Activation of NLRP3 inflammasome by crystalline structures via cell surface contact (original) (raw)
- Article
- Open access
- Published: 02 December 2014
- Yifei Zhang2,3 na1,
- Zhongyuan Tu1 na1,
- Pascal Detampel1 na1,
- Melanie Stenner1 na1,
- Anutosh Ganguly1 na1 &
- …
- Yan Shi1,2,3 na1
Scientific Reports volume 4, Article number: 7281 (2014)Cite this article
- 10k Accesses
- 3 Altmetric
- Metrics details
Subjects
Abstract
Crystalline structures activate the NLRP3 inflammasome, leading to the production of IL-1β, however, the molecular interactions responsible for NLRP3 activation are not fully understood. Cathepsin B release from the ruptured phagolysosome and potassium ion efflux have been suggested to be critical for this activation. Here, we report that Cathepsin B redistribution was not a crucial event in crystal-induced IL-1β production. Silica and monosodium urate crystal-treated macrophages with undisturbed lysosomes demonstrated strong co-localization of ASC and Caspase-1, indicative of NLRP3 inflammasome activation. Importantly, we provided evidence to suggest that macrophage cell membrane binding to immobilized crystals was sufficient to induce IL-1β release and this activation of the NLRP3 inflammasome was inhibited by blocking potassium efflux. Therefore, this work reveals additional complexity in crystalline structure-mediated NLRP3 inflammasome regulations.
Similar content being viewed by others
Introduction
NOD-like receptor inflammasome family members (NLRs)-mediated IL-1β and IL-18 production is a major inflammatory innate defense mechanism1,2. NLRP3, a member of the family, consists of a N terminal PYRIN domain (PYD), a central NACHT domain and C terminal leucine-rich repeats (LRR). The LRR, upon sensing cellular stress, exposes the NACHT domain for homotypic aggregation, triggering a well-characterized cascade leading to NLRP3/ASC/Caspase-1 (Casp-1) complex formation. Pro-Casp-1 in the complex undergoes spontaneous autocleavage, to produce the p10 and p20 subunits that form active Casp-1, which in turn cleaves pro-IL-1β and pro-IL-18 into their mature forms3,4.
Unique among the inflammasomes, NLRP3 has been reported to sense a large variety of stimuli, ranging from bacterial toxins, ATP, membrane-destabilizing agents, to crystalline structures such as monosodium urate (MSU), silica and alum. Since the activating stimuli are diverse, it has been proposed that NLRP3 may detect a set of common cellular changes downstream of initial triggering events. So far, reactive oxygen species (ROS)5, plasma membrane-pore openings/ion channel activities, particularly potassium efflux6,7,8,9, mitochondrial disturbance10,11 and lysosomal rupture12 have been identified as possible intermediates13. Although inflammasome-independent activation by silica and alum crystals exist14, solid structure-mediated NLRP3 activation is thought to involve damage of phagolysosomes12. The release of lysosomal contents, including Cathepsin (Cath) B and L15, may directly activate NLRP3 to induce oligomerization15. This mechanism was suggested to be responsible for alum-mediated adjuvanticity and cholesterol crystal-induced inflammation in atherosclerosis12,16.
Several recent reports, however, have suggested that the lysosomal rupture/Cath B release was not always essential to crystalline/particulate structure-mediated NLRP3 inflammasome activation9,17. Cath B was also found to be non-essential in anthrax lethal toxin mediated-, NLRP1b-dependent cell death18. Interestingly, in situations involving large-scale cell death in vivo, the predominant enzymatic activity was mediated by Cath C being released into the milieu, which presumably cleaves IL-1β19. In the case of alum's adjuvanticity, several groups including ours have reported that alum leads to cell death in vitro and in vivo20,21,22. The dead cells released DNA21,22 that was detected by the host immune system to mediate an adjuvant effect, through pathways independent of NLRP321. Importantly, this observation was corroborated by an in-depth study showing that the lysosomal rupture induced rapid cell death with minimal processing of Casp-123, suggesting that the release of lysosomal contents does not necessarily induce NLRP3 inflammasome activation.
We reported previously that sensing of crystals by a phagocyte plasma membrane, which resulted in sorting of lipids in the contact regions, was sufficient to induce activation of dendritic cells and to enhance their antigen presentation20,24. We found that while the NLRP3 inflammasome was dispensable for alum's adjuvanticity, IL-1β production in response to alum was observed. This raises the question whether cell surface binding by crystals is enough to invoke IL-1β production without internalization. In addition we have also reported that PMMA (methyl methacrylate) beads, with diameters several times larger than phagocytes, robustly activated the NLRP3 inflammasome25, a situation unlikely to involve internalization, lysosomal rupture or Cath B release.
We report here that bone marrow macrophages (BMMAC), activated by MSU or silica crystals, produced IL-1β in the absence of phagocytosis. Crystal-activated BMMAC showed no overt changes in distribution of Cath B, while Casp-1 and ASC co-localization was clearly induced. Cath B- deficient macrophages showed no reduction in IL-1β production. Interestingly, blocking cell surface ion channels and high extracellular potassium ions during the crystal-membrane binding stage severely inhibited IL-1β release. Our work suggests phagocyte membrane ligation by particulate substances may directly modulate ion channel activities and that Cath B is not always essential for NLRP3 inflammasome activation in response to crystalline structures.
Results
Crystal-induced colocalization of ASC and Casp-1 in macrophages without lysosomal disruption
First, the Cath B localization with reference to lysosome in silica-treated or -untreated cells was analyzed. BMMAC were stained with Cath B and Lamp-1, a lysosomal marker26. Although Cath B is known as a vacuolar enzyme with a preferred pH range 5.1–6.227, only a partial association with Lamp-1 was found (Fig. 1A), possibly indicating the presence of this enzyme in the intermediate locations from the ER to the endocytic system28. Silica treatment did not significantly alter the relative positions or their locations (Fig. 1A). To detect more subtle alterations in Cath B localization undetectable by conventional confocal imaging, we turned to SIM (Structured Illumination Microscopy) super resolution microscopy. Similarly, neither difference in Cath B distribution nor its enhanced colocalization with ASC was found, with or without the crystal treatment (Fig. 1B). To ascertain that NLRP3 inflammasome was activated, we searched for ASC speck formation29,30. However, individual specks were detectable only in about 1-5% of crystal-treated cells, which also had a poor association with the amount of IL-1β produced (data not shown). ASC/Casp-1 colocalization was used as an alternative readout. ASC/Casp-1 colocalization was clearly revealed by SIM in silica-treated cells, in comparison with the untreated BMMAC (Fig. 1C. also see the supplemental movies 1 and 2 for 3D rendering of the colocalization obtained from the SIM stacks). Therefore, ASC/Casp-1 colocalization can take place without an overt change in Cath B/Lamp-1 distribution.
Figure 1
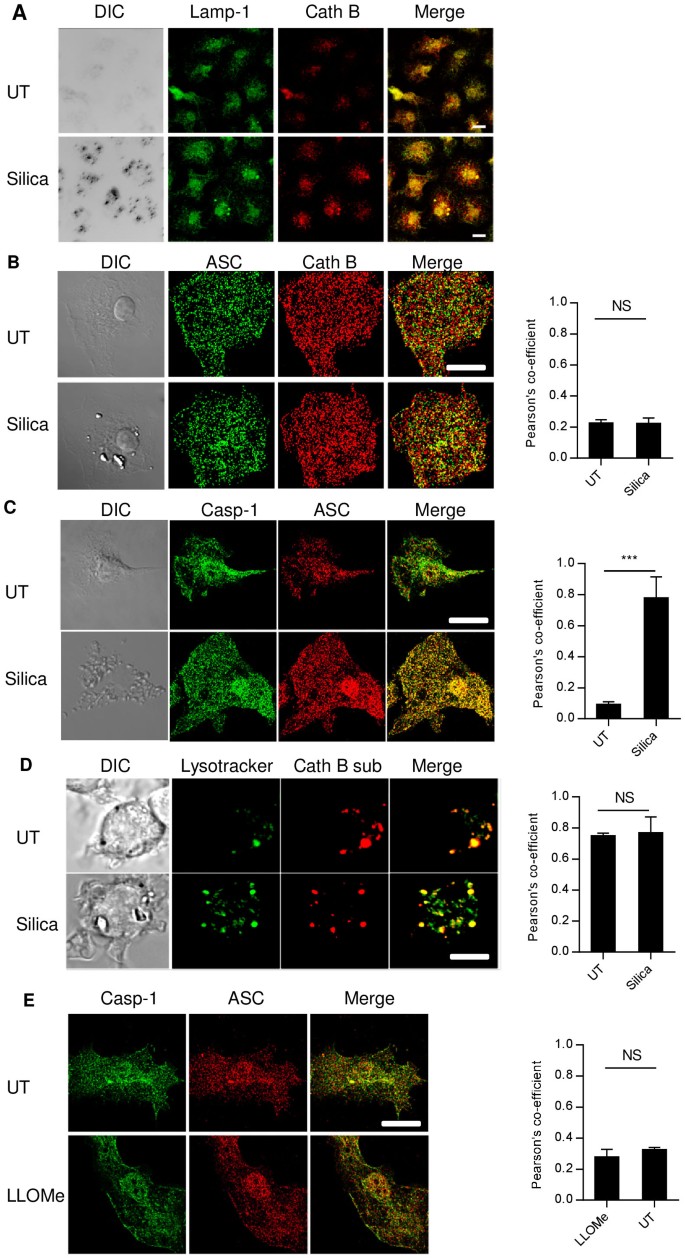
Lysosomal damage is not essential in silica mediated NLRP3 activation.
(A). 7 day, 250 ng/ml ultrapure LPS-primed BMMAC were treated with 100 ug/ml silica 1 hr or left untreated. Cells were fixed and stained with anti-Lamp-1 and Cath B. The association between the two markers was not altered by silica. All bars are 5 um. (B). BMMAC were treated as in A. Structured illumination microscopy (SIM) image of ASC and Cath B was generated after staining with specific antibodies. The right bar graph of Pearson's co-efficient in this and subsequent panels indicate the association between the green and red labels of the images on the left. (C). Identical to B except that Casp-1 antibody was used in place of Cath B. (D). BMMAC were pre-loaded with Lysotracker and Cath B substrate prior to the crystal treatment. 15 minutes later, confocal imaging was performed to determine the colocalization of these two markers. Notice the partial colocalization and the lack of distribution change following the silica treatment. (E). LLOMe-treated cells (20 mM for 1 hr) were stained for ASC/Casp-1 colocalization. Supplemental Fig 3 shows the perfused Lysotracker labelling into the cytosol in LLOMe-treated cells, suggesting lysosomal membrane destabilization. There is no overt cell morphology change or increased association between Casp-1 and ASC in the LLOMe-treated cells.
Intracellular activities of Cath B are often revealed by a fluorescence proteolytic substrate based on a two amino acid Arginine-Arginine sequence12. We found that the digestion product of this substrate was mainly associated with acidic vacuoles labeled by Lysotracker, an amine-based acidotropic probe (Fig. 1D) and its presence was unaltered in Cath B-deficient macrophages (Supp Fig 1), suggesting the digestion sequence of this substrate is non-specifically targeted by other proteolytic activities in low pH vacuoles. In the absence of other proteolytic enzymes, recombinant Cath B digested this substrate, a process inhibited by leupeptin or a neutral pH (Supp Fig 2). LLOMe was then used to destabilize the lysosomal membrane (Supp Fig 3) and Casp-1 and ASC localization was again analyzed by SIM imaging (Fig. 1E)31. No significant ASC/Casp-1 colocalization change was seen at a LLOMe concentration sufficient to trigger lysosomal rupture (Supp Fig 3). This is in line with a previous report that NLRP3/ASC/Casp-1 are dispensable in cell death triggered by lysosomal damage and is instead a consequence of non-specific proteolytic digestion following lysosomal content release23.
Cathepsin B is not essential in silica-induced NLRP3 activation
Results in Figure 1 raise the question as to whether Cath B could somehow participate in the process without being physically present in the cytosol. For instance, Cath B has been found on the plasma membrane32. Therefore, experiments were performed with _Ctsb_−/− BMMAC. Figure 2A upper panel shows that upon silica or MSU stimulation, IL-1β was produced by _Ctsb_−/− BMMAC in amounts similar to WT BMMAC. The lack of Cath B did not change expression levels of IL-1β mRNA in response to LPS priming (Fig. 2A lower panel). _Ctsb_−/− cells are not hyper reactive as their production of TNFα was similar to WT as well (Supp Fig 4). The appearance of mature IL-1β (p17) was also similar to WT (Fig. 2B). Figure 2C shows that cell death following silica and MSU treatment was similar between _Ctsb_−/− and WT BMMAC. To test crystal-induced neutrophil infiltration, MSU was i.p. injected into the peritoneal cavities, the neutrophil infiltrates were gated on population double-positive for CD45/LY6G and counted by FACS. Figure 2D shows that 16 hrs following the injection of MSU crystals, the neutrophil numbers in the peritoneal lavage increased substantially. However, there was no discernible difference in neutrophil infiltration between _Ctsb_−/− and WT or control _Ctss_−/− (Cath S) mice. The cell numbers in IL-1R deficient (_Il1r1_−/−) mice were very low, consistent with previous findings that suggest the parenchymal cells sensed and produced IL-1β thus participated in IL-1β self-amplification loop33. These data clearly demonstrate that NLRP3 activation in response to crystalline structures can take place in the absence of Cath B.
Figure 2
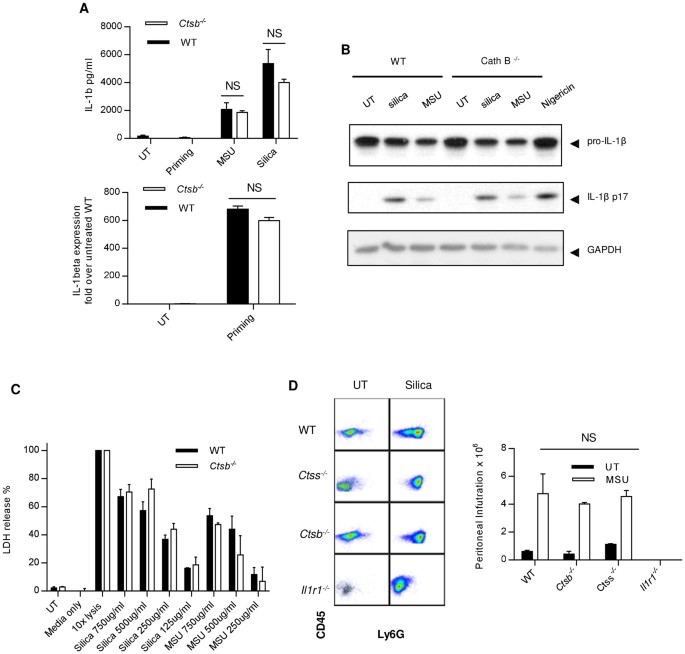
Cath B is not essential in crystal-mediated NLRP3 activation.
(A). _Ctsb_−/− and WT BMMACs were prepared as described in the methods. Cells were treated with 500 ug/ml MSU or silica. The IL-1β production was measured by ELISA after 6 hrs (upper panel). Quantitative PCR was performed on IL-1β mRNA (pro form) and the value was normalized to the level of unprimed WT BMMAC (lower panel). (B). LPS-primed BMMAC were treated with silica or MSU at 500 ug/ml for 6 hrs. The cell lysates and supernatants were blotted with anti IL-1β antibody. GAPDH was used as loading control. For this and the subsequent WB analysis, the assays were run under identical conditions and the blots were cropped from original full-sized images (attached as supplemental data). (C). Ctsb_−/− and WT BMMACs were treated with silica and MSU. A Pierce LDH cytotoxicity kit was used to measure cell death. 10 × Lysis buffer was used as the positive control and all values obtained from other treatments were normalized to this treatment (100%). (D). Ctsb_−/−, Ctss_−/−, Il1r1_−/− and WT mice were i.p. injected with MSU and rested for 16 hrs. Mice were then sacrificed and peritoneal lavages were collected using PBS. Cells were stained with CD45 and Ly6G to identify neutrophils by FACS (left panels). The numbers of infiltrating cells were counted and are shown in the bar graph (right).
Silica and MSU membrane binding without internalization is sufficient to induce IL-1β production
Since cell membrane ligation by crystals substantially increased DC phagocytosis and TNF-α production20, we wondered if crystal binding to the BMMAC membrane was sufficient to induceIL-1β production. To that end, we took advantage of the low solubility of MSU crystals and attached them to the bottom of the culture plates via a thin layer of epoxy (silica crystals cannot be trapped by epoxy). The surface was flushed extensively so that all loosely bound crystals were washed away. The BMMAC were grown on a glass disk to near confluence and the disk was flipped upside down so that the cells were in contact with MSU. After 45 minutes, the disks were removed and transferred to a new well with fresh media. BMMAC contacted by crystals showed morphological changes and displayed more ruffled surface (Fig. 3A). Importantly, very few crystals were attached to the cells, less than the minimal amount needed to trigger IL-1β production in regular experiments (25–50 ug/ml, unpublished results). Although in reduced amounts likely due to fewer cell numbers and the physical disturbance, these cells nonetheless produced IL-1β in the freshly replaced media (Fig. 3B), suggesting crystals can induce IL-1β production in BMMAC by simple surface ligation.
Figure 3
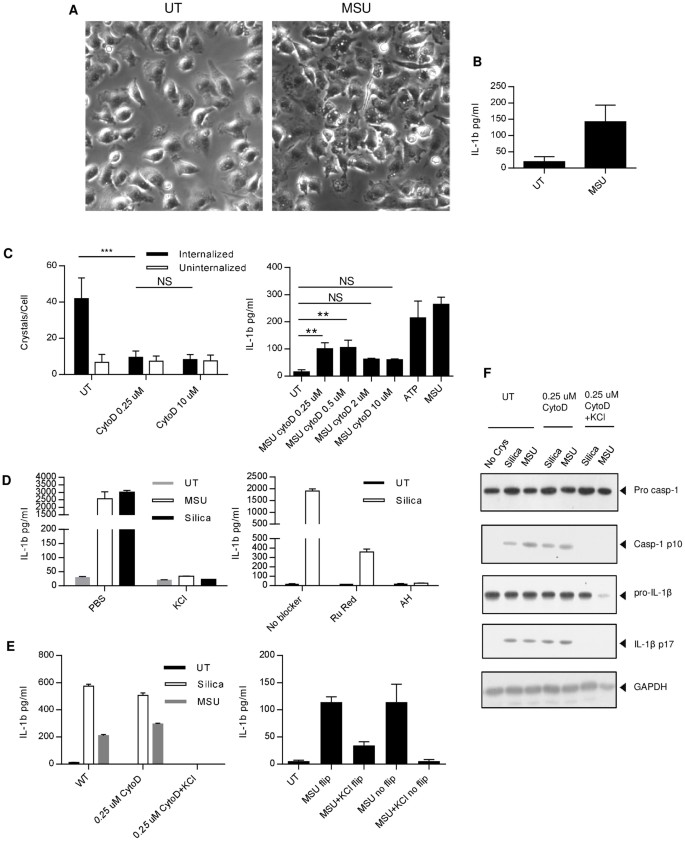
Membrane binding by crystals is sufficient to trigger ion channel activities and IL-1β release.
(A). A layer of MSU crystals were immobilized to the bottom of tissue culture plates with a thin layer of epoxy resin, left to harden and washed. BMMAC were grown on a disk and flipped to be in contact with the crystal layer. 45 minutes later, the disk was removed and the wide field images were taken. Notice very few crystals were attached to the cells. (B). The disks described above were moved to fresh media and the IL-1β production was measured by ELISA after 6 hrs. (C). Left: BMMACs were treated with indicated concentrations of CytoD and followed by addition of silica as described in the methods. Internalization was counted as dark phase crystals and uninternalized crystals as reflective phase. Right panel, IL-1β was measured after 6 hrs. 5 mM ATP was used as the positive control. Notice the apparent production of IL-1β in the presence of CytoD at a concentration (0.25 uM) sufficient to block the internalization of silica. (D). BMMACs were treated with silica in the presence of high extracellular KCl (130 mM, left panel), or ion channel blockers Ruthenium Red (RuRed) and Amiodarone Hydrochloride (AH) (right panel). (E). Left: Identical to C except that KCl was added to low dose CytoD wells, both MSU and silica were used to stimulate BMMAC. Right: Similar to B, except that some disks were not removed from the contact with the immobilized MSU and with the indicated addition of extracellular high KCl. For the flipped disks, the continuing 6 hr assay was performed in the media free of KCl. (F). LPS-primed BMMAC were treated with crystals in the presence of 0.25 uM of CytoD or a combination of CytoD and high extracellular K+; both pro forms of IL-1β and Casp-1 as well as their processed p17 and p10 subunits, respectively, were revealed by Western blotting, GAPDH was used as the loading control.
Cytochalasin D (CytoD) at high concentrations (>2 uM) blocked silica-mediated IL-1β production and inhibited actin microfilament required for cytokinesis34. At lower doses, the blockage of IL-1β in response to silica was proportionally diminished, which was presumed to be due to the inability to block internalization. After careful titration experiments, we found that, low doses of CytoD (0.25 to 0.5 uM) still blocked crystal engulfment to a similar extent as the high dose (10 uM, Fig. 3C, left panel), measured by the number of internalized crystals. At these doses, crystals still attached to the macrophages surface and induced production of IL-1β (Fig. 3C, right panel). Therefore, the binding alone was sufficient in triggering production of IL-1β. This was confirmed by Casp-1/ASC colocalization in the presence of 0.25 uM CytoD (Supp Fig 5). We next determined whether the IL-1β induced in our system was dependent on ion channel activities and K+ efflux. Two ion channel blockers Ruthenium Red (Ru Red) and Amiodarone Hydrochloride (AH)) (Fig. 3D left panel), as well as high K+ concentration in the assay media (Fig. 3D right panel), blocked the IL-1β response to silica. In the presence of high concentration of extracellular K+, we treated BMMAC with MSU and silica in the presence of low dose CytoD (Fig. 3E left panel). The extracellular high K+ completely inhibited the IL-1β production. In addition, when high K+ was present during BMMAC contact with the immobilized epoxy-trapped MSU (Fig. 3E right panel), the subsequent IL-1β release after being transferred to regular media was inhibited. Figure 3F confirms that low dose CytoD, while blocking crystal uptake, did not inhibit Casp-1 processing and IL-1β production. High extracellular K+ however stopped both activities. These results suggest that crystal binding to the cell surface is sufficient to activate the NLRP3 inflammasome with a concomitant K+ outward movement across the plasma membrane.
Discussion
This brief report presents “proof of principle” data aimed at clarifying two issues in the field of inflammasome research: 1. Whether Cath B/lysosomal rupture is absolutely required for crystal-mediated NLRP3 activation; and 2. If the internalization of crystals is essential to activate NLRP3.
An implication rising from this report is that phagocyte membrane engagement by crystals may directly regulate NLRP3 inflammasome activities, potentially via mechanosensing. This activation is accompanied by K+ efflux. The tension generated during crystal binding changes the surface curvature, which is often enough to trigger ion channel opening35,36,37. In addition, the tracking forces exerted on the cytoskeleton during phagocytosis creates strong grip on cortical F-actin at the site of contact and the web of microtubules in the cytoplasm38,39. These events are known to regulate ion channels40,41. A recent paper thoroughly surveyed a list of NLRP3-activation triggers and identified that K+ efflux was the event common to all of them9, while the AIM2 inflammasome activation by intracellular DNA, which involves ASC but not NLRP342, was not affected by K+. However, how ion channel opening is linked to NLRP3 activation remains elusive. In fact, we do not have any direct evidence to suggest K+ efflux is the cause of NLRP3 activation. A recent report indicates that microtubule acetylation in response to MSU, as a result of reduced α tubulin deacetylase, can directly facilitate spatial proximation of NLRP3 and ASC43, suggesting other regulations independent of ion channel activations are also triggered by crystalline structures. It is possible that selective reduction of intracellular ion strength is permissive for one or more critical regulator factors for the activation, oligomerization, or effector functions of some components of the NLRP3 complex.
Lima et al. suggested that the conversion of mature Casp-1 from its pro-form in alum or LLOMe-treated macrophages might be a result of uncontrolled release of proteolytic enzymes, which in comparison with the quantity processed in NLRP3 inflammasome activation triggered by Nigericin, is in a small amount23. The finding is interesting as it mystifies the scenario: if NLRP3/ASC/Casp-1 (required for pyroptosis) are not essential for crystal-induced cell death, then why is IL-1β production stimulated by the crystals severely diminished in NLRP3 inflammasome component deficient cells44? Our results here provide a possible explanation: while cells may die depending on the intensity of phagocytosis, those having only experienced membrane ligation and those with internalized crystals before the lysosomal rupture can mediate NLRP3 inflammasome activation through simple membrane ligation.
The requirement of Cath B in NLRP3 inflammasome activation in response to crystalline structures reported in the literature12,15 is in disagreement with this work and several recent papers9,17. The most likely reason for the disparity is the different experimental settings. For instance, different preparations of MSU crystals greatly affect their ability to activate and kill dendritic cells. It is possible some crystals quickly damage the phagolysosomal membrane and cause rapid release of its contents. It has been recently reported that inflammasome components NLRP3 and ASC are released following their intracellular activation and these components can further assemble to induce the activation of Casp-145,46. The rapid release of large amounts of Cath B may remain enzymatically intact for a brief period and could process Casp-1/IL-1β. Tumor cells are known to use extracellular Cath B to facilitate tissue invasion47. Interestingly, in the reports by Lima et al. and Kono et al. cited above, the authors also found that Cath C is responsible for digestion of cytosolic proteins, including pro Casp-1 and IL-1β23. Cath C, similar to Cath B, prefers low pH for its proteolysis48. The enzymatic activities seen in these two reports are mostly explained by the rapid release of Cath C and its function in a short window prior to inactivation under neutral pH46.
Methods
Mice
C57BL/6 and _Il1r1_−/− mice were bought from Jackson laboratories and housed at University of Calgary Animal Research Centre. _Ctsb_−/− and _Ctss_−/− mice were gifts from Dr. Kenneth Rock of University of Massachusetts Medical School.
Ethics statement
All experimental methods involving mice were approved by the Animal Protocol Committee of University of Calgary and conducted in accordance with the guidelines set forth by Institutional Animal Care and Use Committee (IACUC).
Reagents and antibodies
Uric acid, Cytochalasin D, Cathepsin B, LLOMe, KCl and ultrapure LPS were from Sigma. Nigericin, Ruthenium Red and Amiodarone Hydrochloride were purchased from Tocris. Monosodium urate crystals were made as described in Ng et al49. Silica particles were purchased from US Silica Company. Crystals were added to tissue culture grade endotoxin-free water to get the stock solutions for experiments. Mouse Anti-ASC (SC-22514-R), Mouse Anti-Casp-1 (SC-514), Mouse Anti-Cath B (SC-6493) antibodies were purchased from Santa Cruz. Mouse Anti-Casp-1 P20 subunit (AG-20B-0042) was purchased from Adipogen. Anti Casp-1 antibody for Western blotting was from Abcam (EPR4321). HRP-conjugated secondary antibodies and Anti-Rabbit Dylight 549 were purchased from Jackson ImmunoResearch and eBiosciences. Alexa-conjugated secondary antibodies and ProLong Gold antifade agent were purchased from Invitrogen. Mouse Pan-CD45 (17-0451) and LY6G (11-5931) antibodies were from eBioscience. GAPDH antibody (CST 2118) was from Cell Signaling. Mouse Anti IL-1β antibody (AF-401-NA) was from R&D systems. IL-1β and TNFα ELISA ready-set-go kits were from eBioscience Inc. L cell-conditioned media for BMMAC culture was a gift from Robin Yates of the University of Calgary.
Bone marrow macrophage cultures and assays
BMMAC were grown in complete RPMI medium with 10% FBS and 20% L cell-conditioned media. On day 6, the cells were replated with fresh cell culture media. On day 7, the BMMAC were primed for 2 ½ hrs with 250 ng/ml Ultra-pure LPS (Sigma). After priming, MSU (500 μg/ml) and Silica (500 μg/ml) was added for 6 hrs for the production of IL-1β and TNFα. In some experiments, various concentrations of CytoD were added 30 minutes before the addition of crystals. All treatments to cells were performed in Opti MEM Medium (Invitrogen). The supernatants were collected for ELISA. For Western blot, supernatants were concentrated using a TCA precipitation protocol. Briefly, 90 ul each of 77% TCA and 1% deoxycholate was added to about 1 ml of supernatant and mixed thoroughly. After incubation for 20 minutes on ice, the tubes were centrifuged for 15 minutes at 14000 g. The resulting precipitate was washed with 100% acetone and air dried for a few minutes. The precipitates were resuspended in 1.5 M tris pH 8 before addition of sample buffer and boiled for 10 minutes at 95°C. The cell lysates for western blot were prepared by incubating the samples in Lysis buffer (1% solution containing NP-40, 5 M NaCl, 1 M Tris pH 7.5, 0.5 M EDTA pH 8.0, protease inhibitor cocktail from Sigma) for 20 minutes on ice followed by boiling in the sample buffer containing DTT. Samples were run on 12% SDS-PAGE before transfer to nitrocellulose membrane.
For IL-1β produced from BMMAC in contact with immobilized crystals, dry MSU crystals were layered on premixed Epoxy resin on 12-well plates and allowed to harden overnight. The wells were washed with cell culture-grade water to remove loosely adherent crystals. Opti MEM was added to these wells and left in the incubator at 37°C with 5% CO2 for 15 minutes. BMMACs grown on glass disks were placed on these crystals and allowed to interact for 45 minutes. The disks were carefully removed and placed back into fresh Opti MEM for further incubation of about 6 hrs. Supernatants were collected for ELISA. Phase contrast images were taken to verify that most cells were devoid of crystals.
For ion channel-blocking assays, BMMACs generated as described above were primed with 250 ng/ml LPS (Sigma). After removal of LPS, BMMACs were either left untreated or treated with PBS-10% FBS, 130 mM KCl-10% FBS, 0.5/1/2 uM Ruthenium Red or 10 uM Amiodarone hydrochloride for 30 min. Then BMMACs were stimulated with MSU or silica for 6 hrs; the supernatants were collected ELISA analysis.
LDH assay
BMMACs grown as described above were treated with indicated concentrations of crystals (MSU and Silica) and 10× lysis buffer (positive control). While cells were treated with the crystals for 5 hrs, 10× lysis buffer was given 45 minutes before sample collection. The supernatants were collected and LDH released was measured and calculated using Pierce LDH cytotoxicity kit from Thermo Scientific per manufacturer's instructions.
Cathepsin B enzyme assay
To study Cathepsin B's protease activity50, purified Cathepsin B from bovine spleen (Sigma) (0.5 unit/ml) was incubated with or without general protease inhibitor Leupeptin (25 μM) in buffers of different pH. 25 mM MES with 5 mM DTT was used as buffer for pH 5.1 and 0.1 M PIPES with 5 mM DTT, 1 mM CaCl2, 2.7 mM KCl and 0.5 mM MgCl2 was used as buffer for pH 7.4. Rates of hydrolysis of the fluorogenic substrates: Cath B substrate (CV-RR2, Enzo Lifesciences or ES008, R&D) were measured at 37°C using a FLUOstar Optima fluorescent plate reader (BMG Labtech). Slopes of the initial rate of reaction were determined by curve-fitting applications in Microsoft Excel and expressed relative to untreated controls51.
Immunofluorescence
For fluorescent imaging, LPS-primed cells were allowed to incubate for 1 hr with or without crystals at 37°C. The disks were then washed with PBS twice and were fixed in pre-warmed 2% PFA (Electron Microscopy Sciences) in PBS for 10 minutes, permeabilized in 0.2% Triton X. After permeabilization, the cells were washed in PBS and appropriate primary antibodies were added and allowed to incubate at 37°C for 1 hr. This was followed by secondary antibody incubation and mounting on ProLong Gold antifade agent with DAPI to reveal the nuclei. Whenever the antibodies were from the same host, Xenon direct labeling (Invitrogen) was used to direct label antibodies before addition to cells. For LLOMe treatments, cells were treated for 1 hr with 20 mM LLOMe before fixation. All raw images were incorporated in Image J by LOCI bio-format importer plugin. Colocalization processing: Image J colocalization finder plugin was used. For visualization of Cathepsin B substrate, cells were grown as stated above and the substrate was given to the cells with or without silica (100 μg/ml). Half an hr after incubation, the cells were washed with PBS and imaged using a confocal microscope setting under live conditions. SIM was carried out on an ELYRA super resolution microscopy from Zeiss while confocal imaging was obtained on the same machine under standard settings. All other imaging was done on an Olympus IX-73 microscope followed by deconvolution of the images.
Crystal internalization
BMMAC were seeded onto 24-well plates with glass coverslips at 105 cells/well. Next day, cells were pre-incubated with CytoD or vehicle control at the indicated concentrations for 30 minutes at 37°C before further treated with 30 ug of silica per well for 60 minutes. The internalization was determined as previously described45. Cells were fixed with 4% PFA and permeabilized with 0.5% Triton-X100. Actin was stained with Alexa Fluor 488-conjugated Phalloidin (Invitrogen). Phase contrast microscopy and immunofluorescence microscopy were employed together to determine whether a silica particle was internalized or not. Two criteria were used: 1) internalized particles are phase dark when examined by phase contrast microscopy. 2) actin filament stain disappears around the internalized silica particle but not the uninternalized particles when examined by fluorescence microscopy.
Quantitative PCR
C57BL/6 and _Ctsb_−/− BMMACs were either untreated or primed with ultrapure LPS. Total RNA was extracted using Trizol treatment. 1 ug total RNA was used to generate the first strand cDNA using a RT-kit (TAKARA). The qPCR primers of mouse IL-β were as follows: TTGACGGACCCCAAAAGAT (forward) and GAAGCTGGATGCTCTCATCTG (reverse). SYBR Green I qPCR SuperMixU was used for analysis (TransStart). Assays were performed in triplicate.
In vivo peritonitis
Mice were injected i.p with 2 mg/ml MSU in 500 μL PBS. After 16 hrs the mice were euthanized and the peritonea was lavaged with 9 ml ice-cold PBS. The absolute number of cells obtained was counted in a haemocytometer before staining them with LY6G and CD45 antibodies. The cells were washed with PBS and fixed using 1% PFA to be read using an Attune® Acoustic Focusing Flow Cytometer (Applied Biosystems). All analysis was performed using Treestar Flowjo 10.0.
Statistics
All bar graphs are means with SEM. Statistical analysis for each independent experiment was performed with an unpaired, Student's t test. A P value of less than 0.05 was considered significant. *: <0.05; **: <0.05 and >0.01; ***: 0.01; NS: not significant.
References
- Franchi, L., Eigenbrod, T., Munoz-Planillo, R. & Nunez, G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 10, 241–247 (2009).
Article CAS PubMed PubMed Central Google Scholar - Lamkanfi, M. & Dixit, V. M. Mechanisms and functions of inflammasomes. Cell 157, 1013–1022 (2014).
Article CAS PubMed Google Scholar - Sutterwala, F. S., Haasken, S. & Cassel, S. L. Mechanism of NLRP3 inflammasome activation. Ann N Y Acad Sci 1319, 82–95 (2014).
Article CAS ADS PubMed PubMed Central Google Scholar - Tschopp, J. & Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol 10, 210–215 (2010).
Article CAS PubMed Google Scholar - Zhou, R., Tardivel, A., Thorens, B., Choi, I. & Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol 11, 136–140 (2010).
Article CAS PubMed Google Scholar - Cain, K., Langlais, C., Sun, X. M., Brown, D. G. & Cohen, G. M. Physiological concentrations of K+ inhibit cytochrome c-dependent formation of the apoptosome. J Biol Chem 276, 41985–41990 (2001).
Article CAS PubMed Google Scholar - Arlehamn, C. S., Petrilli, V., Gross, O., Tschopp, J. & Evans, T. J. The role of potassium in inflammasome activation by bacteria. J Biol Chem 285, 10508–10518 (2010).
Article CAS PubMed PubMed Central Google Scholar - Petrilli, V. et al. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 14, 1583–1589 (2007).
Article CAS PubMed Google Scholar - Munoz-Planillo, R. et al. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153 (2013).
Article CAS PubMed PubMed Central Google Scholar - Heid, M. E. et al. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J Immunol 191, 5230–5238 (2013).
Article CAS PubMed Google Scholar - Zhou, R., Yazdi, A. S., Menu, P. & Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 (2011).
Article CAS ADS PubMed Google Scholar - Hornung, V. et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9, 847–856 (2008).
Article CAS PubMed PubMed Central Google Scholar - Franchi, L., Munoz-Planillo, R. & Nunez, G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol 13, 325–332 (2012).
Article CAS PubMed PubMed Central Google Scholar - Kuroda, E. et al. Silica crystals and aluminum salts regulate the production of prostaglandin in macrophages via NALP3 inflammasome-independent mechanisms. Immunity 34, 514–526 (2011).
Article CAS PubMed Google Scholar - Duewell, P. et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 (2010).
Article CAS ADS PubMed PubMed Central Google Scholar - Eisenbarth, S. C., Colegio, O. R., O'Connor, W., Sutterwala, F. S. & Flavell, R. A. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature 453, 1122–1126 (2008).
Article CAS ADS PubMed PubMed Central Google Scholar - Dostert, C. et al. Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS One 4, e6510 (2009).
Article ADS CAS PubMed PubMed Central Google Scholar - Newman, Z. L., Leppla, S. H. & Moayeri, M. CA-074Me protection against anthrax lethal toxin. Infect Immun 77, 4327–4336 (2009).
Article CAS PubMed PubMed Central Google Scholar - Kono, H., Orlowski, G. M., Patel, Z. & Rock, K. L. The IL-1-dependent sterile inflammatory response has a substantial caspase-1-independent component that requires cathepsin C. J Immunol. 189, 3734–3740 (2012).
- Flach, T. L. et al. Alum interaction with dendritic cell membrane lipids is essential for its adjuvanticity. Nat Med 17, 479–487 (2011).
Article CAS PubMed Google Scholar - Marichal, T. et al. DNA released from dying host cells mediates aluminum adjuvant activity. Nat Med 17, 996–1002 (2011).
Article CAS PubMed Google Scholar - Munks, M. W. et al. Aluminum adjuvants elicit fibrin-dependent extracellular traps in vivo. Blood 116, 5191–5199 (2010).
Article CAS PubMed PubMed Central Google Scholar - Lima, H., Jr et al. Role of lysosome rupture in controlling Nlrp3 signaling and necrotic cell death. Cell Cycle 12, 1868–1878 (2013).
Article CAS PubMed Google Scholar - Ng, G. et al. Receptor-independent, direct membrane binding leads to cell-surface lipid sorting and Syk kinase activation in dendritic cells. Immunity 29, 807–818 (2008).
Article CAS PubMed PubMed Central Google Scholar - Malik, A. F. et al. Inflammasome components Asc and caspase-1 mediate biomaterial-induced inflammation and foreign body response. Proc Natl Acad Sci U S A 108, 20095–20100 (2011).
Article CAS ADS PubMed PubMed Central Google Scholar - Chen, J. W., Pan, W., D'Souza, M. P. & August, J. T. Lysosome-associated membrane proteins: characterization of LAMP-1 of macrophage P388 and mouse embryo 3T3 cultured cells. Arch Biochem Biophys 239, 574–586 (1985).
Article CAS PubMed Google Scholar - Guha, S. & Padh, H. Cathepsins: fundamental effectors of endolysosomal proteolysis. Indian J Biochem Biophys 45, 75–90 (2008).
CAS PubMed Google Scholar - Linke, M., Herzog, V. & Brix, K. Trafficking of lysosomal cathepsin B-green fluorescent protein to the surface of thyroid epithelial cells involves the endosomal/lysosomal compartment. J Cell Sci 115, 4877–4889 (2002).
Article CAS PubMed Google Scholar - Bryan, N. B., Dorfleutner, A., Rojanasakul, Y. & Stehlik, C. Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J Immunol 182, 3173–3182 (2009).
Article CAS PubMed Google Scholar - Sagulenko, V. et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ 20, 1149–1160 (2013).
Article CAS PubMed PubMed Central Google Scholar - Thiele, D. L. & Lipsky, P. E. Mechanism of L-leucyl-L-leucine methyl ester-mediated killing of cytotoxic lymphocytes: dependence on a lysosomal thiol protease, dipeptidyl peptidase I, that is enriched in these cells. Proc Natl Acad Sci U S A 87, 83–87 (1990).
Article CAS ADS PubMed PubMed Central Google Scholar - Rempel, S. A. et al. Cathepsin B expression and localization in glioma progression and invasion. Cancer Res 54, 6027–6031 (1994).
CAS PubMed Google Scholar - Chen, C. J. et al. MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest 116, 2262–2271 (2006).
Article CAS PubMed PubMed Central Google Scholar - Shinji, H., Akagawa, K. S. & Yoshida, T. Cytochalasin D inhibits lipopolysaccharide-induced tumor necrosis factor production in macrophages. J Leukoc Biol 54, 336–342 (1993).
Article CAS PubMed Google Scholar - Balleza, D. Mechanical properties of lipid bilayers and regulation of mechanosensitive function: from biological to biomimetic channels. Channels (Austin) 6, 220–233 (2012).
Article CAS Google Scholar - Kumamoto, C. A. Molecular mechanisms of mechanosensing and their roles in fungal contact sensing. Nat Rev Microbiol 6, 667–673 (2008).
Article CAS PubMed PubMed Central Google Scholar - Brohawn, S. G., Su, Z. & MacKinnon, R. Mechanosensitivity is mediated directly by the lipid membrane in TRAAK and TREK1 K+ channels. Proc Natl Acad Sci U S A 111, 3614–3619 (2014).
Article CAS ADS PubMed PubMed Central Google Scholar - Mooren, O. L., Galletta, B. J. & Cooper, J. A. Roles for actin assembly in endocytosis. Annu Rev Biochem 81, 661–686 (2012).
Article CAS PubMed Google Scholar - Harrison, R. E. & Grinstein, S. Phagocytosis and the microtubule cytoskeleton. Biochem Cell Biol 80, 509–515 (2002).
Article CAS PubMed Google Scholar - Sadoshima, J., Takahashi, T., Jahn, L. & Izumo, S. Roles of mechano-sensitive ion channels, cytoskeleton and contractile activity in stretch-induced immediate-early gene expression and hypertrophy of cardiac myocytes. Proc Natl Acad Sci U S A 89, 9905–9909 (1992).
Article CAS ADS PubMed PubMed Central Google Scholar - Martinac, B. The ion channels to cytoskeleton connection as potential mechanism of mechanosensitivity. Biochim Biophys Acta 1838, 682–691 (2014).
Article CAS PubMed Google Scholar - Hornung, V. et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518 (2009).
Article CAS ADS PubMed PubMed Central Google Scholar - Misawa, T. et al. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol 14, 454–460 (2013).
Article CAS PubMed Google Scholar - Martinon, F., Petrilli, V., Mayor, A., Tardivel, A. & Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241 (2006).
Article CAS ADS PubMed Google Scholar - Costantini, L. M., Gilberti, R. M. & Knecht, D. A. The phagocytosis and toxicity of amorphous silica. PLoS One 6, e14647 (2011).
Article CAS ADS PubMed PubMed Central Google Scholar - Baroja-Mazo, A. et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol 15, 738–748 (2014).
Article CAS PubMed Google Scholar - Premzl, A., Zavasnik-Bergant, V., Turk, V. & Kos, J. Intracellular and extracellular cathepsin B facilitate invasion of MCF-10A neoT cells through reconstituted extracellular matrix in vitro. Exp Cell Res 283, 206–214 (2003).
Article CAS PubMed Google Scholar - Dolenc, I., Turk, B., Pungercic, G., Ritonja, A. & Turk, V. Oligomeric structure and substrate induced inhibition of human cathepsin C. J Biol Chem 270, 21626–21631 (1995).
Article CAS PubMed Google Scholar - Ackerman, A. L., Giodini, A. & Cresswell, P. A role for the endoplasmic reticulum protein retrotranslocation machinery during crosspresentation by dendritic cells. Immunity 25, 607–617 (2006).
Article CAS PubMed Google Scholar - Knight, C. G. Human cathepsin B. Application of the substrate N-benzyloxycarbonyl-L-arginyl-L-arginine 2-naphthylamide to a study of the inhibition by leupeptin. Biochem J 189, 447–453 (1980).
Article CAS PubMed PubMed Central Google Scholar - Balce, D. R. et al. Alternative activation of macrophages by IL-4 enhances the proteolytic capacity of their phagosomes through synergistic mechanisms. Blood 118, 4199–4208 (2011).
Article CAS PubMed Google Scholar
Acknowledgements
We would like to thank Leanne Mortimer, Pankaj Tailor, Dale Balce, Akosua Vilasayne and Wei Dong for technical advice and help. Cathepsin L and B knockout mice were gifts from Kenneth Rock of University of Massachusetts Medical School. We acknowledge the support from the University of Calgary Animal Resource Center and Central Flow Cytometry Facility. We thank Dr. Matthias Amrein and Dr. Jim McGee for access to super resolution microscopes. We also thank Dr. Robin Yates for reagents and equipment access. This work was supported by grants from US National Institutes of Health (R01AI098995), Natural Sciences and Engineering Research Council of Canada (RGPIN-355350/396037) and Canadian Institutes for Health Research (MOP-119295) to Y.S.
Author information
Author notes
- Hari Aswin and Zhang Yifei contributed equally to this work.
Authors and Affiliations
- Department of Microbiology, Immunology & Infectious Diseases and Snyder Institute, University of Calgary, Calgary, Alberta, Canada
Aswin Hari, Zhongyuan Tu, Pascal Detampel, Melanie Stenner, Anutosh Ganguly & Yan Shi - Department of Basic Medical Sciences, School of Medicine,
Yifei Zhang & Yan Shi - Tsinghua-Peking Center for Life Sciences, Tsinghua University, Beijing, China
Yifei Zhang & Yan Shi
Authors
- Aswin Hari
You can also search for this author inPubMed Google Scholar - Yifei Zhang
You can also search for this author inPubMed Google Scholar - Zhongyuan Tu
You can also search for this author inPubMed Google Scholar - Pascal Detampel
You can also search for this author inPubMed Google Scholar - Melanie Stenner
You can also search for this author inPubMed Google Scholar - Anutosh Ganguly
You can also search for this author inPubMed Google Scholar - Yan Shi
You can also search for this author inPubMed Google Scholar
Contributions
A.H. performed imaging preparations, neutrophil infiltration, LDH and immobilized crystal- induced IL-1β production assays. A.G. and P.D. performed imaging analyses. Y.Z. performed Western blotting, K+ efflux and ion channel blocking experiments. Z.T. performed the internalization assay. M.S. performed the flow cytometry assays and assisted in cell culture and ELISA assays. Y.S. designed experiments and wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article
Cite this article
Hari, A., Zhang, Y., Tu, Z. et al. Activation of NLRP3 inflammasome by crystalline structures via cell surface contact.Sci Rep 4, 7281 (2014). https://doi.org/10.1038/srep07281
- Received: 13 August 2014
- Accepted: 13 November 2014
- Published: 02 December 2014
- DOI: https://doi.org/10.1038/srep07281