Genomic inference of the metabolism and evolution of the archaeal phylum Aigarchaeota (original) (raw)
Introduction
Recent advancements in metagenomics and single-cell techniques have enabled researchers to obtain a glimpse of the genomic information and genetic diversity of major lineages of Bacteria and Archaea that have eluded microbial cultivation1,2,3,4. One such group is the archaeal lineage Aigarchaeota. Members of the Aigarchaeota are widely distributed in terrestrial and subsurface geothermal systems and marine hydrothermal environments3,5,6,7,8,9. To date, eight non-redundant Aigarchaeota single-amplified genomes (SAGs) and metagenome-assembled genomes (MAGs) exist in the IMG and NCBI RefSeq databases. However, CheckM10 estimates that only one is >90% complete. Phylogenetic analysis of conserved, single-copy core genes showed that Aigarchaeota is closely related to Crenarchaeota, Korarchaeota and Thaumarchaeota3,7, forming the “TACK” superphylum11. As a sister lineage, Thaumarchaeota has been largely studied with respect to their importance in nitrogen cycling, particularly chemolithotrophic ammonia oxidation12,13,14. In contrast, little is known about Aigarchaeota’s ecological role or evolutionary history. It is still controversial whether Aigarchaeota represents a new phylum or a subclade of phylum Thaumarchaeota7,9,15,16.
Here, by integrating genome-resolved metagenomics and comparative genomics, we aim to address the gaps in understanding the origin and evolutionary history of the Aigarchaeota, and their roles in biogeochemical cycling. Our results reveal the facultative or strictly anaerobic lifestyle of Aigarchaeota with the ability to oxidize sulfide to gain energy for growth. Evolutionary genomic analyses of Thaumarchaeota and Aigarchaeota suggest both two phyla evolved from hot habitats and they share a large proportion of gene families with their last common ancestor. Adaptation to different habitats led to the functional differentiation of Aigarchaeota and Thaumarchaeota, with the latter invading a wide diversity of non-thermal environments. Genes acquired horizontally from Euryarchaeota and Firmicutes played a significant role in the functional diversification of Aigarchaeota in hot spring habitats. This study represents a significant advancement in our understanding of the genomic diversity and evolution of Aigarchaeota.
Results and discussion
Aigarchaeota genomes recovered from metagenomes
Metagenome sequencing was performed on two hot spring sediment samples collected from Tengchong county of Yunnan, China (Fig. 1a). Metagenome assembly of sequencing data and genome binning based on tetra-nucleotide frequencies and coverage patterns were conducted, resulting in six near-complete Aigarchaeota genomic bins (Table 1). One bin was obtained from Gumingquan (GMQ, pH 9.3 and temperature 89 °C)17, and five from another pool named Jinze (JZ, pH 6.5 and temperature 75 °C)18 (Supplementary Table 1). The genome sizes of the obtained bins range from 1.09 Mbp to 1.65 Mbp (averagely 1.4 Mbp). They encode an average of 1384 genes with an average gene length of 905 bp. Genomes are well curated and genome quality was evaluated using CheckM10, indicating the high-quality of the genomic bins with genome completeness ranging from 97 to 99% and nearly no contamination of other genome fragments with 16S rRNA and tRNA (>18) are detectable19 (Table 1; Supplementary Fig. 1). A concatenated alignment of 16 ribosomal proteins was used for maximum likelihood tree construction (Fig. 1b). A phylogenetic tree with high bootstrap support reveals that the six genomes are located in distinct lineages. A 16S rRNA-based phylogenetic tree indicates that they represent three groups with 94% as a threshold (Supplementary Fig. 2). GMQ bin_10 and JZ bin_10 are the close neighbors sharing 97% of the amino acid identity (AAI) (Supplementary Figs. 3, 4). JZ bins_28 shows 92% AAI to the published genome of “Candidatus Caldiarchaeum subterraneum” (Supplementary Fig. 3). The only detected Thaumarchaeota genome, DRTY7 bin_36, may be the first genome of the pSL12 lineage, which has been found in hot springs previously20,21,22.
Fig. 1
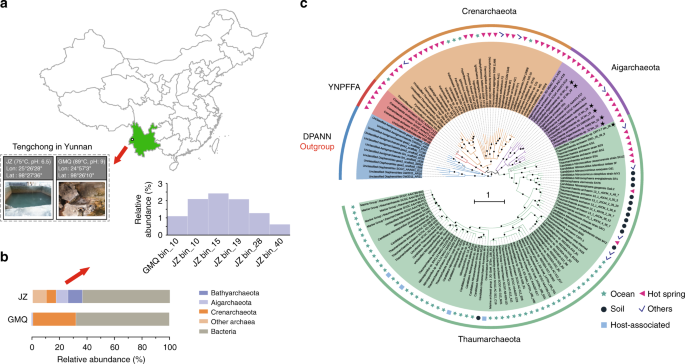
The reconstructed genomes of Aigarchaeota. a Geographic locations of the two hot spring sites, JZ and GMQ, where sediment samples were collected. b Relative abundance of the six aigarchaeal genome bins recovered from metagenomes. c Phylogenetic placement of the bins. The tree was constructed based on the concatenated alignment of 16 ribosomal proteins. Nodes with bootstrap value ≥90 (70) are indicated as solid (hollow) circles
Table 1 General genomic features of the Aigarchaeota and Thaumarchaeota bins reconstructed from the metagenome assembly
Metabolic potential
Most Aigarchaeota genomes contain complete glycolysis pathways and citrate (TCA) cycles (Fig. 2). JZ bin_40 might also degrade cellulose, as it contains endoglucanase and beta-glucosidase. These genes may enable hydrolysis of cellulose to cellobiose and finally to glucose (Fig. 2; Supplementary Data 1). Amino acid sequence analysis showed that the predicted endoglucanase in JZ bin_40 contained a Frv domain, an aminopeptidase belonging to the M42 family. The identified putative endoglucanases share 33~56% identity to currently known endoglucanases, sharin the same structure including the active sites and potential metal-binding sites (Supplementary Fig. 5). Despite the difficulty in distinguishing them based on sequence similarity, previous cellulase assays showed that this might be a new enzyme with hydrolytic activity against cellulose in thermal ecosystems23,24. Additionally, based on the comparison to Carbohydrate-active enzymes (CAZy) database, numerous genes (29) distributed in 15 glycosyl hydrolase (GH) families were observed in JZ bin_40, including genes for alpha-glucosidase (GH31, degradation of starch and disaccharides), beta-glucosidase (disaccharides degradation), alpha-galactosidase (GH36, lactose degradation), alpha-galactosidase (GH4, melibiose degradation), beta-galactosidase (GH1, cellulose or hemicellulose degradation), alpha-mannosidase (GH38, mannose degradation), alpha-amylase (GH57, starch degradation), alpha-L-rhamnosidase (GH78, pectin degradation), and also a glycogen-debranching enzyme (GH63, release of glucose through the breakdown of glycogen) (Fig. 2; Supplementary Table 2). The generated glucose could further support cell growth through oxidation in glycolysis and the TCA cycle. The first-pass KEGG annotation annotated no gene as beta-glucosidase. However, the best hit for one of the genes in JZ bin_40 was identified as beta-glucosidase (GBC70340.1; 99% in query coverage and 86% in amino-acid identity) by BLASTp to the NCBI-nr database manually, underscoring the need for manual curation of annotation results. Additionally, 26 genes involved in oligosaccharide transporters were identified in JZ bin_40 which could be employed to transport mono/polysaccharides into the cell, mainly including 17 and 5 genes involved in multiple and simple sugar transport system, and a complete raffinose/stachyose/melibiose transport system (three genes; Supplementary Data 1).
Fig. 2
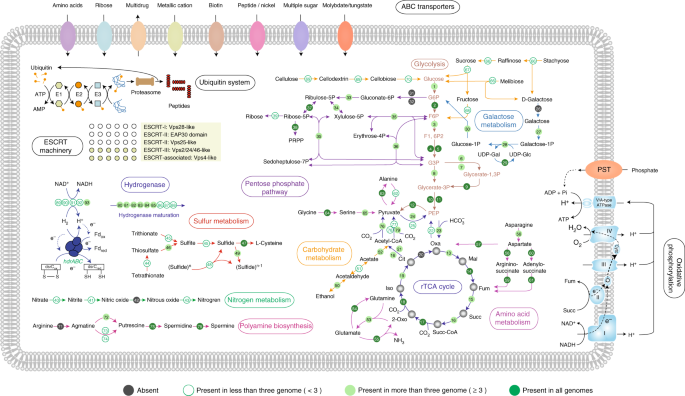
Overview of metabolic potentials in Aigarchaeota. Genes related to glycolysis, galactose metabolism, roTCA cycle, the pentose phosphate pathway, the pyruvate metabolism, beta-oxidation of fatty acids, oxidative phosphorylation chain, protein biosynthesis-related pathway, membrane transporters, and ubiquitin system are shown. Green solid circles represent genes present in all six aigarchaeal genome bins. Green hollow circles represent genes occurring in one to five bins. Gray solid circles represent genes absent in all bins. For full names and copy numbers of the genes in number see Supplementary Data 1. roTCA reversed oxidative tricarboxylic acid cycle; ESCRT the endosomal sorting complexes required for transport, G6p glucose 6-phosphate, F6P fructose 6-phosphate; F1,6P2 fructose 1,6-bisphosphate, PEP phosphoenolpyruvate
Both ATP citrate lyase and citryl-CoA synthase/citryl-CoA lyase were not annotated, indicating the lack of carbon fixation by conventional reductive TCA (rTCA) cycle. Instead, an alternative pathway to generate oxaloacetate, encoding citrate synthase, exists across the Aigarchaeota, suggesting the reversed oxidative TCA (roTCA) pathway for carbon fixation, as proposed recently25. In another recent study, strong evidence was presented that the enzymatic activity for citrate synthase was detectable under chemolithoautotrophic conditions in Thermosulfidibacter takaii ABI70S6, suggesting this reversible enzyme may convert citrate into oxaloacetate and acetyl-CoA26. Previous studies suggested that Aigarchaeota had the potential to fix carbon through the Calvin-Benson-Bassham (CBB) and 3-hydroxypropionate/4-hydroxybutyrate (HP/HB) (dicarboxylate/4-hydroxybuyrate (DC/HB)) cycles3,8. However, no enzymes encoding RubisCO and 4-hydroxybutyryl-CoA dehydratase were observed, suggesting these two pathways are missing from these Aigarchaeota genomes, consistent with a previous study27. Several heme-copper terminal oxidases were identified in all Aigarchaeota except GMQ bin_10 and JZ bin_10, indicating that most of them are aerobes28 (Supplementary Data 1). GMQ bin_10 and JZ bin_10 might be anaerobes because that they do not possess any clearly recognizable terminal oxidases. Despite the anaerobic lifestyle of these two organisms, near-complete complex I of respiratory chains were observed, including nearly all subunits of NADH ubiquinone oxidoreductase (nuo), but with the absence of nuoK and nuoN (Supplementary Fig. 6a, b). Instead, the detected mrpC and mrpD may function as alternatives due to their homology to nuoK and nuoN29. Interestingly, the CXXC motif was identifiable in the nuoD protein in GMQ bin_10 and JZ bin_10, implicating it may function as a Group 4 NiFe hydrogenases (Supplementary Fig. 6c). The absence of acetaldehyde dehydrogenase and acetyl-CoA synthetase (ADP-forming) suggests that these thermophiles cannot generate alcohol, acetate or butyrate through fermentation.
Recent studies suggested that microbes may have exploited carbon monoxide as a supplemental energy source for their growth3,7,30. Pathways for CO metabolism occur frequently among aerobic bacteria31. Most Aigarchaeota bins described here contain at least two copies of all three subunits of the coxLMS complex. However, the absence of cox genes reinforces the anaerobic characteristics of JZ bin_10 and GMQ bin_10. In total, 13 coxL genes were detected among the genomes. Phylogenetic analysis showed that most of them are classified as Form II, a putative CODH, with typical (AYRGAGR) or atypical motifs (PYRGAGR) observed (Supplementary Fig. 7). Only one coxL belonging to JZ bin_19 was identified as Form I with an AYXCSFR signature. This suggests Aigarchaeota could oxidize CO aerobically as an energy supplement when organic substrates are limited. We speculate that the supplied energy might contribute to biomass by coupling carbon fixation through the roTCA cycle.
Sulfur and hydrogen are thought to be crucial in energy cycling in thermal habitats32. It is noteworthy that all bins appear to encode capacity for sulfur utilization. Dissimilatory sulfite reductase encoded by dsrA and dsrB were detected in JZ bin_15, suggesting that this bin could perform dissimilatory sulfite reduction. Aigarchaeota dsrA and dsrB genes are located near the tips of branches with genes from the phylum Firmicutes (Supplementary Fig. 8a), suggesting that JZ bin_15 might have gained this feature by horizontal gene transfer (HGT) from Firmicutes. High amino acid similarity (66 and 63%, respectively) to Thermanaeromonas toyohensis ToBE, within the Firmicutes, supports this observation. Consensus coverage depth shows that the sequence assembly of the _dsrAB_-containing scaffold is reliable (Supplementary Fig. 8b). Previous studies also indicate that thermophiles have undergone frequent HGTs, facilitating their adaptation to the harsh environments33,34,35. The genomic bins GMQ bin_10 and JZ bin_10 harbor genes related to sulfohydrogenase but lack beta and gamma subunits, suggesting a deficiency in sulfur reduction36. The alpha subunit may function as a Ni/Fe hydrogenase in these two organisms, which may be important for redox balance37. Moreover, the gene encoding for sulfide-quinone reductase (sqr) was present in all genome bins except GMQ bin_10 and JZ bin_10. This indicates that most Aigarchaeota are chemolithotrophs, harvesting energy from the oxidation of sulfide to elemental sulfur. In most cases, the oxidation of sulfide by sqr or reduction of sulfite by dsrAB takes place under anaerobic conditions, implicating a facultatively anaerobic lifestyle of these Aigarchaeota. Interestingly, we found several copies of a heterodisulfide reductase (hdr) complex in JZ bin_15 (Supplementary Fig. 9). In particular, one copy clustered with subunits of F420-non-reducing hydrogenase (mvh complex), was also found in some sulfate-reducing microorganisms38,39. The H2 is oxidized by mvhADG to generate protons along with electrons. Due to the absence of methanogensis pathways in this bin, we propose that the released electrons might be transferred to HdrABC to reduce Fdox and DsrC instead of heterodisulfide. Then the reduced DsrC functions as an electron donor by coupling with DsrAB sulfite reductase to perform sulfite reduction39. It was also speculated that the detected Mvh:Hdr complex in Archaeoglobus profundus might be employed to reduce an electron carrier, which in turn functions as an electron donor of the enzymes of sulfite reduction38.
In Aigarchaeota, 19 NiFe hydrogenases and 33 hydrogenase maturation factors are present (Supplementary Data 1), potentially involved redox homeostasis4,36. Most of these hydrogenases form a separate lineage, showing that they are highly divergent from previous reported hydrogenases in other microbes (Supplementary Fig. 10). Phylogenetic analysis reveals that 7, 5, 3, and 4 of the 19 NiFe hydrogenases are type 3a, 3b, 3c, and 4d hydrogenases, respectively, which mainly include F420-reducing hydrogenase (frh), sulfohydrogenase (hyd) and Ni,Fe-hydrogenase III (ech) (Supplementary Fig. 10; Supplementary Data 1). Several of them are Aigarchaeota-specific and are distantly related to other hydrogenases. The mvh complex described above represents a type 3c hydrogenases. These hydrogenases could supply intracellular reducing equivalents needed for various redox reactions40,41.
The presence of Mu-like prophages in JZ bin_28 and bin_40 suggests that Aigarchaeota might be subject to phage infection in hot springs. Prophages impose a fitness burden through DNA insertion in the host genome, but can also provide beneficial functions to their hosts to make them competitive and survive in harsh environments. As in most Archaea, several CRISPR loci consisting of CRISPR arrays, belonging to type I and type III systems, were detected in all the genomes (Supplementary Fig. 11). This illustrates that even under the extreme high temperature, microbes in the community could also be invaded by viruses. This is consistent with previous research showing that these two CRISPR-cas systems are ubiquitous among other Archaea42.
Current available Aigarchaeota genomes were all from hot spring ecosystems, with the exception of “Candidatus Caldiarchaeum subterraneum”, which was from hot fracture water within a subsurface gold mine (Supplementary Data 2)7. Most of them harbor heme-copper oxidases, showing they are aerobes that can use oxygen as an electron acceptor3,30. GMQ bin_10 and JZ bin_10 are anaerobes due to the lack of terminal oxidases. Their phylogenetic position near the tips of the Aigarchaeota illustrates a possible shift from a facultatively anaerobic to a strictly anaerobic lifestyle. In this study, these two bins are divergent from the other four and show dramatic metabolic differences, suggesting that Aigarchaeota is experiencing sympatric evolutionary divergence. Unlike the considerable metabolic diversity within most Aigarchaeota genomes, GMQ bin_10 and JZ_10 are devoid of pathways for carbon monoxide oxidation, aerobic respiration, dissimilatory sulfite reduction, and sulfide oxidation. Carbon fixation is a common trait among most Aigarchaeota genomes by employing either the roTCA cycle (the four genomes in this study) or the 3-hydroxypropionate/4-hydroxybutyrate pathway to fix carbon dioxide8. However, the two anaerobes lack this capability due to the absence of key enzymes. Mostly, rTCA or roTCA cycle are only found in anaerobic organisms, however, some studies show that at least rTCA is functional in microaerobic or aerobic microbes such as “Candidatus Nitrospira defluvii” and Leptospirillum genomes43,44. In “Candidatus Nitrospira defluvii” genome, near complete complex I-V pathways were identified, indicating the aerobic lifestyle of this microbe. Comparative genomics shows that several Aigarchaeota genomes including “Candidatus Caldiarchaeum subterraneum”, “Aigarchaeota archaeon OS1”, and the four genomes in this study have the ability to employ sqr to oxidize reduced sulfide as an electron donor for chemolithotrophy. Other Aigarchaeota lack this capability due to the missing of sqr gene. Closely related neighbors JZ bin_15 and Aigarchaeota archaeon JGI 0000106-J15 were the only two genomes possessing dsrAB genes with high sequence identity (>90%), indicating they inherited sulfite reduction ability vertically, following earlier HGT transfer from Firmicutes.
Eukaryotic signatures in Aigarchaeota genomes
Several eukaryotic signature proteins (ESPs) were identified in Aigarchaeota genomes as described before7 (Supplementary Fig. 12; Supplementary Table 3). All the six genomes harbor an ubiquitin modifier system consisting of Ub-like, E1-like, E2-like, E3-like, and Dub-like proteins, which could be used to degrade or recycle damaged proteins45 (Fig. 2; Supplementary Fig. 13). The whole reaction might be quite similar to the canonical eukaryotic ubiquitylation process. First, pro-ubiquitin could be cleaved by the Rpn11 metalloprotease homolog (IPR000555), leading to the exposure of the di-glycine motif of the modifier at the C-terminus. E1-like enzyme (IPR000594) could subsequently be used to adenylate the residue of the di-glycine motif. The activated modifier could then be transferred to the catalytic cysteine of the E1-like enzyme, forming a covalent thioester intermediate. Next, the transfer of ubiquitin from E1 to the active site cysteine of the E2 could then be triggered by the E2-like conjugating enzyme (IPR000608) via a trans-thioesterification reaction. Finally, E3-like enzymes could catalyze the aminolysis-based transfer of the ubiquitin from the E2-like protein onto a specific target protein, which ultimately results in the covalent attachment of a small ubiquitin modifier to a substrate and further degradation through the proteasomal degradation pathways45. Moreover, a total of 64 ribosomal proteins (RP) were observed among the six genome bins. Over half were identified as eukaryotic-like RPs, including RP-S19e and RP-L30e, which were previously only reported to be present in eukaryotes46 (Supplementary Table 3). RP-L13e and RP-S26e were found in Aigarchaeota, consistent with previous studies suggesting that these two RPs are present in TACK superphylum47. In addition, a wide variety of GTPases were identified among Aigarchaeota (Supplementary Data 1). One genomic bin may encode a member of the Rab family of small GTPases, an enzyme thought to predate the origin of Eukaryotes, and generally presumed to be involved in membrane biogenesis by interacting with proteins48. Like Crenarchaeota, all genomes encode homologs of ESCRT-III and Vps4 components49. Those genes were proposed to play a pivotal role in cell division, and the inactivation of Vps4 leads to the accumulation of enlarged cells50. However, the key genes cdvB and cdvC are present, but a gene for cdvA is absent among all the genomes. Both CdvB and CdvC show low similarity to proteins identified in Eukarya (average ~26% and ~32% identity to the best hits in Eukarya based on the RPS-BLAST). Overall, we found that these archaea are of particular interest because they contain a surprising mixture of bacterial and eukaryotic features. For instance, the sulfite reduction ability in JZ bin_15 is derived from Firmicutes and several genes involved in the roTCA cycle show high amino acid identity to those of the genus Thermus. From Bacteria, they can acquire the abilities related to central metabolism through HGT. Otherwise, they share common ancestry and mechanistic similarities to the functions related to cell division, DNA replication, and transcription in Eukaryotes.
The evolutionary history of Aigarchaeota
The Aigarchaeota represents an evolutionarily diverse group of microbes, which are mainly found in high temperature environments including geothermal springs, the deep subsurface, marine sediments, and marine hydrothermal vents5,6,8,9,51,52,53. It is not yet confirmed whether Aigarchaeota represents a new phylum or represents a deeply branch within the Thaumarchaeota16. To better understand the relationship between the two groups, we carefully selected 94 genomes including 14 Aigarchaeota and 80 Thaumarchaeota from public databases including IMG-M and NCBI (see Methods for the selection criteria), and built phylogenetic and phylogenomic trees (Supplementary Fig. 14; Supplementary Data 2). A 16S rRNA-based phylogenetic tree places Aigarchaeota as the nearest neighbor to Crenarchaeota (Supplementary Fig. 2), consistent with a previous study54, suggesting that Aigarchaeota may represent a new phylum compared to Thaumarchaeota. The phylogenomic tree based on the concatenated alignment of 16 ribosomal proteins shows that Aigarchaeota and Thaumarchaeota come from divergent groups with convincing bootstrap support at the parental node of two lineages, providing further compelling evidence that they are different phyla. However, uncertainties are observed for the affiliation of Aigarchaeota archaeon JGI MDM2 JNZ-1-N15 and Aigarchaeota archaeon JGI MDM2 JNZ-1-K18 due to their uncertain relationship to other Aigarchaeota. Revisiting the phylogenetic placement of these two phyla in an RNA polymerase-based tree even put them into Thaumarchaeota with high bootstrap confidence (>70%; Supplementary Fig. 15). However, we still consider them as Aigarchaeota because they group with Aigarchaeota based on the functional similarity based on KO or COG annotation (Supplementary Fig. 16). To our surprise, we found that one lineage comprised of six genomes (hereafter, we refer to them as “transitional genomes”) has a closer relationship to Aigarchaeota based on KEGG and eggNOG functional annotations, but both the phylogenetic and phylogenomic trees suggest that they represent members of the Thaumarchaeota (Supplementary Fig. 16). In particular, DRTY7 bin_36, which generated in this study, might be the first genome of deep-branching pSL12 group (Supplementary Fig. 2). Principal coordinates analyses (PCoA) based on COGs (KOs) were conducted to visualize the relationship among different members of the Aigarchaeota and Thaumarchaeota. From the PCoA plot, genomes in these two phyla appear to cluster based on their habitat rather than their taxonomic assignment. One lineage consists mainly of Thaumarchaeota genomes inhabiting cold or ambient temperature environments such as marine or soil. The other cluster is mainly composed of the Aigarchaeota and the “transitional genomes”, which are mainly found in high-temperature ecosystems such as terrestrial hot springs (Supplementary Fig. 16). Besides the six transitional genomes, the remaining 71 Thaumarchaeota genomes are all ammonia oxidizers, with six being possible exceptions, possibly because of genome incompleteness (average <62%). Most of them are mesophiles, with only a few from high-temperature environments. “Candidatus Nitrososphaera gargensis” Ga9.2 was proposed to be the first thermophilic ammonium oxidizer, found in the moderate thermal spring in Russia14. The following reported deep-branching AOAs including “Candidatus Nitrosocaldus yellowstonii”, “Candidatus Nitrosocaldus islandicus” isolate 3F, and “Candidatus Nitrosocaldus cavascurensis” strain SCU2 show adaptation to even higher temperature55,56,57. Based on this observation, we propose that the last common ancestor of Aigarchaeota and Thaumarchaeota were thermophiles, and the marine Thaumarchaeota are the descendants of the thermophiles. The detected crenarchaeol, a diagnostic membrane lipid, in “Candidatus Nitrosocaldus yellowstonii” also supports the hot-origin of Thaumarchaeota55. This evolutionary scenario is consistent with the evolution of the destabilizing cyclohexyl ring of crenarchaeol/thaumarchaeol to allow niche invasion of thermophiles into cold environments. Dispersal into cooler habitats would also trigger evolution of the transitional genomes described here. The later genetic interactions with the community members driven by the environmental selection lead to the loss or acquisition of specific genes from organisms in the new microbial communities (e.g., the derived feature of ammonia oxidation) to make them better adapted to the new habitats (Supplementary Fig. 17).
To decipher the evolutionary histories of the Aigarchaeota and Thaumarchaeota, gene gain and loss events were predicted by mapping the inferred orthology of genes to the Bayesian tree. The Bayesian tree is quite robust with all the nodes showing high posterior probability (Supplementary Fig. 18). The Aigarchaeota and Thaumarchaeota are monophyletic and share over 1000 orthologous genes for their common ancestor (Fig. 3). Several terminal oxidases were gained at node 1 (Supplementary Data 1), indicating the ancestor of these two lineages were aerobes. Aigarchaeota are reported to be hyperthermophiles or moderate thermophiles, and are widely distributed in different thermal habitats including terrestrial, marine, and subsurface environments9. Hence, it appears to be reasonable that the ancestor of the Thaumarchaeota likely also originated from high-temperature habitats because little has been changed as revealed by comparing the lineage represented by transitional genomes and the parental nodes of Aigarchaeota (Fig. 3). The evolutionary scenario was also strengthened by a previous study about the “hot” origin of Thaumarchaeota58. Then, we assume that niche invasion from thermal to moderate temperature habitats might have selected for transitional genomes and subsequently canonical Thaumarchaeota. New resources after the colonization of these new habitats might have triggered adaptive radiations, forming the largest branch of the Thaumarchaeota. This finding was further supported by the observation that a large number of gene gain events occurred at nodes 10 and 11 of the tree, taking up ~23 and 24% of gene families at corresponding nodes (Fig. 3). These two steps of gene acquisition largely expanded the genetic diversity of marine Thaumarchaeota, leading to the functional divergence from ancestral thermophilic Thaumarchaeota and Aigarchaeota. Of the total 806 gained genes at nodes 10 and 11 (Supplementary Data 3), further analysis showed that 213 (~62%) and 296 (~64%) of them were identified as hypothetical proteins based on comparison to nogSTRING databases (Fig. 3). In comparison, most of the lost genes at nodes 10 and 11 are related to basic metabolism, including transporters and dehydrogenases (Supplementary Data 3). Additionally, several genes related to nitrogen metabolism including ureABCDEF and amoABC operons were acquired at node 10, indicating that ammonia oxidation is a derived feature of Thaumarchaeota. The amoABC genes in “Candidatus Nitrosocaldus islandicus” 3F and “Candidatus Nitrosocaldus cavascurensis” SCU2 suggest AOAs might originate from thermal habitats, which was also supported by previous findings7,59,60. The inherited ammonia oxidation ability could sustain their growth in nutrient-depleted marine and terrestrial environments with low concentrations of ammonia, allowing them to compete for ammonia as an electron donor and providing a selective advantage relative to AOB, heterotrophs, and phytoplankton61. Urea assimilation is employed by most AOA to supply sufficient ammonia by catalyzing conversion of the urea molecule to two ammonia molecules and one carbon dioxide molecule. Besides the large genetic investment in nitrogen metabolisms, several genes involved in cobalamin (B12) synthesis were observed at node 10, which is necessary to catalyze methyl transfer reactions in amino acid and DNA synthesis62. Previous study revealed a common phenomenon that the B12 biosynthesis pathway was widely distributed among oceanic Thaumarchaeota genomes63, serving as a potential source of multiple B vitamins required by other community members64. However, based on our observation, they might be also inherited from thermophilic AOAs (Fig. 3).
Fig. 3
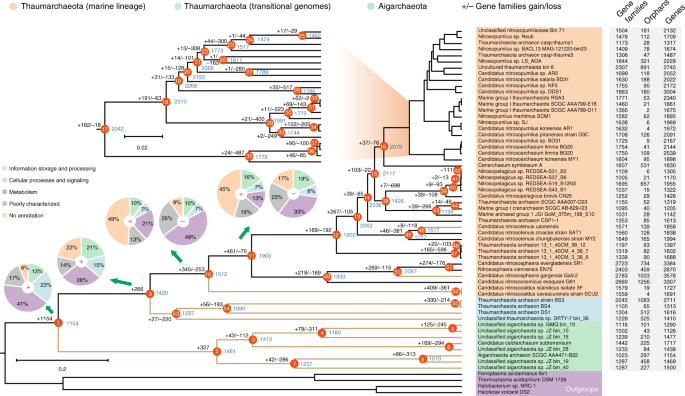
Ancestral genome content reconstruction using COUNT software. The tree topology is based on the Bayesian tree generated from MrBayes78. The numbers of gain and loss events were marked at each lineage of the tree. “+”s represent gain events and “−”s represent loss events. The red star represents the major gene gain event. The pie chart shows the numbers of gained genes by COG categories. Thermophiles are colored as brown branches in the tree. A list of gained and lost genes for the fours key nodes were shown in Supplementary Data 3
We estimated that the ancestor of the Aigarchaeota possessed more gene families than the extant organisms. As time went on, gene loss events occurred frequently in all lineages. To gain a better understanding of this process, we investigated variants of the six genomes to obtain detailed insight into the influence of environmental selection on genomic evolution. Single nucleotide polymorphism (SNP) calling was conducted as described in Materials and Methods. The detected SNPs in the six genomes are comparable except JZ bin_10, which has the smallest genome size, and the most SNPs (23321; Supplementary Table 4). In contrast, GMQ bin_10, the closest neighbor to JZ bin_10 (97% AAI), has a larger genome but fewer SNPs (2386; Supplementary Table 4). More than half of the SNPs in almost all the genomes are synonymous mutations, illustrating that in most cases, those mutations had no effect on their growth in extreme thermal habitats. Almost all the dN/dS values were <1, indicating that purifying selection was acting to remove deleterious mutations.
Genome expansion through HGT
HGT is in important evolutionary process in prokaryotes that has great impact on the diversity of gene repertoires, especially for those microbes in extreme habitats35,65. Putative HGT events have contributed substantially to genome contents of Aigarchaeota (Supplementary Table 4). The transferred genes comprise predominantly basic metabolic functions, with amino acid transport and metabolism (~18.5% of the total HGTs), energy production and conversion (~15.6%), carbohydrate transport and metabolism (~9%), nucleotide transport and metabolism (~5.5%), lipid transport and metabolism (~4.8%), inorganic ion transport and metabolism (~6.7%), secondary metabolites biosynthesis, transport, and catabolism (~2.9%), and coenzyme transport and metabolism (~6.1%) being significantly enriched in this eight functional classifications (Two-tailed Fisher’s exact test with confidence intervals at 99%, P < 0.05; P values were adjusted with the “BH” criteria) (Supplementary Fig. 19). As is typical, informational proteins (e.g., ribosomal proteins, DNA polymerases) underwent fewer HGTs than other gene families (Supplementary Fig. 19). Among the identified potential HGTs, most appear to be acquired from the same domain, including Crenarchaeota and Euryarchaeota (Fig. 4). Bacteria also contributed substantially to generating genetic diversity through HGT, and several genes were transferred from Firmicutes and Proteobacteria (Fig. 4). These results might seem inconsistent with previous studies showing that closely related organisms engage in more frequent genetic exchange than distantly related ones66. However, interdomain gene transfer is highly asymmetric in that Archaea acquire genes much easier from bacteria than vice versa67. Gene acquisition from bacteria appears to have provided the key innovations for Aigarchaeota, facilitating their access to new niches. For example, several CODHs and hydrogenases were identified as HGTs with bacteria serving as donors (Supplementary Data 4). As reported, carbon monoxide and hydrogen are ubiquitous and are believed to be primary substrates and energy provider in hydrothermal habitats68. For most members of Aigarchaeota in this study, they are able to grow via the oxidation of carbon monoxide coupling with oxygen reduction. Several genes related to oxidative phosphorylation in GMQ bin_10 and JZ bin_10 were found in large gene clusters, indicating they were likely acquired in a single event (Supplementary Data 4). The associated genes may be donated by Firmicutes through HGT. This finding provides important clues about the origin of aerobic Aigarchaeota. Firmicutes also endowed JZ bin_15 with sulfite reducing capability with strong evidence as described above (Supplementary Fig. 8a). The roTCA cycle might be a derived feature for most bins except GMQ bin_10 and JZ bin_10. The key enzyme citrate synthase was identified to be imported from Euryarchaeota or Deinococcus–Thermus (Supplementary Data 4). Hence, HGT might be the main driver to provide genetic variability that allows different types of Aigarchaeota to occupy distinct niches, resulting in the functional partitioning and ecological divergence within the same habitat.
Fig. 4
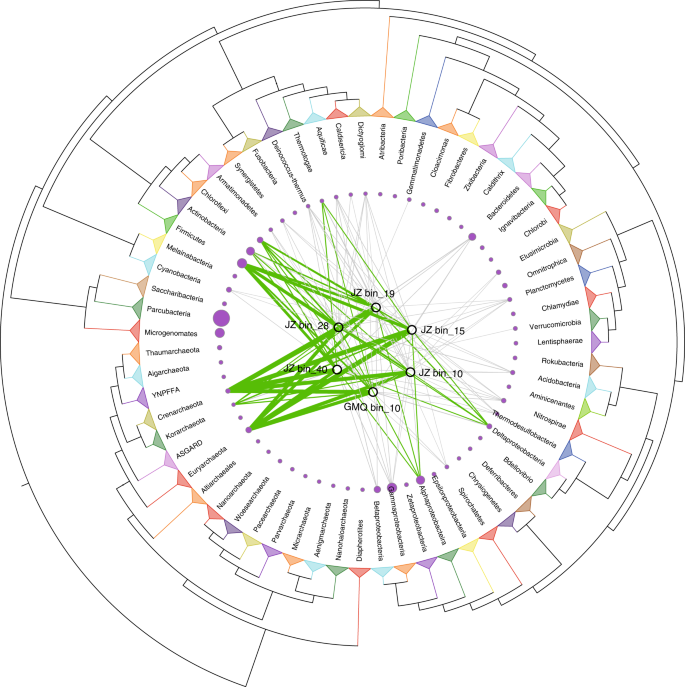
Extensive horizontal gene transfer events detected in Aigarchaeota. The maximum likelihood tree was pruned and redrawn at the phylum level according to the tree of life published by Hug et al.79. Green edges represent potential gene flows from the corresponding phylum involving ≥10 genes, and gray lines are the ones with <10 genes. Edge thickness is proportional to the number of putatively transferred genes. The sizes of red circles are proportional to the number of reference genomes collected for that phylum
Conclusions
Aigarchaeota poorly understood but important lineage that is abundant and widely distributed in thermal habitats. The high-quality genomes described here are diverse both in metabolic pathways and ecological roles, suggesting functional partitioning and ecological divergence within a single geothermal region. To avoid competition among closely related microbes, HGT contributes a lot to their genome innovations and allows them to utilize different kind of substrates. Intriguingly, several of the acquisitions are imported from Bacteria, indicating inter-domain genetic interactions play a key role in shaping the genetic diversity of Aigarchaeota. From the evolutionary perspective, our research supports the hypothesis that the last common ancestor of Aigarchaeota and Thaumarchaeota have a large genome size and both of them are originated from thermal habitats. Overall, this discovery is expected to have a substantial impact on our understanding of their roles in biogeochemical cycling and the evolutionary history of the important but poorly understood phylum Aigarchaeota.
Methods
Sample acquisition, DNA extraction, and metagenomic sequencing
Two hot spring sediment samples including JZ and GMQ were obtained from Tengchong, Yunnan, China. Detailed site description as following: (1) GMQ: A small source pool with a high flow rate (10.4 L/S), with length, width and depth around 98, 79, and 9.5 cm, respectively. Spring water is clear, and silicate sand sediment could be seen at bottom. The spring is surrounded by bush and grass. Leaf litter and other debris were observed in this spring. (2) JZ: Man-made cubic well with side length 400 cm, and depth 150 cm, covered with a ceiling. Water is not clear, bottom is not visible. Mineral deposition was observed on the well walls. The GPS coordinates of the locations from which samples were listed in Fig. 1. Both samples were collected using sterile spatulas and spoons and stored in liquid nitrogen before transporting to the lab. Community genomic DNA was extracted from approximately 20 g of sediment material using PowerSoil DNA Isolation kit (MoBio). DNA concentrations of the extract and constructed libraries (with insert size of 350 bp) were measured with a Qubit fluorometer. Metagenomic sequence data for the two samples are generated using Illumina Hiseq 4000 instruments at Beijing Novogene Bioinformatics Technology Co., Ltd (Beijing, China). The amount of raw sequence data was ~30 Gbp (2 × 150 bp) for each sample.
Metagenome assembly and genome binning
Preprocessing of raw data was carried out as described previously69. The quality metagenomic sequences for each sample were de novo assembled separately using SPAdes (version 3.9.0)70 with the following parameters: −k 33,55,77,99,111-meta. Then, GapCloser (version 1.12; http://soap.genomics.org.cn/) was used with default parameters to fill a proportion of gaps of the assembled scaffolds. Genome binning of the metagenomic assemblies was conducted with a combination of emergent self-organizing maps (ESOM)71 and MetaBAT72. In brief, scaffolds with length <2500 bp in each assembly were removed from the further analysis. Qualified metagenomic data from samples was mapped to each assembly to compute the coverage information using BBMap (version 38.85; http://sourceforge.net/projects/bbmap/) with the parameter as: k = 15 minid = 0.9 build = 1. Then coverage information of the scaffolds and tetranucleotide frequency (TNF) were used to perform the genome binning which conducted by MetaBAT72. All bins were subjected to manual examination to remove contaminations. Specifically, genome bins were sheared into short fragments (5 to 10 kb) and clustered based on the TNF using ESOM71. Then the generated clusters were visualized to judge the situations that if one genome bin has been split into sub-blocks or sub-blocks could be merged into one single bin (Supplementary Fig. 1). To improve the accuracy of genome bins, the obvious discordant points were deleted manually from the clusters. The completeness, contamination and strain heterogeneity of each bin were evaluated using CheckM (version 1.0.5)10. To get the optimal quality of genome bins, clean reads for each genome bin were recruited using BBMap with the parameters listed above. Then, genome bins were reassembled by SPAdes (version 3.9.0)70 with the following parameters: --careful -k 21,33,55,77,99,127. Finally, six genome bins belonging to Aigarchaeota were retained for the further analysis. The subsequent functional annotation, phylogenetic and phylogenomic inference and metabolic pathway reconstruction were conducted as detailed described in the Supplementary Notes 1, 2.
Comparative genomics
All genomes classified under Aigarchaeota or Thaumarchaeota were downloaded from NCBI (https://www.ncbi.nlm.nih.gov/) and IMG-M (https://img.jgi.doe.gov/cgi-bin/m/main.cgi) databases. CheckM10 was used to check the genome quality. Genomes with redundancy and completeness < 50% were removed for the further analysis. Finally, a total of 94 draft genomes were taken into comparison. Seven were from our study including six and one belonged to Aigarchaeota and Thaumarchaeota, respectively. The remaining 87 genomes from the public databases include eight aigarchaeal and 79 thaumarchaeal genomes (Supplementary Data 2). To compute the amino acid identity (AAI) of each pair of genomes, orthologous genes were identified based on reciprocal best BLAST hits (_E_-value < 1e−5) based on their predicted amino acid sequences. AAI was calculated as the mean similarity of all orthologous genes.
For the further comparative analysis, only genomes with completeness >80% were taken into consideration, respectively, which lead to the filtration of extra 38 genomes (Nitrosopumilus sp. AR was also removed due to the high contamination estimated by CheckM). Clusters of homologous protein were reconstructed for the remaining 56 genomes. An all-against-all genomes BLAST were conducted using the thresholds _E_-value <1e-10 and sequence identity ≥30%. MCL (−I 1.4)73 was used to reconstruct protein clusters based on the reciprocal best BLAST hits (rBBHs). This yielded a total of 18,894 protein families with 12,198 were classified as singletons. To address the evolution of life histories of the two phyla, ancestral family sizes were inferred using the program COUNT74 with Dollo parsimony. This approach strictly prohibits multiple gains of genes and allows reconstructing gene gain and loss events at both observed species and potential ancestors (leaves and nodes on the phylogenetic tree).
Putative HGTs were inferred using HGTector75. Homologs of predicted genes were retrieved from the NCBI-nr database using BLASTp as implemented in NCBI BLAST +2.2.28. Quality cutoffs for valid hits were _E_-value ≤1e−20, sequence identity ≥30%, and coverage of query sequence ≥50%. Putatively HGT-derived genes were defined as those with hits from within the Thaumarchaeota (NCBI TaxID 651137, which includes Aigarchaeota) significantly underweighted, and with hits outside the phylum not underweighted.
SNP detection and dN/dS calculation
The quality reads were mapped to the corresponding genomes using BBMap (minid = 0.97). GATK76 was then used to generate the primary high-quality variant calls including SNPs and InDels. Custom Perl scripts were developed to identify and classify all the SNPs as intergenic, intragenic, synonymous (dS) or non-synonymous (dN). Calculation of dN and dS values between GMQ bin_10 and JZ bin_10 were performed using the yn00 program in the PAML package (version 4.9)77. Both the Nei-Gojobori and Yang and Nielsen methods were used. To remove the bias caused by too few dS, genes with dN/dS ratio higher than 5 were excluded from further analysis.
Data availability
The genome bins described in this study have been deposited at JGI IMG-MER under the Study ID Gs0127627 and WGS accessions Ga0180324 (Unclassified Aigarchaeota GMQ bin_10), Ga0180307 (Unclassified Aigarchaeota JZ bin_10), Ga0180308 (Unclassified Aigarchaeota JZ bin_15), Ga0177927 (Unclassified Aigarchaeota JZ bin_19), Ga0180309 (Unclassified Aigarchaeota JZ bin_28), Ga0180310 (Unclassified Aigarchaeota JZ bin_40) and Ga0181444 (Unclassified Thaumarchaeota DRTY7 bin_36). The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- Brown, C. T. et al. Unusual biology across a group comprising more than 15% of domain bacteria. Nature 523, 208–211 (2015).
Article ADS PubMed CAS Google Scholar - Castelle, C. J. et al. Genomic expansion of domain archaea highlights roles for organisms from new phyla in anaerobic carbon cycling. Curr. Biol. 25, 690–701 (2015).
Article PubMed CAS Google Scholar - Rinke, C. et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 499, 431–437 (2013).
Article ADS PubMed CAS Google Scholar - Wrighton, K. C. et al. Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 337, 1661–1665 (2012).
Article ADS PubMed CAS Google Scholar - Nunoura, T. et al. Genetic and functional properties of uncultivated thermophilic crenarchaeotes from a subsurface gold mine as revealed by analysis of genome fragments. Environ. Microbiol. 7, 1967–1984 (2005).
Article PubMed CAS Google Scholar - Nunoura, T. et al. Archaeal diversity and distribution along thermal and geochemical gradients in hydrothermal sediments at the Yonaguni Knoll IV hydrothermal field in the Southern Okinawa Trough. Appl. Environ. Microbiol. 76, 1198–1211 (2010).
Article PubMed CAS Google Scholar - Nunoura, T. et al. Insights into the evolution of Archaea and eukaryotic protein modifier systems revealed by the genome of a novel archaeal group. Nucleic Acids Res. 39, 3204–3223 (2011).
Article PubMed CAS Google Scholar - Beam, J. P. et al. Ecophysiology of an uncultivated lineage of Aigarchaeota from an oxic, hot spring filamentous ‘streamer’community. ISME J. 10, 210–224 (2016).
Article PubMed CAS Google Scholar - Hedlund, B. P. et al. Uncultivated thermophiles: current status and spotlight on ‘Aigarchaeota’. Curr. Opin. Microbiol. 25, 136–145 (2015).
Article PubMed CAS Google Scholar - Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Article PubMed PubMed Central CAS Google Scholar - Guy, L. & Ettema, T. J. The archaeal ‘TACK’ superphylum and the origin of eukaryotes. Trends Microbiol. 19, 580–587 (2011).
Article PubMed CAS Google Scholar - Könneke, M. et al. Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature 437, 543–546 (2005).
Article ADS PubMed CAS Google Scholar - Hallam, S. J. et al. Pathways of carbon assimilation and ammonia oxidation suggested by environmental genomic analyses of marine Crenarchaeota. PLoS Biol. 4, 95 (2006).
Article Google Scholar - Hatzenpichler, R. et al. A moderately thermophilic ammonia-oxidizing crenarchaeote from a hot spring. Proc. Natl Acad. Sci. USA 105, 2134–2139 (2008).
Article ADS PubMed PubMed Central Google Scholar - Brochier-Armanet, C., Forterre, P. & Gribaldo, S. Phylogeny and evolution of the Archaea: one hundred genomes later. Curr. Opin. Microbiol. 14, 274–i281 (2011).
Article PubMed Google Scholar - Bräsen, C., Esser, D., Rauch, B. & Siebers, B. Carbohydrate metabolism in Archaea: current insights into unusual enzymes and pathways and their regulation. Microbiol. Mol. Biol. Rev. 78, 89–175 (2014).
Article PubMed PubMed Central CAS Google Scholar - Hedlund, B. P. et al. A review of the microbiology of the Rehai geothermal field in Tengchong, Yunnan Province, China. Geosci. Front 3, 273–288 (2012).
Article Google Scholar - Hou, W. et al. A comprehensive census of microbial diversity in hot spring of Tengchong, Yunnan Province China using 16S rRNA gene pyrosequencing. PLoS ONE 8, 1–15 (2013).
Google Scholar - Bowers, R. M. et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 35, 725 (2017).
Article PubMed CAS PubMed Central Google Scholar - Barns, S. M., Delwiche, C. F., Palmer, J. D. & Pace, N. R. Perspectives on archaeal diversity, thermophily and monophyly from environmental rRNA sequences. Proc. Natl Acad. Sci. USA 93, 9188–9193 (1996).
Article ADS PubMed CAS PubMed Central Google Scholar - Costa, K. C. et al. Microbiology and geochemistry of Great Boiling and Mud Hot Springs in the United States Great Basin. Extremophiles 13, 447–459 (2009).
Article PubMed CAS Google Scholar - Vick, T. J., Dodsworth, J. A., Costa, K. C., Shock, E. L. & Hedlund, B. P. Microbiology and geochemistry of Little Hot Creek, a hot spring environment in the Long Valley Caldera. Geobiology 8, 140–154 (2010).
Article PubMed CAS Google Scholar - Kobayashi, T., Romaniec, M. P., Barker, P. J., Gerngross, U. T. & Demain, A. L. Nucleotide sequence of gene celM encoding a new endoglucanase (CeIM) of Clostridium thermocellum and purification of the enzyme. J. Ferment. Bioeng. 76, 251–256 (1993).
Article CAS Google Scholar - Zhao, C. et al. High-throughput pyrosequencing used for the discovery of a novel cellulase from a thermophilic cellulose-degrading microbial consortium. Biotechnol. Lett. 39, 123–131 (2017).
Article PubMed CAS Google Scholar - Mall, A. et al. Reversibility of citrate synthase allows autotrophic growth of a thermophilic bacterium. Science 359, 563–567 (2018).
Article ADS PubMed CAS Google Scholar - Nunoura, T. et al. A primordial and reversible TCA cycle in a facultatively chemolithoautotrophic thermophile. Science 359, 559–563 (2018).
Article ADS PubMed CAS Google Scholar - Takami, H., Arai, W., Takemoto, K., Uchiyama, I. & Taniguchi, T. Functional classification of uncultured “Candidatus Caldiarchaeum subterraneum” using the MAPLE system. PLoS ONE 10, e0132994 (2015).
Article PubMed PubMed Central CAS Google Scholar - Le Laz, S. et al. A biochemical approach to study the role of the terminal oxidases in aerobic respiration in Shewanella oneidensis MR-1. PLoS ONE 9, e86343 (2014).
Article ADS PubMed PubMed Central CAS Google Scholar - Mathiesen, C. & Hägerhäll, C. Transmembrane topology of the NuoL, M and N subunits of NADH: quinone oxidoreductase and their homologues among membrane-bound hydrogenases and bona fide antiporters. Biochim. Biophys. Acta 1556, 121–132 (2002).
Article PubMed CAS Google Scholar - Hedlund, B. P., Dodsworth, J. A., Murugapiran, S. K., Rinke, C. & Woyke, T. Impact of single-cell genomics and metagenomics on the emerging view of extremophile “microbial dark matter”. Extremophiles 5, 865–875 (2014).
Article CAS Google Scholar - King, G. A. Molecular and culture-based analyses of aerobic carbon monoxide oxidizer diversity. Appl. Environ. Microbiol. 69, 7257–7265 (2003).
Article PubMed PubMed Central CAS Google Scholar - Spear, J. R., Walker, J. J., McCollom, T. M. & Pace, N. R. Hydrogen and bioenergetics in the Yellowstone geothermal ecosystem. Proc. Natl Acad. Sci. USA 102, 2555–2560 (2005).
Article ADS PubMed CAS PubMed Central Google Scholar - Aravind, L., Tatusov, R. L., Wolf, Y. I., Walker, D. R. & Koonin, E. V. Evidence for massive gene exchange between archaeal and bacterial hyperthermophiles. Trends Genet. 14, 442–444 (1998).
Article PubMed CAS Google Scholar - Nelson, K. E. et al. Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritima. Nature 399, 323–329 (1999).
Article ADS PubMed CAS Google Scholar - Zhaxybayeva, O. et al. On the chimeric nature, thermophilic origin, and phylogenetic placement of the Thermotogales. Proc. Natl Acad. Sci. USA 106, 5865–5870 (2009).
Article ADS PubMed PubMed Central Google Scholar - Ma, K., Schicho, R. N., Kelly, R. M. & Adams, M. W. Hydrogenase of the hyperthermophile Pyrococcus furiosus is an elemental sulfur reductase or sulfhydrogenase: evidence for a sulfur-reducing hydrogenase ancestor. Proc. Natl Acad. Sci. USA 90, 5341–5344 (1993).
Article ADS PubMed CAS PubMed Central Google Scholar - Ma, K., Weiss, R. & Adams, M. W. Characterization of hydrogenase II from the hyperthermophilic archaeon Pyrococcus furiosus and assessment of its role in sulfur reduction. J. Bacteriol. 182, 1864–1871 (2000).
Article PubMed PubMed Central CAS Google Scholar - Mander, G. J., Pierik, A. J., Huber, H. & Hedderich, R. Two distinct heterodisulfide reductase‐like enzymes in the sulfate-reducing archaeon Archaeoglobus profundus. Febs. J. 271, 1106–1116 (2004).
CAS Google Scholar - Ramos, A. R. et al. The FlxABCD-HdrABC proteins correspond to a novel NADH dehydrogenase / heterodisulfide reductase widespread in anaerobic bacteria and involved in ethanol metabolism in Desulfovibrio vulgaris Hildenborough. Environ. Microbiol. 17, 2288–2305 (2015).
Article PubMed CAS Google Scholar - Cramm, R. Genomic view of energy metabolism in Ralstonia eutropha H16. J. Mol. Microb. Biotech. 16, 38–52 (2009).
Article CAS Google Scholar - Peters, J. W. et al. [FeFe]- and [NiFe]-hydrogenase diversity, mechanism, and maturation. Biochim. Biophys. Acta 1853, 1350–1369 (2015).
Article PubMed CAS Google Scholar - Makarova, K. S. et al. Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 9, 467 (2011).
Article PubMed CAS Google Scholar - Goltsman, D. S. A. et al. Community genomic and proteomic analyses of chemoautotrophic iron-oxidizing “_Leptospirillum rubarum_” (Group II) and “_Leptospirillum ferrodiazotrophum_” (Group III) bacteria in acid mine drainage biofilms. Appl. Environ. Microbiol. 75, 4599–4615 (2009).
Article PubMed PubMed Central CAS Google Scholar - Lücker, S. et al. A Nitrospira metagenome illuminates the physiology and evolution of globally important nitrite-oxidizing bacteria. Proc. Natl Acad. Sci. USA 107, 13479–13484 (2010).
Article ADS PubMed PubMed Central Google Scholar - James, R. H. et al. Functional reconstruction of a eukaryotic-like E1/E2/(RING) E3 ubiquitylation cascade from an uncultured archaeon. Nat. Commun. 8, 1120 (2017).
Article ADS CAS Google Scholar - Armache, J. P. et al. Cryo-EM structure and rRNA model of a translating eukaryotic 80S ribosome at 5.5-Å resolution. Proc. Natl Acad. Sci. USA 107, 19748–19753 (2010).
Article ADS PubMed PubMed Central Google Scholar - Spang, A. et al. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 521, 173–179 (2015).
Article ADS PubMed PubMed Central CAS Google Scholar - Surkont, J. & Pereira-Leal, J. B. Are There Rab GTPases in Archaea? Mol. Biol. Evol. 33, 1833–1842 (2016).
Article PubMed PubMed Central CAS Google Scholar - Obita, T. et al. Structural basis for selective recognition of ESCRT-III by the AAA ATPase Vps4. Nature 449, 735–739 (2007).
Article ADS PubMed CAS Google Scholar - Samson, R. Y., Obita, T., Freund, S. M., Williams, R. L. & Bell, S. D. A role for the ESCRT system in cell division in archaea. Science 322, 1710–1713 (2008).
Article ADS PubMed PubMed Central CAS Google Scholar - Meyer-Dombard, D. A. R. et al. Hydrothermal ecotones and streamer biofilm communities in the Lower Geyser Basin, Yellowstone National Park. Environ. Microbiol. 13, 2216–2231 (2011).
Article PubMed CAS Google Scholar - De León, K. B., Gerlach, R., Peyton, B. M. & Fields, M. W. Archaeal and bacterial communities in three alkaline hot springs in Heart Lake Geyser Basin, Yellowstone National Park. Front. Microbiol. 4, 330 (2013).
Google Scholar - Takacs-Vesbach, C. et al. Metagenome sequence analysis of filamentous microbial communities obtained from geochemically distinct geothermal channels reveals specialization of three Aquificales lineages. Front. Microbiol. 4, 84 (2013).
Article PubMed PubMed Central CAS Google Scholar - Pester, M., Schleper, C. & Wagner, M. The Thaumarchaeota: an emerging view of their phylogeny and ecophysiology. Curr. Opin. Microbiol. 14, 300–306 (2011).
Article PubMed PubMed Central CAS Google Scholar - De la Torre, J. R., Walker, C. B., Ingalls, A. E., Könneke, M. & Stahl, D. A. Cultivation of a thermophilic ammonia oxidizing archaeon synthesizing crenarchaeol. Environ. Microbiol. 10, 810–818 (2008).
Article PubMed CAS Google Scholar - Abby, S. S. et al. Candidatus Nitrosocaldus cavascurensis, an ammonia oxidizing, extremely thermophilic archaeon with a highly mobile genome. Front. Microbiol. 9, 28 (2018).
Article PubMed PubMed Central Google Scholar - Daebeler, A. et al. Cultivation and genomic analysis of “Candidatus Nitrosocaldus islandicus,” an Obligately Thermophilic, ammonia-oxidizing thaumarchaeon from a hot spring biofilm in Graendalur Valley, Iceland. Front. Microbiol. 9, 193 (2018).
Article PubMed PubMed Central Google Scholar - Raymann, K., Brochier-Armanet, C. & Gribaldo, S. The two-domain tree of life is linked to a new root for the Archaea. Proc. Natl Acad. Sci. USA 112, 6670–6675 (2015).
Article ADS PubMed CAS PubMed Central Google Scholar - Groussin, M. & Gouy, M. Adaptation to environmental temperature is a major determinant of molecular evolutionary rates in archaea. Mol. Biol. Evol. 28, 2661–2674 (2011).
Article PubMed CAS Google Scholar - Brochier-Armanet, C., Gribaldo, S. & Forterre, P. Spotlight on the Thaumarchaeota. ISME J. 6, 227–230 (2012).
Article PubMed CAS Google Scholar - Martens-Habbena, W., Berube, P. M., Urakawa, H., de La Torre, J. R. & Stahl, D. A. Ammonia oxidation kinetics determine niche separation of nitrifying Archaea and Bacteria. Nature 461, 976 (2009).
Article ADS PubMed CAS Google Scholar - Sañudo-Wilhelmy, S. A., Gómez-Consarnau, L., Suffridge, C. & Webb, E. A. The role of B vitamins in marine biogeochemistry. Ann. Rev. Mar. Sci. 6, 339–367 (2014).
Article PubMed Google Scholar - Doxey, A. C., Kurtz, D. A., Lynch, M. D., Sauder, L. A. & Neufeld, J. D. Aquatic metagenomes implicate Thaumarchaeota in global cobalamin production. ISME J. 9, 461 (2015).
Article PubMed CAS Google Scholar - Santoro, A. E. et al. Genomic and proteomic characterization of “Candidatus Nitrosopelagicus brevis”: an ammonia-oxidizing archaeon from the open ocean. Proc. Natl Acad. Sci. USA 112, 1173–1178 (2015).
Article ADS PubMed CAS PubMed Central Google Scholar - Nelson-Sathi, S. et al. Acquisition of 1,000 eubacterial genes physiologically transformed a methanogen at the origin of Haloarchaea. Proc. Natl Acad. Sci. USA 109, 20537–20542 (2012).
Article ADS PubMed PubMed Central Google Scholar - Caro-Quintero, A. & Konstantinidis, K. T. Inter-phylum HGT has shaped the metabolism of many mesophilic and anaerobic bacteria. ISME J. 9, 958–967 (2015).
Article PubMed CAS Google Scholar - Nelson-Sathi, S. et al. Origins of major archaeal clades correspond to gene acquisitions from bacteria. Nature 517, 77–80 (2015).
Article ADS PubMed CAS Google Scholar - Baker, B. J. et al. Genomic inference of the metabolism of cosmopolitan subsurface Archaea, Hadesarchaea. Nat. Microbiol. 1, 16002 (2016).
Article PubMed CAS Google Scholar - Hua, Z. S. et al. Ecological roles of dominant and rare prokaryotes in acid mine drainage revealed by metagenomics and metatranscriptomics. ISME J. 9, 1280 (2015).
Article PubMed CAS Google Scholar - Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 (2012).
Article MathSciNet PubMed PubMed Central CAS Google Scholar - Dick, G. J. et al. Community-wide analysis of microbial genome sequence signatures. Genome Biol. 10, R85 (2009).
Article PubMed PubMed Central CAS Google Scholar - Kang, D. D., Froula, J., Egan, R. & Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3, e1165 (2015).
Article PubMed PubMed Central CAS Google Scholar - Enright, A. J., Van Dongen, S. & Ouzounis, C. A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30, 1575–1584 (2002).
Article PubMed PubMed Central CAS Google Scholar - Csűös, M. Count: evolutionary analysis of phylogenetic profiles with parsimony and likelihood. Bioinformatics 26, 1910–1912 (2010).
Article CAS Google Scholar - Zhu, Q., Kosoy, M. & Dittmar, K. HGTector: an automated method facilitating genome-wide discovery of putative horizontal gene transfers. BMC Genom. 15, 717 (2014).
Article CAS Google Scholar - DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Article PubMed PubMed Central CAS Google Scholar - Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Article PubMed CAS Google Scholar - Ronquist, F. et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Boil 61, 539–542 (2012).
Article Google Scholar - Hug, L. A. et al. A new view of the tree of life. Nat. Microbiol 1, 16048 (2016).
Article PubMed CAS Google Scholar
Acknowledgements
We thank the Guangdong Magigene Biotechnology Co., Ltd. China for the assistance in data analysis, and the entire staff from Yunnan Tengchong Volcano and Spa Tourist Attraction Development Corporation for strong support. This work was financially supported by the Key Project of International Cooperation of China Ministry of Science and Technology (No. 2013DFA31980), Science and Technology Infrastructure work project of China Ministry of Science and Technology (No. 2015FY110100), the National Natural Science Foundation of China (31600298, U1201233, 31470139 and 31370154), Natural Science Foundation of Guangdong Province, China (No. 2016A030312003), the China Postdoctoral Science Foundation (2016M602567), the Guangdong Province Key Laboratory of Computational Science and the Guangdong Province Computational Science Innovative Research Team. W.J.L. was also supported by Guangdong Province Higher Vocational Colleges and Schools Pearl River Scholar Funded Scheme (2014).
Author information
Author notes
- These authors contributed equally: Zheng-Shuang Hua, Yan-Ni Qu.
Authors and Affiliations
- State Key Laboratory of Biocontrol, Guangdong Key Laboratory of Plant Resources, School of Life Sciences, Sun Yat-Sen University, 510275, Guangzhou, China
Zheng-Shuang Hua, Yan-Ni Qu, En-Min Zhou, Yan-Ling Qi, Yi-Rui Yin, Yang-Zhi Rao, Ye Tian, Yu-Xian Li, Lan Liu & Wen-Jun Li - Department of Pediatrics, University of California San Diego, La Jolla, CA, 92093, USA
Qiyun Zhu & Rob Knight - Department of Earth and Planetary Science, University of California, Berkeley, Berkeley, CA, 94720, USA
Cindy J. Castelle - School of Life Sciences, University of Nevada Las Vegas, Las Vegas, NV, 89154, USA
Brian P. Hedlund - Nevada Institute of Personalized Medicine, University of Nevada Las Vegas, Las Vegas, NV, 89154, USA
Brian P. Hedlund - School of Life Sciences, South China Normal University, 510631, Guangzhou, China
Wen-Sheng Shu - Department of Computer Science and Engineering, University of California San Diego, La Jolla, CA, 92093, USA
Rob Knight - Center for Microbiome Innovation, University of California San Diego, La Jolla, CA, 92093, USA
Rob Knight - College of Fisheries, Henan Normal University, 453007, Xinxiang, China
Wen-Jun Li
Authors
- Zheng-Shuang Hua
- Yan-Ni Qu
- Qiyun Zhu
- En-Min Zhou
- Yan-Ling Qi
- Yi-Rui Yin
- Yang-Zhi Rao
- Ye Tian
- Yu-Xian Li
- Lan Liu
- Cindy J. Castelle
- Brian P. Hedlund
- Wen-Sheng Shu
- Rob Knight
- Wen-Jun Li
Contributions
Z.S.H., Y.N.Q., R.K. W.S.S., and W.J.L. conceived the study. L.L. performed the measurement of physiochemical parameters. Y.R.Y. and Y.T. performed the DNA extraction. Z.S.H. Y.N.Q., Y.X.L., Y.Z.R, C.J.C., E.M.Z., and Y.L.Q. performed the metagenomic analysis, genome binning, and functional annotation. Z.S.H. and Q.Z. performed the detection of horizontal transferred genes and related evolutionary analysis. Z.S.H., Y.N.Q., W.J.L., R.K., B.P.H., and Q.Z. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding author
Correspondence toWen-Jun Li.
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article
Cite this article
Hua, ZS., Qu, YN., Zhu, Q. et al. Genomic inference of the metabolism and evolution of the archaeal phylum Aigarchaeota.Nat Commun 9, 2832 (2018). https://doi.org/10.1038/s41467-018-05284-4
- Received: 13 December 2017
- Accepted: 13 June 2018
- Published: 19 July 2018
- DOI: https://doi.org/10.1038/s41467-018-05284-4