Natively unfolded FG-repeats stabilize the structure of the nuclear pore complex (original) (raw)
. Author manuscript; available in PMC: 2018 Nov 2.
Summary
Nuclear pore complexes (NPCs) are ~100 MDa transport channels assembled from multiple copies of ~30 nucleoporins (Nups). One third of these Nups contain phenylalanine-glycine (FG)-rich repeats forming a diffusion barrier, which is selectively permeable for nuclear transport receptors that interact with these repeats. Here, we identify an additional function of FG-repeats in the structure and biogenesis of the yeast NPC. We demonstrate that GLFG-containing FG-repeats directly bind to multiple scaffold Nups in vitro and act as NPC targeting determinants in vivo. Furthermore, we show that the GLFG repeats of Nup116 function in a redundant manner with Nup188, a non-essential scaffold Nup, to stabilize critical interactions within the NPC scaffold needed for late steps of NPC assembly. Our results reveal a previously unanticipated structural role for natively unfolded GLFG-repeats as velcro to link NPC subcomplexes, and thus add a new layer of connections to current models of the NPC architecture.
eTOC Blurb
In Brief – In addition to forming the permeability barrier, FG repeats in nucleoporins contribute structurally to nuclear pore assembly and function.
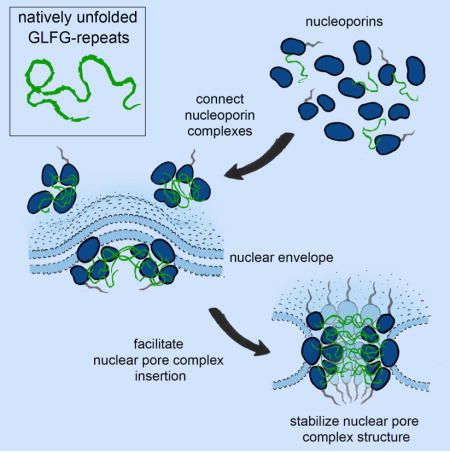
Introduction
The nuclear pore complex (NPC) is a multiprotein channel located in a fusion pore between the outer and inner membranes (ONM and INM) of the nuclear envelope (NE). It serves as a conduit for nucleocytoplasmic transport and is involved in the regulation of gene expression, cell cycle and other cellular processes (Ibarra and Hetzer, 2015). The NPC has a cylindrical shape with a ~100 nm outer and ~40nm inner diameter (Eibauer et al., 2015; von Appen et al., 2015). Whereas each NPC consists of ~500 proteins, called nucleoporins (Nups), there are only ~30 different Nups (Rout et al., 2000), which are present in multiples of 8 copies per NPC (Alber et al., 2007; Ori et al., 2013) and are largely conserved across the whole eukaryotic tree (Cronshaw et al., 2002; Mans et al., 2004; Rout et al., 2000).
The majority of Nups are arranged into subcomplexes, giving rise to the NPC’s overall layered architecture (Alber et al., 2007; Onischenko and Weis, 2011; Strambio-De-Castillia et al., 2010) (Figure 1A). High-resolution structures of multiple individual Nups and Nup subcomplexes have been solved by X-ray crystallography (Brohawn et al., 2008; Brohawn and Schwartz, 2009; Chug et al., 2015; Hurt and Beck, 2015; Kelley et al., 2015; Lin et al., 2016; Schwartz, 2016; Stuwe et al., 2015a; Stuwe et al., 2015b). In addition, the NPC structure has been studied by cryo-electron tomography and biochemical methods, revealing the putative organization of scaffold Nups within the NPC (Alber et al., 2007; Eibauer et al., 2015; Fernandez-Martinez et al., 2016; Kosinski et al., 2016; Lin et al., 2016; von Appen et al., 2015). However, many questions remain about the mechanism organizing multiple Nup subcomplexes and ensuring their assembly into a functional NPC.
Figure 1. Identification of FG-repeat binding proteins.
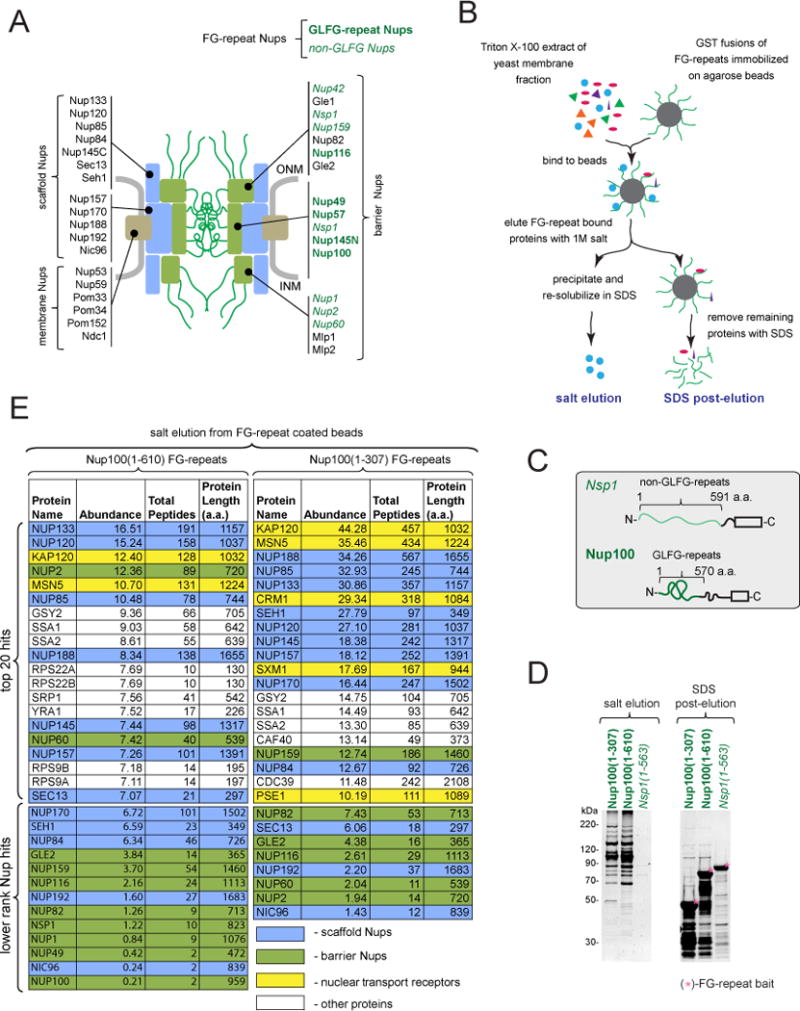
(A) Classification of yeast Nups: membrane Nups physically associate with the NE, scaffold Nups form the structural core of the NPC, and barrier Nups have a primary role in selective nucleocytoplasmic transport (Onischenko and Weis, 2011).
(B) Outline of FG-repeat pulldown procedure in yeast extracts.
(C) Schematic of FG-repeat segments used as baits for yeast extract pulldowns in (B).
(D) SYPRO Ruby stained SDS-PAGE gels showing yeast proteins bound to FG-repeat coated beads. Bound proteins were eluted by 1M salt (left), followed by SDS (right).
(E) Yeast proteins bound to Nup100(1-610) and Nup100(1-307) identified by mass spectrometry and ranked by the abundance factor (Abundance): 100 × (number of peptide hits)/(protein length in amino acids).
A key function of NPCs is to mediate the selective transport of macromolecules across the NE. This selectivity is conferred by FG-repeat-containing segments present in ~1/3 of all Nups (Figure 1A) (Rout et al., 2000), which form a diffusion barrier that blocks free nucleocytoplasmic exchange of particles larger than ~5 nm (Mohr et al., 2009; Ribbeck and Gorlich, 2001). FG-repeats are natively unfolded (Denning et al., 2003), with varying degrees of conformational flexibility (Milles et al., 2015; Yamada et al., 2010). Experiments in Xenopus egg extracts and yeast point to a primary role for a subgroup of FG-repeats enriched in GLFG motifs in establishing the diffusion barrier (Hulsmann et al., 2012; Timney et al., 2016). The GLFG-containing repeats display cohesive properties and can form aggregates with NPC-like selectivity in vitro (Schmidt and Gorlich, 2015a). Although several models for NPC selectivity have been proposed (Lim et al., 2007; Ribbeck and Gorlich, 2002; Rout et al., 2003), it is still unclear how FG-repeats determine the remarkable permeability properties of the NPC.
Selective transport through the NPC requires soluble nuclear transport receptors (NTRs) that interact with both FG-repeats and cargos (Mohr et al., 2009; Ribbeck and Gorlich, 2001). NTRs interact with FG-repeats in a multivalent manner via multiple low affinity (1–10 mM) binding sites scattered on the surface, which is critical for the fast and selective translocation of NTR-cargo complexes through NPCs (Hough et al., 2015; Kapinos et al., 2014; Milles et al., 2015).
In addition to NTRs, several budding yeast scaffold Nups bind FG-repeats in biochemical assays (Allen et al., 2001; Patel et al., 2007; Schrader et al., 2008). This was also observed for human Nup93 (Xu and Powers, 2013) and for the orthologues of Nup188 and Nup192 from thermophilic fungi (Andersen et al., 2013b). However, the functional significance of these interactions remained unclear (Andersen et al., 2013b; Hurt and Beck, 2015; Schmidt and Gorlich, 2015a, b).
In this study, we identify and characterize direct interactions between multiple scaffold Nups and GLFG-type repeats in budding yeast. Our analyses reveal that GLFG-repeats strengthen the interactions between Nups, act as NPC targeting determinants and have a function in NPC biogenesis. This points towards a paradigm shift whereby natively unfolded FG-repeats are not only part of the NPC’s selectivity barrier but also function as velcro that links structural elements within the NPC. This may mechanistically couple the process of NPC assembly with the establishment of a selective transport gateway.
Results
GLFG-repeats directly and specifically bind to a set of scaffold Nups
Yeast NPCs contain eleven FG-repeat Nups, five of which carry GLFG-repeat motifs, hereby referred to as GLFG-repeat Nups. Whereas GLFG-repeat Nups are mainly centrally located within the NPC structure, the rest of FG-Nups (non-GLFG Nups) are localized mostly asymmetrically (Alber et al., 2007; Patel et al., 2007) (Figure 1A).
To better characterize the interactions of FG-repeats, we modified a previously described FG-repeat pulldown protocol (Allen et al., 2001) by utilizing crude membrane extract as the source of prey proteins, which reduces the amount of NTRs that may mediate indirect FG-repeat interactions. Proteins bound to immobilized FG-repeats were eluted using 1M salt. Elution of the remaining proteins with SDS (SDS post-elution step) was used to confirm the efficiency and specificity of the salt elution (Figure 1B).
GLFG-repeats were represented by Nup100(1-610) and Nup100(1-307), and non-GLFG repeats by Nsp1(1-563) (Figure 1C). SDS-PAGE analysis showed that Nup100(1-610) and Nup100(1-307) efficiently bound extract proteins whereas little binding was seen with Nsp1(1-563) (Figure 1D). Surprisingly, mass-spectrometry analysis of the proteins bound to Nup100(1-610) and Nup100(1-307) revealed, in addition to NTRs, many scaffold Nups belonging to the Nup84 and Nic96–subcomplexes amongst the top hits (Figure 1E).
To test whether the binding of the GLFG repeats to the Nup84- and Nic96-subcomplexes in extracts was due to direct interactions, parts of the Nup84- and Nic96–subcomplexes were purified (Figures S1B–D), and their binding to FG-repeats was evaluated individually (Figure S1A). In addition to Nup100 and Nsp1, the tested baits included a portion of Nup116’s GLFG-repeats (Nup116(348-458)) (Figure 4B) and its mutant form in which the phenylalanines within the GLFG motifs were replaced by alanines (Nup116(348-458)FA), preventing its interaction with NTRs (Patel et al., 2007). To control for binding specificity, the preys were pre-mixed with bacterial extract. Salt elution was followed by the elution with SDS to detect salt-resistant interactions (Figure S1A). The procedure was further controlled with a known FG-repeat interactor, the NTR Kap95, and the inert proteins 3xGFP and MBP-GFP-Nup53ΔC (Figures 2A, 2D, S2A).
Figure 4. Effect of Nup116 GLFG-repeats on cell fitness and the connectivity of Nup116 with scaffold Nups. See also Figures S3–S4.
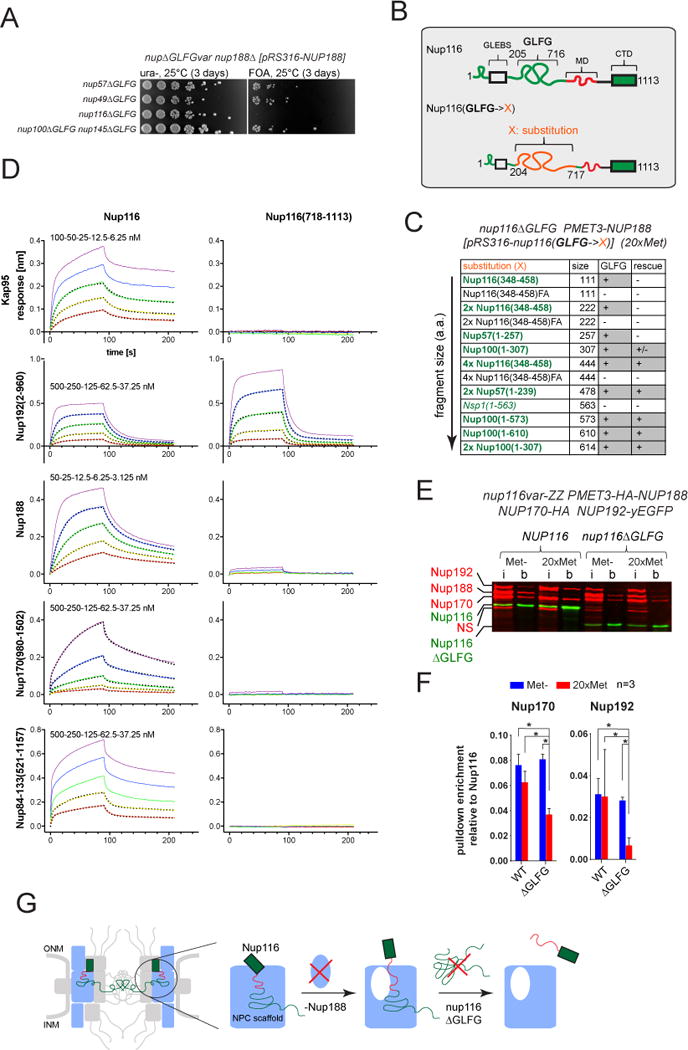
(A) Deletion of NUP188 is synthetically lethal in combination with the deletion of Nup116 GLFG-repeats (nup116ΔGLFG) but not with any other GLFG-repeats (nup57ΔGLFG, nup49ΔGLFG or nup100ΔGLFG nup145NΔGLFG).
(B) Outline of GLFG-repeat segment substitutions in Nup116.
(C) Growth rescue of PMET3-NUP188 nup116ΔGLFG strain by ectopically expressing Nup116 GLFG-repeat substitutions upon repression of Nup188. “+”, “+/−” and “−” - complete rescue, partial rescue and no rescue of cell growth, respectively. See also Figure S3B.
(D) Representative BLI curves showing association and dissociation kinetics for various scaffold Nups and Kap95 with the respective immobilized Nup116 variants. Curves were corrected for buffer background. Five two-fold dilution series of analyte were used for each experiment (purple, blue, green, yellow, red). The highest concentration (purple) was 50 nM (Nup188), 100 nM (Kap95) and 500 nM (Nup84-133, Nup170, Nup192). Dotted lines show fitted curves after global fitting analysis. See also Figures S4A–B.
(E and F) Effects of GLFG-repeats and Nup188 depletion on the connection of Nup116 with scaffold Nups. PMET3-NUP188 strains expressing tagged scaffold Nups (NUP170-HA and NUP192-yEGFP) and either of the ZZ-tagged Nup116 variants (NUP116-ZZ or nup116ΔGLFG-ZZ) were incubated for 12h in Met- or 20xMet media prior to processing for affinity pulldowns with IgG-Dynabeads. (E) Representative Western blot image used to quantify protein amounts in the input (i) and IgG-Dynabeads bound (b) fractions. (NS) – a protein cross-reacting with anti-HA-tag antibody. (F) Plot showing Nup116 co-purification efficiencies of Nup170 and Nup192 based on the corresponding band intensities as (b/i ratio of prey)/(b/i ratio of Nup116). Mean +/− SD from three independent experiments. Asterisks (*) – significant differences, Student t-test p-values (< 0.01). See also Figures S4C–D.
(G) Model describing the connectivity function of Nup116 GLFG-repeats in the absence of Nup188.
Figure 2. Nups of the Nup84- and Nic96-subcomplex directly bind to GLFG-repeats. See also Figures S1–S2.
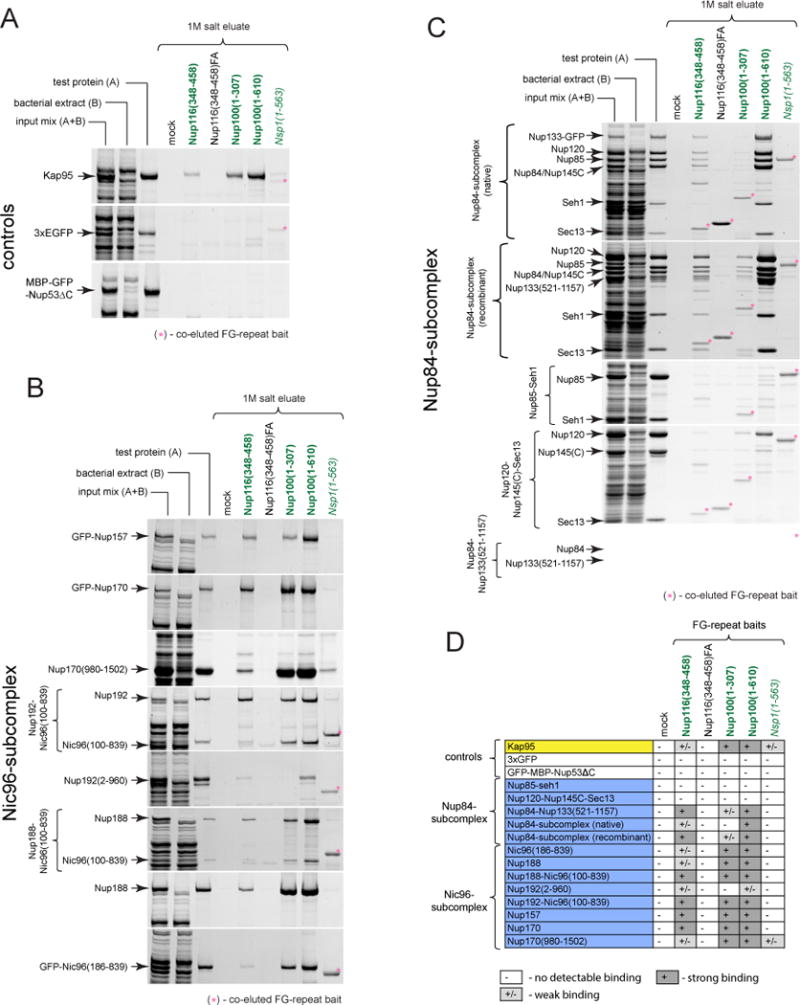
(A–C) Proteins pre-mixed with bacterial extract were subjected to affinity pulldowns with FG-repeats as baits. SYPRO Ruby stained SDS-PAGE gels show (left to right) input mixes, their components (bacterial extract and test proteins, respectively) and proteins eluted with 1M salt from mock or various FG-repeat coated beads. (A) Binding reactions with the NTR Kap95 (positive control) and inert proteins 3xGFP and MBP-GFP-Nup53ΔC (negative controls). Note that only Kap95 can be efficiently purified from the bacterial extract mixtures. (B and C) Similar to (A) FG-repeat binding assays with various portions of (B) Nic96-subcomplex and (C) Nup84-subcomplex.
(D) Results of FG-repeat binding experiments. Binding was considered weak unless the eluted test protein band was comparable to or stronger than the input (test protein).
We observed direct and specific binding to GLFG-repeats for all five members of the Nic96-subcomplex including Nup157, Nup170, the C-terminal stacked α-helical domain of Nup170 (Nup170(980-1502)), Nup188 and truncated versions of Nup192 and Nic96 (Nup192(2-960) and Nic96(186-839), respectively. Consistently, binding was also observed with heterodimeric complexes of Nic96 and Nup188 or Nup192 (Figures 2B, 2D, S2B). Direct binding of the Nup84-subcomplex to GLFG repeats was restricted to the Nup84–Nup133(521-1157) portion (Figures 2C–D, S2C). Thus, direct and specific interactions between GLFG-repeats and a large fraction of scaffold Nups can be detected in vitro.
To test whether these interactions can also be observed within functional NPCs we utilized digitonin-permeabilized HeLa cells, which have been widely used to monitor nucleocytoplasmic transport in vitro (Adam et al., 1990). In this assay, selective NPC translocation occurs when proteins -such as NTRs- bind to FG-repeats within the NPC. YFP-Importin-β and 3xGFP were used as positive and negative controls, respectively (Figures 3A–B). To mimic transport conditions, experiments were performed with or without cytosol and energy (GTP), and with or without wheat germ agglutinin (WGA), which blocks FG-repeat dependent NPC translocation (Mohr et al., 2009)(Figures 3A–B). Unlike MBP-GFP-Nup53ΔC that does not bind FG-repeats (Figure 2D), all tested GLFG-binding scaffold Nups, including GFP-Nup170, GFP-Nup157, GFP-Nup192 (co-purified with Nic96), GFP-Nup188, and GFP-Nic96(186-839), behaved like YFP-Importin-β and displayed efficient nuclear translocation in the absence of cytosol. Nup translocation was inhibited by WGA, whereas addition of cytosol either did not affect or caused varying degrees of inhibition similar to the behavior of YFP-Importin-β (Figures 3A–B). Furthermore, in permeabilized HeLa cells pre-treated with RanGTP to release endogenous NTRs, GFP-Nups not only translocated into the nucleus but also decorated NPCs consistent with their FG-repeat interactions. This was inhibited by the addition of Importin-β (Figures 3C–D). Therefore GLFG-repeat binding scaffold Nups display NTR-like translocation properties and can bind FG-repeats within the intact NPC.
Figure 3. Nuclear translocation and NPC association of GLFG-repeat binding Nups.
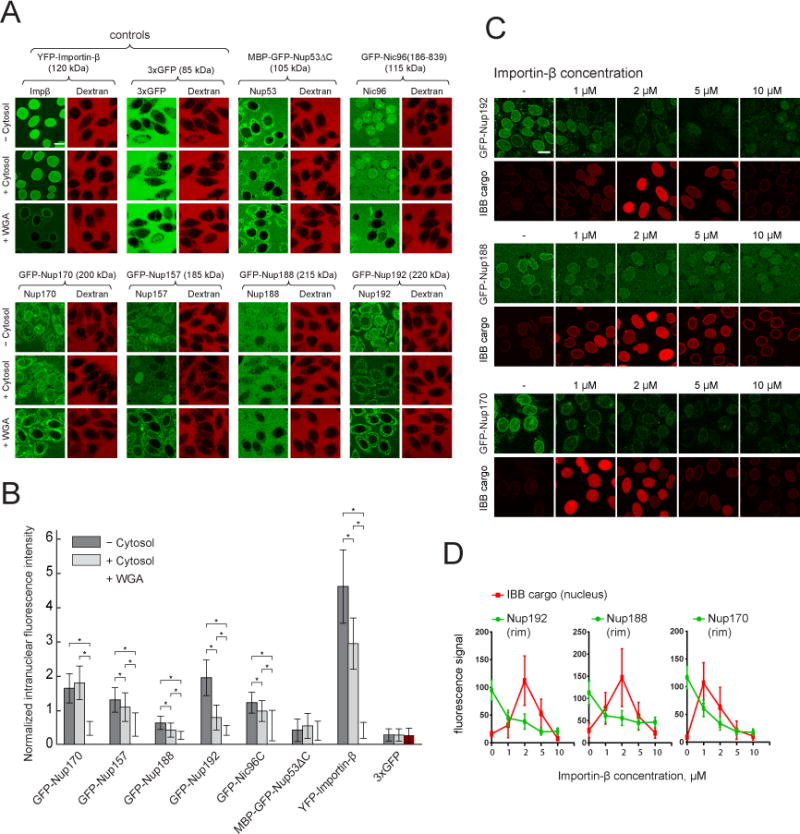
(A) Confocal images showing nuclear accumulation of the indicated proteins (green) 15 minutes after their addition to permeabilized cells. 155 kDa TRITC-dextran (red) was used to control for nuclear intactness.
(B) Plot of the intranuclear fluorescence intensities corresponding to (A) normalized to outside. Mean +−SD (between 26 and 116 intact analyzed nuclei per condition). Only nuclei with intranuclear/outside dextran intensities < 0.3 were considered intact. Asterisks (*) – indicate significant differences, Mann-Whitney p-values (< 0.01).
(C) Confocal images showing nuclear rim localization of Nups (green) and nuclear accumulation of the Importin-β binding cargo (IBB cargo, SnpIBB – Cerulean-biotin/Neutravidin-Dylight549) (red) in Ran-treated permeabilized HeLa cells after15 minutes incubation with the respective analytes pre-mixed with the indicated concentrations of unlabeled Importin-β.
(D) Plots of Nup and IBB cargo signal intensities corresponding to (C). Mean +− SD (n > 30 for each point).
Scale bars: 20 μm.
GLFG-repeats within Nup116 are essential in the absence of scaffold nucleoporin Nup188
We next sought to understand the physiological relevance of the interactions between GLFG-repeats and scaffold Nups. In budding yeast, individual GLFG-repeats are not essential for viability (Adams et al., 2015; Strawn et al., 2004). However, genes that code for physically interacting proteins often interact genetically (Costanzo et al., 2016). We therefore examined the synthetic effects of various GLFG-repeat deletions in combination with deletions of non-essential GLFG-binding scaffold Nups. Intriguingly, we found that the GLFG-repeats of Nup116 are required for viability in the absence of Nup188 (Figure 4A) suggesting that this segment within Nup116 has a redundant function with Nup188, an element of the NPC core.
Within Nup116, the GLFG-repeats are sandwiched between two Nup-interacting domains: the GLEBS domain that binds Gle2 and the middle domain (MD), which binds Nup192 (Bailer et al., 1998; Fischer et al., 2015)(Figure 4B). To examine whether the GLFG-motifs themselves are important for functionality and to exclude that the GLFG domain simply acts as a linker, we constructed variants of Nup116 in which GLFG-repeats were substituted with other sequences (Figure 4B). Non-GLFG substitutions were represented by Nsp1(1-563), Nup116(348-458)FA, 2× Nup116(348-458)FA and 4× Nup116(348-458)FA, spanning a size range of ~100 to ~600 amino acids. GLFG-repeats were represented by Nup100(1-610), Nup100(1-573), Nup100(1-307), 2× Nup100(1-307), Nup57(1-257), 2× Nup57(1-239), Nup116(348-458), 2× Nup116(348-458) and 4× Nup116(348-458), covering a similar size range as for the non-GLFG sequences.
We then tested the fitness of strains with the nup116ΔGLFG mutant background expressing these Nup116 variants when expression of Nup188 was repressed (Figures 4C, S3B). To this end expression of NUP188 was controlled by the methionine-repressible PMET3 promoter (Mao et al., 2002) allowing us to recapitulate the synthetic growth defect of the nup116ΔGLFG nup188Δ mutant in media containing excess methionine (Figure S3A). General functionality of the Nup116 variants was confirmed by their ability to rescue the temperature-sensitive phenotype of the nup116Δ mutant (Doye et al., 1994)(Figure S3B, right).
GLFG-containing repeats of different Nup origin and position potently rescued growth upon depletion of Nup188 including Nup100(1-610), Nup100(1-573), Nup100(1-307), 2× Nup100(1-307), 2× Nup57(1-239), and 4× Nup116(348-458) (Figures 4C, S3B). However, the replacement of phenylanines by alanines within the GLFG-motifs (4× Nup116(348-458)FA) rendered the repeats non-functional (Figures 4C, S3B). In addition, we observed a length dependence since short GLFG-repeats (Nup57(1-257), Nup116(348-458) and 2× Nup116(348-458)) were unable to rescue growth. The functionality could however be regained by increasing the number of repeats (2xNup57(1-239), 4× Nup116(348-458)), and likewise the partial functionality of a Nup100(1-307) GLFG-segment could be enhanced by a tandem version (2× Nup100(1-307)) (Figures 4C, S3B). Again, this functionality required the GLFG-motifs since non-GLFG sequences - even the longest ones - were unable to rescue the growth phenotype (Figures 4C, S3B).
We conclude that the functionality of Nup116 GLFG-repeats critically depends on GLFG-motifs and is determined by generic properties of GLFG-repeats rather than by unique sequence features within the Nup116 GLFG-repeat segment. Furthermore, there is a critical length threshold of ~300 amino acids.
GLFG-repeats connect Nup116 to the NPC scaffold
Our results indicate that in the absence of Nup188, interactions between the GLFG repeats of Nup116 and one or more binding partners are essential for viability. To aid with the identification of such interaction partner(s), we set out to quantitatively analyze the interactions of Nup116 with various GLFG-binding Nups using biolayer interferometry (BLI). We immobilized purified and directionally biotinylated Nup116 and its variant lacking GLFG-repeats on streptavidin-coated sensor tips, and analyzed the interaction with purified binding partners in solution (Figures 4D, S4A). The NTR Kap95 displayed efficient binding to full-length Nup116, but not to a Nup116 variant lacking the GLFG segment, demonstrating that BLI allows for the specific detection of GLFG repeat-mediated interactions (Figures 4D, S4B). Similar to Kap95, binding of the scaffold Nups Nup170, Nup84-Nup133(521-1157) and Nup188 was strictly dependent on the presence of GLFG-repeats in Nup116 (Figures 4D, S4B). In contrast, Nup192(2-960) bound both variants of Nup116, consistent with the previously published observation that Nup192 can also interact with the MD of Nup116 (Fischer et al., 2015) (Figure 4D). However, the presence of the GLFG repeats enhanced the interaction of Nup192(2-960) with Nup116 and lowered the KD about 15-fold to 500 nM, for the dominant of the two binding modes (Figure S4B). These results demonstrate that the GLFG repeats strongly contribute to the interaction between Nup116 and multiple individual scaffold Nups in vitro.
To understand whether GLFG-repeats also affect the interactions of Nup116 with scaffold Nups in vivo, we analyzed the amount of Nup170 and Nup192 that co-purified with either wild-type Nup116 or Nup116ΔGLFG in pulldowns in the presence or absence of Nup188. In contrast to cells expressing wild-type Nup116, the depletion of Nup188 in Nup116ΔGLFG-expressing cells robustly decreased the co-purification efficiency of both Nup170 and Nup192 (Figures 4E–F, S4C–D). Together these results are consistent with a model wherein the GLFG repeats of Nup116 act as velcro connecting Nup116 with several scaffold Nups. In the absence of Nup188, this function of the GLFG repeats becomes essential for the attachment of Nup116 to NPC core elements including Nup192 and Nup170 (Figure 4G).
This model predicts that without Nup188, NPC anchoring of Nup116 becomes dependent on GLFG-repeat mediated interactions. In agreement with this, treatment of Nup188-depleted cells with 5% 1,6-hexanediol, which is known to disrupt GLFG-repeat mediated interactions (Patel et al., 2007), caused rapid and complete mislocalization of Nup116, but this was not observed for the core nucleoporin Nup133 (Figure S4E).
The GLFG-repeats of Nup116 function in NPC biogenesis
To assess the role of the Nup116 GLFG-repeats in the structural organization of the NPC in an unbiased and quantitative manner, we implemented an imaging pipeline to automatically quantify the localization of Nups at the NE in various mutants and experimental conditions. We used the NE/ER marker dsRed-HDEL (Bevis et al., 2002) to trace NE contours, which allowed us to quantify precisely the GFP signal intensities at the NE for various GFP-tagged Nups in thousands of cells (Figure S5).
Taking advantage of this pipeline we examined the localization of various Nups upon Nup188 depletion in cells expressing either full-length Nup116 or the Nup116_Δ_GLFG variant. Whereas depletion of Nup188 in the presence of full-length Nup116 showed no obvious phenotype, in the case of the nup116ΔGLFG mutant, this led to a specific nucleoporin localization defect: both the Nup116ΔGLFG variant itself and its direct interactor Nup82, a component of the NPC cytoplasmic ring (Fernandez-Martinez et al., 2016; Yoshida et al., 2011) showed ~5-fold and ~3-fold decrease in their nuclear rim signal intensities, respectively (Figures 5A–B, S6A, S6D–F). Yet, none of the other tested GLFG-repeat Nups (Nup57 and Nup100) or GLFG-interacting scaffold Nups of the symmetrical NPC core (Nup133, Nup170, Nup157, and Nup192) showed such a localization defect (Figures 5A–C). Furthermore, fluorescence recovery after photobleaching (FRAP) experiments in nup116ΔGLFG cells did not reveal major changes in the NE mobility of the core nucleoporin Nup133 upon Nup188 suppression (Figures S6H–K). Thus, depletion of Nup188 in nup116ΔGLFG cells leads to structural changes in the NPC resulting specifically in the mislocaliztion of Nup116ΔGLFG itself and the cytoplasmic nucleoporin Nup82.
Figure 5. GLFG-repeats of Nup116 are critical for NPC biogenesis in the absence of Nup188. See also Figures S5, S6.
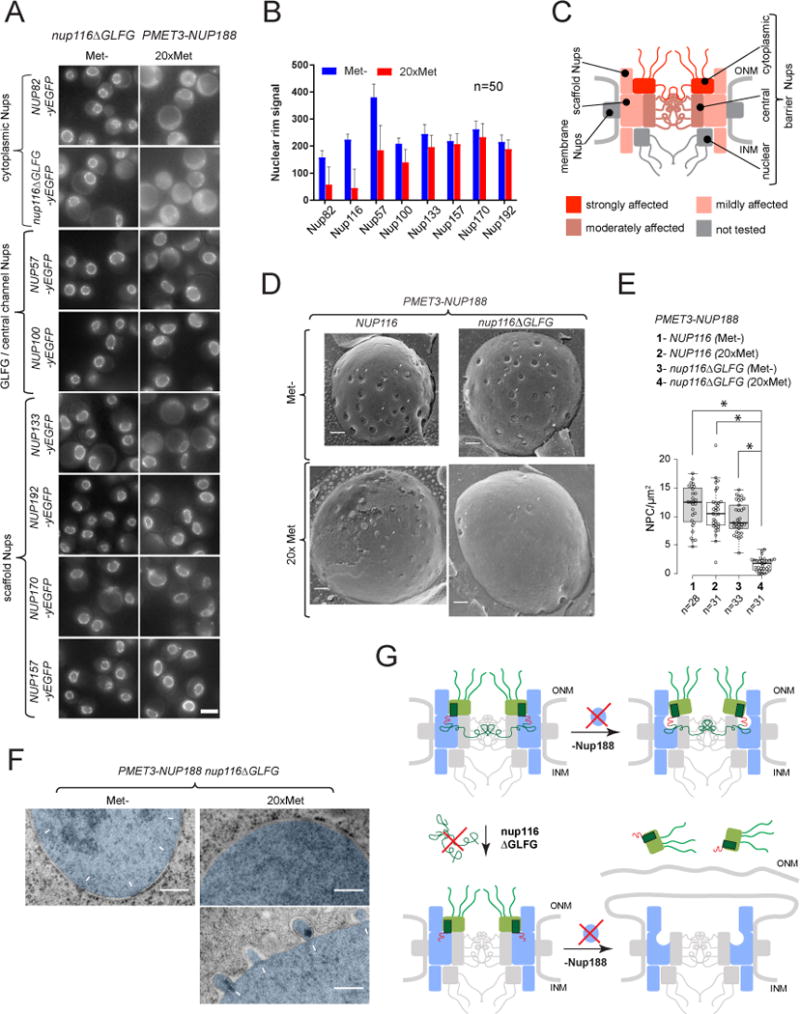
(A) Fluorescence images illustrating localization of GFP-tagged Nups in PMET3-NUP188 nup116ΔGLFG cells after 12h incubation in Met- or 20×Met media. Scale bar – 5 μm.
(B) DsRed-HDEL-based quantification of Nup-GFP signal intensities at the nuclear rim. Mean +/− SD; (n) - number of analyzed image frames per condition.
(C) Graphical summary of Nup mislocalization effects shown in (A and B).
(D) Freeze-fracture SEM images illustrating the NPC morphology at the NE (arrowheads) in PMET3-NUP188 cells expressing either the wild-type or ΔGLFG variant of Nup116, incubated for 12h in Met- or 20× Met media.
(E) Box-plot summarizing densities of NPC structures quantified for the experiments in (C). (n) - number of examined NE fractures. Levels - median values; boxes -interquartile ranges; error bars - upper and lower whiskers; Asterisks (*) – significant differences, Mann-Whitney p-values (< 0.0001).
(F) TEM images of cells treated as described in (D). The nuclear regions (nuc) are pseudo-colored in blue, white arrows – intact NPCs and NE herniations. (G) Model describing the development of NPC defects observed upon deletion of Nup116 GLFG-repeats and depletion of Nup188.
The effects on the localization of Nup116 and Nup82 depended on the GLFG-repeat segment in Nup116 since both Nup116 localization and cell growth could be rescued by substitution of the GLFG repeats with the GLFG repeats from Nup100 (GLFG->Nup100(1-610)-yEGFP) but not by yEGFP (Figures S6B–C). Furthermore, the ability of ectopically expressed Nup116 GLFG-repeat substitutions to restore growth in nup116ΔGLFG cells perfectly correlated with their ability to restore Nup82 localization upon Nup188 suppression (Figures 4C, S6G). Therefore, the GLFG-repeats within Nup116 are needed for the correct structural organization of the NPC and act redundantly with Nup188 to confer NPC incorporation of Nup116 and its interacting partner, the cytoplasmic ring nucleoporin Nup82 (Figure 5C).
The selective localization defect of Nup82 and Nup116 in the absence of the GLFG repeats of Nup116 could be caused either by the formation of NPCs that selectively lack components of the cytoplasmic ring, or by an NPC biogenesis defect that prevents assembly of complete pores. To differentiate between these two possibilities, we complemented our fluorescence microscopy analysis with freeze-fracture and transmission EM. As expected expression of full-length Nup116 did not lead to changes in pore density upon depletion of Nup188. Likewise, Nup116ΔGLFG-expressing cells displayed only a minor decrease in NPC density in the presence of Nup188 (Figures 5D–E). However, upon Nup188 depletion nup116ΔGLFG cells almost entirely lacked detectable pore structures in the NE (Figures 5D–E). Consistently, also few normal pores could be found by transmission EM. Instead we frequently detected herniations of the inner nuclear membrane (Figure 5F), a phenotype often accompanying NPC biogenesis defects. Therefore, the Nup116 GLFG-repeats are critical for NPC biogenesis and their absence leads to the inability of the NPC to fully insert into the NE and incorporate elements of the cytoplasmic ring.
Taken together, these results suggest that Nup188 and Nup116 via its GLFG-repeats function redundantly to connect cytoplasmic and symmetrical core components of the NPC, and that this link is essential for the completion of NPC assembly (Figure 5G).
GLFG-repeats confer efficient NPC targeting
To examine whether GLFG-repeats also impact the organization of intact NPCs, we compared the localization of wild-type Nup116 and its ΔGLFG variants tagged with GFP in cells with an otherwise wild-type background. Removal of the GLFG repeats led to a significant reduction of NE signal of Nup116 (Figures 6A–B). The effect was even more striking when other known Nup116 NPC-binding domains, i.e., the C-terminal domain (CTD) or the middle domain (MD) were deleted (Figures 6C–E). This demonstrates that the GLFG-repeat segment has an additive effect on the Nup116 NE localization functioning together with both the MD and CTD. Consistently, fluorescence loss in photobleaching (FLIP) experiments revealed that the GLFG-repeats enhanced the stability of the Nup116ΔCTD variant at the NPC (Figure 6F). Therefore, in otherwise wild-type NPCs, the GLFG-repeat segment is necessary for stable targeting of Nup116 to the NPC.
Figure 6. Effect of Nup116 GLFG repeats on NPC targeting efficiency and NPC localization stability. See also Figure S7.
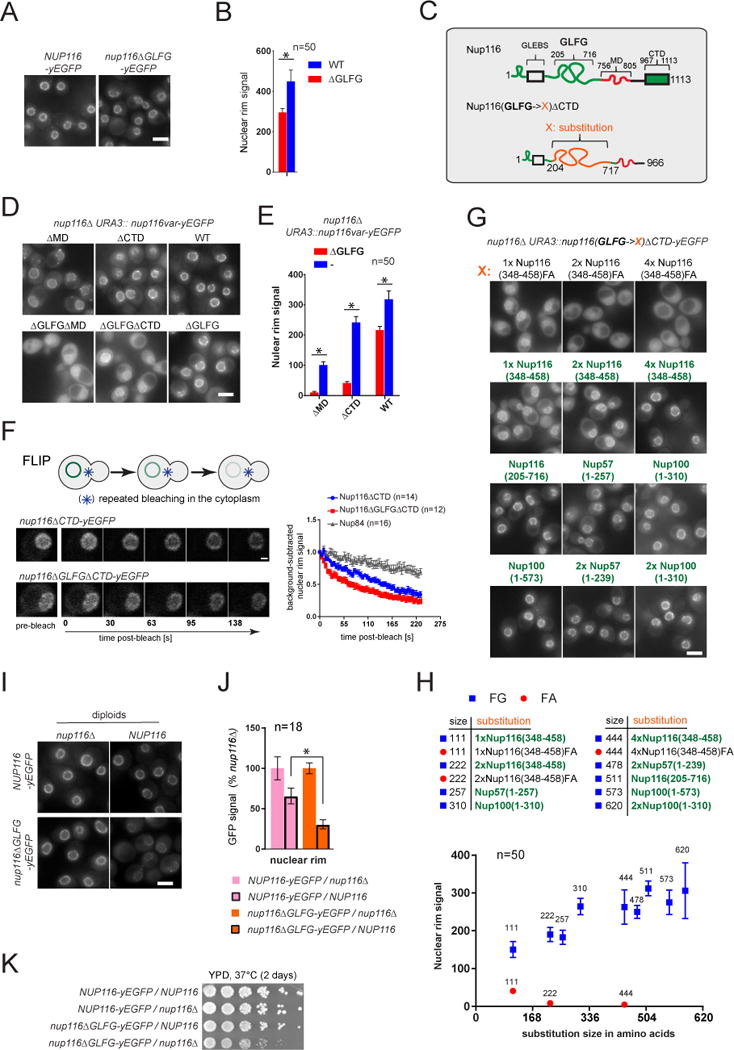
(A, D, G, I) Fluorescence images illustrating intracellular distribution of indicated GFP-labeled Nup116 variants in actively growing yeast cells with the specified genotypes. Scale bars – 5 μm.
(B, E, H) Corresponding (B to A, E to D, H to G) dsRed-HDEL-based quantification of fluorescence signal at the nuclear rim. Mean +/−SD. (n) - number of analyzed image frames. Asterisks (*) – significant differences, Students t-test p-values (< 0.0001).
(C) Schematic of Nup116 and its C-terminally truncated GLFG-substitution variants used in the figure.
(F) Stability of NPC localization analyzed for Nup116 variants and for the symmetric core nucleoporin Nup84 by FLIP. Fluorescence image sequences (left) and the graphs (right) showing loss of background-subtracted nuclear rim signal upon continuous bleaching within the cytosol. Mean +/− SEM. (n) - number of individual traces per condition.
(J) Corresponding to (I) evaluation of differences between the NE incorporation of full-length and ΔGLFG Nup116 variants due to presence of the wild-type NUP116 allele. GFP signal intensities of the tagged Nup116 variants are expressed relative to the values in nup116Δ cells. Note a milder relative drop in the nuclear rim signal for the full-length as compared to ΔGLFG variant conferred by wild-type NUP116 allele. Mean +/−SD. (n) - number of analyzed image frames.
(K) Growth of yeast strains in (I) analyzed on plate at 37°C.
Asterisks (*) – significant differences, Student t-test p-values (< 0.0001).
To understand whether the functions of the Nup116 GLFG-repeats in NPC targeting (Figures 6A–E) and NPC biogenesis (Figures 4C, S6G) are conferred by the same sequence properties we employed GFP-tagged Nup116ΔCTD as reporter and replaced the GLFG-segment with the same sequences that we had utilized for assaying viability and NPC biogenesis in the absence of Nup188. We found an excellent correlation between the ability of individual substitutions to support NPC targeting (Figures 6G–H) and their function in NPC biogenesis (Figures 4C, S6G): efficient NE localization strictly depended on the presence of GLFG motifs and improved when the length of the repeats was increased (Figures 6G–H).
This striking correlation suggests that the Nup116 GLFG-repeats could ensure that functionally intact NPC structures only assemble when they contain GLFG-repeats. To test this, we performed an in vivo competition assay and compared the NE localization of GFP-labeled wild-type and the ΔGLFG variants of Nup116 in diploid cells either in the presence or absence of a wild-type NUP116 allele. Hence, in the same cell two variants of the same protein (labeled and unlabeled) either do or do not compete for NPC incorporation. In comparison to the full-length protein, NPC incorporation of the ΔGLFG variant was significantly suppressed by the competition with the unlabeled wild-type copy (Figures 6I–J). This competition also suppressed a decreased fitness of cells carrying the ΔGLFG variant (Figure 6K). Therefore, the presence of GLFGs repeats confers more efficient NPC incorporation of Nup116.
Finally, we asked whether the NPC targeting property of GLFG-repeats is unique to Nup116 or can also be observed in other GLFG-containing proteins. For this we took advantage of Nup100, the Nup116 paralogue with a similar but distinct domain composition and NPC interactions (Alber et al., 2007; Lin et al., 2016) (Figure S7A). As for Nup116, also the GLFG-repeat segment of Nup100 contributed additively and functioned together with the CTD and MD in NPC targeting (Figures S7B–C). Moreover, NPC localization could be detected for the Nup100 GLFG-repeats alone (Figures S7B–C), further supporting the function of GLFG-repeats as an NPC targeting determinant. Again as for Nup116, the GLFG-repeats in Nup100 also conferred more stable NPC localization and a preferential NPC incorporation in a competition assay (Figures S7D–F). Therefore, the NPC targeting function of GLFG-repeats is not restricted to Nup116 and may be general as it can also be seen with the GLFG repeats of Nup100.
Discussion
FG-repeat sequences are present within ~1/3 of all nuclear pore proteins and their interactions with NTRs are critical in establishing the NPC’s selective diffusion barrier and for nucleocytoplasmic transport (Mohr et al., 2009; Ribbeck and Gorlich, 2001). Here, we identify an additional role of FG-repeats as structural components of the NPC that specifically and directly interact with the scaffold of the NPC and function during NPC assembly.
Whereas Nup/FG-repeat interactions had previously been reported (Allen et al., 2001; Andersen et al., 2013b; Patel et al., 2007; Schrader et al., 2008), their significance and specificity was unclear. Our analyses demonstrate that the family of FG-repeats carrying GLFG-motifs (GLFG-repeats) directly bind to several scaffold nucleoporins including Nup170, Nup157, Nup188, Nup192, Nic96, and the Nup84-Nup133 portion of the Nup84-subcomplex (Figure 2D). Binding of GLFG-repeats to scaffold Nups is dynamic and their interaction strengths are comparable to GLFG/NTR-repeat interactions (Figures 4D, S4B). Furthermore, GLFG-binding Nups –akin to NTRs- are able to translocate across intact NPCs in vitro and compete for NPC binding sites with the NTRs (Figure 3). The significance of these NTR-like translocation properties remains unclear. However, this could aid in the delivery of newly synthesized scaffold subunits to the INM during NPC assembly, which occurs at least partially from the nuclear side (Otsuka et al., 2016). Consistent with such a model, WGA and a dominant-negative fragment of Importin-β were shown to potently block NPC assembly in sealed Xenopus nuclei (D’Angelo et al., 2006; Harel et al., 2003).
Intriguingly, GLFG-binding scaffold Nups also have structural similarities to NTRs. The GLFG-repeat binding domains of Nup170, Nup188, Nup192 and Nic96 are composed of α-helical stacks that share characteristics with the α-helical fold of NTRs (Conti et al., 2006). Moreover, Nup188 and Nup192 are structurally similar to the canonical NTRs importin-α and importin-β (Amlacher et al., 2011; Andersen et al., 2013b; Flemming et al., 2012; Sampathkumar et al., 2013; Stuwe et al., 2014). These similarities may point to a deep evolutionary connection between the membrane-bound and soluble parts of the nucleocytoplasmic transport machinery.
In spite of these parallels, scaffold Nups - unlike NTRs - are stable components of the NPC, and in addition to their GLFG-repeat interactions also bind to other NPC elements. Our results reveal that GLFG-repeats are important to physically connect NPC subunits: (i) the GLFG-repeats of Nup116 are functionally redundant with Nup188, an element of the NPC core (Figure 4A), (ii) in the absence of Nup188, the GLFG-repeats of Nup116 are needed to establish the connection with the GLFG-binding scaffold Nups: Nup170 and Nup192 (Figures 4E–F); and (iii) the GLFG-repeat segments of Nup116 as well as of Nup100 contribute to targeting and stable incorporation of these Nups into intact NPCs (Figures 6, S7).
Mechanistically, the GLFG-repeats are likely to act in concert with other Nup-binding domains, such as the MD and the CTD. This is exemplified by the interaction with Nup192, which can bind to the MD of Nup116 alone, but the interaction is strengthened more than an order of magnitude by GLFG repeats (Figures 4D, S4B). However, unlike interactions mediated by other Nup domains, the binding of GLFG-repeats to scaffold Nups is likely to be promiscuous (Figures 2, 4) and a GLFG segment may bind to several scaffold Nups at the same time. The structural function of GLFG repeats might not be unique to Nup116 but could be a common feature of GLFG-repeat nucleoporins, including Nup100 and Nup57 (Figures 4C, S3B, S6G, 6G–H). In addition, scaffold Nups, like NTRs, likely contain several FG-repeat binding sites (Bednenko et al., 2003; Isgro and Schulten, 2005). Furthermore, GLFG-type repeats also have a propensity to interact homotypically (Patel et al., 2007). Therefore, we propose a model that GLFG-repeats act together with other Nup interactions to form a multivalent network which functions akin to velcro connecting multiple NPC subunits to promote NPC assembly (Figure 7). But other contributions of GLFG-repeats cannot be excluded and additional experiments e.g. in vitro reconstitutions will be important to further investigate role of GLFG-repeats in NPC biogenesis.
Figure 7. Summary model: function of GLFG-type repeats as velcro during NPC biogenesis.
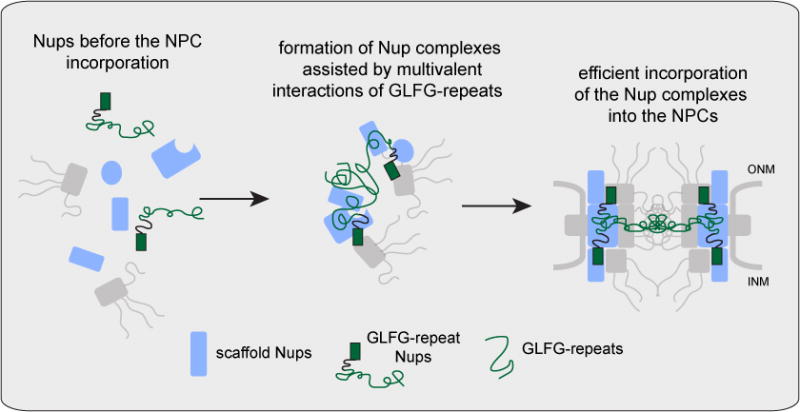
GLFG-type repeats have velcro function to multivalently link scaffold Nups. This function ensures incorporation of GLFG-repeats into the final NPC structure conferring formation of fully functional pores.
The NPC defect associated with the loss of the Nup116 GLFG-repeats (Figure 5) is intriguing, as it points to a malfunction at a late step of NPC biogenesis. Although it is not clear yet whether the structures that accumulate represent true NPC biogenesis intermediates, they nevertheless contain all critical core Nups (Figure 5A–B) already arranged into higher order assemblies as judged by the slow mobility within the NE similar to intact NPCs (Figures S6H–K). But the formation of complete pores that span both nuclear membranes and the incorporation of cytoplasmic NPC components do not occur (Figure 5). A detailed picture of how GLFG-repeats and their physical connections to the NPC scaffold contribute to the ‘hole punch’ mechanism in the NE awaits future studies. However, a dual function of GLFG-repeats in NPC-biogenesis and in the formation of a nucleocytoplamic diffusion barrier in the intact NPC (Figure 7) could ensure that NE fenestration occurs only when all components that form a selective barrier are in place, and would thus prevent the formation of non-selective holes in the NE that allow for an uncontrolled exchange of nuclear and cytoplasmic material. Such a coupling mechanism could also act as a quality-control step since it would promote the preferential incorporation of Nups with intact GLFG-repeat segments, as observed in our allelic competition experiments (Figures 6I–K, S7E–F).
In addition to NPC biogenesis, interactions between GLFG-repeats and scaffold Nups are also likely to contribute to the stability of intact pores (Figure 6). Consistent with this, we and others (Shulga and Goldfarb, 2003) have observed a disruptive effect of 1,6-hexanediol, which inhibits FG-repeat interactions, on the NPC structure in cells with a compromised NPC scaffold (Figure S4E). Furthermore, within the NPC, interactions between GLFG-repeats and the NPC scaffold could help to ‘seal’ the FG-repeat network to the walls of the pore and could thus contribute, together with the cohesive properties of the GLFG repeats, to the establishment of the nucleocytoplasmic diffusion barrier. Being in competition with NTRs (Figure 3), interactions between GLFG-repeats and the NPC scaffold might in addition provide the NPC with a “rubber band”-like structural flexibility, which could be necessary, for example, for the translocation of large substrates or transmembrane proteins. Although further analysis will be required to dissect the global contributions of FG-repeats to NPC function, our results show that FG-repeat segments not only fill the central channel of the NPC as part of the selectivity barrier but also represent an integral part of the NPC structure. Existing structural approaches have been unable to identify the position of natively unfolded FG repeats. Thus, our results extend current architectural models and identify a novel, previously unanticipated layer of connections within the NPC.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Karsten Weis (karsten.weis@bc.biol.ethz.ch).
Experimental Model and Subject Details
Bacterial Strains
The following E. coli strains were used in this study for production of recombinant proteins: BL21 (DE3) (Agilent), BL21(DE3)-RIL (Agilent) and LOBSTR(DE3)-RIL (kerafast) (Andersen et al., 2013a). Strain usage in each case is specified in “Recombinant proteins” section. Cells were cultured in Luria-Bertani (LB) liquid medium supplemented with necessary antibiotics and protein expression was induced, unless otherwise noted, by 0.5 mM IPTG (final concentration (f.c.)).
Yeast Strains
Genotypes of all Saccharomyces cerevisiae strains used in this study and further accompanying information can be found in Table S1. All the strains were derived from W303 (ade2-1 ura3-1 his3-11,15 trp 1-1 leu2- 3,112 can1-100) using standard yeast handling and molecular cloning protocols. Cells were grown either in YPD medium (1% yeast extract, 2% bactopeptone, 2% dextrose), YPGal medium (1% yeast extract, 2% bactopeptone, 2% galactose) or SCD (synthetic complete media containing 2% dextrose) with the indicated amino acid dropouts and/or additives. Unless otherwise noted the cultures were grown at 30°C.
Mammalian Cell Lines
HeLa cells were cultured in DMEM media + 10% fetal bovine serum. Cells were seeded on glass-bottomed poly-lysine coated dishes (MatTek) at a concentration of 5 × 105 cells/dish one day before use.
Methods Details
Common procedures
Recombinant DNA
Names of DNA constructs and further accompanying information can be found in Table S2. Constructs have been produced as outlined below and confirmed by sequencing. For construction of pAU80, pSB142, pNL123, pAU102 and pKW3197 respective yeast protein coding sequences were amplified from genomic DNA, fused with N- or C- terminal affinity tags (see Table S2) and cloned into pETDuet-1 vector (EMD Millipore). For construction of pTS164, pTS165 and pKW2674 the corresponding Nup116 coding sequences or Nup53(1-460) coding sequence were amplified from yeast genomic DNA, fused N-terminally with affinity/labeling tags (see Table S2) and cloned into pSV272 expression vector. pKW3046 and pKW803 were derived from pFA6a-GFP(S65T)-kanMX6 and pFA6a-GFP(S65T)-HIS3MX6 plasmids (Longtine et al., 1998) respectively by cloning the corresponding epitope/affinity tag sequences (see Table S2) via PacI and AscI sites. The same cloning strategy was used to derive pKW2631 and pKW2487 from pFA6a-His3MX6-PGAL1-3HA (Longtine et al., 1998). pKW2740 was constructed by cloning the fusion product between the yeast PGAL1 promoter sequence, tandem affinity tag coding sequence, yeast genome amplified Nic96(100-839) coding fragment and yeast ADH1 terminator sequence into the pRS306 vector (Sikorski and Hieter, 1989). pKW3373 and pKW3638 were constructed by cloning NUP188 and NUP116 genomic loci (ORF + upstream and downstream flanking sequences) amplified from yeast genomic DNA into pRS316 (Sikorski and Hieter, 1989). The pRS16 plasmids encoding for various GLFG repeat substitutions within Nup116 (pKW3657, pKW3659, pKW3660, pKW3661, pKW3662, pKW3663, pKW3728, pKW3729, pKW3731, pKW3732, pKW3775, pKW3776, pKW3779, pKW3780) were derived from pKW3638 by replacing Nup116 GLFG-repeat coding sequence (Nup116(205-715)) with the various GLFG repeat substitution coding sequences (see Table S2).
Preparation of buffers
Buffer recipes frequently used throughout the study are listed in Table S3.
Imaging and analysis of SDS protein gels
Unless otherwise mentioned protein gels were stained with SYPRO Ruby (Thermo Fisher) and imaged using a FluorChem HD2 UV-transilluminator equipped with a CCD-camera (Alpha Innotech). Digital images were analyzed and processed using ImageJ software.
Preparation of RNAse-S coupled beads
0.75g dry CNBr-activated Sepharose 4B beads (GE Healthcare) were swollen for 30 minutes in 10 ml 1 mM HCl, briefly washed 2 times with 10 ml of Coupling Buffer (freshly prepared 100 mM NaHCO3) followed by the addition of 2.5 mg RNAse-S (Sigma-Aldrich) diluted in Coupling Buffer to 1 mg/ml. The mixture was incubated for 2 hours at RT with constant agitation. The beads were then incubated for 1 hour at RT in 1 M ethanolamine pH 9.0, sequentially washed two times/10 ml each with 1 M NaCl, 100 mM glycine pH 2.8, distilled water, and finally equilibrated in PBS pH 7.4 containing 0.01% NaN3. The beads were stored at 4°C.
Preparation of IgG-coupled Sepharose beads
Coupling of rabbit IgG to Sepharose beads was performed essentially as in (Alber et al., 2007) with minor modifications. Specifically, 2.25g dry CNBr-activated Sepharose 4B beads (GE-healthcare) was swollen for 30 minutes in 1 mM HCl at RT and washed briefly two times with 10 ml of Coupling Buffer. The beads were unloaded into a glass beaker, diluted with Coupling Buffer and supplemented while stirring with 25 mg of rabbit IgG powder (Sigma-Aldrich). The stirring was continued over night at 4°C. The beads were then washed at RT with Coupling Buffer followed by agitating 1h each with 1M ethanolamine pH 9.0 and 100 mM glycine pH 2.8. Beads were washed two times/0 ml each with distilled water and PBS pH 7.4, then supplemented with 0.01% NaN3 (f.c.) and stored at 4°C.
Preparation of IgG-coupled Dynabeads
IgG-coupled Dynabeads were prepared essentially as in described in (Alber et al., 2007). 300 mg of magnetic Dynabeads (Thermo Fisher) was re-suspended and vortexed for 30 sec in 16 ml of 0.1M NaPO4 buffer (0.1 M NaH/H2PO4 pH 7.4). Bead suspension was divided into four equal portions (~ 4 ml each) that were agitated in 15 ml screw-cup plastic tubes during preparation of fresh Antibody Mix. For that 100 mg of rabbit IgG powder (Sigma-Aldrich) was solubilized with 3.5 ml distilled water followed by 10 minutes 14K rpm centrifugation in a cooled table top centrifuge at 4°C to pellet insoluble material. 3525 l of supernatant was mixed with 9.85 ml of 0.1M NaPO4 buffer immediately followed by the addition of 6.65 ml 3M Ammonium Sulfate buffer (39.6 g (NH4)2SO4 (MW 132.1) dissolved in 0.1M NaPO4 buffer and adjusted to 100 ml). The Antibody Mix was then filtered through a 0.22 m Millex GP filter (Merck Millipore). The beads were washed by briefly vortexing a bead suspension, settling the beads using an MPC-6 Magnetic Particle Concentrator (Thermo Fisher) and carefully aspirating the buffer, followed by resuspension in 4mL 0.1M NaPO4 buffer per aliquot. After an additional round of 0.1M NaPO4 buffer wash, bead pellets were supplemented with the equal amounts of the Antibody Mix (~ 5 ml per aliquot), vortexed to completely resuspend beads and incubated with constant agitation for ~ 18 h at 30°C. The aliquots of IgG-coupled beads were briefly washed using the Magnetic Particle Concentrator (as described above) one time with 3 ml of each: 100 mM Glycine-HCL pH2.5, 10mM Tris-HCl pH 8.8 and 100 mM freshly prepared triethylamine. This was followed by 4 × 5 min washes with 3 ml PBS pH 7.4, 2× 10 min washes with 3 ml PBS pH 7.4 containing 0.5% Triton X-100 (f.c.). Bead aliquots were pooled, finally equilibrated with PBS pH 7.4 supplemented with 0.02% NaN3 (f.c.) and stored at 4°C.
Purification of proteins
The proteins used in this study were expressed and purified as recombinant proteins in E. coli, from overexpressing yeast cells, or as native proteins from yeast (Nup84-subcomplex).
Recombinant proteins
The GST-FG-repeat fusions: GST-Nup100(1–307) (pKW2959), GST-Nup100(1–610) (pKW2960), GST-Nsp1(1–563) (pKW2958), GST-Nup116(348-458) (pKW2907), GST-Nup116(348-458)FA (pKW2908) were purified essentially as described in (Andersen et al., 2013b). Briefly, the proteins were expressed in E.coli BL21(DE3) and purified on Glutathione Sepharose 4B beads (GE Healthcare) in PBS pH 7.4 supplemented with 10 mM DTT, 0.1 mM PMSF and 0.5% Triton X-100. The beads were washed with PBS. Bound proteins were eluted with glutathione elution buffer (10 mM glutathione, 50 mM Tris-HCl pH 8.0), followed by dialysis against GF1 Buffer (10 mM HEPES pH 7.5, 150 mM NaCl and 1 mM DTT). Protein concentrations were determined using Bio-Rad Protein Assay (Bio-Rad). Protein aliquots were flash-frozen in liquid nitrogen.
_Sc_Kap95 (pKW2470), 3xGFP (pJL71) and YFP-_Hs_Importin-β (pKW1532), _Hs_Importin-β (pKW485) (Chi and Adam, 1997) were expressed in E.coli BL21(DE3) and purified on Ni-affinity resin (Sigma-Aldrich) in Na-phosphate Buffer pH 8.0 (50 mM NaH/H2PO4 pH 8.0, 250 mM NaCl, 5 mM β-mercaptoethanol), followed by dialysis against the GF1 Buffer. The protein aliquots were flash-frozen in liquid nitrogen. Biotin-conjugated His6 – SnpIBB – Cerulean (IBB cargo) was generated as described in (Lowe et al., 2010).
MBP-GFP-Nup53 C (pKW2674) was expressed in E.coli BL21(DE3). Protein expression was induced at OD600 =1.0 at room temperature for 2 h. Cells were lysed with French press, and the protein was purified on Ni-affinity resin (Sigma-Aldrich) in Na-phosphate Buffer pH.7.5 (50 mM NaH/H2PO4 pH 7.5, 300 mM NaCl, 10 mM β-mercaptoethanol), eluted with Na-phosphate Buffer pH.7.5 containing 400 mM Imidazole followed by purification on a Superdex 200 10/300 gel filtration column (GE Healthcare) equilibrated with HEPES-KOAc Buffer (10 mM HEPES pH 7.5, 160 mM KOAc, 1 mM MgOAc2, 1 mM DTT). The peak fractions corresponding to non-degraded and non-aggregated protein were pooled and supplemented with 20% glycerol (f. c.). The protein aliquots were flash-frozen in liquid nitrogen.
Expression of Nup192(2-960) (pKW3197), Nup170(980-1502) (KA18) was performed in E.coli BL21(DE3) cells induced with IPTG at OD600 = 0.7 at 18°C for 16 hours. Cells were lysed using a French press, and the proteins were purified on Ni-affinity resin (Sigma-Aldrich) in Na-phosphate-Imidazole Buffer pH 8.0 (50 mM NaH/H2PO4 pH 8.0, 250 mM NaCl, 5 mM β-mercaptoethanol, 15mM Imidazole pH 8.0). Proteins were then eluted with Na-phosphate-Imidazole Buffer pH 8.0 containing 400 mM Imidazole (f. c.); in the case of Nup192(2-960) the SUMO-tag was cleaved off in solution with human rhinovirus 3C-protease. Unless otherwise mentioned, the proteins were then further purified on a Superdex 200 10/300 gel filtration column (GE Healthcare) equilibrated with GF1 Buffer, pooled fractions were supplemented with 20% glycerol (f. c.), aliquoted and flash-frozen in liquid nitrogen.
Nup120S203-Y220 link3-Nup145C34-712-Sec13 (NL123), Nup85-Seh1 (SB142), and Nup84-Nup133(521-1157) (AU102) were separately expressed in LOBSTR(DE3)-RIL E. coli and initially purified by nickel affinity purification as described by (Kelley et al., 2015). Following nickel purification, N-terminal affinity tags were cleaved with 3C protease and dialyzed overnight. Nup120-Nup145C-Sec13 was dialyzed into 10 mM Tris/HCl pH 8.0, 200 mM NaCl, 0.1 mM EDTA, and 1 mM DTT and prior to loading onto a 5 mL HiTrap Q sepharose fast flow column (GE Healthcare) was diluted 1:1 with Q0 Buffer (10 mM Tris/HCl pH 8.0, 0.1 mM EDTA, and 1mM DTT). Nup120-Nup145C-Sec13 bound to the column and was eluted over a 15 column volume (CV) gradient using Q1 Buffer (10 mM Tris/HCl pH 8.0, 1 M NaCl, 0.1 mM EDTA, and 1 mM DTT). The eluate was pooled and loaded on a Superdex 200 26/60 gel filtration column (GE Healthcare) equilibrated in GF Buffer (10 mM Tris/HCl pH 8.0, 150 mM NaCl, 0.1 mM EDTA, and 1 mM DTT). The resultant peak was pooled and concentrated. Nup85-Seh1 was processed as Nup120-Nup145C-Sec13. Nup84-Nup133(521-1157) was dialyzed into 10 mM potassium phosphate pH 8.0, 150 mM NaCl, 0.1 mM EDTA, and 1 mM DTT and loaded directly onto a 5 mL HiTrap SP sepharose fast flow column (GE Healthcare). Bound Nup84-Nup133(521-1157) was eluted from the column using a 15 CV gradient of 10 mM potassium phosphate pH 8.0, 1 M NaCl, 0.1 mM EDTA, and 1 mM DTT. The pooled eluate was loaded onto a Superdex 200 26/60 gel filtration column equilibrated in GF Buffer. The Nup84-Nup133(521-1157) peak was pooled and concentrated. Purified Nup120-Nup145C-Sec13 was mixed with Nup85-Seh1 in a 1:1.5 ratio for 30 minutes on ice and run over a Superdex 200 26/60 column in GF Buffer. The complex peak, which was separate from excess Nup85-Seh1, was pooled and concentrated. This pentameric complex was mixed with Nup84-Nup133(521-1157) in a 1:1.5 molar ratio for 30 minutes on ice and loaded onto a Superdex 200 26/60 column in GF Buffer. The heptameric Nup84-subcomplex peak ran separately from excess Nup84-Nup133(521-1157) and was subsequently pooled, concentrated, and re-purified on a Superdex 200 26/60 column in GF Buffer. The resulting peak was pooled, concentrated, and used for binding experiments.
For biolayer interferometry (BLI) experiments, Kap95, Nup192(2-960), Nup84-Nup133(521-1157), were prepared as described above. Nup170(980-1502) (KA18) and Nup188 (AU80) were expressed in LOBSTR(DE3)-RIL E.coli cells (kerafast). The cells were grown to OD600 = 0.7 at 37°C, shifted to 18°C and induced with 0.2 mM IPTG for 16 hours. Cell pellets were resuspended in 50 mM potassium phosphate pH 8.0, 500 mM NaCl, 30 mM imidazole, 5 mM BME and lyzed in a Microfluidizer LM20 (Microfluidic). Cleared lysate was applied to Ni-Sepharose, washed, and eluted with 250 mM imidazole pH 8.0, 150 mM NaCl, 5 mM BME. The proteins were purified to homogeneity by anion exchange chromatography over a Mono Q column (GE Healthcare), followed by gel filtration over a Superdex 200 10/300 column (GE Healthcare) equilibrated in GF Buffer.
Nup116 (pTS164) and Nup116(718-1113) (pTS165) were expressed as His6-Avitag-MBP-fusions in LOBSTR(DE3)-RIL E.coli cells. The growth medium was supplemented with 100 μM biotin to boost biotinylation of the Avitag. Otherwise, the purification of the Nup116 constructs was identical to the other BLI probes, except that ion exchange was excluded.
Protein expression in yeast
For purification of GFP-Nup157 (KWY2596) and GFP-Nup170 (KWY2597) yeast pre-cultures were grown overnight at 30°C in 500 ml YPD supplemented with 20 mg/L adenine. Cells were washed with distilled water and cell pellet (re-suspended in a small volume of sterile distilled water) was reinoculated into 2 liters of YPGal. Protein expression was induced for 22 hours at 30°C. Cells were harvested, re-suspended in Lysis Buffer (50 mM HEPES pH 7.5, 150 mM KCl, 10mM MgOAc2, 0.1mM EDTA, 5 mM β-mercaptoethanol) containing 1 mM PMSF (f.c.) and protease inhibitors cocktail (Sigma-Aldrich), and disrupted with 0.5 mm glass beads followed by clearing the lysate from cell debris as described in “Preparation of crude membrane extracts”. The lysate was supplemented with 0.25% Triton-X100 (f. c.) and incubated for 3 hours at 4°C with 2 ml 50% slurry of RNAse-S coupled beads. The beads were washed on a glass filter with Lysis Buffer containing 0.025% Triton-X100, supplemented with 50% glycerol (f. c.) and stored as 50 % slurry at −20°C.
The purification of Nup188 (KWY2836), GFP-Nup188 (KWY2598), Nup188-Nic96(100-839) complex (KWY6292), GFP-Nic96(186-839) (KWY2631), Nup192-Nic96(100-839) complex (KWY6291), and (GFP-Nup192)-Nic96(100-839) complex (KWY3572) from overexpressing yeast cells was performed as described for GFP-Nup157 and GFP-Nup170 with the following exceptions: (1) protein expression was induced for 14 hours instead of 22 hours (2) in all cases except for Nup192-Nic96(100-839) complex and (GFP-Nup192)-Nic96(100-839) complex 0.05% CHAPS instead of Triton-X100 was used during yeast extract preparation, binding to RNAse-S coupled beads, final washes and protein storage. (3) In the cases of Nup192-Nic96(100-839) complex and (GFP-Nup192)-Nic96(100-839) complex 0.2% CHAPS was used during yeast extract preparation and binding to RNAse-S coupled beads, and 0.05% CHAPS was used for the final washes and storage.
Native Nup84-subcomplex
For purification of native yeast Nup84-subcomplex (KWY6349) yeast membranes were prepared and extracted essentially as described in “Preparation of crude membrane extracts” except that 0.05% CHAPS was used as a membrane extraction detergent and the extract was used for protein purification straight after vortexing with glass beads. The extract was incubated with IgG-coupled Sepharose beads for 2 hours at 4°C. The beads were washed with TEV-Elution Buffer (50 mM HEPES pH 7.5, 300 mM KCl, 10mM MgCl2, 0.1mM EDTA, 5mM β-mercaptoethanol, 0.05% CHAPS) in Poly-prep chromatography columns (Bio-Rad). Bound Nup84-subcomplex was cleaved off from the beads with TEV protease and separated from beads by passing through 0.45 μm Ultrafree-MC HV centrifugal filter (Merck Millipore). To analyze the quality of Nup84-subcomplex, the prep was subjected to gel filtration analysis on Superose 6 10/300 GL gel filtration column (GE Healthcare) equilibrated with TEV-Elution Buffer without CHAPS.
Identification of FG-repeat binding proteins
Preparation of crude membrane extracts
For the identification of yeast proteins specifically binding to FG-repeats, crude yeast membrane fraction was used as a source of prey proteins. The membrane fraction was typically prepared from 6 liters of yeast culture (KWY6290) grown in YPD supplemented with 20 mg/L adenine. Cells were harvested at OD600 ~3.0 and washed once with distilled water. All the following procedures unless otherwise noted were performed in ice-cold conditions. Cells were re-suspended in Lysis Buffer (50 mM HEPES pH 7.5, 150 mM KCl, 10mM MgOAc2, 0.1mM EDTA, 5mM β-mercaptoethanol) containing 1 mM PMSF (f.c.) and protease inhibitors cocktail (Sigma-Aldrich) and lysed using 0.5 mm glass beads with BeadBeater homogenizer (BioSpec Products, 1107900-105). Cell debris were removed using low speed centrifugation (5 min, 6000 RPM, SLA1500 rotor) and the crude membrane pellet was obtained by high speed centrifugation of the low speed supernatant (1 h at 18000 RPM, SS-34 rotor). The membrane pellet was re-suspended in Lysis Buffer containing PMSF and the protease inhibitors cocktail, then supplemented with 20% glycerol (f. c.), aliquoted and flash-frozen in liquid nitrogen until further usage. For preparation of membrane extract the membrane prep was thawed at room temperature, diluted 1:2 with Adjustment Buffer (50 mM HEPES pH 7.5, 825 mM KCl, 0.1 mM EDTA, 10 mM MgCl2, 5 mM β-mercaptoethanol), then supplemented with protease inhibitors cocktail and with Triton-X100 (0.03% f. c.). The diluted prep was briefly vortexed with 0.5 mm glass beads and passed through a 2.7 μm syringe filter (Sigma-Aldrich) to remove large insoluble particles. The buffer was then exchanged using PD-10 desalting columns (GE Healthcare) to HEPES-KOAc Buffer (10 mM HEPES pH 7.5, 160 mM KOAc, 1 mM MgOAc2, 1 mM DTT) containing 0.03% Triton-X100.
Preparation of FG-repeat coated beads
FG-repeat coated beads were prepared using 50 μl Glutathione Sepharose 4B beads (GE Healthcare) for each binding reaction, and the following approximate amounts of FG-repeats: 5 nmol GST-Nup100(1–307), 2.5 nmol GST-Nup100(1–610), 3 nmol GST-Nsp1(1–563). The beads were incubated with the FG-repeats for 10 minutes at RT, washed 3 times with 1 ml Salt Elution Buffer (10 mM Tris-HCl pH 8.0, 1 M NaCl, 0.1% Tween-20, 5mM β-mercaptoethanol), and equilibrated with HEPES-KOAc Buffer containing 0.03% Triton-X100.
Binding reactions with membrane extract
For each yeast membrane extract binding reaction, 50 μl of the respective FG-repeat coated beads were pre-mixed with the membrane extract corresponding to ~1 liter of the initial yeast culture. The reactions were agitated for 1 hour at 4°C, briefly washed with HEPES-KOAc Buffer containing 0.03% Triton-X100 and bound proteins were eluted at room temperature 3 times with 100 μl of Salt Elution Buffer. All 3 eluates were pooled, proteins were precipitated at room temperature by methanol-chloroform, washed 3 times with 500 μl ice-cold 90% acetone containing 0.01M HCl and recovered in 75 μl of SDS sample buffer (salt elution). Following salt-elution, remaining proteins - including the FG-repeat bait - were removed from beads with 100 μl SDS sample buffer (SDS post-elution).
Identification of bound proteins
The proteins from salt-elution samples were separated by 6% SDS-PAGE to ~1/8 gel length, stained by GelCode protein stain (Thermo Fisher) and the whole stained areas were processed for in-gel tryptic digestion followed by standard analysis of sample content by LC-MS.
FG-repeat binding assays with purified proteins
Preparation of FG-repeat coated beads
The FG-repeat coated beads were prepared for the binding assays with individually purified proteins (Figures 2, S2) essentially as described in “Identification of FG-repeat binding proteins” with the following exceptions: (1) 25 μl of Glutathione Sepharose beads were used per binding reaction with the following amounts of FG-repeats: 5.0 nmol GST-Nup100(1–307), 2.5 nmol GST-Nup100(1–610), 3.0 nmol GST-Nsp1(1–563), 10 nmol Nup116(348-458), 10 nmol Nup116(348-458)FA; (2) The beads were equilibrated with HEPES-KOAc Buffer containing 0.01% Triton-X100.
Preparation of prey protein mixtures
Yeast-overexpressed proteins were stored immobilized on beads at −20°C (see “Protein overexpression in yeast”) and were freshly purified by gel filtration prior to setting up the FG-repeat binding reactions. To this end the beads were equilibrated with ice-cold Lysis Buffer containing 0.01% Triton-X100 (GFP-Nup157 and GFP-Nup170) or Lysis Buffer containing 0.05% CHAPS (Nup188, Nup188-Nic96(100-839) complex, Nup192-Nic96(100-839) complex and GFP-Nic96(186-839)), cleaved from beads with TEV and filtered through 0.45 μm centrifugal filter (Merck Millipore) followed by purification on a Superdex 200 10/300 gel filtration column (GE Healthcare) equilibrated with HEPES-KCl Buffer (10 mM HEPES pH 7.5, 160 mM KCl, 1 mM MgCl2, 1 mM DTT). The peak fractions were pooled and the protein concentrations were determined using A280 absorbance reads. The native Nup84-subcomplex was freshly purified from the corresponding yeast membrane preps as described in the section “Native Nup84-subcomplex”, and used for FG-repeat binding reactions without further gel filtration purification due to low protein concentrations. All frozen recombinant protein stocks (see “Recombinant proteins”) were quickly thawed and briefly centrifuged prior to usage to remove residual protein aggregates.
FG-repeat binding reactions
Prey protein preps were diluted with HEPES-KOAc Buffer containing 0.01% Triton-X100 and supplemented with bacterial extract to yield following approximate protein amounts in 1 ml volume: 120 μg of bacterial extract proteins and either of 100 pmol _Sc_Kap95, 100 pmol 3xGFP, 100 pmol MBP-GFP-Nup53ΔC, 15 pmol GFP-Nup157, 15 pmol GFP-Nup170, 25 pmol Nup188, 25 pmol Nup192-Nic96(100-839) complex, 25 pmol Nup188-Nic96(100-839) complex, 50 pmol Nup192(2-960), 50 pmol GFP-Nic96(186-839), 200 pmol Nup170(980-1502), 40 pmol of recombinant Nup84-subcomplex, 150 pmol Nup85-Seh1 complex, 150 pmol of Nup145C-Sec13-Nup120 complex, 150 pmol Nup84-Nup133(521-1157) complex, the amount of native Nup84-subcomplex corresponding to 0.7 L of source yeast culture with OD600 = 3.0. Prey protein mixtures were added to the respective FG-repeat coated beads and the binding reactions were gently agitated for 20 min at room temperature. The beads were quickly washed twice with 1 ml of ice-cold HEPES-KOAc Buffer containing 0.01% Triton-X100 and bound proteins were eluted by 100 μl of Salt Elution Buffer followed by 100 μl of SDS-sample buffer (SDS post-elution). Salt eluates were precipitated with methanol-chloroform and re-solubilized in 50 μl of SDS-sample buffer (salt elution). Samples of test proteins (A) and the input mixes (A+B) were made during preparation of prey protein mixtures by methanol-chloroform precipitation of 200 μl of prey protein samples diluted with the HEPES-KOAc Buffer before and after the addition of bacterial extract. The samples of bacterial extract (B) were prepared separately by precipitating 200 μl of bacterial extract diluted with HEPES-KOAc Buffer to 120 μg/ml. All precipitated samples were recovered in 50 μl of SDS-sample buffer. All protein samples were separated by 8% SDS-PAGE.
Biolayer interferometry
Binding of Nup116 variants to scaffold nucleoporins or Kap95 was measured by biolayer interferometry (BLI) using a FortéBio Octet RED96 instrument (Pall Life Sciences). All samples were incubated for at least 10 min at 30°C before the measurement. The biotinylated, Avitagged probes (20–100 nM) were immobilized on streptavidin biosensor tips, pre-incubated in BLI buffer (20 mM HEPES/NaOH pH 7.5, 110 mM K-acetate, 5 mM Na-acetate, 2 mM Mg-acetate, 1% w/v bovine serum albumin, 0.05 % v/v Tween-20, 2 mM DTT) for 5–10 min, to give a loading of ~0.7–0.9 nm after 40–160 sec of incubation. After dipping the sensor in BLI buffer for 60 sec, association with a two-fold dilution series of analyte was measured for 90 sec. Dissociation was measured after dipping the biosensor into BLI buffer for 120 sec. Response values after completion of the association phase were determined using the Octet Analysis Software version 7.0 (Pall Life Sciences); Response values were corrected by subtracting the corresponding value of a negative buffer-only control. For response values above 0.05 nm, kinetic rate constants (kon and koff) were determined for each progress curve by globally fitting the association and dissociation phases to a 2:1 binding model and the dissociation constants (KD values) were calculated as koff/kon.
Quantitative Western blotting
For the assays involving analysis of tagged yeast proteins by quantitative Western blotting (see below), protein samples were separated by 8% SDS-PAGE, transferred to a nitrocellulose membrane and probed with appropriate mixtures of primary antibodies (mouse anti-HA-tag monoclonal antibodies (COVANCE), mouse anti-GFP monoclonal antibodies (Roche), rabbit IgG (Sigma-Aldrich)) followed by a mixture of secondary fluorescently labeled antibodies (anti-mouse-Alexa-680 antibodies (Invitrogen) and anti-rabbit-IR800 antibodies (Rockland Immunochemicals)). Band intensities were quantified with LiCor Odessey CLx system (LI-COR Biosciences) using Image Studio 5.2 Software.
Quantitative affinity pulldowns
For quantitative affinity pulldowns (Figures 4E, 4F, S4C) yeast pre-cultures were grown in Met- liquid media and re-inoculated into 100 ml of either Met- or 20×Met containing media to yield cultures with OD600 ~4.0 after 12 hours incubation at 30°C. Cells were collected by centrifugation, re-suspended in ice-cold HEPES-KOAc Buffer containing 1 mM PMSF and lysed with 0.5 mm glass beads using Mini-Beadbeater-24 (BioSpec Products). Cell debris were removed by centrifugation at 14K rpm for 1 minute in a cooled tabletop centrifuge. 100 μl of supernatant was saved as a total proteins sample. The rest was supplemented with Tween-20 (0.1% f. c.) and mixed with 1.5 mg of rabbit IgG-coupled Dynabeads per reaction. Binding reactions were incubated for 1 hour on a rotating wheel at 4°C, briefly washed with HEPES-KOAc Buffer containing 0.1% Tween-20 and re-suspended in 100 μl HEPES-KOAc Buffer to yield bound fraction. The total and bound fractions were mixed with SDS-PAGE Sample Buffer, pre-heated at 50°C for 3 minutes and, after the removal of beads, the protein samples were completely denatured at 95°C.
Time-course analysis of Nup188 depletion
For quantitative analysis of Nup amounts during the time-course of Nup188 depletion (Figures S6D–F) yeast cultures were grown at 30°C in Met- synthetic medium and methionine (20-fold normal concentration) was added before the cultures reached OD600 =1. Samples were taken in 2h intervals after methionine addition. The culture was periodically diluted with fresh 20×Met medium to keep OD600 below 1.0. Harvested cells were treated for 5 minutes with 0.1M NaOH and re-suspended in 100 μl of SDS-PAGE Sample Buffer, followed by complete denaturation at 95°C.
Nuclear translocation and competition assays
Preparation of protein samples
Yeast-overexpressed protein samples were prepared from bead-immobilized stocks immediately before the nuclear translocation experiments as described in “FG-repeat binding assays with purified proteins” with the following exceptions: (1) for purification of GFP-Nic96(186-839) HEPES-KOAc Buffer (10 mM HEPES pH 7.5, 160 mM KOAc, 1 mM MgOAc2, 1 mM DTT) was used in the gel filtration step. (2) GFP-Nup188 was purified the same way as Nup188 except using HEPES-KOAc Buffer the in gel filtration step. (3) (GFP-Nup192)-Nic96(100-839) complex was purified the same way as Nup192-Nic96(100-839) complex except using HEPES-KOAc Buffer in the gel filtration step. All frozen recombinant protein stocks (see “Recombinant proteins”) were quickly thawed, briefly centrifuged prior to usage to remove residual protein aggregates and diluted in HEPES-KOAc Buffer. In order to match standard buffer conditions used in the in vitro nuclear transport assays the protein samples that were contained in HEPES-KOAc Buffer (GFP-Nic96(186-839), GFP-Nup188, (GFP-Nup192)-Nic96(100-839) complex, MBP-GFP-Nup53ΔC, YFP-Importin-β, 3xGFP) were diluted 1:0.45 with HEPES-KOAc Dilution Buffer (42 mM HEPES pH 7.5, 16 mM NaOAc, 4.2 mM MgOAc2), while the protein samples contained in HEPES-KCl Buffer (10 mM HEPES pH 7.5, 160 mM KCl, 1 mM MgCl2, 1 mM DTT) (GFP-Nup157 and GFP-Nup170) were diluted 1:0.45 with HEPES-KCl Dilution Buffer (42 mM HEPES pH 7.5, 16 mM NaCl, 4.2 mM MgCl).
Setup of nuclear translocation and competition assays
Cell permeabilization was based on the protocol described in (Adam et al., 1990). HeLa cells were washed three times for 2 min each with PBS pH 7.4, followed by a 2 min wash with Permeabilization Buffer (50 mM HEPES pH 7.3, 50 mM KOAc, 8 mM MgCl2), followed by a 5 min permeabilization with digitonin (Sigma Aldrich) at a concentration of 50 μg/mL supplemented with an energy regenerating system of 100 μM ATP (Roche), 100 μM GTP (Roche), 4 mM creatine phosphate (Roche), and 20 U/mL creatine kinase (Roche) in Permeabilization Buffer. Digitonin was subsequently removed by washing three times for 3 min each with HEPES-KOAc Wash Buffer (20 mM HEPES pH 7.3, 110 mM KOAc, 5 mM NaOAc, 2mM MgOAc, 2 mM DTT). After the final wash, excess liquid was removed by aspiration and the appropriate experimental reaction mix was quickly added to the nuclei. Concentrations (approximate) used in the reaction mixes were: 0.4 μM GFP-Nup170, 0.4 μM GFP-Nup157, 0.4 μM GFP-Nup188, 0.3 μM (GFP-Nup192)-Nic96(100-839), 0.9 μM GFP-Nic96(186-839), 1.5 μM MBP-GFP-Nup53ΔC, 1 μM YFP-Importin-β, 1-10 μM Importin-β, 1 μM 3xGFP, 200 μg/mL 155 kDa TRITC-dextran (Sigma-Aldrich), 2 mM DTT, 100 μg/mL WGA (Sigma-Aldrich) and 5% v/v cytosolic extract. Cytosolic extract from Xenopus laevis oocytes was prepared as described in (Levy and Heald, 2010). The cytosolic extract was supplemented with an energy regenerating system consisting of 2 mM GTP (Roche), 100 μM ATP (Roche), 4 mM creatine phosphate (Roche), and 20 U/mL creatine kinase (units of specific activity as stated by Roche). In experiments using WGA, 100 μg/mL WGA was first incubated with the nuclei for 10 min, removed, and then the experimental mix was added to the nuclei. For the Importin-β competition experiments, digitonin-treated nuclei were pre-incubated with active Ran (5 μM RanGDP plus energy regenerating system for 10 min followed by three washes with HEPES-KOAc Wash Buffer). The IBB cargo was pre-mixed for 10 minutes with Neutravidin-Dylight550 (Thermo Scientific) in a molar ratio 4:1 and used in the reactions at final concentration ~ 0.2 μM. All experimental mixes were incubated with the nuclei for 15 min at room temperature before imaging.
Confocal microscope imaging and analysis
Imaging was performed on a Zeiss LSM 700 laser scanning confocal microscope using a 63× oil immersion objective and the Zen 2011 imaging software (Carl Zeiss). Image segmentation and fluorescence quantification were performed using custom software (NuIF_twocolor) in MATLAB R2012a (The MathWorks). For translocation assays the fluorescence intensity inside each nucleus and the average background fluorescence intensity were determined, and the intranuclear to extranuclear fluorescence ratios were then calculated giving normalized fluorescence intensities for both the protein of interest and the dextran control. All nuclei with normalized dextran fluorescence ratios greater than 0.3 were considered to be ‘leaky’ (i.e. overly permeable to large molecules) and rejected. Only intact nuclei were included in subsequent analysis. For Importin-β competition assays the absolute values of GFP-Nup signal at the nuclear rim and IBB cargo signal inside the nucleus were determined automatically using custom software (NuRF) in MATLAB R2012a (The MathWorks) from the confocal images of the competition reactions set with different concentrations of Importin-β and imaged sequentially with the same microscope settings.
Analysis of cell growth
Overnight yeast cultures were grown in permissive conditions in liquid media. For growth assays on agar plates the overnight cultures were diluted with distilled water and plated on specified media in 1:5 dilution series starting with OD600 = 1. Growth curves were acquired using CLARIOstar automated plate reader (BMG Labtech) at 30°C in 24-well plastic dishes (Thermo Fisher) from overnight pre-cultures diluted 1:125 in the specified synthetic liquid media.
Widefield fluorescence microscopy
All imaging experiments in yeast except for FRAP and FLIP (see below) were performed using widefield fluorescence microscopy. Overnight yeast cultures were re-diluted with appropriate synthetic media containing 2× access of adenine and grown at 30°C for 12 h before imaging. 1,6-hexanediol treatment was performed for 10 minutes prior to imaging at 30°C in the respective media containing 5% (f.c.; 1,6-hexanediol (Sigma-Aldrich)). Cells were imaged in 384-well glass bottom plates (MatriPlate) pre-coated with ConA (Sigma-Aldrich) with inverted epifluoresence Ti microscope (Nikon) equipped with a Spectra X LED light source (Lumencore) and Flash 4.0 scMOS camera (Hamamatsu) using a 100× Plan-Apo VC objective NA 1.4 (Nikon) and NIS-Elements AR 4.40 software (Nikon).
Representative images
Representative images were processed using ImageJ software. Brightness and contract were adjusted to the same values for images belonging to the same experiment and were chosen to cover the whole range of signal intensities.
Quantitative image analysis
Cells simultaneously expressing GFP-labeled Nups together with dsRed-HDEL as the nuclear envelope/ER marker (Bevis et al., 2002) were imaged in bright field, and in the dsRed and GFP channels. The pipeline for image segmentation and GFP signal quantification outlined in Figure S5 was implemented in a custom Matlab R2016a script (Average_Nuclear_Envelope_Intensity). The nuclear contours were automatically traced using the bright field and dsRed-HDEL channels. Fully automated segmentation was achieved by edge detection to identify all potential image patches locked to the NE. Each patch was further examined in relation to its neighbors and to strict morphological criteria including region width, intensity and area, and all such patches were used to restore the nuclear contour and create the NE masks. Only masks fulfilling strict quality criteria were retained, reducing the number of NE masks to approximately 40% of all in-focus cells. Further, image areas with high cell density and potential overlap were eliminated from the analysis. Altogether, only 10 to 100 dsRed-HDEL NE masks per image frame survived this selection process. Such masks were used to compute the average GFP signal intensities along the nuclear rim. Final GFP signal readout was represented by median value within the image frame. This pipeline allowed an accurate and unbiased measurement of GFP signal intensities at the NE. For each quantification experiment respective yeast samples were imaged in parallel with the same microscope settings using an automated image acquisition workflow. Typically 25–50 frames were acquired per sample (~ 500–5000 individual nuclear rim contours). Unless otherwise noted the GFP intensity values for each sample were plotted as the mean of frame-derived values +−SD using GraphPad Prism 7 (GraphPad).
Confocal fluorescence microscopy
For fluorescence recovery after photobleaching (FRAP) and fluorescence loss in photobleaching (FLIP) experiments, overnight yeast pre-cultures were re-inoculated into appropriate synthetic media supplemented with 2× access of adenine and incubated at 30°C for 12 h before imaging. Cells were imaged in glass bottom 8-well chambers (IBIDI) pre-coated with ConA (Sigma-Aldrich).
All FRAP and FLIP experiments were performed with a Leica TCS SP8 microscope (Leica DMI6000B-CS) using 63× 1.4NA Oil HC PL APO CS2 objective. The microscope was equipped with a Laser unit for confocal acquisition (AOBS system): 458, 477, 488, 496, 514nm lines Argon laser; 405nm, 561nm, 633nm lasers, and controlled by Leica LAS X SP8 Version 1.0 software.
Fluorescence recovery after photobleaching
Cell images in the GFP channel were recorded with 2.5% of total argon laser power at 14 frames/second using a line accumulation of four. Five pre-bleach and 60 post-bleach frames were acquired. Photobleaching was performed by defining manually a rectangular region comprising ~one-half of yeast cell nucleus and applying 100% of total argon laser power for 0.21 sec. The mobility of GFP-labeled proteins within the NE was evaluated by quantifying signal re-equilibration between the bleached (Ibl) and non-bleached (Inon-bl) halves of the nuclei as 2* Ibl/(Ibl + Inon-bl). The datasets acquired during three consequent days were processed as described above, pooled and expressed as time series with mean +/−SEM at each time point using GraphPad Prism 7 (GraphPad).
Fluorescence loss in photobleaching
Imaging was performed simultaneously in GFP and dsRed channels at 0.75% of total argon laser power for the GFP channel and 0.3% of 561nm laser power for the dsRed channel. The images were acquired at 58 frames/second with line accumulation of three. 30 pre-bleach frames were generated followed by 500 cycles of bleaching and post-bleach frame acquisitions. The bleaching was performed in a cytosolic region positioned far from the nucleus using 100% of total argon laser power for 0.28 seconds per cycle. For each time series the background-subtracted nuclear rim signal has been calculated based on the dsRed-HDEL signal delineating the border of the nuclear envelope region. Loss in photobleaching was calculated as (Ibl/I0bl)/(Inon-bl/I0non-bl) where Ibl - background-subtracted nuclear rim intensity in bleached cell at each respective bleach cycle; I0bl – mean background-subtracted nuclear rim intensity in bleached cell in pre-bleach series; Inon-bl – background-subtracted nuclear region intensity of a non-bleached cell at each respective bleach cycle; I0non-bl – mean background-subtracted nuclear region intensity in pre-bleach series. The datasets acquired during three consequent days were processed as described above, pooled and expressed as time series with mean +/−SEM at each time point using GraphPad Prism 7 (GraphPad).
Electron Microscopy
For all EM experiments yeast pre-cultures were grown in Met- synthetic liquid media and re-inoculated into 100 ml of either Met- or 20×Met containing synthetic media to yield cultures with OD 0.5–1.0 after 12 hours incubation at 30°C.
Freeze-fracture EM
For freeze-fracture EM, cells concentrated by centrifugation were frozen in a Bal-Tec HPM100 high-pressure freezer (Leica, Vienna) sandwiched between 6 mm wide 100 μm deep aluminum carriers (Leica, Vienna) facing each other (giving a total sample thickness of 200 μm). Samples were then transferred to Bal-Tec BAF060 freeze fracture and coating device (Leica, Vienna) using a Bal-Tec VCT100 cryo-vacuum shuttle (Leica, Vienna). Fracturing was performed under vacuum at −120°C, followed by freeze- etching at −110°C for 3 minutes and with tungsten coating at −120°C. Coating was finalized when 2.5 nm coating was deposited at 45° elevation followed by an additional deposition of 1.5 nm at varying elevation angle between 0 and 90°. Coated samples were transferred in VCT100 to Zeiss Leo-1530 SEM (Zeiss, Oberkochen) equipped with a Bal-Tec cryo stage (Leica, Vienna) pre-cooled to −120°C for imaging. Images of cells with fractured nuclear surfaces were acquired at low kV settings with in-lens or Everhardt-Thornley detectors at the magnification of ~98K. NPC densities were evaluated from the SEM images as the number of pore structures found at the nuclear envelope region divided by image area occupied by the nuclear envelope region. Determination of the nuclear envelope areas and counting of the NPC structures in the images was performed manually using ImageJ software.
Transmission EM
Cell cultures were agitated at 100 rpm in a shaker bath just prior to high pressure freezing. Cells were concentrated by suction filtration in a Millipore 15 ml vacuum filtration device (Fisher Scientific) XX1002500) onto a 0.4 μm polycarbonate filter (Fisher Scientific). Concentrated cells were scraped from the filter with a toothpick and loaded into specimen planchettes (Wohlwend Engineering, Sennwald, Switzerland) and frozen in a Bal-Tec HPM 010 high pressure freezer (Technotrade International, Manchester, NH, USA) (McDonald and Muller-Reichert, 2002). Freeze substitution was carried out in a Leica AFS freeze substitution device (Leica Microsystems, Vienna, Austria) in a fixative consisting of 1 % osmium tetroxide plus 0.1% uranyl acetate, plus 5% dH2O in acetone. The AFS was programmed to run at −90°C for 72 hours, warmed to −20°C at a rate of 10°C per hour, held at −20°C for 12 hours then warmed to room temperature (20°C) at a rate of 20°C per hour.
For resin embedding, fixed cells were rinsed 3× for 5 min each in pure acetone, then infiltrated with increasing concentrations of Epon-Araldite resin (Ted Pella Cat no. 180-28) according to the following schedule: 25% for 1 hour, 50% for 2 hours, 75% for 4 hours, then pure resin for 1 hour, followed by pure resin overnight, and a final pure resin rinse for 1 hour. Cells in pure resin were transferred to silicone embedding molds (EMS, Hatfield, PA, USA) and polymerized at 60°C for 2 days. Sections were cut on an Ultracut E ultramicrotome (Leica Microsystems, Vienna, Austria) at 50 nm thickness, picked up on 100 mesh copper grids (EMS, Hatfield, PA, USA), and post-stained for 7 minutes in 2% aqueous uranyl acetate, and 4 minutes in lead citrate. Sections were viewed on an FEI Tecnai (FEI, Hillsboro, OR) electron microscope operating at 120kV, and images recorded on a Gatan (Pleasanton, CA) Ultrascan 1000 digital camera.
Quantification and Statistical Analysis
Details of statistical analysis including the number of acquired data points/independent experiments (n), SD, SE, types of statistical tests used and the corresponding p-values are reported in the figure legends and in “Methods Details”.
Data and Software Availability
Custom image analysis scripts
Automated quantification of intranuclear, cytoplasmic and nuclear rim intensities for the in vitro translocation and competition experiments has been performed using custom MATLAB scripts available at (https://github.com/pascalvallotton/Jeff-scripts-Cell-Onishchenko). dsRed-HDEL based segmentation of yeast nuclear envelopes and quantification GFP signal intensities were performed using custom MATLAB scripts available at (https://github.com/pascalvallotton/Cell_OnishChenko).
Supplementary Material
1
Figure S1. Related to Figure 2; Outline of FG-repeat binding assays with individually purified scaffold Nups and Nup size-exclusion chromatography profiles.
(A) Outline of FG-repeat binding assays with individually purified proteins. Detection of highly enriched test proteins in the 1M salt eluates was used as a criterion for direct binding. Samples from every step (test protein (A), bacterial extract (B), input mix (A +B) and salt elution) were precipitated and re-solubilized in SDS sample buffer. SDS post-elution fraction was also included to control for the efficiency of the 1M salt elution step.
(B–D) Affinity-purified Nups were subjected to size-exclusion chromatography (SEC). Peak fractions corresponding to the non-degraded and non-aggregated protein samples (boxed) were used as inputs for the FG-repeat binding assays outlined in (A), except for the native Nup84-subcomplex that was only analyzed by SEC, and due to low concentrations was used for FG-repeat binding reactions straight after the affinity purification. (B) SEC profiles for the subunits of the Nic96-subcomplex affinity-purified from either overexpressing yeast cells (1,3,4,6,7,8) or bacteria (2,5). (C) SEC profile for the native Nup84-subcomplex affinity-purified from yeast. (D) SEC profiles for the recombinant portions of the Nup84-subcomplex (1,2,3) and the assembled heptameric Nup84-subcomplex (4).
Figure S2. Related to Figure 2; Salt elution and SDS post-elution profiles for FG-repeat binding assays with individually purified test proteins.
(A–C) Non-cropped images shown in (Figures 2A–C), including corresponding SDS post-elution profiles.
Figure S3. Related to Figure 4; Functionality of Nup116 GLFG-repeat substitution sequences.
(A) Growth rates of PMET3-NUP188 cells carrying either wild-type NUP116 or nup116ΔGLFG in liquid culture in the absence (Met-) or presence (20× Met) of methionine in the growth media.
(B) The ability to rescue growth of the nup116ΔGLFG mutant upon repression of Nup188 analyzed for various ectopically expressed GLFG-repeat substitutions on plates (left). General functionality of the same constructs was confirmed by their ability to rescue temperature-sensitive growth defect of the nup116Δ mutant (right).
Figure S4. Related to Figure 4; Role of Nup116 GLFG-repeats in scaffold Nup connectivity in vitro and in vivo.
(A). BLI probes. All proteins that were used in the BLI experiments (Figure 4D) were analyzed on 10% SDS-PAGE and stained with Coomassie Brilliant Blue. Approximately 1 μg protein was loaded per lane. Sizes of protein standards are indicated on the left of the gel.
(B) Parameters describing the binding kinetics determined by globally fitting BLI data (Figure 4D) using a two-site binding model.
(C) Efficiency of Nup188 depletion in affinity pulldown experiments (Figures 4E–F) evaluated by comparing the amount of Nup188 in the input fraction to each of the analyzed Nups (Nup170, Nup192 and Nup116). The values were normalized to wild-type Nup116/Met- conditions. Note that Nup188 amounts are significantly reduced in 20×Met media.
(D) Fluorescence images illustrating Nup192-GFP localization in cell cultures used in the affinity pulldown experiments (Figures 4E–F). Note that in all conditions the core nucleoporin Nup192 displays normal NE localization.
(E) Cells endogenously expressing Nup116-yEGFP or Nup133-yEGFP were incubated in permissive (Met-) or non-permissive (20×Met) media to manipulate expression of Nup188. Fluorescence images (top) illustrate the effect of 5% 1,6-hexanediol assessed after 10 min incubation in the media either with (5% HD) or without (no HD) 1,6-hexanediol. Note that while Nup116 is completely mislocalized in Nup188-depleted cells, the scaffold nucleoporin Nup133 remains localized at the nuclear rim. Model (bottom) suggesting that anchoring of Nup116 to the NPC is contributed by the GLFG-repeats interacting with scaffold Nups. Scale bar – 5 μm.
Figure S5. Related to Figure 5; Pipeline for quantitative analysis of Nup localization in cells expressing dsRed-HDEL as a NE marker.
(A) Nuclear rim contours were automatically generated with a custom Matlab script tracing the dsRed-HDEL channel in cells simultaneously expressing GFP-labeled Nups and the ER luminal marker dsRed-HDEL
(B) For each traced contour, the average Nup-GFP signal intensities at the contour, inside the contour and outside the contour represented Nup-GFP signal at the NE, in the nucleoplasm and in the cytoplasm respectively.
(C) Median values were calculated for the respective intensities for contours within each image frame to account for cell-to-cell variability and minimize the contribution of accidental contour assignment errors.
(D) Population averages and confidence intervals (SD) were evaluated based on the values obtained from ~25 or more individual image frames.
Figure S6. Related to Figure 5; Impact of Nup116 GLFG-repeats on the NPC structural organization analyzed by fluorescence microscopy.
(A) Nup116 GLFG-repeats specifically impact the NPC structural organization (see Figures 5A–B). Representative images (top) and quantitative analysis (bottom) of Nup-GFP localization in PMET3-NUP188 cells expressing wild-type Nup116 after 12h incubation in Met- or 20×Met media. Note that unlike in nup116_Δ_GLFG cells the localization of cytoplasmic Nups (Nup82, Nup116) is not affected upon Nup188 depletion. Mean +/− SD. (n) - number of analyzed image frames.
(B) Growth of PMET3-NUP188 cells endogenously expressing Nup116 variants in which GLFG-repeats were substituted either by yEGFP (nup116(GLFG->yEGFP)) or yEGFP fused with Nup100 GLFG-repeats (nup116(GLFG->nup100(1-610)-yEGFP)) analyzed in Met- and 20×Met media. The strains with wild-type and ΔGLFG variants of Nup116 are used as a reference.
(C) Fluorescence images of cells in (B) after 12h incubation in the respective media.
(D–F) Correlation between the development of Nup82 mislocalization phenotype and depletion of Nup188. Expression of Nup188 was repressed in PMET3-3xHA-NUP188 nup116ΔGLFG NUP82-yEGFP cells by addition of excess methionine. Samples were analyzed at the indicated time points by (D) fluorescence microscopy and (E and F) by quantitative Western blotting. Band intensities corresponding to Nup82-yEGFP and 3xHA-Nup188 were normalized to the initial time point and to a 130kDa cross-reacting protein (NS) used as an internal loading control. Mean +/− SD for each data point is shown.
(G) PMET3-NUP188 nup116ΔGLFG NUP82-yEGFP strains ectopically expressing Nup116 GLFG-repeat substitutions or wild-type Nup116 were incubated for 12h in either Met- or 20×Met media prior to imaging. Fluorescence images illustrate the effect of various Nup116 GLFG-repeat substitutions on the NE localization of Nup82.
(H–K) PMET3-NUP188 nup116ΔGLFG cells endogenously expressing either Nup133-yEGFP (a non-membrane scaffold Nup) or the non-NPC NE/ER membrane protein Sec61-yEGFP (a subunit of the ER membrane translocon complex), were incubated for 12h in Met- or 20×Met media before FRAP analysis. (H) Outline of FRAP assay. Mobility of GFP-labeled proteins (Nup133-yEGFP or Sec61-yEGFP) was evaluated as kinetics of GFP signal re-equilibration in half-bleached nuclei as 2*Ibl/(Ibl+Iref) where Ibl – intensity within bleached region and Iref - intensity within non-bleached (reference) region. (I–K) Plots showing recovery kinetics for Nup133 (I) and Sec61 (J) in the respective media. (K) Nup133 recovery kinetics corrected for differences observed for Sec61-yEGFP in Met- versus 20×Met media. Note that Nup133 does not display significant differences in the mobility between permissive and non-permissive conditions, which are distinct from the non-NPC-related NE/ER protein Sec61.
Figure S7. Related to Figure 6; Effects of Nup100 GLFG-repeats on the NPC targeting efficiency and NPC localization stability.
(A) Schematic showing the domain organization of Nup100. (B and E) Fluorescence images illustrating localization of indicated Nup100 variants in actively growing yeast cells with the indicated genotypes. Scale bars – 5 μm.
(C) Corresponding to (E) dsRed-HDEL based quantification of fluorescence signal at the nuclear rims. Mean +/− SD. (n) - number of analyzed image frames.
(D) NPC localization stability of Nup100 variants analyzed by FLIP as described for Nup116 (Figure 6F).
(F) Corresponding to (E) differences in the NPC incorporation of full-length and GLFG Nup100 variants conferred by the unlabeled wild-type allele, evaluated as described for Nup116 (Figure 6J).
Table S1. List of yeast strains used in this study. Related to STAR Methods
Table S2. List of plasmids used in this study. Related to STAR Methods
Table S3. List of frequently used buffers. Related to STAR Methods
10
2
3
4
5
6
7
8
9
Highlights.
- A subset of nucleoporin FG-repeats with GLFG-motifs bind scaffold nucleoporins
- GLFG-repeats play role in NPC biogenesis and nucleoporin connectivity
- GLFG-repeats act as NPC targeting determinants ensuring assembly of functional NPCs
Acknowledgments
We are grateful to R. Sachdev, E. Dultz, M. Hondele, S. Khawaja, S. Heinrich and other Weis lab members for critical discussions and suggestions, and to O. Posukh (IMCB Novosibirsk, Russia) for graphical work. We are indebted to K. McDonald (UC Berkeley) for his invaluable help with thin-section EM, and we thank F. Lucas, R. Wepf and the Scientific Center for Optical and Electron Microscopy of ETHZ for support with freeze-fracture EM. We acknowledge M. Rexach and S. Wente for providing yeast strains and constructs, and the Vincent J. Coates Proteomics/Mass Spectrometry Laboratory (UC Berkeley) and L. Kohlstaedt for mass spectrometry analyses. This study was supported by grants from NIH/NIGMS (R01GM058065 to K.W. and R01GM077537 to T.U.S.) and the Swiss National Fond (SNF 159731).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceptualization, E.O., K.W., T.U.S.; Methodology, E.O., K.R.A, J.H.T., K.E.K., P.V., C.P.D., T.U.S., K.W.; Software, P.V., J.H.T; Formal Analysis, E.O., J.H.T., and P.V.; Investigation, E.O., K.R.A, J.H.T., T.U.S., K.E.K., C.P.D., A.K., C.F.M., L.Y.C.; Writing – Original Draft, E.O., K.W. Writing – Review & Editing, E.O., K.W., T.U.S., K.R.A., J.H.T., P.V.; Visualization, E.O., J.H.T. and K.R.A.; Funding Acquisition, K.W. and T.U.S.; Supervision, K.W. and T.U.S.;
References
- Adam SA, Marr RS, Gerace L. Nuclear protein import in permeabilized mammalian cells requires soluble cytoplasmic factors. The Journal of cell biology. 1990;111:807–816. doi: 10.1083/jcb.111.3.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams RL, Terry LJ, Wente SR. A Novel Saccharomyces cerevisiae FG Nucleoporin Mutant Collection for Use in Nuclear Pore Complex Functional Experiments. 2015:G3. doi: 10.1534/g3.115.023002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alber F, Dokudovskaya S, Veenhoff LM, Zhang W, Kipper J, Devos D, Suprapto A, Karni-Schmidt O, Williams R, Chait BT, et al. The molecular architecture of the nuclear pore complex. Nature. 2007;450:695–701. doi: 10.1038/nature06405. [DOI] [PubMed] [Google Scholar]
- Allen NP, Huang L, Burlingame A, Rexach M. Proteomic analysis of nucleoporin interacting proteins. The Journal of biological chemistry. 2001;276:29268–29274. doi: 10.1074/jbc.M102629200. [DOI] [PubMed] [Google Scholar]
- Amlacher S, Sarges P, Flemming D, van Noort V, Kunze R, Devos DP, Arumugam M, Bork P, Hurt E. Insight into structure and assembly of the nuclear pore complex by utilizing the genome of a eukaryotic thermophile. Cell. 2011;146:277–289. doi: 10.1016/j.cell.2011.06.039. [DOI] [PubMed] [Google Scholar]
- Andersen KR, Leksa NC, Schwartz TU. Optimized E. coli expression strain LOBSTR eliminates common contaminants from His-tag purification. Proteins. 2013a;81:1857–1861. doi: 10.1002/prot.24364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen KR, Onischenko E, Tang JH, Kumar P, Chen JZ, Ulrich A, Liphardt JT, Weis K, Schwartz TU. Scaffold nucleoporins Nup188 and Nup192 share structural and functional properties with nuclear transport receptors. eLife. 2013b;2:e00745. doi: 10.7554/eLife.00745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailer SM, Siniossoglou S, Podtelejnikov A, Hellwig A, Mann M, Hurt E. Nup116p and nup100p are interchangeable through a conserved motif which constitutes a docking site for the mRNA transport factor gle2p. The EMBO journal. 1998;17:1107–1119. doi: 10.1093/emboj/17.4.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednenko J, Cingolani G, Gerace L. Importin beta contains a COOH-terminal nucleoporin binding region important for nuclear transport. The Journal of cell biology. 2003;162:391–401. doi: 10.1083/jcb.200303085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevis BJ, Hammond AT, Reinke CA, Glick BS. De novo formation of transitional ER sites and Golgi structures in Pichia pastoris. Nature cell biology. 2002;4:750–756. doi: 10.1038/ncb852. [DOI] [PubMed] [Google Scholar]
- Brohawn SG, Leksa NC, Spear ED, Rajashankar KR, Schwartz TU. Structural evidence for common ancestry of the nuclear pore complex and vesicle coats. Science. 2008;322:1369–1373. doi: 10.1126/science.1165886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohawn SG, Schwartz TU. Molecular architecture of the Nup84-Nup145C-Sec13 edge element in the nuclear pore complex lattice. Nature structural & molecular biology. 2009;16:1173–1177. doi: 10.1038/nsmb.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi NC, Adam SA. Functional domains in nuclear import factor p97 for binding the nuclear localization sequence receptor and the nuclear pore. Molecular biology of the cell. 1997;8:945–956. doi: 10.1091/mbc.8.6.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chug H, Trakhanov S, Hulsmann BB, Pleiner T, Gorlich D. Crystal structure of the metazoan Nup62*Nup58*Nup54 nucleoporin complex. Science. 2015;350:106–110. doi: 10.1126/science.aac7420. [DOI] [PubMed] [Google Scholar]
- Conti E, Muller CW, Stewart M. Karyopherin flexibility in nucleocytoplasmic transport. Current opinion in structural biology. 2006;16:237–244. doi: 10.1016/j.sbi.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Costanzo M, VanderSluis B, Koch EN, Baryshnikova A, Pons C, Tan G, Wang W, Usaj M, Hanchard J, Lee SD, et al. A global genetic interaction network maps a wiring diagram of cellular function. Science. 2016;353 doi: 10.1126/science.aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronshaw JM, Krutchinsky AN, Zhang W, Chait BT, Matunis MJ. Proteomic analysis of the mammalian nuclear pore complex. The Journal of cell biology. 2002;158:915–927. doi: 10.1083/jcb.200206106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angelo MA, Anderson DJ, Richard E, Hetzer MW. Nuclear pores form de novo from both sides of the nuclear envelope. Science. 2006;312:440–443. doi: 10.1126/science.1124196. [DOI] [PubMed] [Google Scholar]
- Denning DP, Patel SS, Uversky V, Fink AL, Rexach M. Disorder in the nuclear pore complex: the FG repeat regions of nucleoporins are natively unfolded. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:2450–2455. doi: 10.1073/pnas.0437902100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doye V, Wepf R, Hurt EC. A novel nuclear pore protein Nup133p with distinct roles in poly(A)+ RNA transport and nuclear pore distribution. The EMBO journal. 1994;13:6062–6075. doi: 10.1002/j.1460-2075.1994.tb06953.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eibauer M, Pellanda M, Turgay Y, Dubrovsky A, Wild A, Medalia O. Structure and gating of the nuclear pore complex. Nature communications. 2015;6:7532. doi: 10.1038/ncomms8532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Martinez J, Kim SJ, Shi Y, Upla P, Pellarin R, Gagnon M, Chemmama IE, Wang J, Nudelman I, Zhang W, et al. Structure and Function of the Nuclear Pore Complex Cytoplasmic mRNA Export Platform. Cell. 2016;167:1215–1228 e1225. doi: 10.1016/j.cell.2016.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer J, Teimer R, Amlacher S, Kunze R, Hurt E. Linker Nups connect the nuclear pore complex inner ring with the outer ring and transport channel. Nature structural & molecular biology. 2015;22:774–781. doi: 10.1038/nsmb.3084. [DOI] [PubMed] [Google Scholar]
- Flemming D, Devos DP, Schwarz J, Amlacher S, Lutzmann M, Hurt E. Analysis of the yeast nucleoporin Nup188 reveals a conserved S-like structure with similarity to karyopherins. Journal of structural biology. 2012;177:99–105. doi: 10.1016/j.jsb.2011.11.008. [DOI] [PubMed] [Google Scholar]
- Harel A, Chan RC, Lachish-Zalait A, Zimmerman E, Elbaum M, Forbes DJ. Importin beta negatively regulates nuclear membrane fusion and nuclear pore complex assembly. Molecular biology of the cell. 2003;14:4387–4396. doi: 10.1091/mbc.E03-05-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hough LE, Dutta K, Sparks S, Temel DB, Kamal A, Tetenbaum-Novatt J, Rout MP, Cowburn D. The molecular mechanism of nuclear transport revealed by atomic-scale measurements. eLife. 2015;4 doi: 10.7554/eLife.10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulsmann BB, Labokha AA, Gorlich D. The permeability of reconstituted nuclear pores provides direct evidence for the selective phase model. Cell. 2012;150:738–751. doi: 10.1016/j.cell.2012.07.019. [DOI] [PubMed] [Google Scholar]
- Hurt E, Beck M. Towards understanding nuclear pore complex architecture and dynamics in the age of integrative structural analysis. Current opinion in cell biology. 2015;34:31–38. doi: 10.1016/j.ceb.2015.04.009. [DOI] [PubMed] [Google Scholar]
- Ibarra A, Hetzer MW. Nuclear pore proteins and the control of genome functions. Genes & development. 2015;29:337–349. doi: 10.1101/gad.256495.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isgro TA, Schulten K. Binding dynamics of isolated nucleoporin repeat regions to importin-beta. Structure. 2005;13:1869–1879. doi: 10.1016/j.str.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Kapinos LE, Schoch RL, Wagner RS, Schleicher KD, Lim RY. Karyopherin-centric control of nuclear pores based on molecular occupancy and kinetic analysis of multivalent binding with FG nucleoporins. Biophysical journal. 2014;106:1751–1762. doi: 10.1016/j.bpj.2014.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley K, Knockenhauer KE, Kabachinski G, Schwartz TU. Atomic structure of the Y complex of the nuclear pore. Nature structural & molecular biology. 2015;22:425–431. doi: 10.1038/nsmb.2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosinski J, Mosalaganti S, von Appen A, Teimer R, DiGuilio AL, Wan W, Bui KH, Hagen WJ, Briggs JA, Glavy JS, et al. Molecular architecture of the inner ring scaffold of the human nuclear pore complex. Science. 2016;352:363–365. doi: 10.1126/science.aaf0643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DL, Heald R. Nuclear size is regulated by importin alpha and Ntf2 in Xenopus. Cell. 2010;143:288–298. doi: 10.1016/j.cell.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim RY, Fahrenkrog B, Koser J, Schwarz-Herion K, Deng J, Aebi U. Nanomechanical basis of selective gating by the nuclear pore complex. Science. 2007;318:640–643. doi: 10.1126/science.1145980. [DOI] [PubMed] [Google Scholar]
- Lin DH, Stuwe T, Schilbach S, Rundlet EJ, Perriches T, Mobbs G, Fan Y, Thierbach K, Huber FM, Collins LN, et al. Architecture of the symmetric core of the nuclear pore. Science. 2016;352:aaf1015. doi: 10.1126/science.aaf1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Lowe AR, Siegel JJ, Kalab P, Siu M, Weis K, Liphardt JT. Selectivity mechanism of the nuclear pore complex characterized by single cargo tracking. Nature. 2010;467:600–603. doi: 10.1038/nature09285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mans BJ, Anantharaman V, Aravind L, Koonin EV. Comparative genomics, evolution and origins of the nuclear envelope and nuclear pore complex. Cell cycle. 2004;3:1612–1637. doi: 10.4161/cc.3.12.1316. [DOI] [PubMed] [Google Scholar]
- Mao X, Hu Y, Liang C, Lu C. MET3 promoter: a tightly regulated promoter and its application in construction of conditional lethal strain. Current microbiology. 2002;45:37–40. doi: 10.1007/s00284-001-0046-0. [DOI] [PubMed] [Google Scholar]
- McDonald K, Muller-Reichert T. Cryomethods for thin section electron microscopy. Methods in enzymology. 2002;351:96–123. doi: 10.1016/s0076-6879(02)51843-7. [DOI] [PubMed] [Google Scholar]
- Milles S, Mercadante D, Aramburu IV, Jensen MR, Banterle N, Koehler C, Tyagi S, Clarke J, Shammas SL, Blackledge M, et al. Plasticity of an Ultrafast Interaction between Nucleoporins and Nuclear Transport Receptors. Cell. 2015;163:734–745. doi: 10.1016/j.cell.2015.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr D, Frey S, Fischer T, Guttler T, Gorlich D. Characterisation of the passive permeability barrier of nuclear pore complexes. The EMBO journal. 2009;28:2541–2553. doi: 10.1038/emboj.2009.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onischenko E, Stanton LH, Madrid AS, Kieselbach T, Weis K. Role of the Ndc1 interaction network in yeast nuclear pore complex assembly and maintenance. The Journal of cell biology. 2009;185:475–491. doi: 10.1083/jcb.200810030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onischenko E, Weis K. Nuclear pore complex-a coat specifically tailored for the nuclear envelope. Current opinion in cell biology. 2011;23:293–301. doi: 10.1016/j.ceb.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ori A, Banterle N, Iskar M, Andres-Pons A, Escher C, Khanh Bui H, Sparks L, Solis-Mezarino V, Rinner O, Bork P, et al. Cell type-specific nuclear pores: a case in point for context-dependent stoichiometry of molecular machines. Molecular systems biology. 2013;9:648. doi: 10.1038/msb.2013.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka S, Bui KH, Schorb M, Hossain MJ, Politi AZ, Koch B, Eltsov M, Beck M, Ellenberg J. Nuclear pore assembly proceeds by an inside-out extrusion of the nuclear envelope. eLife. 2016;5 doi: 10.7554/eLife.19071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SS, Belmont BJ, Sante JM, Rexach MF. Natively unfolded nucleoporins gate protein diffusion across the nuclear pore complex. Cell. 2007;129:83–96. doi: 10.1016/j.cell.2007.01.044. [DOI] [PubMed] [Google Scholar]
- Ribbeck K, Gorlich D. Kinetic analysis of translocation through nuclear pore complexes. The EMBO journal. 2001;20:1320–1330. doi: 10.1093/emboj/20.6.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribbeck K, Gorlich D. The permeability barrier of nuclear pore complexes appears to operate via hydrophobic exclusion. The EMBO journal. 2002;21:2664–2671. doi: 10.1093/emboj/21.11.2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout MP, Aitchison JD, Magnasco MO, Chait BT. Virtual gating and nuclear transport: the hole picture. Trends in cell biology. 2003;13:622–628. doi: 10.1016/j.tcb.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Rout MP, Aitchison JD, Suprapto A, Hjertaas K, Zhao Y, Chait BT. The yeast nuclear pore complex: composition, architecture, and transport mechanism. The Journal of cell biology. 2000;148:635–651. doi: 10.1083/jcb.148.4.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampathkumar P, Kim SJ, Upla P, Rice WJ, Phillips J, Timney BL, Pieper U, Bonanno JB, Fernandez-Martinez J, Hakhverdyan Z, et al. Structure, dynamics, evolution, and function of a major scaffold component in the nuclear pore complex. Structure. 2013;21:560–571. doi: 10.1016/j.str.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt HB, Gorlich D. Nup98 FG domains from diverse species spontaneously phase-separate into particles with nuclear pore-like permselectivity. eLife. 2015a;4 doi: 10.7554/eLife.04251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt HB, Gorlich D. Transport Selectivity of Nuclear Pores, Phase Separation, and Membraneless Organelles. Trends in biochemical sciences. 2015b doi: 10.1016/j.tibs.2015.11.001. [DOI] [PubMed] [Google Scholar]
- Schrader N, Stelter P, Flemming D, Kunze R, Hurt E, Vetter IR. Structural basis of the nic96 subcomplex organization in the nuclear pore channel. Molecular cell. 2008;29:46–55. doi: 10.1016/j.molcel.2007.10.022. [DOI] [PubMed] [Google Scholar]
- Schwartz TU. The Structure Inventory of the Nuclear Pore Complex. Journal of molecular biology. 2016;428:1986–2000. doi: 10.1016/j.jmb.2016.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulga N, Goldfarb DS. Binding dynamics of structural nucleoporins govern nuclear pore complex permeability and may mediate channel gating. Molecular and cellular biology. 2003;23:534–542. doi: 10.1128/MCB.23.2.534-542.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strambio-De-Castillia C, Niepel M, Rout MP. The nuclear pore complex: bridging nuclear transport and gene regulation. Nature reviews Molecular cell biology. 2010;11:490–501. doi: 10.1038/nrm2928. [DOI] [PubMed] [Google Scholar]
- Strawn LA, Shen T, Shulga N, Goldfarb DS, Wente SR. Minimal nuclear pore complexes define FG repeat domains essential for transport. Nature cell biology. 2004;6:197–206. doi: 10.1038/ncb1097. [DOI] [PubMed] [Google Scholar]
- Stuwe T, Bley CJ, Thierbach K, Petrovic S, Schilbach S, Mayo DJ, Perriches T, Rundlet EJ, Jeon YE, Collins LN, et al. Architecture of the fungal nuclear pore inner ring complex. Science. 2015a;350:56–64. doi: 10.1126/science.aac9176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuwe T, Correia AR, Lin DH, Paduch M, Lu VT, Kossiakoff AA, Hoelz A. Nuclear pores. Architecture of the nuclear pore complex coat. Science. 2015b;347:1148–1152. doi: 10.1126/science.aaa4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuwe T, Lin DH, Collins LN, Hurt E, Hoelz A. Evidence for an evolutionary relationship between the large adaptor nucleoporin Nup192 and karyopherins. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:2530–2535. doi: 10.1073/pnas.1311081111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timney BL, Raveh B, Mironska R, Trivedi JM, Kim SJ, Russel D, Wente SR, Sali A, Rout MP. Simple rules for passive diffusion through the nuclear pore complex. Journal of Cell Biology. 2016;215:57–76. doi: 10.1083/jcb.201601004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Appen A, Kosinski J, Sparks L, Ori A, DiGuilio AL, Vollmer B, Mackmull MT, Banterle N, Parca L, Kastritis P, et al. In situ structural analysis of the human nuclear pore complex. Nature. 2015;526:140–143. doi: 10.1038/nature15381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Powers MA. In vivo analysis of human nucleoporin repeat domain interactions. Molecular biology of the cell. 2013;24:1222–1231. doi: 10.1091/mbc.E12-08-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada J, Phillips JL, Patel S, Goldfien G, Calestagne-Morelli A, Huang H, Reza R, Acheson J, Krishnan VV, Newsam S, et al. A bimodal distribution of two distinct categories of intrinsically disordered structures with separate functions in FG nucleoporins. Molecular & cellular proteomics : MCP. 2010;9:2205–2224. doi: 10.1074/mcp.M000035-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Seo HS, Debler EW, Blobel G, Hoelz A. Structural and functional analysis of an essential nucleoporin heterotrimer on the cytoplasmic face of the nuclear pore complex. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:16571–16576. doi: 10.1073/pnas.1112846108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1
Figure S1. Related to Figure 2; Outline of FG-repeat binding assays with individually purified scaffold Nups and Nup size-exclusion chromatography profiles.
(A) Outline of FG-repeat binding assays with individually purified proteins. Detection of highly enriched test proteins in the 1M salt eluates was used as a criterion for direct binding. Samples from every step (test protein (A), bacterial extract (B), input mix (A +B) and salt elution) were precipitated and re-solubilized in SDS sample buffer. SDS post-elution fraction was also included to control for the efficiency of the 1M salt elution step.
(B–D) Affinity-purified Nups were subjected to size-exclusion chromatography (SEC). Peak fractions corresponding to the non-degraded and non-aggregated protein samples (boxed) were used as inputs for the FG-repeat binding assays outlined in (A), except for the native Nup84-subcomplex that was only analyzed by SEC, and due to low concentrations was used for FG-repeat binding reactions straight after the affinity purification. (B) SEC profiles for the subunits of the Nic96-subcomplex affinity-purified from either overexpressing yeast cells (1,3,4,6,7,8) or bacteria (2,5). (C) SEC profile for the native Nup84-subcomplex affinity-purified from yeast. (D) SEC profiles for the recombinant portions of the Nup84-subcomplex (1,2,3) and the assembled heptameric Nup84-subcomplex (4).
Figure S2. Related to Figure 2; Salt elution and SDS post-elution profiles for FG-repeat binding assays with individually purified test proteins.
(A–C) Non-cropped images shown in (Figures 2A–C), including corresponding SDS post-elution profiles.
Figure S3. Related to Figure 4; Functionality of Nup116 GLFG-repeat substitution sequences.
(A) Growth rates of PMET3-NUP188 cells carrying either wild-type NUP116 or nup116ΔGLFG in liquid culture in the absence (Met-) or presence (20× Met) of methionine in the growth media.
(B) The ability to rescue growth of the nup116ΔGLFG mutant upon repression of Nup188 analyzed for various ectopically expressed GLFG-repeat substitutions on plates (left). General functionality of the same constructs was confirmed by their ability to rescue temperature-sensitive growth defect of the nup116Δ mutant (right).
Figure S4. Related to Figure 4; Role of Nup116 GLFG-repeats in scaffold Nup connectivity in vitro and in vivo.
(A). BLI probes. All proteins that were used in the BLI experiments (Figure 4D) were analyzed on 10% SDS-PAGE and stained with Coomassie Brilliant Blue. Approximately 1 μg protein was loaded per lane. Sizes of protein standards are indicated on the left of the gel.
(B) Parameters describing the binding kinetics determined by globally fitting BLI data (Figure 4D) using a two-site binding model.
(C) Efficiency of Nup188 depletion in affinity pulldown experiments (Figures 4E–F) evaluated by comparing the amount of Nup188 in the input fraction to each of the analyzed Nups (Nup170, Nup192 and Nup116). The values were normalized to wild-type Nup116/Met- conditions. Note that Nup188 amounts are significantly reduced in 20×Met media.
(D) Fluorescence images illustrating Nup192-GFP localization in cell cultures used in the affinity pulldown experiments (Figures 4E–F). Note that in all conditions the core nucleoporin Nup192 displays normal NE localization.
(E) Cells endogenously expressing Nup116-yEGFP or Nup133-yEGFP were incubated in permissive (Met-) or non-permissive (20×Met) media to manipulate expression of Nup188. Fluorescence images (top) illustrate the effect of 5% 1,6-hexanediol assessed after 10 min incubation in the media either with (5% HD) or without (no HD) 1,6-hexanediol. Note that while Nup116 is completely mislocalized in Nup188-depleted cells, the scaffold nucleoporin Nup133 remains localized at the nuclear rim. Model (bottom) suggesting that anchoring of Nup116 to the NPC is contributed by the GLFG-repeats interacting with scaffold Nups. Scale bar – 5 μm.
Figure S5. Related to Figure 5; Pipeline for quantitative analysis of Nup localization in cells expressing dsRed-HDEL as a NE marker.
(A) Nuclear rim contours were automatically generated with a custom Matlab script tracing the dsRed-HDEL channel in cells simultaneously expressing GFP-labeled Nups and the ER luminal marker dsRed-HDEL
(B) For each traced contour, the average Nup-GFP signal intensities at the contour, inside the contour and outside the contour represented Nup-GFP signal at the NE, in the nucleoplasm and in the cytoplasm respectively.
(C) Median values were calculated for the respective intensities for contours within each image frame to account for cell-to-cell variability and minimize the contribution of accidental contour assignment errors.
(D) Population averages and confidence intervals (SD) were evaluated based on the values obtained from ~25 or more individual image frames.
Figure S6. Related to Figure 5; Impact of Nup116 GLFG-repeats on the NPC structural organization analyzed by fluorescence microscopy.
(A) Nup116 GLFG-repeats specifically impact the NPC structural organization (see Figures 5A–B). Representative images (top) and quantitative analysis (bottom) of Nup-GFP localization in PMET3-NUP188 cells expressing wild-type Nup116 after 12h incubation in Met- or 20×Met media. Note that unlike in nup116_Δ_GLFG cells the localization of cytoplasmic Nups (Nup82, Nup116) is not affected upon Nup188 depletion. Mean +/− SD. (n) - number of analyzed image frames.
(B) Growth of PMET3-NUP188 cells endogenously expressing Nup116 variants in which GLFG-repeats were substituted either by yEGFP (nup116(GLFG->yEGFP)) or yEGFP fused with Nup100 GLFG-repeats (nup116(GLFG->nup100(1-610)-yEGFP)) analyzed in Met- and 20×Met media. The strains with wild-type and ΔGLFG variants of Nup116 are used as a reference.
(C) Fluorescence images of cells in (B) after 12h incubation in the respective media.
(D–F) Correlation between the development of Nup82 mislocalization phenotype and depletion of Nup188. Expression of Nup188 was repressed in PMET3-3xHA-NUP188 nup116ΔGLFG NUP82-yEGFP cells by addition of excess methionine. Samples were analyzed at the indicated time points by (D) fluorescence microscopy and (E and F) by quantitative Western blotting. Band intensities corresponding to Nup82-yEGFP and 3xHA-Nup188 were normalized to the initial time point and to a 130kDa cross-reacting protein (NS) used as an internal loading control. Mean +/− SD for each data point is shown.
(G) PMET3-NUP188 nup116ΔGLFG NUP82-yEGFP strains ectopically expressing Nup116 GLFG-repeat substitutions or wild-type Nup116 were incubated for 12h in either Met- or 20×Met media prior to imaging. Fluorescence images illustrate the effect of various Nup116 GLFG-repeat substitutions on the NE localization of Nup82.
(H–K) PMET3-NUP188 nup116ΔGLFG cells endogenously expressing either Nup133-yEGFP (a non-membrane scaffold Nup) or the non-NPC NE/ER membrane protein Sec61-yEGFP (a subunit of the ER membrane translocon complex), were incubated for 12h in Met- or 20×Met media before FRAP analysis. (H) Outline of FRAP assay. Mobility of GFP-labeled proteins (Nup133-yEGFP or Sec61-yEGFP) was evaluated as kinetics of GFP signal re-equilibration in half-bleached nuclei as 2*Ibl/(Ibl+Iref) where Ibl – intensity within bleached region and Iref - intensity within non-bleached (reference) region. (I–K) Plots showing recovery kinetics for Nup133 (I) and Sec61 (J) in the respective media. (K) Nup133 recovery kinetics corrected for differences observed for Sec61-yEGFP in Met- versus 20×Met media. Note that Nup133 does not display significant differences in the mobility between permissive and non-permissive conditions, which are distinct from the non-NPC-related NE/ER protein Sec61.
Figure S7. Related to Figure 6; Effects of Nup100 GLFG-repeats on the NPC targeting efficiency and NPC localization stability.
(A) Schematic showing the domain organization of Nup100. (B and E) Fluorescence images illustrating localization of indicated Nup100 variants in actively growing yeast cells with the indicated genotypes. Scale bars – 5 μm.
(C) Corresponding to (E) dsRed-HDEL based quantification of fluorescence signal at the nuclear rims. Mean +/− SD. (n) - number of analyzed image frames.
(D) NPC localization stability of Nup100 variants analyzed by FLIP as described for Nup116 (Figure 6F).
(F) Corresponding to (E) differences in the NPC incorporation of full-length and GLFG Nup100 variants conferred by the unlabeled wild-type allele, evaluated as described for Nup116 (Figure 6J).
Table S1. List of yeast strains used in this study. Related to STAR Methods
Table S2. List of plasmids used in this study. Related to STAR Methods
Table S3. List of frequently used buffers. Related to STAR Methods
10
2
3
4
5
6
7
8
9