Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea (original) (raw)
Abstract
We present two standards developed by the Genomic Standards Consortium (GSC) for reporting bacterial and archaeal genome sequences. Both are extensions of the Minimum Information about Any (x) Sequence (MIxS). The standards are the Minimum Information about a Single Amplified Genome (MISAG) and the Minimum Information about a Metagenome-Assembled Genome (MIMAG), including, but not limited to, assembly quality, and estimates of genome completeness and contamination. These standards can be used in combination with other GSC checklists, including the Minimum Information about a Genome Sequence (MIGS), Minimum Information about a Metagenomic Sequence (MIMS), and Minimum Information about a Marker Gene Sequence (MIMARKS). Community-wide adoption of MISAG and MIMAG will facilitate more robust comparative genomic analyses of bacterial and archaeal diversity.
Similar content being viewed by others
Main
The term “uncultivated majority” was coined to denote the fraction of microbes that have not yet been isolated and grown in axenic culture1,2. This diversity was originally identified by sequencing phylogenetically relevant genes, notably the 16S ribosomal RNA gene, and more recently characterized by shotgun metagenomics3,4 and single-cell genomics5,6. Large-scale sequencing efforts that accelerated discovery of this diversity, such as the Human Microbiome Project7, the Earth Microbiome Project8, and the Genomic Encyclopedia of Bacteria and Archaea9 have improved our understanding of microbial diversity and function as it relates to human health, biogeochemical cycling, and the evolutionary relationships that structure the tree of life.
With advances in sequencing technologies, throughput, and bioinformatics approaches, tens to hundreds and even thousands of microbial genomes can be retrieved from complex samples without cultivation of any of the community members10,11,12,13. There are 2,866 single-cell genomes and 4,622 genomes reconstructed from metagenomes, which are already registered in the Genomes OnLine Database (GOLD)14 (Fig. 1). These numbers are increasing rapidly and will soon outpace the rate of sequencing of cultivated microbial isolate genomes10.
Figure 1: Sequencing of bacterial and archaeal genomes3,11,13,37,85,86,87,88,89,90.
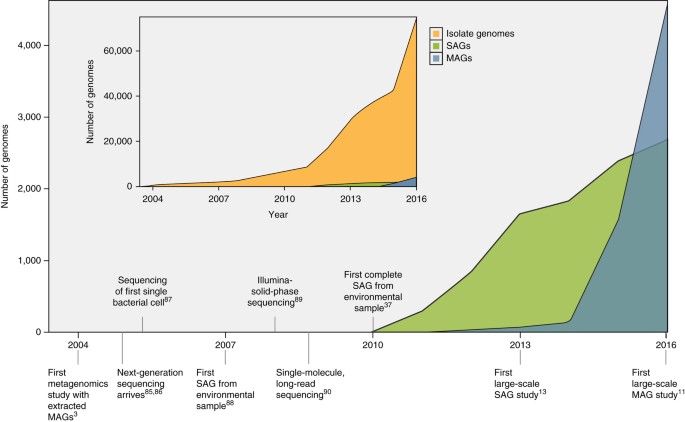
Increase in the number of SAGs and MAGs over time. Inset displays the number of isolate genomes over time for comparison. Data for figure were taken from IMG/GOLD14 in January 2017.
As this field matures, it is crucial to define minimum standards for the generation, deposition, and publication of genomes derived from uncultivated bacteria and archaea and to capture the appropriate metadata in a consistent and standardized manner, in line with previous efforts for cultivated isolate genomes15,16 and marker gene surveys17.
The GSC (http://gensc.org) maintains up-to-date metadata checklists for the MIxS, encompassing MIGS15, MIMS15, and MIMARKS17. Complementing these standards are the Minimum Information about a Biosynthetic Gene Cluster18 and the Minimum Information about Sequence Data and Ecosystem Metadata from the Built Environment19. Here, we develop a set of standards that extend the MIxS checklists. Our standards form a set of recommendations for the generation, analysis, and reporting of bacterial and archaeal single amplified genomes (SAGs) and metagenome-assembled genomes (MAGs; Table 1 and Supplementary Table 1). We hope that these standards will promote the collection and reporting of appropriate contextual metadata necessary to support large-scale comparative studies and assist researchers with retrieving genomes of uncultivated microorganisms from, and depositing them to, the international nucleotide sequence databases.
Table 1 Genome reporting standards for SAGs and MAGs
Our standards feature mandatory requirements, but are flexible enough to accommodate changes over time. For example, as sequence read lengths increase, new methods for assembly and metagenomic binning will likely be devised, and, consequently, sequence databases will need to be updated with metadata that include different sequencing platforms and analysis pipelines. Additionally, as completely new phylogenetic clades are discovered by sequencing, conserved marker gene sets that are used to estimate genome completeness will need to be updated to place new data in the appropriate context.
Minimum information about SAGs and MAGs
SAGs are produced by isolating individual cells, amplifying the genome of each cell using whole genome amplification (WGA), and then sequencing the amplified DNA6,20. MAGs, on the other hand, are produced using computational binning tools that group assembled contigs into genomes from Gbp-level metagenomic data sets21,22,23,24 (Fig. 2 and Supplementary Table 1). Both SAGs and MAGs are often highly fragmented and are sometimes contaminated with non-target sequence. Owing to these challenges, we propose that SAGs and MAGs need to have some shared metadata (Supplementary Table 1). Our standards extend the MIxS checklists by including additional criteria to assess SAG and MAG quality, which will soon become core standards required for submission to suitable databases such as those found at the National Center for Biotechnology Information (NCBI) and the European Molecular Biology Laboratory-European Bioinformatics Institute (EMBL-EBI; Hinxton, UK), the DNA Database of Japan (DDBJ) and GOLD.
Figure 2: Generation of SAGs and MAGs.
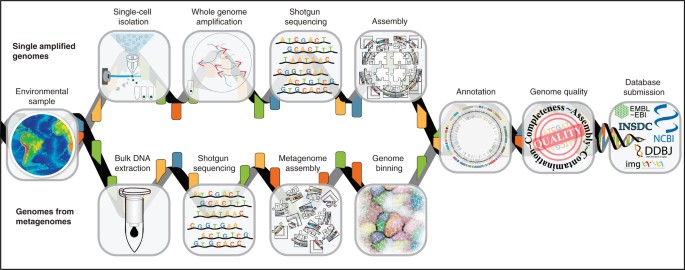
Flow diagram outlining the typical pipeline for the production of both SAGs and MAGs.
Single amplified genomes. Sequencing of genomes from single cells requires specialized instrumentation, such as flow cytometry, microfluidics, or micromanipulators for single-cell isolation, and cleanrooms for downstream handling (Supplementary Table 1)20,25,26,27. Given the extremely low yields of genomic DNA from a single microbial cell (∼1–6 fg)28, DNA must be amplified after cell lysis to generate the quantities required for currently available sequencing technologies. The most commonly used method for WGA is multiple displacement amplification (MDA)29, which relies on the highly processive Phi29 DNA polymerase30. MDA yields significant coverage biases31, alters GC profiles32, and produces chimeric molecules during the amplification reaction33, but remains the primary method for WGA of single cells. Recent advances in assembly algorithms, including single-cell-specific assemblers that use multiple coverage cutoffs (e.g., SPAdes (St. Petersburg Genome Assembler)34 and IDBA-UD (Iterative De Bruijn Graph De Novo Assembler for Short Reads Sequencing Data with Highly Uneven Sequencing Depth)35), along with a number of publicly available k-mer coverage normalization tools36,37, have provided researchers with some tools to tackle the chimeric and biased nature of single-cell sequence data.
Because most bacterial and archaeal cells contain a single or very few genome copies, introducing even trace amounts of contaminant DNA during cell sorting, lysis, or WGA can severely affect downstream SAG data quality. Contamination can originate from multiple sources, including the samples themselves, the laboratory environment, reagents supplied by vendors25,27,38, and library poolmates when multiplexing samples for sequencing. Furthermore, the lack of corresponding laboratory cultures from which genomes could be resequenced and validated using alternative methods presents a fundamental challenge in evaluating the accuracy of SAG assemblies. One way to address this challenge is to benchmark the entire workflow by using mock communities of well-characterized laboratory strains. Comparing the benchmark assemblies to genomes included in a mock sample could provide an estimate of probable errors in novel SAGs from uncultivated microbes. Published benchmark studies have revealed infrequent mismatches (∼9/100 kb), indels (∼2/100 kb), and misassemblies (∼1/Mb) in single-cell genomes39.
The ideal scenario is to produce contaminant-free SAGs20, but as this is not always possible, tools that can detect and eliminate potential contamination at the read and contig (assembly) levels have been developed. Tools for read decontamination, including DeconSeq36, and modules from the BBtools package, such as bbduk.sh (https://sourceforge.net/projects/bbmap/) remove contaminant sequences from query genomes based on user-defined contaminant databases. Quality assurance and/or decontamination of assembled SAGs has primarily been a semi-manual process that scrutinizes a variety of genomic attributes, such as non-target 16S rRNA genes, abnormal k-mer frequencies, and/or variable GC content37. However, more automated tools that identify contaminant contigs in genomic data sets have recently become available, including Anvi'o (Analysis and Visualization Platform for 'Omics Data)40, CheckM41, ProDeGe (Protocol for Fully Automated Decontamination of Genomes)42, and acdc (Automated Contamination Detection and Confidence Estimation)43. Taxonomic assignment of SAGs is generally based on marker gene phylogenies or the 16S rRNA gene sequence20.
There are no definitions and/or guidelines for either the assembly, quality control, and classification of SAGs, or the criteria to assess the final SAG assembly and how to associate the metadata with the assembled genomes.
Metagenome-assembled genomes. Assembly of microbial genomes from metagenomic sequence reads was pioneered in 2004 by Tyson et al.3 by extracting near-complete genomes from a metagenome of an acid mine drainage community that contained only a few bacterial and archaeal taxa. Although assembly of complete microbial genomes was initially restricted to environmental samples with exceptionally low microbial diversity3,44,45, increasing sequencing throughput, read lengths, and improved assembly and binning algorithms have enabled genome-resolved metagenomics to be carried out for communities with high diversity10,11,21,46. To generate a genome, metagenomic sequence reads are assembled into contigs using metagenome-specific algorithms35,47,48,49 and contigs are grouped, and these groups are then assigned to discrete population bins3,4,50.
Criteria used by metagenomic binning software include nucleotide sequence signatures (e.g., GC content and/or tetra-nucleotide frequency), marker gene phylogenies, depth of DNA sequence coverage, and abundance patterns across samples51. If these features are combined, bins of high quality can be produced52. Metagenomic binning has proven powerful for the extraction of genomes of rare community members (<1%). For example, differential coverage binning has been used recently to extract near-complete genomes of the low-abundance candidate phylum TM7 (Saccharibacteria) from wastewater bioreactor samples21. Other approaches have used differential coverage binning to identify species and strains during a time course of gut microbiome development in a newborn infant from 15 to 24 days after delivery53. In a more recent study, >2,500 MAGs were extracted from below-ground sediment and aquifer samples, taking advantage of nucleotide composition signatures, abundance of organisms across samples, and the taxonomic association of metabolic genes10. Tools are available that take advantage of multi-parameter binning, such as GroopM54, MaxBin55, MetaBAT (Metagenome Binning with Abundance and Tetranucleotide Frequencies)[56](/articles/nbt.3893#ref-CR56 "Kang, D.D., Froula, J., Egan, R. & Wang, Z. A robust statistical framework for reconstructing genomes from metagenomic data. Preprint at bioRxiv http://dx.doi.org//10.1101/011460
(2014)."), CONCOCT[57](/articles/nbt.3893#ref-CR57 "Alneberg, J. et al. CONCOCT: Clustering cONtigs on COverage and ComposiTion. Preprint at
https://arxiv.org/abs/1312.4038v1
(2013)."), and MetaWatt[58](/articles/nbt.3893#ref-CR58 "Strous, M., Kraft, B., Bisdorf, R. & Tegetmeyer, H.E. The binning of metagenomic contigs for microbial physiology of mixed cultures. Front. Microbiol. 3, 410 (2012)."). Taxonomic identity of the bins can be assigned by marker gene phylogeny or using the 16S rRNA gene sequence[11](/articles/nbt.3893#ref-CR11 "Brown, C.T. et al. Unusual biology across a group comprising more than 15% of domain Bacteria. Nature 523, 208–211 (2015).").There are no strict definitions and/or guidelines for how to assemble and bin genomes from metagenomes, which parameters to use, how to taxonomically classify and define the end product, or how to include the metadata with the assembled genomes.
Developing MISAG and MIMAG checklists
The three most important criteria for assessing SAG and MAG quality are assembly quality, genome completeness, and a measure of contamination. These criteria are discussed below and their associated standards are summarized in Table 1 (in full in Supplementary Table 1).
For both SAGs and MAGs, assessing assembly quality is non-trivial due to the lack of a 'ground truth'. This is because SAGs and MAGs most often come from organisms that lack a cultivated reference strain. To assist downstream users in the evaluation of assembly quality, we recommend reporting basic assembly statistics from individual SAGs and/or MAGs, including, total assembly size, contig N50/L50, and maximum contig length (Supplementary Table 1). Contigs should not be artificially concatenated before deposition, as the resulting concatenation is not a true representation of the genome. We do not suggest a minimum assembly size, because genomes smaller than 200 kb have been found among symbiotic bacteria59,60,61. Lastly, the presence and completeness of the complement of encoded rRNAs and tRNAs should be used as an additional metric for assembly quality (Table 1). Because these draft genome sequences are not manually curated, the assembly quality standards of Chain et al.16 are not well-suited to SAGs and MAGs. However, in some cases, MAGs are manually curated, sometimes to completion, in which case the standards laid out in Chain et al.16 would be applicable.
The fraction of the genome captured from a SAG and MAG is another important metric because the level of completeness could dictate whether a publicly available genome is suitable for a specific downstream analysis. For example, complete genomes are preferable for pangenome analyses and genetic linkage studies62, whereas partial genomes may be suitable for fragment recruitment analyses26,63, metabolic predictions11, and phylogenetic reconstruction of individual proteins64. There are no established standards for estimating SAG and MAG completeness. The ideal approach might be to map a SAG or MAG to a closely related reference genome sequence. However, this is often not possible given the lack of suitable references for many microbial lineages and high levels of strain heterogeneity65,66,67. Alternatively, researchers have relied on the presence of 'universal' marker genes to estimate completeness. An appropriate marker gene should be present in genomes of nearly all taxa, as a single copy, and not subject to horizontal gene transfer. Although a discussion of approaches to identify such gene sets is beyond the scope of this manuscript, several gene sets have been identified and validated, some of which span both archaeal and bacterial domains68,69,70,71, whereas others are specific to archaeal13 or bacterial13,72,73 genomes. Many of these gene sets are now included in MAG and SAG quality assessment software, such as CheckM41, Anvi'o40, mOTU (Metagenomic Operational Taxonomic Units)74, and BUSCO (Benchmarking Universal Single-Copy Orthologs)71. Because different gene sets can produce different completeness estimates, the set chosen should be based on an established collection, previously validated and published in the literature (any of the above-mentioned sets would be sufficient), or the process of gene selection should be documented. Ribosomal proteins are included in gene sets, but because these genes tend to cluster unevenly across the genome, completeness estimates can be skewed75. To account for this bias, many of the marker sets include housekeeping genes involved in replication and transcription. The CheckM tool takes gene selection a step further by inferring lineage-specific genes based on the position of a query genome in a reference tree using a reduced set of multi-domain markers41. We recommend that MISAG- and MIMAG-compliant submissions use any of the previously mentioned single-copy marker gene sets, or follow a strategy similar to the one used by CheckM to identify gene sets; documentation of the selection process is considered mandatory. Gene sets must also be versioned, so that metadata can clearly indicate the procedure used.
Finally, the fraction of a SAG or MAG that may contain contaminating sequences should be reported. There are many highly recommended tools and techniques that can reduce or remove contaminating DNA in a genome before database submission (see sections on 'Single amplified genomes' and 'Metagenome-assembled genomes', and Supplementary Table 1 under 'decontamination software'). These approaches typically calculate the fraction of single-copy genes used in completeness estimates that are present more than once in a genome21,41,76,77, although contamination can be overestimated when a gene is artificially split at contig ends and scaffolding points. Tools, such as Anvi'o40 and CheckM41, can iteratively scan genomes for contamination to identify contaminant sequences. Both of these tools estimate contamination and provide several functions to enable users to remove contaminating sequences. Finally, we encourage researchers to carry out manual quality control based on nucleotide composition and BLAST-based analyses to identify suspicious contigs. Manual screening can be time consuming, although tools like Anvi'o have enabled interactive decontamination based on relevant parameters, such as GC content, tetranucleotide frequency, coverage, taxonomy, and combinations of these parameters78.
Mandatory standard metrics
We suggest that assembly statistics and estimates of genome completeness and contamination for SAGs and MAGs be mandatory metrics for both reporting in publications and deposition in public databases. Using these simple standards, we recommend that each genome be classified as: finished, high-quality draft, medium-quality draft, or low-quality draft (Table 1 and Supplementary Table 1). Mandatory standards are listed in Table 1, with the full set of standards (including optional and context-dependent) standards listed in Supplementary Table 1. A 'finished' category is reserved for genomes that can be assembled with extensive manual review and editing, into a single, validated, contiguous sequence per replicon, without gaps or ambiguities, having a consensus error rate equivalent to Q50 or better16. This category is reserved for only the highest quality manually curated SAGs and MAGs, and several finished genomes have been produced using these technologies10,11,21,37,79,80,81,82. For MAGs, genomes in this category are to be considered population genomes. 'High-quality draft' will indicate that a SAG or MAG is >90% complete with less than 5% contamination. Genomes in this category should also encode the 23S, 16S, and 5S rRNA genes, and tRNAs for at least 18 of the 20 possible amino acids, as even the reduced genomes of bacterial symbionts typically harbor the full complement of tRNAs83,84. 'Medium-quality draft' SAGs and MAGs are those genomes with completeness estimates of ≥50% and less than 10% contamination (Table 1 and Supplementary Table 1). All other SAGs and MAGs (<50% complete with <10% contamination) should be reported as 'low-quality drafts' (Table 1 and Supplementary Table 1).
All SAG and MAG public database submissions should include, at the very least, the metadata listed as mandatory in Supplementary Table 1. Additional standards include information about the assembly and binning software used and tools to taxonomically identify the genome. Owing to the many experimental and computational challenges associated with the generation of SAGs and MAGs, these minimum standards should be rigorously enforced in future genome submissions.
Conclusions
The GSC standards outlined here are a necessary extension of the MIxS standards, owing to the vast difference between generating genome sequences from cultivated versus uncultivated bacteria and archaea. These recommendations will serve to promote discussion and to generate feedback and subsequent improvements, which is especially relevant in the rapidly changing landscape of genomics technologies. These standards will be incorporated into the current GSC checklists and will complement the MIGS, MIMS, and MIMARKS checklists.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
07 December 2017
In the version of this article initially published, the following acknowledgment was omitted: A.L. was supported by the Russian Science Foundation (grant number 14-50-00069). The error has been corrected in the HTML and PDF versions of the article.
29 November 2017
In the version of this article initially published, the author A. Murat Eren was listed as A.M. Eren. The corresponding affiliation was given as the Knapp Center for Biomedical Discovery, rather than Department of Medicine, University of Chicago, Chicago, Illinois, USA, and Marine Biological Laboratory, Woods Hole, Massachusetts, USA. The errors have been corrected in the HTML and PDF versions of the article.
06 July 2018
A Correction to this paper has been published: https://doi.org/10.1038/nbt0718-660a
References
- Amann, R.I., Ludwig, W. & Schleifer, K.H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59, 143–169 (1995).
CAS PubMed PubMed Central Google Scholar - Rappé, M.S. & Giovannoni, S.J. The uncultured microbial majority. Annu. Rev. Microbiol. 57, 369–394 (2003).
Article CAS PubMed Google Scholar - Tyson, G.W. et al. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 428, 37–43 (2004).
Article CAS PubMed Google Scholar - Venter, J.C. et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 304, 66–74 (2004).
Article PubMed Google Scholar - Lasken, R.S. Single-cell sequencing in its prime. Nat. Biotechnol. 31, 211–212 (2013).
Article CAS PubMed Google Scholar - Stepanauskas, R. Single cell genomics: an individual look at microbes. Curr. Opin. Microbiol. 15, 613–620 (2012).
Article CAS PubMed Google Scholar - Turnbaugh, P.J. et al. The human microbiome project. Nature 449, 804–810 (2007).
Article CAS PubMed PubMed Central Google Scholar - Gilbert, J.A., Jansson, J.K. & Knight, R. The Earth Microbiome project: successes and aspirations. BMC Biol. 12, 69 (2014).
PubMed PubMed Central Google Scholar - Wu, D. et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 462, 1056–1060 (2009).
Article CAS PubMed PubMed Central Google Scholar - Anantharaman, K. et al. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat. Commun. 7, 13219 (2016).
Article CAS PubMed PubMed Central Google Scholar - Brown, C.T. et al. Unusual biology across a group comprising more than 15% of domain Bacteria. Nature 523, 208–211 (2015).
Article CAS PubMed Google Scholar - Eloe-Fadrosh, E.A. et al. Global metagenomic survey reveals a new bacterial candidate phylum in geothermal springs. Nat. Commun. 7, 10476 (2016).
Article CAS PubMed PubMed Central Google Scholar - Rinke, C. et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 499, 431–437 (2013).
Article CAS PubMed Google Scholar - Reddy, T.B.K. et al. The Genomes OnLine Database (GOLD) v.5: a metadata management system based on a four level (meta)genome project classification. Nucleic Acids Res. 43, D1099–D1106 (2015).
Article CAS PubMed Google Scholar - Field, D. et al. The minimum information about a genome sequence (MIGS) specification. Nat. Biotechnol. 26, 541–547 (2008).
Article CAS PubMed PubMed Central Google Scholar - Chain, P.S.G. et al. Genomics. Genome project standards in a new era of sequencing. Science 326, 236–237 (2009).
Article CAS PubMed Google Scholar - Yilmaz, P. et al. Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nat. Biotechnol. 29, 415–420 (2011).
Article CAS PubMed PubMed Central Google Scholar - Medema, M.H. et al. Minimum Information about a Biosynthetic Gene cluster. Nat. Chem. Biol. 11, 625–631 (2015).
Article CAS PubMed PubMed Central Google Scholar - Glass, E.M. et al. MIxS-BE: a MIxS extension defining a minimum information standard for sequence data from the built environment. ISME J. 8, 1–3 (2014).
Article CAS PubMed Google Scholar - Rinke, C. et al. Obtaining genomes from uncultivated environmental microorganisms using FACS-based single-cell genomics. Nat. Protoc. 9, 1038–1048 (2014).
Article CAS PubMed Google Scholar - Albertsen, M. et al. Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol. 31, 533–538 (2013).
Article CAS PubMed Google Scholar - Dick, G.J. et al. Community-wide analysis of microbial genome sequence signatures. Genome Biol. 10, R85 (2009).
Article CAS PubMed PubMed Central Google Scholar - Sharon, I. & Banfield, J.F. Microbiology. Genomes from metagenomics. Science 342, 1057–1058 (2013).
Article CAS PubMed Google Scholar - Nielsen, H.B. et al. Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nat. Biotechnol. 32, 822–828 (2014).
Article CAS PubMed Google Scholar - Stepanauskas, R. & Sieracki, M.E. Matching phylogeny and metabolism in the uncultured marine bacteria, one cell at a time. Proc. Natl. Acad. Sci. USA 104, 9052–9057 (2007).
Article CAS PubMed PubMed Central Google Scholar - Swan, B.K. et al. Prevalent genome streamlining and latitudinal divergence of planktonic bacteria in the surface ocean. Proc. Natl. Acad. Sci. USA 110, 11463–11468 (2013).
Article PubMed PubMed Central Google Scholar - Blainey, P.C. The future is now: single-cell genomics of bacteria and archaea. FEMS Microbiol. Rev. 37, 407–427 (2013).
Article CAS PubMed Google Scholar - Hutchison, C.A. III & Venter, J.C. Single-cell genomics. Nat. Biotechnol. 24, 657–658 (2006).
Article CAS PubMed Google Scholar - Dean, F.B. et al. Comprehensive human genome amplification using multiple displacement amplification. Proc. Natl. Acad. Sci. USA 99, 5261–5266 (2002).
Article CAS PubMed PubMed Central Google Scholar - Lasken, R.S. Single-cell genomic sequencing using Multiple Displacement Amplification. Curr. Opin. Microbiol. 10, 510–516 (2007).
Article CAS PubMed Google Scholar - de Bourcy, C.F. et al. A quantitative comparison of single-cell whole genome amplification methods. PLoS One 9, e105585 (2014).
Article CAS PubMed PubMed Central Google Scholar - Yilmaz, S., Allgaier, M. & Hugenholtz, P. Multiple displacement amplification compromises quantitative analysis of metagenomes. Nat. Methods 7, 943–944 (2010).
Article CAS PubMed Google Scholar - Lasken, R.S. & Stockwell, T.B. Mechanism of chimera formation during the Multiple Displacement Amplification reaction. BMC Biotechnol. 7, 19 (2007).
Article CAS PubMed PubMed Central Google Scholar - Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Article CAS PubMed PubMed Central Google Scholar - Peng, Y., Leung, H.C.M., Yiu, S.M. & Chin, F.Y.L. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428 (2012).
Article CAS PubMed Google Scholar - Schmieder, R. & Edwards, R. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS One 6, e17288 (2011).
Article CAS PubMed PubMed Central Google Scholar - Woyke, T. et al. One bacterial cell, one complete genome. PLoS One 5, e10314 (2010).
Article CAS PubMed PubMed Central Google Scholar - Woyke, T. et al. Decontamination of MDA reagents for single cell whole genome amplification. PLoS One 6, e26161 (2011).
Article CAS PubMed PubMed Central Google Scholar - Clingenpeel, S., Clum, A., Schwientek, P., Rinke, C. & Woyke, T. Reconstructing each cell's genome within complex microbial communities-dream or reality? Front. Microbiol. 5, 771 (2015).
Article PubMed PubMed Central Google Scholar - Eren, A.M. et al. Anvi'o: an advanced analysis and visualization platform for 'omics data. PeerJ 3, e1319 (2015).
Article PubMed PubMed Central Google Scholar - Parks, D.H., Imelfort, M., Skennerton, C.T., Hugenholtz, P. & Tyson, G.W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. PeerJ PrePrints 3, e554v2 (2015).
Google Scholar - Tennessen, K. et al. ProDeGe: a computational protocol for fully automated decontamination of genomes. ISME J. 10, 269–272 (2016).
Article CAS PubMed Google Scholar - Lux, M. et al. acdc - Automated Contamination Detection and Confidence estimation for single-cell genome data. BMC Bioinformatics 17, 543 (2016).
Article PubMed PubMed Central Google Scholar - Woyke, T. et al. Symbiosis insights through metagenomic analysis of a microbial consortium. Nature 443, 950–955 (2006).
Article CAS PubMed Google Scholar - Baker, B.J. et al. Lineages of acidophilic archaea revealed by community genomic analysis. Science 314, 1933–1935 (2006).
Article CAS PubMed Google Scholar - Wrighton, K.C. et al. Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 337, 1661–1665 (2012).
Article CAS PubMed Google Scholar - Boisvert, S., Raymond, F., Godzaridis, E., Laviolette, F. & Corbeil, J. Ray Meta: scalable de novo metagenome assembly and profiling. Genome Biol. 13, R122 (2012).
Article CAS PubMed PubMed Central Google Scholar - Li, D., Liu, C.-M., Luo, R., Sadakane, K. & Lam, T.-W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015).
Article CAS PubMed Google Scholar - Nurk, S., Meleshko, D., Korobeynikov, A. & Pevzner, P.A. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834 (2017).
Article CAS PubMed PubMed Central Google Scholar - Hess, M. et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 331, 463–467 (2011).
Article CAS PubMed Google Scholar - Mande, S.S., Mohammed, M.H. & Ghosh, T.S. Classification of metagenomic sequences: methods and challenges. Brief. Bioinform. 13, 669–681 (2012).
Article PubMed Google Scholar - Nelson, W.C., Maezato, Y., Wu, Y.-W., Romine, M.F. & Lindemann, S.R. Identification and resolution of microdiversity through Metagenomic Sequencing of Parallel Consortia. Appl. Environ. Microbiol. 82, 255–267 (2016).
Article CAS PubMed Google Scholar - Sharon, I. et al. Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization. Genome Res. 23, 111–120 (2013).
Article CAS PubMed PubMed Central Google Scholar - Imelfort, M. et al. GroopM: an automated tool for the recovery of population genomes from related metagenomes. PeerJ 2, e603 (2014).
Article PubMed PubMed Central Google Scholar - Wu, Y.-W., Tang, Y.-H., Tringe, S.G., Simmons, B.A. & Singer, S.W. MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome 2, 26 (2014).
Article CAS PubMed PubMed Central Google Scholar - Kang, D.D., Froula, J., Egan, R. & Wang, Z. A robust statistical framework for reconstructing genomes from metagenomic data. Preprint at bioRxiv http://dx.doi.org//10.1101/011460 (2014).
- Alneberg, J. et al. CONCOCT: Clustering cONtigs on COverage and ComposiTion. Preprint at https://arxiv.org/abs/1312.4038v1 (2013).
- Strous, M., Kraft, B., Bisdorf, R. & Tegetmeyer, H.E. The binning of metagenomic contigs for microbial physiology of mixed cultures. Front. Microbiol. 3, 410 (2012).
Article PubMed PubMed Central Google Scholar - Bennett, G.M., McCutcheon, J.P., MacDonald, B.R., Romanovicz, D. & Moran, N.A. Differential genome evolution between companion symbionts in an insect-bacterial symbiosis. MBio 5, e01697–e14 (2014).
Article CAS PubMed PubMed Central Google Scholar - Nakabachi, A. et al. The 160-kilobase genome of the bacterial endosymbiont Carsonella. Science 314, 267 (2006).
Article CAS PubMed Google Scholar - Venton, D. Highlight: tiniest of the tiny—a new low for genome size. Genome Biol. Evol. 5, 1702–1703 (2013).
Article CAS PubMed PubMed Central Google Scholar - Lasken, R.S. Genomic sequencing of uncultured microorganisms from single cells. Nat. Rev. Microbiol. 10, 631–640 (2012).
Article CAS PubMed Google Scholar - Woyke, T. et al. Assembling the marine metagenome, one cell at a time. PLoS One 4, e5299 (2009).
Article CAS PubMed PubMed Central Google Scholar - Vanwonterghem, I. et al. Methylotrophic methanogenesis discovered in the archaeal phylum Verstraetearchaeota. Nat. Microbiol. 1, 16170 (2016).
Article CAS PubMed Google Scholar - Allen, E.E. & Banfield, J.F. Community genomics in microbial ecology and evolution. Nat. Rev. Microbiol. 3, 489–498 (2005).
Article CAS PubMed Google Scholar - Konstantinidis, K.T., Ramette, A. & Tiedje, J.M. The bacterial species definition in the genomic era. Phil. Trans. R. Soc. Lond. B 361, 1929–1940 (2006).
Article Google Scholar - Zengler, K. Central role of the cell in microbial ecology. Microbiol. Mol. Biol. Rev. 73, 712–729 (2009).
Article CAS PubMed PubMed Central Google Scholar - Darling, A.E. et al. PhyloSift: phylogenetic analysis of genomes and metagenomes. PeerJ 2, e243 (2014).
Article PubMed PubMed Central Google Scholar - Wu, M. & Scott, A.J. Phylogenomic analysis of bacterial and archaeal sequences with AMPHORA2. Bioinformatics 28, 1033–1034 (2012).
Article CAS PubMed Google Scholar - Mende, D.R., Sunagawa, S., Zeller, G. & Bork, P. Accurate and universal delineation of prokaryotic species. Nat. Methods 10, 881–884 (2013).
Article CAS PubMed Google Scholar - Simão, F.A., Waterhouse, R.M., Ioannidis, P., Kriventseva, E.V. & Zdobnov, E.M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Article CAS PubMed Google Scholar - Campbell, J.H. et al. UGA is an additional glycine codon in uncultured SR1 bacteria from the human microbiota. Proc. Natl. Acad. Sci. USA 110, 5540–5545 (2013).
Article CAS PubMed PubMed Central Google Scholar - Wu, M. & Eisen, J.A. A simple, fast, and accurate method of phylogenomic inference. Genome Biol. 9, R151 (2008).
Article CAS PubMed PubMed Central Google Scholar - Sunagawa, S. et al. Metagenomic species profiling using universal phylogenetic marker genes. Nat. Methods 10, 1196–1199 (2013).
Article CAS PubMed Google Scholar - Klappenbach, J.A., Saxman, P.R., Cole, J.R. & Schmidt, T.M. rrndb: the Ribosomal RNA Operon Copy Number Database. Nucleic Acids Res. 29, 181–184 (2001).
Article CAS PubMed PubMed Central Google Scholar - Sekiguchi, Y. et al. First genomic insights into members of a candidate bacterial phylum responsible for wastewater bulking. PeerJ 3, e740 (2015).
Article CAS PubMed PubMed Central Google Scholar - Soo, R.M. et al. An expanded genomic representation of the phylum cyanobacteria. Genome Biol. Evol. 6, 1031–1045 (2014).
Article PubMed PubMed Central Google Scholar - Delmont, T.O. & Eren, A.M. Identifying contamination with advanced visualization and analysis practices: metagenomic approaches for eukaryotic genome assemblies. PeerJ 4, e1839 (2016).
Article CAS PubMed PubMed Central Google Scholar - Chivian, D. et al. Environmental genomics reveals a single-species ecosystem deep within Earth. Science 322, 275–278 (2008).
Article CAS PubMed Google Scholar - Di Rienzi, S.C. et al. The human gut and groundwater harbor non-photosynthetic bacteria belonging to a new candidate phylum sibling to Cyanobacteria. eLife 2, e01102 (2013).
Article CAS PubMed PubMed Central Google Scholar - Castelle, C.J. et al. Extraordinary phylogenetic diversity and metabolic versatility in aquifer sediment. Nat. Commun. 4, 2120 (2013).
Article PubMed Google Scholar - Wrighton, K.C. et al. RubisCO of a nucleoside pathway known from Archaea is found in diverse uncultivated phyla in bacteria. ISME J. 10, 2702–2714 (2016).
Article CAS PubMed PubMed Central Google Scholar - Martinson, V.G., Magoc, T., Koch, H., Salzberg, S.L. & Moran, N.A. Genomic features of a bumble bee symbiont reflect its host environment. Appl. Environ. Microbiol. 80, 3793–3803 (2014).
Article CAS PubMed PubMed Central Google Scholar - Schulz, F. et al. A Rickettsiales symbiont of amoebae with ancient features. Environ. Microbiol. 18, 2326–2342 (2016).
Article CAS PubMed Google Scholar - Shendure, J. et al. Accurate multiplex polony sequencing of an evolved bacterial genome. Science 309, 1728–1732 (2005).
Article CAS PubMed Google Scholar - Margulies, M. et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380 (2005).
Article PubMed PubMed Central Google Scholar - Raghunathan, A. et al. Genomic DNA amplification from a single bacterium. Appl. Environ. Microbiol. 71, 3342–3347 (2005).
Article CAS PubMed PubMed Central Google Scholar - Marcy, Y. et al. Dissecting biological “dark matter” with single-cell genetic analysis of rare and uncultivated TM7 microbes from the human mouth. Proc. Natl. Acad. Sci. USA 104, 11889–11894 (2007).
Article CAS PubMed PubMed Central Google Scholar - Bentley, D.R. et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456, 53–59 (2008).
Article CAS PubMed PubMed Central Google Scholar - Harris, T.D. et al. Single-molecule DNA sequencing of a viral genome. Science 320, 106–109 (2008).
Article CAS PubMed Google Scholar
Acknowledgements
We thank H. Maughan for constructive feedback and editing of the manuscript and Z. Rostomian for support with illustrations. Funding sources: the work conducted by the US Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, is supported under Contract No. DE-AC02-05CH11231. T.W. and D.D. were further supported by the Laboratory Directed Research and Development Program of Lawrence Berkeley National Laboratory under the aforementioned Contract No. R.S. and E.B. were supported by the US National Science Foundation grants DEB-1441717, OCE-1232982, OCE-1136488, and OCE-1335810. T.J.G.E. is supported by grants of the European Research Council (ERC Starting grant 310039) and the Swedish Foundation for Strategic Research (SSF-FFL5). P.H. and D.H.P. are supported by an Australian Laureate Fellowship (FL150100038) from the Australian Research Council, and G.W.T. and C.R. are supported by the Gordon and Betty Moore Foundation (Grant ID:GBMF3801). R.S. supported by NSF grants DEB-1441717 and OCE-1335810. K.D.M. acknowledges funding from the United States National Science Foundation (NSF) Microbial Long Term Ecological Research program (NTL-LTER DEB-1440297), an INSPIRE award (DEB-1344254), and National Institute of Food and Agriculture, US Department of Agriculture Hatch Project 1002996. M.P. acknowledges National Institutes of Health, National Institute of Dental and Craniofacial Research grant 5R01DE024463. A.L. was supported by the Russian Science Foundation (grant number 14-50-00069).
Author information
Author notes
- A list of consortium members appears at the end of the paper.
Authors and Affiliations
- Department of Energy Joint Genome Institute, Walnut Creek, California, USA
Robert M Bowers, Nikos C Kyrpides, Miranda Harmon-Smith, Devin Doud, T B K Reddy, Frederik Schulz, Jessica Jarett, Adam R Rivers, Emiley A Eloe-Fadrosh, Susannah G Tringe, Natalia N Ivanova, Alex Copeland, Alicia Clum, Rex R Malmstrom, Sean P Jungbluth, Nikos C Kyrpides & Tanja Woyke - Bigelow Laboratory for Ocean Sciences, East Boothbay, Maine, USA
Ramunas Stepanauskas & Eric D Becraft - United States Department of Agriculture, Agricultural Research Service, Genomics and Bioinformatics Research Unit, Gainesville, Florida, USA
Adam R Rivers - School of Natural Sciences, University of California Merced, Merced, California, USA
Susannah G Tringe & Tanja Woyke - Broad Institute, Cambridge, Massachusetts, USA
Bruce Birren - Biosciences Division, Oak Ridge National Laboratory, Oakridge, Tennessee, USA
Mircea Podar - Structural and Computational Biology Unit, European Molecular Biology Laboratory, Heidelberg, Germany
Peer Bork - The Jackson Laboratory for Genomic Medicine, Farmington, Connecticut, USA
George M Weinstock - Department of Microbiology & Molecular Genetics, Biomedical Physical Sciences, Michigan State University, East Lansing, Michigan, USA
George M Garrity & George M Garrity - Department of Biology, California State University, San Bernardino, California, USA
Jeremy A Dodsworth - J. Craig Venter Institute, San Diego, California, USA
Shibu Yooseph - J. Craig Venter Institute, Rockville, Maryland, USA
Granger Sutton & Granger Sutton - Microbial Genomics and Bioinformatics Research Group, Max Planck Institute for Marine Microbiology, Bremen, Germany
Frank O Glöckner, Pelin Yilmaz, Frank O Glöckner & Pelin Yilmaz - Biosciences Division, Argonne National Laboratory, Argonne, Illinois, USA
Jack A Gilbert, Folker Meyer, Jack A Gilbert & Folker Meyer - Department of Surgery, University of Chicago, Chicago, Illinois, USA
Jack A Gilbert & Jack A Gilbert - Biological Sciences Division, Earth and Biological Sciences Directorate, Pacific Northwest National Laboratory, Richland, Washington, USA
William C Nelson - Department of Microbiology & Immunology, University of British Columbia, Vancouver, British Columbia, Canada
Steven J Hallam - Center for Dark Energy Biosphere Investigation, University of Southern California, Los Angeles, California, USA
Sean P Jungbluth - Department of Cell and Molecular Biology, Science for Life Laboratory, Uppsala University, Uppsala, Sweden
Thijs J G Ettema - Advanced Genomics Lab, University of Vermont Cancer Center, Burlington, Vermont, USA
Scott Tighe - Georgia Institute of Technology, School of Civil and Environmental Engineering, Atlanta, Georgia, USA
Konstantinos T Konstantinidis - Department of Civil and Environmental Engineering, University of Illinois at Urbana-Champaign, Urbana, Illinois, USA
Wen-Tso Liu - Department of Marine Science, University of Texas-Austin, Marine Science Institute, Austin, Texas, USA
Brett J Baker - Department of Microbiology and Ecosystem Science, University of Vienna, Vienna, Austria
Thomas Rattei - Genome Center, University of California, Davis, California, USA
Jonathan A Eisen - School of Life Sciences, University of Nevada Las Vegas, Las Vegas, Nevada, USA
Brian Hedlund - Nevada Institute of Personalized Medicine, University of Nevada Las Vegas, Las Vegas, Nevada, USA
Brian Hedlund - Department of Civil and Environmental Engineering, University of Wisconsin-Madison, Madison, Wisconsin, USA
Katherine D McMahon - Department of Bacteriology, University of Wisconsin-Madison, Madison, Wisconsin, USA
Katherine D McMahon - Cooperative Institute for Research in Environmental Sciences, University of Colorado, Boulder, Colorado, USA
Noah Fierer - Department of Ecology and Evolutionary Biology, University of Colorado, Boulder, Colorado, USA
Noah Fierer - and Departments of Pediatrics and Computer Science & Engineering, Center for Microbiome Innovation, University of California San Diego, La Jolla, California, USA
Rob Knight & Rob Knight - European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), Welcome Trust Genome Campus, Hinxton, Cambridge, UK
Rob Finn, Guy Cochrane, Rob Finn & Guy Cochrane - National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, Maryland, USA
Ilene Karsch-Mizrachi & Ilene Karsch-Mizrachi - Australian Centre for Ecogenomics, School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, Queensland, Australia
Gene W Tyson, Christian Rinke, Philip Hugenholtz, Donovan H Parks & Philip Hugenholtz - Centre for Algorithmic Biotechnology, ITBM, St. Petersburg State University, St. Petersburg, Russia
Alla Lapidus - Department of Medicine, University of Chicago, Chicago, Illinois, USA
A Murat Eren - Marine Biological Laboratory, Woods Hole, Massachusetts, USA
A Murat Eren - National Cancer Institute, Frederick, Maryland, USA
Lynn Schriml & Lynn Schriml - Department of Earth and Planetary Science, University of California, Berkeley, California, USA
Jillian F Banfield
Authors
- Robert M Bowers
- Nikos C Kyrpides
- Ramunas Stepanauskas
- Miranda Harmon-Smith
- Devin Doud
- T B K Reddy
- Frederik Schulz
- Jessica Jarett
- Adam R Rivers
- Emiley A Eloe-Fadrosh
- Susannah G Tringe
- Natalia N Ivanova
- Alex Copeland
- Alicia Clum
- Eric D Becraft
- Rex R Malmstrom
- Bruce Birren
- Mircea Podar
- Peer Bork
- George M Weinstock
- George M Garrity
- Jeremy A Dodsworth
- Shibu Yooseph
- Granger Sutton
- Frank O Glöckner
- Jack A Gilbert
- William C Nelson
- Steven J Hallam
- Sean P Jungbluth
- Thijs J G Ettema
- Scott Tighe
- Konstantinos T Konstantinidis
- Wen-Tso Liu
- Brett J Baker
- Thomas Rattei
- Jonathan A Eisen
- Brian Hedlund
- Katherine D McMahon
- Noah Fierer
- Rob Knight
- Rob Finn
- Guy Cochrane
- Ilene Karsch-Mizrachi
- Gene W Tyson
- Christian Rinke
- Alla Lapidus
- Folker Meyer
- Pelin Yilmaz
- Donovan H Parks
- A Murat Eren
- Lynn Schriml
- Jillian F Banfield
- Philip Hugenholtz
- Tanja Woyke
Consortia
The Genome Standards Consortium
- Nikos C Kyrpides
- , Lynn Schriml
- , George M Garrity
- , Philip Hugenholtz
- , Granger Sutton
- , Pelin Yilmaz
- , Folker Meyer
- , Frank O Glöckner
- , Jack A Gilbert
- , Rob Knight
- , Rob Finn
- , Guy Cochrane
- & Ilene Karsch-Mizrachi
Corresponding authors
Correspondence toRobert M Bowers or Tanja Woyke.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International (CCCC BY 4.0) license. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article
Cite this article
Bowers, R., Kyrpides, N., Stepanauskas, R. et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea.Nat Biotechnol 35, 725–731 (2017). https://doi.org/10.1038/nbt.3893
- Received: 10 November 2016
- Accepted: 27 April 2017
- Published: 01 August 2017
- Issue Date: August 2017
- DOI: https://doi.org/10.1038/nbt.3893