Metastasis and stem cell pathways (original) (raw)
. Author manuscript; available in PMC: 2011 Nov 14.
Published in final edited form as: Cancer Metastasis Rev. 2007 Jun;26(2):261–271. doi: 10.1007/s10555-007-9053-3
Abstract
Recent studies have described a small population of self-renewing and multipotent cells within tumors termed “cancer stem cells.” These cells share many traits with somatic and embryonic stem cells and are thought to be responsible for driving tumor progression in a growing list of neoplastic diseases. Cells within solid tumors encounter hypoxia due to poor vascular function. Both long-standing and emerging data describe hypoxic effects on somatic and embryonic stem cells, and it is likely that hypoxia also has profound effects on cancer stem cells. These effects include the activation of pathways that induce the dedifferentiation of cancer cells, the maintenance of stem cell identity, and increased metastatic potential. Hypoxia may contribute to tumor progression by specifically impacting these pathways in cancer stem cells.
Keywords: Hypoxia, stem cell, tumor, metastasis
I. Introduction: Hypoxia in tumor development
Solid tumor growth invariably results in the isolation of some areas from the existing vascular network. The poor function of tumor blood vessels further prevents efficient oxygen (O2) delivery and O2-starved (hypoxic) regions expand [1, 2]. Pronounced hypoxia has been clinically correlated with a poor prognosis, and detected in a broad range of tumors such as glioblastoma, head and neck, lung, breast, pancreatic, cervical, prostate and other cancers [3, 4]. Hypoxia contributes directly to the development of more aggressive cancer cells by exerting selective pressure on the tumor cell population which can survive decreased O2 and nutrient levels [5]. Interestingly, parallels exist between the basis of tumor development and embryogenesis, a process in which hypoxia is clearly involved. The developing embryo is exposed to varying O2 levels which likely act to sculpt not only the vascular system but also the tissues themselves [6, 7]. It was observed in the late 1970s that proper neural folding ex vivo required hypoxic exposure [8]. Embryonic exposure to hypoxia acts in part by driving intensively studied gene programs that execute these processes [9]. These programs are responsible for the expression of various genes integral in the formation of proper vasculature [9–12]. The recent flurry of publications describing cancer stem cells offers an attractive additional parallel between embryonic and tumor development. This review will explore the role of hypoxia in shaping, maintaining and potentially even generating cancer stem cells during tumor progression.
Research from a number of laboratories over the past ten years has identified a specific minority of tumor cells which are endowed with extensive potential for cell division. These cells are termed “cancer stem cells” and have recently been identified in a variety different human tumors [13]. The first cancers found to contain a cancer stem cell population were hematopoietic in origin [14, 15]. Early fundamental experiments with these neoplasms established the existence of a cancer initiating cell, with relatively few required to initiate tumors in mice. In addition, these studies established the rarity of cancer stem cells in the tumor population and their ability to proliferate indefinitely. Recently, cancer stem cells have been experimentally isolated in a number of solid tumors, including those of brain [16, 17], breast [18], and colon [19, 20]. These cancer stem cells were identified on the basis of surface markers. In breast cancers these cells are CD44+CD24−/lowLineage− and cells within this population efficiently form tumors in immunocompromised mice [18]. Relatively few stem cells were capable of forming tumors whereas many more cells from the remaining population were required to establish a tumor [18]. Other cancers yielded similar observations (brain tumor stem cells express nestin and CD133, and colon cancer stem cells express CD133) [16, 19, 20]. These studies solidified the recognition that cancer stem cells are a component of solid tumors.
With the concept of cancer stem cells established, studies are now aimed at gaining a better understanding of the molecular basis of how these cells are generated and regulated. It will be critical to determine the signaling pathways that maintain them in the stem cell-state in order to target them therapeutically. Important clues have been gleaned from a comparison between “normal” stem cells (such as embryonic stem [ES] cells and somatic stem cells) and cancer stem cells. This review will examine the role of hypoxia, an important influence in tumor biology, as it specifically impacts cancer stem cell biology. We will first consider the parallels between normal stem cells and cancer stem cells, to illustrate pathways shared between these stem cell types and how hypoxia may promote stem cell maintenance or initiation. We will then examine the role of cancer stem cells in metastasis and potential modifications of this process by hypoxia. Finally, we will consider possible effects of hypoxia on cancer stem cells in therapeutic approaches.
II. Stem cells
Stem cells are characterized by their capacity for self-renewal and multipotency. Specifically, stem cell division generates another stem cell and a daughter which can ultimately become a number of differentiated cell types [21]. This capability is perhaps best understood in the context of hematopoiesis: upon division, a hematopoietic stem cell (HSC) gives rise to one HSC and a mulipotent progenitor which is capable of generating all differentiated blood cell types. This paradigmatic stem cell behavior illustrates what is thought to occur upon cancer stem cell division.
A central issue in stem cell biology is the source of signals that maintain stem cell identity. It is known for example, that HSC reside within specific regions of the bone marrow. Residence in specific locations, or “niches,” appears to be a common feature of stem cells and certainly provides some of the critical stem cell maintenance signals [13, 22–26]. Experiments using C. elegans and Drosophila melanogaster stem cells have demonstrated the need for stem cell association with cells that comprise the niche [25, 26]. Some of the relevant molecular events have been determined in these systems, though in mammals less is known about the niche and the contribution of various signaling pathways [23]. Even less is known about the cancer stem cell niche, and the relevant contribution of niche cells to the cancer stem cell phenotype. The niche is likely comprised of numerous cellular and non-cellular components which contribute to its character [27]. This could be especially true of cancer stem cells, as the environment of solid tumors is certainly more dynamic than that of normal tissues. It is also possible that some cancer stem cells loose dependence on niches, or even create their own niche [27, 28]. The task for cancer stem cell research now includes identifying these niche components and evaluating the niche as a target for therapeutic intervention.
Although specific cells making up the niche have not been identified for a number of normal stem cells, some of the relevant pathways required for stem cell maintenance are known. These include the BMP, Wnt, Notch, PTEN, Shh, and JAK-STAT pathways [23, 25]. Interestingly, at least some of these pathways are affected by hypoxia, and as such, O2 levels may influence their signaling and ultimately stem cell identity. We will return to this point in more detail below.
It is well known that O2 levels regulate the developmental fate of stem cells within the embryo. The mammalian embryo has been experimentally determined to experience low O2 using hypoxia staining dyes and expression of hypoxia induced genes [7]. Interestingly, hypoxia clearly affects the differentiation of human ES cells [29]. In these experiments it was determined that hypoxic culture (3–5% O2) of human ES cells resulted in maintenance of an undifferentiated state, and culture in atmospheric O2 conditions resulted in differentiation. It is tempting to draw parallels between these observations and cancer stem cells, especially given the frequency and extent of hypoxia in tumors.
At least some HSC likely reside in hypoxic microenvironments: bone marrow, which harbors HSC, has been estimated to include microenvironments where O2 concentrations are rather low, near 1–2% [30]. Some controversy exists over the exact location of the HSC niche, and the cells with which HSC make contact [31]. Due to this uncertainty and the technically daunting task of determining the precise O2 levels in the bone marrow, the level of hypoxia that HSC encounter is still unknown, though it is likely that bone marrow cells experience dramatically different levels of O2. Regardless, HSC almost certainly experience some degree of hypoxia. In addition, hypoxic exposure has been shown to affect the biology of HSC. Isolated bone marrow progenitors (Lineage−CD34+CD38−) grown in hypoxic conditions in culture proliferate more than those in atmospheric O2 (21%) [30, 32]. Functionally, HSC grown ex vivo at 1.5% O2 are more readily able to reconstitute the bone marrow of NOD/SCID mice than those grown in atmospheric O2 [32]. Additionally, multiple lines of evidence from our laboratory have demonstrated the important role of O2 levels in embryogenesis. We observed that hematopoietic progenitor proliferation, which occurs in hypoxic conditions in vivo, requires signaling downstream of Hypoxia Inducible Factor (HIF) activation (see below) [33]. In addition, hypoxia (3% O2) drives the specific differentiation program in trophoblast stem cells necessary for proper placentation [34]. Decreased O2 levels also contribute to the differentiation of hemangioblasts from ES cells [35]. Finally, we have observed that low O2 promotes both vasculogenesis and angiogenesis in para-aortic splanchnoplural (P-Sp) explant cultures and that this activity requires HIF [36]. In addition, cytotrophoblasts, cells from the developing placenta, proliferate and maintain an undifferentiated phenotype when cultured at 2% O2 [37]. These cells slow proliferation and differentiate when cultured at 20% O2, indicating that hypoxia is integral in maintaining these traits. Importantly, these observations indicate that O2 levels profoundly influence embryogenesis and contribute to differentiation programs. Cells within the embryo or placenta (for example: hemangioblasts, cytotrophoblasts, and trophoblast stem cells) that typically experience low O2 in vivo are therefore regulated in their proliferation and differentiation by hypoxia. Since these cells share many characteristics with cancer stem cells, hypoxic exposure may encourage cancer stem cell proliferation and survival as well and contribute to their transformed phenotype.
Other stem cells have been described which thrive in hypoxic conditions. Neural crest stem cells or neuronal stem cells respond to hypoxia (O2 levels between 1% and 5%) with increased proliferation when compared to their normoxic counterparts [38, 39]. Interestingly, culture of neural crest stem cells in hypoxic conditions resulted in preferential differentiation into certain cell types [38]. In the context of cancer stem cells, these data indicate that certain cell fates may be more likely to arise from cancer stem cells when they are exposed to low O2. Such skewed differentiation could contribute to the heterogeneity of a tumor population. It is also possible that hypoxia could skew differentiation to more tumorigenic “transit-amplifying cells,” thereby producing a more aggressive tumor. Finally, hypoxic conditions to which these transit-amplifying cells are exposed may drive an undifferentiated phenotype. As cancer cells are frequently more malignant when they are less differentiated, hypoxic maintenance of an undifferentiated state likely contributes to the transformed phenotype. Possible mechanisms behind this assertion await experimental evidence and will be discussed in detail below.
Somatic stem cells are present in adult tissues and can be induced to proliferate under conditions where the tissue needs to increase in size or regenerate [23]. Whether cancer stem cells arise from these somatic stem cells or other more differentiated cells, or even cell-cell fusion is still a matter of debate [40, 41]. However, the parallels between somatic stem cells and cancer stem cells are strong. One important difference, probably lies in the control of their proliferation and differentiation. Whereas somatic stem cells are highly regulated within the tissue microenvironment, by virtue of defined signals within their niche, cancer stem cells are likely to be less restricted [42]. Somatic stem cells have the luxury of a static niche environment by virtue of the relatively unchanging tissue architecture. Conversely, significant heterogeneity of tumor microenvironments coupled with dynamic changes within the tumor certainly impact the regulation of cancer stem cells. These include fluctuations in pH, nutrient, and growth factor levels, as well as O2 concentrations. Fluctuations in the niche environment are likely to have profound effects on the activity of cancer stem cells. While many somatic stem cells traits are shared by cancer stem cells, multiple somatic stem cell pathways are probably subverted by cancer stem cells.
III. Hypoxic Effects on Stem Cells
Several pathways that impact stem cell generation and maintenance are influenced either directly or indirectly by O2 levels. This section will detail these pathways and recent publications which describe the potential role of O2 in their regulation. Hypoxia Inducible Factors (HIFs) are responsible for the bulk of the transcriptional response to hypoxia [9, 43]. In brief, HIFs respond to O2 levels below approximately 3–5%. HIF functions as a heterodimeric transcription factor comprised of a constitutively expressed subunit (HIF–1β or ARNT) and a hypoxically regulated α-subunit. The α-subunits are continually translated and degraded when O2 is available. HIF-α degradation is accomplished by hydroxylation of the subunit by a family of prolyl-hydroxylase enzymes followed by ubiquitylation by the von Hippel-Lindau tumor suppressor. Rapid proteasomal degradation follows. In low O2 conditions, the HIF-α subunits are unhydroxylated and therefore expression is maintained. A recent report identified a mechanism of O2-independent HIF degradation that involves the protein RACK1 in competition with HSP90 [44]. The authors argue that this mechanism of degradation determines the basal state of HIF expression, and may account for different HIF expression levels based on cell type and tissue. When stabilized, HIF-α subunits associate with HIF-1β and activate genes harboring hypoxia response elements (HRE) in their promoters or enhancers. There are multiple HIF-α subunits, which activate the transcription of both common and unique targets [45–47]. HIF activates genes involved in angiogenesis, metabolism, apoptosis, motility and other functions. The evolution of HIFs to respond to tissue hypoxia allows cells to effectively survive and alleviate low O2 tensions. In normal tissues these events are carefully choreographed, enabling the vasculature to fully perfuse the tissue. By virtue of this system, each cell is only a short distance from the nearest capillary.
In contrast to the highly ordered formation of blood vessels in normal tissue, tumor vasculature is highly disorganized [3]. Tumor vessels are poorly formed, tortuous, leaky, and contain frequent dead-ends and occlusions due to dysregulated tumor angiogenesis. Tumors hijack the normal vascular program, but are unable to properly regulate vessel formation. Poorly formed vessels result in frequent and profound hypoxia within tumors and can subject cells to low O2 for relatively significant periods of time. Interestingly, increased severity and extent of hypoxia associated with cancer generally results in a poorer prognosis [3, 4]. The precise mechanisms behind this observation are still under investigation. However, it is well accepted that HIFs contribute to tumor progression [48].
Large areas of hypoxia imply that most cells within a tumor experience some degree of O2 deprivation, including the resident cancer stem cells. The precise effects of hypoxia on these cells are not well characterized, but considering known effects on somatic stem cells hypoxia influences the regulation and perhaps even the generation of these cells. Hypoxic activation of HIF transcription is likely to affect cancer stem cells at multiple levels (Figure 1). In this section we describe specific effects of HIF activity and probable outcomes for cancer stem cells using somatic stem cells as a model.
Figure 1.
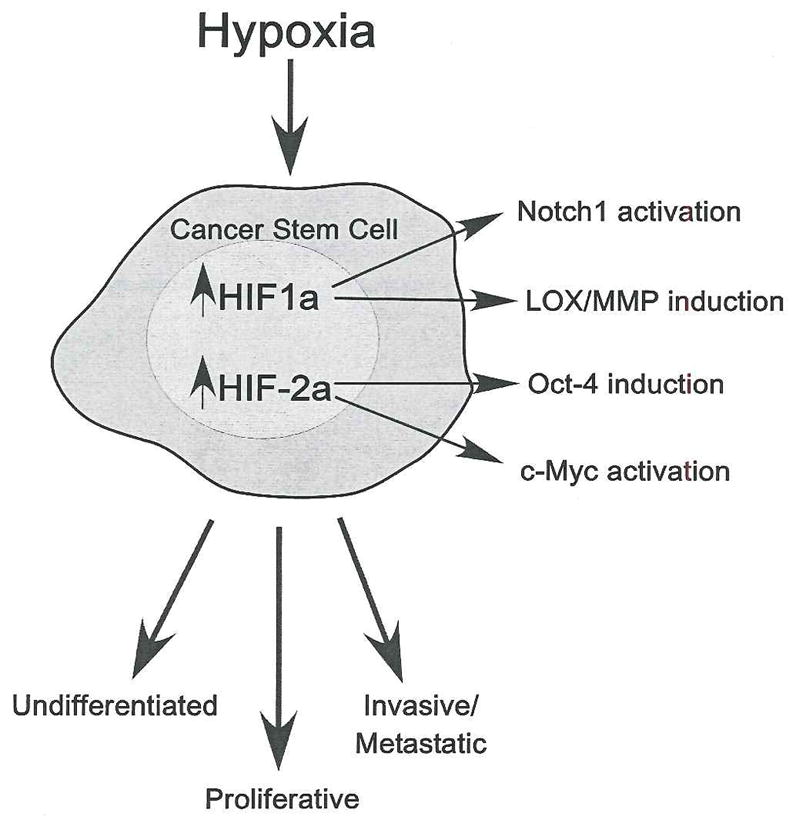
HIF activation in cancer stem cells. Hypoxic exposure within tumors results in the stabilization of HIF-1α and HIF-2α. Increased HIF activity results in the activation of pathways involved in maintaining stem cell identity. HIF-1α can stabilize Notch1, which maintains the undifferentiated state. HIF-1α also induces genes important for invasive and metastatic phenotypes, including lysyl oxidase (LOX) and matrix metalloproteases (MMP). HIF-2α stabilization induces the expression of Oct-4, one of the most important stem cell maintenance factors. HIF-2α also enhances the activity of c-Myc, which enhances proliferation and is also required for stem cell maintenance. Thus, through HIF, hypoxia activates several pathways important for stem cell maintenance. These pathways in turn impact processes such as proliferation, decreased differentiation, invasiveness and metastasis.
Our laboratory has determined that activation of one of the HIF-α subunits, HIF-2α, affects one of the most critical components of stem cell maintenance: the transcription factor Oct-4 [45, 49]. The importance of Oct-4 in ES cells is well documented [50–52]. Oct-4 expression is limited to ES cells, epiblasts, and primordial germ cells (PGCs) and it is not expressed in differentiated adult tissue. However, Oct-4 has been detected in certain tumor tissues and enforced Oct-4 expression in ES cells makes them capable of forming tumors in a dose dependent manner [53]. Interestingly, Oct-4 is a direct target of HIF-2α [49]. ES cell-derived tumors with increased HIF-2α expression exhibit an abundance of undifferentiated tissue, indicating that Oct-4 expression likely contributes to the maintenance of stem cell multipotency [54]. Hypoxic induction of HIF-2α followed by Oct-4 activation could contribute to the formation or maintenance of tumor stem cells.
Recent studies on the well characterized Notch signaling pathway further elucidate mechanisms of stem cell maintenance by hypoxia. The Notch pathway is integral in maintaining stem or progenitor cell identity in a number of contexts including the hematopoietic system and in intestine, skin, [55] and muscle [56]. Notch signaling is also important in the formation of numerous malignancies [57]. The Notch surface receptor is proteolytically cleaved upon binding of its cognate ligand, liberating Notch intracellular domains which travel to the nucleus and associate with a number of DNA-binding proteins. This activates transcription of genes involved in, among other things, inhibiting differentiation. It has recently been shown that hypoxia is an important component of this pathway as HIF–1α binds to the cleaved Notch1 protein and enhances its transcriptional activity [58]. This activity is accomplished by a direct interaction with Notch1, leading to increased stability, and inhibition of neuronal and myogenic cell differentiation. Interestingly, treatment with drugs that inhibit Notch cleavage decrease hypoxic activation of Notch. Additionally, HIF–1α-deficient MEFs exhibit decreased activation of Notch targets under hypoxia. Through HIF-1α-Notch interaction hypoxia could directly maintain myoblasts, HSC, and stem cells in the intestine and skin in an undifferentiated state. Since Notch is critically important for stem cell maintenance, hypoxia may promote Notch signaling in cancer stem cells and maintain them in an undifferentiated state as well.
A recently published study begins to describe hypoxic effects on the Wnt signaling pathway [59]. Wnt activates the transcriptional upregulation of numerous genes involved in proliferation and differentiation and is well known to play an important role in tumor formation. When activated by Wnt signaling, β–catenin binds to its dimerization partner TCF-4/LEF in the nucleus resulting in target gene stimulation. This activity is necessary for the maintenance of stem cell identity, especially in stem cells such as those within the intestinal crypt. Colon cancer cell lines subjected to hypoxia (1% O2) accumulate HIF-1α resulting in HIF-1α/β–catenin interaction, and inhibition of β–catenin/TCF-4 interactions. This coincides with enhanced activation of HIF–1α regulated genes and a block in cell cycle progression. The authors argue that the interplay between these transcription factors allows cancer cells to better withstand tumor hypoxia. Since the Wnt signaling pathway is essential for maintaining stem cells it would appear that these observations contradict the assertion that hypoxia may maintain stem cell identity. However, the authors did not test stem cells specifically. Additionally, the authors did not test the effect of HIF-2α on this β–catenin activity. Finally, the cell lines examined contain alterations in the Wnt pathway, a frequent trait of colon cancer cells. These alterations (loss of APC and β–catenin mutations) could potentially affect the efficiency with which β–catenin and HIF–1α interact. These issues must be addressed experimentally. Importantly however, these data demonstrate that the HIF and Wnt pathways interact and provide a potential mechanism for Wnt-mediated maintenance of the undifferentiated state.
We and others have recently shown that both HIF-α subunits modulate the activity of the well known oncogene c-Myc. HIF–1α antagonizes the activity of c-Myc by competing for binding with the c-Myc partner Sp-1 [60], resulting in cell cycle arrest at low O2 levels. Data from our laboratory have demonstrated that the opposite effect occurs upon expression of HIF-2α [61]. HIF-2α expression results in enhanced c-Myc transcriptional activity, at both c-Myc activated and repressed genes. These observations could have a big impact on cell signaling within solid tumors exposed to hypoxia. The latter study is especially pertinent to the maintenance of stem cells, for which c-Myc is required.
These studies begin to address important questions raised above of the origin of the cancer stem cell. A recent report demonstrated, intriguingly, that the number of genetic alterations required to form pluripotent stem cells from adult fibroblasts is surprisingly small [62]. The authors engineered fibroblasts to express Oct-4, c-Myc, Sox2 and Klf4. Amazingly, these cells appeared morphologically similar to ES cells, and gained stem cell-like proliferative potential. Additionally, they were able to initiate embryogenesis. This fascinating report demonstrates that relatively few mutations would be required to generate a stem-like cell from a differentiated somatic cell. The implications of these studies for tumor formation and especially for cancer stem cells are profound. A small number of affected pathways potentially activate stem-like properties in either normal somatic cells or perhaps more readily, in cancer cells. For cells that have already accumulated multiple mutations, additional changes would generate a stem-like phenotype. Finally, it is interesting that normal hypoxic responses impact at least two of these four alterations. HIF-2α expression, as discussed above, stimulates Oct-4 expression and promotes c-Myc activity. Hypoxic regulation of Oct-4 and c-Myc could powerfully impact cancer stem cell formation, especially in cells expressing HIF-2α. How these pathways interact with different HIF subunits in such cancer stem cells awaits further investigation.
HIF–1α has been demonstrated to inhibit the expression of a component of the DNA repair machinery, MutS through its interaction with c-Myc [63]. Previous work has demonstrated that hypoxia promotes genetic instability, especially in cells able to avoid apoptosis [64]. As genomic instability is recognized as a hallmark of tumor development, frequent hypoxic exposure likely contributes mutations leading to tumor cell transformation. This assertion would be consistent with the “mutator phenotype” associated with more aggressive cancers. Decreased O2 would therefore result in mutations leading to cancer stem cell formation from either resident stem cells or somatic cells.
IV. Hypoxic regulation of dedifferentiation
One model postulates that cancer stem cells are generated from differentiated somatic cells. This event, although certainly rare potentially requires relatively few mutations [62]. However such “dedifferentiation” has been observed in some malignancies including neuroblastoma and breast cancer [65]. Dedifferentiation could begin modestly, with a normal cell obtaining self-renewing capacity. This cell then accumulates additional mutations leading to increased dedifferentiation, finally resulting in a cell exhibiting stem cell-like characteristics. As described in detail above, many stem cell traits can be conferred by low O2, mainly through the inappropriate expression of HIF. Furthermore, increased levels of genomic instability could accelerate the generation of cancer stem cells from differentiated cells. Hypoxia may also play a role in maintaining these stem cell characteristics, through Notch signaling for example (Figure 2). Indeed, evidence for hypoxia-induced cellular dedifferentiation of cells has recently been published for neuroblastoma [66] and breast cancer [67]. In another report, some of the molecular mechanisms underlying neuroblastoma dedifferentiation were characterized [68]. Hypoxic neuroblastoma cells exhibit increased expression of the ID2 protein [68], which has been demonstrated to play an important role in the maintenance of the undifferentiated state of neuroblastoma cells. Interestingly, the ID2 promoter contains multiple HREs, canonical binding sites for HIF. ID2 likely contributes to the dedifferentiation of normal cells to a stem-like phenotype. Again, this event might be an initiating one, followed by further mutations contributing to the overall transformed phenotype of stem cells. ID2 could coordinate with one or more of the events described above to further maintain cells in a dedifferentiated state, such as by increased Notch signaling. These possibilities highlight the potential role for hypoxia in the generation of stem cells from normal cells. Although experimentally demonstrated in neuroblastoma and breast cancer cells, this mechanism potentially accounts for cancer stem cell generation in other tissues as well.
Figure 2.
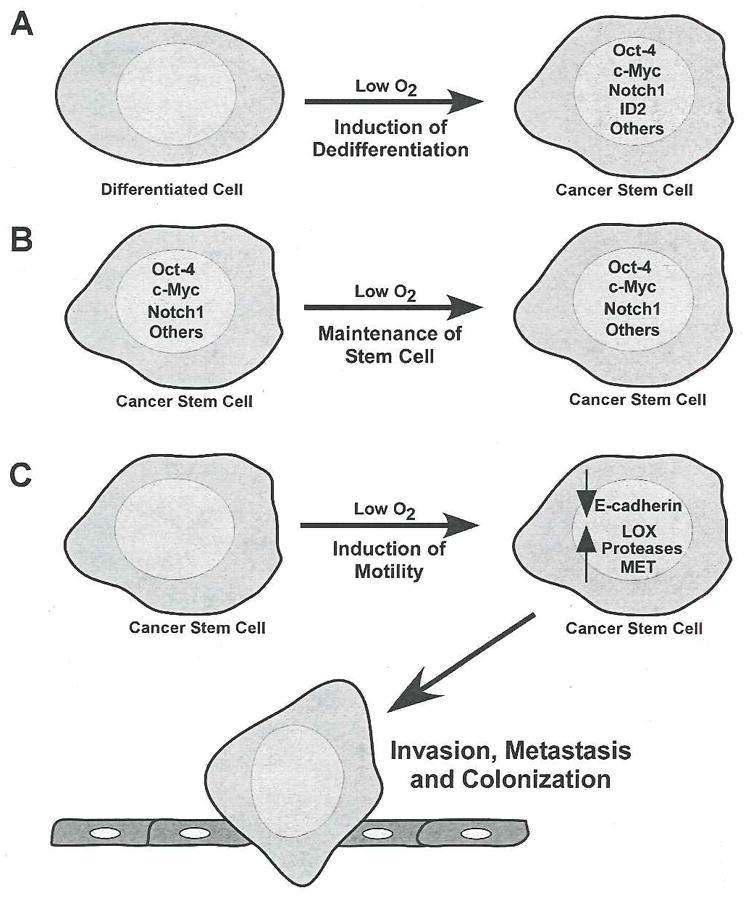
Hypoxic effects on the generation and maintenance of cancer stem cells. Possible mechanisms for the generation and maintenance of cancer stem cells and the role of hypoxia are depicted here. A. Hypoxic exposure activates pathways that ultimately lead to dedifferentiation of a differentiated cell. These include for example, activation of the Notch1 pathway, induction of Oct-4 expression, enhancement of c-Myc activity, ID2 induction (in neuroblastoma), among other alterations. B. Cancer stem cells exposed to hypoxia maintain their stem cell identity by inducing important stem-cell promoting pathways. C. Low O2 increases metastasis in cancer stem cells by reducing E-cadherin levels and increasing pro-metastatic genes such as LOX, MET, and proteases (such as MMP and uPAR). These pathways are regulated by HIF expression. Once the cancer stem cell has left the primary tumor, it colonizes other organs, a process also impacted by hypoxia (see text for details).
Hypoxia also has many effects on cells that are HIF-independent [69], impacting numerous pathways impinging on cellular growth, proliferation, and metabolism. For example, hypoxia negatively regulates the mammalian Target of Rapamycin complex 1 (mTORC1) [70, 71]. Although mechanisms for hypoxic mTORC1 inhibition are still under investigation, this effect has been observed in many cell types. One of the major consequences is a decrease in protein synthesis capacity by the coordinated disabling of p70S6K and translation initiation machinery. These canonical mTORC1 targets are rapidly inhibited by hypoxic exposure, and hypoxia is dominant to many other stimuli that activate mTORC1 [70]. mTORC1 inhibition has been shown to decrease cell cycle progression [72], result in smaller cell size [73], and sensitizes cells to apoptosis, making the kinase a promising therapeutic target [74]. At first glance, this observation seems to contradict the potential advantage that cancer stem cells likely gain from hypoxic exposure. However, it is also known that ES cells thrive under hypoxic conditions and that O2 deprivation induces increased proliferative capacity of many stem cell types (e.g. [30]). Therefore, mTORC1 inhibition by hypoxia may not occur in stem cells. Observations from our laboratory indicate that this may be the case. We have observed that the mTORC1 pathway is regulated differently in ES cells upon hypoxic exposure when compared to mouse embryonic fibroblasts (MEFs) (R. Nayak, B.C.B., M.C.S., unpublished). Alternatively, stem cells may alter the pathway downstream of mTORC1 allowing escape from its inhibitory effects. Finally, other pathways may feed into the mTOR pathway in stem cells, potentially negating the inhibitory effect of hypoxia. In fact, a recent study found that Wnt activates mTOR, which leads to increased protein synthesis and ultimately cell growth [75]. This could be an additional mechanism of tumor cell resistance to the inhibition of mTOR signaling imposed by hypoxia.
V. Metastasis, hypoxia, and cancer stem cells
Recent data indicate that cancer stem cells may be involved in tumor metastasis [76]. Several arguments make this an attractive hypothesis. First, and perhaps simplest is the proliferative potential of cancer stem cells. Since these cells are capable of unlimited division, the arrival of stem cells at distant sites naturally allows them to proliferate sufficiently to establish a new colony. If differentiated cells from the tumor arrived at distant sites and could not proliferate robustly, chances of colony formation would be far lower. Since one cell can initiate a metastatic lesion [77] it is more likely for this event to be accomplished by stem cells than by differentiated cells. Additionally, cancer stem cells are more likely to survive in foreign environments than differentiated cells, given their plasticity. Metastatic cancer stem cells potentially utilize niches present in other tissues, and may be able to recruit components of the niche itself in the new environment. Additionally, by virtue of their multipotency, cancer stem cells generate the heterogeneity in metastatic cancer observed clinically. Through these mechanisms, cancer stem cells are the most likely candidates to successfully establish a metastatic colony (Figure 2).
Several recent reports have described the role of hypoxia in regulating various mechanistic aspects of metastasis. For example, hypoxia results in decreased expression of E-cadherin [78–80]. Loss of E-cadherin is associated with epithelial-mesenchymal transition (EMT), resulting in, among other things, increased cell motility (a requisite step in metastasis). By decreasing E-cadherin expression, hypoxia contributes to the initiation of this process. Hypoxia has also recently been shown to regulate the expression of lysyl oxidase (LOX) [81], which is involved in cell-cell and cell-matrix interactions. LOX expression by metastasizing cells results in an increased ability to attach to foreign sites and establish colonies. Decreased LOX activity in tumors results in decreased ability to productively metastasize. These observations were made in breast and head and neck cancer, and the former has been formally demonstrated to contain cancer stem cells [18]. HIF expression by cancer stem cells could result in increased LOX activity increasing the likelihood of forming new colonies following metastasis.
In addition, HIF-1α activates the expression of the MET proto-oncogene, an important regulator of invasion and metastasis [82]. As MET is normally involved in embryonic development, the inappropriate expression of MET is yet another example of a developmental system subverted by cancer cells. It has been hypothesized that expression of MET in cancer stem cells in particular may contribute to their metastatic phenotype [83]. In addition, MET plays a role in self renewal, and so its expression could confer multiple advantages to cancer stem cells [83]. Importantly, MET is induced by hypoxia in a HIF-1α dependent manner [82], providing another mechanism for the hypoxic induction of both self-renewal and metastasis.
HIF has also been demonstrated to regulate genes dictating ultimate metastatic destination. The chemokine receptor CXCR4, a HIF target in RCC and breast cancer [84], is important in directing metastasis. HIF expression in RCC cells through loss of VHL activity elevates CXCR4 expression, enabling cells to home to specific anatomical locations where CXCR4 ligand SDF1 is expressed. Such homing is thought to be an important aspect of successful metastasis and migration to sites more conducive to growth may help tumor cells survive this process [85]. In this way, cancer stem cells with activated HIF could have an added advantage as metastatic cells.
Finally, several additional HIF targets may also be involved in productive metastasis. These include proteases (MMP2 and uPAR) and structural proteins, both important for the metastatic process [2]. Given the presumed increased mobility of cancer stem cells, exposure to hypoxia could enhance their ability to metastasize.
VI. Hypoxia and cancer stem cells—a therapeutic problem
It has long been recognized that extensive hypoxia within solid tumors correlates with poorer prognosis [3, 4]. Interestingly, as discussed above, hypoxia induces numerous responses in stem cells that confer an advantage to these cells and ultimately result in poor outcomes for the patient. Additionally, cancer stem cells are known to resist therapy aimed at reducing tumor load [4]. Since traditional chemotherapy most readily kills rapidly proliferating cells, cancer stem cells within the population are inherently resistant due to their slow proliferation rate [86]. Additionally, stem cells express high levels of ABC transporters which efficiently exclude drugs [13]. These transporters are also targets of HIF [87]. Cancer stem cells would therefore derive further protection from a hypoxic environment. In addition, hypoxia is known to protect cells from radiation therapy, due to the ineffectiveness of radiation in the absence of O2 [3]. It was recently reported that glioma stem cells are radioresistant due to an increased capacity to activate DNA repair [88]. Again, in the presence of hypoxia, which is a frequent trait of glioblastoma, this radioresistance of the cancer stem cells would be even more significantly enhanced.
The observation that cancer stem cells are resistant to therapy is an especially difficult problem in the clinic. Destruction of differentiated cancer cells but not cancer stem cells preserves those cells most likely to repopulate the tumor following therapy. Therefore, targeting cancer stem cells is of great clinical interest and therefore an active and aggressively pursued area of current research [86, 89]. An exhaustive description of current therapeutic approaches to cancer stem cells is beyond the scope of this review. However, as an illustrative example of a therapeutic approach involving cancer stem cells and their relationship to hypoxia we consider a recently published report. In this study, the authors found that brain tumor stem cells strongly associate with microvessels within brain tumors [17]. These data would suggest that cancer stem cells gain an advantage from proximity to vessels and perhaps to O2. However, the authors determined that the cancer stem cells associated vigorously with the endothelial cells, not necessarily blood vessels to which they contribute. Mixing experiments (of cancer stem cells and endothelial cells) were performed in vitro, where no O2 gradient and indeed no oxygenated blood were present. This indicates that it is likely that the cancer stem cells derive an advantage by proximity to endothelial cells and perhaps surface or secreted proteins associated with these cells. The authors describe this association as a niche for brain tumor stem cells. Even if association with endothelial cells within tumors provides the cancer stem cells with O2, it does not preclude expression of HIF. A report by Pahlman’s group recently determined that HIF-2α can be stabilized at approximately 5% O2 which is the O2 concentration at the ends of capillaries [90]. It is likely that this O2 level is frequently present in tumors due to the poor vasculature. Therapeutically, targeting these vascular niches enabled the researchers to more effectively eliminate the cancer stem cells [17]. Such stem cell targeting therapies hold great promise in fighting the cancers that are known to have cancer stem cells, as well as the many that are likely to be found in the coming years.
VII. Conclusions and future directions
The recognition that hypoxia correlates with poor clinical outcome occurred many years ago. Research currently being conducted is only now allowing us to determine some of the molecular mechanisms that underlie this observation. In addition, the realization that cancer stem cells are involved in a growing number of tumors indicates that these cells will likely be viable targets for chemotherapy in multiple diseases. The work discussed in this review indicates that many of the most troublesome traits of cancer stem cells may be induced or augmented by hypoxic exposure. Targeting cancer stem cells therapeutically will require an assessment of the microenvironment and niche of the stem cells, including the influence of hypoxia on these features.
VIII. Key unanswered questions
The work described in this review raises several important questions. What signaling pathways activated by hypoxia are essential for maintaining the cancer stem cell phenotype? What is the contribution of hypoxia in the dedifferentiation of cancer stem cells and how could this be targeted therapeutically? What role does the niche play in cancer stem cell maintenance, and what role does hypoxia play within this niche? How do we more effectively target cancer stem cells if hypoxia does contribute to their phenotype?
Acknowledgments
We thank the members of the Simon laboratory for helpful discussions. We apologize to our colleagues whose work we could not cite due to space considerations. B.C.B is a Leukemia & Lymphoma Society Fellow. M.C.S. is an Investigator of the Howard Hughes Medical Institute.
Contributor Information
Bryan C. Barnhart, Email: barnhar@mail.med.upenn.edu.
M. Celeste Simon, Email: celeste2@mail.med.upenn.edu.
References
- 1.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 2.Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441:437–443. doi: 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- 3.Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437–447. doi: 10.1038/nrc1367. [DOI] [PubMed] [Google Scholar]
- 4.Hockel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst. 2001;93:266–276. doi: 10.1093/jnci/93.4.266. [DOI] [PubMed] [Google Scholar]
- 5.Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, Giaccia AJ. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature. 1996;379:88–91. doi: 10.1038/379088a0. [DOI] [PubMed] [Google Scholar]
- 6.Rodesch F, Simon P, Donner C, Jauniaux E. Oxygen measurements in endometrial and trophoblastic tissues during early pregnancy. Obstet Gynecol. 1992;80:283–285. [PubMed] [Google Scholar]
- 7.Lee YM, Jeong CH, Koo SY, Son MJ, Song HS, Bae SK, Raleigh JA, Chung HY, Yoo MA, Kim KW. Determination of hypoxic region by hypoxia marker in developing mouse embryos in vivo: a possible signal for vessel development. Dev Dyn. 2001;220:175–186. doi: 10.1002/1097-0177(20010201)220:2<175::AID-DVDY1101>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 8.Morriss GM, New DA. Effect of oxygen concentration on morphogenesis of cranial neural folds and neural crest in cultured rat embryos. J Embryol Exp Morphol. 1979;54:17–35. [PubMed] [Google Scholar]
- 9.Semenza GL. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu Rev Cell Dev Biol. 1999;15:551–578. doi: 10.1146/annurev.cellbio.15.1.551. [DOI] [PubMed] [Google Scholar]
- 10.Maltepe E, Schmidt JV, Baunoch D, Bradfield CA, Simon MC. Abnormal angiogenesis and responses to glucose and oxygen deprivation in mice lacking the protein ARNT. Nature. 1997;386:403–407. doi: 10.1038/386403a0. [DOI] [PubMed] [Google Scholar]
- 11.Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ryan HE, Lo J, Johnson RS. HIF-1 alpha is required for solid tumor formation and embryonic vascularization. Embo J. 1998;17:3005–3015. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer. 2003;3:895–902. doi: 10.1038/nrc1232. [DOI] [PubMed] [Google Scholar]
- 14.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 15.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 16.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 17.Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, Oh EY, Gaber MW, Finklestein D, Allen M, Frank A, Bayazitov IT, Zakharenko SS, Gajjar A, Davidoff A, Gilbertson RJ. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 18.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 20.Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De Maria R. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–115. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 21.Morrison SJ, Shah NM, Anderson DJ. Regulatory mechanisms in stem cell biology. Cell. 1997;88:287–298. doi: 10.1016/s0092-8674(00)81867-x. [DOI] [PubMed] [Google Scholar]
- 22.Moore KA, Lemischka IR. Stem cells and their niches. Science. 2006;311:1880–1885. doi: 10.1126/science.1110542. [DOI] [PubMed] [Google Scholar]
- 23.Joseph NM, Morrison SJ. Toward an understanding of the physiological function of Mammalian stem cells. Dev Cell. 2005;9:173–183. doi: 10.1016/j.devcel.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 24.Li L, Xie T. Stem cell niche: structure and function. Annu Rev Cell Dev Biol. 2005;21:605–631. doi: 10.1146/annurev.cellbio.21.012704.131525. [DOI] [PubMed] [Google Scholar]
- 25.Ohlstein B, Kai T, Decotto E, Spradling A. The stem cell niche: theme and variations. Curr Opin Cell Biol. 2004;16:693–699. doi: 10.1016/j.ceb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 26.Morrison SJ, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006;441:1068–1074. doi: 10.1038/nature04956. [DOI] [PubMed] [Google Scholar]
- 27.Scadden DT. The stem-cell niche as an entity of action. Nature. 2006;441:1075–1079. doi: 10.1038/nature04957. [DOI] [PubMed] [Google Scholar]
- 28.Li L, Neaves WB. Normal stem cells and cancer stem cells: the niche matters. Cancer Res. 2006;66:4553–4557. doi: 10.1158/0008-5472.CAN-05-3986. [DOI] [PubMed] [Google Scholar]
- 29.Ezashi T, Das P, Roberts RM. Low O2 tensions and the prevention of differentiation of hES cells. Proc Natl Acad Sci U S A. 2005;102:4783–4788. doi: 10.1073/pnas.0501283102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cipolleschi MG, Dello Sbarba P, Olivotto M. The role of hypoxia in the maintenance of hematopoietic stem cells. Blood. 1993;82:2031–2037. [PubMed] [Google Scholar]
- 31.Kiel MJ, Yilmaz OH, Iwashita T, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 32.Danet GH, Pan Y, Luongo JL, Bonnet DA, Simon MC. Expansion of human SCID-repopulating cells under hypoxic conditions. J Clin Invest. 2003;112:126–135. doi: 10.1172/JCI17669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adelman DM, Maltepe E, Simon MC. Multilineage embryonic hematopoiesis requires hypoxic ARNT activity. Genes Dev. 1999;13:2478–2483. doi: 10.1101/gad.13.19.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adelman DM, Gertsenstein M, Nagy A, Simon MC, Maltepe E. Placental cell fates are regulated in vivo by HIF-mediated hypoxia responses. Genes Dev. 2000;14:3191–3203. doi: 10.1101/gad.853700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramirez-Bergeron DL, Runge A, Dahl KD, Fehling HJ, Keller G, Simon MC. Hypoxia affects mesoderm and enhances hemangioblast specification during early development. Development. 2004;131:4623–4634. doi: 10.1242/dev.01310. [DOI] [PubMed] [Google Scholar]
- 36.Ramirez-Bergeron DL, Runge A, Adelman DM, Gohil M, Simon MC. HIF-dependent hematopoietic factors regulate the development of the embryonic vasculature. Dev Cell. 2006;11:81–92. doi: 10.1016/j.devcel.2006.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Genbacev O, Zhou Y, Ludlow JW, Fisher SJ. Regulation of human placental development by oxygen tension. Science. 1997;277:1669–1672. doi: 10.1126/science.277.5332.1669. [DOI] [PubMed] [Google Scholar]
- 38.Morrison SJ, Csete M, Groves AK, Melega W, Wold B, Anderson DJ. Culture in reduced levels of oxygen promotes clonogenic sympathoadrenal differentiation by isolated neural crest stem cells. J Neurosci. 2000;20:7370–7376. doi: 10.1523/JNEUROSCI.20-19-07370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Studer L, Csete M, Lee SH, Kabbani N, Walikonis J, Wold B, McKay R. Enhanced proliferation, survival, and dopaminergic differentiation of CNS precursors in lowered oxygen. J Neurosci. 2000;20:7377–7383. doi: 10.1523/JNEUROSCI.20-19-07377.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Polyak K, Hahn WC. Roots and stems: stem cells in cancer. Nat Med. 2006;12:296–300. doi: 10.1038/nm1379. [DOI] [PubMed] [Google Scholar]
- 41.Bjerkvig R, Tysnes BB, Aboody KS, Najbauer J, Terzis AJ. Opinion: the origin of the cancer stem cell: current controversies and new insights. Nat Rev Cancer. 2005;5:899–904. doi: 10.1038/nrc1740. [DOI] [PubMed] [Google Scholar]
- 42.Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, Bourque G, George J, Leong B, Liu J, Wong KY, Sung KW, Lee CW, Zhao XD, Chiu KP, Lipovich L, Kuznetsov VA, Robson P, Stanton LW, Wei CL, Ruan Y, Lim B, Ng HH. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet. 2006;38:431–440. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- 43.Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270:1230–1237. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 44.Liu YV, Baek JH, Zhang H, Diez R, Cole RN, Semenza GL. RACK1 Competes with HSP90 for Binding to HIF-1alpha and Is Required for O(2)-Independent and HSP90 Inhibitor-Induced Degradation of HIF-1alpha. Mol Cell. 2007;25:207–217. doi: 10.1016/j.molcel.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol. 2003;23:9361–9374. doi: 10.1128/MCB.23.24.9361-9374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang V, Davis DA, Haque M, Huang LE, Yarchoan R. Differential gene up-regulation by hypoxia-inducible factor-1alpha and hypoxia-inducible factor-2alpha in HEK293T cells. Cancer Res. 2005;65:3299–3306. doi: 10.1158/0008-5472.CAN-04-4130. [DOI] [PubMed] [Google Scholar]
- 47.Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, Pugh CW, Maxwell PH, Harris AL, Ratcliffe PJ. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol. 2005;25:5675–5686. doi: 10.1128/MCB.25.13.5675-5686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 49.Covello KL, Kehler J, Yu H, Gordan JD, Arsham AM, Hu CJ, Labosky PA, Simon MC, Keith B. HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006;20:557–570. doi: 10.1101/gad.1399906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nichols J, Zevnik B, Anastassiadis K, Niwa H, Klewe-Nebenius D, Chambers I, Scholer H, Smith A. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell. 1998;95:379–391. doi: 10.1016/s0092-8674(00)81769-9. [DOI] [PubMed] [Google Scholar]
- 51.Niwa H, Miyazaki J, Smith AG. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat Genet. 2000;24:372–376. doi: 10.1038/74199. [DOI] [PubMed] [Google Scholar]
- 52.Niwa H, Toyooka Y, Shimosato D, Strumpf D, Takahashi K, Yagi R, Rossant J. Interaction between Oct3/4 and Cdx2 determines trophectoderm differentiation. Cell. 2005;123:917–929. doi: 10.1016/j.cell.2005.08.040. [DOI] [PubMed] [Google Scholar]
- 53.Gidekel S, Pizov G, Bergman Y, Pikarsky E. Oct-3/4 is a dose-dependent oncogenic fate determinant. Cancer Cell. 2003;4:361–370. doi: 10.1016/s1535-6108(03)00270-8. [DOI] [PubMed] [Google Scholar]
- 54.Covello KL, Simon MC, Keith B. Targeted replacement of hypoxia-inducible factor-1alpha by a hypoxia-inducible factor-2alpha knock-in allele promotes tumor growth. Cancer Res. 2005;65:2277–2286. doi: 10.1158/0008-5472.CAN-04-3246. [DOI] [PubMed] [Google Scholar]
- 55.Wilson A, Radtke F. Multiple functions of Notch signaling in self-renewing organs and cancer. FEBS Lett. 2006;580:2860–2868. doi: 10.1016/j.febslet.2006.03.024. [DOI] [PubMed] [Google Scholar]
- 56.Nofziger D, Miyamoto A, Lyons KM, Weinmaster G. Notch signaling imposes two distinct blocks in the differentiation of C2C12 myoblasts. Development. 1999;126:1689–1702. doi: 10.1242/dev.126.8.1689. [DOI] [PubMed] [Google Scholar]
- 57.Weng AP, Aster JC. Multiple niches for Notch in cancer: context is everything. Curr Op in Genet Dev. 2004;14:48–54. doi: 10.1016/j.gde.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 58.Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, Ruas JL, Poellinger L, Lendahl U, Bondesson M. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev Cell. 2005;9:617–628. doi: 10.1016/j.devcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 59.Kaidi A, Williams AC, Paraskeva C. Interaction between beta-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat Cell Biol. 2007 doi: 10.1038/ncb1534. [DOI] [PubMed] [Google Scholar]
- 60.Koshiji M, Kageyama Y, Pete EA, Horikawa I, Barrett JC, Huang LE. HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. Embo J. 2004;23:1949–1956. doi: 10.1038/sj.emboj.7600196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-Myc transcriptional activity. Cancer Cell. 2007 doi: 10.1016/j.ccr.2007.02.006. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 63.Koshiji M, To KK, Hammer S, Kumamoto K, Harris AL, Modrich P, Huang LE. HIF-1alpha induces genetic instability by transcriptionally downregulating MutSalpha expression. Mol Cell. 2005;17:793–803. doi: 10.1016/j.molcel.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 64.Nelson DA, Tan TT, Rabson AB, Anderson D, Degenhardt K, White E. Hypoxia and defective apoptosis drive genomic instability and tumorigenesis. Genes Dev. 2004;18:2095–2107. doi: 10.1101/gad.1204904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Axelson H, Fredlund E, Ovenberger M, Landberg G, Pahlman S. Hypoxia-induced dedifferentiation of tumor cells--a mechanism behind heterogeneity and aggressiveness of solid tumors. Semin Cell Dev Biol. 2005;16:554–563. doi: 10.1016/j.semcdb.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 66.Jogi A, Ora I, Nilsson H, Lindeheim A, Makino Y, Poellinger L, Axelson H, Pahlman S. Hypoxia alters gene expression in human neuroblastoma cells toward an immature and neural crest-like phenotype. Proc Natl Acad Sci U S A. 2002;99:7021–7026. doi: 10.1073/pnas.102660199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Helczynska K, Kronblad A, Jogi A, Nilsson E, Beckman S, Landberg G, Pahlman S. Hypoxia promotes a dedifferentiated phenotype in ductal breast carcinoma in situ. Cancer Res. 2003;63:1441–1444. [PubMed] [Google Scholar]
- 68.Lofstedt T, Jogi A, Sigvardsson M, Gradin K, Poellinger L, Pahlman S, Axelson H. Induction of ID2 expression by hypoxia-inducible factor-1: a role in dedifferentiation of hypoxic neuroblastoma cells. J Biol Chem. 2004;279:39223–39231. doi: 10.1074/jbc.M402904200. [DOI] [PubMed] [Google Scholar]
- 69.Liu L, Simon MC. Regulation of transcription and translation by hypoxia. Cancer Biol Ther. 2004;3:492–497. doi: 10.4161/cbt.3.6.1010. [DOI] [PubMed] [Google Scholar]
- 70.Arsham AM, Howell JJ, Simon MC. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J Biol Chem. 2003;278:29655–29660. doi: 10.1074/jbc.M212770200. [DOI] [PubMed] [Google Scholar]
- 71.Connolly E, Braunstein S, Formenti S, Schneider RJ. Hypoxia inhibits protein synthesis through a 4E-BP1 and elongation factor 2 kinase pathway controlled by mTOR and uncoupled in breast cancer cells. Mol Cell Biol. 2006;26:3955–3965. doi: 10.1128/MCB.26.10.3955-3965.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fingar DC, Richardson CJ, Tee AR, Cheatham L, Tsou C, Blenis J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol Cell Biol. 2004;24:200–216. doi: 10.1128/MCB.24.1.200-216.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002;16:1472–1487. doi: 10.1101/gad.995802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Easton JB, Houghton PJ. mTOR and cancer therapy. Oncogene. 2006;25:6436–6446. doi: 10.1038/sj.onc.1209886. [DOI] [PubMed] [Google Scholar]
- 75.Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, Wang CY, He X, MacDougald OA, You M, Williams BO, Guan KL. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955–968. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 76.Li F, Tiede B, Massague J, Kang Y. Beyond tumorigenesis: cancer stem cells in metastasis. Cell Res. 2007;17:3–14. doi: 10.1038/sj.cr.7310118. [DOI] [PubMed] [Google Scholar]
- 77.Fidler IJ, Talmadge JE. Evidence that intravenously derived murine pulmonary melanoma metastases can originate from the expansion of a single tumor cell. Cancer Res. 1986;46:5167–5171. [PubMed] [Google Scholar]
- 78.Imai T, Horiuchi A, Wang C, Oka K, Ohira S, Nikaido T, Konishi I. Hypoxia attenuates the expression of E-cadherin via up-regulation of SNAIL in ovarian carcinoma cells. Am J Pathol. 2003;163:1437–1447. doi: 10.1016/S0002-9440(10)63501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Esteban MA, Tran MG, Harten SK, Hill P, Castellanos MC, Chandra A, Raval R, O’Brien TS, Maxwell PH. Regulation of E-cadherin expression by VHL and hypoxia-inducible factor. Cancer Res. 2006;66:3567–3575. doi: 10.1158/0008-5472.CAN-05-2670. [DOI] [PubMed] [Google Scholar]
- 80.Krishnamachary B, Zagzag D, Nagasawa H, Rainey K, Okuyama H, Baek JH, Semenza GL. Hypoxia-inducible factor-1-dependent repression of E-cadherin in von Hippel-Lindau tumor suppressor-null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res. 2006;66:2725–2731. doi: 10.1158/0008-5472.CAN-05-3719. [DOI] [PubMed] [Google Scholar]
- 81.Erler JT, Bennewith KL, Nicolau M, Dornhofer N, Kong C, Le QT, Chi JT, Jeffrey SS, Giaccia AJ. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440:1222–1226. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- 82.Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 2003;3:347–361. doi: 10.1016/s1535-6108(03)00085-0. [DOI] [PubMed] [Google Scholar]
- 83.Boccaccio C, Comoglio PM. Invasive growth: a MET-driven genetic programme for cancer and stem cells. Nat Rev Cancer. 2006;6:637–645. doi: 10.1038/nrc1912. [DOI] [PubMed] [Google Scholar]
- 84.Staller P, Sulitkova J, Lisztwan J, Moch H, Oakeley EJ, Krek W. Chemokine receptor CXCR4 downregulated by von Hippel-Lindau tumour suppressor pVHL. Nature. 2003;425:307–311. doi: 10.1038/nature01874. [DOI] [PubMed] [Google Scholar]
- 85.Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- 86.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 87.Krishnamurthy P, Ross DD, Nakanishi T, Bailey-Dell K, Zhou S, Mercer KE, Sarkadi B, Sorrentino BP, Schuetz JD. The stem cell marker Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. J Biol Chem. 2004;279:24218–24225. doi: 10.1074/jbc.M313599200. [DOI] [PubMed] [Google Scholar]
- 88.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 89.Al-Hajj M, Becker MW, Wicha M, Weissman I, Clarke MF. Therapeutic implications of cancer stem cells. Curr Opin Genet Dev. 2004;14:43–47. doi: 10.1016/j.gde.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 90.Holmquist-Mengelbier L, Fredlund E, Lofstedt T, Noguera R, Navarro S, Nilsson H, Pietras A, Vallon-Christersson J, Borg A, Gradin K, Poellinger L, Pahlman S. Recruitment of HIF-1alpha and HIF-2alpha to common target genes is differentially regulated in neuroblastoma: HIF-2alpha promotes an aggressive phenotype. Cancer Cell. 2006;10:413–423. doi: 10.1016/j.ccr.2006.08.026. [DOI] [PubMed] [Google Scholar]