Live to die another way: modes of programmed cell death and the signals emanating from dying cells (original) (raw)
. Author manuscript; available in PMC: 2016 Jun 1.
Published in final edited form as: Nat Rev Mol Cell Biol. 2015 May 20;16(6):329–344. doi: 10.1038/nrm3999
Preface
All life ends in death, but perhaps one of life’s grander ironies is that it also depends on death. Cell-intrinsic suicide pathways, termed programmed cell death (PCD), are crucial for animal development, tissue homeostasis and pathogenesis. Originally, PCD was virtually synonymous with apoptosis, but recently, alternative PCD mechanisms have been reported. Here, we provide an overview of several distinct PCD mechanisms, namely apoptosis, autophagy and necroptosis. In addition, we discuss the complex signals emanating from dying cells, which can either fuel regeneration or instruct additional killing. Further advances in understanding the physiological role of multiple cell death mechanisms and associated signals will be important to selectively manipulate PCD for therapeutic purposes.
“Life and death are one thread, the same line viewed from different sides.”
Lao Tzu (ancient Chinese philosopher)
Introduction
The concept of naturally occurring cell death dates back to 1842, when Karl Vogt showed that the notochord of the midwife toad is removed by cell death during metamorphosis enabling the formation of vertebrae1. However, for many years it was assumed that cell death is no more than an inevitable passive finale of life. This notion was challenged by the observation in the mid 1960s that developmental cell death relies on the expression of endogenous genes, thus giving rise to the term programmed cell death (PCD)2, 3. Another important contribution came from electron microscopy studies carried out by Kerr, Wylie and Currie in the 1970s that defined the ultra-structural hallmarks of different types of cell death4. Although multiple types of cell death had already been described in the 19th century, electron microscopy enabled a clear distinction to be made between these different types of death. Cells that die during normal development and tissue homeostasis have characteristic morphological hallmarks, including cytoplasmic shrinkage, nuclear condensation, and the retention of membrane and organelle integrity — a process termed “apoptosis”4. By contrast, as a result of overwhelming trauma, dying cells swell and rupture, in a mode of cell death named “necrosis”.
Historically, Caenorhabditis elegans, Drosophila melanogaster and Mus musculus have been instrumental in developing our understanding of PCD and its role during animal development (Box 1). The pioneering work in C. elegans from Horvitz and colleagues defined the core apoptotic pathway and revealed the conserved role of caspases in the execution of apoptosis5, 6 (Figure 1A). Since then, additional cell death mechanisms have been reported, indicating that apoptosis is not the only mode of PCD. Here, we provide an overview of several major PCD mechanisms and critically discuss the biological significance of these pathways in vivo. Additional details summarizing cell-based and biochemical studies for individual forms of PCD can be found in several excellent recent reviews7–15. Another rapidly expanding area of research that we cover is signaling by apoptotic cells. Traditionally, it was thought that dying cells have limited signaling capacity being rapidly cleared by phagocytes. However, it is now clear that apoptotic cells release a multitude of signals that profoundly affect their cellular environment. These signals include mitogens to promote proliferation and tissue repair, and death factors to stimulate coordinated cell killing. This extraordinary complexity in the regulation and execution of cell death poses significant experimental challenges, but also presents exciting new opportunities for clinical translation.
Box 1. Programmed cell death in model organisms.
The Caenorhabditis elegans, Drosophila melanogaster and Mus musculus model systems have shaped our understanding of how cells undergo programmed cell death (PCD). C. elegans provides unique opportunities for experimentation due to its defined and invariant cell lineage. In ontogeny of the hermaphrodite worm, 131 of 1090 somatic cells are eliminated by PCD, generating adults with 959 cells172. In loss-of-function mutants for the pro-apoptotic genes egl-1, ced-3 and ced-4, cell death is blocked resulting in survival of these 131 cells; however, the lifespan and behavior of the worm are not affected.
D. melanogaster is considerably more complex, and cell fate and number are not pre-determined but depend on extracellular signals and environmental factors. Therefore, D. melanogaster offers unique opportunities for studying PCD in the context of developmental plasticity and tissue homeostasis. The most prominent form of developmental PCD in the fly is apoptosis, and inhibition of this process causes severe developmental defects, malformations and organismal lethality40–42, 173. However, inhibition of apoptosis does not affect the elimination of specific cells such as nurse cells, indicating that apoptosis is not the only PCD mechanism in flies174.
Consistent with increased organismal complexity, the apoptotic machinery in vertebrates is even more intricate, and is involved in regulating crucial events throughout the organism’s life span. Therefore, it was surprising that mice deleted for key components of the apoptotic machinery only have minor developmental defects and can reach adulthood11. The simplest explanation for the lack of overt phenotypes may be functional redundancy between apoptotic proteins22. However, another possibility is that cells are eliminated by alternative PCD mechanisms when apoptosis is blocked11. Nevertheless, the inhibition of apoptosis in many situations causes embryonic lethality, developmental abnormalities and various pathologies (Table 1).
These developmental studies have been complemented by different models to explain why cells need to die during development: sculpting; deleting structures; supplying nutrients; regulating cell number; and eliminating abnormal cells8, 175.
Figure 1. The core of the apoptotic machinery is conserved.

Functional homologous of apoptotic proteins in C. elegans, Drosophila melanogaster and mammals uncover evolutionary conservation of the apoptotic pathway. A. In C. elegans, EGL-1 (BH3-only protein homolog) binds and inhibits CED-9 (BCL-2-family homolog), resulting in the release of CED-4 (APAF1 homolog) from the CED-9–CED-4 complex. This enables the elimination of cells by CED-3 (caspase). B. In Drosophila, the IAP antagonists Reaper, Hid and Grim (RHG) mediate the degradation of DIAP1, thus liberating Drice and Dcp-1. In addition, this enables Dronc (Caspase-9 homolog) to interact with dArk and form the apoptosome that efficiently activates executioner caspases. The p35 protein is a specific inhibitor of executioner caspases and the activation of the apoptosome might be regulated by the Bcl-2 family members Debcl and Buffy (hence depicted by a dashed line). C. In mammals, the crucial decision as to whether a cell commits to apoptosis is regulated by the fine interplay between the anti-apoptotic BCl-2 subfamily of proteins and the pro-apoptotic BH3-only subfamily of proteins. During apoptosis, BH3-only proteins facilitate a BAX- and BAK-dependent release of cytochrome c from the mitochondria, which binds to APAF1 and gives rise to the apoptosome. In parallel, IAP antagonists including DIABLO, HTRA2 and ARTS translocate from mitochondria and release caspases from their negative regulation by IAPs. In particular, Caspase-9 is liberated from XIAP and activated by the apoptosome, triggering executioner caspases-3 and -7. Homologous proteins are similarly depicted in the three panels.
Type I cell death: apoptosis
Caspases: the cellular executioners
Apoptosis is the most prominent and best-studied mode of PCD during development9, 16. This conserved process, which can be triggered both intrinsically (for example, by DNA damage) or extrinsically (for example, by growth factor withdrawal, steroid hormones, ligation of death receptors), culminates in the activation of caspases, a class of cysteine proteases that are expressed as inactive zymogens in virtually all cells (Figure 1)17, 18. Interestingly, whereas C. elegans is equipped with four caspases, flies and mice contain multiple caspases (7 and 13, respectively), suggesting that higher organismal complexity is matched with a larger number of caspases. Although many of the caspases are instrumental in the execution of apoptosis, these proteins also have non-apoptotic functions in various processes, including immunity, cellular remodeling, learning, memory and differentiation8, 9, 19.
Traditionally, caspases have been subdivided into initiator caspases [G] (Dredd and Dronc in Drosophila; caspase-1, -2, -4, -5, -8, -9, -10, -11, and -12 in mammals) and executioner caspases [G] (Ice and Dcp-1 in Drosophila; caspase-3, -6, -7, and -14 in mammals). Some members of the caspase family can be compensated for whereas others have non-redundant essential functions in vivo (Table 1) 10, 11, 13. Initiator caspases have long N-terminal prodomains enabling the formation of protein platforms that regulate caspase activation. An example is the interaction of the caspase-9 prodomain with apoptotic peptidase-activating factor 1 (APAF1) and cytochrome c, which produces the apoptosome [G] that is thought to initiate apoptosis20. Alternatively, it has been suggested that this platform is responsible for signal amplification rather than initiating the apoptotic casacade, which could explain how executioner caspases can be activated in mice (and flies) mutant for APAF1 (Drosophila Ark) and caspase-9 (Dronc)21, 22, 23. In addition, this provides a potential mechanism for how executioner Caspase -3 and Caspase-7 can regulate upstream events, such as mitochondrial outer membrane permeabilization (MOMP) [G] and the release of cytochrome c24, 25. According to this model, at least in some scenarios, executioner caspases may actually function upstream of initiator caspases.
Table 1.
Physiological function of key cell death genes.
| Key cell death genes | Functional classification | Mutant phenotype | Physiological Function from mutant and RNAi studies |
|---|---|---|---|
| C. elegans | |||
| Egl-1 | BH3 only | Animals are viable and have an additional 131 cells. | Essential for inhibiting CED-9. |
| ced-9 | Bcl-2 like | Developmental lethality due to massive cell death. | Inhibits cell death. |
| ced-4 | Apaf-1 like | As for Egl-1 mutation | Essential for activating CED-3. |
| ced-3 | Caspase-9 like | As for Egl-1 mutation | Essential for cell death. |
| Drosophila | |||
| hid | IAP antagonist | Mutants die as embryos with extensive defects in apoptosis. | Essential for most apoptosis. |
| Reaper, Hid and Grim | IAP antagonists | Mutants die as embryos with extensive defects in apoptosis. | Essential for most apoptosis. |
| Ark | APAF1 like adaptor | Pupal lethality. Defects in developmental death. | Essential for most apoptosis. |
| DIAP1 | IAP | Embryonic lethal due to excessive apoptosis. | Inhibits Dronc, Drice and Dcp-1. |
| Dronc | Caspase-9 like | Pupal lethality. Defects in developmental death. | Essential for most apoptosis. |
| Dcp-1 | Executioner caspase | Mutants are viable. | Can be compensated by Drice similar to mammalian caspases. |
| Ice and Dcp-1 | Executioner caspases | Prepupal lethality. Phenotype is similar to Dronc | Essential for apoptosis. |
| Mammals | |||
| Bcl-2 | BCL-2 subfamily | Abnormal death of renal epithelial progenitors. Fatal kidney disease. | Inhibits apoptotic induction. |
| Puma, Bid and Bim | BH3 subfamily | Similar to Bax and Bak mutants. | Essential for initiation of apoptosis at the mitochondria. |
| Bax and Bak | BAX subfamily | Most mice die during embryogenesis. Neonates display partial syndatcily, imperforated vaginas and an increase in hematopoietic cells | Essential for apoptosis induction. |
| Smac | IAP antagonist | Mice appear normal. | Physiological function found with Caspase-3 (see below). |
| Htra2 | IAP antagonist | Lethal postnatal neurodegenerative disease. | Phenotype does not seem to be related to apoptosis. |
| Sept4/Arts | IAP antagonist | Increased apoptotic resistant stem cell numbers, improved regeneration and increased malignancies. | Important for stem cell apoptosis, tumorgenesis and regeneration. |
| Apaf1 | Capsase-9 adaptor | Similar to Caspase-9−/− mice. Some mice survive adulthood. | Essential for Caspase-9 activation. |
| Xiap | IAP | Mice display impaired wound healing and hair follicle stem cells exhibit increased susceptibility to apoptosis. | Important for stem cell apoptosis and skin regeneration. |
| Caspase-3 | Executioner Caspase | Phenotype depends on genetic background. Perinatal lethal in FVB129 and no phenotype in C57BL/6 mice probably due differences in caspase-7 expression levels. | Essential for neuronal cell death and may have a redundant role in other tissues. |
| Caspase-7 | Executioner Caspase | Mutants appear normal | No apparent apoptotic defects in mutants. Appears to compensate in the absence of caspase-3 |
| Caspase-3 and caspase-7 | Executioner Caspase | Perinatal lethal. Defects in cardiac development. MEFs display apoptotic resistance. | Caspase-3 and -7 are required for execution of apoptosis. |
| Caspase-3 and Smac | Executioner Caspase and IAP antagonist | Perinatal lethality. Somewhat similar to Caspase3−/− and Caspase7−/− mice. | Sensitizing deletion of caspase 3 demonstrates that SMAC modulates Caspase-7 via IAPs |
| Caspase-8 | Initiator caspase | Embryonic lethal. Death receptor-mediated apoptosis is completely impaired | Essential for extrinsic apoptosis mediated by the TNF family and inhibition of necroptosis. |
| Fadd | Caspase-8 adaptor | Similar to Caspase-8−/− mice | Essential for Caspase-8 activation. |
| Caspase-8 and Rip3k | Initiator caspase and Necroptosis factor | Mice develop normally but develop lymphadenopathy by four months of age. | Caspase-8 and RIP3K together seem to be dispensable for mammalian development. |
| Caspase-9 | Initiator caspase | Perinatal lethal, but some animals survive to adulthood. Defects in the CNS. | Required for efficient apoptosis in some cell types. |
Apoptosome formation
A decisive point that determines whether the cell commits to apoptosis is MOMP, which results in the release of cytochrome c from the mitochondria. MOMP is regulated by members of the B-cell lymphoma-2 (BCL-2) family, which contains both anti-apoptotic and pro-apoptotic members (Figure 1)10. The interplay between BCL-2 proteins regulates whether a cell undergoes apoptosis. The assembly of BAX–BAK oligomers within the mitochondrial outer membrane promotes MOMP and the release of intermembrane proteins, such as cytochrome c, to the cytosol. This in turn promotes apoptosome formation and the activation of executioner caspases, thus propagating a proteolytic cascade that leads to the cleavage of large numbers of crucial cellular target proteins, including inactivation of a DNase inhibitor, which causes nuclear DNA fragmentation 17, 22, 26.
The activation of caspases can also occur as a result of death receptor ligation, which results in binding of the initiator caspase-8 to its adaptor FADD (Fas-associated via death domain) and assembly of the death-inducing signaling complex (DISC) [G], as discussed in the necroptosis section below.
Inhibitor of apoptosis proteins (IAPs)
Another important layer of PCD regulation involves the inhibition of caspases by the IAP family (Figure 1)8. IAPs were originally discovered in insect viruses and contain at least one baculovirus inhibitory repeat (BIR) domain [G]27, 28. A large number of cellular IAPs have been described in insects and vertebrates and were grouped into a “family”, but it is important to recognize that these are functionally diverse proteins, many of which have no direct role in apoptosis. Compelling evidence for a crucial role of some IAPs in the regulation of apoptosis came from the analyses of loss-of-function and gain-of-function mutations of Drosophila IAP1 (Diap1)29–31. Inactivation of Diap1 causes large-scale caspase activation and apoptosis, whereas gain-of-function mutations inhibit cell death, indicating that DIAP1 has an essential role in caspase regulation in vivo. IAPs can bind and inhibit caspases in vitro through their BIR domain28. Several IAPs also have a RING (really interesting new gene) motif, enabling them to function as E3 ubiquitin ligases and promote ubiquitylation of key cell death proteins28, 32, 33. In addition, some IAPs contain a ubiquitin-associated (UBA) domain, facilitating interaction with ubiquitylated proteins, and a caspase-recruitment domain (CARD) that mediates homophilic interaction with caspases32, 33. DIAP1 has two important functions by virtue of its RING domain: first, it ubiquitylates caspases thus suppressing apoptosis; second, it auto-ubiquitylates and targets itself for degradation once the cell has committed to apoptosis30, 34–36. Interestingly, DIAP1-mediated ubiquitylation involves both degradative ubiquitylation and non-degradative poly-ubiquitylation that can regulate caspase activity 37, 38.
The mammalian orthologue of DIAP1 is the X-linked inhibitor of apoptosis (XIAP) protein, which also functions as an E3 ligase and is considered to be the most potent caspase inhibitor in vitro39. XIAP-deficient mice are viable and for many years were thought to have only “mild” phenotypes32. However it has recently become clear that XIAP has a crucial role in stem cell apoptosis (see below).
IAP antagonists
For a cell to commit to apoptosis, IAPs must be inactivated by endogenous antagonists (Figure 1). A family of natural IAP antagonists was originally discovered by screening for cell death-defective mutants in Drosophila40. This revealed the essential role of three closely linked genes — reaper, hid and grim (which encode the RHG proteins) — for the induction of apoptosis 40–42. RHG proteins have a common N-terminal IAP-binding motif (IBM) that is structurally conserved between flies and mammals and is essential for IAP binding, IAP inactivation and cell killing43.
Deletion of RHG genes blocks apoptosis, and their ectopic expression is sufficient for the killing of many different cell types40–42, 44. In addition, DIAP1 mutations that reduce RHG protein binding protect against apoptosis, indicating that DIAP1 is a direct and functionally relevant target for the pro-apoptotic activity of RHG proteins (Figure 1B). Furthermore, RHG proteins physically interact with each other to form heteromeric complexes at the mitochondrial outer membrane that are crucial for cell killing in vivo45. Significantly, RHG genes are transcriptionally regulated by embryonic patterning pathways and by many different pro-apoptotic signals14. This indicates that the convergence of different death-inducing signals occurs, at least in part, via a transcriptional mechanism. Finally, the pro-apoptotic activity of the Hid protein is blocked upon phosphorylation by the MAP kinase ROLLED, and Hid is targeted for cell survival by the epidermal growth factor receptor–Ras signaling pathway46, 47. Collectively, these studies established the important physiological role of IAPs and their antagonists and revealed that caspases are controlled by both activating and inhibitory pathways48, 49.
Mammals also have IAP-binding proteins, most notably SMAC (also known as DIABLO) and HTRA2 (also known as Omi and PARK13)50–52. In addition, a large number of mitochondrial proteins that can interact with IAPs through their N-terminal IBM have been identified but remain to be functionally characterized53. SMAC and HTRA2 are localized to the mitochondrial intermembrane and translocate to the cytoplasm upon MOMP (Figure 1C). It is thought that upon cytoplasmic release, these proteins promote cell death by binding IAPs, which promotes caspase activation. However, rigorous genetic evidence to support this model in vivo is still lacking. _HtrA2_−/− animals and cells do not have reduced cell death rates and actually lose a population of neurons in the striatum, resulting in a lethal postnatal neurodegenerative disorder with features of Parkinson disease. This phenotype appears to be due to loss of the protease function of HTRA2 and not its IAP-binding IBM54.
Similarly, _Smac_−/− mice have no overt phenotype, and _Smac_−/− mouse embryonic fibroblasts (MEFs), lymphocytes and hepatocytes respond normally to apoptotic stimuli55. Although a redundancy between SMAC and HTRA2 has been proposed, Smac−/−HtrA2−/− mice have a similar phenotype to their parental single-knockout strains54. An alternative explanation for the physical association of SMAC and HTRA2 with XIAP is that binding functions to promote the ubiquitylation and proteasome-mediated degradation of SMAC and HTRA2 upon release into the cytoplasm (a “cleanup” mechanism)53. Small-molecule IAP antagonists based on the conserved IBM of RHG proteins and SMAC (often referred to as “SMAC-mimetics”) have been developed and are currently under clinical investigation as pro-apoptotic cancer therapeutics56. Initially designed to inhibit XIAP, these compounds target primarily cIAP1 and cIAP2 and sensitize cancer cells to tumour necrosis factor (TNF)-related apoptosis57, 58.
Another mammalian IAP antagonist is ARTS, which is encoded by the SEPT4 gene. ARTS does not contain an IBM but similarly to the RHG proteins, it is localized to the mitochondrial outer membrane59, 60. ARTS functions upstream of MOMP and mitochondrial protein release25. _Sept4_−/− mice are defective in caspase-mediated elimination of bulk cytoplasm during spermiogenesis, have increased numbers of hematopoietic stem and progenitor cells and have increased susceptibility towards developing malignancies61, 62. XIAP is the direct biochemical and physiological target of ARTS, and both proteins are crucial for the regulation of cell death in hair follicle stem cells (HFSCs) [G]60, 63. Loss of ARTS protects HFSCs against apoptosis, increases HFSC number, and leads to marked improvement in wound healing63. Furthermore, _Sept4_−/− mice have improved regeneration of hair follicles from the wound bed and significantly smaller scars63. Conversely, inactivation of XIAP abrogates these phenotypes and impairs wound healing, revealing an in vivo role for XIAP and the importance of apoptosis in regulating HFSC expansion63.
In summary, opposing regulatory pathways simultaneously regulate caspase activity during apoptosis, through either caspase activation (by APAF1) or caspase inhibition (by IAPs), which are coordinately subjected to many layers of upstream regulation. The cell is killed by the apoptotic machinery only after multiple checkpoints have been cleared to enable effector caspase activity that is sufficiently widespread and at a sufficiently high level to cause extensive proteolysis of crucial cellular proteins. Finally, even at a late stage in apoptosis cells can sometimes recover, a phenomenon termed "anastasis" (Greek for “rising to life”)64. This could be a mechanism to preserve cells that are difficult to replace.
Non-apoptotic programmed cell death
Although apoptosis is the most prominent form of PCD during development, emerging evidence indicates that it is not the only mechanism11. In C. elegans, virtually all PCD occurs by apoptosis, except for the linker cell [G], which dies in a caspase-independent manner5, 65. Dying linker cells have unique morphological features, which are also seen in certain neurons that die during spinal cord development of vertebrates, suggesting that this PCD mechanism is conserved throughout evolution (Box 2). In addition, as detailed in the following sections, autophagy and necroptosis have been intensively studied in the context of PCD.
Box 2. Linker cell death.
The linker cell in Caenorhabditis elegans migrates along a predetermined path and completes its migration when positioned between the gonad and cloacal tube, which functions as the sperm’s exit channel. Death of the linker cell prior to completion of its migration results in severe defects in gonadal elongation and impaired male fertility65. The microRNA let-7 and its downstream target, LIN-29, are crucial components regulating linker cell death. In let-7 and LIN-29 mutant animals, the linker cell survives and the connection between gonadal and cloacal tubes fails to form, resulting in sperm accumulation65. However, LIN-29 is also expressed in other cells that are not doomed to die, indicating that LIN-29 is necessary but not sufficient for linker cell death. Recently, it was elegantly shown that linker cell death also requires SEK-1, TIR-1 and the polyglutatamine-repeat protein pqn-41176. However, elucidation of the downstream targets responsible for the demolition phase of the linker cell, as well as of other key death-inducing genes that control linker cell death, is ongoing. Of particular interest, the Drosophila and mouse TIR-1 orthologues (Ect4 and SARM1, respectively) have a role in Wallerian degeneration. Upon injury, loss of Ect4 or SARM1 suppresses axonal degeneration, which indicates that axons can actively promote their own demise and that TIR-1, Ect4 and SARM1 are implicated in non-apoptotic mechanisms of programmed cell death177.
Dying linker cells exhibit non-apoptotic characteristics, such as nuclear envelope crenellation, lack of chromatin condensation, organelle swelling, and increased cytoplasmic membrane-bound structures65, 176. These morphological features resemble cell death during mammalian development, as in the case of chick ciliary ganglion cells and chick spinal cord motor neurons, suggesting that linker cell death is a morphologically conserved form of cell death. Furthermore, nuclear envelope crenellation is implicated in several polyglutatamine expansion diseases, raising the possibility that neurodegeneration is promoted by inappropriately activating a linker cell death like machinery. Future work will shed light on the evolutionary conservation of this intriguing cell death program and its role in development and disease.
Type II cell death: autophagy
Macroautophagy (hereafter referred to as autophagy) is a catabolic process involving the degradation of cytoplasmic components, protein aggregates and organelles by forming autophagosomes, which are degraded by fusion to lysosomes66. This process has been extensively studied in the yeast Saccharomyces cerevisiae in response to starvation, where it functions to protect cells from death by recycling cell contents. Autophagy depends on a large group of evolutionarily conserved ATG (autophagy-related) genes66. In accordance with the protective, pro-survival function of autophagy, silencing and deletion of ATG genes resulted in accelerated cell death12, 67. However, in certain scenarios (and dependent on the organism) it has been suggested that autophagy can lead or contribute to cell death.
Historically, type II cell death was termed autophagic cell death [G], which perhaps misled people to perceive that cell death is achieved by the autophagic machinery12. However, this name simply indicates that cell death is accompanied by large numbers of autophagosomes, thus raising the question of whether autophagy is an unsuccessful attempt at survival rather than a bona fide cell death mechanism.
In Drosophila, dying cells of the salivary glands contain autophagic vacuoles and express ATG genes7, 68. Moreover, deletion of ATG genes attenuates both autophagy and PCD69. Interestingly, the engulfment receptor Draper is required for autophagy during the death of salivary gland cells70. Furthermore, compelling evidence indicates that caspase activity is necessary for the degradation of these cells, indicating the involvement of an apoptotic mechanism. First, anti-apoptotic Diap1 is repressed and pro-apoptotic genes (rpr and hid) are activated prior to the death of salivary gland cells71. Second, dying cells are positive for apoptotic markers66 (such as acridine orange and TUNEL). Third, deletion of Ark (the Drosophila ortholog of Apaf1) or caspase inhibition rescues these cells from death68, 72–76. Thus, autophagy alone is not sufficient for salivary gland cell death.
Autophagic cell death is also associated with the destruction of Drosophila midgut cells. Death of these cells has been characterized by both caspase activation and autophagy; however, recent experiments show that mutations in caspases or their regulators do not rescue these cells from destruction77. By contrast, the inhibition of autophagy genes (Atg1, Atg2 and Atg18) delays midgut degradation without a decrease in caspase activity, which indicates that the death of midgut cells involves a caspase-independent form of PCD that requires autophagy77. Intriguingly, this work also suggests an unknown mechanism for the activation of caspases in these cells, as the executioner caspase Decay was activated in the absence of the initiator caspase Dronc.
In mice, it has also been suggested that autophagy functions as an alternate killing mechanism in situations where apoptosis cannot be executed. Bax−/−Bak−/− MEFs are resistant to various inducers of apoptosis but can undergo cell death in what seems to be an autophagy-mediated manner78, 79. Knockdown of the mammalian ATG gene Beclin 1 (Becn1; ATG6 in yeast) or of Atg5 reduced cell death in Bax−/−Bak−/− MEFs in response to etoposide and starosporin73. Intriguingly, autophagy was not induced when downstream apoptotic processes were inhibited in Casp9−/− or Apaf1−/− MEFs or in response to the addition of the pan caspase inhibitor zVAD-fmk, indicating that BAX and BAK might have a specific role in regulating autophagy. In this regard, it is intriguing that BECN1 has a BH3 domain, which enables the interaction with pro-survival BCL-2 family members (BCL-2, BCL-XL and MCL-1)12, 80. This interaction is conserved and is also seen in C. elegans, where BECN1 binds CED-9 (a BCL-2-like protein)81. The direct interaction between BCL-2, BCL-XL and BECN1 inhibits the pro-autophagy function of BECN1 and is dependent upon subcellular localization80 (Figure 2A). The autophagy-inhibiting BCL-2–BCL-XL–BECN1 complexes localize to the endoplasmic reticulum (ER), indicating that autophagy is regulated in an organelle-specific manner82 (Figure 2A). Interestingly, the BH3 domain of BECN1 also regulates dissociation of this complex. The death-associated protein kinase 1 (DAPK1) can phosphorylate the BH3 domain of BECN1, resulting in reduced affinity for BCL-XL and dissociation of the complex, which leads to the induction of autophagy83. In addition, by competitive interaction, the BH3-only BNIP3 protein can also dissociate the BCL-2–BCL-XL–BECN1 complex84. Finally, BECN1 has been found to be a substrate of caspase-3, and caspase-mediated cleavage of BECN1 abrogated its interaction with BCL-2 (Figure 2A)85. Taken together, the results indicate that the BH3 domain of BECN1 can regulate the association–dissociation kinetics of the BCL-2–BCL-XL–BECN1 complex and thus shift the balance between autophagy and apoptosis85.
Figure 2. Crosstalk between autophagy- and apoptosis-related proteins.

The BH3 domain of the autophagy-related protein Beclin 1 (BECN1) enables the formation of a BCL-2–BCL-XL–BECN1 complex that localizes to the ER and inhibits autophagy. Competitive interaction by the BH3-only protein BNIP3 and phosphorylation by the death-associated protein kinase (DAPK) regulate the dissociation of this complex and drive autophagy. In addition, BECN1 has also been found to be a substrate of caspase-3. B. The autophagy-related protein ATG5 can undergo calpain-dependent cleavage, which switches its function from pro-survival to pro-death. The C-terminal cleavage gives rise to a pro-apoptotic product that translocates to the mitochondria, binds BCL-XL and induces apoptosis. ATG5 can also induce apoptosis by binding to FADD, and it seems that apoptotic features such as phosphatidylserine exposure and membrane blebbing depend upon autophagy for the provision of energy in the form of ATP.
ATG5 may also have a dual role in regulating both autophagy and apoptosis. ATG5 can undergo calpain-dependent cleavage, which switches its function from a pro-survival autophagy protein to death induction. This cleavage removes the C-terminal domain of ATG5 giving rise to a pro-apoptotic product that translocates to the mitochondria where it induces the intrinsic apoptotic program (Figure 2B)86. Interestingly, ATG5 can also bind FADD via its C-terminal domain and stimulate caspase-dependent apoptotic cell death87.
There is some indication that autophagy functions to enable the classic hallmarks of apoptosis, such as phosphatidylserine exposure and membrane blebbing. These processes depend on ATP, and autophagy may help in this regard by generating the metabolic substrates required for the bioenergetics needs of the dying cell. An elegant study demonstrated that ATP produced by the autophagic machinery is responsible for the generation of the find-me and eat-me signals, lysophosphatidylcholine and phosphatidylserine, respectively, in mouse embryoid bodies (Figure 2B)88. Embryoid bodies derived from Becn1−/− or Atg5−/− embryonic stem cells were able to undergo the initial stages of apoptosis, but were unsuccessful in producing the signals required for phagocytic clearance88. Additionally, autophagy might supply the ATP required for membrane blebbing during apoptosis89.
A major unresolved question is to what extent autophagy actually mediates cell death, rather than simply occurs with it. Recently, using a systematic approach to examine the existence of autophagic cell death in mammalian cells, it was shown that of 1,400 death-inducing compounds not one eliminated cells through the induction of autophagy90. Silencing of key autophagy genes was not only ineffective in preventing cell death but actually accelerated it90. As these results are negative in nature they do not exclude the possibility of cell death mediated by autophagy, but they indicate that autophagy does not have a major role in promoting cell death.
Type III cell death: regulated necrosis
For many years necrosis was regarded as an unregulated mode of cell death caused by overwhelming trauma. However, a large number of recent studies point to the existence of multiple modes of regulated necrosis15. Necrosis is characterized by organelle and cellular swelling, rupture of the plasma membrane and release of the intracellular content15. Different modes of regulated necrosis share common morphological features15. Here, we focus on the best-studied form of regulated necrosis, termed necroptosis, which denotes a type of necrotic cell death that depends on RIPK1 (Receptor-interacting protein kinase 1) and/or RIPK313–15. Additional terms have been suggested for necrosis induced by different stimuli, but it remains to be shown that these actually represent different execution mechanisms for programmed necrosis15.
Necroptosis
The term “necroptosis” was coined to specify a non-apoptotic mode of cell death that is elicited by TNF receptor 1 (TNFR1) when caspases are inhibited91. This form of PCD can be inhibited by necrostatin-191 [G]. Although it is just one type of regulated necrosis, necroptosis has often been regarded as synonymous with regulated necrosis91, 92.
Necroptosis can be induced by the ligation of various death receptors, including FAS (CD95), TNFR1 and TNFR2, different TLRs (Toll-like receptors) and intracellular sensors such as DAI and PKR (protein kinase R)93. The TNFR signaling pathway best exemplifies how the necroptotic program can be induced. Upon TNF binding, TNFR1 recruits RIPK1, TRADD (TNFR-associated death domain), cIAP1 and cIAP2, and TRAF2 (TNFR-associated factor 2) and TRAF5, which together form a complex known as complex I (Figure 3A)94. cIAPs mediate Lys63-linked ubiquitylation of RIPK1, enabling the docking of TAK1, TAB2 and TAB3, which results in the activation of the IKK complex. In turn this complex targets IκBα for degradation, liberating NF-κB (nuclear factor kappa B). NF-κB translocates to the nucleus, where it drives transcription of pro-survival genes as well as genes encoding negative feedback proteins, such as IκBα and SEF95–98. Two such pro-survival NF-κB target genes are those encoding A20 and FLIP (Flice-Like-inhibitory-protein). A20 polyubiquitylates RIPK1 with Lys48-linked chains, thus marking it for proteasomal degradation (Figure 3A), whereas the deubiquitylating enzyme cylindromatosis (CYLD) eliminates Lys63-linked ubiquitin chains from RIPK1, leading to the dissociation of RIPK1 from complex I98, 99. This deubiquitylation event changes the function of RIPK1 from promoting survival to promoting death via formation of the death-inducing signaling complex (DISC; also known as complex IIa) comprised of RIPK1, RIPK3, TRADD, FADD, caspase-8 and FLIP15, 94. The DISC has a dual role: it facilitates the cleavage and degradation of CYLD, RIPK1 and RIPK3 to promote survival and it enables the homo-dimerization and catalytic activation of caspase-8 to stimulate apoptosis (Figure 3A). In mice, deletion of the complex IIa components caspase-8 and FLIP results in embryonic lethality100, 101, 102. However, these mice are rescued when RIPK3 is removed (Table 1), suggesting that a key function of complex IIa is to regulate necroptosis (see below)103–105. Furthermore, deletion of Ripk1 results in postnatal lethality that can be prevented by the ablation of Ripk3 together with either Caspase-8 or Fadd (Table 1)106, 107.
Figure 3. TNF-mediated survival, apoptosis and necroptosis.

A. TNF ligation results in recruitment of RIPK1, TRADD, cIAP1 and cIAP2, and TRAF2 and TRAF5 to the TNFR1 to form complex I. cIAP1 and cIAP2 mediate Lys63-linked ubiquitylation of RIPK1, enabling docking of TAK1 (transforming-growth-factor-β-activated-kinase-1) and its binding partners TAB2 and TAB3. The signal is then perpetuated to IKK ((IκBα (inhibitor of kappa B) kinase)), which degrades IκBα, the cytoplasmic inhibitor of canonical NFκB. Once liberated from IκBα, NFκB translocates to the nucleus where it drives the transcription of pro-survival genes as well as feedback antagonists. Two pro-survival NFκB target genes are A20 and FLIP which promote association of RIPK1 with cytoplasmic RIPK3, TNF-receptor-associated-death-domain (TRADD), FADD, caspase-8 and the NF-κB target FLIP to form the death-inducing signaling complex (DISC). FLIPL can hetrodimerize with Caspase-8 and facilitate the cleavage and degradation of CYLD, RIP1K and RIPK3. However, the DISC also enables homo-dimerization and catalytic activation of caspase-8, which activates caspase-3 and caspase-7 to induce apoptosis. When caspase-8 is deleted or inhibited, RIPK1 interacts with RIPK3, resulting in the formation of the necrosome, an interaction that can be inhibited by necrostatin 1 (nec-1). RIPK3 recruits and phosphorylates MLKL, leading to the formation of oligomers that translocate to the plasma membrane. Once at the plasma membrane, MLKL forms membrane-disrupting pores, regulating both Na+ and Ca2+ influx, resulting in membrane rupture. B. cIAPs also function as negative regulators of the non-cannonical NF-kB pathway. In resting cells, a complex encompassing cIAP1, cIAP2, TRAF2 and TRAF3 targets and degrades NIK. Upon ligation of TNFR1, the TRAF2-TRAF3-cIAP1/2 complex is recruited to the receptor, changing the substrate specificity from NIK to the components of the complex. This results in increased NIK levels, phosphorylation of IKKα and p100, proteolytic processing of p100 to p52, and translocation to the nucleus. When cIAPs are depleted the canonical NF_κ_B pathway is inhibited whereas the non-canonical NF_κ_B pathway is not. Under these conditions, the large (~2-MDa) ripoptosome is assembled and, similarly to complex IIa, can stimulate caspase-8-mediated apoptosis.
When Caspase-8 is inactivated, RIPK1 associates with RIPK3, resulting in autophophorylation, transphosphorylation and formation of the necrosome (Figure 3A)108, 109, 110. Accordingly, targeting the kinase domain of RIPK1 with necrostatin 1 inhibits the interaction between RIPK1 and RIPK3 but leaves the pro-survival NF-κB pathway downstream of RIPK1 unaffected91, 111, 112. The phosphorylation of RIPK3 (on Ser227 in humans or Ser232 in mice) enables the recruitment of MLKL (mixed lineage kinase domain like) to initiate necroptosis113, 114. MLKL is phosphorylated by RIPK3, leading to the formation of oligomers that translocate to the plasma membrane, bind phosphatidylinositol phosphates and form membrane-disrupting pores115, 116. The perturbation of membrane integrity seems to induce both Na+ and Ca2+ influx, leading to a rise in osmotic pressure and membrane rupture (Figure 3A)117, 116, 118.
Ripk3−/− or Mlkl−/− mice do not have developmental or homeostatic defects106, 119. However, as many downstream signaling components of necroptosis remain elusive, it is too early to conclude whether necroptosis has a role in developmental or homeostatic processes. By contrast, there is evidence that necroptosis functions in response to bacterial and viral infection104. When Ripk3−/− mice are challenged with vaccinia virus they have increased viral titers and succumb to infection more rapidly than wild-type mice109. This is a result of impaired virus-induced tissue necrosis, inflammation, and control of viral replication. Hence, necroptosis can have two distinct functions. On the one hand it functions as a back-up mechanism to eliminate infected cells when apoptosis is inhibited, and on the other hand it promotes the release of DAMPs (damage associated molecular patterns) [G] to induce the immune response. Importantly, viruses encode specific inhibitors, such as viral inhibitor of caspase activation (v-ICA), viral FLIPs (v-FLIPs) and viral inhibitor of RIPK1 (v-IRA)120, 121, that can affect necroptosis. Interestingly, some viruses (such as murine cytomegalovirus) can block both apoptosis and necroptosis as they encode both a caspase-8 inhibitor and a RIPK3 inhibitor122, 123.
The induction of a pro-inflammatory response upon necroptosis has been observed in various organs, such as the skin and intestine. Epidermal deletion of Fadd and caspase-8 results in skin inflammation, which can be reversed by Ripk3 deletion124, 125. Similarly, mice in which the intestinal epithelial cells were deleted for caspase-8 have ileitis as a result of increased necroptosis within the stem cell crypts126. These data, together with the ability of caspase-8 to cleave RIPK1 and RIPK3, identify caspase-8 as a key regulator of inflammation situated at the crossroads of apoptosis and necroptosis.
Programmed cell death through the ripoptosome
In addition to the function of cIAPs as positive regulators of canonical NF-κB signaling, they also have a key role in regulating the non-canonical NF-κB pathway57, 127, 128. In resting cells, non-canonical NF-κB signaling is repressed by a complex of cIAP1, cIAP2, TRAF2 and TRAF3129. This E3 ligase complex binds to NIK (NF-κB inducing kinase), conjugates Lys48-linked ubiquitin chains and targets NIK for proteasomal degradation. Upon ligation of a subset of TNFR-superfamily members (CD40L, TWEAK and BAFF), the TRAF2–TRAF3–cIAP complex is recruited to the receptor, which changes the substrate specificity of the complex from NIK to any of the components of the complex in a manner that depends on the cellular context and the receptor type. As a result, NIK levels are increased, resulting in phosphorylation of IKKα and the NF-_κ_B precursor p100 (Figure 3B). Sequentially, homodimers of IKKα further phosphorylate p100 to promote its proteolytic processing to p52, which can bind to other NF-_κ_B subunits and translocate to the nucleus. In accordance, when cIAPs are depleted (due to chemotherapeutic drugs, cytokine stimulation or SMAC mimetics) the canonical NF-_κ_B pathway is inhibited whereas the non-canonical NF-_κ_B pathway is activated. Under these conditions, TNF is produced and another death-inducing platform is assembled termed complex IIb or the ripoptosome130, 131. The ripoptosome forms in the cytoplasm independently of TNF, CD95L, TRAIL, death receptors and mitochondrial pathways. This large (~2-MDa) complex is comprised of RIPK1, FADD and caspase-8 and is negatively regulated by cIAP1, cIAP2 and XIAP. Similar to the DISC, the ripoptosome can stimulate caspase-8-mediated apoptosis, as well as necroptosis in a FLIPL- and caspase-8-dependent manner (Figure 3B).
Signaling from dying cells
Apoptotic cells are rarely visible in situ due to efficient clearance mechanisms. When clearance is impaired, uncleared corpses become necrotic and induce inflammation and autoimmunity132. In order to ensure proper cell clearance, apoptotic cells secrete “find me” signals that instruct phagocyte attraction including: lysophosphatidylcholine, sphingosine-1-phosphate, and the nucleotides ATP and UTP132, 133,. Additionally, these cells secrete “eat me” signals of which phosphatidylserine is the best known. In viable cells, phosphatidylserine is exclusively retained on the inner leaflet of the lipid bilayer, but is exposed on the extracellular layer during apoptosis133. Recent work has elegantly shown that in response to apoptotic stimuli, caspase 3 cleaves XKR8, which leads to phosphatidylserine exposure134. Interestingly, CED-8 (the only C. elegans Xk-family homologue) also promotes phosphatidylserine exposure and the engulfment of cell corpses in a caspase-mediated manner135. Although the exposure of phosphatidylserine during apoptosis is conserved in evolution, the exact mechanism by which it facilitates cell clearance remains controversial132. As apoptotic cells are rapidly cleared, it was long thought that they had limited signaling capacity besides secreting “find me” and “eat me” signals. However, recent advances have revealed that apoptotic cells are a rich source of signals that profoundly affect their cellular environment, including mitogens that instruct proliferation and regeneration, and death factors that seed additional apoptosis.
Apoptosis-induced proliferation
Regeneration is a process of regrowth or repair that enables organisms to cope with overwhelming trauma136. Research carried out in a diversity of model systems has now revealed that apoptotic cells can secrete mitogens to directly stimulate cell proliferation. Apoptosis-induced proliferation can aid regeneration and explain how developing tissues can compensate for massive cell loss in response to stress or injury.
The original observation that apoptotic cells can secrete mitogens to stimulate the proliferation of adjacent precursor cells came from studies in Drosophila36, 137, 138 (Figure 4A). A technical problem these studies faced was the rapid clearance of dying cells. The key to overcoming this hurdle was to block the execution of apoptosis by expression of p35, which specifically inhibits executioner caspases. The resulting “undead cells” remain in a prolonged apoptotic state and stimulate tissue overgrowth by releasing mitogenic signals139, 140 (Figure 4A). Candidates for these mitogens include Wingless (Wg, a WNT ortholog) and Decapentaplegic (Dpp, the ortholog of mammalian TGF-β and bone morphogenetic proteins), which are activated under these conditions (Figure 4A)36, 138. Furthermore, Wg signaling is required for cell proliferation in this system36. Finally, the JNK pathway also has a key role in promoting apoptosis-induced proliferation and wound healing in Drosophila36, 141.
Figure 4. Apoptosis-induced proliferation.

Apoptotic cells can secrete mitogens that stimulate growth and regeneration. Caspase targets and mitogenic factors are depicted in pink, and question marks indicate uncertainty. A. In Drosophila, p35 inhibits executioner caspases and retains the cells in an “undead” state. Under these conditions, p53 and JNK are activated and promote the release of Wg (Wnt ortholog) and Dpp (TGFb/BMP homolog), which promotes hyperplastic overgrowth through apoptosis-induced proliferation (AiP). B. In Drosophila, in a natural non p35-dependent system, apoptosis induces tissue regeneration by the secretion of Wg. C. In Drosophila, differentiated neurons induce apoptosis-induced compensatory proliferation via Hedghog (Hh), stimulating non-neuronal cell proliferation. In this scenario, the signal is downstream of Drice and Dcp-1 and not Dronc. D. In Hydra, head regeneration is a caspase-dependent process and apoptotic cells secrete Wnt3 to induce apoptosis-induced compensatory proliferation. E. In newts and Planaria, apoptotic cells and caspase activity are seen at the amputation site; however, it remains to be seen whether the apoptotic cells are responsible for the secretion of Wnt and Hh. F. In Xenopus, tail amputation leads to caspase activity, and inhibition of caspase-3 and caspase-9 inhibits proliferation and regeneration. It is unclear whether this form of compensatory proliferation is mediated by Wnt. G. In zebrafish, fin amputation results in ROS activity, which induces caspase and JNK activation. It remains to be established if FGF20, Wnt and sdf1 are secreted from apoptotic cells to regulate apoptosis-induced compensatory proliferation. H. In mice, liver injury results in ROS production and secretion of IL-11 from hepatocytes, which facilitates apoptosis-induced compensatory proliferation. G. In mice, wound repair and liver regeneration depend on caspase-3 and caspase-7. Caspase-3 mediates the proteolysis of iPLA2, producing PGE2, which is known to stimulate stem cell proliferation, wound repair and regeneration.
Initially, it was not clear whether JNK functions independently of the apoptotic program, is a downstream target of Dronc or is an inducer of pro-apoptotic genes36, 142–144. However, a recent study reconciles these differences by revealing a positive feedback loop in which JNK and p53 function both upstream and downstream of Dronc and RHG genes to amplify the apoptotic signal (Figure 4A)145. Interestingly, disruption of the Drosophila Cdc42-Par6-aPKC epithelial polarity complex stimulates JNK-dependent apoptosis and apoptosis-induced proliferation146.
These observations were further extended in p35-independent systems, demonstrating the requirement of Wg and JNK for proper regeneration147, 148 (Figure 4B). Significantly, in the differentiating Drosophila retina, apoptosis-induced proliferation involves Hedghog (Hh) as the primary mitogenic signal149. Hh secretion is induced in apoptotic photoreceptor neurons, dependent not only on the initiator caspase Dronc but also on the executioner caspases Ice and Dcp-1 (Figure 4C). Therefore, p35 blocks apoptosis-induced proliferation in this tissue (but not in the wing disc), which indicates that distinct mechanisms of apoptosis-induced proliferation might operate in different cell types and tissues.
Subsequently, apoptosis-induced proliferation was also observed in other organisms, including Hydra, Planaria, Xenopus, newts, zebrafish and mice. In the freshwater polyp Hydra, apoptosis is both required and sufficient to induce Wnt3 production for head regeneration after mid-gastric bisection150. The MAPK/CREB pathway triggers apoptosis in Hydra and, reminiscent of the Drosophila apoptosis-induced proliferation mechanism, apoptotic cells release Wnt3 in a caspase-dependent manner to promote cell proliferation and regeneration (Figure 4D)150, 151. Planarians and newts also have large numbers of apoptotic cells at the site of amputation, but whether these cells drive regeneration through apoptosis-induced proliferation is still unknown (Figure 4E)152, 153–156. In the Xenopus tadpole, many apoptotic cells are detected 12 hours after tail amputation and caspase activity is necessary for tail regeneration157. However, it remains to be seen whether Wnt, which is known to have a key role in Xenopus regeneration, is secreted from apoptotic cells (Figure 4F). In zebrafish, reactive oxygen species (ROS) are produced in response to adult fin amputation. ROS stimulate both caspases and JNK in a parallel manner, both of which are required for apoptosis-induced proliferation. Furthermore, the data hint that Fgf20, Sdf1 and Wnts might be secreted from dying cells in this scenario (Figure 4G)158. Similarly, it was recently shown in mice that interleukin-11 (IL-11), a member of the IL-6 pro-inflammatory family of cytokines, is produced by hepatocytes in a ROS-dependent manner. IL-11 is released from apoptotic hepatocytes and induces STAT3 phosphorylation and apoptosis-induced proliferation (Figure 4H)159. Interestingly, ROS can also result in hepatocyte necrosis that releases IL-1, which in turn stimulates and induces apoptosis-induced proliferation 160. In addition, dying mouse MEFs can stimulate the proliferation of stem and progenitor cells in a caspase-dependent manner161. Mice deficient for caspase 3 and caspase 7 have impaired wound healing and liver regeneration. Interestingly, the downstream target of these caspases is prostaglandin E2, a promoter of stem or progenitor cell proliferation and tissue regeneration (Figure 4I)161.
Taken together, there is now a considerable body of evidence indicating that a key function of caspases in dying cells is to facilitate the release of mitogens that stimulate stem/progenitor cell proliferation and thereby mediate regeneration. Given the strong connections between stem cells, regeneration and tumor growth, apoptosis-induced proliferation may also contribute to cancer. Collectively, these findings challenge the simplistic view of apoptosis as a tumor-suppressive or -preventive mechanism. On the one hand, apoptosis suppresses tumor formation by removing damaged and unwanted cells. On the other hand, apoptosis may stimulate tumor growth through apoptosis-induced proliferation. Additional support for this idea comes from the finding that prostaglandin E2 is released by bladder cancer cells and stimulates the proliferation of cancer stem cells, thus leading to tumor repopulation162. Furthermore, deficiency for caspase 3 causes sensitivity towards radiotherapy, suggesting a cell death-induced tumor repopulation mechanism163.
In the future, it will be interesting to examine apoptosis-induced proliferation in different human cancers, and to explore dual-agent therapies designed to kill cancer cells while simultaneously blocking mitogens that might induce proliferation.
Apoptosis-induced apoptosis
Developmental apoptosis often involves the removal of large groups of cells in a cohort8, 16. Examples of “communal suicide” include various morphogenic events in Drosophila, the elimination of the Xenopus tadpole tail and removal of the interdigital webbing in vertebrates. However, how this collective death is achieved remains largely undefined. New insights into this question have come from the recent finding that apoptotic cells have the capacity to instruct additional death in their neighborhood in a phenomenon termed apoptosis-induced apoptosis164. In the D_rosophila_ wing disc, which is comprised of an anterior and a posterior compartment, induction of large-scale apoptosis in the posterior compartment surprisingly resulted in the apoptosis of cells located at a considerable distance in the anterior compartment164.
Apoptosis-induced apoptosis in the wing disc uses Eiger (an orthologue of mammalian TNF) to instruct death from afar. Eiger is produced by dying cells of the posterior compartment and activates JNK in anterior compartment cells to initiate apoptosis (Figure 5A, B)164. Apoptosis-induced apoptosis also operates in vertebrates and has an important physiological function in the coordinated elimination of hair follicle cells. The hair follicle undergoes cycles of rest (telogen), growth (anagen) and degeneration (catagen; where cells located in the lower portion of the hair follicle are eliminated by apoptosis)165. During catagen, apoptotic cells produce TNF that is required for cell death and progression of the hair follicle cycle (Figure 5C)164. Similarly, in situations where DNA damage is too great to repair, TNF is expressed by apoptotic cells. In this scenario, ataxia telangiectasia mutated (ATM) activates NF-κB, which induces TNF expression, thus creating a feed-forward loop that promotes apoptosis and IL-8 secretion166. Interestingly, in contrast to these findings, TNF produced by dying retinal neurons in zebrafish drives glia proliferation during retinal regeneration167.
Figure 5. Apoptosis-induced apoptosis.
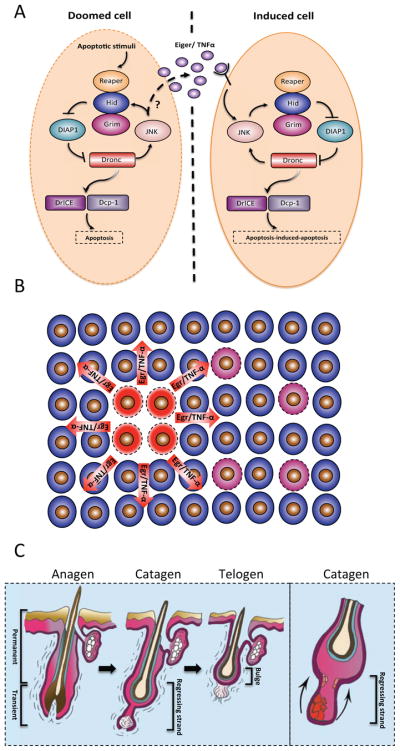
A. Expression of the IAP antagonists Hid or Reaper in ‘doomed’ cells (left panel) can cause apoptosis in distant cells (right panel). The apoptotic signal is propagated by Eiger (a TNF homolog), which can cross compartmental borders (marked by a dashed line), to activate JNK and instruct apoptosis in distant cells. Presumably, the production of Eiger depends on JNK (depicted by a question mark). B. Schematic representation of apoptosis-induced apoptosis. Cells illustrated in red represent the initial apoptotic focal point. These cells emit Eiger in Drosophila and TNF in mice, which result in secondary apoptosis (pink cells). (C right panel) Schematic diagram depicting the hair follicle cycle. Hair follicles cycle between phases of growth (anagen), destruction (catagen) and rest (telogen). During catagen, apoptosis eliminates the lower two-thirds of the hair follicle. Post-catagen, the hair follicle enters the resting telogen phase, followed by a new cycle of hair growth (anagen). (C left panel) Apoptosis-induced apoptosis regulates the hair follicle cycle. During catagen, a primary apoptotic event (red cells), induces a wave of secondary apoptosis (pink cells) that is required for the cohort elimination of transient hair follicle cells.
In Planaria, amputation induces two successive waves of apoptosis: an immediate localized response (peaking after 1–4 hours) and a second systemic response (peaking 3 days post amputation) that can be induced in uninjured organs, presumably to ensure proper remodeling168.
Inter-organ communication can also be seen in Drosophila in response to tissue damage. Wounding the cuticle results in apoptosis in the midgut, an organ distant from the wound site. ROS can mediate the induction of apoptosis in midgut cells, which activates a tissue stem cell regeneration pathway. Intriguingly blocking midgut cell death post wounding leads to fly mortality, suggesting that this mechanism dampens the dangerous systemic wound reaction169.
The observation that apoptotic cells can facilitate additional cell death potentially has many important implications. Apoptosis-induced apoptosis may function in sculpting and deleting tissues where the complete elimination of entire cell groups is required. Additionally, this process might be an efficient means to inhibit viral spreading and might occur in pathologies associated with extensive cell death, such as ischemic stroke, liver failure and neuronal degeneration. Furthermore, apoptosis-induced apoptosis may provide an explanation for the ‘bystander effect”, whereby non-irradiated cells respond in the same manner as radiation-exposed cells170, 171.
In summary, apoptotic cells have the unexpected capacity to stimulate either cell proliferation or further cell death. At this time, it is not clear how the decision between these opposing outcomes is made, and we still have a very incomplete understanding of the role and regulation of cell death in vivo. Further work in this area will likely yield many new surprises, and facilitate the design of rational therapies targeting cell death pathways.
Looking ahead
Since the original proposition of a cellular mechanism of PCD, tremendous progress has been made towards elucidating the biochemical mechanism of apoptosis. In retrospect, it is quite remarkable how the discovery of the C. elegans apoptotic cell death machinery paved the way for unraveling the more intricate systems seen in Drosophila and mammals. However, we are only now beginning to understand the in vivo role of key apoptotic components in mammals_,_ which holds much promise for treating various human disorders.
The synonymy of PCD with apoptosis is now outdated as alternative cell death mechanisms have been described. One prominent process that is often associated with apoptosis is autophagy. However, additional future work is needed to clarify whether autophagy is a bona fide mechanism for cellular killing in vivo.
Another cell death process that is intensely studied is necroptosis. Perhaps the main obstacle limiting our understanding of this process is the challenge to identify necroptotic cells in vivo, and to distinguish whether a cell dies from necroptosis or from necrosis generated in another manner. Also, it remains to be clarified whether necroptosis drives inflammation or is driven by it.
At this time we do not understand why there are so many different ways for a cell to die, how cells are selected in vivo to activate any of these PCD mechanisms, and how survival and death signals are integrated to direct a specific cell fate. Finally, an exciting field of research concerns signals emanating from dying cells. Much remains to be discovered regarding the complex signals that dying cells use to instruct their neighbors, and their role under physiological conditions. Given the importance of cell death during development, homeostasis and disease, deciphering the intriguing “last words” of dying cells promises exciting new discoveries that may have considerable therapeutic value.
Online summary.
- During development programmed cell death performs various functions such as: sculpt and delete structures, supply nutrients, regulate cell number, and eliminate abnormal and dangerous cells.
- Several different PCD mechanisms are used to eliminate cells and we discuss the biological significance of these pathways in vivo.
- Caspase activation is coordinately subjected to many layers of upstream regulation ensuring that the cell is executed only after multiple checkpoints have been cleared.
- Is autophagy a cell death mechanism? Here we carefully review the data supporting autophagy as a bona fide mechanism of cell destruction.
- For many years necrosis was regarded as an unregulated mode of cell death caused by overwhelming trauma. Here, we examine a regulated form of necrosis, termed necroptosis.
- Traditionally, it was thought that dying cells have limited effects on the cellular environment. However, it is now clear that apoptotic cells release signals that can fuel tissue regeneration.
- Cell undergoing apoptosis have the capacity to instruct additional killing in their cellular environment, explaining how “communal suicide” can occur.
Acknowledgments
We thank Y. Fox for help with the illustrations and K. Fox for the endless support. This work was supported in part by NIH grant RO1GM60124 (to H.S.). H.S. is an Investigator with the Howard Hughes Medical Institute. The Deloro career advancement chair supports Y.F.
Glossary
Initiator caspases
Caspases that cleave inactive forms of executioner capases
Executioner caspases
Caspases that cleave various cellular proteins, which in many cases leads to apoptosis
Apoptosome
A protein platform that is formed during apoptosis comprised of cytochrome c that has translocated from the mitochondria to the cytoplasm and APAF1
Mitochondrial outer membrane permeabilization (MOMP)
An event regulated by the BCL-2 protein family that is considered to be the point of no return at which the cell commits to apoptosis
Death-inducing signaling complex (DISC)
A protein platform that is formed by death receptors that can drive apoptosis
Baculovirus inhibitory repeat (BIR) domain
A domain present in IAP proteins that can bind caspases as well as pro-apoptotic factors such as IAP antagonists
Hair follicle stem cells (HFSCs)
Adult stem cells normally situated in a niche called the bulge that function to replenish the hair follicle
Linker cell
A migratory cell of the C. elegans male gonad that dies in a non-apoptotic, caspase-independent manner
Autophagic cell death
A reported mode of PCD that is associated with the appearance of autophagosomes and depends on autophagy-related proteins
Necrostatin 1
A potent and selective inhibitor of necroptosis, originally reported as a selective allosteric inhibitor of the death domain receptor-associated adaptor kinase RIP1 in the necroptosis pathway
DAMPs (damage associated molecular patterns)
Molecules also known as alarmins that are released by stressed cells. They function as endogenous danger signals to initiate and perpetuate the inflammatory response
Biographies
Yaron Fuchs is currently an Assistant Professor and Deloro Chair in the Technion Israel Institute of Technology. He performed a direct Ph.D. track at the Technion Israel Institute of Technology and then carried out his postdoctoral research at Rockefeller University and Howard Hughes Medical Institute (New York, USA). Upon completion, he returned to the Technion where he now heads the Laboratory of Stem Cell Biology and Regenerative Medicine. His research is focused on different modes of cell death and how they regulate diverse aspects in stem cell biology and stem cell-dependent processes.
Hermann Steller is the Strang Professor and Head of the Laboratory of Apoptosis and Cancer Biology at the Rockefeller University and an Investigator of the Howard Hughes Medical Institute. Prior to this he was Professor of Neurobiology at the Massachusetts Institute of Technology in Cambridge, Massachusetts. Hermann Steller received his Diplom (the German equivalent of an M.Sc. degree) in microbiology from the Johann Wolfgang Goethe University, Frankfurt, and his Ph.D. from the European Molecular Biology Laboratory and the University of Heidelberg. His laboratory studies the regulation of apoptosis, how defects in this process contribute to diseases, and how insights into apoptotic pathways can be exploited for the design of new therapies.
Footnotes
Competing interests statement
The authors declare no competing interests.
References
- 1.Vogt C. Untersuchungen über die Entwicklungsgeschichte der Geburtshelferkröte (Alytes obstetricans) Solothurn: Jent und Gassman. 1842:130. [Google Scholar]
- 2.Lockshin RA, Williams CM. Programmed cell death. II Endocrine potentiation of the breakdown of the intersegmental muscles of silkmoths. J Insect Physiol Dev. 1964;10:643–64. [Google Scholar]
- 3.Tata JR. Requirement for RNA and protein synthesis for induced regression of the tadpole tail in organ culture. Dev Biol. 1966;13:77–94. doi: 10.1016/0012-1606(66)90050-9. [DOI] [PubMed] [Google Scholar]
- 4.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–57. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ellis HM, Horvitz HR. Genetic control of programmed cell death in the nematode C. elegans. Cell. 1986;44:817–29. doi: 10.1016/0092-8674(86)90004-8. [DOI] [PubMed] [Google Scholar]
- 6*.Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell. 1993;75:641–52. doi: 10.1016/0092-8674(93)90485-9. The first report of a cellular ‘built-in’ suicide mechanism. [DOI] [PubMed] [Google Scholar]
- 7.Abraham MC, Shaham S. Death without caspases, caspases without death. Trends Cell Biol. 2004;14:184–93. doi: 10.1016/j.tcb.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 8*.Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147:742–58. doi: 10.1016/j.cell.2011.10.033. A comprehensive review that examines the role of apoptosis in development and the non-apoptotic function of caspases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yi CH, Yuan J. The Jekyll and Hyde functions of caspases. Dev Cell. 2009;16:21–34. doi: 10.1016/j.devcel.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nature Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 11.Yuan J, Kroemer G. Alternative cell death mechanisms in development and beyond. Genes Dev. 2010;24:2592–602. doi: 10.1101/gad.1984410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12*.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nature Rev Mol Cell Biol. 2007;8:741–52. doi: 10.1038/nrm2239. This comprehensive review elegantly discusses the intricate relationship between apoptosis and autophagy. [DOI] [PubMed] [Google Scholar]
- 13.Galluzzi L, Kroemer G. Necroptosis: a specialized pathway of programmed necrosis. Cell. 2008;135:1161–3. doi: 10.1016/j.cell.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 14*.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nature Rev Mol Cell Biol. 2010;11:700–14. doi: 10.1038/nrm2970. This review provides a comprehensive analysis of the molecular mechanisms of necroptosis and implications for human pathology. [DOI] [PubMed] [Google Scholar]
- 15.Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nature Rev Mol Cell Biol. 2014;15:135–47. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- 16*.Jacobson MD, Weil M, Raff MC. Programmed cell death in animal development. Cell. 1997;88:347–54. doi: 10.1016/s0092-8674(00)81873-5. A landmark review that focuses on the function of apoptosis in animal development. [DOI] [PubMed] [Google Scholar]
- 17.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–6. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 18.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–6. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 19.Feinstein-Rotkopf Y, Arama E. Can’t live without them, can live with them: roles of caspases during vital cellular processes. Apoptosis. 2009;14:980–95. doi: 10.1007/s10495-009-0346-6. [DOI] [PubMed] [Google Scholar]
- 20*.Rodriguez J, Lazebnik Y. Caspase-9 and APAF-1 form an active holoenzyme. Genes Dev. 1999;13:3179–84. doi: 10.1101/gad.13.24.3179. Elegant work showing that caspase 9 and APAF1 form an active holoenzyme where caspase-9 is the catalytic subunit and APAF1 is its allosteric regulator. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Srivastava M, et al. ARK, the Apaf-1 related killer in Drosophila, requires diverse domains for its apoptotic activity. Cell Death Differ. 2007;14:92–102. doi: 10.1038/sj.cdd.4401931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22*.Nagasaka A, Kawane K, Yoshida H, Nagata S. Apaf-1-independent programmed cell death in mouse development. Cell Death Differ. 2010;17:931–41. doi: 10.1038/cdd.2009.186. A report indicating that in addition to the apoptotic APAF1-dependent mechanism, APAF1-independent death systems also exist. [DOI] [PubMed] [Google Scholar]
- 23.Xu D, Li Y, Arcaro M, Lackey M, Bergmann A. The CARD-carrying caspase Dronc is essential for most, but not all, developmental cell death in Drosophila. Development. 2005;132:2125–34. doi: 10.1242/dev.01790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24*.Lakhani SA, et al. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. 2006;311:847–51. doi: 10.1126/science.1115035. An elegant study indicating that caspase 3 and caspase 7 can regulate what is percieved as an upstream mitochondrial event of apoptosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25*.Edison N, et al. The IAP-antagonist ARTS initiates caspase activation upstream of cytochrome C and SMAC/Diablo. Cell Death Differ. 2012;19:356–68. doi: 10.1038/cdd.2011.112. A report demonstrating that ARTS regulates caspase activation upstream of MOMP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26*.Enari M, et al. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43–50. doi: 10.1038/34112. An elegant study elucidating CAD and its inhibitor, which regulate DNA degradation during apoptosis. [DOI] [PubMed] [Google Scholar]
- 27.Crook NE, Clem RJ, Miller LK. An apoptosis-inhibiting baculovirus gene with a zinc finger-like motif. J Virol. 1993;67:2168–74. doi: 10.1128/jvi.67.4.2168-2174.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vaux DL, Silke J. IAPs, RINGs and ubiquitylation. Nature Rev Mol Cell Biol. 2005;6:287–97. doi: 10.1038/nrm1621. [DOI] [PubMed] [Google Scholar]
- 29*.Wang SL, Hawkins CJ, Yoo SJ, Muller HA, Hay BA. The Drosophila caspase inhibitor DIAP1 is essential for cell survival and is negatively regulated by HID. Cell. 1999;98:453–63. doi: 10.1016/s0092-8674(00)81974-1. A demonstration that DIAP is inhibited by HID and required to block apoptosis-induced caspase activity. [DOI] [PubMed] [Google Scholar]
- 30*.Goyal L, McCall K, Agapite J, Hartwieg E, Steller H. Induction of apoptosis by Drosophila reaper, hid and grim through inhibition of IAP function. EMBO J. 2000;19:589–97. doi: 10.1093/emboj/19.4.589. An in vivo study showing that RHG proteins kill by forming a complex with DIAP1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lisi S, Mazzon I, White K. Diverse domains of THREAD/DIAP1 are required to inhibit apoptosis induced by REAPER and HID in Drosophila. Genetics. 2000;154:669–78. doi: 10.1093/genetics/154.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32*.Schile AJ, Garcia-Fernandez M, Steller H. Regulation of apoptosis by XIAP ubiquitin-ligase activity. Genes Dev. 2008;22:2256–66. doi: 10.1101/gad.1663108. Demonstration of a physiological requirement for XIAP ubiquitin-ligase activity for the inhibition of caspases and for tumor suppression in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Y, Fang S, Jensen JP, Weissman AM, Ashwell JD. Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science. 2000;288:874–7. doi: 10.1126/science.288.5467.874. [DOI] [PubMed] [Google Scholar]
- 34.Ryoo HD, Bergmann A, Gonen H, Ciechanover A, Steller H. Regulation of Drosophila IAP1 degradation and apoptosis by reaper and ubcD1. Nature Cell Biol. 2002;4:432–8. doi: 10.1038/ncb795. [DOI] [PubMed] [Google Scholar]
- 35.Wilson R, et al. The DIAP1 RING finger mediates ubiquitination of Dronc and is indispensable for regulating apoptosis. Nature Cell Biol. 2002;4:445–50. doi: 10.1038/ncb799. [DOI] [PubMed] [Google Scholar]
- 36*.Ryoo HD, Gorenc T, Steller H. Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev Cell. 2004;7:491–501. doi: 10.1016/j.devcel.2004.08.019. This study elucidates a mechanism whereby apoptotic cells activate signaling cascades for compensatory proliferation. [DOI] [PubMed] [Google Scholar]
- 37.Lee TV, et al. Drosophila IAP1-mediated ubiquitylation controls activation of the initiator caspase DRONC independent of protein degradation. PLoS Genet. 2011;7:e1002261. doi: 10.1371/journal.pgen.1002261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ditzel M, et al. Inactivation of effector caspases through nondegradative polyubiquitylation. Mol Cell. 2008;32:540–53. doi: 10.1016/j.molcel.2008.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eckelman BP, Salvesen GS. The human anti-apoptotic proteins cIAP1 and cIAP2 bind but do not inhibit caspases. J Biol Chem. 2006;281:3254–60. doi: 10.1074/jbc.M510863200. [DOI] [PubMed] [Google Scholar]
- 40*.White K, et al. Genetic control of programmed cell death in Drosophila. Science. 1994;264:677–83. doi: 10.1126/science.8171319. Elucidation of the first IAP antagonist, Reaper, which regulates apoptosis in Drosphila. [DOI] [PubMed] [Google Scholar]
- 41.Grether ME, Abrams JM, Agapite J, White K, Steller H. The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev. 1995;9:1694–708. doi: 10.1101/gad.9.14.1694. [DOI] [PubMed] [Google Scholar]
- 42.Chen P, Nordstrom W, Gish B, Abrams JM. grim, a novel cell death gene in Drosophila. Genes Dev. 1996;10:1773–82. doi: 10.1101/gad.10.14.1773. [DOI] [PubMed] [Google Scholar]
- 43.Shi Y. A conserved tetrapeptide motif: potentiating apoptosis through IAP–binding. Cell Death Differ. 2002;9:93–5. doi: 10.1038/sj.cdd.4400957. [DOI] [PubMed] [Google Scholar]
- 44.White K, Tahaoglu E, Steller H. Cell killing by the Drosophila gene reaper. Science. 1996;271:805–7. doi: 10.1126/science.271.5250.805. [DOI] [PubMed] [Google Scholar]
- 45.Sandu C, Ryoo HD, Steller H. Drosophila IAP antagonists form multimeric complexes to promote cell death. J Cell Biol. 2010;190:1039–52. doi: 10.1083/jcb.201004086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46*.Bergmann A, Agapite J, McCall K, Steller H. The Drosophila gene hid is a direct molecular target of Ras-dependent survival signaling. Cell. 1998;95:331–41. doi: 10.1016/s0092-8674(00)81765-1. Elucidation of the IAP antagonist HID, which regulates apoptosis in Drosphila. [DOI] [PubMed] [Google Scholar]
- 47.Bergmann A, Tugentman M, Shilo BZ, Steller H. Regulation of cell number by MAPK-dependent control of apoptosis: a mechanism for trophic survival signaling. Dev Cell. 2002;2:159–70. doi: 10.1016/s1534-5807(02)00116-8. [DOI] [PubMed] [Google Scholar]
- 48.Zhou L, Steller H. Distinct pathways mediate UV-induced apoptosis in Drosophila embryos. Dev Cell. 2003;4:599–605. doi: 10.1016/s1534-5807(03)00085-6. [DOI] [PubMed] [Google Scholar]
- 49.Steller H. Regulation of apoptosis in Drosophila. Cell Death Differ. 2008;15:1132–8. doi: 10.1038/cdd.2008.50. [DOI] [PubMed] [Google Scholar]
- 50.Verhagen AM, et al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 51*.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. References 50 and 51 independently discovered the mammalian IAP antagonist SMAC. [DOI] [PubMed] [Google Scholar]
- 52.Suzuki Y, et al. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell. 2001;8:613–21. doi: 10.1016/s1097-2765(01)00341-0. [DOI] [PubMed] [Google Scholar]
- 53.Verhagen AM, et al. Identification of mammalian mitochondrial proteins that interact with IAPs via N-terminal IAP binding motifs. Cell Death Differ. 2007;14:348–57. doi: 10.1038/sj.cdd.4402001. [DOI] [PubMed] [Google Scholar]
- 54.Martins LM, et al. Neuroprotective role of the Reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol Cell Biol. 2004;24:9848–62. doi: 10.1128/MCB.24.22.9848-9862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Okada H, et al. Generation and characterization of Smac/DIABLO-deficient mice. Mol Cell Biol. 2002;22:3509–17. doi: 10.1128/MCB.22.10.3509-3517.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nature Rev Drug Discov. 2012;11:109–24. doi: 10.1038/nrd3627. [DOI] [PubMed] [Google Scholar]
- 57.Varfolomeev E, et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell. 2007;131:669–81. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 58.Vince JE, et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell. 2007;131:682–93. doi: 10.1016/j.cell.2007.10.037. [DOI] [PubMed] [Google Scholar]
- 59.Larisch S, et al. A novel mitochondrial septin-like protein, ARTS, mediates apoptosis dependent on its P-loop motif. Nature Cell Biol. 2000;2:915–21. doi: 10.1038/35046566. [DOI] [PubMed] [Google Scholar]
- 60*.Gottfried Y, Rotem A, Lotan R, Steller H, Larisch S. The mitochondrial ARTS protein promotes apoptosis through targeting XIAP. EMBO J. 2004;23:1627–35. doi: 10.1038/sj.emboj.7600155. A report demonstrating that XIAP is the biochemical target of ARTS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kissel H, et al. The Sept4 septin locus is required for sperm terminal differentiation in mice. Dev Cell. 2005;8:353–64. doi: 10.1016/j.devcel.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 62*.Garcia-Fernandez M, et al. Sept4/ARTS is required for stem cell apoptosis and tumor suppression. Genes Dev. 2010;24:2282–93. doi: 10.1101/gad.1970110. Work showing that ARTS regulates the survival of haematopeotic stem cells and tumor formation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63*.Fuchs Y, et al. Sept4/ARTS regulates stem cell apoptosis and skin regeneration. Science. 2013;341:286–9. doi: 10.1126/science.1233029. A report showing that ARTS deletion increases hair follicle stem cell numbers and results in improved skin regeneration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tang HL, et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol Biol Cell. 2012;23:2240–52. doi: 10.1091/mbc.E11-11-0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65*.Abraham MC, Lu Y, Shaham S. A morphologically conserved nonapoptotic program promotes linker cell death in Caenorhabditis elegans. Dev Cell. 2007;12:73–86. doi: 10.1016/j.devcel.2006.11.012. This study elegantly demonstrates a non apoptotic cell death program in C. elegans that is morphologically conserved through out evolution. [DOI] [PubMed] [Google Scholar]
- 66.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–41. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 67.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115:2679–88. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gorski SM, et al. A SAGE approach to discovery of genes involved in autophagic cell death. Curr Biol. 2003;13:358–63. doi: 10.1016/s0960-9822(03)00082-4. [DOI] [PubMed] [Google Scholar]
- 69.Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell. 2007;131:1137–48. doi: 10.1016/j.cell.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McPhee CK, Logan MA, Freeman MR, Baehrecke EH. Activation of autophagy during cell death requires the engulfment receptor Draper. Nature. 2010;465:1093–6. doi: 10.1038/nature09127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yin VP, Thummel CS. A balance between the diap1 death inhibitor and reaper and hid death inducers controls steroid-triggered cell death in Drosophila. Proc Natl Acad Sci USA. 2004;101:8022–7. doi: 10.1073/pnas.0402647101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jiang C, Baehrecke EH, Thummel CS. Steroid regulated programmed cell death during Drosophila metamorphosis. Development. 1997;124:4673–83. doi: 10.1242/dev.124.22.4673. [DOI] [PubMed] [Google Scholar]
- 73.Lee CY, Baehrecke EH. Steroid regulation of autophagic programmed cell death during development. Development. 2001;128:1443–55. doi: 10.1242/dev.128.8.1443. [DOI] [PubMed] [Google Scholar]
- 74.Lee CY, et al. Genome-wide analyses of steroid- and radiation-triggered programmed cell death in Drosophila. Curr Biol. 2003;13:350–7. doi: 10.1016/s0960-9822(03)00085-x. [DOI] [PubMed] [Google Scholar]
- 75.Jiang C, Lamblin AF, Steller H, Thummel CS. A steroid-triggered transcriptional hierarchy controls salivary gland cell death during Drosophila metamorphosis. Mol Cell. 2000;5:445–55. doi: 10.1016/s1097-2765(00)80439-6. [DOI] [PubMed] [Google Scholar]
- 76.Akdemir F, et al. Autophagy occurs upstream or parallel to the apoptosome during histolytic cell death. Development. 2006;133:1457–65. doi: 10.1242/dev.02332. [DOI] [PubMed] [Google Scholar]
- 77.Denton D, et al. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol. 2009;19:1741–6. doi: 10.1016/j.cub.2009.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lum JJ, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–48. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 79.Shimizu S, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nature Cell Biol. 2004;6:1221–8. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 80*.Pattingre S, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. A report showing that BCL-2 not only functions as an antiapoptotic protein but also inhibits autophagy by interacting with BECLIN-1. [DOI] [PubMed] [Google Scholar]
- 81.Borsos E, Erdelyi P, Vellai T. Autophagy and apoptosis are redundantly required for C. elegans embryogenesis. Autophagy. 2011;7:557–9. doi: 10.4161/auto.7.5.14685. [DOI] [PubMed] [Google Scholar]
- 82.Maiuri MC, et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–39. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zalckvar E, et al. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009;10:285–92. doi: 10.1038/embor.2008.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009;16:966–75. doi: 10.1038/cdd.2009.33. [DOI] [PubMed] [Google Scholar]
- 85.Zhu Y, et al. Beclin 1 cleavage by caspase-3 inactivates autophagy and promotes apoptosis. Protein Cell. 2010;1:468–77. doi: 10.1007/s13238-010-0048-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yousefi S, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nature Cell Biol. 2006;8:1124–32. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- 87.Pyo JO, et al. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem. 2005;280:20722–9. doi: 10.1074/jbc.M413934200. [DOI] [PubMed] [Google Scholar]
- 88.Qu X, et al. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell. 2007;128:931–46. doi: 10.1016/j.cell.2006.12.044. [DOI] [PubMed] [Google Scholar]
- 89.Inbal B, Bialik S, Sabanay I, Shani G, Kimchi A. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J Cell Biol. 2002;157:455–68. doi: 10.1083/jcb.200109094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shen S, Kepp O, Kroemer G. The end of autophagic cell death? Autophagy. 2012;8:1–3. doi: 10.4161/auto.8.1.16618. [DOI] [PubMed] [Google Scholar]
- 91*.Degterev A, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nature Chem Biol. 2005;1:112–9. doi: 10.1038/nchembio711. This work coined the term necroptosis and identified a chemical inhibitor of this process. [DOI] [PubMed] [Google Scholar]
- 92.Galluzzi L, Kepp O, Krautwald S, Kroemer G, Linkermann A. Molecular mechanisms of regulated necrosis. Semin Cell Dev Biol. 2014;35:24–32. doi: 10.1016/j.semcdb.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 93.Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370:455–65. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94*.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–90. doi: 10.1016/s0092-8674(03)00521-x. This study showed the early biochemical events that are initiated by TNFR1 ligation and the existence of two signaling complexes. [DOI] [PubMed] [Google Scholar]
- 95.Fuchs Y, et al. Sef is an inhibitor of proinflammatory cytokine signaling, acting by cytoplasmic sequestration of NF-kappaB. Dev Cell. 2012;23:611–23. doi: 10.1016/j.devcel.2012.07.013. [DOI] [PubMed] [Google Scholar]
- 96.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–63. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 97.Wertz IE, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–9. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 98.Wright A, et al. Regulation of early wave of germ cell apoptosis and spermatogenesis by deubiquitinating enzyme CYLD. Dev Cell. 2007;13:705–16. doi: 10.1016/j.devcel.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 99*.Hitomi J, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135:1311–23. doi: 10.1016/j.cell.2008.10.044. This study examined necroptosis and apoptosis using a systems biology approach, which enabled the elucidation of the molecular switch between these two processes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Varfolomeev EE, et al. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9:267–76. doi: 10.1016/s1074-7613(00)80609-3. [DOI] [PubMed] [Google Scholar]
- 101.Yeh WC, et al. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science. 1998;279:1954–8. doi: 10.1126/science.279.5358.1954. [DOI] [PubMed] [Google Scholar]
- 102.Yeh WC, et al. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity. 2000;12:633–42. doi: 10.1016/s1074-7613(00)80214-9. [DOI] [PubMed] [Google Scholar]
- 103.Oberst A, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–7. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–23. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 105.Kaiser WJ, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288:31268–79. doi: 10.1074/jbc.M113.462341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Newton K, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343:1357–60. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- 107.Dillon CP, et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell. 2014;157:1189–202. doi: 10.1016/j.cell.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang DW, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–6. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 109.Cho YS, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–23. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110*.He S, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–11. doi: 10.1016/j.cell.2009.05.021. References 108–110 independently revealed the crucial role of RIPK3 in regulating necroptosis. Using various methods, the authors showed that RIPK3 binds RIPK1 and executes the necroptotic program. [DOI] [PubMed] [Google Scholar]
- 111.Degterev A, Yuan J. Expansion and evolution of cell death programmes. Nature Rev Mol Cell Biol. 2008;9:378–90. doi: 10.1038/nrm2393. [DOI] [PubMed] [Google Scholar]
- 112.Christofferson DE, Yuan J. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol. 2010;22:263–8. doi: 10.1016/j.ceb.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113*.Sun L, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–27. doi: 10.1016/j.cell.2011.11.031. An elegant study showing that MLKL lies downstream of RIP3K and is a crucial component of the necroptotic machinary. [DOI] [PubMed] [Google Scholar]
- 114.Xie T, et al. Structural insights into RIP3-mediated necroptotic signaling. Cell Rep. 2013;5:70–8. doi: 10.1016/j.celrep.2013.08.044. [DOI] [PubMed] [Google Scholar]
- 115.Dondelinger Y, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014;7:971–81. doi: 10.1016/j.celrep.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 116.Wang H, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54:133–46. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 117.Chen X, et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014;24:105–21. doi: 10.1038/cr.2013.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cai Z, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nature Cell Biol. 2014;16:55–65. doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wu J, et al. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res. 2013;23:994–1006. doi: 10.1038/cr.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mocarski ES, Kaiser WJ, Livingston-Rosanoff D, Upton JW, Daley-Bauer LP. True grit: programmed necrosis in antiviral host defense, inflammation, and immunogenicity. J Immunol. 2014;192:2019–26. doi: 10.4049/jimmunol.1302426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mack C, Sickmann A, Lembo D, Brune W. Inhibition of proinflammatory and innate immune signaling pathways by a cytomegalovirus RIP1-interacting protein. Proc Natl Acad Sci USA. 2008;105:3094–9. doi: 10.1073/pnas.0800168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Skaletskaya A, et al. A cytomegalovirus-encoded inhibitor of apoptosis that suppresses caspase-8 activation. Proc Natl Acad Sci USA. 2001;98:7829–34. doi: 10.1073/pnas.141108798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe. 2010;7:302–13. doi: 10.1016/j.chom.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bonnet MC, et al. The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity. 2011;35:572–82. doi: 10.1016/j.immuni.2011.08.014. [DOI] [PubMed] [Google Scholar]
- 125.Kovalenko A, et al. Caspase-8 deficiency in epidermal keratinocytes triggers an inflammatory skin disease. J Exp Med. 2009;206:2161–77. doi: 10.1084/jem.20090616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gunther C, et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature. 2011;477:335–9. doi: 10.1038/nature10400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mahoney DJ, et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci USA. 2008;105:11778–83. doi: 10.1073/pnas.0711122105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Darding M, Meier P. IAPs: guardians of RIPK1. Cell Death Differ. 2012;19:58–66. doi: 10.1038/cdd.2011.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zarnegar BJ, et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nature Immunol. 2008;9:1371–8. doi: 10.1038/ni.1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tenev T, et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell. 2011;43:432–48. doi: 10.1016/j.molcel.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 131.Feoktistova M, et al. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell. 2011;43:449–63. doi: 10.1016/j.molcel.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hochreiter-Hufford A, Ravichandran KS. Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb Perspect Biol. 2013;5:a008748. doi: 10.1101/cshperspect.a008748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: signals for a good meal. Nature Rev Immunol. 2007;7:964–74. doi: 10.1038/nri2214. [DOI] [PubMed] [Google Scholar]
- 134.Suzuki J, Denning DP, Imanishi E, Horvitz HR, Nagata S. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science. 2013;341:403–6. doi: 10.1126/science.1236758. [DOI] [PubMed] [Google Scholar]
- 135*.Chen YZ, Mapes J, Lee ES, Skeen-Gaar RR, Xue D. Caspase-mediated activation of Caenorhabditis elegans CED-8 promotes apoptosis and phosphatidylserine externalization. Nature Commun. 2013;4:2726. doi: 10.1038/ncomms3726. The elegant work in references 134 and 135 elucidated that Xk-related protein 8 and CED-8 promote apoptosis and phosphatidylserine exposure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Birnbaum KD, Sanchez Alvarado A. Slicing across kingdoms: regeneration in plants and animals. Cell. 2008;132:697–710. doi: 10.1016/j.cell.2008.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Huh JR, Guo M, Hay BA. Compensatory proliferation induced by cell death in the Drosophila wing disc requires activity of the apical cell death caspase Dronc in a nonapoptotic role. Curr Biol. 2004;14:1262–6. doi: 10.1016/j.cub.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 138*.Perez-Garijo A, Martin FA, Morata G. Caspase inhibition during apoptosis causes abnormal signalling and developmental aberrations in Drosophila. Development. 2004;131:5591–8. doi: 10.1242/dev.01432. This work, together with references 36 and 137, elucidated the mechanism whereby apoptotic cells activate signaling cascades for compensatory proliferation. [DOI] [PubMed] [Google Scholar]
- 139.Bergmann A, Steller H. Apoptosis, stem cells, and tissue regeneration. Sci Signal. 2010;3:re8. doi: 10.1126/scisignal.3145re8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Morata G, Shlevkov E, Perez-Garijo A. Mitogenic signaling from apoptotic cells in Drosophila. Dev Growth Differ. 2011;53:168–76. doi: 10.1111/j.1440-169X.2010.01225.x. [DOI] [PubMed] [Google Scholar]
- 141.Bosch M, Serras F, Martin-Blanco E, Baguna J. JNK signaling pathway required for wound healing in regenerating Drosophila wing imaginal discs. Dev Biol. 2005;280:73–86. doi: 10.1016/j.ydbio.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 142.Perez-Garijo A, Shlevkov E, Morata G. The role of Dpp and Wg in compensatory proliferation and in the formation of hyperplastic overgrowths caused by apoptotic cells in the Drosophila wing disc. Development. 2009;136:1169–77. doi: 10.1242/dev.034017. [DOI] [PubMed] [Google Scholar]
- 143.Kondo S, Senoo-Matsuda N, Hiromi Y, Miura M. DRONC coordinates cell death and compensatory proliferation. Mol Cell Biol. 2006;26:7258–68. doi: 10.1128/MCB.00183-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.McEwen DG, Peifer M. Puckered, a Drosophila MAPK phosphatase, ensures cell viability by antagonizing JNK-induced apoptosis. Development. 2005;132:3935–46. doi: 10.1242/dev.01949. [DOI] [PubMed] [Google Scholar]
- 145.Shlevkov E, Morata G. A dp53/JNK-dependant feedback amplification loop is essential for the apoptotic response to stress in Drosophila. Cell Death Differ. 2012;19:451–60. doi: 10.1038/cdd.2011.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Warner SJ, Yashiro H, Longmore GD. The Cdc42/Par6/aPKC polarity complex regulates apoptosis-induced compensatory proliferation in epithelia. Curr Biol. 2010;20:677–86. doi: 10.1016/j.cub.2010.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bergantinos C, Corominas M, Serras F. Cell death-induced regeneration in wing imaginal discs requires JNK signalling. Development. 2010;137:1169–79. doi: 10.1242/dev.045559. [DOI] [PubMed] [Google Scholar]
- 148.Smith-Bolton RK, Worley MI, Kanda H, Hariharan IK. Regenerative growth in Drosophila imaginal discs is regulated by Wingless and Myc. Dev Cell. 2009;16:797–809. doi: 10.1016/j.devcel.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149*.Fan Y, Bergmann A. Distinct mechanisms of apoptosis-induced compensatory proliferation in proliferating and differentiating tissues in the Drosophila eye. Dev Cell. 2008;14:399–410. doi: 10.1016/j.devcel.2008.01.003. A report indicating that compensatory proliferation is instructed in a distinct manner in differentiating and proliferating cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chera S, et al. Apoptotic cells provide an unexpected source of Wnt3 signaling to drive hydra head regeneration. Dev Cell. 2009;17:279–89. doi: 10.1016/j.devcel.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 151.Chera S, Ghila L, Wenger Y, Galliot B. Injury-induced activation of the MAPK/CREB pathway triggers apoptosis-induced compensatory proliferation in hydra head regeneration. Dev Growth Differ. 2011;53:186–201. doi: 10.1111/j.1440-169X.2011.01250.x. [DOI] [PubMed] [Google Scholar]
- 152.Petersen CP, Reddien PW. A wound-induced Wnt expression program controls planarian regeneration polarity. Proc Natl Acad Sci USA. 2009;106:17061–6. doi: 10.1073/pnas.0906823106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Adell T, Salo E, Boutros M, Bartscherer K. Smed-Evi/Wntless is required for beta-catenin-dependent and -independent processes during planarian regeneration. Development. 2009;136:905–10. doi: 10.1242/dev.033761. [DOI] [PubMed] [Google Scholar]
- 154.Gurley KA, Rink JC, Sanchez Alvarado A. Beta-catenin defines head versus tail identity during planarian regeneration and homeostasis. Science. 2008;319:323–7. doi: 10.1126/science.1150029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Rink JC, Gurley KA, Elliott SA, Sanchez Alvarado A. Planarian Hh signaling regulates regeneration polarity and links Hh pathway evolution to cilia. Science. 2009;326:1406–10. doi: 10.1126/science.1178712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Schnapp E, Kragl M, Rubin L, Tanaka EM. Hedgehog signaling controls dorsoventral patterning, blastema cell proliferation and cartilage induction during axolotl tail regeneration. Development. 2005;132:3243–53. doi: 10.1242/dev.01906. [DOI] [PubMed] [Google Scholar]
- 157.Tseng AS, Adams DS, Qiu D, Koustubhan P, Levin M. Apoptosis is required during early stages of tail regeneration in Xenopus laevis. Dev Biol. 2007;301:62–9. doi: 10.1016/j.ydbio.2006.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Gauron C, et al. Sustained production of ROS triggers compensatory proliferation and is required for regeneration to proceed. Sci Rep. 2013;3:2084. doi: 10.1038/srep02084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Nishina T, et al. Interleukin-11 links oxidative stress and compensatory proliferation. Sci Signal. 2012;5:ra5. doi: 10.1126/scisignal.2002056. [DOI] [PubMed] [Google Scholar]
- 160.Sakurai T, et al. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell. 2008;14:156–65. doi: 10.1016/j.ccr.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Li F, et al. Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci Signal. 2010;3:ra13. doi: 10.1126/scisignal.2000634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kurtova AV, et al. Blocking PGE-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature. 2014;517:209–213. doi: 10.1038/nature14034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163*.Huang Q, et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nature Med. 2011;17:860–6. doi: 10.1038/nm.2385. References 162 and 163 stress the importance of compensatory proliferation in tumorigensis and as a potential target for tumour therapy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164*.Perez-Garijo A, Fuchs Y, Steller H. Apoptotic cells can induce non-autonomous apoptosis through the TNF pathway. Elife. 2013;2:e01004. doi: 10.7554/eLife.01004. This study shows that apoptotic cells can sectrete EIGER/TNF ligands to induce cell death in neighbouring cells, thus enabling cohort cell death. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lindner G, et al. Analysis of apoptosis during hair follicle regression (catagen) Am J Pathol. 1997;151:1601–17. [PMC free article] [PubMed] [Google Scholar]
- 166.Biton S, Ashkenazi A. NEMO and RIP1 control cell fate in response to extensive DNA damage via TNF-alpha feedforward signaling. Cell. 2011;145:92–103. doi: 10.1016/j.cell.2011.02.023. [DOI] [PubMed] [Google Scholar]
- 167.Nelson CM, et al. Tumor necrosis factor-alpha is produced by dying retinal neurons and is required for Muller glia proliferation during zebrafish retinal regeneration. J Neurosci. 2013;33:6524–39. doi: 10.1523/JNEUROSCI.3838-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pellettieri J, et al. Cell death and tissue remodeling in planarian regeneration. Dev Biol. 2010;338:76–85. doi: 10.1016/j.ydbio.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Takeishi A, et al. Homeostatic epithelial renewal in the gut is required for dampening a fatal systemic wound response in Drosophila. Cell Rep. 2013;3:919–30. doi: 10.1016/j.celrep.2013.02.022. [DOI] [PubMed] [Google Scholar]
- 170.Hei TK, Zhou H, Chai Y, Ponnaiya B, Ivanov VN. Radiation induced non-targeted response: mechanism and potential clinical implications. Curr Mol Pharmacol. 2011;4:96–105. doi: 10.2174/1874467211104020096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Prise KM, O’Sullivan JM. Radiation-induced bystander signalling in cancer therapy. Nature Rev Cancer. 2009;9:351–60. doi: 10.1038/nrc2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ellis RE, Yuan JY, Horvitz HR. Mechanisms and functions of cell death. Annu Rev Cell Biol. 1991;7:663–98. doi: 10.1146/annurev.cb.07.110191.003311. [DOI] [PubMed] [Google Scholar]
- 173.Suzanne M, Steller H. Shaping organisms with apoptosis. Cell Death Differ. 2013;20:669–75. doi: 10.1038/cdd.2013.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Peterson JS, Barkett M, McCall K. Stage-specific regulation of caspase activity in drosophila oogenesis. Dev Biol. 2003;260:113–23. doi: 10.1016/s0012-1606(03)00240-9. [DOI] [PubMed] [Google Scholar]
- 175.Monier B, et al. Apico-basal forces exerted by apoptotic cells drive epithelium folding. Nature. 2015;518:245–8. doi: 10.1038/nature14152. [DOI] [PubMed] [Google Scholar]
- 176.Blum ES, Abraham MC, Yoshimura S, Lu Y, Shaham S. Control of nonapoptotic developmental cell death in Caenorhabditis elegans by a polyglutamine-repeat protein. Science. 2012;335:970–3. doi: 10.1126/science.1215156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Osterloh JM, et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. 2012;337:481–4. doi: 10.1126/science.1223899. [DOI] [PMC free article] [PubMed] [Google Scholar]