Role of Histone Acetylation in the Assembly and Modulation of Chromatin Structures (original) (raw)
Abstract
The acetylation of the core histone N-terminal “tail” domains is now recognized as a highly conserved mechanism for regulating chromatin functional states. The following article examines possible roles of acetylation in two critically important cellular processes: replication-coupled nucleosome assembly, and reversible transitions in chromatin higher order structure. After a description of the acetylation of newly synthesized histones, and of the likely acetyltransferases involved, an overview of histone octamer assembly is presented. Our current understanding of the factors thought to assemble chromatin in vivo is then described. Genetic and biochemical investigations of the function the histone tails, and their acetylation, in nucleosome assembly are detailed, followed by an analysis of the importance of histone deacetylation in the maturation of newly replicated chromatin. In the final section the involvement of the histone tail domains in chromatin higher order structures is addressed, along with the role of histone acetylation in chromatin folding. Suggestions for future research are offered in the concluding remarks.
Keywords: Histone, Acetylation, Chromatin, Nucleosome, Assembly, Tails, Structure, Folding
THE assembly of nucleosomes represents the first step in a series of structural transitions that organize newly replicated DNA into the compact yet functionally accessible chromatin fibers that are housed in the eukaryotic nucleus. The protein core of the nucleosome, the histone octamer, possesses a tripartite structure comprising an H32H42 tetramer and two H2A/H2B dimers, arranged to form a rotationally symmetrical particle (14,125). It was once thought that histones serve predominantly as inert structural proteins that repress gene activity in a more or less passive fashion. However, the past few years have provided ample evidence for a contrasting view, that histones are active participants in both the positive and negative regulation of transcription through protein-DNA and protein-protein interactions, under precise yet fluid control.
The regions of the core histones that have been most implicated both in the dynamic regulation of gene expression and in chromatin structure are the N-terminal “tail” domains, stretches of ∼25–40 amino acids (depending on the individual histone) that are strongly positively charged (204,219). The function of the histone tails in chromatin fibers is very complex, both in vivo and in vitro (see below), and the mechanisms through which they act are only now starting to be deciphered (73,125). It has nevertheless been determined that in nucleosomes the tail domains project beyond the gyres of encircling DNA, and are subject to a variety of posttranslational modifications, including acetylation, phosphorylation, methylation, and poly(ADP-ribosylation) (39,189). Acetylation occurs on specific, highly conserved lysine residues (e.g., K5, K8, K12, and K16 of H4, and K9, K14, K18, and K23 of H3) (204,219). As a consequence of their disposition in the chromatin fiber, the tail domains can form contacts with DNA as well as with other histones or nonhistone proteins (73). Not surprisingly, such contacts can be influenced by histone posttranslational modifications, as has been observed for the association of the yeast Tup1 protein with histone H4 (47).
Over the past several years a direct link between histone acetylation and transcription has been established. The observation that a histone acetyltransferase (HAT) activity found in Tetrahymena is a homologue of the yeast transcriptional activator Gcn5p (28) has triggered a series of studies, revealing intrinsic HAT activity in a surprising number of transcriptional activators and coactivators (21,81,139,186,189,190,196). As a complement to these findings, histone deacetylation has been mechanistically tied to gene repression, as evidenced by transcriptional silencing in yeast (24,188), and by the recruitment of histone deacetylase (HDAC) by factors such as the repressor Rb (26,128,131) and the methyl-CpG-binding protein MeCP2 (96,146,221). The involvement of dynamic histone acetylation in regulating gene activity is now known to be both fundamental and pervasive, and as such has been the subject of a host of review articles, covering virtually every facet of the subject (cited above). This aspect of chromatin metabolism will therefore not be extensively treated in the following article. Instead, we will focus on equally important but relatively less explored topics: the role of histone acetylation in nucleosome assembly and chromatin structure.
Over a quarter century ago Dixon and colleagues demonstrated that newly synthesized histone H4 is reversibly acetylated during spermatogenesis (123). Despite the many advances that have been made in our understanding of the regulation of histone acetylation, and of the steps required for chromatin assembly, an explanation for the acetylation of nascent his-tones has yet to emerge. Still, de novo nucleosome assembly pathway(s) and the histone acetyltransferases involved are being defined with increasing precision. The following discussion will open with a description of deposition-related histone acetylation and the enzymes thought to be responsible. Replication-coupled nucleosome assembly in somatic cells will then be examined, with an emphasis on possible functions for histone acetylation in the assembly process. To place acetylation in a operationally relevant context, the participation of the histone tail domains in the formation of chromatin higher order structures will also be addressed, along with the involvement of histone acetylation in chromatin folding. Although seminal contributions will be discussed, in general the following analysis will concentrate on work performed within the past 5 years. Should a more extensive treatment of work on deposition-related histone acetylation be desired, the reader is directed to previous in-depth discussions of the topic (8,51), as well as to a number of additional reviews on the subject of chromatin assembly in somatic and embryonic systems (1,5,19,27,42,68,99,111,114,167).
ACETYLATION OF NEWLY SYNTHESIZED HISTONES
The Sites of Modification
In addition to the observations of Louie et al. (123) cited in the Introduction, the work of two other laboratories provided early evidence for the reversible acetylation of newly synthesized H4. Allfrey and colleagues (168) showed that in duck erythroblasts nascent H4 was found in un-, mono-, and dimodified isoforms, the latter described as a simultaneously monoacetylated/monophosphorylated species. It was also observed that 20 min after chromatin assembly, at least 50% of the new H4 was present in the unmodified form, demonstrating acetate and/or phosphate turnover. Somewhat different results were obtained by Jackson et al. (93) using rat hepatoma tissue culture (HTC) cells: in this case the “dimodification” of new H4 was ascribed almost exclusively to acetylation; however, a similar time-dependent deacetylation of new H4 was also observed. Moreover, although newly synthesized H3 in duck erythroblasts was “acetylated in about the same proportion as the ‘old’ H3” (168), ∼50% of total new H3 in HTC cells was initially mono- (30%), and di- or triacetylated (20%) (93). It must be stressed that all of these early studies were performed prior to the advent of sodium butyrate, trichostatin A, or trapoxin as deacetylase inhibitors; nevertheless, the observed differences in the acetylation of new H3 most likely reflects an actual inherent variability (see below).
The first identification the specific sites acetylated in newly synthesized H4 was made by Allis and colleagues, using the ciliated protozoan Tetrahymena (34). This organism contains a macronucleus that is engaged in both transcription and replication, and a micronucleus that is transcriptionally silent. During replication-coupled nucleosome assembly in both micro- and macronuclei, newly synthesized H4 is diace-tylated exclusively on lysines 4 and 11 (34). These sites correspond to lysines 5 and 12 of H4 in virtually all other organisms, due to a deletion of the arginine at position 3 in Tetrahymena. The “5/12” deposition-related acetylation pattern of newly synthesized H4 appears to be one of the most highly conserved his-tone modifications known, having now been positively identified in Tetrahymena, Drosophila, and human cells (184). This precise acetylation pattern can thus be traced through at least a billion years of eukaryotic evolution.
The acetylation pattern of newly synthesized H3 is considerably more variable. In Tetrahymena, K9 and K14 are predominantly acetylated, while in Drosophila lysines 14 and 23 are the preferred sites (184). In the yeast Saccharomyces cerevisiae, the lysines at positions 9, 14, 23, and 27 all show some acetylation (with K9 and K23 preferred), but most new H3 is monoacetylated (113). While an initial examination of nascent human (HeLa) H3 showed little if any acetylation (184), more recent analyses indicate that 15% or more of new H3 is acetylated on lysines 14, 18, and/or 23. Moreover, prior to deposition acetylation appears to be restricted to the H3.2/ .3 subtypes (C. Matson and A. T. Annunziato, unpublished observations). The reason for this rather striking variability is unknown: the enzyme(s) involved in acetylating new H3 have not yet been positively uncovered.
The “linker” histones H1 and H5 are not reversibly acetylated, but are instead dynamically modified by phosphorylation (204,219). New H1 enters chromatin significantly underphosphorylated, and gains the phosphorylation pattern typical of S phase H1 after approximately 60 min (93). Phosphorylation occurs on serine and threonine residues, in typical cdk target motifs (S/T-P-X-K/R) (219).
Finally, it is noted that nascent H2A and H2B do not appear to exhibit any special patterns of transient modification that distinguish them from the “old” or parental isoforms (36,93). The treatment of these two histones during the discussion of acetylation and chromatin assembly will therefore be limited.
Deposition-Related Histone Acetyltransferases
At least one histone acetyltransferase (or “HAT”) responsible for the acetylation of newly synthesized H4 has been identified with a high degree of certainty. This is the “type B” or HAT-B histone acetyltransferase, which is typically recovered in the cytosol following cellular fractionation. As expected for an enzyme involved in chromatin assembly, early studies revealed that HAT-B is specific for H4, and is able to acetylate free but not nucleosomal H4 (46,61,121,122,138,164,170,191,211). Even more tellingly, analyses of the specific lysines acetylated by HAT-B from a variety of sources have identified either the acetylation of K12 alone [by the _Drosophila_ (183) and yeast enzymes (104,152)], or the production of the complete “5/12” deposition pattern [by HAT-B from _Tetrahymena_ (164), peas (138), humans (33,207), maize (106), and Xenopus (82)]. It is noteworthy that recombinant Hat1p from yeast is capable of acetylating both K5 and K12 of H4, as opposed to the single K12 site seen with the native enzyme; it has therefore been suggested that the native enzyme may be negatively regulated (152). Curiously, in the case of the pea enzyme a third acetyllysine (at K16) was also noted (138), but whether this reflects the activity of HAT-B in vivo, or indeed of newly synthesized pea H4, is uncertain.
The yeast Saccharomyces cerevisiae contains an identifiable HAT-B activity (119,120), and the first isolation of a gene coding for a “cytoplasmic” H4 acetyltransferase (or more specifically for its catalytic subunit) was achieved in this organism (104,152). Termed HAT1, the gene revealed a polypeptide with a molecular weight of ∼42–44 kDa (104), a size that was confirmed using an “in-gel” acetyltransferase assay (152). Hat1p copurified with another protein, termed Hat2p (152), which was identified as the yeast homologue of Rbap46 and Rbap48, two small proteins that bind the retinoblastoma protein, Rb (161,162). Thus, HAT-B “holoenzyme” is a complex, comprising at least two subunits. Subsequent studies of human HAT-B identified a Hat1p catalytic subunit of ∼44–46 kDa and a complex of ∼100 kDa (33,207). Notably, the value of ∼100 kDa corresponds closely to the estimated size of native HAT-B from calf thymus [98 kDa (191)] and maize embryos [90–95 kDa (46)]. Consistent with the data from yeast and human cells, two subunits of the maize HAT-B complex have now been described, a 50-kDa catalytic subunit (HAT-B-p50), and a 46-kDa protein that is homologous to Rbap46/48 (46,129). Significantly, the HAT-B complex in Xenopus oocytes also contains a homologue of Rbap48, along with several members of the 14-3-3 family of phosphoserine binding proteins (82). It is therefore becoming apparent that the enzyme that acetylates nascent H4 is potentially as conserved as the “5/12” deposition pattern of acetylation itself.
In yeast, Hat2p is required for the high-affinity binding of Hat1p to histone H4, as well as for full catalytic activity (152). In a similar fashion, the p46 subunit greatly stimulates acetylation of H4 by human Hat1p (207). Deletion mutantations further demonstrate that p46 recognizes helix 1 of the central domain of histone H4 (207), a region that makes several contacts with DNA in the nucleosome (125,126). Interestingly, the H4 N-terminal tail is not involved in p46 recognition (207). Nucleosome assembly may therefore prevent the p46:H4 interaction, which could account for the inability of HAT-B to acetylate H4 in chromatin. The association of Rbap46/48 homologues with histone acetyltransferases, deacetylases (77,193,226), and chromatin assembly factors (102,201,206) (also see below) suggests that members of the p46/p48 family may act as universal histone docking proteins (167,207).
As noted above, one of the hallmark features of HAT-B is its apparent cytoplasmic localization, at least after cell lysis. To determine if human HAT-B actually resides in the cytoplasm, Verreault et al. (207) employed immunocytochemistry to detect a FLAG-Hat1p fusion protein in S phase cells. Rather surprisingly, most of the fusion protein was located in the nucleus (although trace amounts were also detected in the cytoplasm); a similar distribution was also noted for Hat2p (p46). It had previously been reported that yeast Hat1p was found in both the cytoplasm and the nucleus, following Western analysis of the separated cellular compartments (152). A more recent investigation of yeast acetyltransferases suggests that Hat1p and Hat2p are both present as components of nuclear as well as cytoplasmic HAT complexes, although in this case the biochemical fractionation again relied on cell disruption (169). Studies using Tetrahymena cells (164) and maize embryos (129) also indicate that HAT-B can be present in nuclear and cytoplasmic forms, perhaps shuttling between compartments.
The best indication that HAT-B (or Hat1p) is redistributed in response to cellular signals comes from experiments using Xenopus oocytes and embryos (82). In oocytes an extraordinarily high level of HAT-B activity (∼104 times that found in typical somatic cells) is found in nuclei, whether isolated by manual dissection or by biochemical means; however, during embryogenesis virtually all HAT activity redistributes to the cytosol, presumably to rapidly acetylate the H4 that is synthesized at this time (169). Thus, a picture is emerging of a HAT-B complex that is for the most part nuclear (although easily extractable), and that may under defined circumstances be translocated to the cytoplasm. It has therefore been suggested that HAT-B may be cotransported into the nucleus along with newly synthesized H3 and H4, or indeed may serve as the transport mechanism for these histones (82,207).
The apparently mobile nature of the HAT-B complex raises a question that has not yet been satisfactorily answered, namely, when are newly synthesized H3 and H4 acetylated? In Xenopus oocytes, large pools of free histones are stored in nuclear predeposition complexes (105) along with stored HAT-B (82), but cytoplasmic histones are also detectable (223). Notably, H4 is stored in a diacetylated isoform in Drosophila (65,200) and Xenopus (41,222) oocytes and eggs, and at least in Xenopus the vast majority of diacetylated H4 appears to be nuclear, with trace amounts of monoacetylated H4 seen in the cytoplasm (222). The selective nuclear accumulation of both diacetylated H4 and the HAT-B complex strongly suggests that acetylation is a nuclear event in oocytes. In contrast, the situation during replication-coupled nucleosome assembly in somatic cells is not so readily defined. Here, the rapidity with which nascent histones are transported into the nucleus renders a precise ordering of the acetylation–translocation pathway uncertain. Analysis is further complicated by the likelihood that preassembly histone complexes may leach out of the nucleus when cells are disrupted. Seen in this light, the observation that newly synthesized H3 is found in a preassembly complex that is “cytosolic,” and that contains diacetylated H4 (33,101,156,206), does not demonstrate unequivocally that acetylation occurs in the cytoplasm. A resolution of this question is essential to our understanding of how histones are escorted into the nucleus and assembled into nucleosomes. For example, if acetylation is necessary for histone escort recognition, then one would predict that acetylation in the cytoplasm would be required. If acetylation is not involved in H3/H4 import, nuclear acetylation then becomes feasible. The requirement (or lack thereof) for H4 acetylation during chromatin assembly will be discussed again, when nucleosome assembly is considered in more detail below.
The Hat1p acetyltransferase from yeast has now been crystallized, and its structure determined at 2.3 Å resolution (44,45). The enzyme has an elongated structure, measuring ∼70 Å in its greatest dimension. Hat1p has a curved shape. The acetyl CoA molecule binds a cleft on the concave surface of the protein, with the acetyl group itself marking the active site. Based on the assumptions that the H4 tail has an extended structure, that the readily acetylated lys-12 residue lies in close proximity to the carbonyl group of acetyl CoA, and that Hat1p undergoes no large conformational changes upon H4 binding, a model has been proposed for the enzyme-substrate interaction (44,45). A segment of the H4 tail (between K8 and K16) is thought to bind along a channel that is long enough to accommodate 6–7 amino acid residues. With K12 aligned adjacent to acetyl CoA, K8 and K16 come to lie opposite two acidic regions at the ends of the channel, theoretically engaging in electrostatic interactions that hold H4 in place. If instead K5 is aligned near the acetyl group of acetyl CoA, the fit is not as favorable, although still reasonable. In contrast, aligning K8 adjacent to acetyl CoA causes a steric clash with the channel (44,45). As the authors propose, these considerations may explain why some of the Hat1p enzymes acetylate K12 vigorously while leaving K5 unmodified (at least in vitro), and why K8 and K16 are not modified at all by Hat1p. Consistent with this interpretation, experiments performed using the HAT-B complex from maize suggest that K12 tends to be acetylated prior to K5; however, this preferred order of acetylation is not rigidly maintained (106).
Indirect evidence for the model of H4 binding proposed above has recently been obtained. It was reasoned that, if the charged side chains of K8 and K16 bind Hat1p electrostatically, then neutralization of their positive charges (e.g., by acetylation) would decrease the enzyme-substrate interaction. To test this hypothesis, synthetic H4 tail peptides, either unmodified or variously acetylated on K5, K8, K12, and K16, were used for in vitro HAT-B assays. As predicted, an H4 peptide previously acetylated on K8 and K16 was a very poor substrate for HAT-B (∼15% relative to the unacetylated peptide) (A. Ma-kowski and A. T. Annunziato, unpublished observations). Treating the “8/16” acetylated peptide with histone deacetylase prior to the HAT-B reaction restored acetylation to ∼70% of control levels. These data demonstrate that the site-specific acetylation of histone H4 can regulate the subsequent acetylation of other substrate sites. This may provide a general mechanism for the control of histone acetylation in vivo.
Several groups have examined yeast hat1 and hat2 null mutants in an attempt to elucidate the role of HAT-B, and the function of acetylation during chromatin assembly. Neither hat1, hat2, nor hat1/hat2 double mutants exhibit any obvious phenotype except for the loss of enzyme activity (104,152,169). Moreover, the hat1,gcn5 double HAT mutant (see Introduction) was slow growing but viable, and displayed no phenotype distinct from the gcn5 mutant alone. As these investigators point out, yeast has many histone acetyltransferases with overlapping specificities (120,225), which may account for the absence of synergistic mutational effects. Significantly, the acetylation of newly synthesized H4 has not been directly examined in any of the mutants.
REPLICATION-COUPLED NUCLEOSOME ASSEMBLY
Histone Octamer Assembly
The duplication of chromatin fibers following DNA replication involves two coordinated processes: the transfer of parental histones to new DNA (often termed nucleosome or histone segregation), and the assembly of nucleosomes de novo (summarized in Fig. 1). Both processes have received considerable attention and are becoming increasingly well understood. It is not the purpose of this review to provide a comprehensive analysis of chromatin biosynthesis. Rather, this section will focus on features of chromatin replication that touch upon histone acetylation. Nucleosome assembly de novo will be described first, followed by a description of the relationship between acetylation and nucleosome segregation, and a consideration of chromatin higher order folding. Additional aspects of nucleosome assembly have been outlined in several informative review articles (1,5,19,27,42,69,99,111,114).
Figure 1.
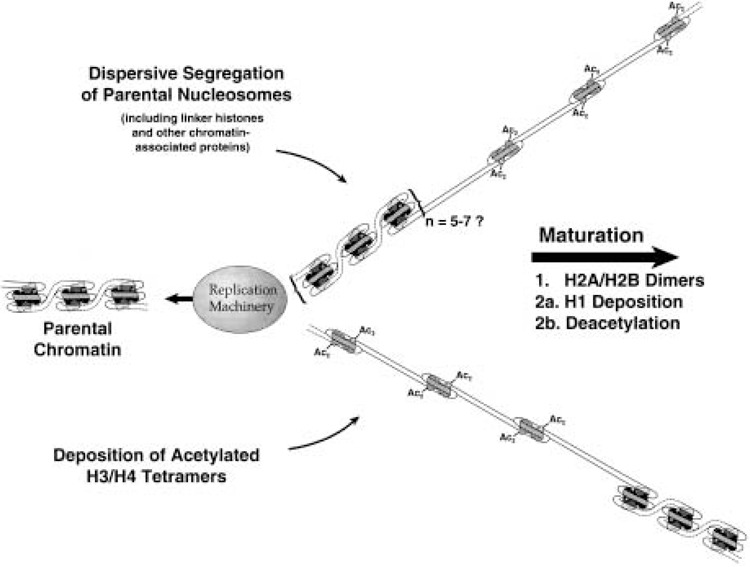
Overview of chromatin replication and assembly. Parental histone octamers segregate in groups to both sides of the replication fork, where their association with histone H1 is quickly reestablished. In the gaps produced by dispersive segregation, de novo nucleosome assembly proceeds in a stepwise fashion. First newly synthesized H3/H4 tetramers, acetylated in specific deposition-related patterns, are deposited onto the nascent DNA. Next, preexisting H2A/H2B dimers (generated through histone exchange in nonreplicating chromatin) associate with the H3/H4 tetramers. Lastly, histone H1 is deposited, and new H3 and H4 are deacetylated. Please see text for details and references.
Histone octamers that are assembled de novo contain old as well as newly synthesized histones, but the assembly of the hybrid octamers is not random. Virtually all newly synthesized H3 and H4 are assembled onto newly replicated DNA; nascent H2A, H2B, and H1 are deposited onto nonreplicating chromatin regions through a process of histone exchange (10,89,90,92). It is therefore clear that the assembly of new DNA requires parental H2A/H2B and H1, displaced from elsewhere in the genome. Consistent with the hypothesis that exchange is driven by transcription (87,88), it has been shown that H2A/H2B exchange occurs selectively on chromatin regions containing acetylated H4 (156). Whether the displaced histones bear any special posttranslational modifications is unknown at this time; nevertheless, in vitro studies have demonstrated that unmodified (nuclear) H2A/H2B can be properly assembled onto replicating plasmid DNA (182). Possibly because the assembly of hybrid octamers runs counter to intuitive predictions, this aspect of histone deposition was relatively slow to emerge. The many lines of evidence leading up to the conclusion that octamer assembly is nonconservative have been previously reviewed (7).
Nucleosome assembly occurs in a stepwise fashion (summarized in Fig. 1). As shown for replication-coupled nucleosome assembly in vitro, first newly synthesized H3 and H4 (acetylated in the respective “deposition type” patterns) are placed onto new DNA, followed by a pair of H2A/H2B dimers (3,53,182). It is almost certain that H3 and H4 are deposited as a preassembled H32H42 tetramer (33,105,156,206). During the 30–60 min following his-tone deposition, H3 and H4 are gradually deacetylated as the nascent chromatin matures (7,8,219). At some point during this process [perhaps immediately after core histone deposition (18)], H1 is added to the newly assembled nucleosome (224). Because parental histones segregate dispersively to both arms of the replication fork (as discussed below), de novo assembly takes place between clusters of “old” parental nucleosomes. Thus, for some period both daughter duplexes contain regions of incompletely assembled DNA.
Of prime importance is the means by which the highly cationic histones are deposited onto DNA in the orderly manner just outlined. Many negatively charged molecules, including RNA (147), are capable of driving nucleosome assembly in vitro (42,114). One feature that has been used to identify bona fide assembly factors is the ability to selectively assemble newly replicated DNA. An in vitro system that has been exploited to detect chromatin assembly factors in somatic cells relies on replicating SV40 DNA, T antigen (required for replication initiation), a cytosolic extract from human 293 cells, and a nuclear extract. The cytosolic extract contains small amounts of soluble, newly synthesized histones, along with cellular proteins that support the replication of double-stranded DNA. Only in the presence of added nuclear extract are nucleosomes assembled onto the replicated products (187).
Fractionation of the nuclear extract revealed a factor that was required to link chromatin assembly to DNA replication. Termed CAF-1 (chromatin assembly factor 1), this factor was initially described as having five subunits, but was subsequently determined to have three: p150, p60, and p48 (180,182). The difference was ascribed to phosphorylation of the p60 subunit (182). Very little p150 is present in cytosolic extracts, while p48 and p60 are abundant in the cytosol; all three are present in nuclear extracts (101,206). Homologues and/or counterparts of the human CAF-1 subunits have been identified in Drosophila and yeast (98,102,201), and CAF-1 activity has also been observed in Xenopus egg extracts (57,97). In somatic cells CAF-1 is concentrated in the interphase nucleus (133,181), and during S phase p150 and p60 colocalize with replication foci (110,133,192).
The interaction of the CAF-1 subunits with each other and with histones has been closely examined. p60 and p150 make direct contact within the CAF-1 complex (101). As noted above, the cytosolic extract contains soluble core histones, including newly synthesized H3 and H4 in the appropriately acetylated isoforms. Nascent H3 and H4 form a cytosolic preassembly complex, and can be coimmunoprecipitated using antibodies that recognize acetylated H4 (33,101,156). However, antiacetylated H4 antibodies immunoprecipitate only a subset of p60 from the cytosolic extract, unless additional recombinant p150 is added (101). It was therefore concluded that p150 is required for p60 to interact with H3/H4 (101). In contrast, H3/H4 plus all three CAF-1 subunits can be extracted from nuclei as members of a single complex, termed “CAC” (i.e., chromatin assembly complex). This suggests that the ultimate assembly of CAF-1 with H3/H4 is a nuclear event.
The p48 subunit of CAF-1 is identical to Rbap48, a WD-repeat protein that binds to the retinoblastoma gene product Rb (161,162), and that is closely related to the p46 subunit of the HAT-B acetyltransferase (please see the discussion of HAT-B). Rbap48 and its homologues are also found to be complexed with histone deacetylase and chromatin remodeling factors, as well as with CAF-1 from other species (134,193,201), and Xenopus Hat1p (82). Consistent with its many roles in chromatin metabolism, p48 binds histone H4 in the absence of p60 or p150, and is a member of the cytosolic H3/H4 complex (206).
The addition of nuclear (mostly unmodified) his-tones to the in vitro chromatin assembly reaction results in the indiscriminate assembly of replicating and nonreplicating plasmids alike, either in the presence or absence of CAF-1 (182). This suggests that there are assembly activities other than CAF-1 in the cytosolic extract (see below), and further, that the CAF-1-mediated assembly of replicating DNA requires the cytosolic (acetylated) histone pool. Moreover, when H3 and H4 are immunodepleted from the cytosolic extract, only nuclear CAC, which contains acetylated histones, can restore assembly activity; the CAF-1 subunits alone are ineffective (206). Of course, a his-tone-deficient extract is not expected to assemble chromatin, and it would be of interest to see if recombinant (unmodified) H3/H4 could effect chromatin assembly in depleted extracts when CAF-1 is present. This leads to the question, “How important is acetylation for CAF-1 to bind and deposit H3/H4?”
Given that none of the CAF-1 subunits have to date been shown to uniquely recognize acetylated H4 (or H3), it may be premature to postulate that selectivity depends on acetylation per se. For example, virtually all newly synthesized, cytosolic H3/H4 sediments as a ∼100-kDa complex, considerably larger than the H3/H4 tetramer alone (53 kDa) (33). As has been discussed, this anomalous sedimentation can be ascribed to the association of p48 with H3/H4 (206). Because p48 can still bind an H4 molecule from which the N-terminal tail has been completely deleted (206), binding is clearly independent of acetylation status. In addition, p48 does not bind histone H3 (206). Thus, the requirement for “cytosolic” H3 and H4 during CAF-1-mediated nucleosome assembly may result, not from their acetylation, but from the fact that these histones are already “primed” for assembly through their association with p48 in the cytosol (see Note Added in Proof). Nevertheless, since p46 (a subunit of the HAT-B holoenzyme) and p48 appear to compete for the same binding site on H4, it is likely that acetylation occurs prior to the assembly of the p48:H3/H4 complex.
The precise role of CAF-1 in assembling cellular chromatin in vivo is still incompletely resolved. Deletion of all three genes (CAC1, CAC2, and CAC3) coding for the CAF-1 subunits in yeast does not prevent nucleosome assembly, or decrease cell viability (102). However, deletion of one or more of the CAF-1 subunits causes increased sensitivity to ultraviolet irradiation, derepression of genes adjacent to telomeres, and partial activation of silent mating loci (48,49,60,100,102,142). Along these lines, the acetylation of H4 on lysine 12 has been linked to transcriptional repression in yeast (25), and K5/K12 H4 acetylation has been observed during the assembly of mammalian heterochromatin domains (192). It has therefore been proposed that the loss of CAF-1 activity results in the assembly of under- or incorrectly acetylated histones, which prevents the establishment of the repressive chromatin structure (49,102,142,192); however, this model has recently been challenged (205). Notably, the p150 subunit of CAF-1 binds directly to HP1 proteins, which are structural components of pericentric and (in Drosophila) telomeric heterochromatin (145). This interaction provides a direct connection between a CAF-1 subunit and the maintenance of heterochromatin domains, which does not specifically involve the K5/K12 acetylation pattern of nascent H4.
As mentioned above, deletion of the CAF-1 subunits also results in a heightened sensitivity to UV light. It has now been demonstrated that CAF-1 functions to reassemble chromatin during DNA repair (57–60,135,140,141). One molecular link between DNA replication and repair involves PCNA (proliferating cell nuclear antigen), which functions as a DNA polymerase sliding clamp (56,107,171). Following replication, PCNA persists on the new DNA until it is unloaded by RFC (replication factor C) [reviewed in (208)]. The CAF-1 p150 subunit recognizes PCNA, providing a mechanism for the recruitment of CAF-1 to the sites of DNA replication and repair (175). The unloading of PCNA by RFC prohibits subsequent nucleosome assembly (175), which accounts for the observed inability of CAF-1 to assemble chromatin more than 120 min after passage of the replication fork (97).
Analysis of the histones associated with CAC purified from the nuclei of human 293 cells yielded somewhat surprising results. H3 present in CAC was predominantly unacetylated (∼60%), the remainder being monomodified (probably by acetylation). CAC-associated H4 was a mixture of unacetylated (33%), monoacetylated (33%), diacetylated (33%), and tri-acetylated (trace) isoforms, with detectable acetylation on lysines 5, 8, and 12 (but not 16) (206). While it is possible that the acetylation of lysine 8 is a normal part of the deposition-related pattern of H4 acetylation, heretofore overlooked, other explanations for this result are possible. For example, the complete deacetylation of as much as one third of CAC-associated H4, as well as the unpredicted acetylation of H4 on K8, may result from postdeposition events—a consequence of the acetylation and deacetylation of newly deposited histones. This, and the considerable evidence that CAF-1 mediates chromatin assembly at the sites of DNA repair, makes it possible that the acetylation patterns of nuclear CAC-associated histones do not entirely result from typical assembly events.
The viability of yeast CAF-1 mutants establishes that multiple chromatin assembly pathways must exist. Recently, a novel factor, termed RCAF (replication-coupling assembly factor) was discovered in Drosophila embryo extracts (200). RCAF mediates chromatin assembly after DNA replication, and after the repair of DNA double-stranded breaks. Cloning of the gene for the largest of the three RCAF poly-peptides revealed it to be the Drosophila homologue of yeast ASF1 (anti-silencing function 1), which de-represses transcriptional silencing when overexpressed (115,178). Strikingly, microsequencing of the two smaller RCAF polypeptides showed that they are histones H3 and H4; moreover, both of these histones are acetylated in specific Drosophila deposition-type patterns: H4 on lysines 5 and 12; H3 on lysine 14 (200). CAF-1 and RCAF assemble chromatin synergistically in vitro, and the simultaneous disruption the CAC1 and ASF1 genes in yeast had a dramatic dere-pressive effect on telomeric and mating-type silencing, greater than that observed in either mutant alone (200). Still, and rather remarkably, the double-mutants remained viable (though less than robust), and so clearly must be able to assemble chromatin through yet another pathway.
The deposition of H2A and H2B in somatic cells has not been as extensively investigated as the H3/H4 assembly pathway. H2A and H2B are not reversibly modified during chromatin assembly, and unlike H3/H4, purified nuclear H2A/H2B can substitute for “cytoplasmic” histones during replication-coupled nucleosome assembly in vitro (182). In Drosophila embryo extracts H2A and H2B are stored complexed to the protein dNAP-1 (86), the Drosophila homologue of human NAP-1. NAP-1 was first described as a potential nucleosome assembly factor by Ishimi et al. (83,85). This ∼60-kDa protein can bind all four histones (with a preference for H2A and H2B), and is able to assemble nucleosomes on nonreplicating DNA (84). NAP-1 can also facilitate transcription factor access to DNA in vitro, by disrupting histone -histone and histone-DNA interactions (210). In Drosophila embryo extracts, NAP-1 acts to chaperone H2A/H2B dimers during ATP-facilitated chromatin assembly (29,86). A yeast homologue of NAP-1 has also been described (55).
NAP-1 is highly abundant in cytosolic extracts prepared from HeLa cells, from which it can be coimmunoprecipitated with H2A (33). Thus, there is evidence for two independent histone complexes in human cytosolic extracts: p48 in association with H3/H4; NAP-1 with H2A (and possibly H2B) (33,156,206). Because it must be recalled that replication-coupled H2A/H2B deposition involves “old” H2A/H2B, which has been displaced from nonreplicating chromatin, it has been suggested that NAP-1 acts as a molecular escort for in vivo H2A/H2B exchange (33). In support of this, a recent comprehensive analysis of protein-protein interactions in yeast demonstrated, through two independent two-hybrid screens, that yeast NAP-1 interacts with H2A in living cells (202). This finding, along with the observation that NAP-1 can shuttle between the cytoplasm and the nucleus (86), would further suggest that NAP-1 is involved in the assembly/exchange of H2A (and H2B).
Histone N-terminal Tails and Nucleosome Assembly
The evolutionary conservation of histone acetylation, and its numerous ties to replication and transcription, have prompted scores of investigations into the function of the histone N-terminal tails [reviewed in (39,73,189,220)]. In this section we will concentrate on the possible role of these histone domains, and the acetylation thereof, in chromatin replication and assembly. Most of this work (but not all) has been performed in yeast [reviewed in (1,8,67,76,179)]. While it is not possible to provide a complete examination of this considerable literature, some points can be highlighted, as follows:
- Yeast cells with deletions of the N-terminus of any one of the core histones are viable, although slow growing (103,132,144,172,209).
- Yeast with simultaneous deletions of the N-termini of both H2A and H2B are inviable (172).
- Similarly, cells lacking both H3 and H4 N-termini are also inviable (118,144).
- Individually replacing any one of lysines 5, 8, 12, and 16 of H4 with positively charged arginine (which cannot be acetylated) has no adverse effects on cell viability or doubling time (43,137); however, altering lysine 16 to an electrostatically neutral amino acid prevents repression of the mating type loci (95,151).
- Altering specific pairs of lysines of H4 (e.g., 5–8, 5–12, 8–12) to arginine—thereby precluding acetylation— slightly increases doubling time; substituting three lysines simultaneously (5–8–12) has a greater effect.
- When all four of the acetylatable lysines of H4 are replaced with arginine the mutant cells either grow very slowly (43,151) or are inviable (137); converting these residues to uncharged amino acids increases doubling time twofold (137,151); corresponding mutations of the acetylatable lysines of H3 have minimal effects (132).
From the above results it can be concluded that, when wild-type H3 is present, it is necessary for H4 to be acetylatable on at least one lysine residue for cells to be viable; that the deacetylation of H4 at lysine 16 is required for normal mating efficiency; and that the specific 5/12 diacetylation of newly synthesized H4 is dispensable for nucleosome assembly. It also appears that the N-terminal tails of H3 and H4 may compensate for each other to some degree. To investigate this further, Ling et al. (118) examined the effects of deleting and/or interchanging the N-terminal domains of H3 and H4.
Exchanging the H3 and H4 tails yielded viable cells; however, neither the H3N-H4C nor the H4N-H3C chimeric proteins were able to support viability when coexpressed with a deletion of the remaining H3 or H4 N-terminus, respectively (118). This is in contrast to the results outlined above in item 1, describing the viability of single tail deletions. When (inviable) mutants with simultaneous deletions of the H3 and H4 tails were examined, some defects in nucleosome assembly were noted in vivo, and with nonreplicating plasmids in vitro using cell extracts. The effect was more pronounced in vivo, although assembly was not completely prevented in either case.
To more precisely define the extent of compensatory acetylation between H3 and H4, site mutations and deletions of these histones have been combined in yeast, and the effects on cell viability and chromatin assembly assessed (130). Mutation of lysines 5 and 12 of H4 (the sites of deposition-related acetylation) to glycine or arginine, coupled with an H3Δ4-30 deletion, permitted cell viability and nucleosome assembly. Similarly, K5, 8, 16→G, K5, 12, 16→G, and K 8, 12, 16→G cells were also viable in the absence of the H3 tail. In contrast, simultaneously changing K5, K8, and K12 to glycine, or all four of the acetylatable lysine residues to glycine, was lethal in combination with H3Δ4-30. Retaining just one lysine at position 5, 8, or 12 conferred viability again.
Nucleosome assembly in vivo was then examined by monitoring the superhelical density of an endogenous 2-μ plasmid present in these strains, based on the principal that each nucleosome adds one negative superhelical turn to covalently closed DNA. In combination with H3Δ4-30, the simultaneous (and lethal) K5, 8, 12→G mutations of H4 significantly reduced, but did not eliminate, plasmid supercoiling in vivo. Extracts prepared from these cells were relatively inefficient at assembling chromatin on nonreplicating plasmids as measured by the supercoiling assay, but were not devoid of assembly activity. The other H4 triple mutations (K5, 12, 16→G; K 8, 12, 16→G; and K5, 8, 16→G) also decreased supercoiling both in vivo and in vitro, but to a lesser degree (130).
K to G mutations mimic the charge neutralizing effect of acetylation, but not the specific structure of lysine or acetyllysine. Also, replacing the first four lysines of H4 with glycine will yield a stretch of 13 consecutive hydrophobic residues, 11 of which are glycine. It is difficult to predict what effect this might have on the structure of the H4 N-terminus, 50% of which may normally be in a-helical subdomains (16). In light of these considerations, suppression of the lethal phenotype in H3Δ4-30 cells by maintaining either K5, K8, or K12 of H4 may suggest either 1) that when the H3 tail is absent acetylation is required at one of these sites; 2) that at least one of these lysines must remain positively charged and unmodified; 3) that the specific structure of acetyllysine is required (at one site); or 4) that at least one hydrophilic residue is needed to preserve tail substructure. Because the triple 5, 8, 12→R H4 mutation was not examined in the H3Δ4-30 background, and K→Q mutations (to preserve tail hydrophilicity) were not attempted, it is not possible at this time to choose between these possibilities. Moreover, the acetylation of H3 and H4 may itself reduce plasmid superhelicity (148,149). It is therefore unclear if the lethality observed is due to insufficient nucleosome assembly, improper chromatin folding, or some other disrupted function.
Zhang et al. (225) have examined the effects of combining mutations of H3 and H4 at the deposition-related acetylation sites. Newly synthesized yeast H3 is preferentially (but not exclusively) acetylated on lysine 9 (113). It was found that H3: K9→R substitution, together with H4: K5, 12→R, extended doubling time only slightly. Similar results were obtained with K→Q mutations. Notably, concurrently altering seven of the nine acetylatable lysines of H3 and H4 (K5, 8, 12 of H4; K9, 14, 18, 23 of H3) to either glycine or arginine yields viable yeast cells, although substantial derepression of telomeres is observed (195). In light of this, it is difficult to argue that the compensatory acetylation of new H3 and H4 is essential for chromatin assembly.
Wolffe and colleagues have asked which domains of H3 and H4 are required for proper deposition and assembly during Xenopus embryogenesis (54). By microinjecting mRNA coding for mutant, epitope-tagged H3 and H4 into fertilized Xenopus eggs, it was determined that the N-terminal tail domain and the C-terminal α-helix of H4 are dispensable. For H3, the N-terminal tail and the N-terminal α-helix were not required. Thus, an individual H3 or H4 molecule without a tail domain, and thus no possibility of being acetylated, can still be assembled into chromatin, in agreement with the data obtained in yeast (118). As the authors point out, in these experiments the mutated histone mRNAs must compete with the naturally occurring, stored histone message present in the Xenopus egg. It is therefore likely that hybrid H3/H4 tetramers will form, containing at least one normal histone molecule. Thus, the ability of the H3 and H4 tails to act redundantly (118) was not specifically tested in these experiments. Nevertheless, minimum requirements for proper chromatin assembly were defined, specifically, those regions involved in H3–H3 and H3–H4 interactions.
What conclusions can be drawn from these genetic explorations of histone acetylation and chromatin assembly? It is safe to say that, in the presence or absence of normal H3, the conserved K5/K12 acetyla tion pattern of newly synthesized H4 is not required to assemble a nucleosome. Even yeast cells that cannot be acetylated on seven of the acetylatable lysines of H3 and H4 (including H4 K5/K12) are viable, although they display severe derepression of telomeric chromatin, possibly the result of an improperly assembled chromatin template. It is also clear that the H3 and H4 tail domains are functionally redundant with respect to cell viability, and to some degree in chromatin assembly. That acetylation may be doing more than merely neutralizing positive charge is further suggested by the observation that at least one of the K5, K8, or K12 sites of H4 must be retained as lysine (and not changed to glycine), when the H3 tail domain is deleted (130); however, a requirement for unmodified lysine cannot be ruled out. Most notably, none of the mutant histones has thus far been tested in replication-coupled nucleosome assembly assays, and there is nothing yet to indicate that the acetylated tails of H3 or H4 specifically interact with any component of the assembly pathway. Other avenues to explore are to combine HAT-B null mutants with H3/ H4 deletions and substitutions, or with deletions of the CAC and RCAF (or ASF1) proteins. In such experiments it may be important to replace the acetylatable lysines of H3 and H4 with hydrophilic residues (e.g., arginine, glutamine) rather than with glycine, to preserve the native structure of the N-termini as much as possible (16).
Even in the presence of a complete H3 N-terminal domain, the K5/K12→R yeast double substitution increases the length of S phase by ∼50%, suggesting that chromatin replication or nucleosome assembly may be relatively inefficient. Cells with such slow doubling times will of course be at a severe selective disadvantage, and will quickly be lost under natural conditions. It may therefore be most useful to consider deposition-related acetylation as an adaptation that facilitates nucleosome assembly, thus accelerating growth rate, but which is not absolutely required by all assembly pathways.
HISTONE ACETYLATION AND CHROMATIN REPLICATION
In this section biochemical inquiries to the involvement of histone acetylation in DNA replication, histone segregation, and chromatin maturation will be addressed.
DNA Replication and Nucleosome Segregation
It has long been known that the histone tails are dispensable for core particle integrity, and that they are not required to reconstitute nucleosomes from purified histones and naked DNA (215,216). Because the core histone tail domains can be digested in situ while leaving the remainder of the proteins intact (213), it is possible to use trypsinized histones to examine tail function [e.g., (15,50,155,197)]. When trypsinized histones were reconstituted onto SV40 DNA templates, it was found that the resulting mini-chromosomes replicated faster in vitro than control SV40 chromatin (approximately twofold) (163). The increase appeared to be due to a faster rate of fork migration. No CAF-1 was present in the replication reaction, so concomitant de novo nucleosome assembly could not occur. Nevertheless, the replicated SV40 contained nucleosomes, derived from the pre-replicated minichromosomes. Thus, the histone tails are not required to transfer parental histones to new DNA. When hyperacetylated SV40 minichromosomes (obtained by treating cells with a deacetylase inhibitor) were similarly tested, only a slight increase in replication rate was observed (1.3- to 1.5-fold increase). It would therefore appear that acetylation has only a minimal effect on fork migration, and is not required to replicate chromatin in vitro.
A powerful approach to the analysis of chromatin metabolism involves the use of antisera that are specific for acetylated histone isoforms (37,112,214). Antibodies that specifically recognize acetylated H4 (117) have been used to study nucleosome assembly and histone exchange in HeLa cells (156). Chromatin replicated and assembled in vivo in the presence of the deacetylase inhibitor sodium butyrate is immuno-precipitated with very high efficiency, consistent with the targeted deposition of new, diacetylated H4 onto newly replicated DNA. Moreover, nascent H2A, H2B, and H3 are coprecipitated with new H4. Because the antibodies used in this study recognize both new and old acetylated H4 molecules, these results do not indicate that all four new core histones are co-deposited to form conservatively assembled octamers. Rather, they demonstrate that new H2A and H2B (which, it may be recalled, are largely deposited onto nonreplicating chromatin) are assembled into nucleosomes that contain parental, acetylated H4. In the same study (156) it was further shown that new H2A and H2B that enter chromatin in the absence of concurrent DNA replication (23,88,124) are preferentially deposited onto acetylated chromatin regions. Acetylated H4 is therefore associated both with replication-coupled nucleosome assembly and with H2A/ H2B exchange.
Given the strong connection that exists between histone acetylation and transcription, it might be predicted that chromatin is highly acetylated in advance of the replication fork, to assist in decondensing chromatin higher order structures. If this were the case, it should be possible to immunoprecipitate segregated parental nucleosomes with acetylation-specific antibodies. To eliminate the contribution of nascent, di-acetylated H4 in such experiments, chromatin is either replicated in vivo in the presence of cycloheximide, or in vitro in isolated nuclei.
Using this approach, it was found that the level of H4 acetylation in segregated nucleosomes was approximately equal to that found in bulk chromatin (153). Thus, native prereplicative chromatin is not obligatorily acetylated, as suggested by the experiments using SV40 cited above. This is also in agreement with an immunohistochemical study of chroma-tin replicated in the protozoan Euplotes, in which it was observed that prereplicative chromatin stained only weakly with anti-(acetylated) H4 antibodies, while postreplicative chromatin (containing newly synthesized H4) stained strongly (150).
As has been suggested (153), the absence of a requirement for histone acetylation (or deacetylation) to generate replication-competent chromatin permits the acetylation status of parental nucleosomes to be preserved, potentially acting as an epigenetic mechanism of inheritance. Because parental nucleosomes (i.e., histones) segregate to both sides of the replication fork (Fig. 1) (7,13,22,30,38,91,159,185), such a mechanism would transfer regulatory information to both daughter cells (94,199,217).
Chromatin Maturation
There is no specific inhibitor of histone acetylation yet available, and so it is not feasible to biochemically preclude the acetylation of newly synthesized H4 in vivo. It is possible, however, to prevent histone deacetylation with sodium butyrate, trichostatin A, etc., and to test the effects on chromatin assembly. As discussed above, under normal conditions newly synthesized H4 is deacetylated within 30–60 min of its deposition. When chromatin is replicated and assembled in vivo in the presence of sodium butyrate, the acetylation of new H3/H4 is preserved. Chromatin assembled in this manner possesses typical nucleosomal structure and repeat length (12,176), yet retains an increased sensitivity to DNase I (12), a feature of nascent chromatin that is rapidly lost under normal replication conditions. Further studies have shown that the increased DNase I sensitivity is not exhibited by mononucleosomes, but instead can be ascribed to a reduction of H1-mediated internucleosomal interactions that depend on the his-tone tail domains (154,155).
Although H1 associates with newly replicated chromatin very rapidly after passage of the replication fork (11,64), its binding is at first highly unstable (18). The labile binding of H1, together with the retention of deposition-type histone acetylation patterns, may have profound effects on the internucleosomal interactions of chromatin replicated in butyrate, thus causing the increased DNase I sensitivity described above. Moreover, chromatin assembled in vitro using hyperacetylated histones also displays increased sensitivity to DNase I (109). Such considerations underscore the importance of reestablishing normal chromatin higher order structures following DNA replication and nucleosome assembly. In the following section the contribution of the histone tails, and of histone acetylation, to chromatin folding will be discussed in detail.
EFFECTS OF ACETYLATION ON THE STRUCTURE AND ACTIVITY OF THE CHROMATIN FIBER
The remainder of the article summarizes and discusses recent advances in our understanding of how the core histone N-termini and histone acetylation modulate the higher order structure, activity, and accessibility of nucleosomal and chromatin arrays. As we define them, nucleosomal arrays are core histone octamer–DNA complexes spaced at physiological intervals, and chromatin arrays (or just “chromatin”) are nucleosomal arrays containing bound linker his-tones. This section is intended to provide a structural foundation for understanding and interpreting functional processes that take place in the context of nucleosomal and chromatin arrays (e.g., replication, transcription). Specifically, we discuss recent evidence indicating that condensation of both nucleosomal and chromatin arrays results directly from the action of the core histone N-termini function, and that acetylation of the N-termini leads to both decondensation and functional activation of nucleosomal arrays.
Functions of the Core Histone N-Termini in Chromatin
Prior to describing the molecular effects of acetylation, we first discuss the role of the unmodified core histone N-termini in chromatin fiber organization. The properties of intact, tailless, and acetylated nucleosome core particles are not discussed in depth, and interested readers are directed to several comprehensive reviews for this information (51,204,219). In general, removal or acetylation of the N-termini of nucleosome core particles has only subtle structural effects, unlike the case for nucleosomal and chromatin arrays.
Solution-State Behavior of Nucleosomal Arrays
The idea that the core histone tail domains play a role in chromatin folding dates back ∼20 years (51,204,219), although historically the in vitro condensation of nucleosomal arrays lacking linker histones has not been considered overly important (51,73). However, during the last 5 years, the unexpectedly complex involvement of the core histone N-termini in nucleosomal array and chromatin condensation has been extensively documented. Virtually all of the recent advances have resulted from systematic in vitro dissection of defined nucleosomal array and chromatin model systems (32,62,158,173,174,177). Figure 2 summarizes the fundamental solution-state behavior of nucleosomal arrays. Nucleosomal arrays in solution are in equilibrium between unfolded, moderately folded, highly folded, and oligomerized conformational states (62,72,158,173,174). In terms of folding, formation of the moderately folded state involves close approach of neighboring nucleosomes (i.e., nucleosome n with nucleosomes n + 1 and/or n + 2) (51,52). Once this folding intermediate has formed, nucleosome n subsequently interacts with nucleosome n + ∼6 to form the extensively folded 30-nm state (J. C. Hansen, unpublished). The details of the extensively folded 30-nm fiber remain unknown, as has been the case for over 20 years (51,204,219). However, it now is generally recognized that the maximally folded 30-nm state is an inherently irregular helix, as opposed to the regular helix that dominated the earlier models [see (51,204,219)].
Figure 2.
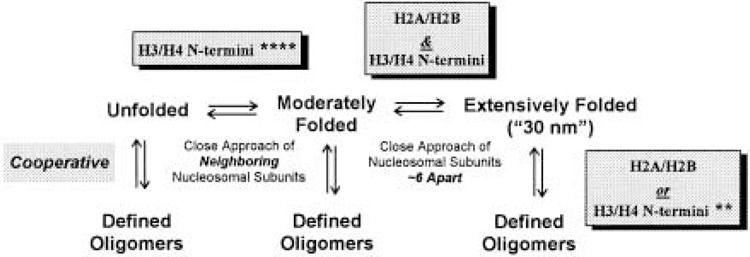
General scheme describing the solution-state behavior of nucleosomal arrays. The number of asterisks (*) represents the degree to which the indicated functions of the N-termini can be replaced by high Mg2+ concentrations. See text for details.
Oligomerization is a reversible, highly cooperative process in which individual nucleosomal arrays self-associate to form very large polymeric structures. Oligomerization is induced at higher Mg concentrations than folding (158,173), is selectively induced by polyamines (158), and occurs independently of whether the nucleosomal array is unfolded, moderately folded, or extensively folded (i.e., oligomerization is not obligatorily coupled to folding) (173). There is strong circumstantial evidence suggesting that in vitro oligomerization is functionally related to long-range transitions that lead to global compaction of interphase chromosomal fibers (51,173). Note that when describing the in vitro behavior of nucleosomal arrays, folding and oligomerization collectively will be referred to as “condensation.”
The Core Histone N-Termini and Chromatin Condensation
By assembling nucleosomal arrays in vitro from selectively proteolyzed histone octamers, it has been demonstrated that tailless nucleosomal arrays i) have unwrapped nucleosomal DNA in very low salt buffer (50,62,197), ii) are unable to form either the moderately (50,62,197) or extensively (197) folded conformation at “physiological” salt concentrations (i.e., 100–200 mM NaCl, 1–2 mM MgCl2), and iii) are unable to oligomerize at even supraphysiological levels of Mg2+ (173). Additional insight into the function of the individual core histone N-termini as well as the mechanisms through which the N-termini mediate array condensation emerged from analyses of “hybrid” trypsinized nucleosomal arrays selectively lacking only the H3/H4 or H2A/H2B N-termini (143,197). These studies revealed for the first time the remarkable degree of complexity of N-termini function in nucleosomal array and chromatin condensation.
In particular, it was shown that the H3/H4 N-termini appeared to mediate formation of the moderately folded state (143,197), while contributions from both the H3/H4 and H2A/H2B N-termini are required to mediate formation of the extensively folded state (197). In distinct contrast, either the H2A/H2B or H3/ H4 N-termini alone were sufficient to mediate oligomerization, although both hybrid arrays oligomerized at higher MgCl2 concentrations than native arrays. Subsequent analysis of the folding of hybrid and completely trypsinized nucleosomal arrays in high MgCl2 (4–10 mM) showed that, even in the absence of the N-termini, the moderately folded conformation, but not the extensively folded state, could be formed in high divalent salts. The ability of high MgCl2 to replace the functions of N-termini in mediating the close approach of neighboring nucleosomes suggests an electrostatic-based mechanism of action is involved in this transition (e.g., tail–DNA interactions), consistent with the highly basic nature of the tails. However, the inability of high MgCl2 to induce formation of the extensively folded conformation of tailless nucleosomal arrays suggests a fundamentally different mechanism mediates this transition. A likely candidate in this case is protein–protein interactions, specifically either internucleosomal tail–tail interactions, and/or binding of the tails to the structured region of another nucleosome (73,127). In this regard, while once thought to be nonstructured coils (51,73,204,219), it is becoming increasingly recognized that the core histone N-termini contain α-helices and perhaps other structural motifs when functioning in a natural chromatin context (73). This idea dates back to the speculations of Grunstein and coworkers (132) and was predicted by even the earliest Chou and Fasman rules (204).
To summarize, the core histone N-termini contribute essential functions to both moderate and extensive folding of the chromatin fiber, and probably the longer range fiber-fiber interactions found in higher chromosomal domains as well (51,71,73,127). In terms of the latter, it recently has been reported that the core histone N-termini are required for metaphase chromatin condensation (40). In addition, each of the N-termini-mediated steps involved in nucleosomal array condensation occur through distinct molecular mechanisms, some of which appear to involve protein-DNA interaction while others involve protein-protein interactions (71,73,197).
N-Termini Rearrangement
Another key observation resulting from studies of trypsinized nucleosomal arrays is that the location of the tails changes in conjunction with folding; specifically, they move from nucleosomal DNA when unfolded in low salt to a nonnucleosomal location while mediating salt-dependent folding (50). Other examples of tail rearrangement have come from studies of the location of the H2A C-terminal tail domain. Although this tail domain could be cross-linked near the dyad of nucleosome core particles (116,203), this location was not observed in either ∼200-bp mononucleosomes (116) or intact nuclei (20). Taken together, these studies reveal that the location of a given tail domain in chromatin is very context dependent. In addition, they indicate that the common practice of digesting native chromatin arrays into nucleosome core particles forces the core histone N-termini to assume locations in the core particle that are not reflective of their location in nucleosomal or chromatin arrays under physiological conditions. This in turn raises the question of whether results obtained in vitro with trypsinized and acetylated nucleosome core particles accurately reflect the behavior of the tail domains in chromatin under physiological conditions.
The Core Histone N-Termini, Linker Histones, and Chromatin Condensation
Linker histones (e.g., H1, H5) are chromatin-associated proteins whose functions and mechanism of action in chromatin have been widely studied (204,219). At the level of chromatin folding, linker histones stabilize the extensively folded 30-nm state(s) of chromatin arrays [see (31,194) and references therein], most likely through linker DNA charge neutralization by the highly basic C-terminus (35). This is consistent with the fact that the folded states of nucleosomal arrays in and of themselves are only moderately stable (31,72,174,194). Nevertheless, trypsinized nucleosomal arrays that contain bound linker histones can neither fold extensively nor oligomerize [(2); L. C. Carruthers and J. C. Hansen, submitted]. The simplest explanation for these results is that the core histone N-termini are absolutely required to specify the correct sequence of internucleosomal interactions that lead to formation of the extensively folded 30-nm fiber, and that linker histones are needed to subsequently stabilize the folded structures. The net result is that at any given salt concentration, linker histones (and probably other chromatin-associated proteins) significantly drive the equilibria shown in Figure 2 toward the extensively folded and oligomerized states, but only after the core histone N-termini have specified the correct internucleosomal interactions.
Structural Consequences of Core Histone Acetylation
There have been surprisingly few studies of the effects of acetylation on the higher order properties of the chromatin fiber, perhaps because the critical roles of the core histone N-termini in chromatin condensation have only recently been documented. However, acetylation now is known to potently disrupt formation of extensively folded nucleosomal arrays in physiological salt, and partially disrupt oligomerization. Also, there is a large body of older data involving effects of acetylation on chromatin “solubility” that in retrospect relates to oligomerization. Unlike the case for nucleosomal arrays, the effects of acetylation on the folding of H1-stabilized chromatin arrays remains to be clearly defined.
Nucleosomal Arrays
The initial model studies of Ausio and coworkers demonstrated that acetylated nucleosomal arrays could not form the moderately folded state in NaCl (63). These experiments subsequently were extended by Tse et al. (198), who examined the Mg2+-dependent condensation of nucleosomal arrays containing different levels of acetylated histone octamers. In these studies, the weight average number of acetates/octamer and distribution of mono-, di-, tri-, and tetraacetylated core histone species were quantitated, although it was not possible to determine the specific amino acid residues that were acetylated. A total of 26 specific lysine residues can be acetylated on the tails of each chicken erythrocyte histone octamer (219). For nucleosomal assays containing an average of six acetates/histone octamer, Mg2+-dependent formation of the moderately and extensively folded conformations was indistinguishable from that of unacetylated arrays. However, quite unexpectedly, an average of only 12 acetates/histone octamer disrupted folding to the same extent as complete removal of the N-termini (62). The fact that complete loss of tail domain function could be achieved under conditions where only 12 out of ∼100 positive charges were neutralized by acetates provides further evidence that the N-terminimediated formation of extensively folded nucleosomal arrays involves a mechanism other than charge neutralization. Of note, the additional six acetate groups that led to array unfolding were distributed among the H2B, H3, and H4 N-termini, consistent with the functions of these tails in mediating folding as defined by the proteolysis studies (see above). Mechanistically, the data collected with differentially acetylated nucleosomal arrays provide further support for the conclusions that tail-mediated formation of extensively folded nucleosomal arrays involves protein-protein interactions, and that acetylation disrupts folding by modulating properties of key secondary structural motifs required for inter-nucleosomal interactions.
In contrast to folding, acetylation only partially disrupts oligomerization of nucleosomal arrays (158,198). Experimentally, this is indicated by the need for increased amounts of MgCl2 to achieve oligomerization as the acetylation content of the arrays is increased. In the one case where it was examined, the extent of inhibition achieved by acetylation was linear over the range of 0–12 acetates/histone octamer (198). Interestingly, polyamines selectively induce oligomerization of nucleosomal arrays in vitro (158), and are necessary for maintenance of the condensed state of isolated chromosomal fibers [see (51)]. Furthermore, the native phenotype of a gcn5 knockout yeast strain can be restored by a mutant in ornithine decarboxylase (spe1), a key enzyme in the biosynthesis of polyamines (158). Together with the biochemical results, these genetic data suggest that one of the functions of the Gcn5-dependent histone acetylation in vivo is to antagonize poly-amine-dependent chromatin condensation.
Chromatin Arrays
There have been very few studies of the structure and stability of acetylated linker histone-containing chromatin, and all have examined endogenous chromatin isolated from butyrate-treated HeLa cells. Initially, McGhee et al. (136) used sedimentation and electric dichroism to study cation-induced folding, and concluded that acetylation had slight destabilizing effects on the extensively folded 30-nm state of chromatin arrays. Subsequent studies using electron microscopy found that the acetylated chromatin formed a moderately folded state but was unable to complete compaction to the extensively folded 30-nm state (9). Very recently, it has been shown that H1 represses acetylation of the tails in chromatin by specific histone acetyltransferases (80). Further insight into the structure/function relationships involving acetylation and chromatin arrays awaits additional experimentation. However, on the basis of all available data, at this point it appears that acetylation may have evolved to regulate the activity of nucleosomal arrays more so than chromatin arrays (also see Transcription section below).
The early chromatin literature contains numerous references to salt-dependent “precipitation” and salt “insolubility” (51). There now is evidence that this literature may be relevant to chromatin oligomerization. Salt insolubility and oligomerization both involve the formation of large aggregated species. In addition, as is the case for oligomerization, formation of insoluble chromatin “precipitants” is easily reversed upon resuspension in low salt buffer [see (154,165) and references therein]. Finally, a major determinant of the salt solubility of endogenous chromatin is the acetylated state of the core histone N-termini; the more acetylated the chromatin, the more MgCl2 required to induce aggregation (6,165,166). Similarly, an insoluble fraction of newly replicated chromatin becomes soluble after tryptic removal of the tail domains (155). Interestingly, it was the differential solubility of native acetylated chromatin that was exploited to isolate the differentially acetylated histone octamers used in the model studies described above (198). In retrospect, it seems almost certain that chromatin “insolubility” is the same phenomenon as N-termini-mediated oligomerization of nucleosomal array and chromatin model systems.
Ultimately, the biochemical and genetic data described above suggest that acetylation is involved in modulating the stability of the internucleosomal interactions responsible for both the local folding and long-range condensation of the chromatin fiber.
Replication, Gene Activation, and Higher Order Chromatin Structure
The realization that a “more open” chromatin structure is involved in gene activation and replication dates back ∼25 years to the studies of Weintraub and co-workers (51,204,212,219). However, deciphering the molecular basis for these observations has proven challenging, befitting the complexity of the processes involved. The ensuing section summarizes recent progress made in these areas. As with the previous sections, the discussion centers around newer results obtained with model system studies. The large body of older literature relevant to this subject has been reviewed extensively [see (219), and references therein].
Replication
The process of replication-linked chromatin assembly has been discussed above and is summarized schematically in Figure 1. It is now possible to accurately describe the higher order structure of the key nucleoprotein intermediates present immediately after DNA replication. Because the core his-tones of the parental nucleosomes are not hyperacetylated and retain (or quickly regain) histone H1 (11,18,64,90,153) (also, see above), the stretches of parentally derived chromatin should be moderately to extensively condensed under physiological ionic conditions, depending on the length of the parental array. Thus, the accessibility of DNA in parental nucleosomes to transcription factors will be greatly reduced. In distinct contrast, the acetylated H3/H4 tetramer arrays sandwiched between the parental arrays will be completely unfolded (5,198) (in this case due to the absence of H2A/H2B dimers rather than because of acetylation), very flexible relative to nucleosomal arrays, and the tetramer-bound DNA will be significantly more accessible to transcription factors and other functionally important chromatin-associated proteins (5). These data provide a structural framework for understanding why there is competition between H2A/H2B dimers, transcription factors, and other functional nonhistone multiprotein complexes for binding to the H3/H4 tetramer arrays present immediately after replication (4,5,17,218,219). Ultimately, due to the combination of the dispersive segregation mechanism and the substantively different physicochemical properties of H3/H4 tetramer arrays compared to parental chromatin arrays, newly replicated chromatin is poised to either mature into bulk inactive chromatin or become programmed into functionally active chromatin depending on the local concentrations of histone and nonhistone proteins in the vicinity of the replication fork, as well as their respective binding affinities for H3/H4 tetramer arrays.
There is no evidence that the chromatin fiber must be decondensed well in advance of the replication fork, and thus the machinery that is responsible for dispersive segregation presumably can deal with condensed chromatin, perhaps by a localized dissociation of histone H1 (64). Higher order structure affects the initiation of replication much in the same way as gene activation (see 51).
Gene Activation
An enormous amount of effort has been spent trying to understand the roles of chromatin in gene activation and repression [reviewed in (219)]. Here we briefly summarize the contributions to gene regulation of nucleosomal array condensation, and its disruption by acetylation. Figure 3 lists the hierarchical series of steps that are involved in the activation of most eukaryotic genes. For the last decade, attention has focused largely on transcription factor/RNA polymerase binding and transcription initiation (steps 5 and 6) (66,70,160), and more recently on nucleosome remodeling (step 4) (108,157). For many years it was thought that conversion of an extensively folded, transcriptionally inactive chromatin array into a potentially active, unfolded nucleosomal array simply required dissociation and/or rearrangement of linker histones (51,219). Notable in this regard, step 2 was not even a component of the activation scheme until it was shown that nucleosomal arrays under physiological conditions were sufficiently folded to repress transcription initiation and elongation by eukaryotic RNA polymerase III (74,75,198).
Figure 3.
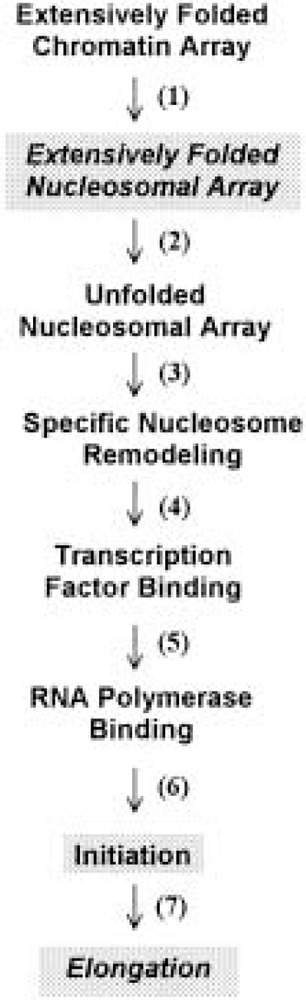
Steps involved in the activation of eukaryotic genes. The steps that are most likely involve acetylation-dependent disruption of nucleosomal array condensation are shaded. See text for details.
Because of its potent ability to decondense nucleosomal arrays, acetylation is likely to have important roles in those steps where disruption of nucleosomal array folding is most crucial (i.e., steps 2, 6, and 7). Step 2 can be thought of as a “preparatory” chromatin transition that poises nucleosomal arrays for transcriptional activation by disrupting the inhibitory folded conformation(s). In this regard, acetylation-induced disruption of the extensive folding of nucleosomal arrays leads to a substantial increase in transcription (10–20-fold) from the same model systems used for the structural studies summarized above. Importantly, whereas previous analogous investigations manipulated the salt concentration (74) or H2A/H2B dimer content (75) to cause array unfolding and transcriptional stimulation, acetylation was capable of achieving the same structural and functional effects even when the arrays remained in physiological salt (198). Thus, acetylation is envisioned as having a key role in preparing chromatin for gene activation (and deacetylation for inactivation) through its ability to disrupt the repressive higher order folding of nucleosomal arrays. Enhancement of transcription initiation also appears to be influenced to the array unfolding that occurs during step 2 (74). Furthermore, array unfolding may be functionally linked to the recently proposed “histone code” hypothesis (189) (i.e., once array unfolding has been induced by acetylation, the modified tails of the unfolded array subsequently may act as target sites for binding specific macromolecules required for transcription initiation).
Once initiation of transcription has occurred, eukaryotic RNA polymerases must be able to elongate through ∼20–100 kb of DNA organized into nucleosomal arrays (219). Given that the extent of nucleosomal array folding present under physiological conditions significantly impedes elongation (74,75,198), it seems likely that large-scale acetylation of entire transcription units may be required to allow efficient elongation in vivo. In this regard, the elegant studies of Hebbes et al. (78,79) have shown that regions of general DNase I sensitivity (indicative of global chromatin decondensation) and histone hyperacetylation precisely co-map with the 33-kb transcription units of several β-globin gene loci in vivo. Also, in this system acetylation occurred several days prior to appearance of transcripts, indicating that the function of acetylation in this case was to prepare the β-globin loci for induction of transcription. The findings of Hebbes et al. are entirely consistent with a role for acetylation-dependent nucleosomal array unfolding in both steps 2 and 7 of the scheme shown in Figure 3.
In conclusion, the structural section of this article has summarized evidence that the core histone N-termini are essential mediators of higher order chromatin condensation, that condensation is capable of potently regulating the activity of the chromatin fiber, and that at least one of the ways that acetylation enhances transcription is through its ability to decondense nucleosomal arrays.
SUMMARY AND PERSPECTIVES
From the foregoing discussion it is clear that great progress has been made toward understanding the structure/function relationships of the core histone N-termini, and their modification by acetylation. Precise descriptions of the patterns, enzymology, and effects of acetylation are now emerging, yielding a picture that is at once conceptually simple yet operationally complex. That acetylation can variously act at the levels of chromatin assembly, histone exchange, transcriptional regulation, and higher order folding is extraordinary, presenting a challenge to those studying these processes as separate phenomena. In this article we have not attempted to address all aspects of acetylation; within those areas considered, nucleosome assembly and fiber dynamics, recent advances still leave many key questions unanswered. Some of these are outlined below.
It is most unlikely that the specific “5/12” diacetylation pattern of newly synthesized H4 would be so rigorously conserved were it to have no function in chromatin biosynthesis. Why then are newly synthesized H3 and H4 acetylated? And does their acetylation function redundantly during chromatin assembly? Thus far, the data suggest that acetylation is not required to assemble nucleosomes, but this remains to be fully tested by combining hat1 (or cac1, asf1) mutations with selected H3/H4 substitutions and deletions. Other important topics include the extent to which HAT-B acts redundantly with other HAT activities, and the ability of CAF-1 to assemble H3/H4 with substitutions and deletions in vitro (see Note Added in Proof).
With respect to chromatin structure and dynamics, it is rather humbling to consider how much remains unknown. To list only a few examples: Does acetylation alter the secondary structure of subdomains of the histone N-termini? Must acetylation be site specific to be effective, and is it coupled to other histone posttranslational modifications? If acetyltransferases are targeted specifically, how is “global” acetylation maintained? Can acetylation act as a “protein code” for the recruitment of additional histone-modifying enzymes and other factors, as proposed by Strahl and Allis (189)? How are the opposing activities of acetylases and deacetylases coordinated? And what attracts deacetylases to the nascent acetylated chromatin fiber? Although such questions must perforce be approached (experimentally) as relatively isolated topics, they naturally reflect common themes: How is acetylation controlled, and how can the neutralization of certain, positively charged lysine residues in the core histone N-termini have such far-reaching effects? Evidence that histone acetylation acts as a fundamental regulatory signal has placed these considerations at the forefront of current inquiries into chromatin structure and function. If the past can serve as prelude, we can expect research efforts to generate at least as many questions as those that will be answered.
ACKNOWLEDGMENTS
The authors were supported by grants from the NIH to A.T.A. (GM 35837) and J.C.H. (GM45916).
NOTE ADDED IN PROOF
It has recently been demonstrated by Stillman and colleagues (Shibahara, K.-i.; Verreault, A.; Stillman, B. The N-terminal tails of H3 and H4 are not necessary for chromatin assembly factor-1-mediated nucleosome assembly onto replicated DNA in vitro. Proc. Natl. Acad. Sci. USA 97:7766–7771; 2000) that CAF-1, and each of the CAF-1 subunits, can stably interact with recombinant H32H42 tetramers lacking the N-terminal tails of both H3 and H4. Moreover, CAF-1 can deposit tailless tetramers onto newly replicated DNA in vitro. Although these observations may eliminate an active role for H3/H4 acetylation during CAF-1-mediated chromatin assembly, it must be recalled that the removal of the histone tails can in some instances mimic the effects of acetylation, and also that acetylation may function in vivo prior to the binding of H3 and H4 to CAF-1. Alternatively, as Shibahara et al. suggest, H3/H4 acetylation may be required by a CAF-1-independent pathway (e.g., by RCAF), or to mark nascent chromatin for recognition by subsequent activities in vivo.
REFERENCES
- 1.Adams C. R.; Kamakaka R. T. Chromatin assembly: Biochemical identities and genetic redundancy. Curr. Opin. Genet. Dev. 9:185–190; 1999. [DOI] [PubMed] [Google Scholar]
- 2.Allan J.; Harborne N.; Rau D. C.; Gould H. Participation of the core histone “tails” in the stabilization of the chromatin solenoid. J. Cell Biol. 93:285–297; 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Almouzni G.; Clark D. J.; Méchali M.; Wolffe A. P. Chromatin assembly on replicating DNA in vitro. Nucleic Acids Res. 18:5767–5774; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Almouzni G.; Mechali M.; Wolffe A. P. Transcription complex disruption caused by a transition in chromatin structure. Mol. Cell. Biol. 11:655–665; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Almouzni G.; Wolffe A. P. Nuclear assembly, structure, and function—the use of Xenopus in vitro systems. Exp. Cell Res. 205:1–15; 1993. [DOI] [PubMed] [Google Scholar]
- 6.Alonso W. R.; Ferris R. C.; Zhang D. E.; Nelson D. A. Chicken erythrocyte beta-globin chromatin: Enhanced solubility is a direct consequence of induced histone hyperacetylation. Nucleic Acids Res. 15: 9325–37; 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Annunziato A. T. Chromatin replication and nucleosome assembly. In: Strauss P. R.; Wilson S. H., eds. The eukaryotic nucleus: Molecular biochemistry and macromolecular assemblies. Caldwell, NJ: The Telford Press; 1990:687–712. [Google Scholar]
- 8.Annunziato A. T. Histone acetylation during chromatin replication and nucleosome assembly. In: Wolffe A. P., ed. The nucleus. Greenwich, CT: JAI Press; 1995:31–55. [Google Scholar]
- 9.Annunziato A. T.; Frado L.-L. Y.; Seale R. L.; Woodcock C. L. F. Treatment with sodium butyrate inhibits the complete condensation of interphase chro-matin. Chromosoma (Berl.) 96:132–138; 1988. [DOI] [PubMed] [Google Scholar]
- 10.Annunziato A. T.; Schindler R. K.; Riggs M. G.; Seale R. L. Association of newly synthesized histones with replicating and nonreplicating regions of chromatin. J. Biol. Chem. 257:8507–8515; 1982. [PubMed] [Google Scholar]
- 11.Annunziato A. T.; Schindler R. K.; Thomas C. A. Jr.; Seale R. L. Dual nature of newly replicated chromatin: Evidence for nucleosomal and non-nucleosomal DNA at the site of native replication forks. J. Biol. Chem. 256:11880–11886; 1981. [PubMed] [Google Scholar]
- 12.Annunziato A. T.; Seale R. L. Histone decetylation is required for the maturation of newly replicated chromatin. J. Biol. Chem. 258:12675–12684; 1983. [PubMed] [Google Scholar]
- 13.Annunziato A. T.; Seale R. L. Presence of nucleosomes within irregularly cleaved fragments of newly replicated chromatin. Nucleic Acids Res. 12:6179–6196; 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arents G.; Burlingame R. W.; Wang B. C.; Love W. E.; Moudrianakis E. N. The nucleosomal core histone octamer at 3.1-a resolution—a tripartite protein assembly and a left-handed superhelix. Proc. Natl. Acad. Sci. USA 88:10148–10152; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ausio J.; Dong F.; van Holde K. E. Use of selectively trypsinized nucleosome core particles to analyze the role of the histone “tails” in the stabilization of the nucleosome. J. Mol. Biol. 206:451–463; 1989. [DOI] [PubMed] [Google Scholar]
- 16.Baneres J. L.; Martin A.; Parello J. The N tails of histones H3 and H4 adopt a highly structured conformation in the nucleosome. J. Mol. Biol. 273:503–508; 1997. [DOI] [PubMed] [Google Scholar]
- 17.Barton M. C.; Emerson B. M. Regulated expression of the beta-globin gene locus in synthetic nuclei. Genes Dev. 8:2453–2465; 1994. [DOI] [PubMed] [Google Scholar]
- 18.Bavykin S.; Srebreva L.; Banchev T.; Tsanev R.; Zlatanova J.; Mirzabekov A. Histone H1 deposition and histone DNA interactions in replicating chromatin. Proc. Natl. Acad. Sci. USA 90:3918–3922; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Becker P. B.; Tsukiyama T.; Wu C. Chromatin assembly extracts from Drosophila embryos. In: Goldstein L. S. B.; Fyrberg E. A., eds., Methods in cell biology, vol. 44 San Diego: Academic Press; 1994:207–223. [DOI] [PubMed] [Google Scholar]
- 20.Belyavsky A. V.; Bavykin S. G.; Goguadze E. G.; Mirzabekov A. D. Primary organization of nucleo-somes containing all five histones and DNA 175 and 165 base-pairs long. J. Mol. Biol. 139:519–536; 1980. [DOI] [PubMed] [Google Scholar]
- 21.Berger S. L. Gene activation by histone and factor acetyltransferases. Curr. Opin. Cell. Biol. 11:336–341; 1999. [DOI] [PubMed] [Google Scholar]
- 22.Bonne-Andrea C.; Wong M. L.; Alberts B. M. In vitro replication through nucleosomes without histone displacement. Nature 343:719–726; 1990. [DOI] [PubMed] [Google Scholar]
- 23.Bonner W. M.; Wu R. S.; Panusz H. T.; Muneses C. Kinetics of accumulation and depletion of soluble newly synthesized histone in the reciprocal regulation of histone and DNA synthesis. Biochemistry 27: 6542–6550; 1988. [DOI] [PubMed] [Google Scholar]
- 24.Braunstein M.; Rose A. B.; Holmes S. G.; Allis C. D.; Broach J. R. Transcriptional silencing in yeast is associated with reduced nucleosome acetylation. Genes Dev. 7:592–604; 1993. [DOI] [PubMed] [Google Scholar]
- 25.Braunstein M.; Sobel R. E.; Allis C. D.; Turner B. M.; Broach J. R. Efficient transcriptional silencing in Saccharomyces cerevisiae requires a heterochromatin histone acetylation pattern. Mol. Cell. Biol. 16: 4349–4356; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brehm A.; Miska E. A.; Mccance D. J.; Reid J. L.; Bannister A. J.; Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 391:597–601; 1998. [DOI] [PubMed] [Google Scholar]
- 27.Brownell J. E.; Allis C. D. Special HATs for special occasions: Linking histone acetylation to chromatin assembly and gene activation. Curr. Opin. Genet. Dev. 6:176–184; 1996. [DOI] [PubMed] [Google Scholar]
- 28.Brownell J. E.; Zhou J. X.; Ranalli T.; Kobayashi R.; Edmondson D. G.; Roth S. Y.; Allis C. D. Tetrahymena histone acetyltransferase A: A homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell 84:843–851; 1996. [DOI] [PubMed] [Google Scholar]
- 29.Bulger M.; Ito T.; Kamakaka R. T.; Kadonaga J. T. Assembly of regularly spaced nucleosome arrays by Drosophila chromatin assembly factor 1 and a 56-kDa histone-binding protein. Proc. Natl. Acad. Sci. USA 92:11726–11730; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burhans W. C.; Vassilev L. T.; Wu J.; Sogo J. M.; Nallaseth F. S.; Depamphilis M. L. Emetine allows identification of origins of mammalian DNA replication by imbalanced DNA synthesis, not through conservative nucleosome segregation. EMBO J. 10: 4351–4360; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carruthers L. M.; Bednar J.; Woodcock C. L.; Hansen J. C. Linker histones stabilize the intrinsic salt-dependent folding of nucleosomal arrays: Mechanistic ramifications for higher-order chromatin folding. Biochemistry 37:14776–14787; 1998. [DOI] [PubMed] [Google Scholar]
- 32.Carruthers L. M.; Tse C.; Walker K. P. 3rd; Hansen J. C. Assembly of defined nucleosomal and chromatin arrays from pure components. Methods Enzymol. 304:19–35; 1999. [DOI] [PubMed] [Google Scholar]
- 33.Chang L.; Loranger S. S.; Mizzen C.; Ernst S. G.; Allis C. D.; Annunziato A. T. Histones in transit: Cytosolic histone complexes and diacetylation of H4 during nucleosome assembly in human cells. Biochemistry 36:469–480; 1997. [DOI] [PubMed] [Google Scholar]
- 34.Chicoine L. G.; Schulman I. G.; Richman R.; Cook R. G.; Allis C. D. Nonrandom utilization of acetylation sites in histones isolated from Tetrahymena: Evidence for functionally distinct H4 acetylation sites. J. Biol. Chem. 261:1071–1076; 1986. [PubMed] [Google Scholar]
- 35.Clark D. J.; Kimura T. Electrostatic mechanism of chromatin folding. J. Mol. Biol. 211:883–96; 1990. [DOI] [PubMed] [Google Scholar]
- 36.Cousens L. S.; Alberts B. M. Accessibility of newly synthesized chromatin to histone acetylase. J. Biol. Chem. 257:3945–3949; 1982. [PubMed] [Google Scholar]
- 37.Crane-Robinson C.; Hebbes T. R.; Clayton A. L.; Thorne A. W. Chromosomal mapping of core histone acetylation by immunoselection. Methods 12:48–56; 1997. [DOI] [PubMed] [Google Scholar]
- 38.Cusick M. F.; DePamphilis M. L.; Wassarman P. M. Dispersive segregation of nucleosomes during replication of simian virus 40 chromosomes. J. Mol. Biol. 178:249–271; 1984. [DOI] [PubMed] [Google Scholar]
- 39.Davie J. R.; Spencer V. A. Control of histone modifications. J. Cell. Biochem. 75(Suppl.):141–148; 1999. [DOI] [PubMed] [Google Scholar]
- 40.de la Barre A. E.; Gerson V.; Gout S.; Creaven M.; Allis C. D.; Dimitrov S. Core histone N-termini play an essential role in mitotic chromosome condensation. EMBO J. 19:379–391; 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dimitrov S.; Almouzni G.; Dasso M.; Wolffe A. P. Chromatin transitions during early Xenopus embryo-genesis—changes in histone-H4 acetylation and in linker histone type. Dev. Biol. 160:214–227; 1993. [DOI] [PubMed] [Google Scholar]
- 42.Dimitrov S.; Wolffe A. P. Chromatin and nuclear assembly: Experimental approaches towards the reconstitution of transcriptionally active and silent states. BBA-Gene Struct. Expr. 1260:1–13; 1995. [DOI] [PubMed] [Google Scholar]
- 43.Durrin L. K.; Mann R. K.; Kayne P. S.; Grunstein M. Yeast histone H4 N-terminal sequence is required for promoter activation in vivo. Cell 65:1023–1031; 1991. [DOI] [PubMed] [Google Scholar]
- 44.Dutnall R. N.; Tafrov S. T.; Sternglanz R.; Ramak-rishnan V. Structure of the histone acetyltransferase Hat1: A paradigm for the GCN5-related N-acetyltransferase superfamily. Cell 94:427–438; 1998. [DOI] [PubMed] [Google Scholar]
- 45.Dutnall R. N.; Tafrov S. T.; Sternglanz R.; Ramak-rishnan V. Structure of the yeast histone acetyltransferase Hat1: Insights into substrate specificity and implications for the Gcn5-related N-acetyltransferase superfamily. Cold Spring Harbor Symp. Quant. Biol. 63:501–507; 1998. [DOI] [PubMed] [Google Scholar]
- 46.Eberharter A.; Lechner T.; Goralikschramel M.; Loidl P. Purification and characterization of the cytoplasmic histone acetyltransferase B of maize embryos. FEBS Lett. 386:75–81; 1996. [DOI] [PubMed] [Google Scholar]
- 47.Edmondson D. G.; Smith M. M.; Roth S. Y. Repression domain of the yeast global repressor Tup1 interacts directly with histones H3 and H4. Genes Dev. 10:1247–1259; 1996. [DOI] [PubMed] [Google Scholar]
- 48.Enomoto S.; Berman J. Chromatin assembly factor I contributes to the maintenance, but not the re-establishment, of silencing at the yeast silent mating loci. Genes Dev. 12:219–232; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Enomoto S.; Mccunezierath P. D.; Geraminejad M.; Sanders M. A.; Berman J. RLF2, a subunit of yeast chromatin assembly factor-I, is required for telomeric chromatin function in vivo. Genes Dev. 11:358–370; 1997. [DOI] [PubMed] [Google Scholar]
- 50.Fletcher T. M.; Hansen J. C. Core histone tail domains mediate oligonucleosome folding and nucleosomal DNA organization through distinct molecular mechanisms. J. Biol. Chem. 270:25359–25362; 1995. [DOI] [PubMed] [Google Scholar]
- 51.Fletcher T. M.; Hansen J. C. The nucleosomal array: Structure/function relationships. Crit. Rev. Eukaryot. Gene Expr. 6:149–188; 1996. [DOI] [PubMed] [Google Scholar]
- 52.Fletcher T. M.; Serwer P.; Hansen J. C. Quantitative analysis of macromolecular conformational changes using agarose gel electrophoresis: Application to chromatin folding. Biochemistry 33:10859–10863; 1994. [DOI] [PubMed] [Google Scholar]
- 53.Fotedar R.; Roberts J. M. Multistep pathway for replication-dependent nucleosome assembly. Proc. Natl. Acad. Sci. USA 86:6459–6463; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Freeman L.; Kurumizaka H.; Wolffe A. P. Functional domains for assembly of histones H3 and H4 into the chromatin of Xenopus embryos. Proc. Natl. Acad. Sci. USA 93:12780–12785; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fujii-Nakata T.; Ishimi Y.; Okuda A.; Kikuchi A. Functional analysis of nucleosome assembly protein, NAP-1—the negatively charged cooh-terminal region is not necessary for the intrinsic assembly activity. J. Biol. Chem. 267:20980–20986; 1992. [PubMed] [Google Scholar]
- 56.Fukuda K.; Morioka H.; Imajou S.; Ikeda S.; Ohtsuka E.; Tsurimoto T. Structure–function relationship of the eukaryotic DNA replication factor, proliferating cell nuclear antigen. J. Biol. Chem. 270: 22527–22534; 1995. [DOI] [PubMed] [Google Scholar]
- 57.Gaillard P. H. L.; Martini E. M. D.; Kaufman P. D.; Stillman B.; Moustacchi E.; Almouzni G. Chromatin assembly coupled to DNA repair: A new role for chromatin assembly factor I. Cell 86:887–896; 1996. [DOI] [PubMed] [Google Scholar]
- 58.Gaillard P. H. L.; Moggs J. G.; Roche D. M. J.; Quivy J. P.; Becker P. B.; Wood R. D.; Almouzni G. Initiation and bidirectional propagation of Chromatin assembly from a target site for nucleotide excision repair. EMBO J. 16:6281–6289; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gaillard P. H. L.; Moggs J. G.; Roche D. M. J.; Quivy J. P.; Becker P. B.; Wood R. D.; Almouzni G. Initiation and bidirectional propagation of chromatin assembly from a target site for nucleotide excision repair. EMBO J. 16:6613; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Game J. C.; Kaufman P. D. Role of Saccharomyces cerevisiae chromatin assembly factor-I in repair of ultraviolet radiation damage in vivo. Genetics 151:485–497; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Garcea R. L.; Alberts B. M. Comparative studies of histone acetylation in nucleosomes, nuclei, and intact cells: Evidence for special factors which modify acetylase action. J. Biol. Chem. 255:11454–11463; 1980. [PubMed] [Google Scholar]
- 62.Garcia-Ramirez M.; Dong F.; Ausio J. Role of the histone tails in the folding of oligonucleosomes depleted of histone-H1. J. Biol. Chem. 267:19587–19595; 1992. [PubMed] [Google Scholar]
- 63.Garcia-Ramirez M.; Rocchini C.; Ausio J. Modulation of chromatin folding by histone acetylation. J. Biol. Chem. 270:17923–17928; 1995. [DOI] [PubMed] [Google Scholar]
- 64.Gasser R.; Koller T.; Sogo J. M. The stability of nucleosomes at the replication fork. J. Mol. Biol. 258: 224–239; 1996. [DOI] [PubMed] [Google Scholar]
- 65.Giancotti V.; Russo E.; Cristini F. d.; Graziosi G.; Micali F.; Crane-Robinson C. Histone modification in early and late Drosophila embryos. Biochem. J. 218:321–329; 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goodrich J. A.; Cutler G.; Tjian R. Contacts in context: Promoter specificity and macromolecular interactions in transcription. Cell 84:825–830; 1996. [DOI] [PubMed] [Google Scholar]
- 67.Grunstein M. Histone acetylation in chromatin structure and transcription. Nature 389:349–352; 1997. [DOI] [PubMed] [Google Scholar]
- 68.Gruss C.; Knippers R. Structure of replicating chromatin. In: Cohn W. E.; Moldave K., eds. Progress in nucleic acid research and molecular biology. San Diego: Academic Press Inc.; 1996:337–365. [DOI] [PubMed] [Google Scholar]
- 69.Gruss C.; Sogo J. M. Chromatin replication. Bioessays 14:1–8; 1992. [DOI] [PubMed] [Google Scholar]
- 70.Hampsey M.; Reinberg D. RNA polymerase II as a control panel for multiple coactivator complexes. Curr. Opin. Genet. Dev. 9:132–9; 1999. [DOI] [PubMed] [Google Scholar]
- 71.Hansen J. C. The core histone N-termini: Combinatorial interaction domains that link chromatin structure with function. Chemtracts Biochem. Mol. Biol. 10:56–59; 1997. [Google Scholar]
- 72.Hansen J. C.; Ausio J.; Stanik V. H.; van Holde K. E. Homogeneous reconstituted oligonucleosomes, evidence for salt-dependent folding in the absence of histone H1. Biochemistry 28:9129–9136; 1989. [DOI] [PubMed] [Google Scholar]
- 73.Hansen J. C.; Tse C.; Wolffe A. P. Structure and function of the core histone N-termini: More than meets the eye. Biochemistry 37:17637–17641; 1998. [DOI] [PubMed] [Google Scholar]
- 74.Hansen J. C.; Wolffe A. P. Influence of chromatin folding on transcription initiation and elongation by RNA polymerase-III. Biochemistry 31:7977–7988; 1992. [DOI] [PubMed] [Google Scholar]
- 75.Hansen J. C.; Wolffe A. P. A role for histones h2a/h2b in chromatin folding and transcriptional repression. Proc. Natl. Acad. Sci. USA 91:2339–2343; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hartzog G. A.; Winston F. Nucleosomes and transcription: Recent lessons from genetics. Curr. Opin. Genet. Dev. 7:192–198; 1997. [DOI] [PubMed] [Google Scholar]
- 77.Hassig C. A.; Fleischer T. C.; Billin A. N.; Schreiber S. L.; Ayer D. E. Histone deacetylase activity is required for full transcriptional repression by mSin3A. Cell 89:341–347; 1997. [DOI] [PubMed] [Google Scholar]
- 78.Hebbes T. R.; Clayton A. L.; Thorne A. W.; Crane-Robinson C. Core histone hyperacetylation co-maps with generalized DNase I sensitivity in the chicken beta-globin chromosomal domain. EMBO J. 13: 1823–1830; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hebbes T. R.; Thorne A. W.; Clayton A. L.; Crane-Robinson C. Histone acetylation and globin gene switching. Nucleic Acids Res. 20:1017–1022; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Herrera J. E.; West K. L.; Schiltz R. L.; Nakatani Y.; Bustin M. Histone H1 is a specific repressor of core histone acetylation in chromatin. Mol. Cell. Biol. 20:523–529; 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Imhof A.; Wolffe A. P. Transcription: Gene control by targeted histone acetylation. Curr. Biol. 8:R422–R424; 1998. [DOI] [PubMed] [Google Scholar]
- 82.Imhof A.; Wolffe A. P. Purification and properties of the Xenopus Hat1 acetyltransferase: Association with the 14-3-3 proteins in the oocyte nucleus. Biochemistry 38:13085–13093; 1999. [DOI] [PubMed] [Google Scholar]
- 83.Ishimi Y.; Hirosumi J.; Sato W.; Sugasawa K.; Yokota S.; Hanoaoka F.; Yamada M.-a. Purification and initial characterization of a protein which facilitates assembly of nucleosome-like structure from mammalian cells. Eur. J. Biochem. 142:431–439; 1984. [DOI] [PubMed] [Google Scholar]
- 84.Ishimi Y.; Kojima M.; Yamada M.; Hanaoka F. Binding mode of nucleosome-assembly protein (AP-I) and histones. Eur. J. Biochem. 162:19–24; 1987. [DOI] [PubMed] [Google Scholar]
- 85.Ishimi Y.; Yasuda H.; Hirosumi J.; Hanaoka F.; Yamada M.-a. A protein which facilitates the assembly of nucleosome-like structures in vitro in mammalian cells. J. Biochem. 94:735–744; 1983. [DOI] [PubMed] [Google Scholar]
- 86.Ito T.; Bulger M.; Kobayashi R.; Kadonaga J. T.Drosophila NAP-1 is a core histone chaperone that functions in ATP-facilitated assembly of regularly spaced nucleosomal arrays. Mol. Cell. Biol. 16:3112–3124; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jackson S.; Brooks W.; Jackson V. Dynamics of the interactions of histones H2A,H2B and H3,H4 with torsionally stressed DNA. Biochemistry 33:5392–5403; 1994. [DOI] [PubMed] [Google Scholar]
- 88.Jackson V. In vivo studies on the dynamics of histone–DNA interaction: Evidence for nucleosome dissolution during replication and transcription and a low level of dissolution independent of both. Biochemistry 29:719–731; 1990. [DOI] [PubMed] [Google Scholar]
- 89.Jackson V.; Chalkley R. A new method for the isolation of replicative chromatin: Selective deposition of histone on both new and old DNA. Cell 23:121–134; 1981. [DOI] [PubMed] [Google Scholar]
- 90.Jackson V.; Chalkley R. A reevaluation of new histone deposition on replicating chromatin. J. Biol. Chem. 256:5095–5103; 1981. [PubMed] [Google Scholar]
- 91.Jackson V.; Chalkley R. Histone segregation on replicating chromatin. Biochemistry 24:6930–6938; 1985. [DOI] [PubMed] [Google Scholar]
- 92.Jackson V.; Marshall S.; Chalkley R. The sites of deposition of newly synthesized histone. Nucleic Acids Res. 9:4563–4581; 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jackson V.; Shires A.; Tanphaichitr N.; Chalkley R. Modifications to histones immediately after synthesis. J. Mol. Biol. 104:471–483; 1976. [DOI] [PubMed] [Google Scholar]
- 94.Jeppesen P. Histone acetylation: A possible mechanism for the inheritance of cell memory at mitosis. Bioessays 19:67–74; 1997. [DOI] [PubMed] [Google Scholar]
- 95.Johnson L. M.; Kayne P. S.; Kahn E. S.; Grunstein M. Genetic evidence for an interaction between SIR3 and histone H4 in the repression of the silent mating loci in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 87:6286–6290; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jones P. L.; Veenstra G. J. C.; Wade P. A.; Vermaak D.; Kass S. U.; Landsberger N.; Strouboulis J.; Wolffe A. P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 19:187–191; 1998. [DOI] [PubMed] [Google Scholar]
- 97.Kamakaka R. T.; Bulger M.; Kaufman P. D.; Still-man B.; Kadonaga J. T. Postreplicative chromatin assembly by Drosophila and human chromatin assembly factor 1. Mol. Cell. Biol. 16:810–817; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kamakaka R. T.; Kaufman P. D.; Stillman B.; Mit-sis P. G.; Kadonaga J. T. Simian virus 40 origin-and T-antigen-dependent DNA replication with Drosophila factors in vitro. Mol. Cell. Biol. 14:5114–5122; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kaufman P. D. Nucleosome assembly: The CAF and the HAT. Curr. Opin. Biol. 8:369–373; 1996. [DOI] [PubMed] [Google Scholar]
- 100.Kaufman P. D.; Cohen J. L.; Osley M. A. Hir proteins are required for position-dependent gene silencing in Saccharomyces cerevisiae in the absence of chromatin assembly factor I. Mol. Cell. Biol. 18: 4793–4806; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kaufman P. D.; Kobayashi R.; Kessler N.; Stillman B. The p150 and p60 subunits of chromatin assembly factor I: A molecular link between newly synthesized histones and DNA replication. Cell 81:1105–1114; 1995. [DOI] [PubMed] [Google Scholar]
- 102.Kaufman P. D.; Kobayashi R.; Stillman B. Ultraviolet radiation sensitivity and reduction of telomeric silencing Saccharomyces cerevisiae cells lacking chromatin assembly factor-I. Genes Dev. 11:345–357; 1997. [DOI] [PubMed] [Google Scholar]
- 103.Kayne P. S.; Kim U.-J.; Han M.; Mullen J. R.; Yoshizaki F.; Grunstein M. Extremely conserved histone H4 N-terminus is dispensable for growth but essential for repressing the silent mating loci in yeast. Cell 55:27–39; 1988. [DOI] [PubMed] [Google Scholar]
- 104.Kleff S.; Andrulis E. D.; Anderson C. W.; Sternglanz R. Identification of a gene encoding a yeast histone H4 acetyltransferase. J. Biol. Chem. 270: 24674–24677; 1995. [DOI] [PubMed] [Google Scholar]
- 105.Kleinschmidt J. A.; Fortkamp E.; Krohne G.; Zentgraf H.; Franke W. W. Co-existence of two different types of soluble histone complexes in nuclei of Xeno-pus laevis oocytes. J. Biol. Chem. 260:1166–1176; 1985. [PubMed] [Google Scholar]
- 106.Kolle D.; Sarg B.; Lindner H.; Loidl P. Substrate and sequential site specificity of cytoplasmic histone acetyltransferases of maize and rat liver. FEBS Lett. 421:109–114; 1998. [DOI] [PubMed] [Google Scholar]
- 107.Kong X. P.; Onrust R.; O’Donnell M.; Kuriyan J. Three-dimensional structure of the beta subunit of E. coli DNA polymerase III holoenzyme: A sliding DNA clamp. Cell 69:425–37; 1992. [DOI] [PubMed] [Google Scholar]
- 108.Kornberg R. D.; Lorch Y. Chromatin-modifying and -remodeling complexes. Curr. Opin. Genet. Dev. 9:148–151; 1999. [DOI] [PubMed] [Google Scholar]
- 109.Krajewski W. A.; Becker P. B. Reconstitution of hyperacetylated, DNase I-sensitive chromatin characterized by high conformational flexibility of nucleosomal DNA. Proc. Natl. Acad. Sci. USA 95:1540–1545; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Krude T. Chromatin assembly factor 1 (CAF-1) colocalizes with replication foci in HeLa cell nuclei. Exp. Cell Res. 220:304–311; 1995. [DOI] [PubMed] [Google Scholar]
- 111.Krude T. Chromatin assembly during DNA replication in somatic cells. Eur. J. Biochem. 263:1–5; 1999. [DOI] [PubMed] [Google Scholar]
- 112.Kuo M. H.; Allis C. D. In vivo cross-linking and immunoprecipitation for studying dynamic protein: DNA associations in chromatin environment. Methods 19:425–433; 1999. [DOI] [PubMed] [Google Scholar]
- 113.Kuo M. H.; Brownell J. E.; Sobel R. E.; Ranalli T. A.; Cook R. G.; Edmondson D. G.; Roth S. Y.; Allis C. D. Transcription-linked acetylation by Gcn5p of histones H3 and H4 at specific lysines. Nature 383:269–272; 1996. [DOI] [PubMed] [Google Scholar]
- 114.Laskey R. A.; Mills A. D.; Philpot A.; Leno G. H.; Dilworth S. M.; Dingwall C. The role of nucleo-plasmin in chromatin assembly and disassembly. Philos. Trans. R. Soc. Lond. B 339:263–269; 1993. [DOI] [PubMed] [Google Scholar]
- 115.Le S.; Davis C.; Konopka J. B.; Sternglanz R. Two new S-phase-specific genes from Saccharomyces cerevisiae. Yeast 13:1029–1042; 1997. [DOI] [PubMed] [Google Scholar]
- 116.Lee K. M.; Hayes J. J. Linker DNA and H1-dependent reorganization of histone–DNA interactions within the nucleosome. Biochemistry 37:8622–8628; 1998. [DOI] [PubMed] [Google Scholar]
- 117.Lin R.; Leone J. W.; Cook R. G.; Allis C. D. Antibodies specific to acetylated histones document the existence of deposition- and transcription-related histone acetylation in Tetrahymena. J. Cell Biol. 108: 1577–1588; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ling X. F.; Harkness T. A. A.; Schultz M. C.; Fish-eradams G.; Grunstein M. Yeast histone H3 and H4 amino termini are important for nucleosome assembly in vivo and in vitro: Redundant and position-independent functions in assembly but not in gene regulation. Genes Dev. 10:686–699; 1996. [DOI] [PubMed] [Google Scholar]
- 119.Lopez-Rodas G.; Perez-Ortin J. E.; Tordera V.; Salvador M. L.; Franco L. Partial purification and properties of two histone acetyltransferases from the yeast, Saccharomyces cerevisiae. Arch. Biochem. Biophys. 239:184–90; 1985. [DOI] [PubMed] [Google Scholar]
- 120.Lopez-Rodas G.; Tordera V.; Sanchez del Pino M. M.; Franco L. Yeast contains multiple forms of histone acetyltransferase. J. Biol. Chem. 264:19028–19033; 1989. [PubMed] [Google Scholar]
- 121.Lopezrodas G.; Georgieva E. I.; Sendra R.; Loidl P. Histone acetylation in Zea-Mays. 1. Activities of histone acetyltransferases and histone deacetylases. J. Biol. Chem. 266:18745–18750; 1991. [PubMed] [Google Scholar]
- 122.Lopezrodas G.; Tordera V.; Delpino M. M. S.; Franco L. Subcellular localization and nucleosome specificity of yeast histone acetyltransferases. Biochemistry 30:3728–3732; 1991. [DOI] [PubMed] [Google Scholar]
- 123.Louie A. J.; Candido E. P. M.; Dixon G. Enzymatic modifications and their possible roles in regulating the binding of basic proteins to DNA and in controlling chromosomal structure. Cold Spring Harbor Symp. Quant. Biol. 38:808–819; 1974. [DOI] [PubMed] [Google Scholar]
- 124.Louters L.; Chalkley R. Exchange of histones H1, H2A, and H2B in vivo. Biochemistry 24:3080–3085; 1985. [DOI] [PubMed] [Google Scholar]
- 125.Luger K.; Mader A. W.; Richmond R. K.; Sargent D. F.; Richmond T. J. Crystal structure of the nucleosome core particle at 2.8 angstrom resolution. Nature 389:251–260; 1997. [DOI] [PubMed] [Google Scholar]
- 126.Luger K.; Richmond T. J. DNA binding within the nucleosome core. Curr. Opin. Struct. Biol. 8:33–40; 1998. [DOI] [PubMed] [Google Scholar]
- 127.Luger K.; Richmond T. J. The histone tails of the nucleosome. Curr. Opin. Genet. Dev. 8:140–146; 1998. [DOI] [PubMed] [Google Scholar]
- 128.Luo R. X.; Postigo A. A.; Dean D. C. Rb interacts with histone deacetylase to repress transcription. Cell 92:463–473; 1998. [DOI] [PubMed] [Google Scholar]
- 129.Lusser A.; Eberharter A.; Loidl A.; Goralik-Schramel M.; Horngacher M.; Haas H.; Loidl P. Analysis of the histone acetyltransferase B complex of maize embryos. Nucleic Acids Res. 27:4427–4435; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ma X. J.; Wu J. S.; Altheim B. A.; Schultz M. C.; Grunstein M. Deposition-related sites K5/K12 in histone H4 are not required for nucleosome deposition in yeast. Proc. Natl. Acad. Sci. USA 95:6693–6698; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Magnaghijaulin L.; Groisman R.; Naguibneva I.; Robin P.; Lorain S.; Levillain J. P.; Troalen F.; Trouche D.; Harel-Bellan A. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature 391:601–605; 1998. [DOI] [PubMed] [Google Scholar]
- 132.Mann R. K.; Grunstein M. Histone H3 N-Terminal mutations allow hyperactivation of the yeast GAL1 gene in vivo. EMBO J. 11:3297–3306; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Marheineke K.; Krude T. Nucleosome assembly activity and intracellular localization of human CAF-1 changes during the cell division cycle. J. Biol. Chem. 273:15279–15286; 1998. [DOI] [PubMed] [Google Scholar]
- 134.Martinez-Balbas M. A.; Tsukiyama T.; Gdula D.; Wu C.Drosophila NURF-55, a WD repeat protein involved in histone metabolism. Proc. Natl. Acad. Sci. USA 95:132–137; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Martini E.; Roche D. M. J.; Marheineke K.; Verreault A.; Almouzni G. Recruitment of phosphorylated chromatin assembly factor 1 to chromatin after UV irradiation of human cells. J. Cell Biol. 143:563–575; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.McGhee J. D.; Nickol J. M.; Felsenfeld G.; Rau D. C. Histone hyperacetylation has little effect on the higher order folding of chromatin. Nucleic Acids Res. 11:4065–4075; 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Megee P. C.; Morgan B. A.; Mittman B. A.; Smith M. M. Genetic analysis of histone H4: Essential role of lysines subject to reversible acetylation. Science 247:841–845; 1990. [DOI] [PubMed] [Google Scholar]
- 138.Mingarro I.; Sendra R.; Salvador M. L.; Franco L. Site specificity of pea histone acetyltransferase-B in vitro. J. Biol. Chem. 268:13248–13252; 1993. [PubMed] [Google Scholar]
- 139.Mizzen C. A.; Allis C. D. Linking histone acetylation to transcriptional regulation. Cell. Mol. Life Sci. 54:6–20; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Moggs J. G.; Almouzni G. Chromatin rearrangements during nucleotide excision repair. Biochimie 81:45–52; 1999. [DOI] [PubMed] [Google Scholar]
- 141.Moggs J. G.; Grandi P.; Quivy J. P.; Jonsson Z. O.; Hubscher U.; Becker P. B.; Almouzni G. A CAF-1-PCNA-mediated chromatin assembly pathway triggered by sensing DNA damage. Mol. Cell. Biol. 20: 1206–1218; 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Monson E. K.; Debruin D.; Zakian V. A. The yeast Cac1 protein is required for the stable inheritance of transcriptionally repressed chromatin at telomeres. Proc. Natl. Acad. Sci. USA 94:13081–13086; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Moore S. C.; Ausio J. Major role of the histones H3-H4 in the folding of the chromatin fiber. Biochem. Biophys. Res. Commun. 230:136–139; 1997. [DOI] [PubMed] [Google Scholar]
- 144.Morgan B. A.; Mittman B. A.; Smith M. M. The highly conserved N-terminal domains of histone-H3 and histone-H4 are required for normal cell cycle progression. Mol. Cell. Biol. 11:4111–4120; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Murzina N.; Verreault A.; Laue E.; Stillman B. Heterochromatin dynamics in mouse cells: Interaction between chromatin assembly factor 1 and HP1 proteins. Mol. Cell 4:529–540; 1999. [DOI] [PubMed] [Google Scholar]
- 146.Nan X. S.; Ng H. H.; Johnson C. A.; Laherty C. D.; Turner B. M.; Eisenman R. N.; Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393:386–389; 1998. [DOI] [PubMed] [Google Scholar]
- 147.Nelson T.; Wiegand R.; Brutlag D. Ribonucleic acid and other polyanions facilitate chromatin assembly in vitro. Biochemistry 20:2594–2601; 1981. [DOI] [PubMed] [Google Scholar]
- 148.Norton V. G.; Imai B. S.; Yau P.; Bradbury E. M. Histone acetylation reduces nucleosomal core particle linking number change. Cell 57:449–457; 1989. [DOI] [PubMed] [Google Scholar]
- 149.Norton V. G.; Marvin K. W.; Yau P.; Bradbury E. M. Nucleosome linking number change controlled by acetylation of histones H3 and H4. J. Biol. Chem. 265:19848–19852; 1990. [PubMed] [Google Scholar]
- 150.Olins D. E.; Olins A. L.; Herrmann A.; Lin R. L.; Allis C. D.; Robert-Nicoud M. Localization of acetylated histone H4 in the macronucleus of euplotes. Chromosoma 100:377–385; 1991. [Google Scholar]
- 151.Park E.-C.; Szostak J. W. Point mutations in the yeast histone h4 gene prevent silencing of the silent mating type locus HML. Mol. Cell. Biol. 10:4932–4934; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Parthun M. R.; Widom J.; Gottschling D. E. The major cytoplasmic histone acetyltransferase in yeast: Links to chromatin replication and histone metabolism. Cell 87:85–94; 1996. [DOI] [PubMed] [Google Scholar]
- 153.Perry C. A.; Allis C. D.; Annunziato A. T. Parental nucleosomes segregated to newly replicated chromatin are underacetylated relative to those assembled de novo. Biochemistry 32:13615–13623; 1993. [DOI] [PubMed] [Google Scholar]
- 154.Perry C. A.; Annunziato A. T. Influence of histone acetylation on the solubility, H1 content and DNase I sensitivity of newly assembled chromatin. Nucleic Acids Res. 17:4275–4291; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Perry C. A.; Annunziato A. T. Histone acetylation reduces H1-mediated nucleosome interactions during chromatin assembly. Exp. Cell Res. 196:337–345; 1991. [DOI] [PubMed] [Google Scholar]
- 156.Perry C. A.; Dadd C. A.; Allis C. D.; Annunziato A. T. Analysis of nucleosome assembly and histone exchange using antibodies specific for acetylated H4. Biochemistry 32:13605–13614; 1993. [DOI] [PubMed] [Google Scholar]
- 157.Pollard K. J.; Peterson C. L. Chromatin remodeling: A marriage between two families? Bioessays 20:771–780; 1998. [DOI] [PubMed] [Google Scholar]
- 158.Pollard K. J.; Samuels M. L.; Crowley K. A.; Hansen J. C.; Peterson C. L. Functional interaction between GCN5 and polyamines: A new role for core histone acetylation. EMBO J. 18:5622–5633; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Pospelov V.; Russev G.; Vassilev L.; Tsanev R. Nucleosome segregation in chromatin replicated in the presence of cycloheximide. J. Mol. Biol. 156:79–91; 1982. [DOI] [PubMed] [Google Scholar]
- 160.Ptashne M.; Gann A. Transcriptional activation by recruitment. Nature 386:569–577; 1997. [DOI] [PubMed] [Google Scholar]
- 161.Qian Y. W.; Lee E. Y. Dual retinoblastoma-binding proteins with properties related to a negative regulator of ras in yeast. J. Biol. Chem. 270:25507–13; 1995. [DOI] [PubMed] [Google Scholar]
- 162.Qian Y. W.; Wang Y. C.; Hollingsworth R. E. Jr.; Jones D.; Ling N.; Lee E. Y. A retinoblastoma-binding protein related to a negative regulator of Ras in yeast. Nature 364:648–52; 1993. [DOI] [PubMed] [Google Scholar]
- 163.Quintini G.; Treuner K.; Gruss C.; Knippers R. Role of amino-terminal histone domains in chromatin replication. Mol. Cell. Biol. 16:2888–2897; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Richman R.; Chicoine L. G.; Collini C. M. P.; Cook R. G.; Allis C. D. Micronuclei and the cytoplasm of growing Tetrahymena contain a histone acetylase activity which is highly specific for free histone H4. J. Cell Biol. 106:1017–1026; 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ridsdale J. A.; Hendzel M. J.; Delcuve G. P.; Davie J. R. Histone acetylation reduces the capacity of H1 histones to condense transcriptionally active/competent chromatin. J. Biol. Chem. 265:5150–5156; 1990. [PubMed] [Google Scholar]
- 166.Ridsdale J. A.; Rattner J. B.; Davie J. R. Erythroid-specific gene chromatin has an altered association with linker histones. Nucleic Acids Res. 16:5915–5926; 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Roth S. Y.; Allis C. D. Histone acetylation and chromatin assembly: A single escort, multiple dances? Cell 87:5–8; 1996. [DOI] [PubMed] [Google Scholar]
- 168.Ruiz-Carrillo A.; Wangh L. J.; Allfrey V. G. Processing of newly synthesized histone molecules. Science 190:117–128; 1975. [DOI] [PubMed] [Google Scholar]
- 169.Ruiz-Garcia A. B.; Sendra R.; Galiana M.; Pamblanco M.; Perezortin J. E.; Tordera V. HAT1 and HAT2 proteins are components of a yeast nuclear histone acetyltransferase enzyme specific for free histone H4. J. Biol. Chem. 273:12599–12605; 1998. [DOI] [PubMed] [Google Scholar]
- 170.Salvador M. L.; Sendra R.; López-Rodas G.; Tordera V.; Franco L. On the ubiquitous presence of histone acetyltransferase B in eukaryotes. FEBS Lett. 191:55–58; 1985. [Google Scholar]
- 171.Schurtenberger P.; Egelhaaf S. U.; Hindges R.; Maga G.; Jonsson Z. O.; May R. P.; Glatter O.; Hubscher U. The solution structure of functionally active human proliferating cell nuclear antigen determined by small-angle neutron scattering. J. Mol. Biol. 275:123–132; 1998. [DOI] [PubMed] [Google Scholar]
- 172.Schuster T.; Han M.; Grunstein M. Yeast histone H2A and H2B amino termini have interchangeable functions. Cell 45:445–451; 1986. [DOI] [PubMed] [Google Scholar]
- 173.Schwarz P. M.; Felthauser A.; Fletcher T. M.; Hansen J. C. Reversible oligonucleosome self-association: Dependence on divalent cations and core histone tail domains. Biochemistry 35:4009–4015; 1996. [DOI] [PubMed] [Google Scholar]
- 174.Schwarz P. M.; Hansen J. C. Formation and stability of higher order chromatin structures—Contributions of the histone octamer. J. Biol. Chem. 269:16284–16289; 1994. [PubMed] [Google Scholar]
- 175.Shibahara K.; Stillman B. Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell 96:575–585; 1999. [DOI] [PubMed] [Google Scholar]
- 176.Shimamura A.; Worcel A. The assembly of regularly spaced nucleosomes in the Xenopus oocyte S-150 extract is accompanied by deacetylation of histone H4. J. Biol. Chem. 264:14524–14530; 1989. [PubMed] [Google Scholar]
- 177.Simpson R. T.; Thoma F.; Brubaker J. M. Chromatin reconstituted from tandemly repeated cloned DNA fragments and core histones: A model system for study of higher order structure. Cell 42:799–808; 1985. [DOI] [PubMed] [Google Scholar]
- 178.Singer M. S.; Kahana A.; Wolf A. J.; Meisinger L. L.; Peterson S. E.; Goggin C.; Mahowald M.; Gottschling D. E. Identification of high-copy disrupters of telomeric silencing in Saccharomyces cerevisiae. Genetics 150:613–632; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Smith M. M.; Santisteban M. S. Genetic dissection of histone function. Methods 15:269–281; 1998. [DOI] [PubMed] [Google Scholar]
- 180.Smith S.; Stillman B. purification and characterization of CAF-I, a human cell factor required for chromatin assembly during DNA replication in vitro. Cell 58:15–25; 1989. [DOI] [PubMed] [Google Scholar]
- 181.Smith S.; Stillman B. Immunological characterization of chromatin assembly factor-I, a human cell factor required for chromatin assembly during DNA replication in vitro. J. Biol. Chem. 266:12041–12047; 1991. [PubMed] [Google Scholar]
- 182.Smith S.; Stillman B. Stepwise assembly of chromatin during DNA replication in vitro. EMBO J. 10: 971–980; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sobel R. E.; Cook R. G.; Allis C. D. Non-random acetylation of histone H4 by a cytoplasmic histone acetyltransferase as determined by novel methodology. J. Biol. Chem. 269:18576–18582; 1994. [PubMed] [Google Scholar]
- 184.Sobel R. E.; Cook R. G.; Perry C. A.; Annunziato A. T.; Allis C. D. Conservation of deposition-related acetylation sites in newly synthesized histones H3 and H4. Proc. Natl. Acad. Sci. USA 92:1237–1241; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sogo J. M.; Stahl H.; Koller T.; Knippers R. Structure of replicating simian virus chromosomes. J. Mol. Biol. 189–204; 1986. [DOI] [PubMed] [Google Scholar]
- 186.Spencer V. A.; Davie J. R. Role of covalent modifications of histones in regulating gene expression. Gene 240:1–12; 1999. [DOI] [PubMed] [Google Scholar]
- 187.Stillman B. Chromatin assembly during DNA replication in vitro. Cell 45:555–565; 1986. [DOI] [PubMed] [Google Scholar]
- 188.Stone E. M.; Pillus L. Silent chromatin in yeast: An orchestrated medley featuring sir3p. Bioessays 20: 30–40; 1998. [DOI] [PubMed] [Google Scholar]
- 189.Strahl B. D.; Allis C. D. The language of covalent histone modifications. Nature 403:41–45; 2000. [DOI] [PubMed] [Google Scholar]
- 190.Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 12:599–606; 1998. [DOI] [PubMed] [Google Scholar]
- 191.Sures I.; Gallwitz D. Histone specific acetyltransferases from calf thymus. Isolation, properties, and substrate specificity of three different enzymes. Biochemistry 19:943–951; 1980. [DOI] [PubMed] [Google Scholar]
- 192.Taddei A.; Roche D.; Sibarita J. B.; Turner B. M.; Almouzni G. Duplication and maintenance of heterochromatin domains. J. Cell Biol. 147:1153–1166; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Taunton J.; Hassig C. A.; Schreiber S. L. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 272:408–411; 1996. [DOI] [PubMed] [Google Scholar]
- 194.Thoma F.; Koller T.; Klug A. Involvement of histone H1 in the organization of the nucleosome and of the salt-dependent superstructures of chromatin. J. Cell Biol. 83:403–427; 1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Thompson J. S.; Ling X. F.; Grunstein M. Histone H3 amino terminus is required for telomeric and silent mating locus repression in yeast. Nature 369: 245–247; 1994. [DOI] [PubMed] [Google Scholar]
- 196.Travers A. Chromatin modification: How to put a HAT on the histones. Curr. Biol. 9:R23–R25; 1999. [DOI] [PubMed] [Google Scholar]
- 197.Tse C.; Hansen J. C. Hybrid trypsinized nucleosomal arrays: Identification of multiple functional roles of the H2A/H2B and H3/H4 N-termini in chromatin fiber compaction. Biochemistry 36:11381–11388; 1997. [DOI] [PubMed] [Google Scholar]
- 198.Tse C.; Sera T.; Wolffe A. P.; Hansen J. C. Disruption of higher-order folding by core histone acetylation dramatically enhances transcription of nucleosomal arrays by RNA polymerase III. Mol. Cell. Biol. 18:4629–4638; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Turner B. M. Histone acetylation as an epigenetic determinant of long-term transcriptional competence. Cell. Mol. Life Sci. 54:21–31; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Tyler J. K.; Adams C. R.; Chen S. R.; Kobayashi R.; Kamakaka R. T.; Kadonaga J. T. The RCAF complex mediates chromatin assembly during DNA replication and repair. Nature 402:555–560; 1999. [DOI] [PubMed] [Google Scholar]
- 201.Tyler J. K.; Bulger M.; Kamakaka R. T.; Kobayashi R.; Kadonaga J. T. The p55 subunit of Drosophila chromatin assembly factor 1 is homologous to a histone deacetylase-associated protein. Mol. Cell. Biol. 16:6149–6159; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Uetz P.; Giot L.; Cagney G.; Mansfield T. A.; Judson R. S.; Knight J. R.; Lockshon D.; Narayan V.; Srinivasan M.; Pochart P.; Qureshi-Emili A.; Li Y.; Godwin B.; Conover D.; Kalbfleisch T.; Vijayadamodar G.; Yang M.; Johnston M.; Fields S.; Rothberg J. M. A comprehensive analysis of protein–protein interactions in Saccharomyces cerevisiae. Nature 403:623–627; 2000. [DOI] [PubMed] [Google Scholar]
- 203.Usachenko S. I.; Bavykin S. G.; Gavin I. M.; Bradbury E. M. Rearrangement of the histone H2A C-terminal domain in the nucleosome. Proc. Natl. Acad. Sci. USA 91:6845–6849; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.van Holde K. E. Chromatin. New York: Springer-Verlag; 1988. [Google Scholar]
- 205.Venditti S.; VegaPalas M. A.; DiMauro E. Heterochromatin organization of a natural yeast telomere— recruitment of Sir3p through interaction with histone H4 M terminus is required for the establishment of repressive structures. J. Biol. Chem. 274:1928–1933; 1999. [DOI] [PubMed] [Google Scholar]
- 206.Verreault A.; Kaufman P. D.; Kobayashi R.; Stillman B. Nucleosome assembly by a complex of CAF-1 and acetylated histones H3/H4. Cell 87:95–104; 1996. [DOI] [PubMed] [Google Scholar]
- 207.Verreault A.; Kaufman P. D.; Kobayashi R.; Stillman B. Nucleosomal DNA regulates the core-histone-binding subunit of the human Hat1 acetyltransferase. Curr. Biol. 8:96–108; 1997. [DOI] [PubMed] [Google Scholar]
- 208.Waga S.; Stillman B. The DNA replication fork in eukaryotic cells. Annu. Rev. Biochem. 67:721–751; 1998. [DOI] [PubMed] [Google Scholar]
- 209.Wallis J. W.; Rykowski M.; Grunstein M. Yeast histone H2B containing large amino terminus deletions can function in vivo. Cell 35:711–719; 1983. [DOI] [PubMed] [Google Scholar]
- 210.Walter P. P.; Owenhughes T. A.; Cote J.; Workman J. L. Stimulation of transcription factor binding and histone displacement by nucleosome assembly protein 1 and nucleoplasmin requires disruption of the histone octamer. Mol. Cell. Biol. 15:6178–6187; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Weigand R. C.; Brutlag D. L. Histone acetylase from Drosophila melanogaster specific for H4. J. Biol. Chem. 256:4578–4583; 1981. [PubMed] [Google Scholar]
- 212.Weintraub H.; Groudine M. Chromosomal subunits in active genes have an altered conformation. Science 193:848–856; 1976. [DOI] [PubMed] [Google Scholar]
- 213.Weintraub H.; Van Lente F. Dissection of chromosome structure with trypsin and nucleases. Proc. Natl. Acad. Sci. USA 71:4249–4253; 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.White D. A.; Belyaev N. D.; Turner B. M. Preparation of site-specific antibodies to acetylated histones. Methods 19:417–424; 1999. [DOI] [PubMed] [Google Scholar]
- 215.Whitlock J. P. Jr.; Simpson R. T. Localization of the sites along nucleosome DNA which interact with NH2-terminal histone regions. J. Biol. Chem. 252: 6516–6520; 1977. [PubMed] [Google Scholar]
- 216.Whitlock J. P. Jr.; Stein A. Folding of DNA by histones which lack their NH2-terminal regions. J. Biol. Chem. 253:3857–3861; 1978. [PubMed] [Google Scholar]
- 217.Wolffe A. P. Inheritance of chromatin states. Dev. Genet. 15:463–470; 1994. [DOI] [PubMed] [Google Scholar]
- 218.Wolffe A. P. Transcription—in tune with the histones. Cell 77:13–16; 1994. [DOI] [PubMed] [Google Scholar]
- 219.Wolffe A. P. Chromatin: Structure and function, 3rd ed. San Diego: Academic Press, Inc.; 1999. [Google Scholar]
- 220.Wolffe A. P.; Hayes J. J. Chromatin disruption and modification. Nucleic Acids Res. 27:711–720; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Wolffe A. P.; Matzke M. A. Epigenetics: Regulation through repression. Science 286:481–486; 1999. [DOI] [PubMed] [Google Scholar]
- 222.Woodland H. R. The modification of stored histones H3 and H4 during the oogenesis and early development of Xenopus laevis. Dev. Biol. 68:360–370; 1979. [DOI] [PubMed] [Google Scholar]
- 223.Woodland H. R.; Adamson E. D. The synthesis and storage of histones during the oogenesis of Xenopus laevis. Dev. Biol. 57:118–135; 1977. [DOI] [PubMed] [Google Scholar]
- 224.Worcel A.; Han S.; Wong M. L. Assembly of newly replicated chromatin. Cell 15:969–977; 1978. [DOI] [PubMed] [Google Scholar]
- 225.Zhang W. Z.; Bone J. R.; Edmondson D. G.; Turner B. M.; Roth S. Y. Essential and redundant functions of histone acetylation revealed by mutation of target lysines and loss of the Gcn5p acetyltransferase. EMBO J. 17:3155–3167; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Zhang Y.; Iratni R.; Erdjument-Bromage H.; Tempst P.; Reinberg D. Histone deacetylases and SAP18, a novel polypeptide, are components of a human Sin3 complex. Cell 89:357–364; 1997. [DOI] [PubMed] [Google Scholar]