Chromosome-level genome assembly of golden pompano (Trachinotus ovatus) in the family Carangidae (original) (raw)
Abstract
Golden pompano (Trachinotus ovatus), a marine fish in the Carangidae family, has a wide geographical distribution and adapts to severe environmental rigours. It is also an economically valuable aquaculture fish. To understand the genetic mechanism of adaption to environmental rigours and improve the production in aquaculture, we assembled its genome. By combination of Illumina and Pacbio reads, the obtained genome sequence is 647.5 Mb with the contig N50 of 1.80 Mb and the scaffold N50 of 5.05 Mb. The assembly covers 98.9% of the estimated genome size (655 Mb). Based on Hi-C data, 99.4% of the assembled bases are anchored into 24 pseudo-chromosomes. The annotation includes 21,915 protein-coding genes, in which 95.7% of 2,586 BUSCO vertebrate conserved genes are complete. This genome is expected to contribute to the comparative analysis of the Carangidae family.
| Measurement(s) | DNA • chromosome conformation capture assay • transcription profiling assay |
|---|---|
| Technology Type(s) | DNA sequencing • Hi-C • RNA sequencing |
| Factor Type(s) | organism part |
| Sample Characteristic - Organism | Trachinotus ovatus |
| Sample Characteristic - Environment | ocean biome |
Machine-accessible metadata file describing the reported data: https://doi.org/10.6084/m9.figshare.9933515
Similar content being viewed by others
Background & Summary
The golden pompano, Trachinotus ovatus (Linnaeus 1758), belongs to Carangiformes and is widely distributed in tropical and subtropical oceans[1](/articles/s41597-019-0238-8#ref-CR1 "FishBase consortium, https://www.fishbase.in/Summary/SpeciesSummary.php?ID=1773&AT=pompano
(2019)."). From a biogeographic perspective, this fish readily tolerates different environments. In addition, this fish has been one of the most importantly economic marine fish in China[2](/articles/s41597-019-0238-8#ref-CR2 "Zhenzhen, X. et al. Transcriptome analysis of the Trachinotus ovatus: identification of reproduction, growth and immune-related genes and microsatellite markers. PLoS One 9, e109419 (2014)."). However, overfishing, diseases, and degeneration of genetic diversity have caused serious economic losses in _T_. _ovatus_ production[3](/articles/s41597-019-0238-8#ref-CR3 "Tu, Z., Li, H., Zhang, X., Sun, Y. & Zhou, Y. Complete genome sequence and comparative genomics of the golden pompano (Trachinotus ovatus) pathogen, Vibrio harveyi strain QT520. PeerJ 5, e4127 (2017)."). Many solutions, including selective breeding[4](/articles/s41597-019-0238-8#ref-CR4 "Sun, L., Zhang, D., Jiang, S., Guo, H. & Zhu, C. Isolation and characterization of 21 polymorphic microstatellites in golden pompano Trachinotus ovatus. Conservation Genetics Resources 5, 1107–1109 (2013)."), identification of trait-associated genes[5](/articles/s41597-019-0238-8#ref-CR5 "Zhu, K.-C. et al. Identification of Fatty Acid Desaturase 6 in Golden Pompano Trachinotus Ovatus (Linnaeus 1758) and Its Regulation by the PPARαb Transcription Factor. International journal of molecular sciences 20, 23 (2018)."), and dietary supplementation[6](/articles/s41597-019-0238-8#ref-CR6 "Zhou, C. et al. The Effects of dietary soybean isoflavones on growth, innate immune responses, hepatic antioxidant abilities and disease resistance of juvenile golden pompano Trachinotus ovatus. Fish & Shellfish Immunology 43, 158–166 (2015)."), are adopted to overcome these problems and improve the production.The golden pompano is a marine fish in the Carangidae family. One characteristic of this family is the indistinguishable sex chromosomes7. It is speculated that sex chromosomes in this family has not been largely differentiated, distinct from those with well-differentiated sex chromosomes8. Therefore, fish in this family could be used to analyse the initial evolution status of the sex-determination system. Another characteristic of this family is tolerance to high turbidity, rapid pH changes and low dissolved oxygen concentrations and crowding9. The Carangidae fish are potential candidates to study resistance to stress.
A high-quality genome assembly is necessary to understand the functional, ecological and evolutional genomics of this species and other fish in the Carangidae family. In the present study, we presented a chromosome-level genome assembly of pompano using Illumina sequencing, Pacbio sequencing, and Hi-C technology (Fig. 1). We produced 105 Gb of cleaned Illumina reads of genomic DNA, 16.9 Gb Pacbio long reads, and 114.8 Gb cleaned data from a Hi-C library. The genome size was estimated to be 655 Mb (Fig. 2). A 647.5 Mb assembly of pompano was generated. The contig N50 length and scaffold N50 length were 1.80 Mb and 5.05 Mb, respectively. Based on 114.8 Gb Hi-C data, 99.4% of the assembly were anchored into 24 pseudo-chromosomes. The annotation includes 21,915 protein-coding genes.
Fig. 1
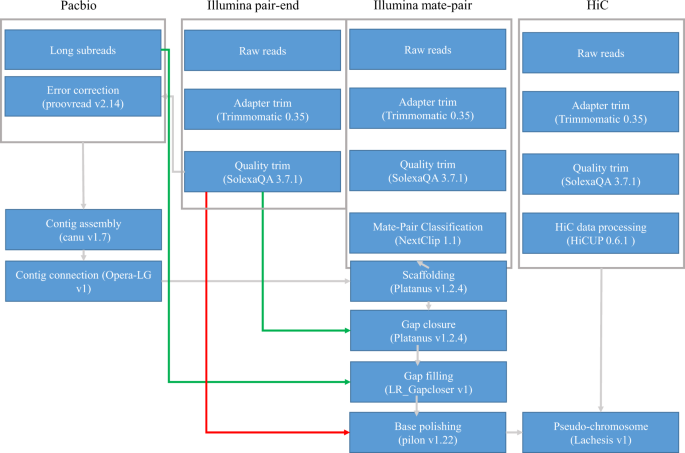
The pipelines of the chromosome-level pompano genome assembly.
Fig. 2
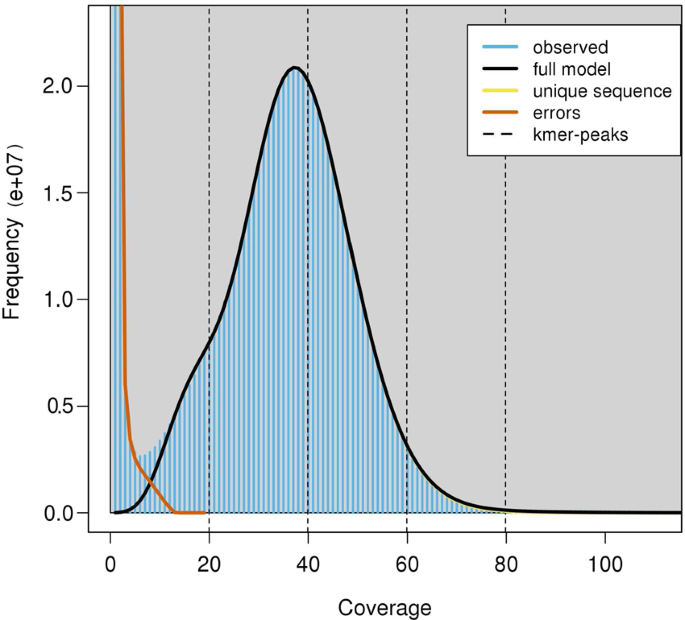
The K-mer distribution of Illumina paired-end reads using GenomeScope based on k value of 31. Frequency distribution of k-mers of different occurrences in two pair-end libraries. K-mer occurrences (x axis) were plotted against their frequencies (y axis).
The coverage of the estimated genome size (98.9%), the proportion of bases anchored to the pseudo-chromosomes (99.4%), the RNA-seq alignment ratio (90.5%), the proportion of pompano protein-coding genes having homologues (97.5%), and the ratio of complete BUSCO vertebrate genes (95.7%) all indicate that this genome assembly is of high quality. The genome assembly and its annotated information would be useful for studies on environmental adaptions, resistance to disease and sex determination. This genome has already proven to be useful to mine functional genes underlying resistance to disease10,11. It is the first chromosome-level genome in the Carangidae family and is expected to contribute to the study of the diversity, speciation, and evolution of this family.
Methods
Ethics statement
The sampled fish in this study was permitted by the Animal Care and Use Committee of South China Sea fisheries Research Institute, Chinese Academy of fishery Sciences (No. SCSFRI96-253) and performed by the regulations and guidelines established with this committee.
Sampling and sequencing
A female pompano was collected in Xincun Bay, Hainan, China. Total genomic DNA was extracted using a DNA Extraction Kit (MAGEN Company, Guangdong, China) following the manufacturer’s protocols. The quality and quantity of total DNA were determined by a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA). We constructed two paired-end libraries (insert sizes of 500 and 700 bp) and three mate-pair libraries (insert sizes of 3, 5, and 14 kb) according to Illumina standard procedures (Illumina, San Diego, CA, USA). The libraries were sequenced on a HiSeq 2500 system with 250 bp PE mode or 100 bp PE mode (Table 1).
Table 1 Data statistics of whole genome sequencing reads of pompano.
The extracted DNA molecules were also used to construct two 20 kb libraries following the PacBio manufacturing protocols (Pacific Biosciences, CA, USA). The libraries were then sequenced with two cells on PacBio Sequel platform (Table 1).
The Hi-C technique has been applied into constructing chromosome-level assemblies12,13. We prepared a Hi-C library for the chromosome assembly of pompano following the strategy of Rao et al.14. Briefly, the blood sample was fixed with fresh formaldehyde and then DNA-protein bonds were created. The restriction enzyme of Mbo I digested the DNA and the overhanging 5′ ends of the DNA fragments were repaired with a biotinylated residue. The fragments close to each other in the nucleus during fixation were ligated. The Hi-C fragments were further sheared by sonication into smaller fragments of ~350 bp in size, which were then pulled-down with streptavidin beads. The Hi-C library for Illumina sequencing was prepared according to the manufacturer’s standard procedures. The library was sequenced on the Illumina HiSeq X Ten platform with 150 bp PE mode.
Eight tissues (blood, liver, muscle, brain, spleen, fin, ovary and stomach) were collected. Total RNA from each tissue was extracted and treated with DNase I (Thermo Fisher Scientific, Wilmington, DE, USA) to remove genomic DNA. The RNA integrity of each tissue was confirmed with a Bioanalyzer 2100 (Agilent Technologies, Santa Clara, USA). For each tissue, we constructed two RNA-sequencing libraries with an insert size of 300 bp and then sequenced them on the Illumina HiSeq platform with 150 bp PE mode.
Read filtration and genome size estimation
The genomic sequencing reads from five Illumina libraries were first cleaned to remove the adapters using Trimmomatic-0.3515. Then the quality trimming was performed using SolexaQA v3.7.116 to filter the low-quality bases and short reads <25 bp. We produced 105 Gb of cleaned Illumina reads of genomic DNA. Especially, the reads from the mate-pair libraries, were further subjected to classification of the mate pairs using the Nextclip v1.117.
Pacbio sequencing generated ~16.9 Gb long reads (Table 1). The mean and N50 length were 7.4 and 12.2 kb, respectively. We corrected the Pacbio long reads with reads from two Illumina paired-end libraries using proovread v2.1418. Additionally, the paired-end reads of the Hi-C library were trimmed by filtering adapters and removing reads of low quality with Trimmomatic-0.3515 and SolexaQA v3.7.116, respectively. 382 million cleaned reads with the total bases of 114.8 Gb were generated from the Hi-C library.
Before genome assembly and gene annotation, we estimated the genome size by the k-mer analysis using 35.58 Gb filtered reads from the two paired-end Illumina libraries (500 bp and 700 bp libraries). The number of effective k-mers and the peak depth of a series of k values (17, 19, 21, 23, 25, 27, 29, and 31) were produced using Jeffyfish (v2.2)19 with the C-setting. The genome size was estimated following the formula Genome_Size = (Total k-mers - Erroneous k-mers)/Peak20. The maximal genome size was calculated to be 655 Mb when a k-mer size was 31 (Table 2). The estimated genome size was within the range of previously reported sizes of other Carangidae fish (614.2 Mb~716.4 Mb, Table 3). Hence, the sequencing coverages of the cleaned Illumina reads, Pacbio reads, and Hi-C data were 114.5, 25.7, and 175.1-fold, respectively. The rate of genome heterozygosity estimated by GenomeScope (v1.0.0)21 was around 0.31% (Fig. 2). The low heterozygosity indicated this genome to be homozygous.
Table 2 Estimation of genome size of pompano by k-mer analysis.
Table 3 Comparisons of other published Carangiformes assemblies.
Hybrid assembly, scaffolding, and chromosome anchoring
The error-corrected long reads were assembled using Canu v1.722 with the default parameters of correctedErrorRate as 0.039. The contigs were further connected into longer contigs with the error-corrected long reads using Opera-LG23. The contigs were further scaffolded using mate-pair libraries, and the gaps in the scaffolds were closed with reads from the paired-end libraries using Platanus v1.2.424. The gaps in the assemblies were further filled with the raw long reads using LR_Gapcloser v1.025. The final genome sequences were polished by pilon v1.2226 using cleaned Illumina short reads to correct errors in base level. A ~647.5 Mb genome assembly of pompano with 373 scaffolds was constructed. The assembly covered 98.9% of estimated genome regions. The contig N50 length and scaffold N50 length were 1.80 Mb and 5.05 Mb, respectively. A total of 137 scaffolds, longer than 1.26 Mb, covered over 90% of the assembly (Table 2).
To anchor scaffolds into pseudo-chromosomes, HiCUP v0.6.127 was firstly used to map and process the reads from the Hi-C library. Two reads of pairs were mapped to the polished scaffolds using Bowtie 228 with the default parameters. If both two reads from one pair were uniquely mapped to the assembly, this pair was retained for the downstream filtration. HiCUP removed invalid pairs which were generated from contiguous sequences, circularization, dangling ends, internal fragments, re-ligation, PCR duplication, and fragments of wrong size. Based on the refined alignments, we clustered 321 scaffolds into pseudo-chromosomes using Lachesis v1.029. It is reported that pompano genome consists of 24 chromosomes by linkage group analysis30 and karyotyping31. Therefore, the pseudo-chromosome number was set as 24. Finally, Lachesis ordered and oriented 259 scaffolds into 24 pseudo-chromosomes, corresponding to 69.4% and 99.4% of the assembly by sequence number and base count, respectively. The average pseudo-chromosome length was 26.84 Mb. The unanchored 114 scaffolds were much short with an average length of 33.3 kb, covering only 0.6% of the assembly. To validate the correction of the Hi-C scaffolding to pseudo-chromosome level, we constructed an interaction matrix with cleaned reads from the Hi-C library using HiC-Pro32 (default parameters and LIGATION_SITE = GATC). The genome was divided into bins of equal size of 100 Kb, and the number of contacts was determined between each pair of reported bins. A contact map plotted with HiCPlotter33 confirmed the genome structure and quality (Fig. 3). Compared with other Carangidae fish, it is the first chromosome-level assembly in this family (Table 3).
Fig. 3
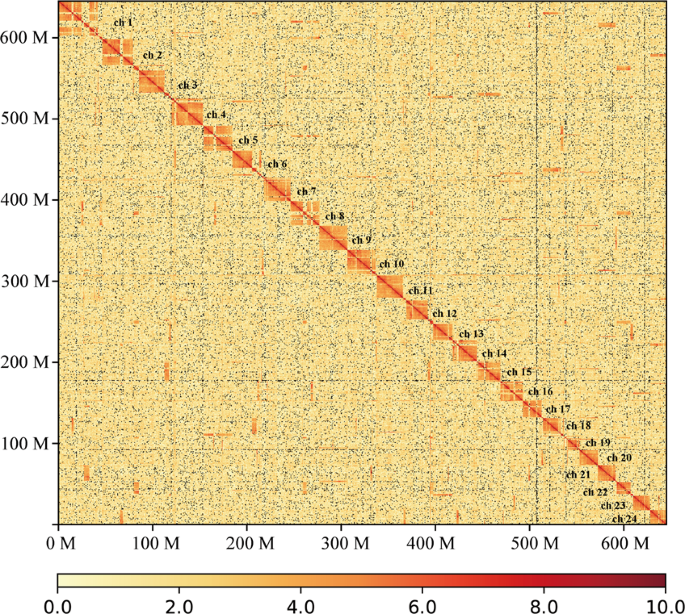
Hi-C chromosome contact map. Each block represents a Hi-C contact between two genomic loci within a 100-kb window. Darker color of a block indicates higher contact intensity.
De novo repeat prediction and classification
Before predicting protein-coding genes, we masked the repetitive regions of the assembly using a combination of ab initio and homology-based approaches. RepeatModeler v1.0.11 (http://www.repeatmasker.org/RepeatModeler/) was used to construct a pompano-specific repeat library consisting of 1,134 consensus repeats. By using RepeatMasker v4.0.7 (http://repeatmasker.org/cgi-bin/WEBRepeatMasker), the repeat regions of this assembly were masked first with the Repbase teleost repeat library34 and then with the pompano-specific library. The results from the stepwise method identified 131.22 Mb of repeat sequences, included 109.9 Mb of interspersed repeats and 21.1 Mb of tandem repeats. Among classified interspersed repeats, DNA transposons were more abundant than retrotransposons. The repeats accounted for 20.25% of the assembly (Table 4), close to that of published diploid fish genomes35,36,37.
Table 4 Repeat content in pompano genome.
Gene prediction and functional annotation
Based on the repeat-masked assembly, we predicted gene models by integrating ab initio predictions, homologue prediction, and RNA-seq models. First, Fgenesh38 was used to construct de novo gene models. Second, we aligned fish proteins from the Ensembl database39 to the assembly using BLAT40. All fish proteins annotated in Ensembl database were downloaded to construct an Ensembl fish protein set. The proteins having alignments with over 70% coverage were re-aligned to the assembly using GeneWise41 for accurately spliced alignments. Third, a total of 32 Gb of clean RNA-seq reads from eight tissues trimmed by Trimmomatic-0.3516 and SolexaQA v3.7.116 were used to construct RNA-seq based gene models. RNA-seq reads were mapped to the genome using HISAT242, and the alignments were input to Cufflinks43 to predict transcripts. All three sets of gene models were merged to form a comprehensive consensus gene set using Cuffmerge43. For each model, the longest transcript was selected as the representative transcript. The coding region and protein sequence of the representative transcript were predicted using Transdecoder (https://transdecoder.github.io/). A consensus pompano gene set consisted of 21,915 protein-coding genes. The protein-coding gene number and structures were comparable with that of published Carangiformes genomes (Table 3).
Then we searched for homologues of pompano proteins by aligning them against the Swiss-Prot database, TrEMBL database44 and Ensembl fish protein set with Blastp (e value of 10−5). Homologue searches found that 21,365 of pompano genes had homologues in at least one database (Table 5). The KEGG biological pathways and Gene Ontology terms of each gene were annotated using the KEGG Automatic Annotation Server45 and Blast2GO46, respectively. Among the identified protein-coding genes, 20,594 genes were annotated to have at least one Gene Ontology (GO) term, and 7,956 genes were mapped to KEGG pathways. Finally, 21,365 genes (97.5%) were assigned to at least one of five databases (Table 5).
Table 5 Annotation of pompano genes to different databases.
Quality assessment of genome assembly and gene annotation
The quality of the assembly was evaluated using multiple indicators. (1) To estimate the quality value (QV) of the assembly, the cleaned reads from two paired-end libraries were mapped to the assembly with BWA47 and then the pipeup file produced by SAMtools48 were input to Referee49 to calculate a quality score for every position. Referee provided a higher scoring base to an erroneous position and this reference base was considered to be an error. We estimated that this genome had one error per 1000 base pairs with a quality value of 30. (2) We validated the assembly by comparing the cleaned read spectrum from two paired-end libraries with the copy number in the assembly using KAT toolkit50. The k-mer showed the homozygous distribution without a heterozygous peak (Fig. 4), consistent with the low heterozygosity observed by GenomeScope (Fig. 2). The main content occurred once, suggesting that the homozygous regions were not expanded. Furthermore, the absent k-mers (black) at the frequency of average sampling depth was low (Fig. 4), suggesting a high level of assembly completeness. The assembly correctly represented kmer spectrum from the cleaned Illumina reads. (3) We aligned Pacbio long reads to the repeat-masked assembly using Minimap251 and retained those alignments having read coverages over 90%. Almost 98.9% of long reads were uniquely aligned, suggesting that few homozygous contents were duplicated (Fig. 5). The cleaned Illumina reads were aligned to the repeat-masked assembly using BWA47. With the coverage threshold of 90%, over 96.3% of reads were uniquely aligned, also supporting few duplicated homozygous contents (Fig. 5). These two distributions were consistent with the main unique content in the KAT analysis. (4) The insert size distributions of paired-end/mate-pair libraries by aligning reads to the genome using BWA47 were consistent with the estimated insert sizes (Fig. 6). (5) The clean RNA-seq reads from multiple tissues had an average alignment ratio of 90.5% to the assembly using HISAT242 (Table 6). All the indicators suggested a high-quality genomic resource for the further analysis. The indistinguishable sex chromosome is one characteristic of this family. This chromosome-level assembly would provide a reference to identify sex chromosome and study the evolution of sex chromosome.
Fig. 4
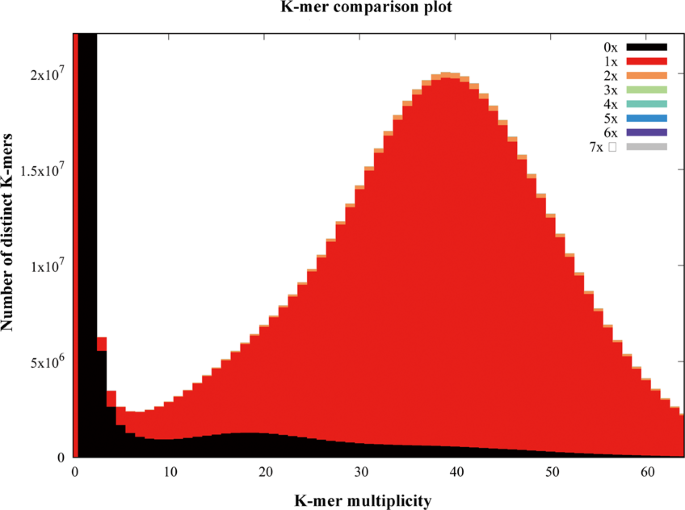
K-mer spectra copy number plot. Different color on the stacked bars represents copy number on the assembly. Frequency counts (spectral distribution) are computed on the Illumina paired-end reads.
Fig. 5
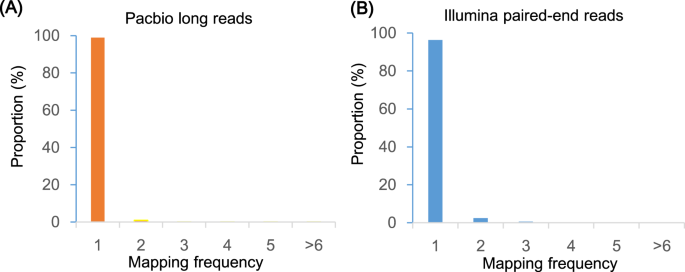
Alignment frequency distribution of Pacbio long reads and Illumina short reads.
Fig. 6
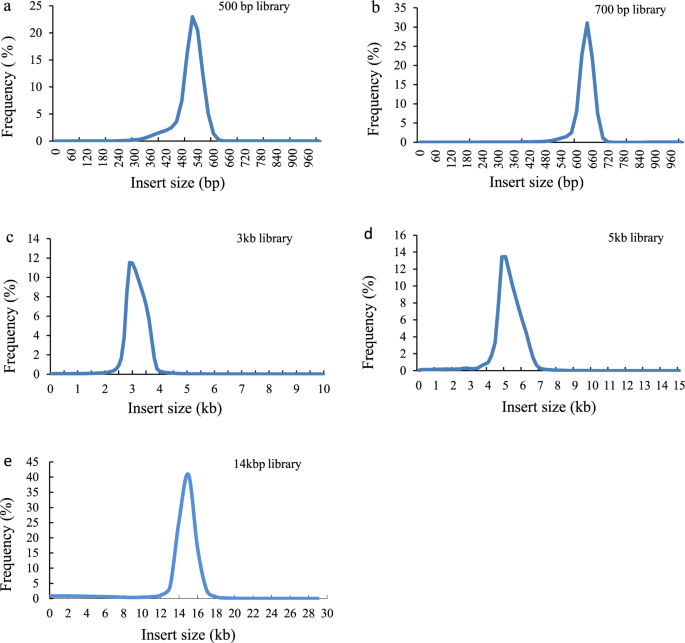
Distribution of insert sizes of sequencing reads in five libraries.
Table 6 Mapping ratio of RNA-seq reads from eight tissues.
The completeness of pompano genes was evaluated by using BUSCO software52. The pompano genes were compared with the 2,586 BUSCO vertebrate conserved gene set. Comparing pompano genes with the vertebrate gene set revealed that 95.7% of the vertebrate genes were identified as complete. The ‘complete and single-copy BUSCOs’ genes accounted for 94.3% of the total genes, and the ‘complete and duplicated BUSCOs’ genes represented 1.4% (Table 7).
Table 7 BUSCO evaluation of the pompano genes compared with the vertebrate gene set.
Comparison of pompano genome with other Carangiformes genomes
We then compared the pompano genome with other four Carangiformes genomes, including three Carangidae genomes (Seriola quinqueradiata, Seriola dumerili, and Seriola rivoliana) and one Echeneidae genome (Echeneis naucrates) using Mashmap253 (mapping segment length = 500 Kb, and perc_identity = 75). The genomic sequences of three Carangidae fish showed synteny to pompano genome (Fig. 7a–c). We found that the 24 pseudo-chromosomes of Echeneis naucrates had clear one-to-one relationship to pompano pseudo-chromosomes (Fig. 7d), suggesting that these two genomes did not experience chromosome fission and fusion events. These results revealed that the pompano genome will contribute to the study of the genome evolution of the Carangidae family and the Carangiformes order.
Fig. 7
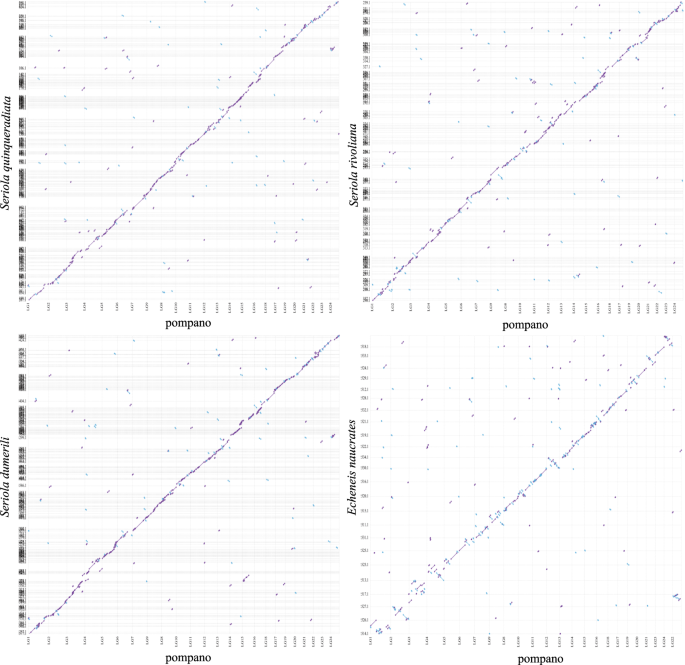
Whole genome plot of four Carangiformes genomes compared to pompano genome. Alignment dot plots show the genome comparisons between four Carangiformes assemblies (_y_-axis) and pompano assembly (_x_-axis). Dotted lines (vertical and horizontal, respectively) are the boundaries of chromosome and of scaffolds in the assemblies. (a) Plot between the assemblies of Seriola quinqueradiata and pompano. (b) Plot between Seriola rivoliana assembly and pompano assembly. (c) Plot between Seriola dumerili assembly and pompano assembly. (d) Plot between Echeneis naucrates assembly and pompano assembly.
Data Records
All sequencing data, genome assembly, predicted gene models and functional annotation were deposited in public repositories. The Illumina genomic sequencing reads, Pacbio long reads, Hi-C data, and RNA-seq reads of eight tissues were deposited in Sequence Read Archive at NCBI SRP136697[54](/articles/s41597-019-0238-8#ref-CR54 " NCBI Sequence Read Archive, https://identifiers.org/ncbi/insdc.sra:SRP136697
(2017)."). The chromosome-level assembly was available in the GenBank at NCBI UWUD01000000[55](/articles/s41597-019-0238-8#ref-CR55 "Guo, L. Trachinotus ovatus, whole genome shotgun sequencing project. GenBank,
https://identifiers.org/ncbi/insdc:UWUD01000000
(2018)."). The assembled contig, scaffolds, gene structure, homologs, and functional annotations were stored in Figshare[56](/articles/s41597-019-0238-8#ref-CR56 "Zhang., D.-C. Whole genome sequencing of female pompano (Trachinotus ovatus). figshare.
https://doi.org/10.6084/m9.figshare.7570727.v3
(2019).").Technical Validation
Three metrics, including peak length, total amount, and concentration were used to estimate the degradation level and quality of DNA samples. To construct Illumina libraries, the peak length of the isolated DNA was ≥20 kb and total DNA ≥5 μg with minimum 50 ng/μL. For PacBio libraries, the peak length was ≥40 kb and total DNA ≥7 μg with minimum 70 ng/μL. To construct the RNA-seq library of each tissue, the RNA integrity was ≥7.0 and total RNA ≥10 μg with rRNA ratio ≥1.5.
Code availability
Canu in the genome assembly and BLAT alignment in the gene prediction were utilized with specific parameters, described in Methods. The other bioinformatics tools were run with the default parameters. There were no any custom specific codes.
References
- FishBase consortium, https://www.fishbase.in/Summary/SpeciesSummary.php?ID=1773&AT=pompano (2019).
- Zhenzhen, X. et al. Transcriptome analysis of the Trachinotus ovatus: identification of reproduction, growth and immune-related genes and microsatellite markers. PLoS One 9, e109419 (2014).
Article ADS CAS PubMed PubMed Central Google Scholar - Tu, Z., Li, H., Zhang, X., Sun, Y. & Zhou, Y. Complete genome sequence and comparative genomics of the golden pompano (Trachinotus ovatus) pathogen, Vibrio harveyi strain QT520. PeerJ 5, e4127 (2017).
Article CAS PubMed PubMed Central Google Scholar - Sun, L., Zhang, D., Jiang, S., Guo, H. & Zhu, C. Isolation and characterization of 21 polymorphic microstatellites in golden pompano Trachinotus ovatus. Conservation Genetics Resources 5, 1107–1109 (2013).
Article Google Scholar - Zhu, K.-C. et al. Identification of Fatty Acid Desaturase 6 in Golden Pompano Trachinotus Ovatus (Linnaeus 1758) and Its Regulation by the PPARαb Transcription Factor. International journal of molecular sciences 20, 23 (2018).
- Zhou, C. et al. The Effects of dietary soybean isoflavones on growth, innate immune responses, hepatic antioxidant abilities and disease resistance of juvenile golden pompano Trachinotus ovatus. Fish & Shellfish Immunology 43, 158–166 (2015).
Article CAS Google Scholar - Chai, X., Li, X., Lu, R. & Clarke, S. Karyotype analysis of the yellowtail kingfish Seriola lalandi lalandi (Perciformes: Carangidae) from South Australia. Aquaculture Research 40, 1735–1741 (2009).
Article Google Scholar - Chen, S. et al. Whole-genome sequence of a flatfish provides insights into ZW sex chromosome evolution and adaptation to a benthic lifestyle. Nat Genet 46, 253–260 (2014).
Article CAS PubMed Google Scholar - Jory, D. E., Iversen, E. S. & Lewis, R. H. Culture of fishes of the genus Trachinotus (Carangidae) in the western Atlantic: prospects and problems. Journal of the World Mariculture Society 16, 87–94 (1985).
Article Google Scholar - Wu, M. et al. Genomic structure and molecular characterization of Toll-like receptors 1 and 2 from golden pompano Trachinotus ovatus (Linnaeus, 1758) and their expression response to three types of pathogen-associated molecular patterns. Dev Comp Immunol 86, 34–40 (2018).
Article CAS PubMed Google Scholar - Zhu, K. et al. Genomic structure, expression pattern and polymorphisms of GILT in golden pompano Trachinotus ovatus (Linnaeus 1758). Gene 665, 18–25 (2018).
Article CAS PubMed Google Scholar - Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92 (2017).
Article ADS CAS PubMed PubMed Central Google Scholar - Shao, C. et al. Chromosome-level genome assembly of the spotted sea bass, Lateolabrax maculatus. GigaScience 7,11 (2018).
- Rao, S. S. P. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Article CAS PubMed PubMed Central Google Scholar - Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Article CAS PubMed PubMed Central Google Scholar - Cox, M. P., Peterson, D. A. & Biggs, P. J. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC bioinformatics 11, 485 (2010).
Article PubMed PubMed Central Google Scholar - Leggett, R. M., Clavijo, B. J., Clissold, L., Clark, M. D. & Caccamo, M. NextClip: an analysis and read preparation tool for Nextera Long Mate Pair libraries. Bioinformatics 30, 566–568 (2014).
Article CAS PubMed Google Scholar - Hackl, T., Hedrich, R., Schultz, J. & Förster, F. proovread: large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics 30, 3004–3011 (2014).
Article CAS PubMed PubMed Central Google Scholar - Marçais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Article CAS PubMed PubMed Central Google Scholar - Xia, Q. et al. The genome assembly of asparagus bean, Vigna unguiculata ssp. sesquipedialis. Scientific Data 6, 124 (2019).
Article CAS PubMed PubMed Central Google Scholar - Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Article CAS PubMed PubMed Central Google Scholar - Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27, 722–736 (2017).
Article CAS PubMed PubMed Central Google Scholar - Gao, S., Bertrand, D., Chia, B. K. H. & Nagarajan, N. OPERA-LG: efficient and exact scaffolding of large, repeat-rich eukaryotic genomes with performance guarantees. Genome biology 17, 102 (2016).
Article CAS PubMed PubMed Central Google Scholar - Kajitani, R. et al. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res 24, 1384–1395 (2014).
Article CAS PubMed PubMed Central Google Scholar - Xu, G.-C. et al. LR_Gapcloser: a tiling path-based gap closer that uses long reads to complete genome assembly. Giga Science, 8, y157 (2018).
- Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS one 9, e112963–e112963 (2014).
Article ADS CAS PubMed PubMed Central Google Scholar - Wingett, S. et al. HiCUP: pipeline for mapping and processing Hi-C data. F1000 Research 4, 1310–1310 (2015).
Article CAS PubMed PubMed Central Google Scholar - Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods 9, 357 (2012).
Article CAS PubMed PubMed Central Google Scholar - Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nature biotechnology 31, 1119 (2013).
Article CAS PubMed PubMed Central Google Scholar - Zhang, G. et al. Construction of high-density genetic linkage maps and QTL mapping in the golden pompano. Aquaculture 482, 90–95 (2018).
Article Google Scholar - Hu, S., Min-lian, H. E., Hai-fa, Z., Yun-xin, W. & Yong-zhong, L. Study on the karyotyre in the Trachinotus ovatus. Journal of GuangZhou university (natural science edition) 6, 23–25 (2007).
ADS Google Scholar - Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biology 16, 259 (2015).
Article CAS PubMed PubMed Central Google Scholar - Akdemir, K. C. & Chin, L. HiCPlotter integrates genomic data with interaction matrices. Genome biology 16, 198 (2015).
Article CAS PubMed PubMed Central Google Scholar - Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11 (2015).
Article PubMed PubMed Central Google Scholar - Star, B. et al. The genome sequence of Atlantic cod reveals a unique immune system. Nature 477, 207–210 (2011).
Article ADS CAS PubMed PubMed Central Google Scholar - Vij, S. et al. Chromosomal-Level Assembly of the Asian Seabass Genome Using Long Sequence Reads and Multi-layered Scaffolding. PLoS Genet 12, e1005954 (2016).
Article CAS PubMed PubMed Central Google Scholar - Wu, C. et al. The draft genome of the large yellow croaker reveals well-developed innate immunity. Nature Communications 5, 5227 (2014).
Article ADS PubMed Google Scholar - Salamov, A. A. & Solovyev, V. V. Ab initio gene finding in Drosophila genomic DNA. Genome research 10, 516–522 (2000).
Article CAS PubMed PubMed Central Google Scholar - Aken, B. L. et al. Ensembl 2017. Nucleic Acids Res 45, D635–D642 (2017).
Article CAS PubMed Google Scholar - Kent, W. J. BLAT–the BLAST-like alignment tool. Genome Res 12, 656–664 (2002).
Article CAS PubMed PubMed Central Google Scholar - Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res 14, 988–995 (2004).
Article CAS PubMed PubMed Central Google Scholar - Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nature methods 12, 357–360 (2015).
Article CAS PubMed PubMed Central Google Scholar - Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols 7, 562–578 (2012).
Article CAS PubMed PubMed Central Google Scholar - The UniProt, C. UniProt: a hub for protein information. Nucleic Acids Research 43, D204–D212 (2015).
Article CAS Google Scholar - Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C. & Kanehisa, M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic acids research 35, W182–W185 (2007).
Article PubMed PubMed Central Google Scholar - Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Article CAS PubMed Google Scholar - Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Article CAS PubMed PubMed Central Google Scholar - Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Article CAS PubMed PubMed Central Google Scholar - Thomas, G. W. C. & Hahn, M. W. Referee: Reference Assembly Quality Scores. Genome Biol Evol 11, 1483–1486 (2019).
Article PubMed PubMed Central Google Scholar - Mapleson, D., Garcia Accinelli, G., Kettleborough, G., Wright, J. & Clavijo, B. J. KAT: a K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics 33, 574–576 (2016).
PubMed Central Google Scholar - Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Article CAS PubMed PubMed Central Google Scholar - Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Article CAS PubMed Google Scholar - Jain, C., Koren, S., Dilthey, A., Phillippy, A. M. & Aluru, S. A fast adaptive algorithm for computing whole-genome homology maps. Bioinformatics 34, i748–i756 (2018).
Article CAS PubMed PubMed Central Google Scholar - NCBI Sequence Read Archive, https://identifiers.org/ncbi/insdc.sra:SRP136697 (2017).
- Guo, L. Trachinotus ovatus, whole genome shotgun sequencing project. GenBank, https://identifiers.org/ncbi/insdc:UWUD01000000 (2018).
- Zhang., D.-C. Whole genome sequencing of female pompano (Trachinotus ovatus). figshare. https://doi.org/10.6084/m9.figshare.7570727.v3 (2019).
- Ozaki, A. & Araki, K. Seriola quinqueradiata isolate Squ1, whole genome shotgun sequencing project. GenBank, https://identifiers.org/ncbi/insdc:BDMU00000000 (2017).
- Araki, K., Ozaki, A., Aoki, J. & Kawase, J. Seriola dumerili isolate Sdu_G_001, whole genome shotgun sequencing project. GenBank, https://identifiers.org/ncbi/insdc:BDQW01000000 (2018).
- Purcell, C. M. et al. Seriola lalandi dorsalis isolate HSWRI2012SDOR001, whole genome shotgun sequencing project. GenBank, https://identifiers.org/ncbi/insdc:PEQF00000000 (2017).
- Seetharam, A. S. et al. Seriola rivoliana isolate HWSR04, whole genome shotgun sequencing project. GenBank, https://identifiers.org/ncbi/insdc:PVUN00000000 (2018).
- Megens, H.-J. et al. Seriola lalandi isolate YTK006, whole genome shotgun sequencing project. GenBank, https://identifiers.org/ncbi/insdc:MAII01000000 (2018).
- Sharing, W. S. I. D. Echeneis naucrates, whole genome shotgun sequencing project. GenBank, https://identifiers.org/ncbi/insdc:CAAHFO010000000 (2019).
Acknowledgements
This work was supported by grants from National Key Research and Development Program of China (2018YFD0900301 and 2018YFD0900102), China-ASEAN Maritime Cooperation Fund (00-201620821), China Agriculture Research System (CARS-47), National Science & Technology Infrastructure platform (2019DKA30470), and Financial Fund of Ministry of Agriculture and Rural affairs of China (NFZX2013).
Author information
Author notes
- These authors contributed equally: Dian-Chang Zhang, Liang Guo, Hua-Yang Guo and Ke-Cheng Zhu.
Authors and Affiliations
- Key Laboratory of South China Sea Fishery Resources Exploitation & Utilization, Ministry of Agriculture and Rural Affairs, South China Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, 231 Xingang Road West, Haizhu District, Guangzhou, 510300, China
Dian-Chang Zhang, Liang Guo, Hua-Yang Guo, Ke-Cheng Zhu, Nan Zhang, Bao-Suo Liu & Shi-Gui Jiang - Key Laboratory of Aquatic Genomics, Ministry of Agriculture and Rural Affairs, CAFS Key Laboratory of Aquatic Genomics and Beijing Key Laboratory of Fishery Biotechnology, Chinese Academy of Fishery Sciences, Beijing, 100141, China
Shang-Qi Li, Yan Zhang & Jiong-Tang Li
Authors
- Dian-Chang Zhang
- Liang Guo
- Hua-Yang Guo
- Ke-Cheng Zhu
- Shang-Qi Li
- Yan Zhang
- Nan Zhang
- Bao-Suo Liu
- Shi-Gui Jiang
- Jiong-Tang Li
Contributions
Zhang D.C., Li J.T. and Jiang S.G. conceived the project. Li J.T., Zhang D.C., Guo H.Y. and Zhu K.C., Xiao J., Li S.Q. and Zhang Y. participated in genome assembly and data analysis. Zhang N., Liu B.S. and Guo L. extracted the genomic DNA and performed genome sequencing. Li J.T., Zhang D.C., Jiang S.G., Guo H.Y. and Zhu K.C. prepared the manuscript.
Corresponding authors
Correspondence toDian-Chang Zhang or Jiong-Tang Li.
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files associated with this article.
About this article
Cite this article
Zhang, DC., Guo, L., Guo, HY. et al. Chromosome-level genome assembly of golden pompano (Trachinotus ovatus) in the family Carangidae.Sci Data 6, 216 (2019). https://doi.org/10.1038/s41597-019-0238-8
- Received: 04 April 2019
- Accepted: 11 September 2019
- Published: 22 October 2019
- DOI: https://doi.org/10.1038/s41597-019-0238-8