Connections between Epigenetic Gene Silencing and Human Disease (original) (raw)
. Author manuscript; available in PMC: 2007 Jun 17.
Abstract
Alterations in epigenetic gene regulation are associated with human disease. Here, we discuss connections between DNA methylation and histone methylation, providing examples in which defects in these processes are linked with disease. Mutations in genes encoding DNA methyltransferases and proteins that bind methylated cytosine residues cause changes in gene expression and alterations in the patterns of DNA methylation. These changes are associated with cancer and congenital diseases due to defects in imprinting. Gene silencing is also controlled through histone methylation. Altered levels of methyltransferases that modify lysine 27 of histone H3 (K27H3) and lysine 9 of histone H3 (K9H3) correlate with changes in Rb signaling and disruption of the cell cycle in cancer cells. The K27H3 mark recruits a Polycomb complex that is involved in regulating stem cell pluripotency, silencing of developmentally regulated genes, and controlling cancer progression. The K9H3 methyl mark recruits HP1, a structural protein that plays a role in heterochromatin formation, gene silencing, and viral latency. Cells exhibiting altered levels of HP1 are predicted to show a loss of silencing at genes regulating cancer progression. Gene silencing through K27H3 and K9H3 can involve histone deacetylation and DNA methylation, suggesting cross talk between epigenetic silencing systems through direct interactions among the various players. The reversible nature of these epigenetic modifications offers therapeutic possibilities for a wide spectrum of disease.
Keywords: chromatin, DNA methylation, epigenetic gene silencing, histone methylation, human disease
1. DNA methylation
Altered gene expression can play a causal role in human disease. In many cases, altered expression results from genetic lesions within the gene or regulatory sequences. However, in some cases genetic lesions are absent from the gene. In such instances, aberrant epigenetic modifications of the chromatin surrounding the gene are the cause of altered expression. There are two major epigenetic gene silencing mechanisms that account for a growing number of diseases: cytosine DNA methylation and covalent histone modification.
The 5' cytosine of CpG dinucleotides within mammalian genomes can be methylated by de novo DNA methyltransferases such as DNMT3A and DNMT3B [1]. Maintenance of DNA methylation is performed by DNMT1, utilizing hemimethylated DNA as a substrate. This provides a mechanism to propagate the epigenetic mark following DNA replication. The methyl groups serve as docking sites for gene silencing proteins [2]. In general, DNA methylation correlates with increased chromatin condensation and gene silencing [1].
There are several ways in which altered patterns of DNA methylation lead to disease (Table 1). CpG dinucleotides are generally methylated in normal cells, with the exception of hypomethylation at CpG “islands” located upstream of many active genes [3]. In contrast, cancer cells exhibit a global hypomethylation and CpG island hypermethylation [3]. This shift in the pattern of DNA methylation frequently results in inappropriate silencing of genes, especially tumor suppressor genes, leading to numerous types of cancer. For example, expression of the serine protease inhibitor family member maspin is reduced due to methylation of promoter sequences in many advanced forms of cancer [4-6].
Table 1.
Gene silencing proteins and disease.
| Protein | Cellular Defect / Disease | References |
|---|---|---|
| DNMT1 | Developmental abnormalities | [110-112] |
| Igf2 imprinting | [9] | |
| Colon cancer | [113-115] | |
| Lymphoma | [116-118] | |
| Pancreatic cancer | [119] | |
| DNMT3B | Developmental abnormalities | [120] |
| ICF | [1,121-123] | |
| Bladder cancer | [124] | |
| Breast cancer | [124,125*] | |
| Colon cancer | [124] | |
| Hepatocellular carcinoma | [126*,127*] | |
| Lung cancer | [124,128*,129*] | |
| MeCP2 | Chromosome instability/ cell cycle defects | [49,54,55,57, 130] |
| Breast cancer | [131] | |
| Rett syndrome | [132, 133], RETTBase | |
| EZH2 | Cell cycle defects | [35-37] |
| Barrett's esophagus | [134] | |
| Bladder cancer | [135,136] | |
| Breast cancer | [41,42,45,137] | |
| Colorectal cancer | [138] | |
| Melanoma | [137] | |
| Myeloma/ lymphoma | [29,43] [44,46,47,139-141] | |
| Hepatocellular carcinoma | [142] | |
| Prostate cancer | [34,39,40,137] | |
| Wilms tumor | [143] | |
| Suv39h1 | Blood cell defects (RBC and WBC) | [54,56,59] |
| Chromosome instability/ cell cycle defects | [49,54,55,57] [130] | |
| Chromosome instability | [68-73,75-77] | |
| HP1 | Breast cancer | [78,81,82,144] |
| Medulloblastoma | [86] | |
| Papillary thyroid carcinoma | [85] | |
| Viral latency | [87-92] |
Alterations in methylation patterns are responsible for several congenital diseases that affect growth through the misregulation of imprinted genes. Mammalian genomes contain dispersed clusters of genes in which the expression state of each allele is determined by the parent of origin [7]. Transcription within these clusters is regulated by Imprinting Centers (ICRs), DNA regions that are typically 1-2 kb in size and enriched with CpG dinucleotides [8]. ICRs exhibit allele-specific DNA methylation and histone modifications. An ICR positioned between the insulin-like growth factor IGF2 and H19 genes is methylated only on the paternal allele, presumably by DNMT1 [9]. This methylation blocks the association of the zinc finger protein CTCF [10,11]. On the unmethylated maternally derived allele, ICR is bound by CTCF, which functions as an insulator by blocking interactions between IGF2 enhancers located upstream of the H19 gene. Altered expression of IGF2 due to changes in the imprinted status at ICR result in two diseases with different clinical characteristics, Beckwith-Weidemann Syndrome (BWS, OMIM I30650) and Silver-Russel Syndrome (SRS, OMIM 180860) [7] (Table 1). BWS is primarily identified by macroglossia, umbilical abnormalities and gigantism. In a subset of BWS individuals, DNA methylation at the ICR occurs on both the maternal and paternal alleles, resulting in loss of H19 expression and activation of IGF2 on both alleles. SRS is identified by low birth weight, slow postnatal growth, characteristic facial abnormalities and body asymmetry. In a subset of SRS individuals, DNA methylation does not occur within the ICR on either allele, resulting in the expression of H19 from both alleles and complete loss of IGF2 expression.
In addition to alterations in the patterns of DNA methylation, loss of DNA methyl transferase also leads to disease (Table 1). Immunodeficiency-centromeric instability-facial anomalies (ICF, OMIM 24860) is an autosomal recessive disorder caused by defects within the catalytic domain of DNMT3B [1]. Phenotypes of ICF include instability of chromosomes 1, 9 and 16, which are enriched for pericentric satellite II and III sequences containing CpG dinucleotides. In addition, ICF is associated with immune system malfunctions, facial abnormalities, and short life expectancy. Given that ICF individuals lack the function of a de novo DNA methyltransferase, it was anticipated that changes in patterns of DNA methylation would lead to alterations in gene expression. Expression profiling studies showed alterations in gene expression as expected, however, changes in the DNA methylation patterns at these genes were minor [1]. In contrast, the satellite sequences were found to be hypomethylated [12]. Thus, methylation of repetitive sequences, such as the satellites, might be important for establishing the spatial positioning of chromosomes within the nucleus that is necessary for proper gene regulation [13].
Given that mutations within DNA methyltransferase genes are associated with disease, it follows that mutations within genes encoding proteins that bind to methylated cytosines also result in disease. MeCP2 (methyl-CpG-binding protein 2) is a member of a class of DNA methyl binding proteins (MBDs) that specifically recognize methylated cytosine residues [14]. These binding proteins function by recruiting histone deacetylases (HDACs) to silence target genes. Mutations in the X-linked gene encoding MeCP2 are responsible for approximately 95% of classic Rett syndrome cases (RTT, OMIM 312750) [15] (Table 1). RTT is the second most common form of mental retardation in females, estimated to affect 1 in 10,000 females. Overt disease phenotypes are not observed until after 6 – 8 months of age, at which time mental abnormalities and motor function impairment become obvious. The delayed onset of the syndrome suggests problems with neuronal differentiation. One curious aspect of the disease is that MeCP2 is broadly expressed, yet only neuronal function appears to be affected. Thus, it was hypothesized that loss of MeCP2 would result in alterations of gene expression within the brain. To address this hypothesis, transcriptional profiling experiments were performed using brain tissue from a Mecp2 knock-out mouse model in comparison to control mice [16]. Surprisingly, very subtle alterations in brain-specific gene expression were observed. However, changes in gene expression within a subset of brain cells might not have been detected, as a mixture of cell types was analyzed. The lack of global changes in gene expression might also be due to redundancy in function among the DNA methyl binding proteins as suggested by MBD knock-out studies [14]. Thus, the phenotypic consequences resulting from RTT might be due to subtle changes in gene expression or mis-regulation of gene expression in a limited number of cells within the brain. To date, several MeCP2 target genes have been identified, including brain-derived neurotrophic factor (Bdnf) [15]. Interestingly, BDNF protein levels are decreased in the Mecp2 mouse knock-out model, rather than an anticipated increase if MeCP2 were playing a silencing role. This decrease could reflect complex issues related to neuronal differentiation [17]. Satisfying features that support Bdnf as a key gene involved in the RTT phenotype are a role in neural differentiation, a knock-out mouse that exhibits overlapping phenotypes with RTT syndrome mouse models, and the fact that over-expression of BDNF in the forebrain rescues some of the phenotypes associated with the Mecp2 mutant mice [17]. While these data argue for a connection between MeCP2 and BDPF, questions as to the mechanistic defects associated with RTT remain unresolved [18]. Complicating this issue, MeCP2 appears to play a role in mRNA splicing, through interactions with the Y box binding protein [19], suggesting post-transcriptional events could contribute to the RTT phenotype. Collectively, it is clear that defects in multiple steps of the DNA methylation pathway cause disease, however, the molecular defects that contribute to disease development remain to be elucidated.
2. Histone methylation
Modifications such as phosphorylation, acetylation and methylation frequently occur on histones tails that extend from the nucleosome core [20]. These modifications serve to alter charge interactions of the histone tails with DNA, thereby influencing chromatin packaging. In addition, these modifications serve as binding sites for specific factors that “read” a proposed histone code [21]. In most cases, specific modifications correlate with biological functions such as chromatin condensation, transcriptional regulation and DNA replication. Disruption of the epigenetic modifications on histones throughout the genome is a universal feature observed in cancer cells [22]. Here, we will focus on connections between two histone methyltransferases involved in gene silencing and cancer.
2.1 E(z) and disease
Enhancer of Zeste (EZH2) is one of two mammalian homologues of Drosophila E(Z), a SET domain protein that methylates K27H3 [23]. E(Z) is a Polycomb group (PcG) protein involved in homeotic gene repression in Drosophila [24]. Two Polycomb complexes have been elucidated, Polycomb Repressor Complex 1 and 2 (PRC1 and PRC2) [25]. While components differ among species, the core PRC1 components in mammals are BMI-1, Ring-1, HPH and HPC. The core PRC2 components are EED, EZH2, SU(Z)12. PRC2 is recruited to target genes for the initiation of silencing. PRC2 associates with Type I HDACs, such as HDAC1 and 2, through interactions with EED [26] (Figure 1a). In addition, immunoprecipitation of EZH2 and EED show interactions with DNMT1, 3A and 3B, suggesting a link between histone methylation and DNA methylation [27] (Figure 1a). This was supported by experiments in which cells treated with 5'-aza-deoxycytidine or RNAi knock-down of any of the DNMTs, showed loss of silencing at EZH2 target genes [27]. Following PRC2 silencing, PRC1 is recruited for maintenance of the silent state. Collectively, these data support a model whereby Polycomb silencing occurs through histone deacetylation, subsequent histone methylation and DNA methylation initiated by PRC2, and maintained by PRC1[28] (Figure 1a).
Figure 1.
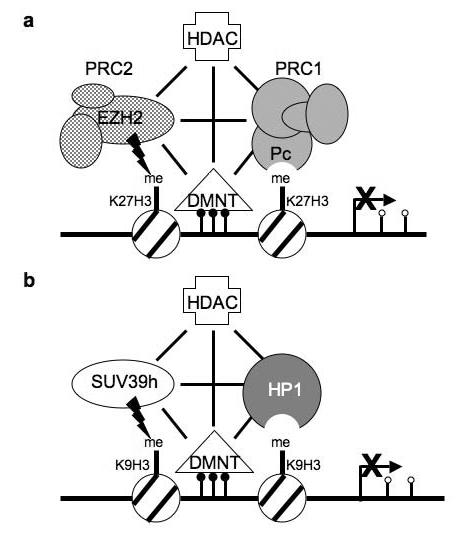
Diagram showing the connections between epigenetic gene silencing factors. (a) Gene silencing associated with K27H3. HDACs deacetylate nucleosomes, allowing for methylation of K27H3 by EZH2, providing a bindings site for Polycomb. Components of Polycomb complexes interact with DMNTs involved in methylating cytosine residues (small filled circles). Lines indicate interactions between the gene silencing factors. (b) Gene silencing associated with K9H3 methylation. HDACs deacetylate nucleosomes, allowing for methylation of K9H3 by SUV39h, providing a binding site for HP1. DNA methyltransferases modify cytosine residues (small filled circles) to repress gene expression. Lines indicate interactions between the gene silencing factors. In cancer cells hypermethylation typically occurs within CpG islands of promoter regions, while isolated CpGs at other locations are hypomethylated (small open circles).
Homozygous EZH2 knock-out mice die during embryogenesis, while tissue-specific knock-out of EZH2 caused defects in B cell maturation [29,30]. A role for EZH2 in developmentally regulated gene expression has been corroborated by chromatin immunoprecipitation experiments coupled with microarray analysis [31]. PRC1 and PRC2 components were mainly associated with silenced genes involved in differentiation in embryonic stem cells; upon differentiation, epigenetic marks associated with these genes changed [31,32]. Interestingly, both methylated K4 (an activating mark) and K27 (a silencing mark) were found in “bivalent domains” of the promoters of the embryonic silenced genes encoding transcription factors that govern pluripotency [32,33]. This duality of epigenetic marking was resolved upon differentiation, as the appropriate expression or silencing marks on cell lineage genes were retained upon commitment to a particular cellular fate. Surprisingly, this combination of marks results in silencing similar to that mediated by methylated K27 in differentiated cells. This suggests that Polycomb silencing is initiated in embryogenesis, and that retention of the epigenetic mark upon differentiation results in a continuation of silencing later in development [32]. Perhaps it is not surprising that defects of this gene silencing system within differentiated cells (see below) correlate with a poorer prognosis for many cancers, as loss of silencing can provide increased plasticity for cancer cells.
Levels of EZH2 are important for control of cell cycle regulation, with increased levels leading to cancer [34]. One mechanism for controlling EZH2 levels is through transcriptional regulation by p53 [35]. EZH2 and Rb potentially compete for the binding of HDAC1, a component in E2F-mediated gene silencing [36]. Activated p53 silences EZH2 transcription, allowing for repression of E2F-regulated cell cycle genes by Rb through HDACs [37]. In general, EZH2 up-regulation through increased transcription or gene amplification correlates with cancer [38]. EZH2 over-expression is observed in many types of cancer including prostate, breast, and lymphomas (Table 1). In prostate cancer, EZH2 is up-regulated with metastasis, while EED levels remain unchanged [39]. Cellular proliferation is dependent on the SET domain of EZH2, and RNAi knock-down of EZH2 led to cellular growth inhibition and arrest in G2/M. In fact, a combination of high EZH2 expression with moderate to low E-cadherin is one of the best markers for prostate cancer progression [38,40].
Similar data for EZH2 exists for breast cancer and lymphoma (Table 1), where increased levels of expression correlate with tumor proliferation and poor prognosis. Importantly, the methyltransferase activity of the SET domain plays an important role in cell invasion [41-44]. In normal, differentiated cells an almost mutually exclusive expression pattern exists for BMI-1 (a component of PRC1) and EZH2 (a component of PRC2). It appears that BMI-1 and EZH2 are only highly co-expressed in cancer cells [45,46]. While the pattern may be slightly different in the germinal center, in general, B cells lack EzH2 expression and highly expressed BMI-1 in resting or well-differentiated cells. In cycling or poorly differentiated cells the opposite is true [46,47]. One possible model generated by these data is that PRC1 and PRC2 exchange silencing duties when cells enter and exit the proliferation stage during differentiation and cancer formation. These transitions are accomplished through deacetylation of histones at target genes via HDACs, recruitment of PRC2 with subsequent histone and DNA methylation, with final localization of PRC1 to maintain the silenced state [28,47,48]. This model is supported by studies in Drosophila and may be a general paradigm of epigenetic regulation by Polycomb [25].
2.2 SUV39h and disease
The SUV39h1 and SUV39h2 proteins are homologues of Drosophila SU(VAR)3-9, a SET domain histone methyltransferase involved in heterochromatin formation and gene silencing [49]. In mice, these two proteins are co-expressed during embryogenesis, but are differentially expressed in the adult, where SUV39h1 is present in multiple tissues and SUV39h2 is restricted to the testes [50]. The methylation of K9H3 provides a binding site for Heterochromatin Protein 1 (HP1), a structural protein enriched in heterochromatin (see below) [51,52] (Figure 1b). Loss of both SUV39h1 and SUV39h2 or HP1 leads to reduction of methylation at K20H4, a conserved hallmark of heterochromatin, suggesting sequential histone modifications occur in the process of heterochromatin formation [53].
The role of SUV39h proteins in development has been studied using mouse knock-out models. Mice lacking either Suv39h1 or Su39vh2 showed normal viability and fertility, suggesting redundancy of function between the two histone methyltransferases during the embryonic stage [54]. In Suv39h1 and Suv39h2 double knock-out mice, decreased viability was observed with death at embryonic day 12.5. Surviving mice exhibited slow growth and developed B cell lymphomas similar to non-Hodgkin's lymphoma [54]. In addition, males were infertile with a delay in meiosis. At the cellular level, chromosomal segregation defects and abnormally long telomeres containing reduced levels of trimethylated K9H3 and HP1 were observed. Similar phenotypes were apparent in mice upon over-expression of SUV39h1, including slow growth, cell cycle progression defects, chromosome missegregation, and mislocalization of HP1 [55,56]. Taken together, these studies demonstrate a critical role for proper levels of SUV39h1 in chromosome dynamics and development.
On a cellular level, increased K9H3 methylation is observed in differentiated, but not cycling cells [57]. In addition, over-expression of SUV39h1 in red blood cell progenitors caused abnormal cell cycle profiles and immortalization similar to that caused by loss of p53 [56]. Interestingly, p53 loss is normally accompanied by changes in karyotype. Despite the fact that SUV39h has been implicated in chromosome segregation, immortalized Suv39h1 over-expressing cells possessed normal karyotypes. However, these cells did show changes in the Retinoblastoma protein (Rb) signaling pathway, including down-regulation of p21 and up-regulation of Rb, accompanied by phosphorylation [56]. Rb is involved in transcriptional control of E2F-regulated cell cycle genes. Rb binds to SUV39h1 and HP1 to mediate gene silencing of target genes [58]. Importantly, Rb mutants that disrupt interactions with or cause mislocalization of SUV39h1 give rise to cancer [59].
Rb is regulated by oncogenic Ras [60]. Suv39h1 knock-out mice over-expressing a constitutively active oncogenic form of Ras in blood progenitor cells developed lymphoma [59]. Analysis of cells from these mice showed increased levels of p16INK4a, consistent with activation of the Rb pathway [60]. Interestingly, loss of SUV39h1 appeared to bypass the requirement for the loss of p53 in the formation of lymphoma, without evidence of aneuploidy due to chromosomal segregation defects. These results broadly suggest an anti-oncogenic role for SUV39h1 as an early barrier in the prevention of cancer in cases of unregulated Ras signaling [59]. Simultaneous treatment of cells expressing oncogenic Ras with the histone deacetylase inhibitor TSA and the DNA demethylation inhibitor 5'-aza-deoxycytodine resulted in increased cancer and early death, similar to the effects caused by the loss of SUV39h1 [59]. These findings suggest a connection between histone modifications and DNA methylation (Figure 1b), and suggest the possibility of adverse outcomes upon treatment with such inhibitors.
3. HP1 and disease
One function of K9H3 methylation is to serve as a binding site for HP1 (Figure 1b). HP1 is conserved among species, with mice and humans each possessing three genes encoding HP1-like proteins [61]. In humans these are referred to as HP1Hsα, HP1Hsβ and HP1Hsγ. These proteins share significant amino acid sequence identity, yet have distinct chromosomal localization patterns [61-63]. HP1 proteins contain a chromo domain that binds methylated K9H3, and a chromo shadow domain that dimerizes [64]. Dimerization establishes a platform in which nuclear proteins containing the PxVxL pentapeptide motif interact [65]. Association of HP1 with a target gene causes alterations in chromatin structure and gene silencing by mechanisms that are not completely understood [66,67].
Understanding the function of HP1 in chromosome dynamics and gene regulation has implications for determining disease mechanisms. A reduction in HP1 levels causes kinetochore defects, loss of chromosome cohesion and condensation, as well as aberrant chromosome segregation [68-73]. In addition to these defects associated with centromere function, telomere function is also impaired. In Drosophila, telomeric fusions are observed in mutants lacking HP1 [74]. In mammals, over-expression of HP1Hsα or HP1Hsβ, but not HP1Hsγ, results in telomere fusions and telomere shortening, due to reduced interactions with hTERT, the catalytic component of telomerase [75-77]. Thus, modulation of HP1 levels could contribute to aneuploidy and telomere fusion events that occur during cancer progression.
Taking into account the multiple roles HP1 proteins play in many aspects of nuclear function, it is easy to understand how a reduction in HP1 could impact processes related to cell growth and cancer progression. Connections between HP1 and breast cancer were derived from studies comparing breast cancer cell lines with different invasive/metastatic potential [78]. Invasion properties are based on an in vitro assay [79], whereas the ability to metastasize is based on behavior of cells following injection into an immune compromised mouse [80]. HP1Hsα was found to be down-regulated in highly invasive/metastatic cells compared with poorly invasive/non-metastatic breast cancer cells and metastasis tissue from breast cancer patients [81] (Table 1). A causal role in invasion has been demonstrated by modulation of HP1Hsα levels [82]. Increased levels of HP1Hsα in highly invasive/metastatic cells decreased invasion. In contrast, knock-down of HP1Hsα with RNAi in poorly invasive/non-metastatic cells increased invasion. Alterations in invasion were not accompanied by changes in cell growth rate [82]. However changes in gene expression were observed, suggesting HP1Hsα regulates genes required for an invasive phenotype (T.J.M and L.L.W, unpublished data). Collectively, these findings suggest that HP1Hsα is a member of a small class of proteins known as metastasis suppressors. Unlike tumor suppressors that alter growth of a primary tumor, metastasis suppressors regulate metastasis without affecting cancer cell growth [83,84]. Studies in which HP1Hsα levels are altered in human breast cancer cells and assayed for metastasis in a mouse model are needed to determine whether HP1Hsα is a metastasis suppressor. In addition to changes in the levels of HP1Hsα in breast cancer cells, decreased HP1Hsα correlates with tumor progression in multiple human cancers. HP1 _Hs_α mRNA levels are reduced in advanced forms of papillary thyroid carcinoma [85]. HP1Hsα mRNA is also down-regulated in medulloblastoma; this down-regulation correlates with treatment failure [86]. Thus, HP1Hsα might play a role in the progression of many types of cancer.
In addition to the connections between HP1 and cancer, viruses have usurped HP1 for functions related to replication within host cells (Table 1). In virally permissive cells, human cytomegalovirus lytic genes are expressed; in non-permissive cells HP1Hsβ association with viral promoter sequences and correlates with viral latency [87]. Upon differentiation into macrophages, HP1Hsβ association is lost, and gene activation occurs [87-89]. Similarly, in the case of Kaposi's sarcoma-associated herpes virus, an interaction of the viral latency-associated nuclear antigen with exogenously supplied mouse HP1α and HP1β caused transcriptional inhibition [90]. For HIV-1, transcriptional inhibition occurs by a different mechanism in which a complex consisting of CTIP2 and HP1 sequesters the viral _trans_-activator Tat [91]. In addition to these examples in which HP1 governs viral transcriptional control, the polyoma JC virus has co-opted HP1 by an independent mechanism. The viral encoded protein Agno subverts the association of HP1 with the nuclear envelope protein lamin B receptor (LBR) causing increased nuclear envelope flexibility and more efficient escape of viral particles [92]. Thus, viruses utilize HP1 for both transcriptional control and regulation of nuclear architecture.
4. Connections among epigenetic gene silencing systems
Through the course of investigating the role of epigenetic modifications involved in disease, connections among DNA methylation, histone acetylation and histone methylation have become apparent (Figure 1). Studies in model organisms have specifically revealed a connection between DNA methylation and histone methylation. In Neurospora, a screen for mutants that lacked CpG DNA methylation identified a K9H3 histone methyltransferase [93]. In Arabidopsis, mutants lacking a histone H3 methyltransferase show altered CpNpG methylation and reactivation of endogenous retrotransposons [94,95]. The reverse situation can also exist in which methylated CpG sites in Arabidopsis direct sites of K9H3 methylation [96-98]. Similarly, in mammals, methylation of CpG islands within pericentric repetitive DNA elements is directed by histone methylation [99-102]. Conversely, in some instances DNA methylation precedes histone modifications, as methyl-CpG binding proteins such as MeCP2 recruit histone deacetylases and histone methyltransferases [103-105]. In fact, HDACs interact with many factors involved in epigenetic silencing [106] (Figure 1). Thus, the interplay between DNA methylation and histone modifications appears to serve in reinforcing the transcriptionally silent state.
5. Dynamics of epigenetic modifications and therapy
The dynamic nature of epigenetic gene regulation is important to consider in the context of disease. Both the loss and gain of gene silencing at target genes can potentially be reversed through drug treatment [106]. Drugs that inhibit DNA methylation can reactivate silenced genes in cancer cells, possibly re-establishing cell cycle control [107]. Drugs that inhibit histone deacetylation block cell cycle progression and cause apoptosis by unknown mechanisms [107]. While HDAC inhibitors alter the abundance of acetylated histones, other possible targets that could account for the observed affects include acetylated chromatin proteins such as E2F and p53 that regulate cell cycle. This network of epigenetic modifications might favor transient drug treatment, as initial reversal of the transcriptional status of a gene could be propagated by other epigenetic modifiers in the absence of prolonged drug treatment. Alternatively, the connections among epigenetic modifications might hinder successful drug treatment. Evidence for this comes from a recent study in which drug treatment resulted in gene activation, yet a subset of the epigenetic gene silencing marks remained [108]. Such marks could serve as memory to seed subsequent silencing events in the absence of continual drug treatment. The recent discovery of histone demethylases provides additional drug targets for reversing the transcriptionally silent state [109]. A combination of drugs that alter DNA methylation, histone acetylation, and histone methylation might ultimately be the most effective means of combating disease.
Acknowledgements
We apologize to the many investigators whose research could not be cited due to space limitations. We would like to thank Al Klingelhutz and members of the Wallrath lab for comments on the manuscript ,and Judith Kassis for discussions. Research is supported by an NIH grant (GM61513) to L.L.W., a grant from the Department of Defense Breast Cancer Research Foundation (DAMD17-02-1-0424) to L.L.W. and a Susan G. Komen Dissertation Research Award (DISS0403121) to T.J.M.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ehrlich M, Buchanan KL, Tsien F, Jiang G, Sun B, Uicker W, Weemaes CM, Smeets D, Sperling K, Belohradsky BH, Tommerup N, Misek DE, Rouillard JM, Kuick R, Hanash SM. DNA methyltransferase 3B mutations linked to the ICF syndrome cause dysregulation of lymphogenesis genes. Hum Mol Genet. 2001;10:2917–2931. doi: 10.1093/hmg/10.25.2917. [DOI] [PubMed] [Google Scholar]
- 2.Wade PA. Methyl CpG-binding proteins and transcriptional repression. Bioessays. 2001;23:1131–1137. doi: 10.1002/bies.10008. [DOI] [PubMed] [Google Scholar]
- 3.Lund AH, van Lohuizen M. Epigenetics and cancer. Genes Dev. 2004;18:2315–2335. doi: 10.1101/gad.1232504. [DOI] [PubMed] [Google Scholar]
- 4.Boltze C, Schneider-Stock R, Quednow C, Hinze R, Mawrin C, Hribaschek A, Roessner A, Hoang-Vu C. Silencing of the maspin gene by promoter hypermethylation in thyroid cancer. Int J Mol Med. 2003;12:479–484. [PubMed] [Google Scholar]
- 5.Futscher BW, O'Meara MM, Kim CJ, Rennels MA, Lu D, Gruman LM, Seftor RE, Hendrix MJ, Domann FE. Aberrant methylation of the maspin promoter is an early event in human breast cancer. Neoplasia. 2004;6:380–389. doi: 10.1593/neo.04115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yatabe Y, Mitsudomi T, Takahashi T. Maspin expression in normal lung and non-small-cell lung cancers: cellular property-associated expression under the control of promoter DNA methylation. Oncogene. 2004;23:4041–4049. doi: 10.1038/sj.onc.1207557. [DOI] [PubMed] [Google Scholar]
- 7.Delaval K, Wagschal A, Feil R. Epigenetic deregulation of imprinting in congenital diseases of aberrant growth. Bioessays. 2006;28:453–459. doi: 10.1002/bies.20407. [DOI] [PubMed] [Google Scholar]
- 8.Lewis A, Reik W. How imprinting centres work. Cytogenet Genome Res. 2006;113:81–89. doi: 10.1159/000090818. [DOI] [PubMed] [Google Scholar]
- 9.Biniszkiewicz D, Gribnau J, Ramsahoye B, Gaudet F, Eggan K, Humpherys D, Mastrangelo MA, Jun Z, Walter J, Jaenisch R. Dnmt1 overexpression causes genomic hypermethylation, loss of imprinting, and embryonic lethality. Mol Cell Biol. 2002;22:2124–2135. doi: 10.1128/MCB.22.7.2124-2135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bell AC, Felsenfeld G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature. 2000;405:482–485. doi: 10.1038/35013100. [DOI] [PubMed] [Google Scholar]
- 11.Schonherr N, Meyer E, Eggermann K, Ranke MB, Wollmann HA, Eggermann T. (Epi)mutations in 11p15 significantly contribute to Silver-Russell syndrome: but are they generally involved in growth retardation? Eur J Med Genet. 2006 doi: 10.1016/j.ejmg.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 12.Hassan AH, Neely KE, Vignali M, Reese JC, Workman JL. Promoter targeting of chromatin-modifying complexes. Front Biosci. 2001;6:D1054–1064. doi: 10.2741/hassan. [DOI] [PubMed] [Google Scholar]
- 13.Bickmore WA, van der Maarel SM. Perturbations of chromatin structure in human genetic disease: recent advances. Hum Mol Genet. 2003;12(Spec No 2):R207–213. doi: 10.1093/hmg/ddg260. [DOI] [PubMed] [Google Scholar]
- 14.Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet. 2002;3:662–673. doi: 10.1038/nrg887. [DOI] [PubMed] [Google Scholar]
- 15.Moretti P, Zoghbi HY. MeCP2 dysfunction in Rett syndrome and related disorders. Curr Opin Genet Dev. 2006 doi: 10.1016/j.gde.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 16.Tudor M, Akbarian S, Chen RZ, Jaenisch R. Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc Natl Acad Sci U S A. 2002;99:15536–15541. doi: 10.1073/pnas.242566899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 18.Sun YE, Wu H. The ups and downs of BDNF in Rett syndrome. Neuron. 2006;49:321–323. doi: 10.1016/j.neuron.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 19.Young JI, Hong EP, Castle JC, Crespo-Barreto J, Bowman AB, Rose MF, Kang D, Richman R, Johnson JM, Berget S, Zoghbi HY. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc Natl Acad Sci U S A. 2005;102:17551–17558. doi: 10.1073/pnas.0507856102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peterson CL, Laniel MA. Histones and histone modifications. Curr Biol. 2004;14:R546–551. doi: 10.1016/j.cub.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 21.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 22.Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, Iyer NG, Perez-Rosado A, Calvo E, Lopez JA, Cano A, Calasanz MJ, Colomer D, Piris MA, Ahn N, Imhof A, Caldas C, Jenuwein T, Esteller M. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 23.Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16:2893–2905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, Miller EL, O'Connor MB, Kingston RE, Simon JA. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002;111:197–208. doi: 10.1016/s0092-8674(02)00976-5. [DOI] [PubMed] [Google Scholar]
- 25.Levine SS, King IF, Kingston RE. Division of labor in polycomb group repression. Trends Biochem Sci. 2004;29:478–485. doi: 10.1016/j.tibs.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 26.van der Vlag J, Otte AP. Transcriptional repression mediated by the human polycomb-group protein EED involves histone deacetylation. Nat Genet. 1999;23:474–478. doi: 10.1038/70602. [DOI] [PubMed] [Google Scholar]
- 27.Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, Bollen M, Esteller M, Di Croce L, de Launoit Y, Fuks F. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439:871–874. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- 28.Taghavi P, van Lohuizen M. Developmental biology: two paths to silence merge. Nature. 2006;439:794–795. doi: 10.1038/439794a. [DOI] [PubMed] [Google Scholar]
- 29.Su IH, Basavaraj A, Krutchinsky AN, Hobert O, Ullrich A, Chait BT, Tarakhovsky A. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat Immunol. 2003;4:124–131. doi: 10.1038/ni876. [DOI] [PubMed] [Google Scholar]
- 30.O'Carroll D, Erhardt S, Pagani M, Barton SC, Surani MA, Jenuwein T. The polycomb-group gene Ezh2 is required for early mouse development. Mol Cell Biol. 2001;21:4330–4336. doi: 10.1128/MCB.21.13.4330-4336.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006;20:1123–1136. doi: 10.1101/gad.381706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 33.Buszczak M, Spradling AC. Searching chromatin for stem cell identity. Cell. 2006;125:233–236. doi: 10.1016/j.cell.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 34.Sellers WR, Loda M. The EZH2 polycomb transcriptional repressor--a marker or mover of metastatic prostate cancer? Cancer Cell. 2002;2:349–350. doi: 10.1016/s1535-6108(02)00187-3. [DOI] [PubMed] [Google Scholar]
- 35.Tang X, Milyavsky M, Shats I, Erez N, Goldfinger N, Rotter V. Activated p53 suppresses the histone methyltransferase EZH2 gene. Oncogene. 2004;23:5759–5769. doi: 10.1038/sj.onc.1207706. [DOI] [PubMed] [Google Scholar]
- 36.Tonini T, Bagella L, D'Andrilli G, Claudio PP, Giordano A. Ezh2 reduces the ability of HDAC1-dependent pRb2/p130 transcriptional repression of cyclin A. Oncogene. 2004;23:4930–4937. doi: 10.1038/sj.onc.1207608. [DOI] [PubMed] [Google Scholar]
- 37.Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22:5323–5335. doi: 10.1093/emboj/cdg542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saramaki OR, Tammela TL, Martikainen PM, Vessella RL, Visakorpi T. The gene for polycomb group protein enhancer of zeste homolog 2 (EZH2) is amplified in late-stage prostate cancer. Genes Chromosomes Cancer. 2006;45:639–645. doi: 10.1002/gcc.20327. [DOI] [PubMed] [Google Scholar]
- 39.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, Rubin MA, Chinnaiyan AM. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 40.Rhodes DR, Sanda MG, Otte AP, Chinnaiyan AM, Rubin MA. Multiplex biomarker approach for determining risk of prostate-specific antigen-defined recurrence of prostate cancer. J Natl Cancer Inst. 2003;95:661–668. doi: 10.1093/jnci/95.9.661. [DOI] [PubMed] [Google Scholar]
- 41.Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, Sabel MS, Livant D, Weiss SJ, Rubin MA, Chinnaiyan AM. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A. 2003;100:11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ding L, Erdmann C, Chinnaiyan AM, Merajver SD, Kleer CG. Identification of EZH2 as a molecular marker for a precancerous state in morphologically normal breast tissues. Cancer Res. 2006;66:4095–4099. doi: 10.1158/0008-5472.CAN-05-4300. [DOI] [PubMed] [Google Scholar]
- 43.Hobert O, Sures I, Ciossek T, Fuchs M, Ullrich A. Isolation and developmental expression analysis of Enx-1, a novel mouse Polycomb group gene. Mech Dev. 1996;55:171–184. doi: 10.1016/0925-4773(96)00499-6. [DOI] [PubMed] [Google Scholar]
- 44.Fukuyama T, Otsuka T, Shigematsu H, Uchida N, Arima F, Ohno Y, Iwasaki H, Fukuda T, Niho Y. Proliferative involvement of ENX-1, a putative human polycomb group gene, in haematopoietic cells. Br J Haematol. 2000;108:842–847. doi: 10.1046/j.1365-2141.2000.01914.x. [DOI] [PubMed] [Google Scholar]
- 45.Raaphorst FM, Meijer CJ, Fieret E, Blokzijl T, Mommers E, Buerger H, Packeisen J, Sewalt RA, Otte AP, van Diest PJ. Poorly differentiated breast carcinoma is associated with increased expression of the human polycomb group EZH2 gene. Neoplasia. 2003;5:481–488. doi: 10.1016/s1476-5586(03)80032-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dukers DF, van Galen JC, Giroth C, Jansen P, Sewalt RG, Otte AP, Kluin-Nelemans HC, Meijer CJ, Raaphorst FM. Unique polycomb gene expression pattern in Hodgkin's lymphoma and Hodgkin's lymphoma-derived cell lines. Am J Pathol. 2004;164:873–881. doi: 10.1016/S0002-9440(10)63175-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Galen JC, Dukers DF, Giroth C, Sewalt RG, Otte AP, Meijer CJ, Raaphorst FM. Distinct expression patterns of polycomb oncoproteins and their binding partners during the germinal center reaction. Eur J Immunol. 2004;34:1870–1881. doi: 10.1002/eji.200424985. [DOI] [PubMed] [Google Scholar]
- 48.Sewalt RG, Lachner M, Vargas M, Hamer KM, den Blaauwen JL, Hendrix T, Melcher M, Schweizer D, Jenuwein T, Otte AP. Selective interactions between vertebrate polycomb homologs and the SUV39H1 histone lysine methyltransferase suggest that histone H3-K9 methylation contributes to chromosomal targeting of Polycomb group proteins. Mol Cell Biol. 2002;22:5539–5553. doi: 10.1128/MCB.22.15.5539-5553.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 50.O'Carroll D, Scherthan H, Peters AH, Opravil S, Haynes AR, Laible G, Rea S, Schmid M, Lebersorger A, Jerratsch M, Sattler L, Mattei MG, Denny P, Brown SD, Schweizer D, Jenuwein T. Isolation and characterization of Suv39h2, a second histone H3 methyltransferase gene that displays testis-specific expression. Mol Cell Biol. 2000;20:9423–9433. doi: 10.1128/mcb.20.24.9423-9433.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 52.Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–120. doi: 10.1038/35065132. [DOI] [PubMed] [Google Scholar]
- 53.Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G, Reinberg D, Jenuwein T. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004;18:1251–1262. doi: 10.1101/gad.300704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peters AH, O'Carroll D, Scherthan H, Mechtler K, Sauer S, Schofer C, Weipoltshammer K, Pagani M, Lachner M, Kohlmaier A, Opravil S, Doyle M, Sibilia M, Jenuwein T. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell. 2001;107:323–337. doi: 10.1016/s0092-8674(01)00542-6. [DOI] [PubMed] [Google Scholar]
- 55.Melcher M, Schmid M, Aagaard L, Selenko P, Laible G, Jenuwein T. Structure-function analysis of SUV39H1 reveals a dominant role in heterochromatin organization, chromosome segregation, and mitotic progression. Mol Cell Biol. 2000;20:3728–3741. doi: 10.1128/mcb.20.10.3728-3741.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Czvitkovich S, Sauer S, Peters AH, Deiner E, Wolf A, Laible G, Opravil S, Beug H, Jenuwein T. Over-expression of the SUV39H1 histone methyltransferase induces altered proliferation and differentiation in transgenic mice. Mech Dev. 2001;107:141–153. doi: 10.1016/s0925-4773(01)00464-6. [DOI] [PubMed] [Google Scholar]
- 57.Ait-Si-Ali S, Guasconi V, Fritsch L, Yahi H, Sekhri R, Naguibneva I, Robin P, Cabon F, Polesskaya A, Harel-Bellan A. A Suv39h-dependent mechanism for silencing S-phase genes in differentiating but not in cycling cells. EMBO J. 2004;23:605–615. doi: 10.1038/sj.emboj.7600074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nielsen SJ, Schneider R, Bauer UM, Bannister AJ, Morrison A, O'Carroll D, Firestein R, Cleary M, Jenuwein T, Herrera RE, Kouzarides T. Rb targets histone H3 methylation and HP1 to promoters. Nature. 2001;412:561–565. doi: 10.1038/35087620. [DOI] [PubMed] [Google Scholar]
- 59.Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, Stein H, Dorken B, Jenuwein T, Schmitt CA. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–665. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- 60.Palmero I, Pantoja C, Serrano M. p19ARF links the tumour suppressor p53 to Ras. Nature. 1998;395:125–126. doi: 10.1038/25870. [DOI] [PubMed] [Google Scholar]
- 61.Li Y, Kirschmann DA, Wallrath LL. Does heterochromatin protein 1 always follow code? Proc Natl Acad Sci U S A. 2002;99(Suppl 4):16462–16469. doi: 10.1073/pnas.162371699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nielsen AL, Oulad-Abdelghani M, Ortiz JA, Remboutsika E, Chambon P, Losson R. Heterochromatin formation in mammalian cells: interaction between histones and HP1 proteins. Mol Cell. 2001;7:729–739. doi: 10.1016/s1097-2765(01)00218-0. [DOI] [PubMed] [Google Scholar]
- 63.Minc E, Allory Y, Worman HJ, Courvalin JC, Buendia B. Localization and phosphorylation of HP1 proteins during the cell cycle in mammalian cells. Chromosoma. 1999;108:220–234. doi: 10.1007/s004120050372. [DOI] [PubMed] [Google Scholar]
- 64.Cowieson NP, Partridge JF, Allshire RC, McLaughlin PJ. Dimerisation of a chromo shadow domain and distinctions from the chromodomain as revealed by structural analysis. Curr Biol. 2000;10:517–525. doi: 10.1016/s0960-9822(00)00467-x. [DOI] [PubMed] [Google Scholar]
- 65.Thiru A, Nietlispach D, Mott HR, Okuwaki M, Lyon D, Nielsen PR, Hirshberg M, Verreault A, Murzina NV, Laue ED. Structural basis of HP1/PXVXL motif peptide interactions and HP1 localisation to heterochromatin. EMBO J. 2004;23:489–499. doi: 10.1038/sj.emboj.7600088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Danzer JR, Wallrath LL. Mechanisms of HP1-mediated gene silencing in Drosophila. Development. 2004;131:3571–3580. doi: 10.1242/dev.01223. [DOI] [PubMed] [Google Scholar]
- 67.Wallrath LL, Elgin SC. Position effect variegation in Drosophila is associated with an altered chromatin structure. Genes Dev. 1995;9:1263–1277. doi: 10.1101/gad.9.10.1263. [DOI] [PubMed] [Google Scholar]
- 68.Bernard P, Maure JF, Partridge JF, Genier S, Javerzat JP, Allshire RC. Requirement of heterochromatin for cohesion at centromeres. Science. 2001;294:2539–2542. doi: 10.1126/science.1064027. [DOI] [PubMed] [Google Scholar]
- 69.Ekwall K, Javerzat JP, Lorentz A, Schmidt H, Cranston G, Allshire R. The chromodomain protein Swi6: a key component at fission yeast centromeres. Science. 1995;269:1429–1431. doi: 10.1126/science.7660126. [DOI] [PubMed] [Google Scholar]
- 70.Nonaka N, Kitajima T, Yokobayashi S, Xiao G, Yamamoto M, Grewal SI, Watanabe Y. Recruitment of cohesin to heterochromatic regions by Swi6/HP1 in fission yeast. Nat Cell Biol. 2002;4:89–93. doi: 10.1038/ncb739. [DOI] [PubMed] [Google Scholar]
- 71.Obuse C, Iwasaki O, Kiyomitsu T, Goshima G, Toyoda Y, Yanagida M. A conserved Mis12 centromere complex is linked to heterochromatic HP1 and outer kinetochore protein Zwint-1. Nat Cell Biol. 2004;6:1135–1141. doi: 10.1038/ncb1187. [DOI] [PubMed] [Google Scholar]
- 72.Ainsztein AM, Kandels-Lewis SE, Mackay AM, Earnshaw WC. INCENP centromere and spindle targeting: identification of essential conserved motifs and involvement of heterochromatin protein HP1. J Cell Biol. 1998;143:1763–1774. doi: 10.1083/jcb.143.7.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vagnarelli PB, Earnshaw WC. INCENP loss from an inactive centromere correlates with the loss of sister chromatid cohesion. Chromosoma. 2001;110:393–401. doi: 10.1007/s004120100163. [DOI] [PubMed] [Google Scholar]
- 74.Fanti L, Giovinazzo G, Berloco M, Pimpinelli S. The heterochromatin protein 1 prevents telomere fusions in Drosophila. Mol Cell. 1998;2:527–538. doi: 10.1016/s1097-2765(00)80152-5. [DOI] [PubMed] [Google Scholar]
- 75.Cenci G, Ciapponi L, Gatti M. The mechanism of telomere protection: a comparison between Drosophila and humans. Chromosoma. 2005;114:135–145. doi: 10.1007/s00412-005-0005-9. [DOI] [PubMed] [Google Scholar]
- 76.Sharma GG, Hwang KK, Pandita RK, Gupta A, Dhar S, Parenteau J, Agarwal M, Worman HJ, Wellinger RJ, Pandita TK. Human heterochromatin protein 1 isoforms HP1(Hsalpha) and HP1(Hsbeta) interfere with hTERT-telomere interactions and correlate with changes in cell growth and response to ionizing radiation. Mol Cell Biol. 2003;23:8363–8376. doi: 10.1128/MCB.23.22.8363-8376.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Song K, Jung Y, Jung D, Lee I. Human Ku70 interacts with heterochromatin protein 1alpha. J Biol Chem. 2001;276:8321–8327. doi: 10.1074/jbc.M008779200. [DOI] [PubMed] [Google Scholar]
- 78.Kirschmann DA, Seftor EA, Nieva DR, Mariano EA, Hendrix MJ. Differentially expressed genes associated with the metastatic phenotype in breast cancer. Breast Cancer Res Treat. 1999;55:127–136. doi: 10.1023/a:1006188129423. [DOI] [PubMed] [Google Scholar]
- 79.Gehlsen KR, Wagner HN, Jr, Hendrix MJ. Membrane invasion culture system (MICS) Med Instrum. 1984;18:268–271. [PubMed] [Google Scholar]
- 80.Khanna C, Hunter K. Modeling metastasis in vivo. Carcinogenesis. 2005;26:513–523. doi: 10.1093/carcin/bgh261. [DOI] [PubMed] [Google Scholar]
- 81.Kirschmann DA, Lininger RA, Gardner LM, Seftor EA, Odero VA, Ainsztein AM, Earnshaw WC, Wallrath LL, Hendrix MJ. Down-regulation of HP1Hsalpha expression is associated with the metastatic phenotype in breast cancer. Cancer Res. 2000;60:3359–3363. [PubMed] [Google Scholar]
- 82.Norwood LE, Moss TJ, Margaryan NV, Cook SL, Wright L, Seftor EA, Hendrix MJ, Kirschmann DA, Wallrath LL. A requirement for dimerization of HP1Hsalpha in suppression of breast cancer invasion. J Biol Chem. 2006 doi: 10.1074/jbc.M512454200. [DOI] [PubMed] [Google Scholar]
- 83.Berger JC, Vander Griend DJ, Robinson VL, Hickson JA, Rinker-Schaeffer CW. Metastasis Suppressor Genes: From Gene Identification to Protein Function and Regulation. Cancer Biol Ther. 2005;4 doi: 10.4161/cbt.4.8.1865. [DOI] [PubMed] [Google Scholar]
- 84.Steeg PS, Ouatas T, Halverson D, Palmieri D, Salerno M. Metastasis suppressor genes: basic biology and potential clinical use. Clin Breast Cancer. 2003;4:51–62. doi: 10.3816/cbc.2003.n.012. [DOI] [PubMed] [Google Scholar]
- 85.Wasenius VM, Hemmer S, Kettunen E, Knuutila S, Franssila K, Joensuu H. Hepatocyte growth factor receptor, matrix metalloproteinase-11, tissue inhibitor of metalloproteinase-1, and fibronectin are up-regulated in papillary thyroid carcinoma: a cDNA and tissue microarray study. Clin Cancer Res. 2003;9:68–75. [PubMed] [Google Scholar]
- 86.Pomeroy SL, Tamayo P, Gaasenbeek M, Sturla LM, Angelo M, McLaughlin ME, Kim JY, Goumnerova LC, Black PM, Lau C, Allen JC, Zagzag D, Olson JM, Curran T, Wetmore C, Biegel JA, Poggio T, Mukherjee S, Rifkin R, Califano A, Stolovitzky G, Louis DN, Mesirov JP, Lander ES, Golub TR. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature. 2002;415:436–442. doi: 10.1038/415436a. [DOI] [PubMed] [Google Scholar]
- 87.Murphy JC, Fischle W, Verdin E, Sinclair JH. Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J. 2002;21:1112–1120. doi: 10.1093/emboj/21.5.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Reeves MB, Lehner PJ, Sissons JG, Sinclair JH. An in vitro model for the regulation of human cytomegalovirus latency and reactivation in dendritic cells by chromatin remodelling. J Gen Virol. 2005;86:2949–2954. doi: 10.1099/vir.0.81161-0. [DOI] [PubMed] [Google Scholar]
- 89.Reeves MB, MacAry PA, Lehner PJ, Sissons JG, Sinclair JH. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc Natl Acad Sci U S A. 2005;102:4140–4145. doi: 10.1073/pnas.0408994102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lim C, Lee D, Seo T, Choi C, Choe J. Latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus functionally interacts with heterochromatin protein 1. J Biol Chem. 2003;278:7397–7405. doi: 10.1074/jbc.M211912200. [DOI] [PubMed] [Google Scholar]
- 91.Rohr O, Lecestre D, Chasserot-Golaz S, Marban C, Avram D, Aunis D, Leid M, Schaeffer E. Recruitment of Tat to heterochromatin protein HP1 via interaction with CTIP2 inhibits human immunodeficiency virus type 1 replication in microglial cells. J Virol. 2003;77:5415–5427. doi: 10.1128/JVI.77.9.5415-5427.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Okada Y, Suzuki T, Sunden Y, Orba Y, Kose S, Imamoto N, Takahashi H, Tanaka S, Hall WW, Nagashima K, Sawa H. Dissociation of heterochromatin protein 1 from lamin B receptor induced by human polyomavirus agnoprotein: role in nuclear egress of viral particles. EMBO. Rep. 2005;6:452–457. doi: 10.1038/sj.embor.7400406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tamaru H, Selker EU. A histone H3 methyltransferase controls DNA methylation in Neurospora crassa. Nature. 2001;414:277–283. doi: 10.1038/35104508. [DOI] [PubMed] [Google Scholar]
- 94.Jackson JP, Lindroth AM, Cao X, Jacobsen SE. Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature. 2002;416:556–560. doi: 10.1038/nature731. [DOI] [PubMed] [Google Scholar]
- 95.Malagnac F, Bartee L, Bender J. An Arabidopsis SET domain protein required for maintenance but not establishment of DNA methylation. EMBO J. 2002;21:6842–6852. doi: 10.1093/emboj/cdf687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Johnson L, Cao X, Jacobsen S. Interplay between two epigenetic marks. DNA methylation and histone H3 lysine 9 methylation. Curr Biol. 2002;12:1360–1367. doi: 10.1016/s0960-9822(02)00976-4. [DOI] [PubMed] [Google Scholar]
- 97.Tariq M, Saze H, Probst AV, Lichota J, Habu Y, Paszkowski J. Erasure of CpG methylation in Arabidopsis alters patterns of histone H3 methylation in heterochromatin. Proc Natl Acad Sci U S A. 2003;100:8823–8827. doi: 10.1073/pnas.1432939100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Soppe WJ, Jasencakova Z, Houben A, Kakutani T, Meister A, Huang MS, Jacobsen SE, Schubert I, Fransz PF. DNA methylation controls histone H3 lysine 9 methylation and heterochromatin assembly in Arabidopsis. EMBO J. 2002;21:6549–6559. doi: 10.1093/emboj/cdf657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lehnertz B, Ueda Y, Derijck AA, Braunschweig U, Perez-Burgos L, Kubicek S, Chen T, Li E, Jenuwein T, Peters AH. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr Biol. 2003;13:1192–1200. doi: 10.1016/s0960-9822(03)00432-9. [DOI] [PubMed] [Google Scholar]
- 100.Yan Q, Cho E, Lockett S, Muegge K. Association of Lsh, a regulator of DNA methylation, with pericentromeric heterochromatin is dependent on intact heterochromatin. Mol Cell Biol. 2003;23:8416–8428. doi: 10.1128/MCB.23.23.8416-8428.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yan Q, Huang J, Fan T, Zhu H, Muegge K. Lsh, a modulator of CpG methylation, is crucial for normal histone methylation. EMBO J. 2003;22:5154–5162. doi: 10.1093/emboj/cdg493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Freitag M, Selker EU. Controlling DNA methylation: many roads to one modification. Curr Opin Genet Dev. 2005;15:191–199. doi: 10.1016/j.gde.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 103.Bird AP, Wolffe AP. Methylation-induced repression--belts, braces, and chromatin. Cell. 1999;99:451–454. doi: 10.1016/s0092-8674(00)81532-9. [DOI] [PubMed] [Google Scholar]
- 104.Fuks F, Hurd PJ, Deplus R, Kouzarides T. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 2003;31:2305–2312. doi: 10.1093/nar/gkg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem. 2003;278:4035–4040. doi: 10.1074/jbc.M210256200. [DOI] [PubMed] [Google Scholar]
- 106.Inche AG, La Thangue NB. Chromatin control and cancer-drug discovery: realizing the promise. Drug Discov Today. 2006;11:97–109. doi: 10.1016/S1359-6446(05)03691-3. [DOI] [PubMed] [Google Scholar]
- 107.Suzuki T, Miyata N. Epigenetic control using natural products and synthetic molecules. Curr Med Chem. 2006;13:935–958. doi: 10.2174/092986706776361067. [DOI] [PubMed] [Google Scholar]
- 108.McGarvey KM, Fahrner JA, Greene E, Martens J, Jenuwein T, Baylin SB. Silenced tumor suppressor genes reactivated by DNA demethylation do not return to a fully euchromatic chromatin state. Cancer Res. 2006;66:3541–3549. doi: 10.1158/0008-5472.CAN-05-2481. [DOI] [PubMed] [Google Scholar]
- 109.Trojer P, Reinberg D. Histone lysine demethylases and their impact on epigenetics. Cell. 2006;125:213–217. doi: 10.1016/j.cell.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 110.Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 111.Lyko F, Ramsahoye BH, Kashevsky H, Tudor M, Mastrangelo MA, Orr-Weaver TL, Jaenisch R. Mammalian (cytosine-5) methyltransferases cause genomic DNA methylation and lethality in Drosophila. Nat Genet. 1999;23:363–366. doi: 10.1038/15551. [DOI] [PubMed] [Google Scholar]
- 112.Howell CY, Bestor TH, Ding F, Latham KE, Mertineit C, Trasler JM, Chaillet JR. Genomic imprinting disrupted by a maternal effect mutation in the Dnmt1 gene. Cell. 2001;104:829–838. doi: 10.1016/s0092-8674(01)00280-x. [DOI] [PubMed] [Google Scholar]
- 113.el-Deiry WS, Nelkin BD, Celano P, Yen RW, Falco JP, Hamilton SR, Baylin SB. High expression of the DNA methyltransferase gene characterizes human neoplastic cells and progression stages of colon cancer. Proc Natl Acad Sci U S A. 1991;88:3470–3474. doi: 10.1073/pnas.88.8.3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Paz MF, Wei S, Cigudosa JC, Rodriguez-Perales S, Peinado MA, Huang TH, Esteller M. Genetic unmasking of epigenetically silenced tumor suppressor genes in colon cancer cells deficient in DNA methyltransferases. Hum Mol Genet. 2003;12:2209–2219. doi: 10.1093/hmg/ddg226. [DOI] [PubMed] [Google Scholar]
- 115.Rhee I, Jair KW, Yen RW, Lengauer C, Herman JG, Kinzler KW, Vogelstein B, Baylin SB, Schuebel KE. CpG methylation is maintained in human cancer cells lacking DNMT1. Nature. 2000;404:1003–1007. doi: 10.1038/35010000. [DOI] [PubMed] [Google Scholar]
- 116.Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, Perez-Melgosa M, Sweetser MT, Schlissel MS, Nguyen S, Cherry SR, Tsai JH, Tucker SM, Weaver WM, Kelso A, Jaenisch R, Wilson CB. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 2001;15:763–774. doi: 10.1016/s1074-7613(01)00227-8. [DOI] [PubMed] [Google Scholar]
- 117.Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, Leonhardt H, Jaenisch R. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300:489–492. doi: 10.1126/science.1083558. [DOI] [PubMed] [Google Scholar]
- 118.Mizuno S, Chijiwa T, Okamura T, Akashi K, Fukumaki Y, Niho Y, Sasaki H. Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood. 2001;97:1172–1179. doi: 10.1182/blood.v97.5.1172. [DOI] [PubMed] [Google Scholar]
- 119.Peng DF, Kanai Y, Sawada M, Ushijima S, Hiraoka N, Kosuge T, Hirohashi S. Increased DNA methyltransferase 1 (DNMT1) protein expression in precancerous conditions and ductal carcinomas of the pancreas. Cancer Sci. 2005;96:403–408. doi: 10.1111/j.1349-7006.2005.00071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tate P, Skarnes W, Bird A. The methyl-CpG binding protein MeCP2 is essential for embryonic development in the mouse. Nat Genet . 1996;12:205–208. doi: 10.1038/ng0296-205. [DOI] [PubMed] [Google Scholar]
- 121.Xu GL, Bestor TH, Bourc'his D, Hsieh CL, Tommerup N, Bugge M, Hulten M, Qu X, Russo JJ, Viegas-Pequignot E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 1999;402:187–191. doi: 10.1038/46052. [DOI] [PubMed] [Google Scholar]
- 122.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 123.Jiang YL, Rigolet M, Bourc'his D, Nigon F, Bokesoy I, Fryns JP, Hulten M, Jonveaux P, Maraschio P, Megarbane A, Moncla A, Viegas-Pequignot E. DNMT3B mutations and DNA methylation defect define two types of ICF syndrome. Hum Mutat. 2005;25:56–63. doi: 10.1002/humu.20113. [DOI] [PubMed] [Google Scholar]
- 124.Beaulieu N, Morin S, Chute IC, Robert MF, Nguyen H, MacLeod AR. An essential role for DNA methyltransferase DNMT3B in cancer cell survival. J Biol Chem. 2002;277:28176–28181. doi: 10.1074/jbc.M204734200. [DOI] [PubMed] [Google Scholar]
- 125.Girault I, Tozlu S, Lidereau R, Bieche I. Expression analysis of DNA methyltransferases 1, 3A, and 3B in sporadic breast carcinomas. Clin Cancer Res. 2003;9:4415–4422. [PubMed] [Google Scholar]
- 126.Saito Y, Kanai Y, Sakamoto M, Saito H, Ishii H, Hirohashi S. Expression of mRNA for DNA methyltransferases and methyl-CpG-binding proteins and DNA methylation status on CpG islands and pericentromeric satellite regions during human hepatocarcinogenesis. Hepatology. 2001;33:561–568. doi: 10.1053/jhep.2001.22507. [DOI] [PubMed] [Google Scholar]
- 127.Saito Y, Kanai Y, Sakamoto M, Saito H, Ishii H, Hirohashi S. Overexpression of a splice variant of DNA methyltransferase 3b, DNMT3b4, associated with DNA hypomethylation on pericentromeric satellite regions during human hepatocarcinogenesis. Proc Natl Acad Sci U S A. 2002;99:10060–10065. doi: 10.1073/pnas.152121799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lee SJ, Jeon HS, Jang JS, Park SH, Lee GY, Lee BH, Kim CH, Kang YM, Lee WK, Kam S, Park RW, Kim IS, Cho YL, Jung TH, Park JY. DNMT3B polymorphisms and risk of primary lung cancer. Carcinogenesis. 2005;26:403–409. doi: 10.1093/carcin/bgh307. [DOI] [PubMed] [Google Scholar]
- 129.Shen H, Wang L, Spitz MR, Hong WK, Mao L, Wei Q. A novel polymorphism in human cytosine DNA-methyltransferase-3B promoter is associated with an increased risk of lung cancer. Cancer Res. 2002;62:4992–4995. [PubMed] [Google Scholar]
- 130.Garcia-Cao M, O'Sullivan R, Peters AH, Jenuwein T, Blasco MA. Epigenetic regulation of telomere length in mammalian cells by the Suv39h1 and Suv39h2 histone methyltransferases. Nat Genet. 2004;36:94–99. doi: 10.1038/ng1278. [DOI] [PubMed] [Google Scholar]
- 131.Muller HM, Fiegl H, Goebel G, Hubalek MM, Widschwendter A, Muller-Holzner E, Marth C, Widschwendter M. MeCP2 and MBD2 expression in human neoplastic and non-neoplastic breast tissue and its association with oestrogen receptor status. Br J Cancer. 2003;89:1934–1939. doi: 10.1038/sj.bjc.6601392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 133.Christodoulou J, Grimm A, Maher T, Bennetts B. RettBASE: The IRSA MECP2 variation database-a new mutation database in evolution. Hum Mutat. 2003;21:466–472. doi: 10.1002/humu.10194. [DOI] [PubMed] [Google Scholar]
- 134.Merola E, Mattioli E, Minimo C, Zuo W, Rabitti C, Cicala M, Caviglia R, Pollice L, Gabbrielli A, Giordano A, Claudio PP. Immunohistochemical evaluation of pRb2/p130, VEGF, EZH2, p53, p16, p21waf-1, p27, and PCNA in Barrett's esophagus. J Cell Physiol. 2006;207:512–519. doi: 10.1002/jcp.20590. [DOI] [PubMed] [Google Scholar]
- 135.Arisan S, Buyuktuncer ED, Palavan-Unsal N, Caskurlu T, Cakir OO, Ergenekon E. Increased expression of EZH2, a polycomb group protein, in bladder carcinoma. Urol Int. 2005;75:252–257. doi: 10.1159/000087804. [DOI] [PubMed] [Google Scholar]
- 136.Raman JD, Mongan NP, Tickoo SK, Boorjian SA, Scherr DS, Gudas LJ. Increased expression of the polycomb group gene, EZH2, in transitional cell carcinoma of the bladder. Clin Cancer Res . 2005;11:8570–8576. doi: 10.1158/1078-0432.CCR-05-1047. [DOI] [PubMed] [Google Scholar]
- 137.Bachmann IM, Halvorsen OJ, Collett K, Stefansson IM, Straume O, Haukaas SA, Salvesen HB, Otte AP, Akslen LA. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol. 2006;24:268–273. doi: 10.1200/JCO.2005.01.5180. [DOI] [PubMed] [Google Scholar]
- 138.Mimori K, Ogawa K, Okamoto M, Sudo T, Inoue H, Mori M. Clinical significance of enhancer of zeste homolog 2 expression in colorectal cancer cases. Eur J Surg Oncol. 2005;31:376–380. doi: 10.1016/j.ejso.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 139.van Kemenade FJ, Raaphorst FM, Blokzijl T, Fieret E, Hamer KM, Satijn DP, Otte AP, Meijer CJ. Coexpression of BMI-1 and EZH2 polycomb-group proteins is associated with cycling cells and degree of malignancy in B-cell non-Hodgkin lymphoma. Blood. 2001;97:3896–3901. doi: 10.1182/blood.v97.12.3896. [DOI] [PubMed] [Google Scholar]
- 140.Visser HP, Gunster MJ, Kluin-Nelemans HC, Manders EM, Raaphorst FM, Meijer CJ, Willemze R, Otte AP. The Polycomb group protein EZH2 is upregulated in proliferating, cultured human mantle cell lymphoma. Br J Haematol. 2001;112:950–958. doi: 10.1046/j.1365-2141.2001.02641.x. [DOI] [PubMed] [Google Scholar]
- 141.Croonquist PA, Van Ness B. The polycomb group protein enhancer of zeste homolog 2 (EZH 2) is an oncogene that influences myeloma cell growth and the mutant ras phenotype. Oncogene. 2005;24:6269–6280. doi: 10.1038/sj.onc.1208771. [DOI] [PubMed] [Google Scholar]
- 142.Sudo T, Utsunomiya T, Mimori K, Nagahara H, Ogawa K, Inoue H, Wakiyama S, Fujita H, Shirouzu K, Mori M. Clinicopathological significance of EZH2 mRNA expression in patients with hepatocellular carcinoma. Br J Cancer. 2005;92:1754–1758. doi: 10.1038/sj.bjc.6602531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zirn B, Hartmann O, Samans B, Krause M, Wittmann S, Mertens F, Graf N, Eilers M, Gessler M. Expression profiling of Wilms tumors reveals new candidate genes for different clinical parameters. Int J Cancer. 2006;118:1954–1962. doi: 10.1002/ijc.21564. [DOI] [PubMed] [Google Scholar]
- 144.Norwood LE, Grade SK, Cryderman DE, Hines KA, Furiasse N, Toro R, Li Y, Dhasarathy A, Kladde MP, Hendrix MJ, Kirschmann DA, Wallrath LL. Conserved properties of HP1(Hsalpha) Gene. 2004;336:37–46. doi: 10.1016/j.gene.2004.04.003. [DOI] [PubMed] [Google Scholar]