Evolutionary Genomics of Defense Systems in Archaea and Bacteria (original) (raw)
. Author manuscript; available in PMC: 2018 Apr 13.
Abstract
Evolution of bacteria and archaea involves an incessant arms race against an enormous diversity of genetic parasites. Accordingly, a substantial fraction of the genes in most bacteria and archaea are dedicated to antiparasite defense. The functions of these defense systems follow several distinct strategies, including innate immunity; adaptive immunity; and dormancy induction, or programmed cell death. Recent comparative genomic studies taking advantage of the expanding database of microbial genomes and metagenomes, combined with direct experiments, resulted in the discovery of several previously unknown defense systems, including innate immunity centered on Argonaute proteins, bacteriophage exclusion, and new types of CRISPR-Cas systems of adaptive immunity. Some general principles of function and evolution of defense systems are starting to crystallize, in particular, extensive gain and loss of defense genes during the evolution of prokaryotes; formation of genomic defense islands; evolutionary connections between mobile genetic elements and defense, whereby genes of mobile elements are repeatedly recruited for defense functions; the partially selfish and addictive behavior of the defense systems; and coupling between immunity and dormancy induction/programmed cell death.
Keywords: antivirus defense, mobile genetic elements, innate immunity, adaptive immunity, dormancy, programmed cell death, restriction-modification, toxins-antitoxins, CRISPR-Cas
INTRODUCTION
It is well established that the most abundant biological entities in the biosphere are viruses, in particular, tailed bacteriophages. In most environments, the ratio of the number of virus particles to the number of cells is between 10 and 100 (140, 154, 155). Furthermore, genomes of most cellular life-forms, including the compact genomes of bacteria and archaea, contain numerous integrated mobile genetic elements (MGEs), such as transposons and proviruses, that make up the vast prokaryotic mobilome (47, 164). Strikingly, transposases are the most common genes detected in metagenomes (6). This enormous abundance is paralleled by the remarkable genetic diversity of viruses and other mobile elements that could be the principal reservoir of new genes on earth. Both theory and observation indicate that persistence of MGEs of different kinds is virtually inevitable throughout the evolution of life (68, 78, 160). Indeed, viruses and/or mobile elements have been found in association with virtually every cellular organism that has been studied in sufficient detail, with the possible exception of some intracellular parasitic and symbiotic bacteria. Thus, the entire history of life is the story of host-parasite coevolution, in which a key element is the perennial arms race (45, 85, 151). Driven by the arms race with parasites, cellular organisms have evolved extremely elaborate and diverse defense systems that function via several distinct strategies, and the great majority of organisms deploy more than one such strategy.
Based on their principles of action, the defense mechanisms of bacteria and archaea can be classified into three broad groups: (a) resistance based on variation of virus receptors, (b) immunity, and (c) dormancy induction and programmed cell death (101, 109). Resistance involves programmed mechanisms of receptor change, such as phase variation, and in some cases, physical masking of receptors, such that virus binding and penetration are precluded (25, 65, 94). The immunity systems’ function depends on the ability to distinguish genomes of invaders (nonself) from the host (self) genome and protect the latter while inactivating the former. The immune systems themselves are naturally divided into the relatively nonspecific innate immunity and the highly specific adaptive (acquired) immunity. The best-characterized innate immunity systems are the extremely numerous and diverse restriction-modification (RM) modules that employ methylation to label and thus protect the self genomic DNA while cleaving any unmodified nonself DNA (89, 126, 175). A distinct variant of RM is DNA phosphorothioation (known as the DND system), which labels self DNA by phosphorothioation instead of methylation and destroys unmodified, nonself DNA (38). Recently, two additional bacterial and archaeal innate immunity systems were characterized. One, bacteriophage exclusion, can be considered yet another major variation on the RM theme, in which, however, the target foreign DNA is not degraded (8, 52). The second newly discovered type of innate immunity, centered on the prokaryotic Argonaute proteins, generates guide RNA or DNA molecules from the invader genomes and utilizes them to recognize and destroy the targets (63, 158).
Unlike the innate immunity systems, which attack nonself invaders (more or less) indiscriminately, the adaptive immunity systems, represented by CRISPR (clustered regularly interspaced short palindromic repeats)-Cas (CRISPR-associated genes), memorize the encounters with infectious agents by incorporating pieces of foreign genetic information into the host genome and attack invaders specifically upon new encounters using the cognate guide RNAs (7, 72, 105, 166, 174).
The second major type of defense system functions through induction of dormancy or programmed cell death in response to infection. Numerous and enormously diverse toxin-antitoxin (TA) systems belong in this category. In this type of defense, infection disrupts the balance between toxin and antitoxin, resulting in activation of the former and abrogation of central cellular functions, such as translation, hence precluding virus reproduction (50, 94, 108). In addition to the TA systems, the functionally similar abortive infection (ABI) systems often employ the mechanism of programmed cell death or dormancy (28).
Recent comparative genomic studies taking advantage of the rapidly growing amount and diversity of genomic and metagenomics sequences, and in some cases focused specifically on the search for new defense systems, have substantially expanded the range of identified defense functions. In addition, previously unknown connections between different types of defense systems have been revealed, and several general trends in the evolution of bacterial and archaeal defense have been elucidated.
In this review article, we present a current snapshot of the diversity of bacterial and archaeal immune and dormancy induction systems, highlight new discoveries, and discuss emerging generalizations on the evolution of antivirus defense in prokaryotes.
A COMPREHENSIVE CENSUS OF DEFENSE SYSTEMS IN BACTERIAL AND ARCHAEAL GENOMES
The fraction of bacterial and archaeal genes that encode defense systems varies broadly across microbial diversity, from complete absence, in intracellular parasitic bacteria with the smallest known genomes, to about 10% (Figure 1). All types of defense systems show bell-shaped distributions that are at first approximation compatible with random scatter and suggest that there are no distinct classes of defense-rich and defense-poor microbes. Compared to our previous similar census (109), the distributions have shifted to the left owing to the discovery of numerous genomes with few identifiable defense systems, the so-called candidate phyla radiation (CPR) of bacteria, and novel phyla of archaea with small genomes (17, 62). Clearly, these distributions represent the low bound for each type of defense system because, given the characteristic rapid evolution of defense genes, many additional variants most likely remain to be identified; moreover, completely new defense strategies are expected to be discovered, adding to the overall count.
Figure 1.
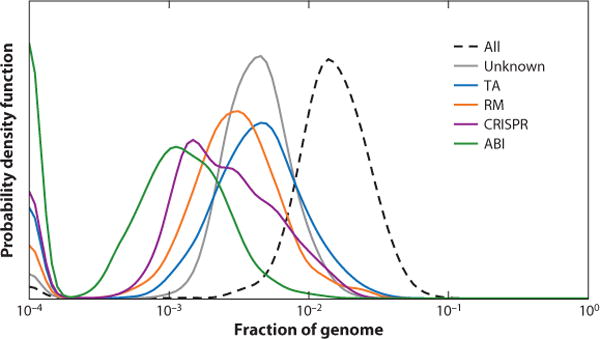
Abundance of the major types of defense systems in bacterial and archaeal genomes. Smoothed probability density for the distributions across 4,961 complete genomes of bacteria and archaea is shown. The number of genes in each category was calculated as described earlier (109). Abbreviations: ABI, abortive infection; CRISPR, clustered regularly interspaced short palindromic repeats; RM, restriction modification; TA, toxin-antitoxin.
The overall occurrence of defense systems shows nearly perfect linear scaling with the total number of genes in microbial genomes (109). However, this linear scaling results from significantly different trends for distinct types of defense cancelling out. The number of TA genes scales superlinearly, as a power of ∼1.3 of the total number of genes, and ABI system genes take an approximately constant fraction of the genome (∼1 per 1,000 genes), whereas RM and cas genes scale sublinearly (exponents of 0.65 and 0.73, respectively) with the genome size (Figure 2). The superlinear scaling of TA genes has been observed previously (109) and remains consistent. In contrast, our previous analysis has failed to show significant scaling with genome size for CRISPR-Cas systems (exponent indistinguishable from zero). The change in the observed trend, again, can be linked to the discovery of many small bacterial genomes, most of which conspicuously lack CRISPR-Cas systems (17, 18). The differential scaling of defense systems with genome size implies that the most informative approach for comparative analysis requires comparing the observed occurrence of defense genes relative to the expected occurrence given the genome size in the respective microbes. The biological underpinnings of the class-specific scaling factors for defense systems remain difficult to infer; some of these trends might have to do with additional, nondefense functions of defense genes (172).
Figure 2.
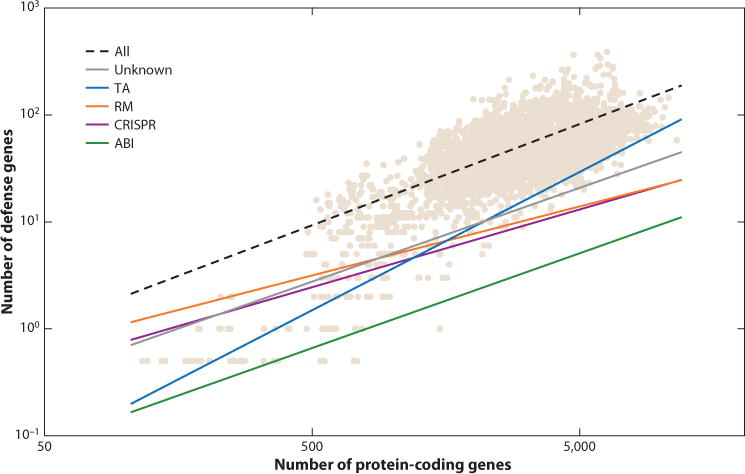
Scaling of the major types of defense systems with the total number of genes. Tan data points represent the total abundance of the respective defense systems in individual genomes; lines show the power law scaling relative to the genome size. The number of genes in each category and the power law scaling parameters were calculated as described earlier (109). Abbreviations: ABI, abortive infection; CRISPR, clustered regularly interspaced short palindromic repeats; RM, restriction modification; TA, toxin-antitoxin.
In our previous survey of prokaryotic defense systems, principal component analysis led to the delineation of four seemingly discrete classes that apparently corresponded to distinct defense strategies (109). This distinct structure disappeared from the current analysis of the substantially expanded genome collection, indicating that there is neither strong association nor antagonism between the representation of different classes of defense systems in bacterial and archaeal genomes (Figure 3). Nevertheless, the observed smoothing of the distribution does not imply absence of significant biases in the representation of defense systems in particular bacterial or archaeal taxa, or in microbes with a specific lifestyle. The main trends reported previously remain in place, namely, the pronounced enrichment of defense systems in archaea compared to bacteria and in thermophiles (especially hyperthermophiles) compared to mesophiles and psychrophiles (109). These two trends, the connections of defense system representation with domain-level taxonomy and growth temperature, seem to be independent of each other. Temperature dependence is particularly dramatic in the case of the CRISPR-Cas systems that are virtually ubiquitous among hyperthermophiles but are only found in about one-third of mesophiles (105, 106).
Figure 3.
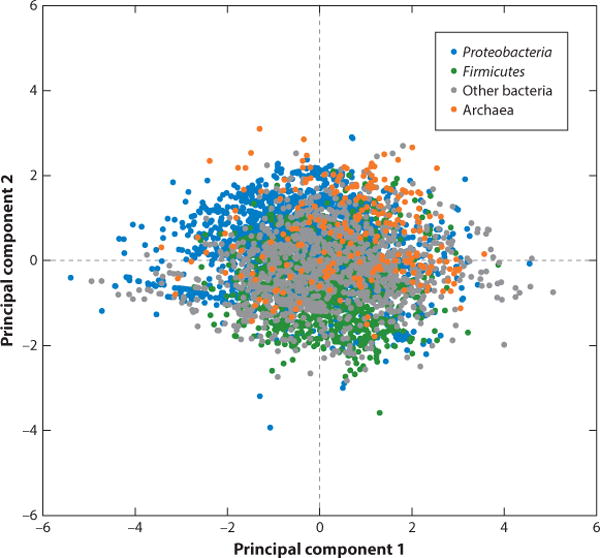
Principal component analysis of bacterial and archaeal defense systems (abortive infection, CRISPR, restriction modification, toxin-antitoxin). Excess or deficit (in natural log scale) of the five groups of defense systems relative to the expectations, derived from the genome size, was projected into the space of the first two principal components, as described previously (109).
At the next level of granularity, significant enrichment or depletion of defense systems (relative to the expectation derived from the genome size and the trends in Figure 2) is noticeable in several bacterial and archaeal phyla (Table 1). Observations that stand out include the substantial enrichment of all classes of defense systems in Chlorobi, dramatic enrichment of CRISPR-Cas in Thermotogae in contrast to a paucity of other defense mechanisms, and major enrichment of CRISPR-Cas in Crenarchaeota. In contrast, the apparent depletion of all classes of defense systems in Thaumarchaeota implies the existence of unrecognized defense machineries awaiting discovery; the same most likely applies to the CPR bacteria (not included in Table 1 because only a few genomes are currently complete).
Table 1.
Over- and underrepresentation of defense systems in bacterial and archaeal phyla
| ABI | CRISPR | RM | TA | Unknown | |
|---|---|---|---|---|---|
| Archaea | |||||
| Crenarchaeota | −0.640 | 1.106 | −0.413 | 0.459 | −0.900 |
| Euryarchaeota | −0.286 | 0.343 | 0.103 | −0.069 | −0.185 |
| Thaumarchaeota | −0.815 | −0.944 | −0.319 | −1.643 | −0.975 |
| Bacteria | |||||
| Acidobacteria | −0.038 | −0.678 | −0.385 | 0.326 | −0.387 |
| Actinobacteria | −0.249 | −0.179 | −0.286 | −0.394 | −0.103 |
| Aquificae | −1.025 | 0.604 | −0.719 | −0.192 | −0.078 |
| Bacteroidetes | 0.165 | −0.449 | −0.086 | −0.642 | −0.056 |
| Chlamydiae | −0.079 | −0.802 | −1.563 | −1.093 | 0.174 |
| Chlorobi | 0.745 | 0.696 | 0.348 | 0.978 | 0.488 |
| Chloroflexi | −0.464 | 0.298 | 0.307 | −0.094 | −0.065 |
| Cyanobacteria | 0.114 | −0.150 | 0.730 | −0.169 | −0.172 |
| Deinococcus-Thermus | −0.821 | 0.192 | −0.193 | 0.026 | −0.147 |
| Firmicutes | 0.333 | −0.233 | −0.349 | −0.336 | −0.144 |
| Fusobacteria | −0.046 | 0.083 | 0.048 | 0.122 | 0.350 |
| Nitrospirae | −0.593 | 0.160 | 0.398 | 0.785 | −0.030 |
| Peregrinibacteria | −0.982 | −0.778 | −0.562 | −0.326 | −2.405 |
| Planctomycetes | 0.125 | 0.105 | 0.247 | −0.923 | −0.193 |
| Proteobacteria | −0.783 | −0.682 | −0.440 | −0.309 | −0.132 |
| Spirochaetes | −0.161 | −0.345 | −0.193 | −0.204 | 0.133 |
| Tenericutes | −0.201 | −1.313 | 0.352 | −1.219 | −0.209 |
| Thermotogae | −0.105 | 0.839 | −0.596 | −0.564 | −0.722 |
| Verrucomicrobia | −0.261 | −0.632 | −0.241 | −0.830 | −0.341 |
| Unclassified | −0.189 | −0.604 | 0.080 | −0.132 | −0.319 |
INNATE IMMUNITY
Restriction Modification
RM systems are the first type of bacterial defense against foreign DNA that were discovered and studied in great detail, thanks primarily to the enormous utility of restriction endonucleases (REases) (89, 126, 132, 175).
The latest classification structure divides RM systems into four types (I–IV) on the basis of the subunit composition, ATP (GTP) requirement, and cleavage mechanism (132, 138, 139). type II RM systems are the simplest and by far the most common and are mostly used for experimental applications because they cleave target DNA at highly specific sites. Subtypes of the type II systems are primarily based on cleavage specificity (138). The type II systems consist solely of the methyltransferase-REase pair that is typically encoded within the same operon, although cases of apparent disjointed localization of the two genes have been reported (40). The most complex, ATP-dependent type I RM systems encompass three genes that encode the R (restriction), M (modification), and S (specificity) subunits of the RMS complex; the R subunit, in addition to REase, also contains a distinct ATPase domain that belongs to helicase superfamily II (15, 99, 163). Type III RM systems resemble type II systems in that they consist of only R and M subunits, but they are similar to type I systems in that the R subunit also contains the helicase domain and the reaction is ATP dependent (19, 136). Type IV RM systems are distinct two-subunit complexes that consist of a AAA+ family GTPase and an endonuclease (15, 100). Their mode of action is fundamentally different from the other three types in that they nonspecifically cleave modified phage DNA containing 5-hydroxymethylcytosine or 5-hydroxymethyluracil. In fact, type IV systems are best denoted R, because modification enzymes in this case are not parts of the defense machinery. These modifications protect phage DNA from cleavage by the conventional REases of types I, II, and III, and apparently type IV R systems have evolved on multiple occasions during the bacterial-phage arms race, as a counter-counterdefense mechanism that overcomes such protection (15, 100).
A distinct variation on the RM theme, more recently discovered defense systems function through site-specific DNA backbone modification, namely, phosphorothioation, and cleavage of unmodified DNA. The genes required for modification (dndABCDE; after DNA damage) and restriction (dndFGH) have been identified in several hundred bacterial and archaeal genomes (59). The structures and biochemical activities of the DndA (cysteine desulfurase), DndC (phosphoadenosine phosphosulfate reductase), and DndE (DNA-binding protein with a distinct fold) that are directly involved in phosphorothioation have been thoroughly characterized (26, 61, 178, 184). Additionally, it has been shown that DndB is a transcriptional regulator of the dnd operons (57), but the functions of the other genes associated with this system are less clear (177).
Coevolution of the RM systems with the prokaryotic genomes is an intriguing yet understudied subject. An early genomic analysis has shown that RM recognition sites are avoided in some bacterial genomes at a statistically significant level (48). Recently, however, it has been demonstrated that type II recognition sites are avoided in about half of the bacterial and archaeal genomes whereas sites for other RM types are not avoided (141). Avoidance of type II sites apparently depends on the lifespan of RM systems in the host genome; i.e., it evolves over relatively long periods of coevolution (141). Recent extensive genomic analysis has revealed a strong connection between the abundance of RM modules and MGEs in microbial genomes, suggesting that although RM systems are rarely encoded by plasmids or prophages, they nevertheless disseminate with the help of MGEs (123). Conversely, the abundance of RM modules also strongly positively correlates with the estimated rate of horizontal gene transfer (HGT), suggesting a major role of RM in this process (124). These findings indicate that the roles of RM in prokaryotes go beyond defense functions (41).
Phage Growth Limitation and Bacteriophage Exclusion
An early study has reported a unique bacteriophage-resistant phenotype, denoted Pgl (phage growth limitation), in Streptomyces coelicolor (27). Strains that carried the Pgl locus supported one cycle of phage growth, but the released phage was not infectious (27, 153). The Pgl system consists of the adenine-specific DNA methylase PglX, serine-threonine kinase PglW, predicted P-loop ATPase PglY, and predicted alkaline phosphatase PglZ (186). Comparative analysis of the neighborhoods of the pglZ gene (the hallmark of the Pgl systems found in all its variants but not in other genomic contexts) revealed substantial complexity of genetic organization of this system. Judging by the presence of the pglZ gene, different variants of this system are encoded in about 10% of the analyzed complete genomes that represent most of the major bacterial lineages as well as several methanogenic and halophilic archaea. The unusual properties of the Pgl system prompted the hypothesis that it functions via a reverse RM mechanism, i.e., by methylating the DNA of the phage progeny rather than the host DNA, so that upon reinfection, the surviving cells in the same colony activate the Pgl system and prevent phage growth (27, 153). Recent work has validated the prediction of activities of the Pgl proteins and additionally demonstrated cellular toxicity of PglX that is mitigated by PglZ (60) (see below for discussion of the connections between immunity and toxicity in bacteria and archaea). The new findings appear compatible with the reverse RM mechanism, although direct evidence remains to be garnered.
The recently discovered BREX (bacteriophage exclusion) system shares two genes with the Pgl systems (pglX and pglZ) but appears to employ a different defense strategy (8, 52). The BREX system protects bacteria from even a single-cycle phage burst. The abrogation of the phage DNA replication by BREX involves methylation of the host DNA at nonpalindromic sites, whereby only one strand is methylated, but not cleavage of the phage DNA. The exact mechanism of action remains unknown, but it has been shown that DNA methylation by PglX is essential for protection (52).
Phylogenetic analysis of PglZ identified six major branches in which PglZ is associated with different, although overlapping, sets of additional genes (52). Accordingly, it has been proposed that these defense systems be denoted BREX types 1 to 6. The two most common types are BREX, to be renamed BREX type 1, and the originally discovered Pgl, which will become BREX type 2.
Although key details remain to be elucidated, the BREX/Pgl class of defense mechanisms seems to follow the general principle of discrimination between self and nonself that is characteristic of the RM systems; i.e., discrimination between modified and unmodified DNA. However, downstream of the discrimination step, the mechanisms of RM and BREX/Pgl diverge, as the latter do not cleave the foreign DNA but rather inactivate it via unknown mechanisms.
Argonaute-Centered RNA/DNA-Guided Defense
The Argonaute-centered defense systems represent the branch of innate immunity that relies on guide RNA or DNA molecules for self versus nonself discrimination. This principle of nonself recognition fundamentally differentiates Argonaute-based defense from RM-type systems, which function by recognizing modifications that tag self or, less commonly, nonself DNA. The Argonaute proteins (Ago) have been initially characterized as the key players in eukaryotic RNA interference (RNAi). The first function of Ago characterized at the molecular level was that of a slicer; i.e., the RNase that cleaves the target RNA base paired with a small interfering (si)RNA (96, 97, 150). Shortly thereafter, it was shown that enzymatically inactive members of the Ago family complexed with microRNAs (miRNAs) reversibly suppress the translation of the target mRNAs without cleaving them (64).
Argonautes are large proteins of about 800 to 1,200 amino acids that contain, in addition to the PIWI endonuclease domain (RNase H fold), noncatalytic domains, namely, the PAZ (PIWI-Argonaute-Zwille), MID (middle), and N domains, as well as two linkers, L1 and L2 (21, 22, 129, 130, 157). The MID domain is essential for binding the 5′ end of the guide and is present in all Ago proteins. The PAZ domain, which adopts an oligosaccharide-binding (OB)-fold core typical of diverse nucleic acid–binding proteins, is not essential for guide binding but stabilizes the guide from the 3′ end. The N domain is not required for guide loading but contributes to the dissociation of the second, passenger strand of the loaded double-stranded RNA and to the target cleavage. Only the PIWI and MID domains are present throughout the Ago family, whereas the PAZ and N domains are missing in some family members.
Initially, Argonautes were described as highly conserved eukaryote-specific proteins (14, 161), but within a short time, prokaryotic homologs of eukaryotic Ago (henceforth, pAgo and eAgo, respectively) were discovered in many bacteria and archaea (5). However, the spread of pAgo across the diversity of prokaryotes is limited, with about one-third of the archaeal genomes and about 10% of the bacterial genomes encoding a member of this family (158). The structures of several pAgos have been solved, establishing the identities of the PIWI, PAZ, and MID domains and unexpectedly demonstrating that at least some pAgos preferentially bind guide DNA rather than RNA molecules (170, 185). For several years after the discovery of pAgos, their biological functions remained obscure. However, comparative analysis of the genomic neighborhoods of the pAgo genes has strongly suggested a role in defense (112). Many of the pAgo genes are embedded in defense islands, the regions of bacterial and archaeal genomes that are enriched for genes involved in various defense functions. Thus, it has been proposed that pAgos are DNA- and/or RNA-guided nucleases that recognize and cleave cognate (foreign) nucleic acids. Furthermore, genes encoding pAgo variants with inactivated PIWI domains are often adjacent to genes encoding other nucleases, leading to the hypothesis that these enzymatically inactive pAgos mediate target recognition via the guide DNA or RNA, after which the target is cleaved by the associated active nuclease.
The hypothesis on the defense function of pAgo has been experimentally validated, although the scope of the experiments has been limited. In vitro guide-dependent endonuclease activity has been demonstrated for pAgos from the bacteria Aquifex aeolicus (185) and Thermus thermophilus (144) and the archaea Methanocaldococcus jannaschii (176) and Pyrococcus furiosus (156). All three catalytically active pAgos employ single-stranded (ss)DNA guides but differ in their ability to cleave RNA or DNA. In contrast, the RNA-specific pAgo of the bacterium Rhodobacter sphaeroides, in which the catalytic center of the PIWI domain is disrupted (125), displayed no nuclease activity.
Defense functions have been demonstrated for the pAgo from R. sphaeroides (125) and T. thermophilus (157). The T. thermophilus pAgo restricts plasmid replication by cleaving the plasmid DNA using plasmid-derived small ssDNA guides. The mechanism of the guide generation is not understood in detail, but the involvement of the catalytic residues of the PIWI domain has been demonstrated (157). Thus, pAgo probably first shreds the plasmid DNA in a guide-independent (and, presumably, sequence-independent) manner and then becomes a target-specific nuclease after acquiring the guides. It is unclear what determines self/nonself discrimination at this first stage of the pAgo defense. For the R. sphaeroides pAgo, association with short RNAs that represent much of the bacterial transcriptome has been demonstrated (125). In addition, this pAgo is associated with ssDNA molecules complementary to the small RNAs, and this DNA population is enriched in foreign sequences, those from plasmids as well as mobile elements integrated into the bacterial chromosome. Apparently, in R. sphaeroides, pAgo samples degradation products of the bacterial transcriptome and then, via unknown mechanisms, preferentially generates complementary DNAs for foreign sequences that are used to repress the expression of the cognate elements. Whether or not the function of this catalytically inactive pAgo requires other nucleases remains to be determined. Nevertheless, the presence of pAgo within evolutionarily conserved operons with genes for nucleases and helicases (112, 158) implies complex organization of the pAgo-centered defense systems that remains to be investigated. In particular, such experiments should clarify the mechanisms employed by the pAgo-centered defense systems to generate the guide RNA and DNA molecules and discriminate between self and nonself sequences.
ADAPTIVE IMMUNITY: THE CRISPR-CAS SYSTEM
The CRISPR-Cas system employs a unique defense mechanism that involves incorporation of foreign DNA fragments into CRISPR arrays and subsequent utilization of processed transcripts of these inserts (spacers) as guide RNAs to cleave the cognate genome (54, 69, 83, 104, 114). Thus, CRISPR-Cas is a bona fide adaptive (acquired) immunity system, a function that had not been known to exist in prokaryotes. Moreover, CRISPR-Cas mediates inheritance of acquired characters, i.e., resistance to a virus or plasmid, and hence appears to be the most compelling demonstrated case of Lamarckian evolution (84). Apart from their role in antiviral defense that by now has been demonstrated in numerous experiments, CRISPR-Cas systems gave rise to the new generation of tools for genome editing and regulation. Thanks to this enormous practical utility, CRISPR research has turned into a burgeoning, highly dynamic field of microbiology and biotechnology that is covered, from different angles, in numerous recent reviews (11, 12, 20, 42, 43, 135, 173, 174); here we only briefly outline the functional and architectural diversity and comparative genomics of CRISPR-Cas and discuss likely scenarios for the evolution of the different types of CRISPR-Cas.
CRISPR-Cas systems show remarkable diversity of gene composition, genomic loci organization, and Cas protein sequences (106). Nevertheless, comprehensive comparative analysis has revealed major unifying themes in their evolution. These common trends include multiple, major contributions of MGEs; duplications of cas genes yielding functionally versatile effector complexes; and modular organization, with frequent recombination of the modules (106, 110, 113). The two main modules of the CRISPR-Cas systems comprise the suites of genes encoding proteins involved in adaptation (spacer acquisition) and effector functions, i.e., pre–CRISPR RNA (pre-crRNA) processing, and target recognition and cleavage. Additionally, various proteins involved in ancillary roles such as regulation of the CRISPR response and probably CRISPR-associated programmed cell death, can be assigned to a third, accessory module.
The CRISPR-Cas systems comprise two classes that differ with respect to the composition and complexity of the effector modules: Class 1 systems possess multisubunit effector complexes, whereas the effector modules of class 2 consist of a single, large protein, such as Cas9, Cas12, and Cas13 (106, 147). In contrast to the effector module, the composition of the adaptation module is nearly uniform across the diverse CRISPR-Cas systems. The adaptation module consists of Cas1 and Cas2, although in some CRISPR-Cas variants, additional proteins, such as the effectors themselves (e.g., Cas9) and accessory proteins (e.g., Cas4), interact or even form fusions with Cas1 or Cas2 and are also required for adaptation (2). Cas1 is the active integrase that catalyzes the protospacer excision from the target DNA and insertion into the CRISPR array, whereas Cas2 forms the structural scaffold of the adaptation complex (120, 121).
Comparative genomic analysis has revealed the likely ancestry of Cas1. Examination of the genomic context of cas1 homologs that are not associated with CRISPR-cas loci led to the discovery of a novel superfamily of self-synthesizing transposons, the casposons, so named because the Cas1 homolog they encode was predicted to function as the transposase (integrase) (91, 92). The integrase activity of the casposon-encoded Cas1 (dubbed the casposase) subsequently has been validated experimentally (58), and similar target site specificities of casposon integration and CRISPR spacer incorporation have been demonstrated (9). Although the currently identified casposons do not encode Cas2, some encode Cas4 and additional nucleases (92). It seems likely that the entire adaptation module and perhaps even additional Cas proteins have been contributed to the emerging CRISPR-Cas system by the ancestral casposon (82). Furthermore, the prototype CRISPR repeats and the leader sequence could have originated from a duplicated target site of the casposon (90). The origin of the CRISPR-Cas adaptation module from the integration machinery of a transposon indicates that the adaptive immunity systems in prokaryotes and eukaryotes evolved along parallel trajectories, through recruitment of unrelated MGEs (81).
The ancestry of the effector module is far less clear. Given that class 1 CRISPR-Cas systems are almost universally present in archaea and common in bacteria, whereas class 2 systems are an order of magnitude less abundant, the multisubunit effector complexes of class 1 are the most likely ancestral form (146). Despite the high diversity of Cas proteins, the core subunits of the class 1 effector complexes largely consist of multiple variants of the same domain, the RNA recognition motif (RRM) (110). Some of the RRM domains possess nuclease activity, whereas others are nonenzymatic RNA-binding proteins. This construction of the effector complexes from ultimately homologous, even if highly diverged, building blocks implies evolution by gene duplication, with subsequent extensive diversification driven by the host-parasite arms race. Conceivably, the ultimate ancestor of the core Cas proteins could have been an RRM domain–containing nuclease, such as Cas10, that gave rise to the extant multitude of active and inactivated versions (110). Subsequent evolution of the CRISPR-Cas systems also involved recruitment of additional proteins, such as the helicase-nuclease Cas3 in the type I systems. What was the function of the original effector CRISPR-Cas module, before the fusion with the adaptation module, supposedly brought about by a casposon? The previously proposed possibility is that the effector module evolved from an ancestral innate immunity system that acquired the adaptation capability following the integration of a casposon next to the innate immunity locus (82). So far, however, innate immunity systems homologous to CRISPR-Cas effector complexes have not been identified. Therefore, an alternative scenario would derive the class 1 effector module from within the ancestral casposon, which in this case would be postulated to have encoded at least one RRM domain–containing protein endowed with nuclease activity.
The provenance of class 2 effector modules has been established with much greater confidence (146, 147). The type II and type V effectors (Cas9 and Cas12, respectively) appear to derive from the extremely abundant but poorly characterized transposon genes known as tnpB, which encode nucleases that belong to the RuvC-like family of RNase H fold nucleases. The role of TnpB in the transposon life cycle is unclear, as it is not required for transposition (131). In the type II and type V-A effectors, this nuclease cleaves the nontarget DNA strand, whereas the target strand (complementary to the crRNA) is cleaved by an additional nuclease, the identity of which differs between Cas9 and Cas12a (34, 181). Remarkably, in the case of the type V-B effectors, both target and nontarget DNA strands are cleaved by the RuvC-like endonuclease domain, which undergoes a major conformational change triggered by the initial, nontarget strand cleavage (98, 182). Notably, the effector nucleases of type V-A and type VI are also responsible for the processing of pre-crRNA yielding mature crRNA guides; the catalytic domains and sites responsible for crRNA maturation remain poorly characterized but are clearly distinct from those involved in target cleavage (37, 44, 98). These findings are in sharp contrast to processing in type II, which involves RNase III, a ubiquitous bacterial enzyme (24), and emphasize the striking diversity of CRISPR-Cas molecular mechanisms.
For the type II CRISPR-Cas effectors, Cas9, a distinct family of TnpB homologs, denoted IscB, has been identified as the direct ancestor, as indicated by the high level of sequence similarity and the presence of an HNH nuclease domain inserted into the RuvC-like domain (71). For the type V effectors, the direct ancestors are difficult to identify, but different subfamilies of TnpB appear to have given rise to different subtypes, as indicated by sequence similarity and phylogenetic analysis (146, 147). The type VI effectors, Cas13, are unrelated to those in other CRISPR-Cas types and contain two HEPN (higher eukaryotes and prokaryotes nucleotide-binding) domains, which cleave RNA targets (1, 3, 149). As with type V effectors, the exact ancestors of these proteins are difficult to pinpoint, but it appears likely that either HEPN domain–containing Cas proteins of class 1 CRISPR-Cas systems, such as Csx6 and Csn1, or a distinct HEPN domain–containing toxin could be implicated (147). The current evolutionary scenario posits that class 2 CRISPR-Cas systems evolved when mobile elements encoding ancestors of class 2 effectors, most commonly TnpB nucleases, integrated near orphan CRISPR arrays or displaced class 1 effector operons (147). Type II, type V, and type VI systems (and most likely different types and subtypes of type V) evolved independently on several occasions, as indicated by their distinct evolutionary affinities with different groups of TnpB or HEPN domain–containing proteins (147). Thus, the evolutionary history of class 2 systems is largely a story of the second major contribution (after the casposons) of mobile elements to the evolution of CRISPR-Cas adaptive immunity.
CRISPR-Cas systems are present in nearly all archaeal genomes but only in about 30–40% of bacterial genomes (18, 106). The fate of CRISPR-Cas systems, i.e., maintenance or loss from a microbial population, appears to be determined by the balance between the benefits of adaptive immunity for efficient host defense against viruses and other MGEs, and the cost incurred by these systems that is thought to be associated with both autoimmunity and abrogation of HGT by CRISPR-Cas (86). Mathematical models of virus-host coevolution suggest that this balance hinges on the diversity of viruses encountered by a host population, with the greatest benefit of adaptive immunity associated with moderate diversity (171) as well as the host population size, because CRISPR-Cas systems are predicted to be more efficacious in smaller populations (67). It appears likely that both conditions are met in microbial communities that thrive under extreme (in particular, hyperthermal) conditions, resulting in the ubiquitous presence of CRISPR-Cas in archaeal hyperthermophiles (67, 171). However, definitive study of virus-host coevolution requires detailed analysis of the dynamics of both populations in nature.
DORMANCY INDUCTION AND PROGRAMMED CELL DEATH: TOXINS-ANTITOXINS AND ABORTIVE INFECTION
Prokaryotic TA systems were originally described as addictive modules that are carried by plasmids and ensure their persistence in microbial cell lineages (49, 50). TA systems currently are partitioned into six types, with type I and type II systems the most abundant; the latter are by far the best characterized (23). The toxin component of all TA systems is a protein that kills cells if expressed above a certain level; the antitoxin reversibly inactivates the toxin and/or regulates its expression, thereby preventing cell killing (127). In type I and type III TA systems, the antitoxin is a small RNA that downregulates expression of the respective toxin gene (16), whereas in type II TA systems, the antitoxin is a protein that forms a complex with the toxin, in which the toxin is reversibly inactivated. Unlike the toxins, the antitoxins typically are metabolically unstable; unless the antitoxin is continuously expressed, the free toxin can accumulate in amounts sufficient to kill a cell (50, 51, 55, 168). After division, a daughter cell that fails to receive a copy of a plasmid carrying a TA gene module will die because the antitoxin will be depleted before the toxin (115). Given that tight coregulation of the toxin and antitoxin resulting in stoichiometric production of the two components of the TA systems is required for cell survival, these systems most often form two-gene operons; typically, the toxins (and in type II TA systems, the antitoxins as well) are small, highly compact proteins, apparently of the minimum size required to carry out the respective biochemical activities (108) (see below).
Analysis of multiple bacterial and archaeal genomes has shown that numerous TA systems are encoded not only on plasmids but also on chromosomes (50, 159). This surprising finding triggered a lively debate on potential cellular functions of the chromosomal TA systems and prompted comparative genomic and experimental studies that resulted in the discovery of numerous new TA systems. These discoveries and the current understanding of the biological roles of TA systems are summarized in several recent reviews (49, 55, 159, 180). Although the biology of the TA systems is far from fully understood, the prevailing view is that they provide a mechanism for cell persistence to cope with various stress conditions, in particular virus infection (128, 167, 168). A more contentious issue is whether or how often the TA systems cause programmed cell death, or altruistic cell suicide (see below).
The type I toxins are small membrane proteins that permeabilize cell membranes (35, 46). In contrast, the majority of type II toxins target different components of the translation systems, especially mRNA that is cleaved by toxin nucleases, known as interferases (29, 179). However, other targets of type II toxins have been identified as well, such as DNA gyrase (30) and the cell division GTPase FtsZ (162). Because type I toxins have never been implicated in virus resistance or other defense functions and are not frequently observed in defense islands, we do not discuss them further in this review. Instead, we focus on type II TA systems and discuss the results of the recent efforts to identify new TA families using comparative genomic approaches.
Three computational approaches for prediction of new TA systems have been developed: (a) guilt by association, that is, prediction of new toxins or antitoxins based on linkage, in bacterial and archaeal genomes, to genes that belong to known antitoxin or toxin families (95, 108); (b) identification of gene pairs with characteristic features of TA systems, such as tight linkage of genes encoding small proteins, propensity for HGT, and presence on plasmids or within genomic islands with other defense genes (108, 111); and (c) statistical analysis of whole-genome sequencing clones aimed at identification of genes that are unclonable (toxic) in E. coli (74).
Recent comprehensive studies revealed numerous genes that are unclonable in E. coli but do not meet the definition of TA systems, including many metabolic enzymes and informational genes, such as ribosomal proteins (74, 142). Although not all of these genes form two-gene operons that are typical of TA systems, these findings imply that dosage imbalance or toxicity of an intermediate metabolite can result in apparent toxicity of a gene that can be offset by tight regulation or coexpression of an enzyme utilizing the toxic product, thus masquerading as a TA system. Thus, gene toxicity is a phenomenon that extends beyond the typical TA systems, so that identification of the latter requires additional experimentation and/or analysis of the domain architectures of the respective proteins. Conversely, searches of genome sequences for novel TA systems identified numerous stand-alone homologs of toxins (108, 109). Such solo toxins might belong to still uncharacterized TA systems in which the toxin and the antitoxin are tightly coregulated despite the lack of the typical operon organization, or are tightly regulated to minimize expression, or perhaps lack toxicity owing to some subtle structural changes.
Systematic computational and experimental search for TA systems resulted in the identification of many abundant but experimentally uncharacterized variants (74, 108, 109, 142). One of these consists of a pair of genes that are among the most common genes in hyperthermophilic archaea and encode a HEPN domain–containing protein and a small, so-called minimal nucleotidyltransferase (MNT) (4). The HEPN domain–containing protein is predicted to function as an RNA-cleaving toxin, whereas the MNT is the predicted antitoxin (108). The HEPN-MNT module shows all the typical features of TA systems (108), and the toxic effect of the HEPN protein along with the antitoxin activity of the MNT indeed have been experimentally demonstrated in the bacterium Shewanella oneidensis (183). Nevertheless, the molecular mechanism of this system—particularly, the role of the nucleotidyltransferase activity of the antitoxin—remains unclear.
ABI involves a distinct variety of TA systems. The ABI mechanisms abrogate virus infection at different stages, often by causing death of infected cells and thus precluding virus spread (28, 94). Some of the ABI systems are two-component modules that display the typical features of TA systems, in particular those of type III systems. As with other TA systems, although numerous ABI systems have been identified by genetic approaches, molecular mechanisms for only a small minority of these are understood (94). Many of the ABI systems share domains with TA systems, and the HEPN domain in particular is among the most prominent (109). These ABI systems are predicted to function as TA targeting translation. Notably, several other, membrane-associated, ABI systems cause membrane leakage similar to type I TA systems (33, 36). Several ABI systems, including AbiU1, AbiL, and AbiR, are often associated and might interact with RM modules (109, 111). Another frequent component of ABI systems is reverse transcriptase, a hallmark protein of MGEs. In the case of ABI, the reverse transcriptase catalyzes nontemplated synthesis of random sequence DNA that remains covalently attached to the protein and contributes to abortive infection (169).
In summary, TA and related ABI modules are among the most common and versatile defense systems in bacteria and archaea. These modules are often carried by plasmids and themselves represent a distinct type of mobile element (see below). TA systems are nearly ubiquitous in bacteria and archaea, but they have not been detected in most bacterial endosymbionts or, among archaea, in Thermoplasmatales; several methanotrophs with small genomes; and the only known group of archaeal symbionts, the Nanoarchaeota (95, 108). The distribution of TA systems across phyla is clearly nonuniform, with many systems significantly over- or underrepresented in various taxa (95, 108) (Table 1). Unlike other defense systems, the occurrence of TA systems in genomes scales superlinearly with genome size (Figure 2), possibly reflecting the comparatively high rate of plasmid assimilation by large genomes. Genomic occurrence of TA systems shows exceptional variability even in closely related genomes, presumably owing to plasmid-mediated transfer (95, 108). Because of the combinatorial reassortment of toxins and antitoxins, the TA systems form a strongly connected network in which the main hubs, i.e., common domains linked to many partners, are the RNase toxins PIN and RelE and antitoxins containing the ribbon-helix-helix (RHH) and helix-turn-helix (HTH) DNA-binding domains (109). Genome analysis has led to the identification of numerous stand-alone toxin and antitoxin genes that account for over 50% of the genes in the largest families. These findings suggest that in many cases, the interaction between toxins and antitoxins occurs in trans and the required tight coregulation of the respective genes is secured by still unknown mechanisms (95, 108).
Virtually all currently characterized ABI systems come from one group of model organisms, the lactococci (28). In all likelihood, this is only the tip of the proverbial iceberg, whereas the true diversity of this type of defense module in bacteria and archaea remains to be revealed. Indeed, analysis of defense islands leads to the identification of numerous uncharacterized gene families that could be new ABI-like defense systems (111).
GENERAL TRENDS IN THE EVOLUTION OF DEFENSE SYSTEMS: EXTENSIVE LOSS, GAIN AND TRANSFER, AND CLUSTERING OF DEFENSE GENES IN GENOMIC ISLANDS
Genome-wide analysis of defense systems reveals notable general trends. Reconstruction of gene loss and gain history in many groups of closely related bacterial and archaeal genomes has shown that, after the MGEs, the defense genes are the most evolutionarily dynamic functional class of genes (133, 134). Both gain and loss rates of defense genes are significantly higher than the respective mean rates across all gene categories, although they generally follow the overall patterns of microbial gene dynamics. Thus, gene loss rates are typically two to three times greater than gain rates, most likely because genes are lost in a clock-like manner, whereas gene gain appears to occur in spurts. There are, however, notable exceptions, such as bacteria in the genus Shewanella that appear to be actively gaining defense systems (134). A detailed analysis of individual cases of gain and loss of defense systems has shown that such events are often associated with and possibly mediated by concomitant loss or gain of MGEs (134).
A second, perhaps related major trend in the evolution of defense systems is their frequent clustering in distinct genomic regions that have been denoted defense islands, by analogy with pathogenicity and symbiotic islands (Figures 4, 5) (111). The trend for island formation has been found to be statistically highly significant for all classes of defense systems, with the exception of CRISPR-Cas. The majority of the defense islands are small and only include a few genes, but some reach over 100 (Figure 4). Many islands combine diverse defense systems, such as various RM and TA modules, and might also include genes involved in novel mechanisms, as illustrated in Figure 5 by the gene pair encoding a predicted ATPase and an HNH family nuclease.
Figure 4.
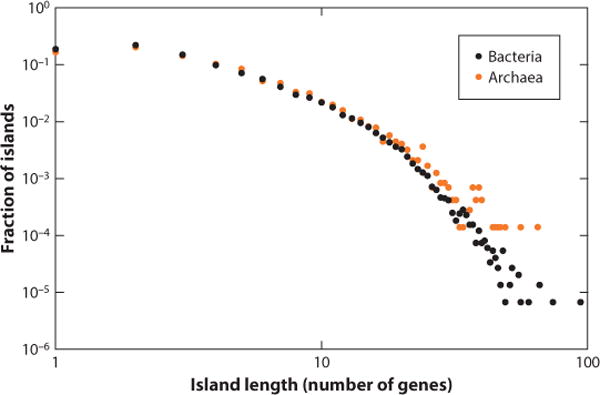
Length distributions of defense islands in bacterial and archaeal genomes.
Figure 5.
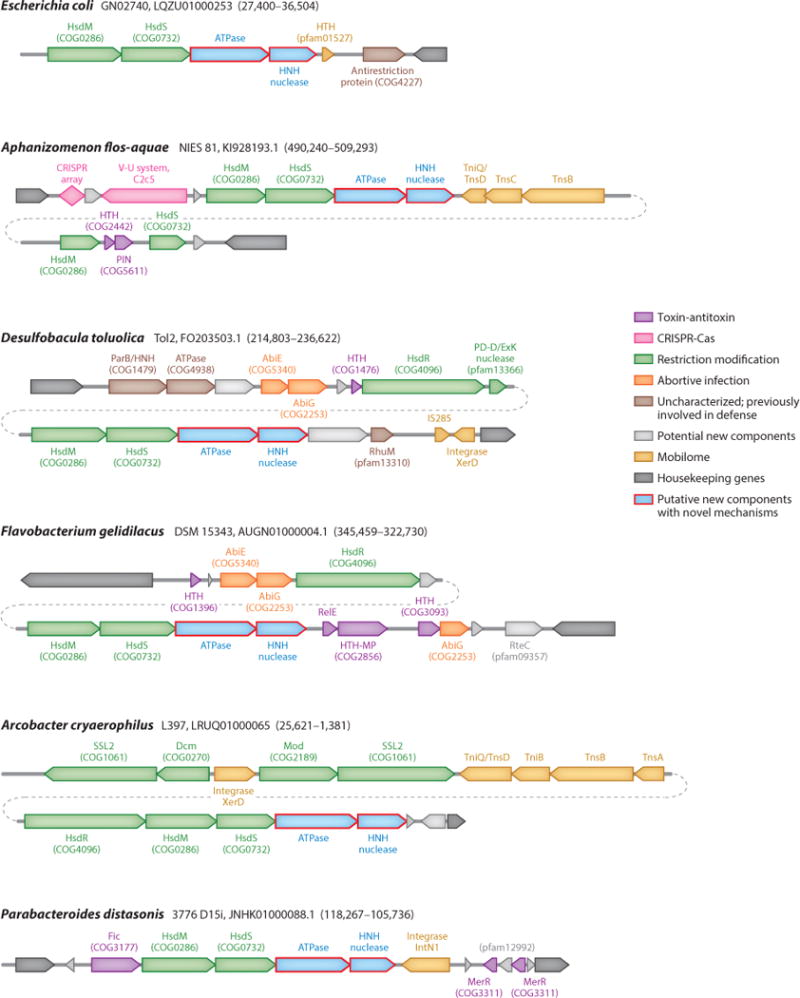
Defense islands as a potential source of novel defense systems. For each island, the genome name, the respective nucleotide sequence ID, and genomic coordinates are provided. Block arrows indicate the direction of tAranscription, roughly to scale. PIN, HNH, and PD-D/ExK are distinct nuclease families. Abbreviations: HTH, helix-turn-helix; HTH-MP, HTH domain fused to Zn-dependent metalloprotease.
The clustering of defense genes most likely reflects two evolutionary factors that are quite different in character but have similar effects (109). The first is a garbage pile effect, whereby defense genes and MGEs are often attracted to the same regions in genomes simply because these genes are nonessential, such that gain or loss of genetic material there is unlikely to be strongly deleterious. However, a different cause behind the emergence and especially persistence of defense islands is likely to be selection for colocalization of functionally interacting defense systems, as discussed in the next section. Regardless of the relative contributions of different evolutionary forces, the wide spread of defense islands in microbial genomes creates potential for prediction of novel defense systems via the guilt by association principle.
INTERACTIONS BETWEEN DEFENSE STRATEGIES: COUPLING BETWEEN IMMUNITY AND DORMANCY INDUCTION/PROGRAMMED CELL DEATH
The genomic loci encoding immune systems often also include dedicated programmed cell death modules, such as TA modules, and some proteins are shared by the two types of defense systems. The CRISPR-Cas systems, the most complex form of prokaryotic defense, present the most remarkable cases. One of the key proteins in the first, adaptation, phase of the CRISPR response, Cas2, is homologous to the toxins of the VapD family of mRNA interferases (103, 104). The primary role of Cas2 in CRISPR-Cas is that of a structural scaffold of the adaptation complex, in which Cas1 is the endonuclease component (2, 120, 121, 152). The interferase catalytic site is conserved in some, though not all, Cas2 proteins but is not required for adaptation (120). Thus, at least in some CRISPR-Cas systems, Cas2 might play a secondary role as an RNase, possibly a toxin (101). Indeed, non-sequence-specific nuclease activity of several Cas2 proteins against both DNA and RNA, but typically with a preference for RNA substrates, has been demonstrated although catalytically active Cas2 proteins do not appear to be toxic when overexpressed in E. coli (10, 32, 53, 70, 118). The conservation of the catalytic site of Cas2 implies that the RNase activity of this protein is functional in a subset of CRISPR-Cas systems, and interferase activity remains a distinct possibility.
Many CRISPR-Cas systems also encompass additional nucleases, in particular (predicted) RNases of the HEPN superfamily (3, 102). The RNase activity of two of these proteins, Csm6 and Csx1, has been demonstrated experimentally (39, 119, 145). Most of the HEPN-containing Cas proteins additionally contain the CARF (CRISPR-associated Rossmann fold) that is predicted to bind ligands, most likely nucleotides, and perform signaling functions (102). Notably, the Csm6 protein, which consists of a CARF and a HEPN domain, is not required for the type III-B CRISPR-Cas interference (39), suggesting a different, accessory function for this protein. As pointed out above, the HEPN domain–containing RNases are the most abundant among the toxin components of TA modules in archaea and are common in bacteria as well (3, 108). Thus, the HEPN domain–containing Cas proteins also might possess toxin activity that could be masked by another domain of the same protein or by a distinct Cas protein serving as the antitoxin. In some CRISPR-Cas systems, the CARF domain is fused to predicted nucleases not containing the HEPN domain, in particular, Cas4 homologs (102). An intriguing possibility is that these diverse CARF-linked nucleases are toxins regulated by the CARF domain through ligand binding.
CRISPR-associated toxin activity has been experimentally demonstrated for the Csa5 protein of the type I-A CRISPR-Cas system from the archaeon Sulfolobus solfataricus. Infection of S. solfataricus with the SIRV2 virus induced expression of Csa5 to the toxic level and resulted in cell death, suggesting that the toxicity of this protein represents a programmed cell death response to virus infection (56). The Csa5 protein is the _α_-helical small subunit of the Cascade CRISPR RNA-processing complex of type I-A and appears to lack nuclease activity (31). Thus, as noted above for the TA modules in general, CRISPR-associated toxicity might involve different, still uncharacterized mechanisms.
Apart from the CRISPR-Cas systems, genomic analysis demonstrates association of TA modules with innate immunity (in particular, RM) loci (101, 109) (Figure 5). These observations have prompted the hypothesis on functional coupling between immunity and programmed cell death/dormancy (101). Two versions of such coupling were considered. First, programmed cell death can be viewed as the strategy of last resort, to which an infected cell turns when it senses the impending failure of defense systems to stop virus reproduction. An alternative, albeit arguably less plausible, scenario is that, faced with intense virus reproduction, the immune system turns on the dormancy induction machinery, not only protecting the surrounding cells but also giving the infected cell itself a chance to recover once the virus clears. The two strategies might merge, considering that there is never a guarantee that a cell will reemerge from dormancy. The common presence of genes encoding CARF fused with diverse nucleases in CRISPR-cas loci (102) (Figure 2) implies that the CARF domain functions as a sensor of the imminent defeat of the immune system in the battle against the virus, in response to a hypothetical alarmone (most likely, a nucleotide derivative) that remains to be identified (88). Although the immunity-suicide coupling hypothesis was conjured largely on indirect evidence, an experimentally well-characterized case of such coupling is presented by a bacterial antiphage system which includes two HEPN domain–containing RNases, RloC and PrrC, that exert a toxic effect through tRNA cleavage (13, 73, 75, 165).
The recent comprehensive search for genomic loci that encode large proteins containing putative nuclease domains that could function as class 2 CRISPR-Cas effectors has revealed, arguably, the most direct links between microbial immunity and programmed cell death so far discovered (1, 146, 147, 149). Type VI effector proteins contain two HEPN domains predicted to possess RNase activity (146, 147). An RNase activity that requires both HEPN domains indeed has been demonstrated for the type VI-A and VI-B effectors (Cas13a, Cas13b) (1, 149). The type VI effectors provide efficient protection against the RNA bacteriophage MS2, but in addition, when primed with a cognate RNA they turn into a promiscuous RNase that cleaves any RNA molecules present in the reaction mix with little sequence specificity (1, 149). A significant decrease in bacterial viability was observed when Cas13a was coexpressed with the cognate RNA, most likely indicative of dormancy induction (1). Given that RNA bacteriophages are minor components of the bacterial virosphere (80), the principal function of type VI CRISPR-Cas systems is most likely defense against DNA phages through the toxic effect triggered by the recognition of a cognate phage transcript and resulting in dormancy or programmed cell death.
Whether the cell that turns on the self-afflicting program kills itself or goes into dormancy, the decision it faces is the same: The cell has to employ a built-in sensor to predict the outcome of infection and act accordingly (88). If the sensor module predicts that the viral onslaught is manageable, the immune system is mobilized to full capacity. In contrast, a dire prognosis activates self-destruction. The sensors and the signals they register differ between defense systems. The antiphage HEPN-containing RNases RloC and PrrC sense cell damage directly, through double-stranded DNA break, and the concentration of dTTP, which serves as an alarmone during phage infection (75, 76, 93). The ligand(s) sensed by the CARF domain in the case of CRISPR-Cas systems remain to be identified, but it is highly likely that CARF domains (102) function as toggles between the immune and self-afflicting responses. The nature and modes of the switching signals, their threshold values and what determines these, and whether these features specifically depend on the character of virus-host interaction are all intriguing directions for further study.
Type VI CRISPR-Cas systems are a special and so far the most obvious case of immunity-suicide coupling. These systems appear to short-circuit the typical defense relay by effectively skipping the damage-sensing step and employing the main immune effector as the suicide weapon as well (Figure 3, Table 1). For the Cas13 proteins to switch to the promiscuous RNA cleavage mode, the only signal required seems to be the target recognition (1, 149). Still, sensing the target RNA concentration, which would reflect multiplicity of infection and/or the expression rate of the virus genome, by the Cas13 proteins themselves could play a role even in this case. Conceivably, more elaborate defense strategies that involve dedicated sensors, such as class 1 CRISPR-Cas, outcompete the simple ones where the self-destruction program is activated at the first alarm signal, perhaps explaining the relative rarity of type VI systems among bacteria (147).
MOBILE GENETIC ELEMENTS AND DEFENSE SYSTEMS IN BACTERIA AND ARCHAEA
A major theme emerging in the studies on the evolution of defense systems is their close link to MGEs. This connection might at first seem paradoxical in that defense machineries share components and have similar properties with the very elements against which they defend the host. However, there seems to be a clear evolutionary logic behind these relationships. First, major classes of bacterial and archaeal defense systems, in particular RM and TA modules, themselves possess properties of MGEs (77, 116, 117). These defense systems do not encode proteins involved in their own replication or transposition and therefore do not qualify as aggressive MGEs, such as viruses or transposons. Nevertheless, the extensive horizontal mobility of these modules is immediately apparent from phylogenetic analysis. Furthermore, the RM and especially TA modules often reside on plasmids and thus employ a piggyback mode of dissemination. Most importantly, although these defense modules lack means for active replication and/or transposition, they do possess the addiction mechanisms that ensure their own persistence (66, 122).
Such mechanisms are outlined above and involve cell killing by the toxin when a cell does not receive a copy or in other situations when the antitoxin gene is lost or inactivated. Given the tight coregulation of the toxin and antitoxin genes, along with the specific properties of the proteins themselves, such as the instability of type II antitoxins, it appears clear that addiction mechanisms are adaptations for a distinct type of persistence strategy, and accordingly, these defense systems qualify as selfish MGEs. From this perspective, the capacity of the RM and TA modules to kill virus-infected cells can be viewed as a kin selection strategy that rescues neighboring host cells in the population that are likely to carry the same or a closely related MGE. The host defense, then, comes across as a by-product of MGE evolution. Importantly, although RM systems are traditionally discussed in the context of defense whereas TA systems are more often viewed as MGEs, the mechanisms and lifestyles of the two classes of elements are very similar, to the extent that they can be justifiably analyzed within the same toxin-antitoxin framework (88, 115).
The second aspect of the defense-MGE connection is the recurrent recruitment of MGEs, or more typically, individual MGE genes, for defense functions. As discussed above, CRISPR-Cas systems are the best case in point. Casposons apparently gave rise to the adaptation module, whereas TnpB-encoding transposons are the ancestors of the effector genes of most of class 2 systems (82, 90, 147). Additionally, some type III CRISPR-Cas loci have recruited reverse transcriptases, most likely from group II introns, providing for spacer acquisition by reverse transcription of RNA molecules (148). The recruitment of MGEs by defense systems seems to stem from two factors. First, the very mobility of these elements, combined with the existence of defense islands that are usually tolerant to insertion of genetic material, facilitates utilization of MGEs. Second, enzymes that are central to the lifestyle of MGEs, such as transposases and various other nucleases, are readily utilizable for defense function. Thus, such enzymes serve as “guns for hire” that are alternately employed for offense, defense, and counterdefense, depending on the highest bid (81). Indeed, plasmid-encoded TA modules are effectively counterdefense devices that prevent the host population from purging the plasmid, and counterdefense functions of CRISPR-Cas systems recruited by phages have been demonstrated as well (143).
CONCLUDING REMARKS
Defense mechanisms in bacteria, in particular RM systems, have been studied for decades. However, the expanding comparative genomic analysis followed by intensive experimental testing of predictions has led to both quantitative and qualitative expansion of the prokaryotic defense repertoire. It has become clear that, in addition to systems that function via protein–nucleic acid recognition, such as RM and TA systems, bacteria and archaea widely employ small guide RNAs and DNAs, as demonstrated by the discovery of pAgo-based and CRISPR-based immune mechanisms (79).
Discovery of the CRISPR-Cas mechanism led to the fundamental realization that adaptive (acquired) immunity is not a prerogative of “higher” animals, such as vertebrates, but rather an ancient mode of defense that is widespread in prokaryotes. Moreover, CRISPR-Cas presents the best known case for heritable acquired immunity that appears to qualify as a Lamarckian mode of evolution (84, 87). Thus, it has been speculated that all organisms possess some forms of both innate and acquired immunity (137).
Certain general principles of defense evolution are becoming apparent. The defense systems can be naturally partitioned on a plane bounded by the principle of target recognition on one axis and the existence of immune memory (innate versus acquired immunity) on the other (Table 2). An intriguing question, then, is whether acquired immunity systems based on protein–nucleic acid or protein-protein recognition remain to be discovered. Taken together, the findings discussed here show that the versatility of prokaryotic defense mechanisms matches that in eukaryotes. The analysis of defense islands, and more generally, of the dark matter in bacterial and archaeal genomes (8, 107), has led to the identification of many gene clusters, some of which contain proteins or domains that likely participate in defense. However, the mechanisms of such defense cannot be easily predicted by analogy with the well-characterized defense systems, suggesting that novel principles or at least major variations on known ones remain to be discovered.
Table 2.
The prokaryotic defense space
| Interaction defining specificity | |||
|---|---|---|---|
| RNA/DNA–DNA/RNA | Protein–RNA/DNA | Protein–protein | |
| Innate immunity | pAgo-centered defense | RM | Receptor escape |
| Adaptive immunity | CRISPR-Cas | ? | Receptor phase variation? |
| Dormancy/PCD | ? | TA/ABI | TA with protease, kinase toxins |
The defense strategy adopted by infected cells hinges on a life-or-death decision that is made on the basis of information gathered and processed by dedicated sensors. Depending on the damage level measured by such sensors, either the immunity program or dormancy/programmed cell death is turned on (88). Admittedly, this perspective on antivirus defense is speculative, and the study of damage sensors and the signals they recognize, in particular alarmones, can be expected to develop into a rich research program.
Another major theme in the evolution of defense is the close and multifaceted relationship between defense systems and MGEs. Some of them, such as RM and TA systems, possess defining features of MGEs, whereas others, such as CRISPR-Cas, evolved via recruitment of MGEs. The notion of guns for hire that can be employed for either offense (in MGEs) or defense (81) implies that additional cases of such recruitment exist and merit systematic investigation.
Finally, the very nature of the enzymatic activities (such as those of transposases and other sequence-specific nucleases) employed by both MGEs and defense systems makes them top candidates for the development of genome editing tools. Two generations of such tools, based first on RM and then on CRISPR-Cas, have sequentially transformed laboratory practice. Further study of microbial defense systems might eventually lead to new breakthroughs in biotechnology.
SUMMARY POINTS.
- Bacteria and archaea are engaged in an incessant arms race with parasitic genetic elements, and nearly all encode multiple defense systems.
- Prokaryotic defense systems can be classified as innate immunity, adaptive immunity, and dormancy or programmed cell death induction.
- Defense systems are often encoded in genomic islands.
- Some defense systems show features of mobile genetic elements, whereas others recruit genes from mobile elements for defense functions.
- There seems to be a coupling between immunity and dormancy/programmed cell death, so that a cell chooses one of the two defense strategies by sensing the degree of DNA damage caused by infection.
Acknowledgments
The authors thank Sergey Shmakov for expert technical assistance. The authors’ research is supported by intramural funds of the US Department of Health and Human Services (to National Library of Medicine).
Footnotes
*
This is a work of the U.S. Government and is not subject to copyright protection in the United States.
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Abudayyeh OO, Gootenberg JS, Konermann S, Joung J, Slaymaker IM, et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016;353:aaf5573. doi: 10.1126/science.aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amitai G, Sorek R. CRISPR-Cas adaptation: insights into the mechanism of action. Nat Rev Microbiol. 2016;14:67–76. doi: 10.1038/nrmicro.2015.14. [DOI] [PubMed] [Google Scholar]
- 3.Anantharaman V, Makarova KS, Burroughs AM, Koonin EV, Aravind L. Comprehensive analysis of the HEPN superfamily: identification of novel roles in intra-genomic conflicts, defense, pathogenesis and RNA processing. Biol Direct. 2013;8:15. doi: 10.1186/1745-6150-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aravind L, Koonin EV. DNA polymerase beta-like nucleotidyltransferase superfamily: identification of three new families, classification and evolutionary history. Nucleic Acids Res. 1999;27:1609–18. doi: 10.1093/nar/27.7.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aravind L, Watanabe H, Lipman DJ, Koonin EV. Lineage-specific loss and divergence of functionally linked genes in eukaryotes. PNAS. 2000;97:11319–24. doi: 10.1073/pnas.200346997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aziz RK, Breitbart M, Edwards RA. Transposases are the most abundant, most ubiquitous genes in nature. Nucleic Acids Res. 2010;38:4207–17. doi: 10.1093/nar/gkq140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barrangou R. CRISPR-Cas systems and RNA-guided interference. Wiley Interdiscip Rev RNA. 2013;4:267–78. doi: 10.1002/wrna.1159. [DOI] [PubMed] [Google Scholar]
- 8.Barrangou R, van der Oost J. Bacteriophage exclusion, a new defense system. EMBO J. 2015;34:134–35. doi: 10.15252/embj.201490620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beguin P, Charpin N, Koonin EV, Forterre P, Krupovic M. Casposon integration shows strong target site preference and recapitulates protospacer integration by CRISPR-Cas systems. Nucleic Acids Res. 2016;44:10367–76. doi: 10.1093/nar/gkw821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beloglazova N, Brown G, Zimmerman MD, Proudfoot M, Makarova KS, et al. A novel family of sequence-specific endoribonucleases associated with the clustered regularly interspaced short palindromic repeats. J Biol Chem. 2008;283:20361–71. doi: 10.1074/jbc.M803225200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhaya D, Davison M, Barrangou R. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet. 2011;45:273–97. doi: 10.1146/annurev-genet-110410-132430. [DOI] [PubMed] [Google Scholar]
- 12.Bikard D, Marraffini LA. Innate and adaptive immunity in bacteria: mechanisms of programmed genetic variation to fight bacteriophages. Curr Opin Immunol. 2012;24:15–20. doi: 10.1016/j.coi.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 13.Blanga-Kanfi S, Amitsur M, Azem A, Kaufmann G. PrrC-anticodon nuclease: functional organization of a prototypical bacterial restriction RNase. Nucleic Acids Res. 2006;34:3209–19. doi: 10.1093/nar/gkl415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bohmert K, Camus I, Bellini C, Bouchez D, Caboche M, Benning C. AGO1 defines a novel locus of Arabidopsis controlling leaf development. EMBO J. 1998;17:170–80. doi: 10.1093/emboj/17.1.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bourniquel AA, Bickle TA. Complex restriction enzymes: NTP-driven molecular motors. Biochimie. 2002;84:1047–59. doi: 10.1016/s0300-9084(02)00020-2. [DOI] [PubMed] [Google Scholar]
- 16.Brantl S, Jahn N. sRNAs in bacterial type I and type III toxin-antitoxin systems. FEMS Microbiol Rev. 2015;39:413–27. doi: 10.1093/femsre/fuv003. [DOI] [PubMed] [Google Scholar]
- 17.Brown CT, Hug LA, Thomas BC, Sharon I, Castelle CJ, et al. Unusual biology across a group comprising more than 15% of domain Bacteria. Nature. 2015;523:208–11. doi: 10.1038/nature14486. [DOI] [PubMed] [Google Scholar]
- 18.Burstein D, Sun CL, Brown CT, Sharon I, Anantharaman K, et al. Major bacterial lineages are essentially devoid of CRISPR-Cas viral defence systems. Nat Commun. 2016;7:10613. doi: 10.1038/ncomms10613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Butterer A, Pernstich C, Smith RM, Sobott F, Szczelkun MD, Toth J. Type III restriction endonucleases are heterotrimeric: comprising one helicase-nuclease subunit and a dimeric methyltransferase that binds only one specific DNA. Nucleic Acids Res. 2014;42:5139–50. doi: 10.1093/nar/gku122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carroll D. A CRISPR approach to gene targeting. Mol Ther. 2012;20:1658–60. doi: 10.1038/mt.2012.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cerutti H, Casas-Mollano JA. On the origin and functions of RNA-mediated silencing: From protists to man. Curr Genet. 2006;50:81–99. doi: 10.1007/s00294-006-0078-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cerutti L, Mian N, Bateman A. Domains in gene silencing and cell differentiation proteins: the novel PAZ domain and redefinition of the Piwi domain. Trends Biochem Sci. 2000;25:481–82. doi: 10.1016/s0968-0004(00)01641-8. [DOI] [PubMed] [Google Scholar]
- 23.Chan WT, Espinosa M, Yeo CC. Keeping the wolves at bay: antitoxins of prokaryotic type II toxin-antitoxin systems. Front Mol Biosci. 2016;3:9. doi: 10.3389/fmolb.2016.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Charpentier E, Richter H, van der Oost J, White MF. Biogenesis pathways of RNA guides in archaeal and bacterial CRISPR-Cas adaptive immunity. FEMS Microbiol Rev. 2015;39:428–41. doi: 10.1093/femsre/fuv023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chaturongakul S, Ounjai P. Phage-host interplay: examples from tailed phages and gram-negative bacterial pathogens. Front Microbiol. 2014;5:442. doi: 10.3389/fmicb.2014.00442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen F, Zhang Z, Lin K, Qian T, Zhang Y, et al. Crystal structure of the cysteine desulfurase DndA from Streptomyces lividans which is involved in DNA phosphorothioation. PLOS ONE. 2012;7:e36635. doi: 10.1371/journal.pone.0036635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chinenova TA, Mkrtumian NM, Lomovskaia ND. Genetic characteristics of a new phage resistance trait in Streptomyces coelicolor A3(2) Genetika. 1982;18:1945–52. In Russian. [PubMed] [Google Scholar]
- 28.Chopin MC, Chopin A, Bidnenko E. Phage abortive infection in lactococci: variations on a theme. Curr Opin Microbiol. 2005;8:473–79. doi: 10.1016/j.mib.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 29.Christensen-Dalsgaard M, Overgaard M, Winther KS, Gerdes K. RNA decay by messenger RNA interferases. Methods Enzymol. 2008;447:521–35. doi: 10.1016/S0076-6879(08)02225-8. [DOI] [PubMed] [Google Scholar]
- 30.Dao-Thi MH, Van Melderen L, De Genst E, Afif H, Buts L, et al. Molecular basis of gyrase poisoning by the addiction toxin CcdB. J Mol Biol. 2005;348:1091–102. doi: 10.1016/j.jmb.2005.03.049. [DOI] [PubMed] [Google Scholar]
- 31.Daume M, Plagens A, Randau L. DNA binding properties of the small Cascade subunit Csa5. PLOS ONE. 2014;9:e105716. doi: 10.1371/journal.pone.0105716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dixit B, Ghosh KK, Fernandes G, Kumar P, Gogoi P, Kumar M. Dual nuclease activity of a Cas2 protein in CRISPR-Cas subtype I-B of Leptospira interrogans. FEBS Lett. 2016;590:1002–16. doi: 10.1002/1873-3468.12124. [DOI] [PubMed] [Google Scholar]
- 33.Domingues S, McGovern S, Plochocka D, Santos MA, Ehrlich SD, et al. The lactococcal abortive infection protein AbiP is membrane-anchored and binds nucleic acids. Virology. 2008;373:14–24. doi: 10.1016/j.virol.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 34.Dong D, Ren K, Qiu X, Zheng J, Guo M, et al. The crystal structure of Cpf1 in complex with CRISPR RNA. Nature. 2016;532:522–26. doi: 10.1038/nature17944. [DOI] [PubMed] [Google Scholar]
- 35.Durand S, Jahn N, Condon C, Brantl S. Type I toxin-antitoxin systems in Bacillus subtilis. RNA Biol. 2012;9:1491–97. doi: 10.4161/rna.22358. [DOI] [PubMed] [Google Scholar]
- 36.Durmaz E, Klaenhammer TR. Abortive phage resistance mechanism AbiZ speeds the lysis clock to cause premature lysis of phage-infected Lactococcus lactis. J Bacteriol. 2007;189:1417–25. doi: 10.1128/JB.00904-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.East-Seletsky A, O’Connell MR, Knight SC, Burstein D, Cate JH, et al. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature. 2016;538:270–73. doi: 10.1038/nature19802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eckstein F. Phosphorothioation of DNA in bacteria. Nat Chem Biol. 2007;3:689–90. doi: 10.1038/nchembio1107-689. [DOI] [PubMed] [Google Scholar]
- 39.Elmore JR, Sheppard NF, Ramia N, Deighan T, Li H, et al. Bipartite recognition of target RNAs activates DNA cleavage by the Type III-B CRISPR-Cas system. Genes Dev. 2016;30:447–59. doi: 10.1101/gad.272153.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ershova AS, Karyagina AS, Vasiliev MO, Lyashchuk AM, Lunin VG, et al. Solitary restriction endonucleases in prokaryotic genomes. Nucleic Acids Res. 2012;40:10107–15. doi: 10.1093/nar/gks853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ershova AS, Rusinov IS, Spirin SA, Karyagina AS, Alexeevski AV. Role of restriction-modification systems in prokaryotic evolution and ecology. Biochemistry. 2015;80:1373–86. doi: 10.1134/S0006297915100193. [DOI] [PubMed] [Google Scholar]
- 42.Fabre L, Zhang J, Guigon G, Le Hello S, Guibert V, et al. CRISPR typing and subtyping for improved laboratory surveillance of Salmonella infections. PLOS ONE. 2012;7:e36995. doi: 10.1371/journal.pone.0036995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fineran PC, Charpentier E. Memory of viral infections by CRISPR-Cas adaptive immune systems: acquisition of new information. Virology. 2012;434:202–9. doi: 10.1016/j.virol.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 44.Fonfara I, Richter H, Bratovic M, Le Rhun A, Charpentier E. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature. 2016;532:517–21. doi: 10.1038/nature17945. [DOI] [PubMed] [Google Scholar]
- 45.Forterre P, Prangishvili D. The great billion-year war between ribosome- and capsid-encoding organisms (cells and viruses) as the major source of evolutionary novelties. Ann N Y Acad Sci. 2009;1178:65–77. doi: 10.1111/j.1749-6632.2009.04993.x. [DOI] [PubMed] [Google Scholar]
- 46.Fozo EM, Hemm MR, Storz G. Small toxic proteins and the antisense RNAs that repress them. Microbiol Mol Biol Rev. 2008;72:579–89. doi: 10.1128/MMBR.00025-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frost LS, Leplae R, Summers AO, Toussaint A. Mobile genetic elements: the agents of open source evolution. Nat Rev Microbiol. 2005;3:722–32. doi: 10.1038/nrmicro1235. [DOI] [PubMed] [Google Scholar]
- 48.Gelfand MS, Koonin EV. Avoidance of palindromic words in bacterial and archaeal genomes: a close connection with restriction enzymes. Nucleic Acids Res. 1997;25:2430–39. doi: 10.1093/nar/25.12.2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gerdes K, editor. Prokaryotic Toxin-Antitoxins. Berlin: Springer; 2012. [Google Scholar]
- 50.Gerdes K, Christensen SK, Lobner-Olesen A. Prokaryotic toxin-antitoxin stress response loci. Nat Rev Microbiol. 2005;3:371–82. doi: 10.1038/nrmicro1147. [DOI] [PubMed] [Google Scholar]
- 51.Goeders N, Van Melderen L. Toxin-antitoxin systems as multilevel interaction systems. Toxins. 2014;6:304–24. doi: 10.3390/toxins6010304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goldfarb T, Sberro H, Weinstock E, Cohen O, Doron S, et al. BREX is a novel phage resistance system widespread in microbial genomes. EMBO J. 2015;34:169–83. doi: 10.15252/embj.201489455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gunderson FF, Mallama CA, Fairbairn SG, Cianciotto NP. Nuclease activity of Legionella pneumophila Cas2 promotes intracellular infection of amoebal host cells. Infect Immun. 2015;83:1008–18. doi: 10.1128/IAI.03102-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haft DH, Selengut J, Mongodin EF, Nelson KE. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLOS Comput Biol. 2005;1:e60. doi: 10.1371/journal.pcbi.0010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hayes F, Van Melderen L. Toxins-antitoxins: diversity, evolution and function. Crit Rev Biochem Mol Biol. 2011;46:386–408. doi: 10.3109/10409238.2011.600437. [DOI] [PubMed] [Google Scholar]
- 56.He F, Chen L, Peng X. First experimental evidence for the presence of a CRISPR toxin in Sulfolobus. J Mol Biol. 2014;426:3683–88. doi: 10.1016/j.jmb.2014.09.016. [DOI] [PubMed] [Google Scholar]
- 57.He W, Huang T, Tang Y, Liu Y, Wu X, et al. Regulation of DNA phosphorothioate modification in Salmonella enterica by DndB. Sci Rep. 2015;5:12368. doi: 10.1038/srep12368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hickman AB, Dyda F. The casposon-encoded Cas1 protein from Aciduliprofundum boonei is a DNA integrase that generates target site duplications. Nucleic Acids Res. 2015;43:10576–87. doi: 10.1093/nar/gkv1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ho WS, Ou HY, Yeo CC, Thong KL. The dnd operon for DNA phosphorothioation modification system in Escherichia coli is located in diverse genomic islands. BMC Genom. 2015;16:199. doi: 10.1186/s12864-015-1421-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hoskisson PA, Sumby P, Smith MC. The phage growth limitation system in Streptomyces coelicolor A(3)2 is a toxin/antitoxin system, comprising enzymes with DNA methyltransferase, protein kinase and ATPase activity. Virology. 2015;477:100–9. doi: 10.1016/j.virol.2014.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hu W, Wang C, Liang J, Zhang T, Hu Z, et al. Structural insights into DndE from Escherichia coli B7A involved in DNA phosphorothioation modification. Cell Res. 2012;22:1203–6. doi: 10.1038/cr.2012.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hug LA, Baker BJ, Anantharaman K, Brown CT, Probst AJ, et al. A new view of the tree of life. Nat Microbiol. 2016;1:16048. doi: 10.1038/nmicrobiol.2016.48. [DOI] [PubMed] [Google Scholar]
- 63.Hur JK, Olovnikov I, Aravin AA. Prokaryotic Argonautes defend genomes against invasive DNA. Trends Biochem Sci. 2014;39:257–59. doi: 10.1016/j.tibs.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hutvagner G, Simard MJ. Argonaute proteins: key players in RNA silencing. Nat Rev Mol Cell Biol. 2008;9:22–32. doi: 10.1038/nrm2321. [DOI] [PubMed] [Google Scholar]
- 65.Hyman P, Abedon ST. Bacteriophage host range and bacterial resistance. Adv Appl Microbiol. 2010;70:217–48. doi: 10.1016/S0065-2164(10)70007-1. [DOI] [PubMed] [Google Scholar]
- 66.Ichige A, Kobayashi I. Stability of EcoRI restriction-modification enzymes in vivo differentiates the EcoRI restriction-modification system from other postsegregational cell killing systems. J Bacteriol. 2005;187:6612–21. doi: 10.1128/JB.187.19.6612-6621.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Iranzo J, Lobkovsky AE, Wolf YI, Koonin EV. Evolutionary dynamics of the prokaryotic adaptive immunity system CRISPR-Cas in an explicit ecological context. J Bacteriol. 2013;195:3834–44. doi: 10.1128/JB.00412-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Iranzo J, Puigbo P, Lobkovsky AE, Wolf YI, Koonin EV. Inevitability of genetic parasites. Genome Biol Evol. 2016;8:2856–69. doi: 10.1093/gbe/evw193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jansen R, Embden JD, Gaastra W, Schouls LM. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol. 2002;43:1565–75. doi: 10.1046/j.1365-2958.2002.02839.x. [DOI] [PubMed] [Google Scholar]
- 70.Ka D, Kim D, Baek G, Bae E. Structural and functional characterization of Streptococcus pyogenes Cas2 protein under different pH conditions. Biochem Biophys Res Commun. 2014;451:152–7. doi: 10.1016/j.bbrc.2014.07.087. [DOI] [PubMed] [Google Scholar]
- 71.Kapitonov VV, Makarova KS, Koonin EV. ISC, a novel group of bacterial and archaeal DNA transposons that encode Cas9 homologs. J Bacteriol. 2015;198:797–807. doi: 10.1128/JB.00783-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Karginov FV, Hannon GJ. The CRISPR system: small RNA-guided defense in bacteria and archaea. Mol Cell. 2010;37:7–19. doi: 10.1016/j.molcel.2009.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kaufmann G. Anticodon nucleases. Trends Biochem Sci. 2000;25:70–74. doi: 10.1016/s0968-0004(99)01525-x. [DOI] [PubMed] [Google Scholar]
- 74.Kimelman A, Levy A, Sberro H, Kidron S, Leavitt A, et al. A vast collection of microbial genes that are toxic to bacteria. Genome Res. 2012;22:802–9. doi: 10.1101/gr.133850.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Klaiman D, Kaufmann G. Phage T4-induced dTTP accretion bolsters a tRNase-based host de-fense. Virology. 2011;414:97–101. doi: 10.1016/j.virol.2011.03.022. [DOI] [PubMed] [Google Scholar]
- 76.Klaiman D, Steinfels-Kohn E, Kaufmann G. A DNA break inducer activates the anticodon nuclease RloC and the adaptive immunity in Acinetobacter baylyi ADP1. Nucleic Acids Res. 2014;42:328–39. doi: 10.1093/nar/gkt851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kobayashi I. Behavior of restriction-modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res. 2001;29:3742–56. doi: 10.1093/nar/29.18.3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Koonin EV. Viruses and mobile elements as drivers of evolutionary transitions. Philos Trans R Soc Lond B Biol Sci. 2016;371:20150442. doi: 10.1098/rstb.2015.0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Koonin EV. Evolution of RNA- and DNA-guided antivirus defense systems in prokaryotes and eukaryotes: common ancestry vs convergence. Biol Direct. 2017;12:5. doi: 10.1186/s13062-017-0177-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Koonin EV, Dolja VV, Krupovic M. Origins and evolution of viruses of eukaryotes: the ultimate modularity. Virology. 2015;479–480:2–25. doi: 10.1016/j.virol.2015.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Koonin EV, Krupovic M. Evolution of adaptive immunity from transposable elements combined with innate immune systems. Nat Rev Genet. 2015;16:184–92. doi: 10.1038/nrg3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Koonin EV, Krupovic M. A movable defense. The Scientist. 2015 Jan 1; [Google Scholar]
- 83.Koonin EV, Makarova KS. CRISPR-Cas: an adaptive immunity system in prokaryotes. F1000 Biol Rep. 2009;1:95. doi: 10.3410/B1-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Koonin EV, Wolf YI. Is evolution Darwinian or/and Lamarckian? Biol Direct. 2009;4:42. doi: 10.1186/1745-6150-4-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Koonin EV, Wolf YI. Evolution of microbes and viruses: a paradigm shift in evolutionary biology? Front Cell Infect Microbiol. 2012;2:119. doi: 10.3389/fcimb.2012.00119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Koonin EV, Wolf YI. Evolution of the CRISPR-Cas adaptive immunity systems in prokaryotes: models and observations on virus-host coevolution. Mol Biosyst. 2015;11:20–27. doi: 10.1039/c4mb00438h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Koonin EV, Wolf YI. Just how Lamarckian is CRISPR-Cas immunity: the continuum of evolvability mechanisms. Biol Direct. 2016;11:9. doi: 10.1186/s13062-016-0111-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Koonin EV, Zhang F. Coupling immunity and programmed cell suicide in prokaryotes: life-or-death choices. BioEssays. 2017;39:1–9. doi: 10.1002/bies.201600186. [DOI] [PubMed] [Google Scholar]
- 89.Kovall RA, Matthews BW. Type II restriction endonucleases: structural, functional and evolutionary relationships. Curr Opin Chem Biol. 1999;3:578–83. doi: 10.1016/s1367-5931(99)00012-5. [DOI] [PubMed] [Google Scholar]
- 90.Krupovic M, Beguin P, Koonin EV. Casposons: mobile genetic elements that gave rise to the CRISPR-Cas adaptation machinery. Curr Opin Microbiol. 2017;38:36–43. doi: 10.1016/j.mib.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Krupovic M, Koonin EV. Self-synthesizing transposons: unexpected key players in the evolution of viruses and defense systems. Curr Opin Microbiol. 2016;31:25–33. doi: 10.1016/j.mib.2016.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Krupovic M, Makarova KS, Forterre P, Prangishvili D, Koonin EV. Casposons: a new superfamily of self-synthesizing DNA transposons at the origin of prokaryotic CRISPR-Cas immunity. BMC Biol. 2014;12:36. doi: 10.1186/1741-7007-12-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Krutkina E, Klaiman D, Margalit T, Jerabeck-Willemsen M, Kaufmann G. Dual nucleotide specificity determinants of an infection aborting anticodon nuclease. Virology. 2016;487:260–72. doi: 10.1016/j.virol.2015.10.012. [DOI] [PubMed] [Google Scholar]
- 94.Labrie SJ, Samson JE, Moineau S. Bacteriophage resistance mechanisms. Nat Rev Microbiol. 2010;8:317–27. doi: 10.1038/nrmicro2315. [DOI] [PubMed] [Google Scholar]
- 95.Leplae R, Geeraerts D, Hallez R, Guglielmini J, Dreze P, Van Melderen L. Diversity of bacterial type II toxin-antitoxin systems: a comprehensive search and functional analysis of novel families. Nucleic Acids Res. 2011;39:5513–25. doi: 10.1093/nar/gkr131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lingel A, Izaurralde E. RNAi: finding the elusive endonuclease. RNA. 2004;10:1675–79. doi: 10.1261/rna.7175704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, et al. Argonaute2 is the catalytic engine of mammalian RNAi. Science. 2004;305:1437–41. doi: 10.1126/science.1102513. [DOI] [PubMed] [Google Scholar]
- 98.Liu L, Li X, Wang J, Wang M, Chen P, et al. Two distant catalytic sites are responsible for C2c2 RNase activities. Cell. 2017;168:121–34. doi: 10.1016/j.cell.2016.12.031. [DOI] [PubMed] [Google Scholar]
- 99.Loenen WA, Dryden DT, Raleigh EA, Wilson GG. Type I restriction enzymes and their relatives. Nucleic Acids Res. 2014;42:20–44. doi: 10.1093/nar/gkt847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Loenen WA, Raleigh EA. The other face of restriction: modification-dependent enzymes. Nucleic Acids Res. 2014;42:56–69. doi: 10.1093/nar/gkt747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Makarova KS, Anantharaman V, Aravind L, Koonin EV. Live virus-free or die: Coupling of antivirus immunity and programmed suicide or dormancy in prokaryotes. Biol Direct. 2012;7:40. doi: 10.1186/1745-6150-7-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Makarova KS, Anantharaman V, Grishin NV, Koonin EV, Aravind L. CARF and WYL domains: ligand-binding regulators of prokaryotic defense systems. Front Genet. 2014;5:102. doi: 10.3389/fgene.2014.00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Makarova KS, Aravind L, Wolf YI, Koonin EV. Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol Direct. 2011;6:38. doi: 10.1186/1745-6150-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV. A putative RNA-interference-based immune system in prokaryotes: Computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct. 2006;1:7. doi: 10.1186/1745-6150-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;9:467–77. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, et al. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol. 2015;13:722–36. doi: 10.1038/nrmicro3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Makarova KS, Wolf YI, Forterre P, Prangishvili D, Krupovic M, Koonin EV. Dark matter in archaeal genomes: a rich source of novel mobile elements, defense systems and secretory complexes. Extremophiles. 2014;18:877–93. doi: 10.1007/s00792-014-0672-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Makarova KS, Wolf YI, Koonin EV. Comprehensive comparative-genomic analysis of type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol Direct. 2009;4:19. doi: 10.1186/1745-6150-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Makarova KS, Wolf YI, Koonin EV. Comparative genomics of defense systems in archaea and bacteria. Nucleic Acids Res. 2013;41:4360–77. doi: 10.1093/nar/gkt157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Makarova KS, Wolf YI, Koonin EV. The basic building blocks and evolution of CRISPR-Cas systems. Biochem Soc Trans. 2013;41:1392–400. doi: 10.1042/BST20130038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Makarova KS, Wolf YI, Snir S, Koonin EV. Defense islands in bacterial and archaeal genomes and prediction of novel defense systems. J Bacteriol. 2011;193:6039–56. doi: 10.1128/JB.05535-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Makarova KS, Wolf YI, van der Oost J, Koonin EV. Prokaryotic homologs of Argonaute proteins are predicted to function as key components of a novel system of defense against mobile genetic elements. Biol Direct. 2009;4:29. doi: 10.1186/1745-6150-4-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mohanraju P, Makarova KS, Zetsche B, Zhang F, Koonin EV, van der Oost J. Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science. 2016;353:aad5147. doi: 10.1126/science.aad5147. [DOI] [PubMed] [Google Scholar]
- 114.Mojica FJ, Diez-Villasenor C, Garcia-Martinez J, Soria E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol. 2005;60:174–82. doi: 10.1007/s00239-004-0046-3. [DOI] [PubMed] [Google Scholar]
- 115.Mruk I, Kobayashi I. To be or not to be: regulation of restriction-modification systems and other toxin-antitoxin systems. Nucleic Acids Res. 2014;42:70–86. doi: 10.1093/nar/gkt711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Naito T, Kusano K, Kobayashi I. Selfish behavior of restriction-modification systems. Science. 1995;267:897–99. doi: 10.1126/science.7846533. [DOI] [PubMed] [Google Scholar]
- 117.Nakayama Y, Kobayashi I. Restriction-modification gene complexes as selfish gene entities: Roles of a regulatory system in their establishment, maintenance, and apoptotic mutual exclusion. PNAS. 1998;95:6442–47. doi: 10.1073/pnas.95.11.6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nam KH, Ding F, Haitjema C, Huang Q, DeLisa MP, Ke A. Double-stranded endonuclease activity in Bacillus halodurans clustered regularly interspaced short palindromic repeats (CRISPR)-associated Cas2 protein. J Biol Chem. 2012;287:35943–52. doi: 10.1074/jbc.M112.382598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Niewoehner O, Jinek M. Structural basis for the endoribonuclease activity of the type III-A CRISPR-associated protein Csm6. RNA. 2016;22:318–29. doi: 10.1261/rna.054098.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nunez JK, Kranzusch PJ, Noeske J, Wright AV, Davies CW, Doudna JA. Cas1-Cas2 complex formation mediates spacer acquisition during CRISPR-Cas adaptive immunity. Nat Struct Mol Biol. 2014;21:528–34. doi: 10.1038/nsmb.2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nunez JK, Lee AS, Engelman A, Doudna JA. Integrase-mediated spacer acquisition during CRISPR-Cas adaptive immunity. Nature. 2015;519:193–98. doi: 10.1038/nature14237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ohno S, Handa N, Watanabe-Matsui M, Takahashi N, Kobayashi I. Maintenance forced by a restriction-modification system can be modulated by a region in its modification enzyme not essential for methyltransferase activity. J Bacteriol. 2008;190:2039–49. doi: 10.1128/JB.01319-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Oliveira PH, Touchon M, Rocha EP. The interplay of restriction-modification systems with mobile genetic elements and their prokaryotic hosts. Nucleic Acids Res. 2014;42:10618–31. doi: 10.1093/nar/gku734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Oliveira PH, Touchon M, Rocha EP. Regulation of genetic flux between bacteria by restriction-modification systems. PNAS. 2016;113:5658–63. doi: 10.1073/pnas.1603257113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Olovnikov I, Chan K, Sachidanandam R, Newman DK, Aravin AA. Bacterial Argonaute samples the transcriptome to identify foreign DNA. Mol Cell. 2013;51:594–605. doi: 10.1016/j.molcel.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Orlowski J, Bujnicki JM. Structural and evolutionary classification of Type II restriction enzymes based on theoretical and experimental analyses. Nucleic Acids Res. 2008;36:3552–69. doi: 10.1093/nar/gkn175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Otsuka Y. Prokaryotic toxin-antitoxin systems: novel regulations of the toxins. Curr Genet. 2016;62:379–82. doi: 10.1007/s00294-015-0557-z. [DOI] [PubMed] [Google Scholar]
- 128.Page R, Peti W. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat Chem Biol. 2016;12:208–14. doi: 10.1038/nchembio.2044. [DOI] [PubMed] [Google Scholar]
- 129.Parker JS, Barford D. Argonaute: a scaffold for the function of short regulatory RNAs. Trends Biochem Sci. 2006;31:622–30. doi: 10.1016/j.tibs.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 130.Parker JS, Roe SM, Barford D. Structural insights into mRNA recognition from a PIWI domain-siRNA guide complex. Nature. 2005;434:663–66. doi: 10.1038/nature03462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pasternak C, Dulermo R, Ton-Hoang B, Debuchy R, Siguier P, et al. ISDra2 transposition in Deinococcus radiodurans is downregulated by TnpB. Mol Microbiol. 2013;88:443–55. doi: 10.1111/mmi.12194. [DOI] [PubMed] [Google Scholar]
- 132.Pingoud A, Wilson GG, Wende W. Type II restriction endonucleases—a historical perspective and more. Nucleic Acids Res. 2014;42:7489–527. doi: 10.1093/nar/gku447. Erratum. 2016. Nucleic Acids Res. 44:8011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Puigbò P, Lobkovsky AE, Kristensen DM, Wolf YI, Koonin EV. Genomes in turmoil: quantification of genome dynamics in prokaryote supergenomes. BMC Biol. 2014;12:66. doi: 10.1186/s12915-014-0066-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Puigbò P, Makarova KS, Kristensen DM, Wolf YI, Koonin EV. Reconstruction of the evolution of microbial defense systems. BMC Evol Biol. 2017;17:94. doi: 10.1186/s12862-017-0942-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Qi L, Haurwitz RE, Shao W, Doudna JA, Arkin AP. RNA processing enables predictable programming of gene expression. Nat Biotechnol. 2012;30:1002–6. doi: 10.1038/nbt.2355. [DOI] [PubMed] [Google Scholar]
- 136.Rao DN, Dryden DT, Bheemanaik S. Type III restriction-modification enzymes: a historical perspective. Nucleic Acids Res. 2014;42:45–55. doi: 10.1093/nar/gkt616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rimer J, Cohen IR, Friedman N. Do all creatures possess an acquired immune system of some sort? BioEssays. 2014;36:273–81. doi: 10.1002/bies.201300124. [DOI] [PubMed] [Google Scholar]
- 138.Roberts RJ, Belfort M, Bestor T, Bhagwat AS, Bickle TA, et al. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 2003;31:1805–12. doi: 10.1093/nar/gkg274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE–enzymes and genes for DNA restriction and modification. Nucleic Acids Res. 2007;35:D269–70. doi: 10.1093/nar/gkl891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rohwer F, Thurber RV. Viruses manipulate the marine environment. Nature. 2009;459:207–12. doi: 10.1038/nature08060. [DOI] [PubMed] [Google Scholar]
- 141.Rusinov I, Ershova A, Karyagina A, Spirin S, Alexeevski A. Lifespan of restriction-modification systems critically affects avoidance of their recognition sites in host genomes. BMC Genom. 2015;16:1084. doi: 10.1186/s12864-015-2288-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sberro H, Leavitt A, Kiro R, Koh E, Peleg Y, et al. Discovery of functional toxin/antitoxin systems in bacteria by shotgun cloning. Mol Cell. 2013;50:136–48. doi: 10.1016/j.molcel.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Seed KD, Lazinski DW, Calderwood SB, Camilli A. A bacteriophage encodes its own CRISPR/Cas adaptive response to evade host innate immunity. Nature. 2013;494:489–91. doi: 10.1038/nature11927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Sheng G, Zhao H, Wang J, Rao Y, Tian W, et al. Structure-based cleavage mechanism of Thermus thermophilus Argonaute DNA guide strand-mediated DNA target cleavage. PNAS. 2014;111:652–57. doi: 10.1073/pnas.1321032111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sheppard NF, Glover CV, 3rd, Terns RM, Terns MP. The CRISPR-associated Csx1 protein of Pyrococcus furiosus is an adenosine-specific endoribonuclease. RNA. 2016;22:216–24. doi: 10.1261/rna.039842.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Shmakov S, Abudayyeh OO, Makarova KS, Wolf YI, Gootenberg JS, et al. Discovery and functional characterization of diverse class 2 CRISPR-Cas systems. Mol Cell. 2015;60:385–97. doi: 10.1016/j.molcel.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Shmakov S, Smargon A, Scott D, Cox D, Pyzocha N, et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nat Rev Microbiol. 2017;15:169–82. doi: 10.1038/nrmicro.2016.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Silas S, Mohr G, Sidote DJ, Markham LM, Sanchez-Amat A, et al. Direct CRISPR spacer acquisition from RNA by a natural reverse transcriptase-Cas1 fusion protein. Science. 2016;351:aad4234. doi: 10.1126/science.aad4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Smargon AA, Cox DB, Pyzocha NK, Zheng K, Slaymaker IM, et al. Cas13b is a type VI-B CRISPR-associated RNA-guided RNase differentially regulated by accessory proteins Csx27 and Csx28. Mol Cell. 2017;65:618–30.e7. doi: 10.1016/j.molcel.2016.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Song JJ, Smith SK, Hannon GJ, Joshua-Tor L. Crystal structure of Argonaute and its implications for RISC slicer activity. Science. 2004;305:1434–37. doi: 10.1126/science.1102514. [DOI] [PubMed] [Google Scholar]
- 151.Stern A, Sorek R. The phage-host arms race: shaping the evolution of microbes. BioEssays. 2011;33:43–51. doi: 10.1002/bies.201000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sternberg SH, Richter H, Charpentier E, Qimron U. Adaptation in CRISPR-Cas systems. Mol Cell. 2016;61:797–808. doi: 10.1016/j.molcel.2016.01.030. [DOI] [PubMed] [Google Scholar]
- 153.Sumby P, Smith MC. Genetics of the phage growth limitation (Pgl) system of Streptomyces coelicolor A3(2) Mol Microbiol. 2002;44:489–500. doi: 10.1046/j.1365-2958.2002.02896.x. [DOI] [PubMed] [Google Scholar]
- 154.Suttle CA. Viruses in the sea. Nature. 2005;437:356–61. doi: 10.1038/nature04160. [DOI] [PubMed] [Google Scholar]
- 155.Suttle CA. Marine viruses—major players in the global ecosystem. Nat Rev Microbiol. 2007;5:801–12. doi: 10.1038/nrmicro1750. [DOI] [PubMed] [Google Scholar]
- 156.Swarts DC, Hegge JW, Hinojo I, Shiimori M, Ellis MA, et al. Argonaute of the archaeon Pyrococcus furiosus is a DNA-guided nuclease that targets cognate DNA. Nucleic Acids Res. 2015;43:5120–29. doi: 10.1093/nar/gkv415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Swarts DC, Jore MM, Westra ER, Zhu Y, Janssen JH, et al. DNA-guided DNA interference by a prokaryotic Argonaute. Nature. 2014;507:258–61. doi: 10.1038/nature12971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Swarts DC, Makarova K, Wang Y, Nakanishi K, Ketting RF, et al. The evolutionary journey of Argonaute proteins. Nat Struct Mol Biol. 2014;21:743–53. doi: 10.1038/nsmb.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Syed MA, Levesque CM. Chromosomal bacterial type II toxin-antitoxin systems. Can J Microbiol. 2012;58:553–62. doi: 10.1139/w2012-025. [DOI] [PubMed] [Google Scholar]
- 160.Szathmary E, Maynard Smith J. From replicators to reproducers: the first major transitions leading to life. J Theor Biol. 1997;187:555–71. doi: 10.1006/jtbi.1996.0389. [DOI] [PubMed] [Google Scholar]
- 161.Tabara H, Sarkissian M, Kelly WG, Fleenor J, Grishok A, et al. The rde-1 gene, RNA interference, and transposon silencing in C. elegans. Cell. 1999;99:123–32. doi: 10.1016/s0092-8674(00)81644-x. [DOI] [PubMed] [Google Scholar]
- 162.Tan Q, Awano N, Inouye M. YeeV is an Escherichia coli toxin that inhibits cell division by targeting the cytoskeleton proteins, FtsZ and MreB. Mol Microbiol. 2011;79:109–18. doi: 10.1111/j.1365-2958.2010.07433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Tock MR, Dryden DT. The biology of restriction and anti-restriction. Curr Opin Microbiol. 2005;8:466–72. doi: 10.1016/j.mib.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 164.Toussaint A, Chandler M. Prokaryote genome fluidity: toward a system approach of the mobilome. Methods Mol Biol. 2012;804:57–80. doi: 10.1007/978-1-61779-361-5_4. [DOI] [PubMed] [Google Scholar]
- 165.Uzan M. RNA processing and decay in bacteriophage T4. Prog Mol Biol Transl Sci. 2009;85:43–89. doi: 10.1016/S0079-6603(08)00802-7. [DOI] [PubMed] [Google Scholar]
- 166.van der Oost J, Jore MM, Westra ER, Lundgren M, Brouns SJ. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends Biochem Sci. 2009;34:401–7. doi: 10.1016/j.tibs.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 167.Van Melderen L. Toxin-antitoxin systems: why so many, what for? Curr Opin Microbiol. 2010;13:781–85. doi: 10.1016/j.mib.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 168.Van Melderen L, Saavedra De Bast M. Bacterial toxin-antitoxin systems: more than selfish entities? PLOS Genet. 2009;5:e1000437. doi: 10.1371/journal.pgen.1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wang C, Villion M, Semper C, Coros C, Moineau S, Zimmerly S. A reverse transcriptase-related protein mediates phage resistance and polymerizes untemplated DNA in vitro. Nucleic Acids Res. 2011;39:7620–29. doi: 10.1093/nar/gkr397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Wang Y, Juranek S, Li H, Sheng G, Tuschl T, Patel DJ. Structure of an Argonaute silencing complex with a seed-containing guide DNA and target RNA duplex. Nature. 2008;456:921–26. doi: 10.1038/nature07666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Weinberger AD, Wolf YI, Lobkovsky AE, Gilmore MS, Koonin EV. Viral diversity threshold for adaptive immunity in prokaryotes. mBio. 2012;3:e00456–12. doi: 10.1128/mBio.00456-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Westra ER, Buckling A, Fineran PC. CRISPR-Cas systems: beyond adaptive immunity. Nat Rev Microbiol. 2014;12:317–26. doi: 10.1038/nrmicro3241. [DOI] [PubMed] [Google Scholar]
- 173.Westra ER, Swarts DC, Staals RH, Jore MM, Brouns SJ, van der Oost J. The CRISPRs, they are a-changin’: how prokaryotes generate adaptive immunity. Annu Rev Genet. 2012;46:311–39. doi: 10.1146/annurev-genet-110711-155447. [DOI] [PubMed] [Google Scholar]
- 174.Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331–38. doi: 10.1038/nature10886. [DOI] [PubMed] [Google Scholar]
- 175.Williams RJ. Restriction endonucleases: classification, properties, and applications. Mol Biotechnol. 2003;23:225–43. doi: 10.1385/mb:23:3:225. [DOI] [PubMed] [Google Scholar]
- 176.Willkomm S, Zander A, Grohmann D, Restle T. Mechanistic insights into archaeal and human Argonaute substrate binding and cleavage properties. PLOS ONE. 2016;11:e0164695. doi: 10.1371/journal.pone.0164695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Xiong W, Zhao G, Yu H, He X. Interactions of Dnd proteins involved in bacterial DNA phosphorothioate modification. Front Microbiol. 2015;6:1139. doi: 10.3389/fmicb.2015.01139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Xu T, Liang J, Chen S, Wang L, He X, et al. DNA phosphorothioation in Streptomyces lividans: mutational analysis of the dnd locus. BMC Microbiol. 2009;9:41. doi: 10.1186/1471-2180-9-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Yamaguchi Y, Inouye M. mRNA interferases, sequence-specific endoribonucleases from the toxin-antitoxin systems. Prog Mol Biol Transl Sci. 2009;85:467–500. doi: 10.1016/S0079-6603(08)00812-X. [DOI] [PubMed] [Google Scholar]
- 180.Yamaguchi Y, Park JH, Inouye M. Toxin-antitoxin systems in bacteria and archaea. Annu Rev Genet. 2011;45:61–79. doi: 10.1146/annurev-genet-110410-132412. [DOI] [PubMed] [Google Scholar]
- 181.Yamano T, Nishimasu H, Zetsche B, Hirano H, Slaymaker IM, et al. Crystal structure of Cpf1 in complex with guide RNA and target DNA. Cell. 2016;165:949–62. doi: 10.1016/j.cell.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Yang H, Gao P, Rajashankar KR, Patel DJ. PAM-dependent target DNA recognition and cleavage by C2c1 CRISPR-Cas endonuclease. Cell. 2016;167:1814–28.e12. doi: 10.1016/j.cell.2016.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Yao J, Guo Y, Zeng Z, Liu X, Shi F, Wang X. Identification and characterization of a HEPN-MNT family type II toxin-antitoxin in Shewanella oneidensis. Microb Biotechnol. 2015;8:961–73. doi: 10.1111/1751-7915.12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.You D, Wang L, Yao F, Zhou X, Deng Z. A novel DNA modification by sulfur: DndA is a NifS-like cysteine desulfurase capable of assembling DndC as an iron-sulfur cluster protein in Streptomyces lividans. Biochemistry. 2007;46:6126–33. doi: 10.1021/bi602615k. [DOI] [PubMed] [Google Scholar]
- 185.Yuan YR, Pei Y, Ma JB, Kuryavyi V, Zhadina M, et al. Crystal structure of A. aeolicus Argonaute, a site-specific DNA-guided endoribonuclease, provides insights into RISC-mediated mRNA cleavage. Mol Cell. 2005;19:405–19. doi: 10.1016/j.molcel.2005.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zalatan JG, Fenn TD, Herschlag D. Comparative enzymology in the alkaline phosphatase super-family to determine the catalytic role of an active-site metal ion. J Mol Biol. 2008;384:1174–89. doi: 10.1016/j.jmb.2008.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]