Oligomeric Aβ-induced synaptic dysfunction in Alzheimer’s disease (original) (raw)
Abstract
Alzheimer’s disease (AD) is a devastating disease characterized by synaptic and neuronal loss in the elderly. Compelling evidence suggests that soluble amyloid-β peptide (Aβ) oligomers induce synaptic loss in AD. Aβ-induced synaptic dysfunction is dependent on overstimulation of _N_-methyl-D-aspartate receptors (NMDARs) resulting in aberrant activation of redox-mediated events as well as elevation of cytoplasmic Ca2+, which in turn triggers downstream pathways involving phospho-tau (p-tau), caspases, Cdk5/dynamin-related protein 1 (Drp1), calcineurin/PP2B, PP2A, Gsk-3β, Fyn, cofilin, and CaMKII and causes endocytosis of AMPA receptors (AMPARs) as well as NMDARs. Dysfunction in these pathways leads to mitochondrial dysfunction, bioenergetic compromise and consequent synaptic dysfunction and loss, impaired long-term potentiation (LTP), and cognitive decline. Evidence also suggests that Aβ may, at least in part, mediate these events by causing an aberrant rise in extrasynaptic glutamate levels by inhibiting glutamate uptake or triggering glutamate release from glial cells. Consequent extrasynaptic NMDAR (eNMDAR) overstimulation then results in synaptic dysfunction via the aforementioned pathways. Consistent with this model of Aβ-induced synaptic loss, Aβ synaptic toxicity can be partially ameliorated by the NMDAR antagonists (such as memantine and NitroMemantine). PSD-95, an important scaffolding protein that regulates synaptic distribution and activity of both NMDA and AMPA receptors, is also functionally disrupted by Aβ. PSD-95 dysregulation is likely an important intermediate step in the pathological cascade of events caused by Aβ. In summary, Aβ-induced synaptic dysfunction is a complicated process involving multiple pathways, components and biological events, and their underlying mechanisms, albeit as yet incompletely understood, may offer hope for new therapeutic avenues.
Keywords: Alzheimer’s disease, Synaptic loss, Aβ oligomers, Cognitive decline, Calcium, NMDA receptors, PSD-95, Mitochondrial dysfunction, Tau hyperphosphorylation, Aberrant neuronal network activity
Introduction
Alzheimer’s disease (AD) is the most common form of dementia among the elderly. It is clinically characterized by progressive memory loss and cognitive dysfunction, with the eventual inability to perform activities of daily living (ADLs). According to the Alzheimer’s Association (http://www.alz.org), 5.4 million Americans are currently living with Alzheimer’s disease, and one in eight older Americans will develop AD. While drugs are available to temporarily improve memory and cognitive function or delay the progress of dementia, AD remains a devastating neurodegenerative disorder without any effective cure or disease-modifying therapy [1, 2]. Following diagnosis, AD patients generally survive from several years to 20 years, depending on age and other health conditions. Although AD is pathologically characterized by the presence of extracellular deposition of plaques comprised of Aβ peptide and neurofibrillary tangles (NFTs) comprised of hyperphosphorylated-tau protein, accumulating evidence suggests that these abnormal protein deposits are unlikely the causative events in AD as Aβ plaque or NFT volume poorly correlate with the severity of dementia. Instead, the degree of dementia in premortem patients correlates more closely with the level of soluble oligomers of Aβ species in postmortem brains, especially in hippocampal and cortex regions associated with learning and memory function [3–5]. Aβ peptides are proteolytic products of the amyloid precursor protein (APP) and are sequentially cleaved by β- and γ-secretases [6]. Although Aβ peptides of varying length are produced, Aβ1-42 is considered to be comparatively more amyloidogenic and readily assembles into soluble oligomers and consequent fibril deposits. Aβ oligomers, also termed as Aβ-derived diffusible ligands (ADDLs), are thought to induce synaptic loss and progressive cognitive decline in AD, whereas monomers and fibrillary aggregates may be more inconsequential to pathogenesis [7]. In addition, synaptic protein depletion [8–10] and synaptic loss [11, 12] are also found in the same regions in human AD or mild cognitive impairment (MCI) postmortem brains, and the level of synaptic reduction closely parallels the degree of premortem cognitive impairment. These studies suggest that oligomeric Aβ elevation and synaptic loss, rather than Aβ plaque load, may represent the best indicators of the severity of dementia or cognitive impairment in AD. Moreover, these findings imply that rescue of synapses could prove to be disease modifying in AD.
Using transgenic animal models of AD, additional studies suggest that synaptic loss is induced by pathological Aβ elevation [13]. Although the molecular mechanism is still not fully understood, it is generally believed that Aβ oligomers at pathological concentrations trigger (most likely via an indirect pathway) overstimulation of extrasynaptic NMDA receptors (eNMDARs), leading to aberrant redox events and Ca2+ upregulation. Subsequent activation of downstream signal transduction pathways trigger a cascade of pathological events leading to synaptic disruption and neuronal loss. These include increased oxidative/nitrosative stress and mitochondrial dysfunction with consequent bioenergetic compromise, leading to dysregulation of synaptic neurotransmission and abnormal neuronal network activity [7, 14]. A number of synaptic proteins have been proposed as potential Aβ-binding partners under pathological conditions and their interactions are believed to mediate Aβ-induced synaptic dysfunction. These proteins include, but are not limited to, α7-nicotinic acetylcholine receptors (α7nAChRs) [15], NMDARs [16, 17], mGluR5 [18], neurotrophin receptor p75NTR[19], cellular prion protein (PrPC) [20], PSD-95 [21], glutamate transporter [22], ephrin type-B receptor 2 (EphB2) [23], and ephrin type-A receptor 4 (EphA4) [24, 25]. The involvement of some of these proteins in Aβ-mediated neurotoxicity will be discussed in the following sections.
Oligomeric Aβ induces synaptic dysfunction in AD mice
AD or amnestic MCI patients usually have trouble with spatial orientation in their daily routine and perform poorly in clinically-designed, hippocampus-dependent memory and navigation tests [26–28]. Studies using animal models of AD have suggested that soluble oligomeric Aβ species are critical in initiating a pathogenic cascade leading to synaptic dysfunction, neuronal loss, and AD-like cognitive impairment [7]. The association between amyloidogenic Aβ species and age-dependent memory loss was first described in Tg2576 mice expressing a human APP695 transgene containing the Swedish mutation (K670N/M671L) [29]. These mice show elevated brain Aβ1-42/43 levels by ELISA and perform poorly in Morris water maze tests of spatial memory [29, 30]. The association between Aβ and cognitive impairment has also been documented in other transgenic (Tg) animal models of AD including 3XTg-AD mice, which express three human mutant AD gene variants (PS1M146V, APPSwe, and tauP301L) and develop progressive plaques and tangles [31, 32] and the human amyloid precursor protein-overexpressing (hAPP) J20 mouse model [33]. Synaptic dysfunction was also observed in these AD transgenic mice with aberrantly elevated levels of oligomeric Aβ and deficits in learning and memory (see selected reviews: [34–36]). Moreover, these studies showed that synaptic and cognitive impairments are associated with the elevation of soluble oligomeric Aβ species and usually evident prior to the appearance of plaques and tangles in the relevant brain regions.
NMDAR-dependent long-term potentiation (LTP) in hippocampus has attracted broad attention as an electrophysiological measurement of synaptic strength and plasticity in various AD models. LTP induction requires activation of NMDARs, which triggers a signaling cascade that induces the recruitment of AMPARs into the postsynaptic membrane [37]. On the other hand, NMDAR-dependent long-term depression (LTD) is an activity-dependent reduction in the efficacy of neuronal synapses, which is mediated, at least in part, by AMPAR endocytosis [37]. Multiple studies have shown that LTP is impaired in AD or Aβ-exposed wild-type hippocampus, and this impairment is dependent on NMDARs and downstream pathways [38–44]. In contrast to LTP, Aβ application enhances LTD [22, 45, 46], consistent with the notion that Aβ causes synaptic depression.
Emerging evidence suggests that Aβ-induced synaptic dysfunction is dependent on NMDAR-mediated activity and occurs via aberrant redox events as well as elevation of cytoplasmic Ca2+ and activation of downstream pathways involving Ca2+-dependent protein phosphatase calcineurin/PP2B and protein phosphatase 2A (PP2A) (Figure 1) [47–49]. Concerning the Ca2+-dependent events, upon activation, calcineurin further activates or inactivates its target proteins via dephosphorylation. For example, dephosphorylation and activation of the actin filament severing protein cofilin by calcineurin result in dendritic spine loss, which can be rescued by overexpression of the inactive cofilin phosphomimetic S3D [48]. Interestingly, a recent study reported that Aβ oligomers interact with murine PirB (paired immunoglobulin-like receptor B) and its human ortholog LilrB2 (leukocyte immunoglobulin-like receptor B2) with nanomolar affinity. This interaction enhanced cofilin signaling and contributed to memory loss in AD transgenic mice [50]. Aβ-induced synaptic degeneration also involves surface removal and endocytosis of AMPARs [45]. In support of this, surface AMPARs are downregulated through endocytosis in wild-type neurons rapidly upon Aβ-application [51] and in AD transgenic mice [52]. Concurrently, AMPAR-mediated synaptic currents are also downregulated in AD double knock-in (mutant APP and PS1) transgenic mice [53]. Aβ-induced AMPAR endocytosis or surface removal is dependent on the activation of calcineurin/PP2B [54] and requires downregulation of Ca2+/calmodulin-dependent protein kinase II (CaMKII) [55]. Similar to AMPARs, Aβ can induce surface removal or endocytosis of NMDARs, which is mediated by dephosphorylation of NR2B (GluN2B), an NMDAR subunit, at p-Tyr1472 by the tyrosine phosphatase STEP [56, 57]. Interestingly, the apoptotic effector component caspase-3 has also been suggested to play a role in synaptic plasticity and its activation is required in AMPAR removal and consequent LTD induction [58]. Activation of caspase-3 has been further shown to trigger early synaptic dysfunction in AD transgenic mice [59]. This is consistent with the observation that caspase-3 is enriched in postsynaptic densities [60]. In addition to ionic glutamate receptor NMDARs and AMPARs, metabotropic glutamate receptors (mGluRs) at extrasynaptic or perisynaptic sites have also been shown to play an important role in Aβ-induced synaptic dysfunction [46], likely activated by pathologically elevated glutamate at extrasynaptic sites [22].
Figure 1.
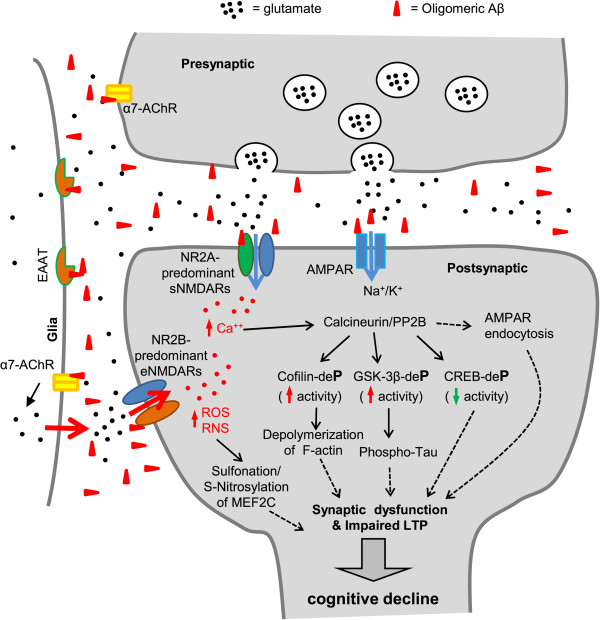
Schematic diagram outlining mechanisms of oligomeric Aβ-induced synaptic dysfunction. At pathological concentrations, Aβ oligomers may interact with multiple astrocytic, microglial, and neuronal synaptic proteins, including α7-AChRs and NMDARs, triggering a series of toxic synaptic events. These events include aberrant activation of NMDARs (especially NR2B-containing extrasynaptic NMDARs), elevated neuronal calcium influx, calcium-dependent activation of calcineurin/PP2B and its downstream signal transduction pathways, involving cofilin, GSK-3β, CREB, and MEF2. This results in aberrant redox reactions and severing/depolymerizing F-actin, tau-hyperphosphorylation, endocytosis of AMPARs, and eventually leads to synaptic dysfunction and cognitive impairment.
It has also been reported that pathological concentrations of Aβ oligomers disrupt glutamate uptake, thus increasing glutamate levels to enhance LTD [22] or impair LTP [61]. Consistent with these results, the levels of both glutamate transporters EAAT1 and EAAT2, which are responsible for the majority of glutamate uptake in glial cells, are downregulated in the hippocampus of AD patients [62]. Due to impaired glutamate uptake, glutamate spills out of synapses and accumulates in the extrasynaptic spaces, thereby inducing overactivation of NR2B-predominant eNMDARs [61]. In addition to attenuated glutamate uptake, oligomeric Aβ increases glutamate levels by triggering aberrant astrocyte glutamate release and accumulation within the extrasynaptic space thereby overactivating eNMDARs, which results in synaptic damage via a variety of aberrant transcriptional cascades and redox-mediated posttranslational modifications [63–69]. These eNMDAR-mediated pathways include hyperactivation of neuronal nitric oxide synthase (nNOS) and Ca2+-overload of mitochondria, generating excessive reactive nitrogen and oxygen species (RNS/ROS). This excess results in aberrant oxidation reactions, such as S-nitrosylation and sulfonation, on various proteins, thereby disrupting their normal activity. One example is illustrated by excessive S-nitrosylation or sulfonation of the transcription factor MEF2C, thereby adversely affecting downstream effectors of neuronal survival and adult neurogenesis [68, 70]. Importantly, memantine, an FDA-approved, uncompetitive NMDAR open-channel blocker, at proper concentration, preferentially blocks pathologically-stimulated eNMDARs over physiologically-activated synaptic NMDARs (sNMDARs) [71]. Indeed, application of memantine, and to a greater degree its improved derivative NitroMemantine [72], has been shown to mitigate Aβ-induced synaptic dysfunction and cognitive deficits [63, 73]. Taken together, these studies depict an alternative mechanism underlying Aβ-induced synaptic dysfunction based on glutamate-mediated hyperexcitability and synaptic excitotoxicity: Aβ-induced accumulation of excessive glutamate in the extrasynaptic space can occur through disruption of the glutamate uptake system or by triggering astrocytic glutamate release, which in turn aberrantly activates eNMDARs and induces synaptic dysfunction (Figure 1).
NMDAR and AMPAR function at postsynaptic sites are influenced by interaction with PSD-95, an important postsynaptic scaffolding protein that plays a critical role in protein assembly, synaptic development and neural plasticity [74, 75]. Under pathological conditions however, synaptic PSD-95 levels are decreased in AD postmortem brains, and the degree of reduction correlates with both the level of Aβ oligomers and the severity of dementia [21, 76–78]. Similarly, PSD-95 is also reduced in AD transgenic mouse neurons or neurons exposed to Aβ oligomers, concomitant with dendritic spine loss and surface AMPAR removal [51, 52, 79]. These results suggest that synaptic disruption of PSD-95 may play a role in the pathogenesis of AD. Consistent with this idea, it has been shown by co-immunoprecipitation that Aβ interacts with PSD-95 and is co-localized with PSD-95 specifically at excitatory synapses in human postmortem AD brain as well as in cultured murine neurons exposed to Aβ oligomers [21, 80]. Therefore, it is possible that Aβ may interact with PSD-95 directly to cause synaptic damage under pathological conditions. Interestingly, overexpression of α1-takusan, a PSD-95-binding and synaptic-stabilizing protein [81], decreases Aβ-induced synaptic damage, protecting from dendritic spine loss, decreased synaptic expression of PSD-95, and downregulated AMPAR-mediated synaptic currents in cultured neurons [82]. Further evidence has shown that increased PSD-95 expression in hippocampus after treatment with the Hsp90 inhibitor 17-AAG can improve cognitive function in an animal model of AD, apparently via synaptic enhancement [83]. Therefore, molecules that modulate the integrity of PSD-95 and improve synaptic functions may have therapeutic potential for reducing Aβ-induced synaptic injury and cognitive impairment in AD.
A number of other synaptic proteins have been proposed to be putative receptors for Aβ oligomers, and play important roles in oligomeric Aβ-induced synaptic dysfunction and cognitive impairment. For example, soluble Aβ oligomers can bind with nanomolar affinity to PrPC, and PrP knockout or anti-PrP antibodies can rescue oligomeric Aβ-induced synaptic dysfunction and spatial memory. These results suggest that prion proteins play an important role in AD pathogenesis [20]. The α7nAChR modulates calcium homeostasis and release of the neurotransmitter glutamate, two important parameters involved in learning and memory. Endogenous Aβ species have been reported to bind to α7nAChRs with nanomolar affinity in co-immunoprecipitation experiments on human AD postmortem brain extracts [15]. This interaction consequently triggers α7nAChR-dependent NMDAR endocytosis leading to synaptic and cognitive dysfunction [15, 56]. The ephrin family of receptor tyrosine kinases has also been found to be a potential receptor that interacts with Aβ oligomers under pathological conditions. It has been shown that Aβ oligomer binding to EphB2 induces its degradation, leading to impairments in NMDAR-mediated synaptic activity and cognitive function [23]. Conversely, EphB2 overexpression reverses deficits in NMDAR-dependent LTP and cognitive impairments in AD Tg mice [23]. Recently, ephrin A4 (EphA4), another ephrin receptor family member, was also identified as a putative Aβ receptor [24, 25]. In contrast to EphB2 degradation, interaction with Aβ activates EphA4, leading to suppression of LTP and spine loss in AD transgenic mice. Application of either EphA4 shRNA or EphA4 inhibitors/antagonists has been reported to rescue these deficits, suggesting that EphA4 activation plays a critical role in Aβ-induced synaptic dysfunction [24, 25].
Alternative pathways contribute to Aβ-induced synaptic and neuronal loss in AD
Aβ-induced synaptic dysfunction can also be mediated by molecules and their downstream pathways that are not directly associated with NMDA receptor-mediated activities. It has been shown that Aβ can bind to the low-affinity p75 neurotrophin receptor (p75NTR) and activate its death domain to induce apoptosis [19]. Moreover, surface expression level of p75 is upregulated in SH-SY5Y neuroblastoma cells after exposure to Aβ oligomers and in hippocampal neurons in AD transgenic mice [84]. Consistent with these animal studies, the level of membrane-associated p75 in hippocampus was significantly higher in human postmortem AD brains compared to age-matched controls [85]. Interestingly, APP cleavage is differentially regulated by the neurotrophin high-affinity receptor TrkA and the low-affinity receptor p75NTR; p75NTR promotes whereas TrkA decreases APP β-cleavage [86]. Therefore, aberrantly upregulated p75NTR together with TrkA downregulation in aged brains results in increased Aβ generation [87]. In addition, p75NTR may also enhance Aβ production via its ability to stabilize BACE1 or β-secretase through the activation of sphingomyelinase and consequent ceramide production [87, 88].
In addition to neurotrophin receptors, insulin and insulin-like growth factor receptors and their cognate signaling pathways play a critical role in synaptic plasticity and cognitive function by affecting both excitatory and inhibitory synaptic activity [89, 90]. Growing evidence suggests that AD may represent a metabolic disease of the brain associated with brain insulin and insulin-like growth factor-I (IGF-I) resistance and deficiency. Impaired insulin signaling may contribute to dysregulation of downstream pro-survival pathways, including decreased signaling mediated by PI3K, Akt, and Wnt/β-catenin; moreover, disrupted insulin-related signaling may enhance pathogenic pathways such as GSK-3β to trigger tau hyperphosphorylation [91, 92]. Therefore, disrupted components of brain insulin signaling pathways may represent potential therapeutic targets in AD [91, 92]. It has been suggested that both extracellular and intracellular Aβ oligomers contribute to neuronal dysfunction. Intracellular Aβ inhibits insulin receptor signaling by interfering with the interaction between phosphoinositide-dependent kinase (PDK) and Akt, thus inhibiting Akt activation and abolishing insulin-mediated neuroprotection [93]. Extracellular Aβ oligomers or ADDLs bound to synaptic sites can induce removal of surface insulin receptors and contribute to synaptic loss, which can be rescued by insulin treatment [94]. Encouraging results obtained from intranasal insulin therapy in aged adults [95] as well as in AD and MCI patients [96] support the idea that insulin signaling is disrupted during normal aging and in clinical cases of dementia.
Furthermore, Wnt family members promote synaptic formation and regulate synaptic function by binding to receptors of the Frizzled (Fz) and low-density lipoprotein-related protein (LRP) families on the cell surface to activate either β-catenin-dependent canonical signaling (Wnt/β-catenin) or β-catenin-independent non-canonical signaling pathways; the latter pathways include the Wnt/PCP and Wnt/Ca2+ cascades [97]. Multiple Wnt signaling components are dysregulated in AD and such impairments are likely to contribute to synaptic dysfunction and cognitive decline in AD. Wnt co-receptor LRP6 variants suppress Wnt signaling activity and are associated with late-onset AD [98]. An endogenous Wnt inhibitor, Dickkopf-1 (Dkk1), which disrupts Wnt-induced Fz/LRP complex formation [99], is increased in human postmortem AD brains [100] and mouse model AD brains [101], where Dkk1 has been found to co-localize with active GSK-3 and phospho-tau. Additional studies suggest that Dkk1 levels increase after Aβ oligomer exposure [102]. Dkk1 upregulation appears to be required for Aβ-induced synaptic loss since synaptic damage as well as tau phosphorylation are abolished by either Dkk1 knockdown [100] or Dkk1 blocking antibodies [102]. Interestingly, it has been shown that Aβ may directly interact with Fz receptors of Wnt ligands [103] although the significance of this interaction remains to be determined. Taken together, these studies suggest that dysregulated Wnt signaling contributes to Aβ-induced synaptic loss in AD, which raises the possibility that Wnt signaling components may represent potential therapeutic targets in AD.
Aberrant neuronal network activity and seizures in AD
Epileptic seizures were once considered to be rare or an epiphenomenon in AD. However, accumulating evidence suggests that increased seizure activity may be the consequence of a disrupted neuronal network, contributing to cognitive decline and the onset of dementia [104]. It has been estimated that 10 to 22% of AD patients experience at least one episode of an unprovoked seizure [105]. However, it is likely that these numbers are underestimates for the following reasons: (1) AD patients with dementia may not recall having seizures if they were unwitnessed, and (2) some types of seizure activity, including complex partial seizures, manifest symptoms such as confusion or delirium, which are similar to those normally seen in AD patients, and therefore may go undetected. Prior observational studies have shown that the occurrence of a first unprovoked seizure in patients 55 years or older is significantly greater in AD and other dementias compared to the general population [106]. The increased incidence can be as high as 87-fold in AD patients with early-onset FAD [107]. Consistent with this result, a more recent study found that FAD mutations in APP, PSEN1, and PSEN2 are all associated with higher risk for a first unprovoked seizure [108]. These results suggest a tight association between epileptic seizures and genetic mutations that cause aberrant expression of Aβ oligomers and induce early-onset AD.
Mechanistic insight into this abnormal excitatory phenomenon was obtained in studies using AD transgenic mice expressing human mutant APP and presenilin genes. Several early studies showed that AD transgenic mice expressing human Aβ fragments [109] or APP mutations [110, 111] exhibit increased spontaneous seizure activity, although these unprovoked seizure events are rare and often ignored. Detection of subtle seizure phenotypes (or nonconvulsive seizures) and aberrant neuronal network activities have been made possible through the use of video-electroencephalography (EEG) monitoring in AD transgenic mice expressing FAD mutations [112–114]. Acute application of levetiracetam (LEV or Keppra®), an antiepileptic drug, can effectively suppress abnormal EEG spike activity, and chronic treatment with LEV can even reverse AD-like phenotypes including synaptic dysfunction, hippocampal remodeling, and learning and memory deficits in human APP transgenic mice [115]. Therefore, antiepileptic drugs, such as LEV, that suppress the aberrant electrical activity that contributes to seizures may provide an alternative approach to the treatment of AD.
Aβ-induced tau hyperphosphorylation and its role in synaptic loss
The presence of hyperphosphorylated tau-enriched neurofibrillary tangles is one of the classical pathological hallmarks of AD. Tau is a microtubule-associated protein (MAP) that was originally identified as an important protein for microtubule (MT) assembly [116] and for stabilization of the MT network [117]. Under pathological conditions, tau becomes hyperphosphorylated and disassociated from microtubules, subsequently forming soluble aggregates, insoluble filaments, and eventually neurofibrillary tangles (NFTs) in affected brain regions. This pathology occurs not only in AD but also in several other neurological disorders, which are collectively termed tauopathies [118, 119]. Phosphorylated tau (p-tau) colocalizes with Aβ in synaptic terminals from both postmortem AD brain [120, 121] and transgenic mouse AD brain [122]. These prior studies have shown that expression of p-tau in synaptic terminals correlates with Aβ levels, and increased p-tau expression also correlates with a reduction in total synapse number. A causal association between oligomeric Aβ exposure and p-tau formation has been demonstrated in several studies. For example, Talantova et al. [63] reported that oligomeric Aβ caused astrocytic glutamate release, which in turn activated extrasynaptic NMDARs, resulting in increased p-tau levels. Additional studies have shown that Aβ-induced synaptic loss is tau-dependent since tau deletion or reduction can rescue Aβ-induced synaptic loss and cognitive impairment in AD transgenic mice [123–126]. Moreover, a recent study characterized interactions between oligomeric Aβ and p-tau in both human and animal AD brains by co-immunoprecipitation and immunohistology, and this interaction progressively increased with disease progression [127]. It is thus possible that pathological interactions between oligomeric Aβ and p-tau are important intermediate steps in Aβ-induced synaptic loss and neuronal damage. Although previously known as an axonal protein, tau is also expressed in dendrites and in the postsynaptic density, albeit at much lower levels [124]. Depletion of dendritic or postsynaptic tau prevents abnormal postsynaptic targeting of the tyrosine kinase Fyn and rescues impaired learning and memory function in AD transgenic mice, suggesting a vital role of dendritic and postsynaptic tau in Aβ-induced synaptic loss [124]. In cultured neurons, Aβ-induced tau hyperphosphorylation and dendritic disruption can be attenuated by anti-Aβ antibodies or tau reduction using RNAi-targeting strategies [128–130]. Therefore, it is possible that Aβ-induced tau hyperphosphorylation is an important intermediate event that leads to synaptic dysfunction. Indeed, only pseudohyperphosphorylated tau, which mimics hyperphosphorylated tau, but not phosphorylation-deficient tau, which mimics regular tau, is mislocalized and accumulated in dendritic spines [131]. Consequently, hyperphosphorylated tau that is mislocalized to dendritic spines has been reported to induce synaptic dysfunction by impairing AMPAR surface expression and synaptic transmission [131]. Consistent with this finding, tau deletion or inhibition of tau hyperphosphorylation using a glycogen synthase kinase 3β (GSK-3β) inhibitor can prevent Aβ-induced impairment of LTP [132]. GSK-3β-mediated tau phosphorylation and Aβ production can also be reduced by RPS23R1 protein via activation of the adenylate cyclase/cAMP/PKA pathway. In turn, this pathway leads to synaptic enhancement and improved AD pathology [133]. Tau-dependent synaptic dysfunction may also involve the tyrosine kinase Fyn [124]. Pseudohyperphosphorylated tau binds Fyn more tightly than wild-type tau, thus increasing Fyn activity and leading to synaptic damage [134]. Intriguingly, a study using human AD induced pluripotent stem cell (iPSC)-derived neurons has suggested that increased tau phosphorylation at Thr231 is mediated by β-secretase activity [135]. This finding raises the possibility that Aβ-induced tau pathology and synaptic damage can be mediated by an alternative, non-Aβ-mediated pathway. Thus, future in-depth studies will determine the role of dendritic/postsynaptic tau and its hyperphosphorylation in Aβ-induced synaptic loss.
Aβ-induced mitochondrial dysfunction and synaptic loss in AD
Under physiological conditions, mitochondria provide much of the energy that is required to maintain normal synaptic activity and plasticity [136]. However, under pathological conditions, mitochondrial impairment has been suggested to be an important early event contributing to synaptic loss and neurodegeneration in AD [137–139]. In human postmortem brains, Aβ has been reported to accumulate aberrantly in mitochondria with abnormal morphology, suggesting that Aβ may influence mitochondrial morphogenesis [140–143]. Wild-type neurons exposed in vitro to oligomeric Aβ or AD transgenic neurons in vivo exhibit excessive mitochondrial fission (mitochondrial fragmentation), a sign of impairment in mitochondrial dynamics, further implicating Aβ in mitochondrial dysregulation [144–147]. Therefore, it is possible that Aβ at pathological concentrations can trigger mitochondrial impairment, which in turn leads to bioenergetics compromise, synaptic starvation and damage in AD. Consistent with this hypothesis, mitochondrial impairment caused by oligomeric Aβ or toxic factors generated downstream of Aβ, such as RNS/NO, occurs prior to synaptic and neurite injury [148] and precedes AD pathology [149]. It has been suggested that abnormal interactions between Aβ and the mitochondrial fission protein dynamin-related protein 1 (Drp1) play an important role in mitochondrial dysfunction and synaptic damage in AD [150]. Additional evidence suggests that Aβ-induced mitochondrial impairment is likely mediated by abnormal interaction between oligomeric Aβ and mitochondrial matrix protein ABAD [151] or cyclophilin D (CypD) [152]. It has also been suggested that appoptosin, a glycine/5-amino-levulinic acid transporter mediating heme synthesis in mitochondria, is involved in Aβ-induced neurodegeneration by inducing ROS release and resultant apoptosis under pathological conditions; these pathogenic processes can be prevented by appoptosin downregulation [133].
Synaptic mitochondria, or mitochondria residing in synapses, are usually older, and thus much more vulnerable to persistent insults such as Aβ compared to nonsynaptic mitochondria. Aβ species reportedly also accumulate at much higher levels in synaptic mitochondria compared to nonsynaptic mitochondria [153, 154]. Accordingly, synaptic mitochondria show greater susceptibility to damage compared to nonsynaptic mitochondria in AD transgenic mice [153, 154]. Mitochondrial function such as respiratory rate, ROS production, membrane potential, and cytochrome c oxidase activity are compromised more severely in synaptic mitochondria than in nonsynaptic mitochondria [153, 154]. The degree of mitochondrial impairment is also region-specific. Greater damage is found in areas related to learning and memory such as the hippocampus and cortex, while only moderate damage is found in other brain regions [154].
Tau pathology strongly correlates with mitochondrial impairment [125, 130, 155–158], suggesting that tau may play a role in Aβ-induced mitochondrial dysfunction. It has been shown using immunoprecipitation and immunofluorescence that hyperphosphorylated tau abnormally interacts and colocalizes with the mitochondrial fission protein Drp1 in postmortem AD brains [158]. In the same brain specimens, elevated levels of Drp1 and mitochondrial fragmentation have also been identified [150]. These studies suggest that hyperphosphorylated tau is associated with Aβ-induced mitochondrial dysfunction, likely through its abnormal interaction with Drp1 in AD neurons. Consistent with this notion, Aβ application in cultured neurons results in elevated expression of mitochondrial fission proteins Drp1 and Fis1 and reduced expression of fusion proteins Mfn1, Mfn2, and Opa1, consistent with mitochondrial fragmentation phenotypes observed in AD [146]. Additional recent evidence also supports a role for hyperphosphorylated tau in mitochondrial impairment. For example, overexpression of the postsynaptic protein α1-takusan inhibits Aβ-induced tau hyperphosphorylation and prevents Aβ-induced mitochondrial fragmentation in cultured neurons [82].
Moreover, pathological redox reactions of mitochondrial proteins triggered by oligomeric Aβ, in part mediated by aberrant eNMDAR stimulation as discussed above, have been described. These reactions include S-nitrosylation of Drp1 (to form SNO-Drp1), resulting in excessive mitochondrial fission with consequent bioenergetic compromise and hence synaptic damage [64]. Interestingly, Cdk5 has been shown to act as a transnitrosylase, transferring NO from Cdk5 to Drp1 (as opposed to the classical role of Cdk5 as a kinase). Transnitrosylation hyperactivates Drp1 under these disease conditions, and, importantly, S-nitrosylation of Cdk5 is triggered by oligomeric Aβ peptide [67]. Thus, the initial nitrosylation of Cdk5 may represent in inciting event in mitochondrial fragmentation, with resulting bioenergetic failure and synaptic loss. Figure 2 summarizes our current understanding of Aβ-induced mitochondrial impairment as discussed above.
Figure 2.
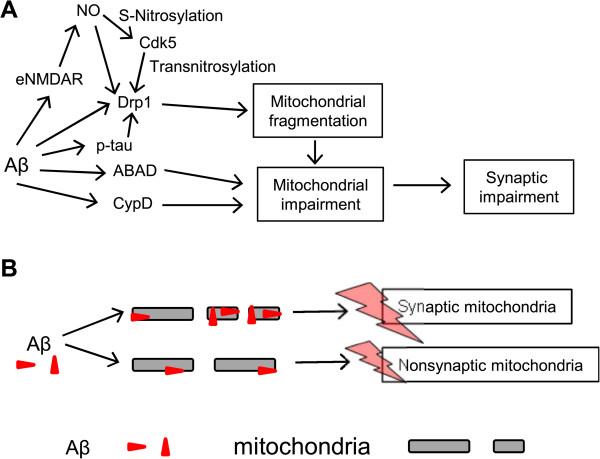
Mitochondrial impairment and synaptic dysfunction in AD. (A) Under pathological conditions, Aβ oligomers directly or indirectly affect mitochondrial fission proteins such as Drp1 and mitochondrial matrix proteins, including ABAD and CypD. Such interactions may mediate Aβ-induced mitochondrial fragmentation with consequent bioenergetics failure and resulting synaptic loss. (B) Synaptic mitochondria are more vulnerable to pathological toxins than non-synaptic mitochondria. Aβ is also more likely accumulated in synaptic mitochondria than in non-synaptic mitochondria, thus enhancing damage on synaptic mitochondria.
Conclusions
Accumulating evidence suggests that oligomeric Aβ plays a central role in synaptic dysfunction and cognitive decline in AD. Studies using animal models have revealed that Aβ-induced synaptic dysfunction involves multiple pathological events and various integral signaling systems, including glutamate receptors and their downstream pathways, abnormal elevation of extrasynaptic glutamate levels and subsequent eNMDAR-mediated excitotoxicity, tau hyperphosphorylation, and impaired mitochondria. However, the relevance of these studies to AD in human remains debatable. Furthermore, the detailed molecular mechanisms underlying these events are still not fully understood. Several fundamental questions remain, such as the role of tau phosphorylation in Aβ-induced synaptic dysfunction, the physiological relevance of Aβ-binding partners or receptors in neuronal degeneration, and even the direct role of Aβ itself in AD. Since a large variety proteins and distinctive pathways may be involved in the pathogenesis of AD, there may be no definitive treatment that can ubiquitously treat all AD patients. Indeed, the complexity of AD is exemplified by a diverse set of genetic mutations associated with AD. If this holds true, personalized drug candidates may need to be developed to cater to various genetic profiles, severity in cognitive decline, and other environmental factors. These challenges remain daunting. However, development of new technologies to treat dysregulated molecular pathways downstream of Aβ and phospho-tau may enable us to utilize our emerging knowledge of these pathways in order to develop novel strategies in the treatment and prevention of AD.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (R01AG038710, R01AG021173, R01AG044420 and R01NS046673), the Alzheimer’s Association (to HX), NIH grants (P01 HD29587, R01 NS086890, R21 NS080799 and P30 NS076411), Department of Defense (Army) grant W81XWH-13-0053, the Brain & Behavior Research Foundation (to S.A.L.), Shiley-Marcos Alzheimer’s Disease Research Center (UCSD) Pilot Award, and NIH grant R21 MH102672 (to S.-I.O.).
Abbreviations
ADDLs
Aβ-derived diffusible ligands
ADLs
Activities of daily living
α7nAChRs
α7-nicotinic acetylcholine receptors
AD
Alzheimer’s disease
AMPARs
AMPA receptors
Aβ
Amyloid-β peptide
APP
Amyloid Precursor Protein
CaMKII
Ca2+/calmodulin-dependent protein kinase II
Cdk5
Cyclin-dependent kinase 5
CypD
Cyclophilin D
Dkk1
Dickkopf-1
Drp1
Dynamin-related protein 1
EEG
Electroencephalography
eNMDARs
Extrasynaptic NMDARs
EAAT1
Excitatory Amino Acid Transporter 1
EAAT2
Excitatory Amino Acid Transporter 2
GSK-3β
Glycogen synthase kinase 3β
hAPP
Human amyloid precursor protein-overexpressing
iPSC
Induced pluripotent stem cell
LTD
Long-term depression
LilrB2
Leukocyte immunoglobulin-like receptor B2
IGF-I
Insulin-like growth factor-I
LTP
Long-term potentiation
LRP
Low-density lipoprotein-related protein
mGluRs
Metabotropic glutamate receptors
MT
Microtubule
MCI
Mild cognitive impairment
NFTs
Neurofibrillary tangles
nNOS
Neuronal nitric oxide synthase
NMDARs
_N_-methyl-D-aspartate receptors
PirB
Paired immunoglobulin-like receptor B
p-tau
Phospho-tau
PDK
Phosphoinositide-dependent kinase
PrPC
Prion protein
PKA
Protein kinase A
PP2A
Protein phosphatase 2A
PP2B
Protein phosphatase 2B
RNS
Reactive nitrogen species
ROS
Reactive oxygen species
sNMDARs
Synaptic NMDARs
Tg
Transgenic.
Footnotes
Competing interests
The authors declare that S.A.L. is the inventor on world-wide patents for the use of memantine and NitroMemantine for neurodegenerative disorders. Per Harvard University guidelines, S.A.L. participates in a royalty-sharing agreement with his former institution Boston Children’s Hospital/Harvard Medical School, which licensed the drug memantine (Namenda®) to Forest Laboratories, Inc. The other authors declare no financial conflicts of interest.
Authors’ contributions
ST and HX wrote the manuscript. SO and SAL revised the manuscript for intellectual content. All authors read and approved the final manuscript.
Contributor Information
Shichun Tu, Email: shichuntu@sanfordburnham.org.
Shu-ichi Okamoto, Email: sokamoto@sanfordburnham.org.
Stuart A Lipton, Email: slipton@sanfordburnham.org.
Huaxi Xu, Email: xuh@sanfordburnham.org.
References
- 1.Patel L, Grossberg GT. Combination therapy for Alzheimer’s disease. Drugs Aging. 2011;28:539–546. doi: 10.2165/11591860-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 2.Delrieu J, Piau A, Caillaud C, Voisin T, Vellas B. Managing cognitive dysfunction through the continuum of Alzheimer’s disease: role of pharmacotherapy. CNS Drugs. 2011;25:213–226. doi: 10.2165/11539810-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 3.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 4.Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/S0002-9440(10)65184-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Vbeyreuther K, Bush AI, Masters CL. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::AID-ANA8>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 6.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 7.Selkoe DJ. Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT, Irizarry MC. Early Aβ accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–931. doi: 10.1212/01.WNL.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- 9.Reddy PH, Geethalakshmi M, Byung SP, Joline J, Geoffrey M, William W, Jr, Jeffrey K, Maria M. Differential loss of synaptic proteins in Alzheimer’s disease: implications for synaptic dysfunction. J Alzheimers Dis. 2005;7:103–117. doi: 10.3233/jad-2005-7203. [DOI] [PubMed] [Google Scholar]
- 10.Masliah E, Mallory M, Alford M, DeTeresa R, Hansen LA, McKeel DW, Jr, Morris JC. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology. 2001;56:127–129. doi: 10.1212/WNL.56.1.127. [DOI] [PubMed] [Google Scholar]
- 11.Scheff SW, Price DA, Schmitt FA, Mufson EJ. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging. 2006;27:1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 12.Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007;68:1501–1508. doi: 10.1212/01.wnl.0000260698.46517.8f. [DOI] [PubMed] [Google Scholar]
- 13.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 14.Palop JJ, Mucke L. Amyloid-β-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat Neurosci. 2010;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang HY, Lee DHS, D’Andrea MR, Peterson PA, Shank RP, Reitz AB. β-Amyloid1–42 binds to α7 nicotinic acetylcholine receptor with high affinity: implications for Alzheimer’s disease pathology. J Biol Chem. 2000;275:5626–5632. doi: 10.1074/jbc.275.8.5626. [DOI] [PubMed] [Google Scholar]
- 16.De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL. Aβ oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–11601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- 17.Decker H, Jürgensen S, Adrover MF, Brito-Moreira J, Bomfim TR, Klein WL, Epstein AL, De Felice FG, Jerusalinsky D, Ferreira ST. N-Methyl-d-aspartate receptors are required for synaptic targeting of Alzheimer’s toxic amyloid-β peptide oligomers. J Neurochem. 2010;115:1520–1529. doi: 10.1111/j.1471-4159.2010.07058.x. [DOI] [PubMed] [Google Scholar]
- 18.Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL, Triller A. Deleterious effects of amyloid β oligomers acting as an extracellular scaffold for mGluR5. Neuron. 2010;66:739–754. doi: 10.1016/j.neuron.2010.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yaar M, Zhai S, Pilch PF, Doyle SM, Eisenhauer PB, Fine RE, Gilchrest BA. Binding of β-amyloid to the p75 neurotrophin receptor induces apoptosis: a possible mechanism for Alzheimer’s disease. J Clin Invest. 1997;100:2333–2340. doi: 10.1172/JCI119772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pham E, Crews L, Ubhi K, Hansen L, Adame A, Cartier A, Salmon D, Galasko D, Michael S, Savas JN, Yates JR, Glabe C, Masliah E. Progressive accumulation of amyloid-β oligomers in Alzheimer’s disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FASEB J. 2010;277:3051–3067. doi: 10.1111/j.1742-4658.2010.07719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble oligomers of amyloid β protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cisse M, Halabisky B, Harris J, Devidze N, Dubal DB, Sun B, Orr A, Lotz G, Kim DH, Hamto P, Ho K, Yu G-Q, Mucke L. Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature. 2011;469:47–52. doi: 10.1038/nature09635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vargas LM, Leal N, Estrada LD, Gonzalez A, Serrano F, Araya K, Gysling K, Inestrosa NC, Pasquale EB, Alvarez AR. EphA4 activation of c-Abl mediates synaptic loss and LTP blockade caused by amyloid-β oligomers. PLoS One. 2014;9:e92309. doi: 10.1371/journal.pone.0092309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu AK, Hung KW, Huang H, Gu S, Shen Y, Cheng EY, Ip FC, Huang X, Fu WY, Ip NY. Blockade of EphA4 signaling ameliorates hippocampal synaptic dysfunctions in mouse models of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2014;111:9959–9964. doi: 10.1073/pnas.1405803111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pai MC, Jacobs WJ. Topographical disorientation in community-residing patients with Alzheimer’s disease. Int J Geriatr Psychiatry. 2004;19:250–255. doi: 10.1002/gps.1081. [DOI] [PubMed] [Google Scholar]
- 27.Hort J, Laczo J, Vyhnalek M, Bojar M, Bures J, Vlcek K. Spatial navigation deficit in amnestic mild cognitive impairment. Proc Natl Acad Sci U S A. 2007;104:4042–4047. doi: 10.1073/pnas.0611314104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Monacelli AM, Cushman LA, Kavcic V, Duffy CJ. Spatial disorientation in Alzheimer’s disease: the remembrance of things passed. Neurology. 2003;61:1491–1497. doi: 10.1212/WNL.61.11.1491. [DOI] [PubMed] [Google Scholar]
- 29.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–103. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 30.Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, Carlson GA, Younkin SG, Ashe KH. The relationship between Aβ and memory in the Tg2576 mouse model of Alzheimer’s disease. J Neurosci. 2002;22:1858–1867. doi: 10.1523/JNEUROSCI.22-05-01858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. TTriple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/S0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 32.Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Aβ causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 33.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McGowan E, Eriksen J, Hutton M. A decade of modeling Alzheimer’s disease in transgenic mice. Trends Genet. 2006;22:281–289. doi: 10.1016/j.tig.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 35.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 36.Yamin G. NMDA receptor-dependent signaling pathways that underlie amyloid β-protein disruption of LTP in the hippocampus. J Neurosci Res. 2009;87:1729–1736. doi: 10.1002/jnr.21998. [DOI] [PubMed] [Google Scholar]
- 37.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 38.Cullen WK, Suh Y-H, Anwyl R, Rowan MJ. Block of LTP in rat hippocampus in vivo by amyloid precursor protein fragments. Neuroreport. 1997;8:3213–3217. doi: 10.1097/00001756-199710200-00006. [DOI] [PubMed] [Google Scholar]
- 39.Chen QS, Kagan BL, Hirakura Y, Xie CW. Impairment of hippocampal long-term potentiation by Alzheimer amyloid β-peptides. J Neurosci Res. 2000;60:65–72. doi: 10.1002/(SICI)1097-4547(20000401)60:1<65::AID-JNR7>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 40.Chen Q-S, Wei W-Z, Shimahara T, Xie C-W. Alzheimer amyloid β-peptide inhibits the late phase of long-term potentiation through calcineurin-dependent mechanisms in the hippocampal dentate gyrus. Neurobiol Learn Mem. 2002;77:354–371. doi: 10.1006/nlme.2001.4034. [DOI] [PubMed] [Google Scholar]
- 41.Stephan A, Laroche S, Davis S. Generation of aggregated β-amyloid in the rat hippocampus impairs synaptic transmission and plasticity and causes memory deficits. J Neurosci. 2001;21:5703–5714. doi: 10.1523/JNEUROSCI.21-15-05703.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 43.Knobloch M, Farinelli M, Konietzko U, Nitsch RM, Mansuy IM. Aβ oligomer-mediated long-term potentiation impairment involves protein phosphatase 1-dependent mechanisms. J Neurosci. 2007;27:7648–7653. doi: 10.1523/JNEUROSCI.0395-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao D, Watson JB, Xie CW. Amyloid β prevents activation of calcium/calmodulin-dependent protein kinase II and AMPA receptor phosphorylation during hippocampal long-term potentiation. J Neurophysiol. 2004;92:2853–2858. doi: 10.1152/jn.00485.2004. [DOI] [PubMed] [Google Scholar]
- 45.Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Aβ-Induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/S0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- 48.Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang X, Takata T, Bai X, Ou F, Yokono K, Sakurai T. Pyruvate prevents the inhibition of the long-term potentiation induced by amyloid-β through protein phosphatase 2A inactivation. J Alzheimers Dis. 2012;30:665–673. doi: 10.3233/JAD-2012-101869. [DOI] [PubMed] [Google Scholar]
- 50.Kim T, Vidal GS, Djurisic M, William CM, Birnbaum ME, Garcia KC, Hyman BT, Shatz CJ. Human LilrB2 Is a β-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer’s model. Science. 2013;341:1399–1404. doi: 10.1126/science.1242077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roselli F, Tirard M, Lu J, Hutzler P, Lamberti P, Livrea P, Morabito M, Almeida OFX. Soluble β-amyloid1-40 induces NMDA-dependent degradation of postsynaptic density-95 at glutamatergic synapses. J Neurosci. 2005;25:11061–11070. doi: 10.1523/JNEUROSCI.3034-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Almeida CG, Tampellini D, Takahashi RH, Greengard P, Lin MT, Snyder EM, Gouras GK. β-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis. 2005;20:187–198. doi: 10.1016/j.nbd.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 53.Chang EH, Savage MJ, Flood DG, Thomas JM, Levy RB, Mahadomrongkul V, Shirao T, Aoki C, Huerta PT. AMPA receptor downscaling at the onset of Alzheimer’s disease pathology in double knockin mice. Proc Natl Acad Sci U S A. 2006;103:3410–3415. doi: 10.1073/pnas.0507313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao W-Q, Santini F, Breese R, Ross D, Zhang XD, Stone DJ, Ferrer M, Townsend M, Wolfe AL, Seager MA, Kinney GG, Shughrue PJ, Ray WJ. Inhibition of calcineurin-mediated endocytosis and and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors prevents amyloid β oligomer-induced synaptic disruption. J Biol Chem. 2010;285:7619–7632. doi: 10.1074/jbc.M109.057182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gu Z, Liu W, Yan Z. β-Amyloid impairs AMPA receptor trafficking and function by reducing Ca2+/calmodulin-dependent protein kinase II synaptic distribution. J Biol Chem. 2009;284:10639–10649. doi: 10.1074/jbc.M806508200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-β. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- 57.Kurup P, Zhang Y, Xu J, Venkitaramani DV, Haroutunian V, Greengard P, Nairn AC, Lombroso PJ. Aβ-mediated NMDA receptor endocytosis in Alzheimer’s disease involves ubiquitination of the tyrosine phosphatase STEP61. J Neurosci. 2010;30:5948–5957. doi: 10.1523/JNEUROSCI.0157-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li Z, Jo J, Jia J-M, Lo S-C, Whitcomb DJ, Jiao S, Cho K, Sheng M. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell. 2010;141:859–871. doi: 10.1016/j.cell.2010.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.D’Amelio M, Cavallucci V, Middei S, Marchetti C, Pacioni S, Ferri A, Diamantini A, De Zio D, Carrara P, Battistini L, Moreno S, Bacci A, Ammassari-Teule M, Marie H, Cecconi F. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat Neurosci. 2011;14:69–76. doi: 10.1038/nn.2709. [DOI] [PubMed] [Google Scholar]
- 60.Louneva N, Cohen JW, Han LY, Talbot K, Wilson RS, Bennett DA, Trojanowski JQ, Arnold SE. Caspase-3 is enriched in postsynaptic densities and increased in Alzheimer’s disease. Am J Pathol. 2008;173:1488–1495. doi: 10.2353/ajpath.2008.080434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci. 2011;31:6627–6638. doi: 10.1523/JNEUROSCI.0203-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jacob CP, Koutsilieri E, Bartl J, Neuen-Jacob E, Arzberger T, Zander N, Ravid R, Roggendorf W, Riederer P, Grunblatt E. Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer’s disease. J Alzheimers Dis. 2007;11:97–116. doi: 10.3233/jad-2007-11113. [DOI] [PubMed] [Google Scholar]
- 63.Talantova M, Sanz-Blasco S, Zhang X, Xia P, Akhtar MW, Okamoto S, Dziewczapolski G, Nakamura T, Cao G, Pratt AE, Kang YJ, Tu S, Molokanova E, McKercher SR, Hires SA, Sason H, Stouffer DG, Buczynski MW, Solomon JP, Michael S, Powers ET, Kelly JW, Roberts A, Tong G, Fang-Newmeyer T, Parker J, Holland EA, Zhang D, Nakanishi N, Chen HS. Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci U S A. 2013;110:E2518–E2527. doi: 10.1073/pnas.1306832110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA. S-nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nakamura T, Wang L, Wong CC, Scott FL, Eckelman BP, Han X, Tzitzilonis C, Meng F, Gu Z, Holland EA, Clemente AT, Okamoto S, Salvesen GS, Riek R, Yates JR, III, Lipton SA. Transnitrosylation of XIAP regulates caspase-dependent neuronal cell death. Mol Cell. 2010;39:184–195. doi: 10.1016/j.molcel.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nakamura T, Tu S, Akhtar MW, Sunico CR, Okamoto S, Lipton SA. Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron. 2013;78:596–614. doi: 10.1016/j.neuron.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qu J, Nakamura T, Cao G, Holland EA, McKercher SR, Lipton SA. S-Nitrosylation activates Cdk5 and contributes to synaptic spine loss induced by β-amyloid peptide. Proc Natl Acad Sci U S A. 2011;108:14330–14335. doi: 10.1073/pnas.1105172108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Okamoto S-I, Nakamura T, Cieplak P, Chan Shing F, Kalashnikova E, Liao L, Saleem S, Han X, Clemente A, Nutter A, Sances S, Brechtel C, Haus D, Haun F, Sanz-Blasco S, Huang X, Li H, Zaremba JD, Cui J, Gu Z, Nikzad R, Harrop A, McKercher SR, Godzik A, Yates JR, III, Lipton SA. S-Nitrosylation-mediated redox transcriptional switch modulates neurogenesis and neuronal cell death. Cell Rep. 2014;8:1–12. doi: 10.1016/j.celrep.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Molokanova E, Akhtar MW, Sanz-Blasco S, Tu S, Piña-Crespo JC, McKercher SR, Lipton SA. Differential effects of synaptic and extrasynaptic NMDA receptors on Aβ-induced nitric oxide production in cerebrocortical neurons. J Neurosci. 2014;34:5023–5028. doi: 10.1523/JNEUROSCI.2907-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ryan SD, Dolatabadi N, Chan SF, Zhang X, Akhtar MW, Parker J, Soldner F, Sunico CR, Nagar S, Talantova M, Lee B, Lopez K, Nutter A, Shan B, Molokanova E, Zhang Y, Han X, Nakamura T, Masliah E, Yates JR, III, Nakanishi N, Andreyev AY, Okamoto S, Jaenisch R, Ambasudhan R, Lipton SA. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1α transcription. Cell. 2013;155:1351–1364. doi: 10.1016/j.cell.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Xia P, Chen HS, Zhang D, Lipton SA. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J Neurosci. 2010;30:11246–11250. doi: 10.1523/JNEUROSCI.2488-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang Y, Eu J, Washburn M, Gong T, Chen HS, James WL, Lipton SA, Stamler JS, Went GT, Porter S. The pharmacology of aminoadamantane nitrates. Curr Alzheimer Res. 2006;3:201–204. doi: 10.2174/156720506777632808. [DOI] [PubMed] [Google Scholar]
- 73.Figueiredo CP, Clarke JR, Ledo JH, Ribeiro FC, Costa CV, Melo HM, Mota-Sales AP, Saraiva LM, Klein WL, Sebollela A, De Felice FG, Ferreira ST. Memantine rescues transient cognitive impairment caused by high-molecular-weight Aβ oligomers but not the persistent impairment induced by low-molecular-weight oligomers. J Neurosci. 2013;33:9626–9634. doi: 10.1523/JNEUROSCI.0482-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- 75.Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- 76.Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM. Synaptic changes in Alzheimer’s disease: increased amyloid-β and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am J Pathol. 2004;165:1809–1817. doi: 10.1016/S0002-9440(10)63436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Proctor DT, Coulson EJ, Dodd PR. Reduction in post-synaptic scaffolding PSD-95 and SAP-102 protein levels in the Alzheimer inferior temporal cortex is correlated with disease pathology. J Alzheimers Dis. 2010;21:795–811. doi: 10.3233/JAD-2010-100090. [DOI] [PubMed] [Google Scholar]
- 78.Sultana R, Banks WA, Butterfield DA. Decreased levels of PSD95 and two associated proteins and increased levels of BCl2 and caspase 3 in hippocampus from subjects with amnestic mild cognitive impairment: insights into their potential roles for loss of synapses and memory, accumulation of Aβ, and neurodegeneration in a prodromal stage of Alzheimer’s disease. J Neurosci Res. 2010;88:469–477. doi: 10.1002/jnr.22227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, Hyman BT. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25:7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer’s-related amyloid β oligomers. J Neurosci. 2004;24:10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tu S, Shin Y, Zago WM, States BA, Eroshkin A, Lipton SA, Tong GG, Nakanishi N. Takusan: a large gene family that regulates synaptic activity. Neuron. 2007;55:69–85. doi: 10.1016/j.neuron.2007.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nakanishi N, Ryan SD, Zhang X, Khan A, Holland T, Cho E-G, Huang X, Liao F-F, Xu H, Lipton SA, Tu S. Synaptic protein α1-takusan mitigates amyloid-β-induced synaptic loss via interaction with tau and postsynaptic density-95 at postsynaptic sites. J Neurosci. 2013;33:14170–14183. doi: 10.1523/JNEUROSCI.4646-10.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen Y, Wang B, Liu D, Li JJ, Xue Y, Sakata K, Zhu LQ, Heldt SA, Xu H, Liao FF. Hsp90 chaperone inhibitor 17-AAG attenuates Aβ-induced synaptic toxicity and memory impairment. J Neurosci. 2014;34:2464–2470. doi: 10.1523/JNEUROSCI.0151-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chakravarthy B, Gaudet C, Menard M, Atkinson T, Brown L, Laferla FM, Armato U, Whitfield J. Amyloid-β peptides stimulate the expression of the p75(NTR) neurotrophin receptor in SHSY5Y human neuroblastoma cells and AD transgenic mice. J Alzheimers Dis. 2010;19:915–925. doi: 10.3233/JAD-2010-1288. [DOI] [PubMed] [Google Scholar]
- 85.Chakravarthy B, Menard M, Ito S, Gaudet C, Dal Pra I, Armato U, Whitfield J. Hippocampal membrane-associated p75NTR levels are increased in Alzheimer’s disease. J Alzheimers Dis. 2012;30:675–684. doi: 10.3233/JAD-2012-120115. [DOI] [PubMed] [Google Scholar]
- 86.Costantini C, Weindruch R, Della Valle G, Puglielli L. A TrkA-to-p75NTR molecular switch activates amyloid β-peptide generation during aging. Biochem J. 2005;391:59–67. doi: 10.1042/BJ20050700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Costantini C, Scrable H, Puglielli L. An aging pathway controls the TrkA to p75NTR receptor switch and amyloid β peptide generation. EMBO J. 2006;25:1997–2006. doi: 10.1038/sj.emboj.7601062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Puglielli L, Ellis BC, Saunders AJ, Kovacs DM. Ceramide stabilizes β-site amyloid precursor protein-cleaving enzyme 1 and promotes amyloid β-peptide biogenesis. J Biol Chem. 2003;278:19777–19783. doi: 10.1074/jbc.M300466200. [DOI] [PubMed] [Google Scholar]
- 89.Zhao W-Q, Alkon DL. Role of insulin and insulin receptor in learning and memory. Mol Cell Endocrinol. 2001;177:125–134. doi: 10.1016/S0303-7207(01)00455-5. [DOI] [PubMed] [Google Scholar]
- 90.Zhao W-Q, Chen H, Quon MJ, Alkon DL. Insulin and the insulin receptor in experimental models of learning and memory. Eur J Pharmacol. 2004;490:71–81. doi: 10.1016/j.ejphar.2004.02.045. [DOI] [PubMed] [Google Scholar]
- 91.Liao FF, Xu H. Insulin signaling in sporadic Alzheimer’s disease. Sci Signal. 2009;2:pe36. doi: 10.1126/scisignal.274pe36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.de la Monte SM. Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Curr Alzheimer Res. 2012;9:35–66. doi: 10.2174/156720512799015037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee H-K, Kumar P, Fu Q, Rosen KM, Querfurth HW. The insulin/Akt signaling pathway is targeted by intracellular β-amyloid. Mol Biol Cell. 2009;20:1533–1544. doi: 10.1091/mbc.E08-07-0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.De Felice FG, Vieira MN, Bomfim TR, Decker H, Velasco PT, Lambert MP, Viola KL, Zhao WQ, Ferreira ST, Klein WL. Protection of synapses against Alzheimer’s-linked toxins: insulin signaling prevents the pathogenic binding of Aβ oligomers. Proc Natl Acad Sci U S A. 2009;106:1971–1976. doi: 10.1073/pnas.0809158106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Reger MA, Watson GS, Green PS, Baker LD, Cholerton B, Fishel MA, Plymate SR, Cherrier MM, Schellenberg GD, Frey Ii WH, Craft S. Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-β in memory-impaired older adults. J Alzheimers Dis. 2008;13:323–331. doi: 10.3233/jad-2008-13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, Arbuckle M, Callaghan M, Tsai E, Plymate SR, Green PS, Leverenz J, Cross D, Gerton B. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol. 2012;69:29–38. doi: 10.1001/archneurol.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Inestrosa NC, Arenas E. Emerging roles of Wnts in the adult nervous system. Nat Rev Neurosci. 2010;11:77–86. doi: 10.1038/nrn2755. [DOI] [PubMed] [Google Scholar]
- 98.De Ferrari GV, Papassotiropoulos A, Biechele T, Wavrant De-Vrieze F, Avila ME, Major MB, Myers A, Saez K, Henriquez JP, Zhao A, Wollmer MA, Nitsch RM, Hock C, Morris CM, Hardy J, Moon RT. Common genetic variation within the low-density lipoprotein receptor-related protein 6 and late-onset Alzheimer’s disease. Proc Natl Acad Sci U S A. 2007;104:9434–9439. doi: 10.1073/pnas.0603523104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Semënov MV, Zhang X, He X. DKK1 antagonizes Wnt signaling without promotion of LRP6 internalization and degradation. J Biol Chem. 2008;283:21427–21432. doi: 10.1074/jbc.M800014200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Caricasole A, Copani A, Caraci F, Aronica E, Rozemuller AJ, Caruso A, Storto M, Gaviraghi G, Terstappen GC, Nicoletti F. Induction of Dickkopf-1, a negative modulator of the Wnt pathway, is associated with neuronal degeneration in Alzheimer’s brain. J Neurosci. 2004;24:6021–6027. doi: 10.1523/JNEUROSCI.1381-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rosi MC, Luccarini I, Grossi C, Fiorentini A, Spillantini MG, Prisco A, Scali C, Gianfriddo M, Caricasole A, Terstappen GC, Casamenti F. Increased Dickkopf-1 expression in transgenic mouse models of neurodegenerative disease. J Neurochem. 2010;112:1539–1551. doi: 10.1111/j.1471-4159.2009.06566.x. [DOI] [PubMed] [Google Scholar]
- 102.Purro SA, Dickins EM, Salinas PC. The secreted Wnt antagonist Dickkopf-1 is required for Amyloid β-mediated synaptic loss. J Neurosci. 2012;32:3492–3498. doi: 10.1523/JNEUROSCI.4562-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Magdesian MH, Carvalho MMVF, Mendes FA, Saraiva LM, Juliano MA, Juliano L, Garcia-Abreu J, Ferreira ST. Amyloid-β binds to the extracellular cysteine-rich domain of Frizzled and inhibits Wnt/β-Catenin signaling. J Biol Chem. 2008;283:9359–9368. doi: 10.1074/jbc.M707108200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Larner AJ. Epileptic seizures in AD patients. Neuromolecular Med. 2010;12:71–77. doi: 10.1007/s12017-009-8076-z. [DOI] [PubMed] [Google Scholar]
- 105.Mendez M, Lim G. Seizures in elderly patients with dementia: epidemiology and management. Drugs Aging. 2003;20:791–803. doi: 10.2165/00002512-200320110-00001. [DOI] [PubMed] [Google Scholar]
- 106.Hesdorffer DC, Hauser WA, Annegers JF, Kokmen E, Rocca WA. Dementia and adult-onset unprovoked seizures. Neurology. 1996;46:727–730. doi: 10.1212/WNL.46.3.727. [DOI] [PubMed] [Google Scholar]
- 107.Amatniek JC, Hauser WA, DelCastillo-Castaneda C, Jacobs DM, Marder K, Bell K, Albert M, Brandt J, Stern Y. Incidence and predictors of seizures in patients with Alzheimer’s disease. Epilepsia. 2006;47:867–872. doi: 10.1111/j.1528-1167.2006.00554.x. [DOI] [PubMed] [Google Scholar]
- 108.Noebels J. A perfect storm: converging paths of epilepsy and Alzheimer’s dementia intersect in the hippocampal formation. Epilepsia. 2011;52:39–46. doi: 10.1111/j.1528-1167.2010.02909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.LaFerla FM, Tinkle BT, Bieberich CJ, Haudenschild CC, Jay G. The Alzheimer’s Aβ peptide induces neurodegeneration and apoptotic cell death in transgenic mice. Nat Genet. 1995;9:21–30. doi: 10.1038/ng0195-21. [DOI] [PubMed] [Google Scholar]
- 110.Moechars D, Lorent K, De Strooper B, Dewachter I, Van Leuven F. Expression in brain of amyloid precursor protein mutated in the α-secretase site causes disturbed behavior, neuronal degeneration and premature death in transgenic mice. EMBO J. 1996;15:1265–1274. [PMC free article] [PubMed] [Google Scholar]
- 111.Kumar-Singh S, Dewachter I, Moechars D, Lübke U, De Jonghe C, Ceuterick C, Checler F, Naidu A, Cordell B, Cras P, Van Broeckhoven C, Van Leuven F. Behavioral disturbances without amyloid deposits in mice overexpressing human amyloid precursor protein with Flemish (A692G) or Dutch (E693Q) mutation. Neurobiol Dis. 2000;7:9–22. doi: 10.1006/nbdi.1999.0272. [DOI] [PubMed] [Google Scholar]
- 112.Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu G-Q, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fülöp L, Penke B, Zilberter Y, Harkany T, Pitkänen A, Tanila H. Amyloid β-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 2009;29:3453–3462. doi: 10.1523/JNEUROSCI.5215-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Harris JA, Devidze N, Verret L, Ho K, Halabisky B, Thwin MT, Kim D, Hamto P, Lo I, Yu G-Q, Palop JJ, Masliah E, Mucke L. Transsynaptic progression of amyloid-β-induced neuronal dysfunction within the entorhinal-hippocampal network. Neuron. 2010;68:428–441. doi: 10.1016/j.neuron.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR, Devidze N, Ho K, Yu G-Q, Palop JJ, Mucke L. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc Natl Acad Sci U S A. 2012;109:E2895–E2903. doi: 10.1073/pnas.1121081109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975;72:1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lee G, Rook SL. Expression of tau protein in non-neuronal cells: microtubule binding and stabilization. J Cell Sci. 1992;102:227–237. doi: 10.1242/jcs.102.2.227. [DOI] [PubMed] [Google Scholar]
- 118.Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 119.Mazanetz MP, Fischer PM. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat Rev Drug Discov. 2007;6:464–479. doi: 10.1038/nrd2111. [DOI] [PubMed] [Google Scholar]
- 120.Fein JA, Sokolow S, Miller CA, Vinters HV, Yang F, Cole GM, Gylys KH. Co-localization of amyloid β and tau pathology in Alzheimer’s disease synaptosomes. Am J Pathol. 2008;172:1683–1692. doi: 10.2353/ajpath.2008.070829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sokolow S, Henkins KM, Bilousova T, Miller CA, Vinters HV, Poon W, Cole GM, Gylys KH. AD synapses contain abundant Aβ monomer and multiple soluble oligomers, including a 56-kDa assembly. Neurobiol Aging. 2012;33:1545–1555. doi: 10.1016/j.neurobiolaging.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Takahashi RH, Capetillo-Zarate E, Lin MT, Milner TA, Gouras GK. Co-occurrence of Alzheimer’s disease β-amyloid and tau pathologies at synapses. Neurobiol Aging. 2010;31:1145–1152. doi: 10.1016/j.neurobiolaging.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu G-Q, Mucke L. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 124.Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer’s disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 125.Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L. Tau reduction prevents Aβ-induced defects in axonal transport. Science. 2010;330:198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu G-Q, Palop JJ, Noebels JL, Mucke L. Amyloid-β/Fyn–induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J Neurosci. 2011;31:700–711. doi: 10.1523/JNEUROSCI.4152-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Manczak M, Reddy PH. Abnormal interaction of oligomeric amyloid-β with phosphorylated tau: implications to synaptic dysfunction and neuronal damage. J Alzheimers Dis. 2013;36:285–295. doi: 10.3233/JAD-130275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, Bigio EH, Jerecic J, Acton PJ, Shughrue PJ, Chen-Dodson E, Kinney GG, Klein WL. Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by Aβ oligomers. Neurobiol Aging. 2008;29:1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ. Soluble amyloid β-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci U S A. 2011;108:5819–5824. doi: 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zempel H, Thies E, Mandelkow E, Mandelkow EM. Aβ oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30:11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan L-L, Ashe KH, Liao D. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–1081. doi: 10.1016/j.neuron.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shipton OA, Leitz JR, Dworzak J, Acton CEJ, Tunbridge EM, Denk F, Dawson HN, Vitek MP, Wade-Martins R, Paulsen O, Vargas-Caballero M. Tau protein is required for amyloid β-induced impairment of hippocampal long-term potentiation. J Neurosci. 2011;31:1688–1692. doi: 10.1523/JNEUROSCI.2610-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhang H, Zhang Y-W, Chen Y, Huang X, Zhou F, Wang W, Xian B, Zhang X, Masliah E, Chen Q, Han J-DJ, Bu G, Reed JC, Liao F-F, Chen Y-G, Xu H. Appoptosin is a novel pro-apoptotic protein and mediates cell death in neurodegeneration. J Neurosci. 2012;32:15565–15576. doi: 10.1523/JNEUROSCI.3668-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bhaskar K, Yen S-H, Lee G. Disease-related modifications in tau affect the interaction between Fyn and Tau. J Biol Chem. 2005;280:35119–35125. doi: 10.1074/jbc.M505895200. [DOI] [PubMed] [Google Scholar]
- 135.Israel MA, Yuan SH, Bardy C, Reyna SM, Mu YL, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, Carson CT, Laurent LC, Marsala M, Gage FH, Remes AM, Koo EH, Goldstein LSB. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012;482:216–220. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mattson MP, Gleichmann M, Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron. 2008;60:748–766. doi: 10.1016/j.neuron.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wang X, Su B, Zheng L, Perry G, Smith MA, Zhu X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J Neurochem. 2009;109:153–159. doi: 10.1111/j.1471-4159.2009.05867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci. 2008;9:505–518. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Reddy PH, Beal MF. Amyloid β, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol Med. 2008;14:45–53. doi: 10.1016/j.molmed.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci. 2001;21:3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Maurer I, Zierz S, Möller HJ. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol Aging. 2000;21:455–462. doi: 10.1016/S0197-4580(00)00112-3. [DOI] [PubMed] [Google Scholar]
- 142.Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD. Mitochondrial Aβ: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005;19:2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 143.Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of Aβ accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 144.Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, Casadesus G, Zhu X. Amyloid-β overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A. 2008;105:19318–19323. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wang X, Su B, Lee H-G, Li X, Perry G, Smith MA, Zhu X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Manczak M, Mao P, Calkins MJ, Cornea A, Reddy AP, Murphy MP, Szeto HH, Park B, Reddy PH. Mitochondria-targeted antioxidants protect against amyloid-β toxicity in Alzheimer’s disease neurons. J Alzheimers Dis. 2010;20:609–631. doi: 10.3233/JAD-2010-100564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum Mol Genet. 2011;20:4515–4529. doi: 10.1093/hmg/ddr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Graber S, Kovacs I, Lee WD, Waggoner J, Cui J, White AD, Bossy B, Martinou J-C, Youle RJ, Lipton SA, Ellisman MH, Perkins GA, Bossy-Wetzel E. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006;25:3900–3911. doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2009;106:14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Manczak M, Calkins MJ, Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid β with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum Mol Genet. 2011;20:2495–2509. doi: 10.1093/hmg/ddr139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue L-F, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H. ABAD directly links Aβ to mitochondrial toxicity in Alzheimer’s disease. Science. 2004;304:448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 152.Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Du H, Guo L, Yan S, Sosunov AA, McKhann GM, ShiDu Yan S. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc Natl Acad Sci U S A. 2010;107:18670–18675. doi: 10.1073/pnas.1006586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Dragicevic N, Mamcarz M, Zhu Y, Buzzeo R, Tan J, Arendash GW, Bradshaw PC. Mitochondrial amyloid-β levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer’s transgenic mice. J Alzheimers Dis. 2010;20:535–550. doi: 10.3233/JAD-2010-100342. [DOI] [PubMed] [Google Scholar]
- 155.Mandelkow EM, Stamer K, Vogel R, Thies E, Mandelkow E. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol Aging. 2003;24:1079–1085. doi: 10.1016/j.neurobiolaging.2003.04.007. [DOI] [PubMed] [Google Scholar]
- 156.Shahpasand K, Uemura I, Saito T, Asano T, Hata K, Shibata K, Toyoshima Y, Hasegawa M, Hisanaga S-I. Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer’s disease. J Neurosci. 2012;32:2430–2441. doi: 10.1523/JNEUROSCI.5927-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kopeikina KJ, Carlson GA, Pitstick R, Ludvigson AE, Peters A, Luebke JI, Koffie RM, Frosch MP, Hyman BT, Spires-Jones TL. Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer’s disease brain. Am J Pathol. 2011;179:2071–2082. doi: 10.1016/j.ajpath.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Manczak M, Reddy PH. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum Mol Genet. 2012;21:2538–2547. doi: 10.1093/hmg/dds072. [DOI] [PMC free article] [PubMed] [Google Scholar]