic determination of gene regulatory networks in human cells. [DOI] [PubMed] [Google Scholar]
- 32.Lemon B, Tjian R. Orchestrated response: a symphony of transcription factors for gene control. Genes Dev. 2000;14:2551–2569. doi: 10.1101/gad.831000. [DOI] [PubMed] [Google Scholar]
- 33.Ptashne M, Gann A. Transcriptional activation by recruitment. Nature. 1997;386:569–577. doi: 10.1038/386569a0. [DOI] [PubMed] [Google Scholar]
- 34.Kingston RE, Narlikar GJ. ATP-dependent remodeling and acetylation as regulators of chromatin fluidity. Genes Dev. 1999;13:2339–2352. doi: 10.1101/gad.13.18.2339. [DOI] [PubMed] [Google Scholar]
- 35.Lomvardas S, Thanos D. Modifying gene expression programs by altering core promoter chromatin architecture. Cell. 2002;110:261–271. doi: 10.1016/s0092-8674(02)00822-x. [DOI] [PubMed] [Google Scholar]
- 36.Cosma MP, Tanaka T, Nasmyth K. Ordered recruitment of transcription and chromatin remodeling factors to a cell cycle-and developmentally regulated promoter. Cell. 1999;97:299–311. doi: 10.1016/s0092-8674(00)80740-0. [DOI] [PubMed] [Google Scholar]
- 37.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 38.Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006;20:1405–1428. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- 39.Myers LC, Kornberg RD. Mediator of transcriptional regulation. Annu Rev Biochem. 2000;69:729–749. doi: 10.1146/annurev.biochem.69.1.729. [DOI] [PubMed] [Google Scholar]
- 40.Soutoglou E, Talianidis I. Coordination of PIC assembly and chromatin remodeling during differentiation-induced gene activation. Science (New York, NY) 2002;295:1901–1904. doi: 10.1126/science.1068356. [DOI] [PubMed] [Google Scholar]
- 41.Hatzis P, Talianidis I. Dynamics of enhancer–promoter communication during differentiation-induced gene activation. Mol Cell. 2002;10:1467–1477. doi: 10.1016/s1097-2765(02)00786-4. [DOI] [PubMed] [Google Scholar]
- 42.Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 43••.Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. This study demonstrated that mammalian enhancers could be accurately and efficiently identified by experimentally mapping the genomic binding sites of p300, a histone acetyltransferase and transcriptional coactivator protein. The authors performed a large number of transgenic mice assays to rigorously prove the enhancer activities of the identified binding sites in mouse embryos. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44••.Heintzman ND, Stuart RK, Hon G, Fu YT, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu CX, Ching KA, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. In this study, the authors identified a chromatin modification signature for transcriptional enhancers, and then showed that this pattern of chromatin modifications could be used to predict enhancers in the human genome. The work formed the basis for genome-wide prediction of enhancers as described in Ref. [22•] [DOI] [PubMed] [Google Scholar]
- 45.Lomvardas S, Barnea G, Pisapia DJ, Mendelsohn M, Kirkland J, Axel R. Interchromosomal interactions and olfactory receptor choice. Cell. 2006;126:403–413. doi: 10.1016/j.cell.2006.06.035. [DOI] [PubMed] [Google Scholar]
- 46.Miele A, Dekker J. Long-range chromosomal interactions and gene regulation. Mol bioSyst. 2008;4:1046–1057. doi: 10.1039/b803580f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dekker J. A closer look at long-range chromosomal interactions. Trends Biochem Sci. 2003;28:277–280. doi: 10.1016/S0968-0004(03)00089-6. [DOI] [PubMed] [Google Scholar]
- 48.Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, Rubio ED, Krumm A, Lamb J, Nusbaum C, et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006;16:1299–1309. doi: 10.1101/gr.5571506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, de Laat W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C) Nat Genet. 2006;38:1348–1354. doi: 10.1038/ng1896. [DOI] [PubMed] [Google Scholar]
- 50.Wurtele H, Chartrand P. Genome-wide scanning of HoxB1-associated loci in mouse ES cells using an open-ended Chromosome Conformation Capture methodology. Chromosome Res. 2006;14:477–495. doi: 10.1007/s10577-006-1075-0. [DOI] [PubMed] [Google Scholar]
- 51.Zhao Z, Tavoosidana G, Sjolinder M, Gondor A, Mariano P, Wang S, Kanduri C, Lezcano M, Sandhu KS, Singh U, et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat Genet. 2006;38:1341–1347. doi: 10.1038/ng1891. [DOI] [PubMed] [Google Scholar]
- 52.Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K, et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sandelin A, Alkema W, Engstrom P, Wasserman WW, Lenhard B. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32:D91–94. doi: 10.1093/nar/gkh012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Newburger DE, Bulyk ML. UniPROBE: an online database of protein binding microarray data on protein-DNA interactions. Nucleic Acids Res. 2009;37:D77–82. doi: 10.1093/nar/gkn660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, Chan ET, Metzler G, Vedenko A, Chen X, et al. Diversity and complexity in DNA recognition by transcription factors. Science. 2009;14:14. doi: 10.1126/science.1162327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berger MF, Badis G, Gehrke AR, Talukder S, Philippakis AA, Pena-Castillo L, Alleyne TM, Mnaimneh S, Botvinnik OB, Chan ET, et al. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell. 2008;133:1266–1276. doi: 10.1016/j.cell.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Noyes MB, Christensen RG, Wakabayashi A, Stormo GD, Brodsky MH, Wolfe SA. Analysis of homeodomain specificities allows the family-wide prediction of preferred recognition sites. Cell. 2008;133:1277–1289. doi: 10.1016/j.cell.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Berger MF, Philippakis AA, Qureshi AM, He FS, Estep PW, Bulyk ML. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat Biotechnol. 2006;24:1429–1435. doi: 10.1038/nbt1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mukherjee S, Berger MF, Jona G, Wang XS, Muzzey D, Snyder M, Young RA, Bulyk ML. Rapid analysis of the DNA-binding specificities of transcription factors with DNA microarrays. Nat Genet. 2004;36:1331–1339. doi: 10.1038/ng1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bulyk ML, Huang X, Choo Y, Church GM. Exploring the DNA-binding specificities of zinc fingers with DNA microarrays. Proc Natl Acad Sci U S A. 2001;98:7158–7163. doi: 10.1073/pnas.111163698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Osborne CS, Chakalova L, Brown KE, Carter D, Horton A, Debrand E, Goyenechea B, Mitchell JA, Lopes S, Reik W, et al. Active genes dynamically colocalize to shared sites of ongoing transcription. Nat Genet. 2004;36:1065–1071. doi: 10.1038/ng1423. [DOI] [PubMed] [Google Scholar]
- 62.Hu Q, Kwon Y-S, Nunez E, Cardamone MD, Hutt KR, Ohgi KA, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG, et al. Enhancing nuclear receptor-induced transcription requires nuclear motor and LSD1-dependent gene networking in interchromatin granules. Proc Natl Acad Sci U S A. 2008;105:19199–19204. doi: 10.1073/pnas.0810634105. [DOI] [PMC free article] [PubMed] [Google Scholar]